
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA ruthenium–oligonucleotide bioconjugated photosensitizing aptamer for cancer cell specific photodynamic therapy†
Luke K.
McKenzie‡
 ab,
Marie
Flamme‡
abc,
Patrick S.
Felder
b,
Johannes
Karges
b,
Frederic
Bonhomme
d,
Albert
Gandioso
b,
Christian
Malosse
e,
Gilles
Gasser
*b and
Marcel
Hollenstein
*a
ab,
Marie
Flamme‡
abc,
Patrick S.
Felder
b,
Johannes
Karges
b,
Frederic
Bonhomme
d,
Albert
Gandioso
b,
Christian
Malosse
e,
Gilles
Gasser
*b and
Marcel
Hollenstein
*a
aInstitut Pasteur, Department of Structural Biology and Chemistry, Laboratory for Bioorganic Chemistry of Nucleic Acids, CNRS UMR3523, 28 rue du Docteur Roux, 75724 Paris Cedex 15, France. E-mail: marcel.hollenstein@pasteur.fr; Web: https://research.pasteur.fr/en/team/bioorganic-chemistry-of-nucleic-acids/ Tel: +33 1 44 38 94 66
bChimie ParisTech, PSL University, CNRS, Institute of Chemistry for Life and Health Sciences, Laboratory for Inorganic Chemical Biology, 75005 Paris, France. E-mail: gilles.gasser@chimieparistech.psl.eu; Web: http://www.gassergroup.com Tel: +33 1 85 78 41 51
cUniversité de Paris, 12 rue de l'École de Médecine, 75006 Paris, France
dInstitut Pasteur, Department of Structural Biology and Chemistry, Unité de Chimie Biologique Epigénétique, UMR CNRS 3523, 28 rue du Docteur Roux, 75724 Paris Cedex 15, France
eInstitut Pasteur, Mass Spectrometry for Biology Unit, 28 rue du Docteur Roux, 75724 Paris Cedex 15, France
First published on 2nd November 2021
Abstract
Ruthenium complexes have emerged as a promising class of compounds for use as photosensitizers (PSs) in photodynamic therapy (PDT) due to their attractive photophysical properties and relative ease of chemical alteration. While promising, they generally are not inherently targeting to disease sites and may therefore be prone to side effects and require higher doses. Aptamers are short oligonucleotides that bind specific targets with high affinity. One such aptamer is AS1411, a nucleolin targeting, G-quadruplex forming, DNA aptamer. Here we present the first example of direct conjugation of a Ru(II) polypyridyl complex-based PS to an aptamer and an assessment of its in vitro cancer cell specific photosensitization including discussion of the challenges faced.
Introduction
The exploration of ruthenium (Ru) complexes for use as photosensitizers (PSs) for photodynamic therapy (PDT) has exploded in recent years1–13 and, with the recent entrance into clinical trials of the Ru(II)-based PS TLD-1433, this looks only likely to intensify.7 PDT combines a PS, activating light and cellular oxygen to produce 1O2/reactive oxygen species (ROS) resulting in localised cell death.3 While PDT of cancer promises tumour specificity through localised irradiation at the tumour site, the PS has generally only a low cancer cell specificity. A number of strategies have been investigated to enhance the cancer specificity and/or organelle specific targeting of Ru-based PDT PSs such as bioconjugation to proteins11,14–16 and nanobodies17 as well as through nanoparticle incorporation.18–22 This would allow for the use of lower doses of PS and the reduction of side effects.One targeting modality, little explored in combination with ruthenium complexes, is the use of aptamers.23 Aptamers are short oligonucleotides with high binding affinity to a specific target, such as proteins or entire cells, and are usually selected through the Systematic Evolution of Ligands by Exponential enrichment (SELEX) process.24–26 Aptamers are considered to be nucleic acid analogues of antibodies but offer a number of advantages such as synthetic reproducibility and comparatively low-cost, high scale, synthesis. One such aptamer is AS1411, unusually discovered accidentally rather than through the traditional SELEX process.27AS1411 is an aptamer for nucleolin, a protein generally expressed in the nucleoli of cells, though overexpressed in a wide number of cancer cell lines with atypical cell surface expression.28AS1411 has been shown to be internalised in many cancer cells lines such as breast (MCF7) and prostate (DU145), among many others, while normal cell lines typically do not incorporate the aptamer.29 The G-rich oligonucleotide forms a G-quadruplex in the presence of certain metal ions such as K+ and this formation is believed to be required for uptake. AS1411 has been explored as a potential cancer specific drug delivery system with drug loading via intercalation30 as well as through covalent linking.31,32 Kim et al. recently conjugated the known PS Ce6 to the 3′-end of AS1411via a PEG chain. Photosensitization in the nanomolar range was observed for the resulting aptamer in three cancer cell lines (MCF-7, HCT 116, and SKOV-3) while no photosensitization was observed in a normal cell line (L-929).33 A number of papers have been released exploring the use of AS1411 for targeted PDT including both direct conjugation33,34 and intercalation.30 Previously, the ruthenium complex [Ru(bpy)2(tip)]2+ was loaded into nanoparticles adorned with AS1411 for cell specific release with the system improving survival in a murine glioma model.35 The direct use of Ruthenium complexes with AS1411 in this context poses some previously unexplored questions. Certain ruthenium complexes are known to interact with G-quadruplexes, for example a series of dinuclear Ruthenium complexes were shown to bind to and stabilise telomeric G-quadruplexes.36 While many Ruthenium complexes have been demonstrated to specifically target, and usually stabilize, G-quadruplex DNA over standard DNA duplexes.36–44 As such we faced the question of whether to approach the system via intercalation, as demonstrated previously with the non-ruthenium PS TMPyP30 or by conjugation, eventually settling with the latter due to questions of complete intercalation would be achieved and whether intercalation may result in quenching and reduction of 1O2 yields.
We recently published our findings of a series of rationally designed Ru(II) polypyridine complexes based on the [Ru(phen)2(bpy)]2+ scaffold of which one complex (Ru) was demonstrated to be capable of remarkable photosensitization in the nanomolar range at 595 nm (Fig. 1a).1 Capitalizing on the excellent results obtained by Kim et al., we decided to investigate whether the coupling of Ru to this aptamer would indeed allow for selective targeting of cancer cells. Through addition of azide functionality the resulting complex (RuN3) may be suitable for use in a diverse range of applications beyond those explored in this study such as dye sensitized solar cells (DSSCs) and catalysis.45 In this article, we describe the design, synthesis, biophysical characterization, and in vitro evaluation of a series of ruthenium–AS1411 conjugates (Ru–AS1411) utilizing Ru in combination with the cancer-targeting aptamer AS1411.1
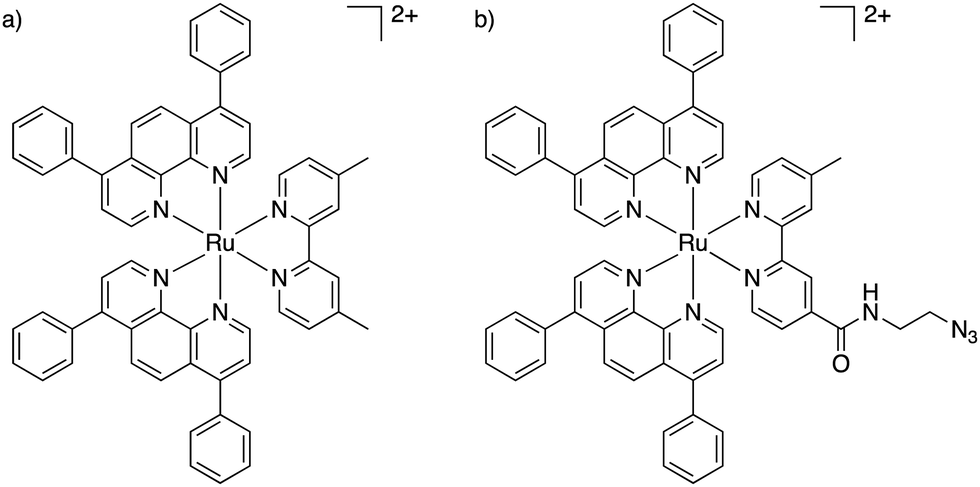 | ||
| Fig. 1 Chemical structures of (a) Ru;1 (b) RuN3. | ||
Results and discussion
Synthesis and characterisation
The previously reported Ru (Fig. 1a) was chosen for conjugation to AS1411 due to its remarkable photophysical and photosensitizing properties at wavelengths up to 595 nm.1 Through the addition of an azide functionality on the bipyridine ligand, the resulting Ru(II) complex (RuN3) (Fig. 1b) is primed to form the ruthenium-containing aptamer conjugates (Ru–AS1411s, Fig. S1, ESI†) through the standard Cu(I)-catalyzed azide–alkyne cycloaddition (click) reaction. The AS1411 aptamer consists of a 26 mer sequence (template T1, Table 1). With the rationale of reduced G-quadruplex disruption, three spacer Ts were appended at the 3′-end followed by 3′ alkyne modification resulting in the 29 mer sequence T2 (Table 1). We also designed sequences T3 and T4 where the alkyne moiety is placed at the 5′ rather than at the 3′ position of AS1411 aptamer to probe the effect on PDT efficiency and G-quadruplex formation capacity and with shorter (T3) and longer (T4) connecting linker moieties.RuN3 synthesis
The synthesis of RuN3 was achieved by adapting previously published procedures.46–51 The synthesis of [RuBphen2Cl2] was found to be more efficient via the intermediate [Ru(dmso)4Cl2] (Fig. 2) than by direct synthesis from commercially available RuCl3, a finding that has been hinted at in the literature.46 LiCl is used as an additive during the synthesis of [RuBphen2Cl2] to prevent the formation of the tri-substituted [Ru(Bphen)3]Cl2 as an undesired side product (Bphen = 4,7-diphenyl-1,10-phenanthroline, also known as bathophenanthroline). The synthesis of bpyN3 was performed via EDCI-based amide coupling. 4′-Methyl-[2,2′-bipyridine]-4-carboxylic acid is commercially available while 2-azidoethan-1-amine (N3EtNH2) was produced by nucleophilic substitution of 2-chloroethylamine with sodium azide.52 The ligand bpyN3 was directly coordinated to the Ru(II) precursor [RuBphen2Cl2] to obtain RuN3.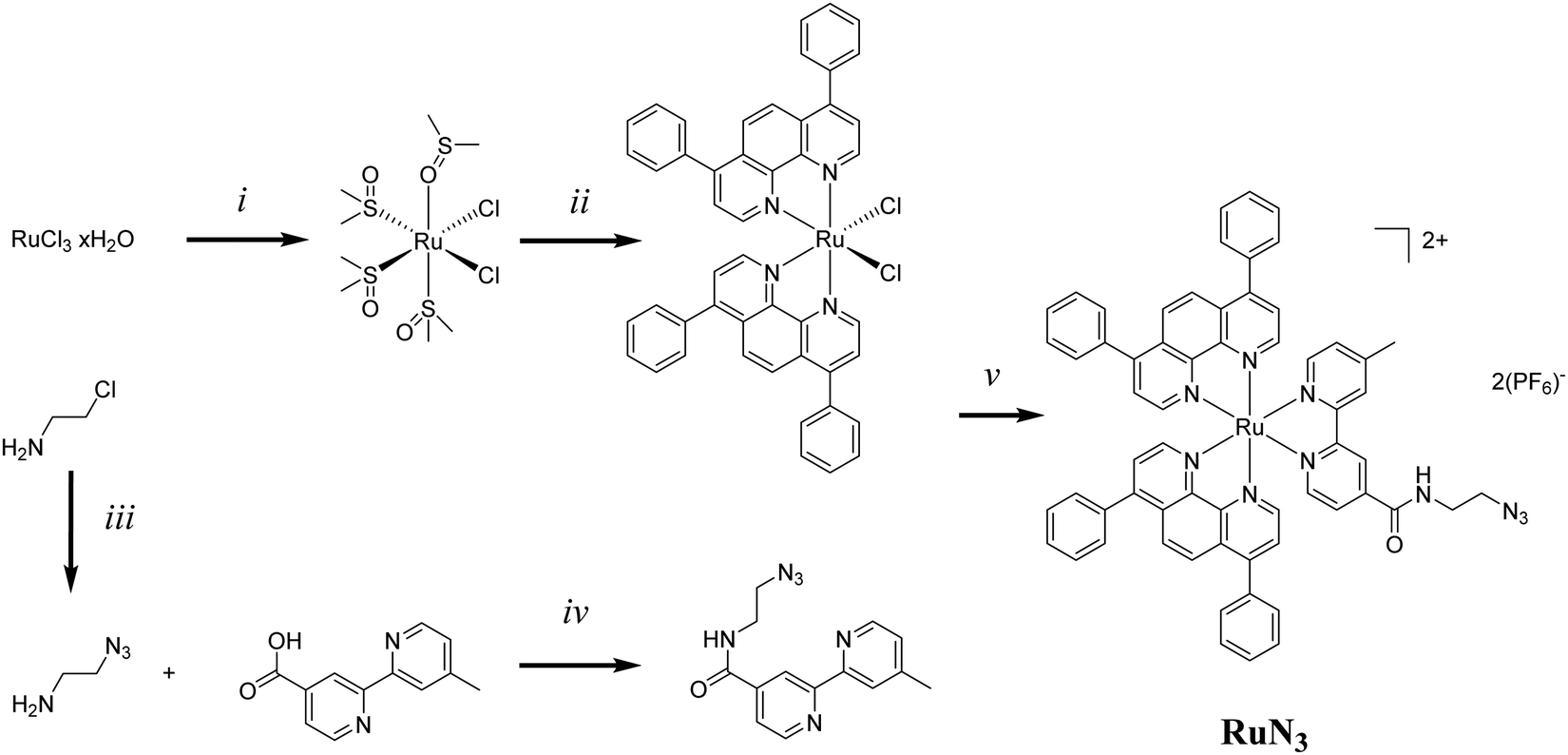 | ||
| Fig. 2 Synthetic procedure for RuN3; (i) EtOH DMSO, reflux, 4 h; DMSO, 125 °C, 1 h, 85%; (ii) Bphen, LiCl, DMF, reflux, 5 h, 72%; (iii) NaN3, H2O, 80 °C, 12 h, 82%; (iv) EDCI, NHS, DIPEA, CH2Cl2, r.t., 12 h, 56%; (v) MeOH, H2O, reflux, 15 h, 64%. | ||
RuN3 was fully characterized by NMR (Fig. S3–S8, ESI†), HR-MS ESI (Fig. S9, ESI†) and IR spectroscopy (Fig. S10, ESI†). Present are the characteristic patterns of the disubstituted phen and bpy ligands (8.0–9.0 ppm), as well as the rotating phen groups (7.5 ppm, broad), the four protons of the aliphatic EtN3 end (3.5–3.6 ppm) and the three methyl protons (2.6 ppm), the structure was successfully identified by 1H NMR (Fig. S3, ESI†). The molecular formula of RuN3 was confirmed by HR-MS (C62H46N10ORu, expected: 524.1453, found: 524.1447) in positive mode as the [M]2+ ion (Fig. S9, ESI†). The characteristic azide peak in the IR spectrum was identified at 2100 cm−1, interestingly, it was observed that the IR spectrum is strongly dominated by the large amount of delocalized C–C signal (Fig. S10, ESI†).
RuN3 Photophysical and biological analyses
The absorption spectrum of RuN3 (Fig. S11a, ESI†) exhibits the two Soret bands typical for this type of Bphen-containing Ru(II) complex.4,49,53–55 The local maximum at 470 nm, with bathochromic off-tailing up to 600 nm,49,54 is associated with the PDT-active metal-to-ligand charge transfer (MLCT) band. RuN3 has a large Stokes shift (0.74 eV) with an emission maximum at 640 nm (in acetonitrile, Fig. S11b, ESI†) and a luminescence quantum yield of Φ = 2.6% in comparison to [Ru(bpy)3]Cl2 (Φ = 5.9%).56 The excited state lifetime was determined to be 220 ns in an air-saturated and 1076 ns in a degassed environment, which is comparable with [Ru(bpy)3]Cl2.2,49,54 The change of the excited state lifetime is indicative of an interaction of the metal complex with molecular oxygen. The singlet oxygen yield (ΦΔ) of RuN3 was measured by both direct measurement of singlet oxygen phosphorescence and by indirect measurement using 1O2 scavengers as previously described.57RuN3 was found with a value of 68% using the direct method (λexc = 450 nm, acetonitrile), similarly to Ru under the same conditions (61%1). The ΦΔ upon excitation at 595 nm was still impressive (51% in acetonitrile, Table 2) indicating suitability for use as a PS over a range of activation wavelengths.| Spectroscopic properties | Singlet oxygen yield (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| UV/vis absorption λ, nm (ε, M−1 cm−1 × 103) | λ em (nm) | Φ em (%) | τ air saturated (μs) | τ degassed (μs) | 450 nm direct | 450 nm indirect | 540 nm indirect | 595 nm indirect | |
| RuN3 | 278a (176); 433a (40); 463a (45) | 640a | 2.6a | 0.220a | 1.076a | 68a | 71a, 7b | 64a, 5b | 51a, 3b |
Next, RuN3 was tested in MCF-7 (breast cancer) and RPE-1 (normal retina) cell lines to determine its light and dark toxicities with irradiation at 480 nm (10 min, 3.21 J cm−2) and 595 nm (2 h, 22.47 J cm−2) (Table 3). The results indicate that addition of the linker does not hinder the photosensitizing action of RuN3 while addition of the azide linker appears to generally reduce the dark toxicity of the molecule compared to Ru.1
| RuN3 | ||||||
|---|---|---|---|---|---|---|
| 480 nm | 595 nm | |||||
| Light | Dark | PI | Light | Dark | PI | |
| MCF-7 | 0.574 (±0.060) | >100 | >174 | 2.081 (±0.050) | >100 | >48.05 |
| RPE-1 | 0.015 (±0.069) | 58.58 (±0.086) | 3905 | 0.946 (±0.150) | 69.31 (±0.166) | 73.3 |
Ru–AS1411 conjugates
Click reactions were chosen for conjugation due to perceived ease and accessibility.58–60 We initially followed the reaction conditions set out in a paper describing the ideal click conditions for use with oligonucleotides.61 The initial click reactions were between RuN3 and T2 aiming to yield the system AS1411-3′-TTT-Ru. To avoid intercalation of RuN3 during the reaction the phosphate-buffered saline (PBS) in the published protocol was replaced with tris-buffered saline to avoid K+ and hence G-quadruplex formation. Similarly, the ‘Monarch® PCR & DNA Cleanup Kit’ was chosen for desalting the sample prior to HPLC purification as the binding buffer does not contain potassium. While PAGE of the click product confirmed conjugation (Fig. S12, ESI†), no G-quadruplex formation was observed by CD spectroscopy (Fig. S13a, ESI†). G-quadruplexes are known to produce distinctive peaks in their CD spectra when formed, as seen for AS1411 (Fig. S13b, ESI†), and CD spectroscopy is used as a means to validate their presence.62–64 In the case of AS1411, the distinctive CD signature includes a positive peak at around 260![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) nm and a negative peak at around 240nm63,65 which were not observed when Ru was attached at the 3′ end under these experimental conditions regardless on the concentration of K+. Nevertheless, an in vitro PDT assay was performed using MCF-7 (breast cancer), HT-29 (colorectal adenocarcinoma) and RPE-1 (retinal pigment epithelial) cell lines. MCF-7 cells are cell surface nucleolin expressing cells and are widely used in in vitro tests of AS1411. An immunofluorescence assay confirmed cell surface nucleolin expression in HT-29 while no cell surface nucleolin expression was detected in RPE-1 cells (Fig. S14, ESI†). No PDT effect was observed following light treatment at 480 nm in any cell line up to 2 μM (data not included).
nm and a negative peak at around 240nm63,65 which were not observed when Ru was attached at the 3′ end under these experimental conditions regardless on the concentration of K+. Nevertheless, an in vitro PDT assay was performed using MCF-7 (breast cancer), HT-29 (colorectal adenocarcinoma) and RPE-1 (retinal pigment epithelial) cell lines. MCF-7 cells are cell surface nucleolin expressing cells and are widely used in in vitro tests of AS1411. An immunofluorescence assay confirmed cell surface nucleolin expression in HT-29 while no cell surface nucleolin expression was detected in RPE-1 cells (Fig. S14, ESI†). No PDT effect was observed following light treatment at 480 nm in any cell line up to 2 μM (data not included).
We hypothesised that this inability to form G-quadruplexes was either due to the position of the conjugated Ru or due to guanine oxidation issues arising from the click reaction.66,67 In order to investigate the Ru hypothesis, we decided to conjugate the Ru at the 5′ end with both a two T or five T spacers T3 and T4.
For the click reaction conditions in the outlined protocol,61 10 molar equivalents of CuSO4 and 50 molar equivalents of sodium ascorbate are used in thoroughly degassed solution. To investigate the effect of these click conditions on G-quadruplex formation, T2 alone (without RuN3) was incubated under the same conditions. Post purification no G-quadruplex melting curve was detectable, indicating potential oxidation of guanine and loss of G-quadruplex formation (Fig. S15a, ESI†). T2 was subsequently incubated with one molar equivalent of CuSO4 and two molar equivalents of TBTA for 1, 2 and 18 hours with no loss of G-quadruplex melting curve (Fig. S15a, ESI†). Considering the complication encountered using click reactions in the context of conjugation of Ru(II) complexes to G-quadruplex-forming systems, we would advise the use of alternative coupling methods, or at least careful consideration of the click conditions prior to use.
The revised click conditions were used with RuN3 and T3, T4 and T2 to yield AS1411-5′-TT-Ru, AS1411-5′-TTTTT-Ru and AS1411-3′-TTT-Ru as monitored and purified by HPLC with confirmation of product formation by LCMS (Fig. S16, ESI†). G-quadruplex formation was confirmed for all three samples by CD spectroscopy (Fig. S17, ESI†), thermal difference spectra (TDS) (Fig. S18, ESI†) and Tm-melting experiments (Fig. S15b, ESI†). TDS spectra can be used to analyse types of G-quadruplex formed.65AS1411-5′-TT-Ru, AS1411-5′-TTTTT-Ru and AS1411-3′-TTT-Ru have Tm values of 54.6, 51.5 and 61.6 °C respectively, measured in 0.1 M KCl, compared to 50.0 °C for unmodified AS1411 (T1). It is interesting to note that position of the Ru relative to the AS1411 core sequence has quite profound effects in the resulting Tm values with a very large increase of 11.6 °C for conjugation at the 3′-end. This perhaps indicates formation of different G-quadruplex structures or the predominance of one particular structures of the at least 8 different monomeric structures63 known to constitute AS1411. Hence, conjugation of Ru or possibly other substituents might represent a means to modulate the amount and the nature of G-quadruplex structures adopted by AS1411.
All three Ru–AS1411 are prone to forming insoluble ‘crystals’ if dried to completion by speed vac. We hypothesise that this is due to intrastrand interactions whereby G-quadruplex formation is initiated by the conjugated ruthenium complexes. Partial solubility can be recovered by heating to 95 °C in a 100 mM KCl solution with vigorous shaking.
Biological evaluation
All three Ru–AS1411 were tested in MCF-7 (breast cancer) and RPE-1 (normal retina) cell lines as model cell surface nucleolin positive and negative cell lines. It is clear from the CD spectrum of AS1411 that the cation concentration in the cell media used (5 mM KCl, 154 mM NaCl (DMEM)) is insufficient for complete G-quadruplex formation (Fig. S13b, ESI†). As such all Ru–AS1411 were kept in stock solutions at 20 μM in 50 mM KCl. The additional KCl was kept constant across all wells including control wells. Following a 2 hour incubation and light treatment (λexc = 480 nm, 3.21 J cm−2, 10 min) AS1411-5′-TTTTT-Ru reduced cell viability to a greater extent in MCF7 cells compared to RPE-1 cells, demonstrating the potential for cell specific PDT (Table 4 and Fig. 3, Fig. S20a, ESI†). Interestingly AS1411-5′-TTTTT-Ru has a G-quadruplex Tm closest to the original sequence (51.5 °C vs. 50.0 °C) perhaps suggesting that AS1411-5′-TTTTT-Ru maintains the same form of G-quadruplex. Confocal microscopy confirmed uptake in MCF7 and RPE-1 cells (Fig. S19, ESI†).68 After a 4 hour incubation and light treatment (λexc = 480 nm, 3.21 J cm−2, 10 min) all three Ru–AS1411s are phototoxic and cell line specificity is lost while only a mild reduction in cell viability is seen in the dark (Table 4 and Fig. S20b, ESI†). RuN3 alone is highly phototoxic after a 4 hour incubation in both cell lines (Table 3). Following a 2 hour incubation and light irradiation at 540 nm (40 min, 9.5 J cm−2), AS1411-5′-TTTTT-Ru maintains phototoxicity in MCF-7 cells though at 595 nm (2 h, 22.47 J cm−2) we were unable to detect phototoxicity within our concentration range (up to 1 μM). We believe this is due to the low range of concentrations tested coupled with the longer irradiation times.| AS1411-5′-TTTTT-Ru | AS1411-5′-TT-Ru | AS1411-3′-TTT-Ru | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 480 nm | 540 nm | 595 nm | 480 nm | 480 nm | |||||||||
| Light | Dark | PI | Light | Dark | Light | Dark | Light | Dark | PI | Light | Dark | PI | |
| MCF-7 2 h | 0.340 (±0.077) | >1 | >2.9 | 0.990 (±0.061) | >1 | >1 | >1 | 0.524 (±0.070) | >1 | >1.9 | 0.370 (±0.084) | >1 | >2.7 |
| RPE-1 2 h | 0.735 (±0.084) | >1 | >1.36 | >1 | >1 | >1 | >1 | 0.582 (±0.065) | >1 | >1.7 | 0.474 (±0.084) | >1 | >2.1 |
| MCF-7 4 h | 0.120 (±0.043) | >1 | >8.33 | nd | nd | nd | nd | 0.274 (±0.032) | >1 | >3.6 | 0.134 (±0.032) | >1 | >7.5 |
| RPE-1 4 h | 0.082 (±0.058) | >1 | >12.19 | nd | nd | nd | nd | 0.231 (±0.047) | >1 | >4.3 | 0.181 (±0.059) | >1 | >5.5 |
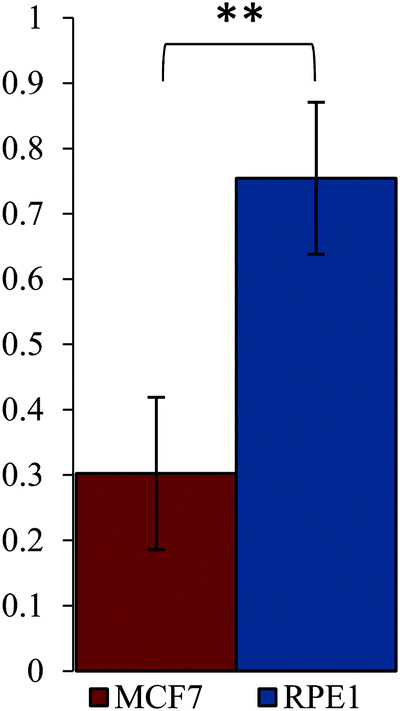 | ||
| Fig. 3 Relative cell survival in MCF-7 and RPE1 cell lines following treatment with AS1411-5′-TTTTT-Ru (0.5 μM, 2 hours) and light (480 nm, 10 min, 3.21 J cm−2). Significance evaluated by Student's T-Test ** p < 0.01. | ||
It was hypothesized that loss of specificity at 4 hours may be due to nuclease-mediated degradation of the oligonucleotides in cell media. As such a stability test was performed in cell medium (DMEM, 10% Foetal calf serum) to investigate the degradation, if any, of the oligonucleotides (Fig. S21, ESI†). It is clear that AS1411 alone (with 5′-FAM, T5) is susceptible to degradation with a significant laddering in the PAGE gel from the 30 minute incubation. Where Ru conjugation is at the 3′ end, in AS1411-3′-TTT-Ru, degradation appears lessen significantly, while both AS1411-5′-TT-Ru and AS1411-5′-TTTTT-Ru, where conjugation is at the 5′ end appear susceptible to degradation, though perhaps to a lesser extent when compared to AS1411.
These data may indicate that only AS1411-5′-TTTTT-Ru achieves specific uptake in the target cells, while formation of differing G-Quadruplex structures by AS1411-5′-TT-Ru and AS1411-3′-TTT-Ru (as indicated by their Tm values) may result in only non-specific uptake. Conjugation of the RuN3 is likely to increase the lipophilicity of the Ru–AS1411s and may increase their non-specific uptake. Non-specific uptake and photodynamic effect have been previously reported with AS1411.30
Conclusions
In conclusion, we have successfully synthesised an azide functionalized Ruthenium complex RuN3 and obtained three Ru–AS1411 by click reaction. A thorough biophysical investigation revealed that the covalently linked Ru complexes did not interfere with G-quadruplex formation, especially when located at the 5′-end of AS1411. At a 2 hour incubation timepoint AS1411-5′-TTTTT-Ru was selectively photosensitizing towards a cell surface nucleolin expressing cell line (MCF-7), indicating the potential for targeting offered by this system.Overall, we demonstrate the usefulness of conjugating Ru-based PSs to aptamers to enhance their therapeutic usefulness by conveying specificity to Ru-mediated PDT. Such an approach can readily be expanded to other aptamers by application of this straightforward method described herein and the presence of Ru-based PSs at 5′ termini is not expected to negatively impact the binding efficiency of the resulting aptamer-drug conjugates. A combination of chemically modified aptamers such as XNAs69–72 that enhance their nuclease stability with potent PSs such as Ru complexes is expected to improve the efficiency of the PDT treatment modality in the near future.
Experimental
Synthesis of RuN3
Solvents for reactions were of pro analysis (p.a.) grade or distilled prior to use. Ruthenium trichloride x-hydrate was provided by I2CNS (Zurich), 4,7-diphenyl-1,10-phenanthroline, lithium chloride (anhydrous, 99%), and ammonium hexafluorophosphate by Alfa Aesar, sodium azide by Sigma-Aldrich, and 4′-methyl-[2,2′-bipyridine]-4-carboxylic acid by FluoroChem.Instrumentation and methods
Amber glass or clear glassware wrapped in tin foil was used when protection from light was necessary. Reactions were carried out under N2 and monitored for completion by HPLC or thin-layer chromatography. Column chromatography: Merck silica gel 60 (40–63 μm) with the indicated solvent system. HPLC: 2× Agilent G1361 1260 Prep Pump system with Agilent G7115A 1260 DAD WR Detector equipped with an Agilent Pursuit XRs 5C18 (Analytic: 100 Å, C18 5 μm 250 × 4.6 mm) Column. The solvents (HPLC grade) were millipore water (0.1% trifluoroacetic acid (TFA)), solvent A) and acetonitrile (MeCN) (0.1% TFA, solvent B). The HPLC gradients used are as follow (S1): 0–3 min: isocratic 95% A (5% B); 3–17 min: linear gradient from 95% A (5% B) to 0% A (100% B); 17–23 min: isocratic 0% A (100% B), 23–25 min: linear gradient from 0% A (100% B) to 95% A (5% B). The flow rate was 1 ml min−1. Detection was performed at 215 nm, 250 nm, 350 nm, 450 nm, 550 nm and 650 nm with a slit of 4 nm. IR spectra: spectrumTwo FT-IR Spectrometer (PerkinElmer) equipped with a Specac Golden GateTM ATR (attenuated total reflection) accessory; applied as neat samples; 1/λ in cm−1. NMR-data: deuterated NMR solvents were obtained from Eurisotop (France). 1H NMR spectra in CD3OD or CD3CN; BrukerAV-400 (400 MHz); δ in ppm relative to solvent. (CD3OD (p, 3.31 ppm) and CD3CN (p, 1.94 ppm)), J in Hz. 13C NMR spectra in CD3OD or CD3CN; Bruker AV-400 (100.6 MHz); δ in ppm rel. to solv. ((CD3OD (49.00 ppm) and CD3CN (118.26 ppm)); multiplicities from DEPT-135 and DEPT-90 experiments. 19F NMR spectra in CD3OD or CD3CN. electrospray ionization mass spectrometry (ESI-MS): experiments were carried out using an LTQ-Orbitrap XL from Thermo Scientific (Thermo Fisher Scientific, Courtaboeuf, France) and operated in positive ionization mode, with a spray voltage of 3.6 kV. No sheath and auxiliary gas were used. Applied voltages were 40 and 100 V for the ion transfer capillary and the tube lens, respectively. The ion transfer capillary was held at 275 °C. Detection was achieved in the Orbitrap with a resolution set to 100.000 (at m/z 400) and a m/z range between 150–2000 in profile mode. Spectrum was analyzed using the acquisition software XCalibur 2.1 (Thermo Fisher Scientific, Courtaboeuf, France). The automatic gain control (AGC) allowed the accumulation of up to 21 × 105 ions for FTMS scans, maximum injection time was set to 300 ms and 1 μscan was acquired. 10 μl was injecting using a Thermo Finnigan Surveyor HPLC system (Thermo Fisher Scientific, Courtaboeuf, France) with a continuous infusion of methanol (MeOH) at 100 μl min−1.[Ru(dmso)4Cl2]
[Ru(dmso)4Cl2] was synthesized by adapting a given procedure.46 Ruthenium(III)-trichloride-x-hydrate (9.83 g, 37.6 mmol, assuming x = 3) was suspended in EtOH (125 ml, dry) and heated to reflux for 3 h, while a color change from dark brown to dark green was observed. The mixture was filtrated and the solv. was reduced in vacuo to afford a deep green paste. The residue was suspended in DMSO (20 ml, dry) and refluxed for 2 h. The heating bath was turned off, and the mixture was left cooling slowly to r.t. in the oil bath while slowly stirring. Cold acetone (200 ml, dry) was added while stirring and the mixture was left overnight at −25 °C for crystallization before filtration. The solid residue was washed several times with acetone to afford [Ru(dmso)4Cl2] as yellow solid (15.5 g, 31.9 mmol, 85%). Spectroscopic data (1H NMR) were in agreement with the literature.461H NMR (400 MHz, D2O) δ 3.58–3.31 (m, 18H), 2.71 (s, 6H). 13C NMR (101 MHz, D2O) δ 46.73, 46.51, 45.70, 45.11, 44.67, 44.32, 38.67.[RuBphen2Cl2]
[RuBphen2Cl2] was synthesized by adapting a given procedure.73 A mixture of [Ru(dmso)2Cl2] (3.48 g, 7.17 mmol), 4,7-diphenyl-1,10-phenanthroline (Bphen, 5.00 g, 15.0 mmol) and LiCl (2.13 g, 50.3 mmol) was dissolved in DMF (150 ml) and refluxed for 24 h. After cooling to r.t., the solvent was reduced in vacuo and acetone (500 ml) was slowly added while stirring. The mixture was then stored at −25 °C overnight for crystallization before filtration. The solid residue was washed with water, acetone and Et2O to afford [RuBphen2Cl2] as a black-purple solid (3.25 g, 3.88 mmol, 72%). Spectroscopic data (1HNMR) were in agreement with the literature.731H NMR (400 MHz, CD2Cl2) δ 10.55 (s, 2H), 8.50 (d, J = 5.5 Hz, 1H), 8.15 (s, 1H), 8.09 (s, 1H), 8.07 (s, 1H), 7.96 (s, 2H), 7.94 (t, J = 2.8 Hz, 3H), 7.92 (s, 1H), 7.73 (s, 1H), 7.69 (d, J = 6.9 Hz, 3H), 7.63–7.47 (m, 7H), 7.46–7.34 (m, 7H), 7.12 (d, J = 5.6 Hz, 2H). 13C NMR (101 MHz, CD2Cl2) δ 154.89, 153.04, 152.57, 151.49, 150.88, 149.67, 146.37, 144.96, 137.08, 130.63, 130.18, 129.52, 129.40, 126.10, 125.85, 125.63, 124.90. HPLC: S1 TR = 7.542 min.N3EtNH2
N3EtNH2 was synthesized following a previously published method.74 NaN3 (2.55 g, 39.2 mmol) was added to a solution of 2-chloroethanamine hydrochloride (1.5 g, 12.9 mmol) in H2O (10 ml). The resulting mixture was heated with stirring at 80 °C overnight before the reaction was quenched by addition of aqueous KOH (dropwise until a pH of around 12). Following extraction by diethyl ether (3 × 20 ml) and washing with brine (20 ml) the organic layers were combined and dried with anhydrous MgSO4. The solvent was removed carefully in vacuo (N3EtNH2 is volatile) to give N3EtNH2 as a colourless oil (1.54 g, 82%). 1H NMR (400 MHz, MeOD) δ 3.38 (t, 2H, J = 5.94 Hz), δ 2.77 (m, 2H).bpyN3
The compound N-(2-azidoethyl)-4′-methyl-[2,2′-bipyridine]-4-carboxamide (bpyN3) was synthesized by suspending 4′-methyl-[2,2′-bipyridine]-4-carboxylic acid (85.70 mg, 0.40 mmol), EDCI-HCl (82.4 mg, 0.43 mmol), NHS (49.10 mg, 0.43 mmol) and in CH2Cl2 (20 ml). After addition of DIPEA (72.0 μl, 0.45 mmol), the solution is getting clear while left stirring for 30 min. N3EtNH2 (90 μl, 0.45 mmol) was added and the mixture was left stirring overnight. The solv. was removed in vacuo and the crude residue was purified on alumina (20:1 CH2Cl2/iPrOH) to deliver a colourless oil. After washing with pentane, spontaneous induced crystallization delivered bpyN3 as a colorless solid (63.2 mg, 0.224 mmol, 56%). 1H NMR (400 MHz, methanol-d4) δ 8.79 (d, J = 5.3 Hz, 1H arom.), 8.68 (s, 1H arom.), 8.53 (d, J = 5.1 Hz, 1H arom.), 8.22 (s, 1H arom.), 7.82–7.68 (m, 1H arom.), 7.32 (d, J = 5.0 Hz, 1H arom.), 3.62 (t, J = 5.8 Hz, 2H), 3.55 (d, J = 5.8 Hz, 2H), 2.48 (s, 3H).
RuN3
The complex [Ru(Bphen)2(bpyN3)][PF6]2 (RuN3) was synthesized by adapting a given procedure.49[Ru(Bphen)2Cl2] (69 mg, 82 μmol) and bpyN3 (31 mg, 0.108 mmol) were suspended in a water/MeOH mixture (1:1, 20 ml, degassed) and refluxed for 15 h resulting in a deep red solution The solution was cooled to r.t. and a few drops of saturated NH4PF6 solution (sat.) were added while stirring to form a red precipitate. After filtration, the crude solid was purified by column chromatography on silica 60 using a system of MeCN-KNO3aq. (20:1, 0.24 M). The product-containing phases were combined and the solv.s were removed in vacuo. The redish residue was dissolved in MeOH and a few drops of saturated NH4PF6 solution and water were added while stirring, to afford a clear solution with a red precipitate. Filtration delivered complex RuN3 as a red solid (70 mg, 52 μmol, 64%). IR (cm−1): 2020w, 1890s, 1620w, 1550w, 1420w, 1310w, 1220w, 1020w, 1100m, 1030w, 830s, 770s, 730m, 700m. 1H NMR (400 MHz, CD3CN) δ 9.85 (dd, J = 16.9, 5.7 Hz, 1H), 8.89 (d, J = 1.4 Hz, 1H arom.), 8.61 (s, 1H arom.), 8.29 (dd, J = 18.9, 5.5 Hz, 2H arom.), 8.20 (dd, J = 12.0, 9.5 Hz, 2H arom.), 8.19 (s, 2H), 8.10 (dd, J = 5.5, 2.8 Hz, 2H arom.), 8.01 (dd, J = 5.9, 0.6 Hz, 1H arom.), 7.75 (t, J = 5.5 Hz, 2H arom.), 7.72–7.55 (m, 24H arom.), 7.26 (ddd, J = 5.8, 1.8, 0.8 Hz, 1H arom.), 3.61 (q, J = 5.3 Hz, 2H), 3.53 (t, J = 5.7 Hz, 2H), 2.59 (s, 3H). 13C NMR (101 MHz, CD3CN) δ 153.16, 153.02, 152.34, 150.12, 143.13, 136.69, 136.63, 130.81, 130.75, 130.69, 130.65, 130.15, 130.11, 129.99, 127.05, 126.49, 125.13, 122.55, 51.03, 40.30, 21.28. 19F NMR (376 MHz, CD3CN) δ −72.74 (d, J = 707.1 Hz, PF6). HR-MS (ESI+): m/z 524.1447 [M – 2(PF6)]2+ (calculated: 524.1453). HPLC: S1 TR = 16.271 min.
Click reactions
Oligonucleotides T1, T2 and T5 were purchased from Microsynth AG, oligonucleotides T3 and T4 were ordered from Integrated DNA Technologies and were stored at 1 mM in H2O. RuN3 was stored at 5 mM in DMF, Sodium ascorbate was made fresh at 50 mM in H2O, CuSO4 was stored at 5 mM in H2O, TBTA was stored at 10 mM in DMSO. All solutions were thoroughly degassed by argon bubbling prior and post mixing of appropriate quantities of stock solutions. The reactions were performed in Eppendorf tubes at 30 °C with gentle shaking for 2 hours. The Monarch® PCR & DNA Cleanup Kit was used to desalt the reactions before HPLC purification. The quantities of reagents used is specified in Table 5. Confirmation of product formation was obtained by LCMS (m/z found: 10082.7427, 10994.8808 and 10560.8611 for AS1411-5′-TT-Ru, AS1411-5′-TTTTT-Ru and AS1411-3′-TTT-Ru respectively).HPLC
HPLC purification was performed using an Äkta™ pure system (GE Healthcare) equipped with Kinetex® semi preparative Reversed Phase C18 column (5 μm, 250 × 10.0 mm). Buffer A: 20 mM Triethylammonium acetate (TEAA) buffer; buffer B: 30% 20 mM TEAA/70% Acetonitrile. A flow rate of 1.5 ml min−1 was used with UV/Vis detection at 280 and 430 nm.Spectroscopic measurements
The absorption spectra of the sample was measured with a SpectraMax M2 Spectrometer (Molecular Devices). For measurement of the emission, the sample was irradiated at 450 nm with a NT342B Nd-YAG pumped optical parametric oscillator (Ekspla). The emission was focused at right angle to the excitation pathway and directed to a Princeton Instruments Acton SP-2300i monochromator. The signal was detected with a XPI-Max 4 CCD camera (Princeton Instruments).LC-MS
LC-MS analyses were performed on a Q exactive mass spectrometer (Thermo Fisher Scientific) equipped with an electrospray ionisation source (H-ESI II Probe) coupled with an Ultimate 3000 RS HPLC (Thermo Fisher Scientific). Compounds were injected onto a ThermoFisher Hypersil Gold aQ chromatography column (100 mm × 2.1 mm, 1.9 μm particle size) heated at 30 °C. The flow rate was set at 0.3 ml min−1 and the mobile phase consisted of (A) water + 0.1% formic acid and (B) acetonitrile + 0.1% formic acid. The gradient used was: 5% B during 0–3 minutes then 5% to 100% B linear during 3–8 minutes. Ions were analysed in negative ion mode. MS resolution was 70000 with an AGC target of 1e6 and a maximum injection time of 240 ms. Multicharged ions were processed using Xtract software. A UV detector set at 270 nm was also used as a control.
Luminescence quantum yield
The sample was prepared in an acetonitrile solution with an absorption of 0.1 at 450 nm. The sample was irradiated at 450 nm with a NT342B Nd-YAG pumped optical parametric oscillator (Ekspla). The emission was focused at right angle to the excitation pathway and directed to a Princeton Instruments Acton SP-2300i monochromator. The signal was detected with a XPI-Max 4 CCD camera (Princeton Instruments). The luminescence quantum yields were determined by comparison with the reference [Ru(2,2′-bipyridine)3]Cl2 in acetonitrile (Φem = 5.9%56) applying the following formula:| Φem,sample = Φem,reference × (Freference/Fsample) × (Isample/Ireference) × (nsample/nreference)2 | (1) |
| F = 1 − 10−A | (2) |
Lifetime
The sample was prepared in an air saturated as well as a degassed acetonitrile solution with an absorption of 0.2 at 450 nm. The sample was irradiated at 450 nm with a NT342B Nd-YAG pumped optical parametric oscillator (Ekspla). The emission was focused at right angle to the excitation pathway and directed to a Princeton Instruments Acton SP-2300i monochromator. The signal was detected with a R928 photomultiplier tube (Hamamatsu).Singlet oxygen – direct evaluation
The sample was prepared in an air saturated acetonitrile or D2O solution with an absorption of 0.2 at 450 nm. The sample was irradiated at 450 nm with a mounted M450LP1 LED (Thorlabs) whose light was focused with aspheric condenser lenses. Using a T-Cube LED Driver (Thorlabs), the intensity of the irradiation was varied and monitored with an optical power and energy meter. The emission was focused at right angle to the excitation pathway and directed to a Princeton Instruments Acton SP-2300i monochromator. To cut off light at wavelengths shorter than 850 nm, a longpass glass filter was placed in front of the monochromator entrance slit. The signal was detected with an EO-817L IR-sensitive liquid nitrogen cooled germanium diode detector (North Coast Scientific Corp.). The luminescence signal, centered at 1270 nm, was measured from 1100 to 1400 nm. The obtained data was analyzed upon plotting the integrated luminescence peaks against the percentage of the irradiation intensity. The slope of the linear regression was calculated and compared with the reference Rose Bengal (Φ = 76%75). The absorption of the sample was corrected with an absorption correction factor. The singlet oxygen quantum yields were calculated using the following formula:| Φsample = Φreference × (Ssample/Sreference) × (Ireference/Isample) | (3) |
| I = I0 × (1 − 10−A) | (4) |
Singlet oxygen – indirect evaluation
Measurement in acetonitrile: the sample was prepared in an air-saturated acetonitrile solution with an absorption of 0.2 at the irradiation wavelength, N,N-dimethyl-4-nitrosoaniline aniline (RNO, 24 μM) and imidazole (12 mM). Measurement in PBS buffer: The sample was prepared in an air-saturated PBS solution containing the complex with an absorption of 0.2 at the irradiation wavelength, N,N-dimethyl-4-nitrosoaniline aniline (RNO, 20 μM) and histidine (10 mM). The samples were irradiated for various time points with an Atlas Photonics LUMOS BIO irradiator. The absorption of the samples was constantly monitored with a SpectraMax M2 Microplate Reader (Molecular Devices). The difference in absorption (A0 − A) at 420 nm for the measurement in acetonitrile or at 440 nm for the measurement in PBS was determined. The difference in absorption was then plotted against the irradiation times and the slope of the linear regression calculated. The absorption of the sample was corrected with an absorption correction factor. The singlet oxygen quantum yields were calculated using the same formulas as used for the direct evaluation.Gel electrophoresis
Acrylamide/bisacrylamide (29:1, 40%) was obtained from Fisher Scientific. Visualization of PAGE gels was performed by fluorescence imaging using a Typhoon Trio phosphorimager with the ImageQuant software from GE Healthcare. Samples were loaded in blue loading dye (70% formamide, EDTA 50 mM, 0.1% bromophenol blue, 0.1% xylene cyanol, H2O).
CD spectroscopy
Circular dichroism experiments were performed on a Aviv 215 spectropolarimeter at 37 °C. Concentration of AS1411 and Ru–AS1411s was kept constant (10 μM in water) and CD measured using a 1 cm pathlength cuvette. Stock salt solutions (1 M KCl and NaCl) were used. CD spectra were recorded at the Molecular Biophysics platform at Institut Pasteur.Thermal difference spectra
TDS were performed on an Agilent Cary UV-Vis Compact Peltier machine in 60 μl volume quartz cuvettes. The UV/Vis absorption spectra of concentrations of around 3 μM were measured between 335 nm and 220 nm at 20 °C and 90 °C. The TDS was generated by subtracting the spectra at 20 °C from those at 90 °C following a previously published protocol.65Cell culture experiments
Cells lines were treated in appropriate cell culture media of DMEM (Gibco, LifeTechnologies, USA) supplemented with 10% foetal calf serum (FCS) for the HT29 and MCF-7 cell lines (Gibco) and DMEM/F-12 (Gibco) supplemented with 10% foetal calf serum (Gibco) for the RPE-1 cell line. All media was also supplemented with 100 U ml−1 penicillin–streptomycin mixture (Gibco). Cells were incubated at 37 °C in 5% CO2. Cells were passaged when 80% confluency was reached and used within 15 passages from initial purchase. Ru–AS1411s were stored at 20 μM in sterile 50 mM KCl. RuN3 was stored in DMSO at 10 mM.Cytotoxicity experiments
96 well dishes were seeded with MCF-7 cells (6000 cells per well) or RPE-1 cells (4000 cells per well) and incubated overnight. Cell media was replaced with treatment solutions prepared to the concentrations specified and incubated for 2 or 4 hours. Concentrations of additional KCl (5 mM) and DMSO (0.01%) were kept constant across treatments. Following the incubation, wells were washed (2 × PBS) and the media replaced (100 μl) before either being treated with light (λexc = 480 nm, 3.21 J cm−2, 10 min) or kept in the dark. After 48 h incubation the cells were treated with resazurin (0.2 mg mL−1 final concentration in appropriate media) and incubated a further 4 h. The plates were read by fluorescence plate reader SpectraMax M5 micro plate reader (λex, 540 nm; λem, 590 nm).Confocal microscopy
Sterilized 12 mm Menzel–Gläser coverslips were added to 6 well dishes (3 coverslips per well) and seeded with cells (2 × 105 cells per well for HT-29 and MCF-7 cell lines and 1.5 × 105 for RPE-1) and incubated overnight. For Ru–AS1411s imaging, coverslips were transferred to separate wells in a 12-well dish and treatment solutions added. Following a 2 hour time point the wells were washed (3 × PBS) and treated with paraformaldehyde (4% in PBS, 15 min) before being washed (3 × PBS) and mounted to microscope slides (Prolong Glass Antifade Mountant). For anti-nucleolin staining coverslips were transferred to separate wells in a 12-well dish and incubated with Nucblue (2 drops ml−1 in media, 20 min) before being washed (3 × PBS) and treated with paraformaldehyde (4% in PBS, 15 min) before being washed again (3 × PBS). Blocking solution was added (0.2% BSA, 0,05% Saponin in PBS) for 15 min at RT. The primary antibody anti-nucleolin (ZN004, Thermofisher) was added (5 μg ml−1, 1 h) before the cover slips were washed (3 × 0.2% BSA, 0.05% Saponin in PBS) and the secondary antibody added Alexa 488 conjugated secondary antibodies (Jackson ImmunoResearch Laboratory, 1:400 dilution, 30 min). The coverslips were then mounted to microscope slides (Prolong Glass Antifade Mountant). The slides were then imaged. For the Ru–AS1411sλexc = 405 nm and λem = 600–750 nm. The microscope DAPI imaging settings were used for Nucblue and λexc = 488 nm, λem = 510–540 nm used to image the secondary antibody. Images were recorded at the Cellular and Molecular Imaging Technical Platform, INSERMUMS025–CNRSUMS3612, Faculty of Pharmacy of Paris, Paris Descartes University, Paris, France.
T m measurements
The melting experiments were performed on an Agilent Cary UV-Vis Compact 3500 Peltier machine in 60 μl volume quartz cuvettes. AS1411 samples were prepared in 0.1 M KCl to yield a solution with OD295 between 0.3 and 0.6. Paraffin oil (100 μl) was added. A control cell (0.1 M KCl) was prepared into which the temperature probe was placed. The Tm values were measured with a total of three heating and cooling ramps each (1 °C minute−1) with data processed from the heating ramps.Media stability tests
Samples of all three Ru–AS1411s (20 pmol each, 1 μL from 20 μM sample in 50 mM KCl) were incubated in cell culture medium (DMEM with 10% FCS, 10 μl each) and incubated for the stated time at 37 °C. Blue loading dye was added (10 μl) and sampled heated (95 °C, 5 min) before being loaded on PAGE gel.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We thank Dr Philippe Goldner for access to state-of-the-art laser apparatus. We thank financial support from the Technology Transfer and Industrial Partnership Department (DARRI) of Institut Pasteur (grant INNOV 17-19) and the Joe W. and Dorothy Doresett Brown Foundation (exploratory and advanced grants). This work was financially supported by an ERC Consolidator Grant PhotoMedMet to G. G. (GA 681679), has received support under the program “Investissements d’ Avenir” launched by the French Government and implemented by the ANR with the reference ANR-10-IDEX-0001-02 PSL (G. G.). L. K. M. thanks the ARC Foundation for cancer research for a postdoctoral Research Fellowship (grant number: S-FB18006). A. G also thanks the ARC Foundation for cancer research for a postdoctoral Research Fellowship. The authors acknowledge a DIM1Health 2019 grant from the Région Ile de France to the project EpiK (coordinated by P. B. Arimondo) for the LCMS equipment. M. F. gratefully acknowledges a fellowship from the doctoral school MTCI, Université de Paris.References
- J. Karges, F. Heinemann, M. Jakubaszek, F. Maschietto, C. Subecz, M. Dotou, R. Vinck, O. Blacque, M. Tharaud, B. Goud, E. Viñuelas-Zahinos, B. Spingler, I. Ciofini and G. Gasser, J. Am. Chem. Soc., 2020, 142, 6578–6587 CrossRef CAS.
- S. Monro, K. L. Colón, H. Yin, J. Roque III, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2018, 119, 797–828 CrossRef.
- L. K. McKenzie, H. E. Bryant and J. A. Weinstein, Coord. Chem. Rev., 2019, 379, 2–29 CrossRef CAS.
- F. Heinemann, J. Karges and G. Gasser, Acc. Chem. Res., 2017, 50, 2727–2736 CrossRef CAS.
- J. D. Knoll and C. Turro, Coord. Chem. Rev., 2015, 282, 110–126 CrossRef PubMed.
- C. Mari, V. Pierroz, S. Ferrari and G. Gasser, Chem. Sci., 2015, 6, 2660–2686 RSC.
- S. A. McFarland, A. Mandel, R. Dumoulin-White and G. Gasser, Curr. Opin. Chem. Biol., 2020, 56, 23–27 CrossRef CAS PubMed.
- M. G. Walker, P. J. Jarman, M. R. Gill, X. Tian, H. Ahmad, P. A. Reddy, L. McKenzie, J. A. Weinstein, A. J. Meijer and G. Battaglia, Chem. – Eur. J., 2016, 22, 5996–6000 CrossRef CAS PubMed.
- F. E. Poynton, S. A. Bright, S. Blasco, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2017, 46, 7706–7756 RSC.
- L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z.-S. Chen, Chem. Soc. Rev., 2017, 46, 5771–5804 RSC.
- C. S. Burke, A. Byrne and T. E. Keyes, J. Am. Chem. Soc., 2018, 140, 6945–6955 CrossRef CAS PubMed.
- P. S. Felder, S. Keller and G. Gasser, Adv. Therapeutics, 2020, 3, 1900139 CrossRef.
- M. Jakubaszek, B. Goud, S. Ferrari and G. Gasser, Chem. Commun., 2018, 54, 13040–13059 RSC.
- S. Chakrabortty, B. K. Agrawalla, A. Stumper, N. M. Vegi, S. Fischer, C. Reichardt, M. Kögler, B. Dietzek, M. Feuring-Buske and C. Buske, J. Am. Chem. Soc., 2017, 139, 2512–2519 CrossRef CAS.
- D. Cullinane, K. S. Gkika, A. Byrne and T. E. Keyes, J. Inorg. Biochem., 2020, 207, 111032 CrossRef CAS PubMed.
- A. Martin, A. Byrne, C. S. Burke, R. J. Forster and T. E. Keyes, J. Am. Chem. Soc., 2014, 136, 15300–15309 CrossRef CAS PubMed.
- J. Karges, M. Jakubaszek, C. Mari, K. Zarschler, B. Goud, H. Stephan and G. Gasser, ChemBioChem, 2020, 21, 531–542 CrossRef CAS.
- W. Sun, S. Li, B. Häupler, J. Liu, S. Jin, W. Steffen, U. S. Schubert, H. J. Butt, X. J. Liang and S. Wu, Adv. Mater., 2017, 29, 1603702 CrossRef.
- N. Soliman, L. K. McKenzie, J. Karges, E. Bertrand, M. Tharaud, M. Jakubaszek, V. Guérineau, B. Goud, M. Hollenstein and G. Gasser, Chem. Sci., 2020, 11, 2657–2663 RSC.
- E. Villemin, Y. C. Ong, C. M. Thomas and G. Gasser, Nat. Rev. Chem., 2019, 3, 261–282 CrossRef CAS.
- Y. Ellahioui, M. Patra, C. Mari, R. Kaabi, J. Karges, G. Gasser and S. Gómez-Ruiz, Dalton Trans., 2019, 48, 5940–5951 RSC.
- J. Karges, J. Li, L. Zeng, H. Chao and G. Gasser, ACS Appl. Mater. Interfaces, 2020, 12, 54433–54444 CrossRef CAS.
- J. Yan, T. Gao, Z. Lu, J. Yin, Y. Zhang and R. Pei, ACS Appl. Mater. Interfaces, 2021, 13, 27749–27773 CrossRef CAS.
- P. Röthlisberger and M. Hollenstein, Adv. Drug Delivery Rev., 2018, 134, 3–21 CrossRef.
- L. K. McKenzie, R. El-Khoury, J. D. Thorpe, M. J. Damha and M. Hollenstein, Chem. Soc. Rev., 2021, 50, 5126–5164 RSC.
- L. Li, S. Xu, H. Yan, X. Li, H. S. Yazd, X. Li, T. Huang, C. Cui, J. Jiang and W. Tan, Angew. Chem., Int. Ed., 2021, 60, 2221–2231 CrossRef CAS.
- P. J. Bates, D. A. Laber, D. M. Miller, S. D. Thomas and J. O. Trent, Exp. Mol. Pathol., 2009, 86, 151–164 CrossRef CAS PubMed.
- C. M. Berger, X. Gaume and P. Bouvet, Biochimie, 2015, 113, 78–85 CrossRef CAS PubMed.
- C. R. Ireson and L. R. Kelland, Mol. Cancer Ther., 2006, 5, 2957–2962 CrossRef CAS PubMed.
- Y.-A. Shieh, S.-J. Yang, M.-F. Wei and M.-J. Shieh, ACS Nano, 2010, 4, 1433–1442 CrossRef CAS.
- Q. Liu, L. Xu, X. Zhang, N. Li, J. Zheng, M. Guan, X. Fang, C. Wang and C. Shu, Chem. – Asian J., 2013, 8, 2370–2376 CrossRef CAS.
- J. Ai, Y. Xu, B. Lou, D. Li and E. Wang, Talanta, 2014, 118, 54–60 CrossRef CAS.
- J. Kim, W. Park, D. Kim, E. S. Lee, D. H. Lee, S. Jeong, J. M. Park and K. Na, Adv. Funct. Mater., 2019, 29, 1900084 CrossRef.
- Y. Yang, J. He, W. Zhu, X. Pan, H. S. Yazd, C. Cui, L. Yang, X. Li, L. Li and L. Cheng, Theranostics, 2020, 10, 4030 CrossRef CAS PubMed.
- X. Zhu, H. Zhou, Y. Liu, Y. Wen, C. Wei, Q. Yu and J. Liu, Acta Biomater., 2018, 82, 143–157 CrossRef CAS PubMed.
- L. Xu, X. Chen, J. Wu, J. Wang, L. Ji and H. Chao, Chem. – Eur. J., 2015, 21, 4008–4020 CrossRef CAS.
- D. Sun, Y. Liu, D. Liu, R. Zhang, X. Yang and J. Liu, Chem. – Eur. J., 2012, 18, 4285–4295 CrossRef CAS.
- G.-L. Liao, X. Chen, L.-N. Ji and H. Chao, Chem. Commun., 2012, 48, 10781–10783 RSC.
- E. Wachter, D. Moyá, S. Parkin and E. C. Glazer, Chem. – Eur. J., 2016, 22, 550–559 CrossRef CAS.
- G. Piraux, L. Bar, M. Abraham, T. Lavergne, H. Jamet, J. Dejeu, L. Marcélis, E. Defrancq and B. Elias, Chem. – Eur. J., 2017, 23, 11872–11880 CrossRef CAS.
- L. Xu, D. Zhang, J. Huang, M. Deng, M. Zhang and X. Zhou, Chem. Commun., 2010, 46, 743–745 RSC.
- K. McQuaid, H. Abell, S. P. Gurung, D. R. Allan, G. Winter, T. Sorensen, D. J. Cardin, J. A. Brazier, C. J. Cardin and J. P. Hall, Angew. Chem., Int. Ed., 2019, 58, 9881–9885 CrossRef CAS.
- Q. Yu, Y. Liu, C. Wang, D. Sun, X. Yang, Y. Liu and J. Liu, PLoS One, 2012, 7, e50902 CrossRef CAS PubMed.
- J. Weynand, A. Diman, M. Abraham, L. Marcélis, H. Jamet, A. Decottignies, J. Dejeu, E. Defrancq and B. Elias, Chem. – Eur. J., 2018, 24, 19216–19227 CrossRef CAS.
- N. Zabarska, A. Stumper and S. Rau, Dalton Trans., 2016, 45, 2338–2351 RSC.
- I. Brastos, E. Alessio, M. E. Ringenberg and T. B. Rauchfuss, Inorg. Synth., 2010, 35, 148–163 Search PubMed.
- R. Caspar, C. Cordier, J. B. Waern, C. Guyard-Duhayon, M. Gruselle, P. Le Floch and H. Amouri, Inorg. Chem., 2006, 45, 4071–4078 CrossRef CAS.
- C. A. Panetta, H. J. Kumpaty, N. E. Heimer, M. C. Leavy and C. L. Hussey, J. Org. Chem., 1999, 64, 1015–1021 CrossRef CAS.
- J. Karges, F. Heinemann, F. Maschietto, M. Patra, O. Blacque, I. Ciofini, B. Spingler and G. Gasser, Bioorg. Med. Chem., 2019, 27, 2666–2675 CrossRef CAS PubMed.
- J. Iley and R. Tolando, J. Chem. Soc., Perkin Trans. 2, 2000, 2328–2336, 10.1039/b001756f.
- V. G. Correia, J. C. Abreu, C. A. Barata and L. H. Andrade, Org. Lett., 2017, 19, 1060–1063 CrossRef CAS PubMed.
- Q. Sun, Z. Kang, L. Xue, Y. Shang, Z. Su, H. Sun, Q. Ping, R. Mo and C. Zhang, J. Am. Chem. Soc., 2015, 137, 6000–6010 CrossRef CAS.
- G. Lemercier, M. Four and S. Chevreux, Coord. Chem. Rev., 2018, 368, 1–12 CrossRef CAS.
- J. Karges, O. Blacque, P. Goldner, H. Chao and G. Gasser, Eur. J. Inorg. Chem., 2019, 3704–3712 CrossRef CAS.
- J. Karges, D. Díaz-García, S. Prashar, S. Gómez-Ruiz and G. Gasser, ACS Appl. Bio Mater., 2021, 4, 4394–4405 CrossRef CAS.
- K. Nakamaru, Bull. Chem. Soc. Jpn., 1982, 55, 1639–1640 CrossRef CAS.
- J. Karges, S. Kuang, F. Maschietto, O. Blacque, I. Ciofini, H. Chao and G. Gasser, Nat. Commun., 2020, 11, 1–13 Search PubMed.
- Y. W. Cheung, P. Röthlisberger, A. E. Mechaly, P. Weber, F. Levi-Acobas, Y. Lo, A. W. C. Wong, A. B. Kinghorn, A. Haouz, G. P. Savage, M. Hollenstein and J. A. Tanner, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 16790–16798 CrossRef CAS PubMed.
- N. Z. Fantoni, A. H. El-Sagheer and T. Brown, Chem. Rev., 2021, 121(12), 7122–7154 CrossRef CAS PubMed.
- D. Ganz, D. Harijan and H.-A. Wagenknecht, RSC Chem. Biol., 2020, 1, 86–97 RSC.
- S. Mack, M. F. Fouz, S. K. Dey and S. R. Das, Curr. Protocols Chem. Biol., 2016, 8, 83–95 CrossRef.
- I. Kejnovska, D. Renciuk, J. Palacky and M. Vorlickova, Methods Mol. Biol., 2019, 2035, 25–44 CrossRef CAS.
- M. M. Dailey, M. C. Miller, P. J. Bates, A. N. Lane and J. O. Trent, Nucleic Acids Res., 2010, 38, 4877–4888 CrossRef CAS PubMed.
- Z. Bagheri, B. Ranjbar, H. Latifi, M. I. Zibaii, T. T. Moghadam and A. Azizi, Int. J. Biol. Macromol., 2015, 72, 806–811 CrossRef CAS.
- J. Carvalho, A. Paiva, M. P. C. Campello, A. Paulo, J.-L. Mergny, G. F. Salgado, J. A. Queiroz and C. Cruz, Sci. Rep., 2019, 9, 1–12 CAS.
- G. R. Abel Jr, Z. A. Calabrese, J. Ayco, J. E. Hein and T. Ye, Bioconjugate Chem., 2016, 27, 698–704 CrossRef.
- K. Liu, P. K. Lat, H.-Z. Yu and D. Sen, Nucleic Acids Res., 2020, 48, 7356–7370 CAS.
- Y. Jing, M. Cai, L. Zhou, J. Jiang, J. Gao and H. Wang, Talanta, 2020, 217, 121037 CrossRef CAS.
- M. R. Dunn, C. M. McCloskey, P. Buckley, K. Rhea and J. C. Chaput, J. Am. Chem. Soc., 2020, 142, 7721–7724 CrossRef CAS PubMed.
- E. Eremeeva, A. Fikatas, L. Margamuljana, M. Abramov, D. Schols, E. Groaz and P. Herdewijn, Nucleic Acids Res., 2019, 47, 4927–4939 CrossRef CAS.
- K. M. Rose, I. A. Ferreira-Bravo, M. Li, R. Craigie, M. A. Ditzler, P. Holliger and J. J. DeStefano, ACS Chem. Biol., 2019, 14, 2166–2175 CAS.
- A. E. Rangel, Z. Chen, T. M. Ayele and J. M. Heemstra, Nucleic Acids Res., 2018, 46, 8057–8068 CrossRef CAS.
- A. Notaro, M. Jakubaszek, N. Rotthowe, F. Maschietto, R. Vinck, P. S. Felder, B. Goud, M. Tharaud, I. Ciofini, F. Bedioui, R. F. Winter and G. Gasser, J. Am. Chem. Soc., 2020, 142, 6066–6084 CrossRef CAS PubMed.
- J. Dash, Z. A. Waller, G. D. Pantoş and S. Balasubramanian, Chem. – Eur. J., 2011, 17, 4571–4581 CrossRef CAS.
- I. E. Kochevar and R. W. Redmond, Methods Enzymol., 2000, 319, 20–28 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cb00146a |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |