
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvanced antifouling and antibacterial hydrogels enabled by controlled thermo-responses of a biocompatible polymer composite†
Fei
Pan‡
 *a,
Sixuan
Zhang
a,
Stefanie
Altenried
a,
Flavia
Zuber
a,
Qian
Chen
b and
Qun
Ren
*a
*a,
Sixuan
Zhang
a,
Stefanie
Altenried
a,
Flavia
Zuber
a,
Qian
Chen
b and
Qun
Ren
*a
aEmpa, Swiss Federal Laboratories for Materials Science and Technology, Laboratory for Biointerfaces, Lerchenfeldstrasse 5, 9014 St. Gallen, Switzerland. E-mail: Fei.Pan@empa.ch; Qun.Ren@empa.ch
bInstitute of Functional Nano & Soft Materials (FUNSOM), Jiangsu Key Laboratory for Carbon-Based Functional Materials & Devices, Soochow University, 215123 Suzhou, China
First published on 23rd September 2022
Abstract
To optimally apply antibiotics and antimicrobials, smart wound dressing conferring controlled drug release and preventing adhesions of biological objects is advantageous. Poly(N-isopropylacrylamide) (PNIPAAm), a conventional thermo-responsive polymer, and poly(2-methacryloyloxyethyl phosphorylcholine) (PMPC), a typical antifouling polymer, have therefore potential to be fabricated as copolymers to achieve dual functions of thermo-responsiveness and antifouling. Herein, a hydrogel made of PNIPAM–co-PMPC was designed and loaded with octenidine, a widely applied antimicrobial agent for wound treatment, to achieve both antifouling and triggered drug release. The thermo-switch of the fabricated hydrogel allowed 25-fold more octenidine release at 37 °C (infected wound temperature) than at 30 °C (normal skin temperature) after 120 minutes, which led to at least a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lg reduction of the viable bacteria at 37 °C on artificially infected wounds. Furthermore, we pioneeringly assessed the antifouling property of the material in PBS buffer using single molecule/cell/bacterial force spectroscopy, and revealed that the fabricated hydrogel displayed distinctive antifouling properties against proteins, mammalian cells, and bacteria. This work demonstrated a promising design of a hydrogel applicable for preventing and treating wound infections. The concept of dual-functional materials can be envisaged for other clinical applications related to the prevention of biofilm-associated infections, such as urinary catheters, stents, and dental implants.
lg reduction of the viable bacteria at 37 °C on artificially infected wounds. Furthermore, we pioneeringly assessed the antifouling property of the material in PBS buffer using single molecule/cell/bacterial force spectroscopy, and revealed that the fabricated hydrogel displayed distinctive antifouling properties against proteins, mammalian cells, and bacteria. This work demonstrated a promising design of a hydrogel applicable for preventing and treating wound infections. The concept of dual-functional materials can be envisaged for other clinical applications related to the prevention of biofilm-associated infections, such as urinary catheters, stents, and dental implants.
Chronic wounds have emerged as one of the most challenging problems for clinical treatment due to bacterial infections and biofilm formation.1–4 Consequently, they can generate painful suffering in the patients and cause a substantial burden to the global health care.5,6 Treatment with antibiotics is one of the often-used means for chronic wounds. However, the prevailing antibiotic treatment of infections usually accompanies the overuse and misuse of antibiotics, which are the leading causes of the emergence of antimicrobial resistance.7 Therefore, it is urgent to find alternatives to avoid the overuse of antimicrobials.8,9 In this regard, it is highly desired to develop wound dressings exhibiting prominent antifouling and antibacterial properties.10,11 Since the local temperature difference between infected wounds and normal skin can be as high as 5 °C,12 the thermo-difference can be exploited to design a thermo-responsive wound-dressing material to achieve a triggered release of the loaded antimicrobial agents upon a thermal stimulus.
Poly(N-isopropylacrylamide) (PNIPAAm), widely applied as a thermo-responsive polymer,13 demonstrates a sharp and reversible phase transition between hydrophobicity and hydrophilicity at a lower critical solution temperature (LCST) of 32 ± 1–2 °C.14 The temperature switch across LCST of PNIPAAm can thereby yield a sol–gel transformation due to the existence of hydrophilic amide (–CONH–) groups and hydrophobic isopropyl (–CH(CH3)2) side chains.15,16 PNIPAAm has hence been intensively utilized as a biomaterial for drug delivery, scaffolds, and wound dressing.17,18 It is known that the desired wound dressing should be hardly adhesive to wound tissues to avoid bleeding and damage to the new epithelial tissues.19 Poly(2-methacryloyloxyethyl phosphorylcholine) (PMPC) is a biocompatible zwitterionic polymer that displays unique hydration and electrolytic behaviors, and biocompatibility.20 The exceptional biocompatibility of PMPC originates from its phospholipid polar group on the side chain, which inhibits non-specific interactions with serum proteins, bacteria, mammalian cells, and platelets.21,22 Moreover, PMPC, after hydrogel formation, displays high gas and solute permeability.23,24 Therefore, a combination of the advantages of PMPC and PNIPAAm can probably yield a dual-functional material with thermo-response and antifouling. Hydrogels are widely applied in soft tissue engineering of skin, blood vessels, muscle, and fat,25 as hydrogels display three-dimensional (3D) networks containing physically or chemically cross-linked bonds of hydrophilic polymers. Therefore, hydrogels can accelerate wound healing by absorbing wound exudates and permitting oxygen diffusion.26,27 Moreover, hydrogels can maintain a high moisture level of the wound bed as their highly hydrated 3D polymeric network contains several-fold more water than the polymeric network itself.28 Additionally, hydrogels can be adjusted into varying sizes and shapes,29,30 and have the capacity to be loaded with cells, antimicrobial agents, growth factors, and biomacromolecules.31 Therefore hydrogel-derived wound dressings are highly desired to treat skin wounds.32 Hence a hydrogel is an ideal form to take advantage of PNIPAAm and PMPC.
In this work, a thermo-responsive and antifouling wound dressing was designed (Fig. 1), and its antibacterial activity and antifouling property were subsequently investigated against different bio-organisms. PNIPAAm was exploited to form a random copolymer with PMPC, an antifouling material, to develop a poly(NIPAAm-co-MPC) hydrogel. The copolymer properties were well-tuned to achieve controlled drug release through the anticipated recognition of the thermo-difference between infected wounds (close to a physiological temperature of 37 °C33) and normal skin (in the range of 30–35 °C34). Hence, octenidine (OCT, an efficacious antiseptic35,36) was utilized as a model drug to evaluate the controlled drug release. Furthermore, we pioneeringly utilized a BioAFM to evaluate the antifouling property of the fabricated hydrogel by employing single molecule force spectroscopy (SMFS), single cell force spectroscopy (SCFS), and single bacterial force spectroscopy (SBFS) under aqueous conditions to obtain a better understanding of the underlying interactions. The conventional antifouling analysis was also performed for the empirical results under macroscopic conditions. Moreover, the antibacterial efficacy was evaluated under in vitro conditions and further analyzed using artificially generated wounds on ex vivo human skin. Thereby, the designed poly(NIPAAm-co-MPC) hydrogel can be a powerful tool in combating wound infections, through the demonstrated dual functions of antifouling property and triggered release. The concept of the proposed dual-functional hydrogel also manifests magnificent clinical potential for broad applications to improve biomedical devices.
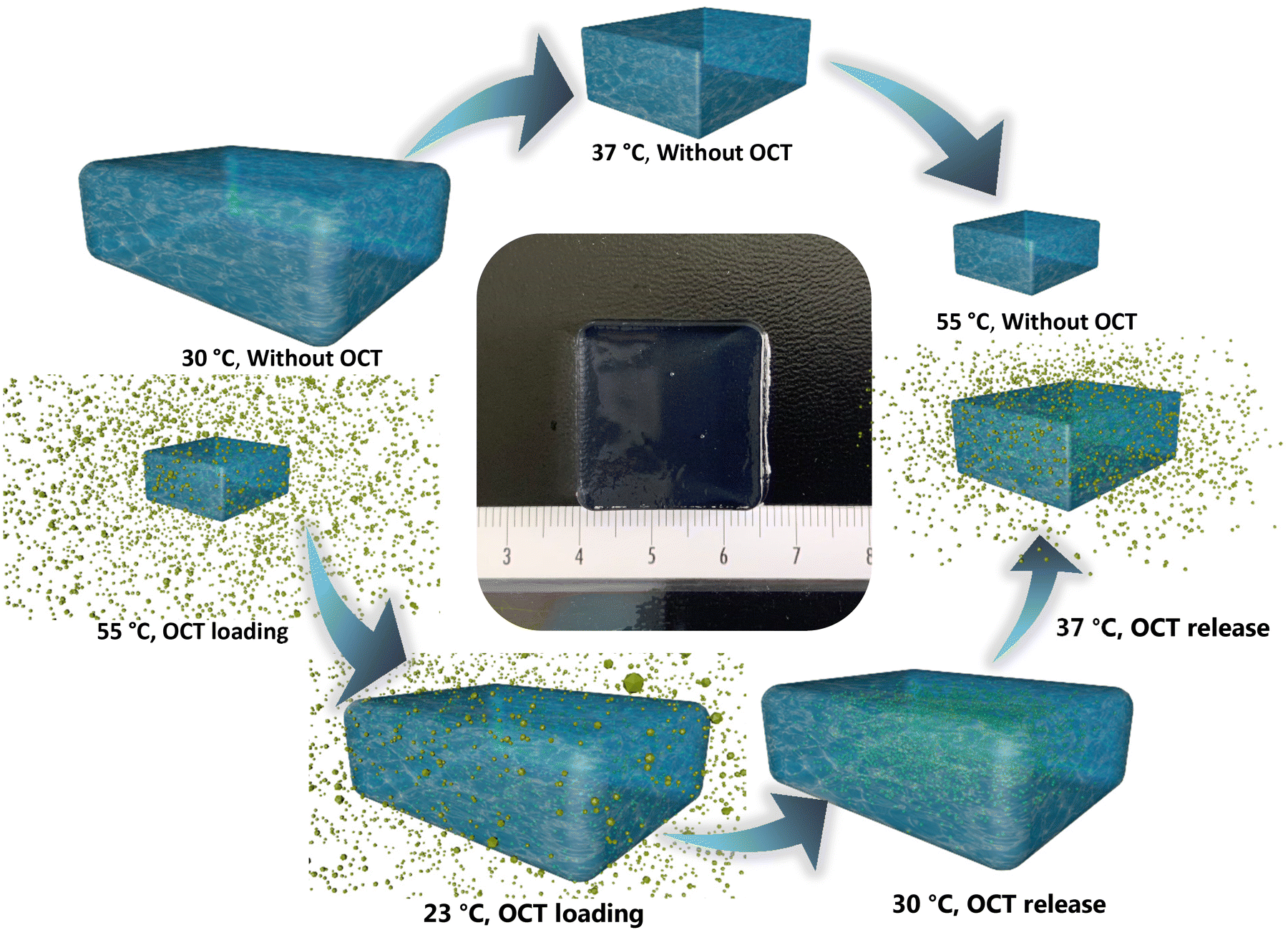 | ||
| Fig. 1 The schematic hypothesis of thermo-responsive hydrogel for a controlled OCT release. Once the temperature rises above the lower critical solution temperature (LCST), the hydrogel would begin to contract due to the sol–gel transformation. The volume of the designed hydrogel of well-tuned LCST (above 30 °C but lower than 37 °C) would decrease when the temperature rises from 30 °C to 37 °C and 55 °C. In contrast, the hydrogel would expand its volume when the temperature reduces from 55 °C to room temperature (23 °C). We can therefore use this property to load OCT inside the hydrogel. We can subsequently retain OCT inside the hydrogel at 30 °C, but trigger the OCT release at 37 °C. Therefore, a controlled antimicrobial dosage can be achieved through temperature moderation. The center is hydrogel 5:5 of the well-tuned LCST. | ||
Results and discussion
To obtain antifouling and antibacterial materials, the poly(NIPAAm-co-MPC) hydrogel was proposed to display a low critical solution temperature (LCST) within 30 °C–37 °C, which could permit the proposed hydrogel to shrink (above LCST) and expand reversibly (below LCST). Thereby, the anticipated drug could be loaded into the hydrogel matrix when the hydrogel immersed into a drug solution experienced a temperature decrease from 55 °C to room temperature (23 °C). Furthermore, the hydrogels loaded with the drug can hypothetically restrain the drug release at 30 °C (the normal skin temperature) but trigger the drug release at 37 °C (the temperature of an infected wound) (Fig. 1). Additionally, thanks to the contribution of the MPC components, the proposed hydrogel could manifest anti-adhesive property. Hence poly(NIPAAm-co-MPC) hydrogels of 4 different monomer ratios (MPC to NIPAAm: 4:6, 5:5, 6:4, and 7:3) described in Materials and Methods were designed and evaluated towards selection for the optimized dual-function.
Controlled drug release and characterization
The cumulative release of OCT from hydrogels 4:6, 5:5, 6:4, and 7:3 was assessed as reported4 by UV-vis spectroscopy in PBS buffer at 30 °C and 37 °C, respectively, to simulate the temperatures of normal skin and infected wounds (Fig. 2a and Tables S1 and S2†). Hydrogel 4:6 allowed the OCT release at 30 °C of 4.84 ± 0.34 and 5.07 ± 0.44 mg L−1, respectively, for a release time of 120 and 600 minutes, and at 37 °C of 5.12 ± 0.42 and 5.24 ± 0.77 mg L−1. Hydrogel 5:5 exhibited a release at 30 °C of 0.41 ± 0.13 and 1.52 ± 0.31 mg L−1, respectively, for a release time of 120 and 600 minutes. Interestingly, hydrogel 5:5 permitted a much higher OCT release at 37 °C of 10.77 ± 0.88 and 21.96 ± 2.88 mg L−1, for a release time of 120 and 600 minutes. Hydrogel 6:4 and 7:3 showed a similar release profile to that of hydrogel 5:5, suggesting that thermo-difference can regulate OCT release from hydrogels 5:5, 6:4, and 7:3, namely, when the temperature reached 37 °C, the thermo-switch was automatically turned on, whereas when the temperature remained less than 30 °C, the switch stayed persistently off. Since a similar effect of the thermo-switch was observed for hydrogels 5:5, 6:4, and 7:3, hydrogel 5:5 was selected for further analysis due to its high content of the PMPC component, the antifouling contributor. DSC was then performed to identify the thermal switch button, the LCST, of hydrogel 5:5, which can thermally regulate the OCT release. The phase transition (sol–gel transformation) temperatures (LCSTs) were respectively identified as 34.3–35.3 °C and 34.2–35.2 °C for hydrogel 5:5 with and without the loading of OCT, according to the reported method4 (Fig. 2b). The LCST of the fabricated hydrogel 5:5 located between 30 °C and 37 °C indicates that OCT release can be restrained at 30 °C and propelled at 37 °C. Additionally, the phase transition was impacted by LCST, leading to a large change in the average relative size (aligned to the mold size, 0.3 mL) of hydrogel 5:5 at different temperatures. Briefly, the average relative sizes at room temperature and 30 °C were respectively 8.3 and 7.9 but decreased to 5.4 at 37 °C and 2.4 at 55 °C (Fig. 2c). Furthermore, thermogravimetric analysis (TGA) showed a smaller (0.7%) weight loss in the measured temperature range (21–80 °C) for hydrogel 5:5 with and without an OCT loading implying that these two thermo-sensitive hydrogels were also thermally stable (Fig. S1†).
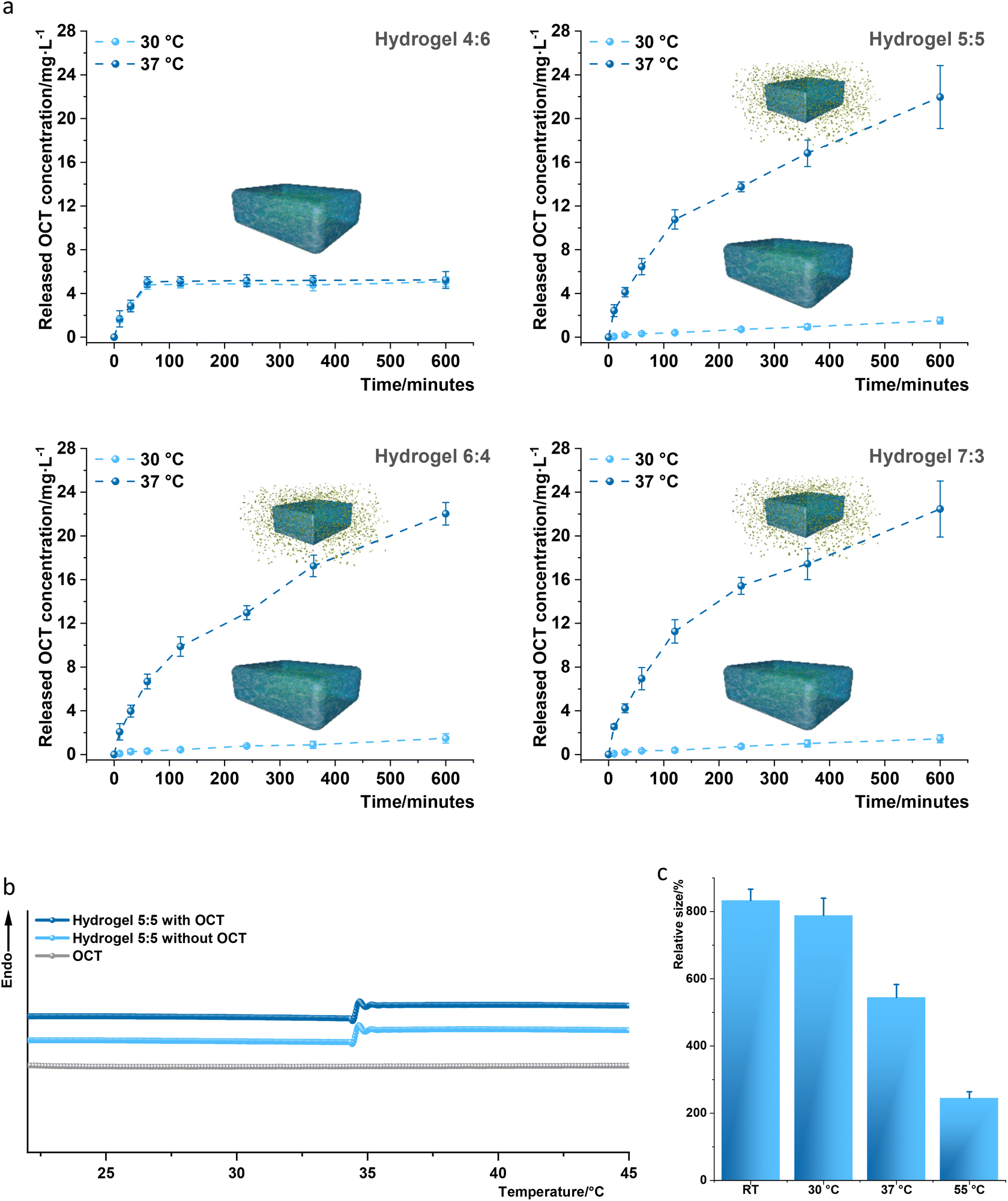 | ||
| Fig. 2 Characterization of the fabricated hydrogels. (a) Analysis of thermo-regulated OCT release from OCT-loaded hydrogels 4:6, 5:5, 6:4, and 7:3 samples through a UV-visible spectrophotometer as reported.4 Cumulative OCT in vitro release from hydrogels 4:6, 5:5, 6:4, and 7:3 samples loaded with OCT was assessed at 30 °C (light blue dashed lines) and 37 °C (dark blue dashed lines). The total OCT amount loaded in hydrogels 4:6, 5:5, 6:4, and 7:3 was determined to be 7.8 ± 0.7 mg L−1, 28.9 ± 1.1 mg L−1, 27.7 ± 1.9 mg L−1 and 28.4 ± 0.9 mg L−1, respectively, after a thorough OCT release from the corresponding hydrogel in PBS at 50 °C for 3 days. The in vitro release analysis was conducted three times with three replicates, and every replicate was measured three times. One set of measurements has been displayed together with standard deviations (error bars) derived from 9 data. (b) The sol–gel transformation of the designed hydrogel was herein analyzed by differential scanning calorimetry (DSC) in PBS buffer (pH 7.4) to characterize the LSCT in a consecutive heating process. The identified LCSTs were respectively 34.3–35.3 °C and 34.2–35.2 °C for hydrogel 5:5 with and without loading of OCT. The LCST was measured five times for every sample, and one data set was displayed. (c) The relative size of hydrogel 5:5 after completely swelling. The sizes of hydrogel 5:5 were respectively measured three times by a graduated cylinder at room temperature (RT) after swelling for 5 days in deionized water and at 30 °C, 37 °C, and 55 °C after incubation for 4 hours. All the measured sizes were aligned to the original mold size (0.3 mL) to fabricate the hydrogel. | ||
The surface mechanical properties of hydrogel 5:5 with and without OCT were further analyzed with AFM.3 Young's moduli of hydrogel 5:5 with and without OCT were respectively 179.3 ± 46.8 and 180.2 ± 45.4 kPa, within the range for most hydrogels37 (Table S3†). However, the surface adhesion forces of both samples were respectively 118.1 ± 35.2 and 112.2 ± 31.6 pN, which was significantly lower than that for the mPEG surface 475.7 ± 23.5 pN (Table S3†). Hydrogel 5:5 with and without OCT therefore displayed a distinctive repulsive property.
Antifouling analysis
Single molecule force spectroscopy (SMFS) was used to investigate the antifouling properties of the fabricated hydrogel 5:5 with and without OCT towards biomolecules, i.e., proteins. To this end, human serum albumin (HSA), the most abundant protein in human blood plasma, was coated on an AFM cantilever based on the previously reported method38 (Fig. 3a). Single cell force spectroscopy (SCFS) and single bacteria force spectroscopy (SBFS) were performed using normal human dermal fibroblasts (nHDFs, Fig. 3b) and the bacteria Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus, and S. epidermidis (Fig. 3c–f). Before measurements, all the human and bacterial cells were immobilized on AFM cantilevers in PBS (pH 7.4). The modified AFM cantilevers, by either HSA or cells, were applied to the hydrogel sample surface, including control samples (mPEG as the positive control for weak adhesion and PDMS 40:1 as the negative control for strong adhesion), and the adhesion force was measured. The SMFS, SCFS, and SBFS analysis showed that the fabricated hydrogel 5:5 with and without OCT allowed lower adhesion force, similar to the antifouling mPEG, for all tested bio-organisms ranging from 237 pN to 537 pN (Fig. 3). In contrast, PDMS 40:1 exhibited the adhesion force for all tested bio-organisms of 1187–5165 pN (Fig. 3). The antifouling analysis of hydrogel 5:5 with/without OCT was aligned to mPEG, a conventional antifouling material,39 whereas a significant difference was noted compared to PDMS 40:1, a fouling material.3 Therefore, the antifouling property of hydrogel 5:5 with/without OCT was confirmed at the levels of single molecule and single cell/bacterium.
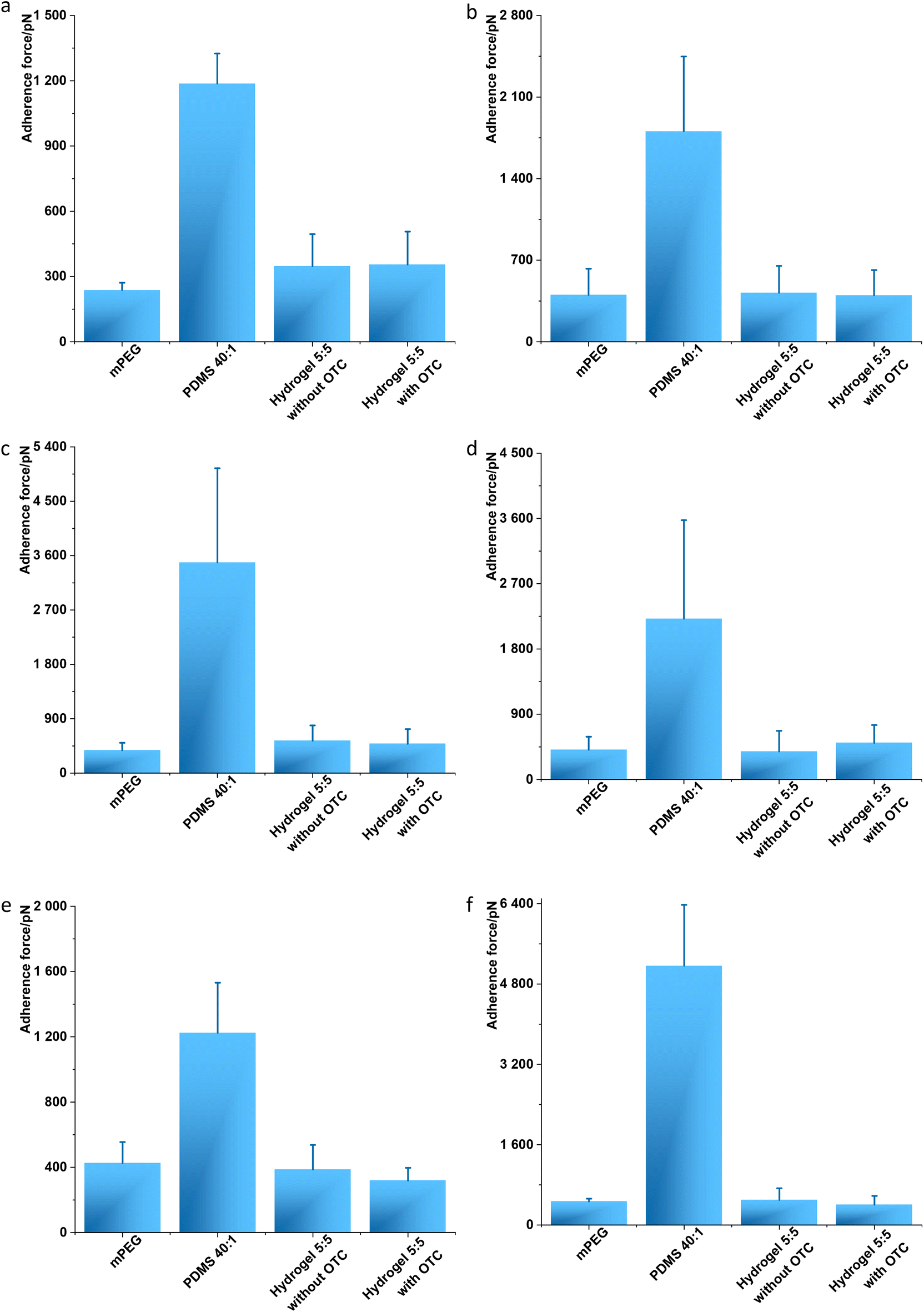 | ||
| Fig. 3 Antifouling analysis through BioAFM. (a) Single molecular adhesion force was measured between human serum albumin (HSA) and material surfaces. N = 10. (b) Single cell adhesion force was measured based on normal human dermal fibroblasts (nHDFs). N ≥ 10. All the surfaces were additionally assessed by single bacterial adhesion force through E. coli (c), P. aeruginosa (d), S. aureus (e), and S. epidermidis (f). N ≥ 10 for every measurement. The adhesion force in every graph measured on PDMS 40:1 was significantly different from the other samples, and there was no significant difference between the other samples. Student's t-test (p < 0.05) was performed for statistical analysis. | ||
To investigate the antifouling property of hydrogel 5:5 at the macroscopic level, a conventional adhesion assay was performed with E. coli and P. aeruginosa (Fig. 4a and b) and S. aureus and S. epidermidis (Fig. 4c and d). Gold substrates coated with mPEG were applied as a positive control, and PDMS 40:1 was exploited as a negative control.3 The adherent E. coli cells on mPEG and hydrogel 5:5 with and without OCT loading displayed no significant difference and in amounts (cells per mm2) of 71 ± 26, 53 ± 22, and 40 ± 12, respectively (Fig. 4a). However, PDMS 40:1 allowed more than 100 times adherent bacterial cells (8641 ± 11 cells per mm2) compared to the other samples (Fig. 4a). A similar trend regarding the adhered amounts (cells per mm2) of P. aeruginosa on sample surfaces was noticed: 115 ± 24, 113 ± 38, 85 ± 11, and 3134 ± 668, respectively, on hydrogel 5:5 with and without OCT, mPEG, and PDMS 40:1 (Fig. 4b). This phenomenon was also observed when analyzing adhered Gram-positive bacteria on sample surfaces. The amounts (cells per mm2) of adherent S. aureus on hydrogel 5:5 with and without OCT, mPEG, and PDMS 40:1 were respectively 49 ± 12, 43 ± 22, 64 ± 23, and 3830 ± 599 (Fig. 4c). Analogously, those of S. epidermidis on hydrogel 5:5 with and without OCT, mPEG, and PDMS 40:1 were respectively 49 ± 21, 48 ± 15, 66 ± 25, and 8121 ± 842 (Fig. 4c). The amounts showed no significant difference in every assessed bacterial strain on hydrogel 5:5 with/without OCT compared to mPEG (the positive control for antifouling evaluation) but displayed a significant difference when aligned to PDMS 40:1 (the negative control regarding the antifouling analysis). Therefore, a clear antifouling property was demonstrated for hydrogel 5:5 with and without an OCT loading (Fig. 4).
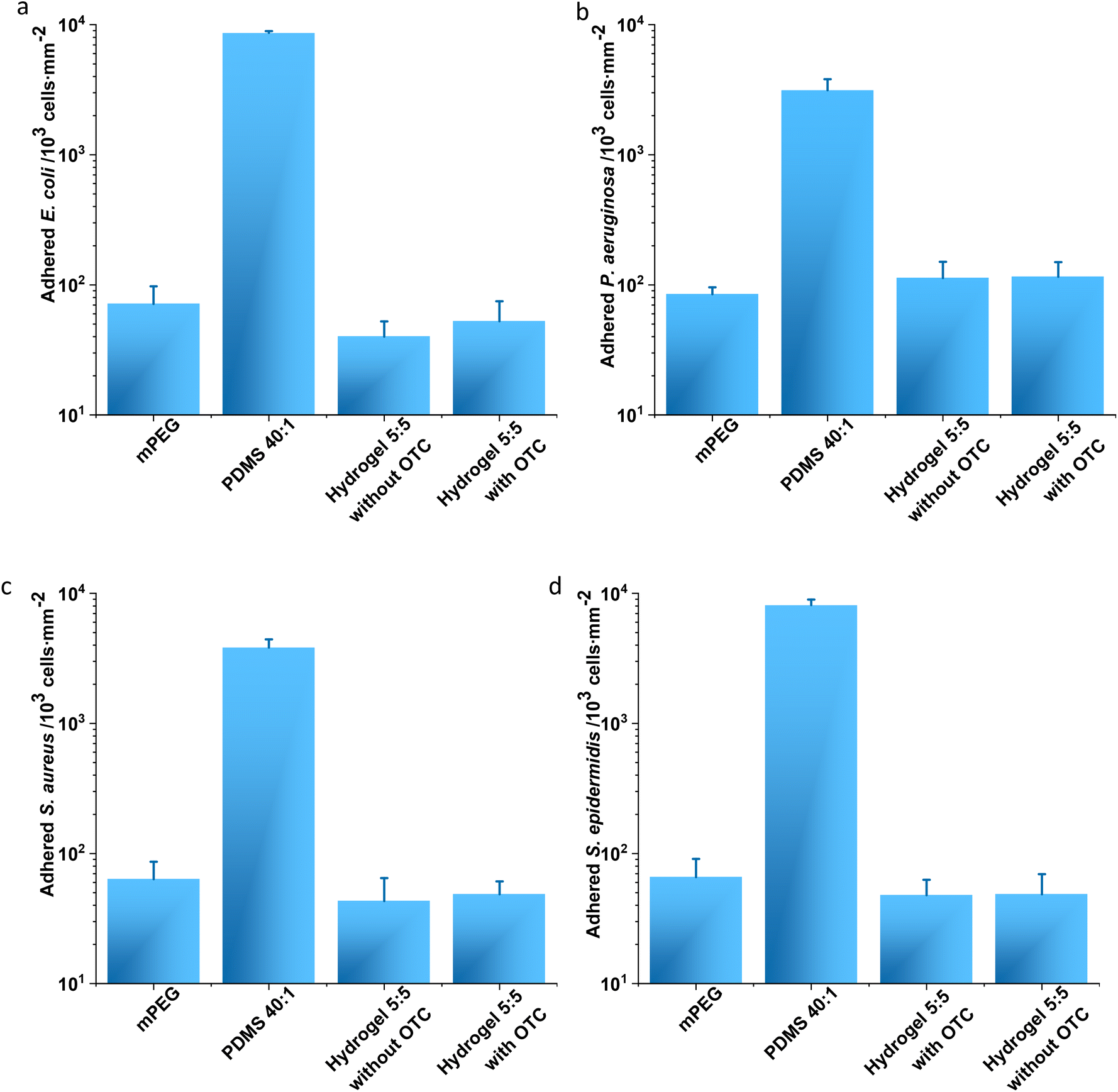 | ||
| Fig. 4 Analysis of bacterial adhesion. E. coli DSMZ 22312 (a) and P. aeruginosa ATCC 43390 (b), two typical Gram-negative bacterial pathogens, and S. aureus ATCC 6538 (c) and S. epidermidis 49461 (d), two typical Gram-positive bacterial pathogens, were tested. No significant difference was noticed in every adhesion analysis between hydrogel 5:5 and mPEG samples, but a significant difference was noticed when comparing mPEG and hydrogel 5:5 without/with OCT to PDMS 40:1. Every analysis was performed with three replicates. Student's t-test (p < 0.05) was performed for statistical analysis. | ||
The endowed antifouling property, confirmed by both SMFS, SCFS, and SBFS and conventional bacterial adhesion assays, most likely originated from the component of PMPC in the copolymerized hydrogel. The monomer of PMPC is MPC, a zwitterionic molecule mimetic of cell membranes, comprising a hydrophilic polar headgroup of phospholipids.40 Thus the zwitterionic phosphorylcholine surface of hydrogel 5:5 can strongly bind water and consequently generate a hydration layer preventing the attachment of biological organisms.41
Antibacterial efficacy and cytotoxicity
The antibacterial efficacy of hydrogel 5:5 against two typical bacterial pathogens on human wounds,42P. aeruginosa and S. aureus, were evaluated at 30 °C and 37 °C. Both pathogens were loaded on hydrogel surfaces and subsequently incubated for 2 hours. The contaminated hydrogels were placed on top of the BHI agar for 2 seconds. The bacterial growth on BHI agar for 12 hours displayed distinctive differences (Fig. 5a): hydrogel 5:5 incubated at 37 °C with bacteria did not lead to any colony formation on BHI agar, whereas clear bacterial growth was observed for hydrogel 5:5 incubated at 30 °C. For a quantitative analysis, hydrogel 5:5 with OCT was incubated with bacterial suspensions (P. aeruginosa and S. aureus) at both 30 °C and 37 °C for 2 hours, and viable bacterial cells were subsequently counted on agar plates. For P. aeruginosa, the number of the viable cells was 1.05 ± 0.09 × 107 colony forming units (CFU) per mL for the sample incubated at 30 °C, but the number fell to 1007 ± 106 CFU mL−1 for that at 37 °C. Similar results were observed for S. aureus: 1.10 ± 0.10 × 106 CFU mL−1 for incubation at 30 °C but 221 ± 34 CFU mL−1 for incubation at 37 °C. Namely, at least a 3lg reduction in the viable cells was observed after incubation with hydrogel 5:5 containing OCT at 37 °C compared to that at 30 °C (Fig. 5b). Given the accumulative OCT release from hydrogel 5:5 within 2 hours at 0.41 ± 0.13 mg L−1 for 30 °C and 10.77 ± 0.88 mg L−1 for 37 °C (Fig. 2a and Tables S1 and S2†), it is not surprising to obtain a dramatic reduction in CFUs at 37 °C, compared with that at 30 °C. Hence, hydrogel 5:5 loaded with OCT demonstrated an effectively triggered antibacterial activity against P. aeruginosa and S. aureus.
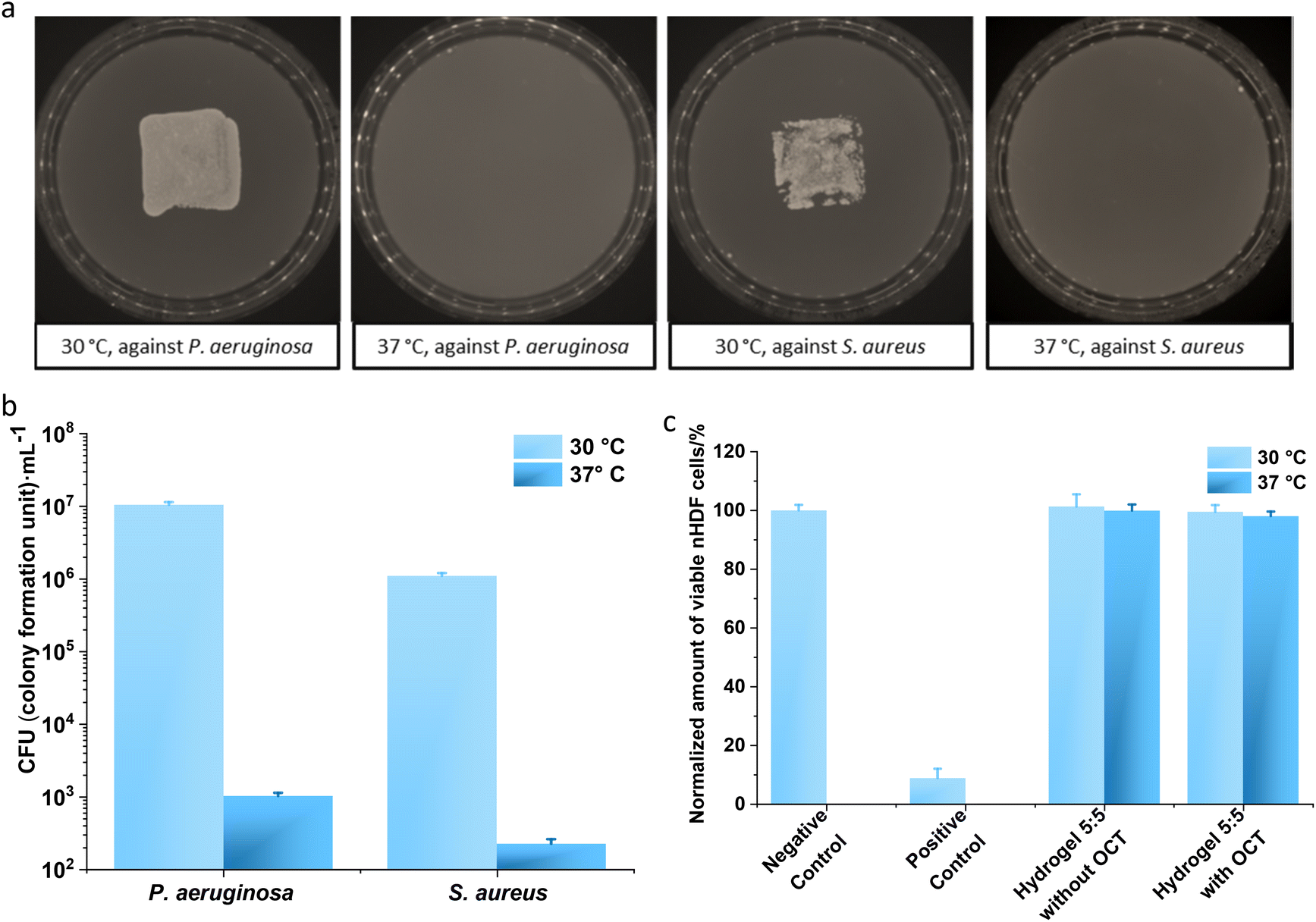 | ||
| Fig. 5 Thermo-regulated antibacterial efficacy and cytotoxicity. To evaluate the regulated antibacterial efficacy by the designed thermo-switch, hydrogel 5:5 loaded with OCT respectively interacted with P. aeruginosa and S. aureus at 30 °C and 37 °C for 2 hours (a). The infected hydrogel surfaces were brought to touch the BHI agar for 2 seconds, and the agar plate was then incubated at 37 °C for 12 hours. Bacterial growth was observed on agar plates touched with hydrogel 5:5 loaded with OCT at 30 °C. In contrast, no bacterial growth was observed on agar plates touched with hydrogel 5:5 loaded with OCT at 37 °C. The evaluation of antibacterial efficacy was assessed six times, and one set of representative images was displayed. Moreover, a quantitative evaluation of the antibacterial activity of hydrogel 5:5 loaded with OCT was performed as well at 30 °C and 37 °C against P. aeruginosa and S. aureus pathogens (b). A significant difference was noticed at 30 °C and 37 °C for each bacterial pathogen. The analysis was performed three times and every time with three replicates. (c) The cytotoxicity assay of hydrogel 5:5 without and with OCT loading was performed with the sample extracts towards normal human dermal fibroblasts (nHDFs). The hydrogel samples were placed in DMEM containing 1% penicillin/streptomycin (PS) at 30 °C and 37 °C for 24 h to generate extracts. No significant difference was noticed between all the extracts of hydrogel samples and the negative control except between hydrogel extracts and the positive control. Error bars denote the standard deviations of 9 measurements. Student's t-test (p < 0.05) was employed for statistical analysis. | ||
The cytotoxicity of hydrogel 5:5 with and without OCT loading was evaluated using normal human dermal fibroblasts (nHDFs) (Fig. 4d). Extract solutions were collected after immersing hydrogel samples into DMEM (Dulbecco's Modified Eagle's Medium) with 1% penicillin/streptomycin (PS) for 24 h at room temperature and 37 °C, respectively. The viability of nHDFs was consequently measured by applying an MTS [(3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] assay, as reported.4 The cytotoxicity of hydrogels was evaluated and compared with the negative control. The cytotoxic cut-off was defined at 70% viability of cells in the negative control. The extracts of samples displayed over 95% cell viability (Fig. 5c). Therefore, cytotoxicity was not detected in the extracts from hydrogel 5:5 with and without OCT loading after incubation of 24 hours at 30 °C and 37 °C.
To simulate a clinical infection, wounds were artificially generated on ex vivo human skin and subsequently intentionally infected by P. aeruginosa and S. aureus for 24 hours. Then the infected wounds were treated at 30 °C and 37 °C with hydrogel 5:5 with and without an OCT loading, respectively, alongside infected wounds without hydrogel treatment (negative control). All the wound samples except wound surfaces were kept in DMEM containing 10% fetal calf serum (FCS) during the assays (Fig. 6a and b). When treated with hydrogel 5:5 with OCT at 37 °C, the number of viable bacteria on the P. aeruginosa infected wounds was dramatically reduced to 4122 ± 308 CFU mL−1, whereas at 30 °C, the number was 8.36 ± 0.90 × 106 CFU mL−1. The reduction of viable cells was also not observed for treatment with hydrogel 5:5 without OCT, leading to 9.24 ± 0.74 × 106 CFU mL−1 and 8.39 ± 0.81 × 106 CFU mL−1 at 30 °C and 37 °C, respectively. A similar phenomenon was also observed for S. aureus. The treatment with hydrogel 5:5 with OCT at 37 °C led to 2055 ± 802 CFU mL−1 but at 30 °C to 7.01 ± 0.60 × 106 CFU mL−1. Through the treatment with hydrogel 5:5 without OCT at 30 °C and 37 °C, the viable S. aureus on the artificial wounds were 7.00 ± 0.35 × 106 CFU mL−1 and 8.28 ± 0.89 × 106 respectively. Briefly, a 3lg reduction in viable bacterial cells on the infected ex vivo skin wounds was observed for both the applied Gram-negative and -positive pathogens after treatment with OCT-loaded hydrogel 5:5 at 37 °C. Histological staining was applied to evaluate the antibacterial efficacy of hydrogel 5:5 loaded with OCT (Fig. 6c) on artificially infected wounds. S. aureus suspension was loaded onto the artificial wounds and grown for three days. Subsequently, hydrogel 5:5 loaded with OCT was applied to the infected wounds for two hours at 30 °C and 37 °C. All the wound samples except wound surfaces were kept in DMEM containing 10% FCS during the assays. Histological staining was then performed to evaluate the antibacterial efficacy qualitatively. Bacterial clusters were observed on infected wounds treated by PBS buffer (negative control) and treated by OCT-loaded hydrogel 5:5 at 30 °C. However, no bacterial clusters were observed on the infected wounds treated with OCT-loaded hydrogel 5:5 at 37 °C (Fig. 6c). Hydrogel 5:5 loaded with OCT hence displayed a thermo-regulated antibacterial efficacy.
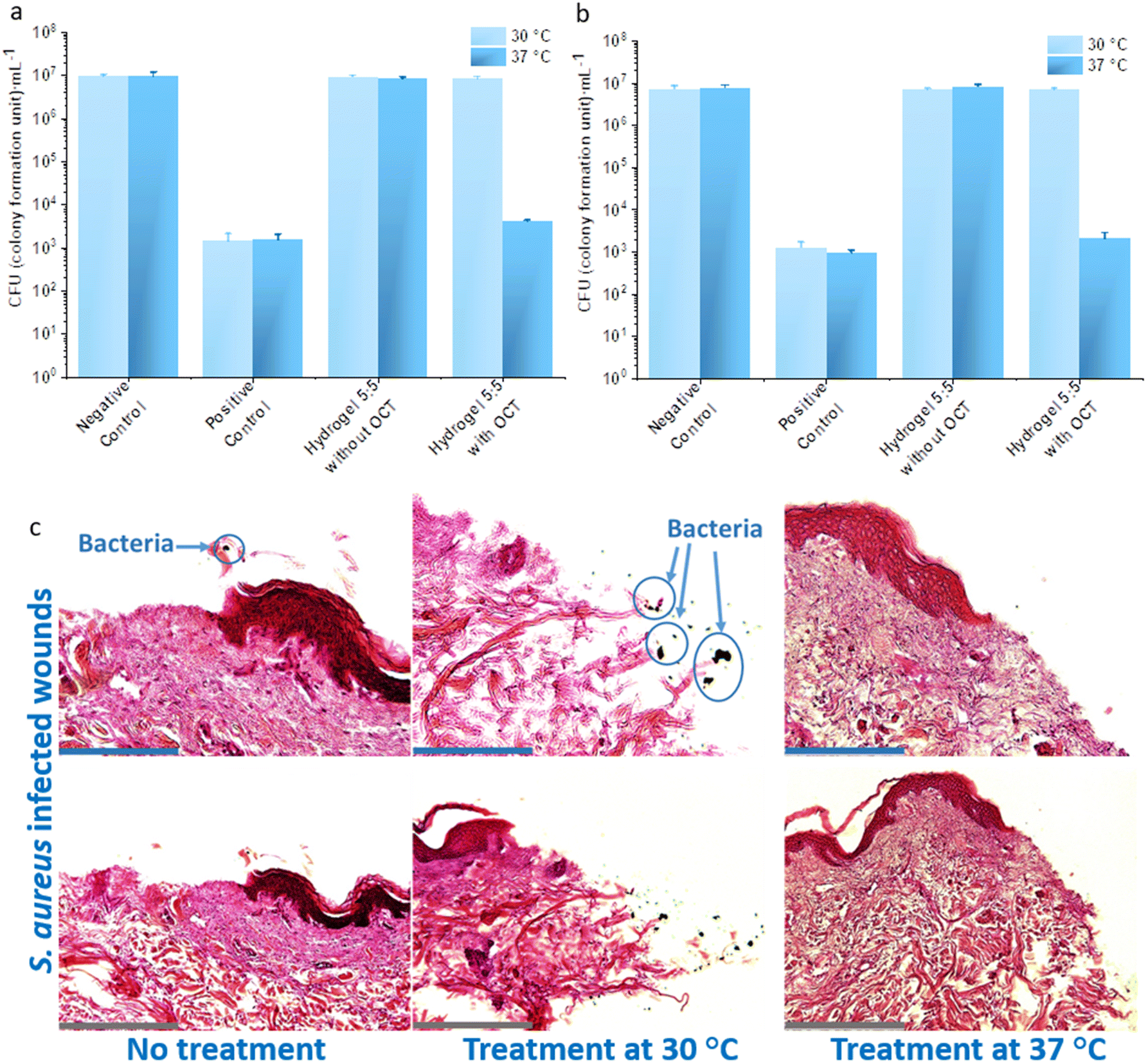 | ||
| Fig. 6 Bacterial killing analysis on artificially infected wounds. Antibacterial efficacy of hydrogel 5:5 with an OCT loading on artificially infected wounds was evaluated at 30 °C and 37 °C respectively towards P. aeruginosa (a) and S. aureus (b) pathogens. P. aeruginosa and S. aureus were loaded on artificial wounds for 24 hours to generate bacterial infections. Hydrogel 5:5 samples without and with OCT loading were placed on the infected wound for 2 hours at 30 °C and 37 °C. All the wound samples except the wound surface were kept in DMEM containing 10% FCS during the assays. Artificial wounds without hydrogel treatment were analyzed as a negative control, but those treated with 20 μL 500 mg L−1 OCT were positive control. The initial loaded amount (20 μL) of P. aeruginosa and S. aureus on artificial wounds had a respective concentration of 8.55 ± 0.90 × 105 CFU mL−1 and 9.80 ± 0.62 × 105 CFU mL−1. There was no significant difference noticed at 30 °C between hydrogel 5:5 samples and the negative control except with the positive control. A significant difference was noticed at 37 °C between hydrogel 5:5 with OCT and the rest samples. The analysis was performed with three replicates, and every replicate was analyzed three times to yield standard deviations. Student's t-test (p < 0.05) was performed for statistical analysis. (c) Histological staining analysis of thermo-responsive treatment efficacy. Mechanical wounds were artificially generated and then infected by S. aureus for three days. The infected wounds were subsequently treated with hydrogel 5:5 loaded with OCT at 30 °C and 37 °C. All the wound samples except wound surface remained in DMEM containing 10% FCS during the assays. Scale bar: 145 μm for blue ones and 290 μm for grey ones. | ||
Conclusions
In this proof of concept study, a hydrogel with a well-tuned LCST was successfully designed and fabricated to achieve a triggered release of the loaded drug regulated by the thermo-difference between normal skin and infected wounds. A prominent antibacterial efficacy was noticed at 37 °C against Gram-negative and -positive pathogens. Furthermore, the well-designed hydrogel displayed excellent antifouling property at the scales of single molecule and single cell/bacterium towards human serum albumin, normal human dermal fibroblasts, Gram-negative and -positive bacterial cells. Additionally, bacterial adhesion assays were performed to empirically demonstrate the fabricated hydrogel's antifouling property. Moreover, hydrogel 5:5 loaded with OCT led to a nearly 3lg reduction in viable bacterial cells at 37 °C on the artificially infected ex vivo skin compared with those treated at 30 °C. This work herein demonstrated an antifouling hydrogel, which simultaneously displayed a triggered antibacterial function through the non-invasive self-regulated mechanism to avoid the overuse of antimicrobial agents. Furthermore, we proved the potential to use a BioAFM to evaluate the antifouling properties of the material. The developed dual-functional hydrogel manifests a prominent application potential in treating infections on chronic wounds. Moreover, the concept proposed in this work can be further exploited to develop other advanced medical devices (e.g., urinary catheters, stents, and dental implants) to prevent bacterial infections.
Methods
Materials
All chemicals and reagents were purchased with analytical purity from Sigma-Aldrich (Buchs, Switzerland) and applied as received unless otherwise noted. Octenidine dihydrochloride (OCT) was purchased from TCI Chemicals, Japan. Phosphate-buffered saline (PBS) at pH 7.4 was prepared by dissolving 8 g L−1 NaCl, 0.2 g L−1 KH2PO4, and 1.44 g L−1 Na2PO4 in distilled water. Bacterial growth medium (LB broth) was prepared as follows: 10 g L−1 tryptone, 5 g L−1 yeast extract, and 5 g L−1 NaCl in distilled water. Ex vivo human skin samples were obtained from Cantonal Hospital St Gallen, with anonymous consent given by the donors and exempted from ethical approval.Fabrication of PDMS 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 and drug-loaded hydrogels
:1 and drug-loaded hydrogels
PDMS 40:1 samples were prepared as reported.3 Briefly, a Sylgard184 silicone elastomer kit (Dow Corning Inc., USA) was used to mix with a curing agent in a weight ratio of 40:1. After vigorous mixing, the viscous mixtures were vacuumed for half an hour, and a 15 mL mixture was then poured into plastic Petri dishes (Greiner Bio-One GmbH, Austria, diameter of 9.4 cm) to achieve PDMS samples with roughly 1.8 mm thickness. Afterward, all the Petri dishes were horizontally placed inside a vacuum drying oven (SalvisLab Vacucenter, Switzerland) and vacuumed for 30 min, and then kept at 60 °C for 24 hours. All samples, unless otherwise mentioned, were immersed into 70% ethanol for 20 min and dried in a vacuum before further analysis.
The hydrogels were prepared through a photo-induced copolymerization of 2-methacryloyloxyethyl phosphorylcholine (MPC) and N-isopropylacrylamide (NIPAAm) with molar ratios of 4:6, 5:5, 6:4, and 7:3 together with ethylene dimethacrylate (EDMA) as the cross-linker. A solution containing 0.01% Irgacure D-2959 as a photo-initiator was prepared with a total monomer (MPC and NIPAAm) concentration of 2.86 mol L−1 and a cross-linker concentration of 0.1 mol L−1. The solution was then sonicated for 5 min and afterward was pipetted into a multiwell Teflon mold (L × W × H: 1 cm × 1 cm × 0.3 cm) with coverslips. Subsequently, a UV curing of 10 minutes was applied to obtain gels which were transferred into PBS (pH 7.4) for swelling of 120 hours to reach equilibrium.
To prepare octenidine (OCT)-loaded hydrogels, every piece of hydrogel was placed in a 50 mL TPP tube with 45 mL of 500 mg L−1 OCT solution in a 55 °C water bath for 4 hours to maximize the contraction due to the PNIPAAm component. After the incubation, the hydrogel samples were cooled to room temperature to allow swelling, encapsulating the OCT solution. The hydrogel samples were immersed into the OCT solution after cooling and stored in the fridge (4 °C) for 2 days before further analysis.
In vitro drug release kinetics
The hydrogels were rinsed with PBS buffer (pH 7.4), placed inside a TPP tube containing 45 mL of PBS buffer, and incubated at 30 °C or 37 °C with shaking at 30 rpm. At specified time intervals within 600 minutes, 50 μL of the suspension was collected and replaced with fresh PBS of the same volume. The total OCT amount loaded inside the hydrogel was measured after a release of OCT at 50 °C for 3 days. A UV-visible spectrophotometer (PowerWave HT, BioTek Instruments Inc., USA) was used to quantify the OCT release at the wavelength of 282 nm,37 using a standard reference curve.37DSC measurement
Hydrogel 5:5 without and with OCT loading, after removing excess liquid on the sample surface, was analyzed for the LCST (around 6 mg sample sealed inside aluminum crucibles) by differential scanning calorimetry (DSC, Netzsch Polyma 214, Netzsch, Selb, Germany). The heating rate set for the measurement was 10 °C min−1, and the data were recorded from 22 °C to 45 °C at a flow rate of nitrogen gas of 40 mL min−1.
Relative size measurement
A piece of hydrogel, well-swollen at the respective measuring temperature, was delicately cleaned by Kimtech science precision wipers (KIMBERLY CLARK, Surrey, England) to only remove the water on the hydrogel. Subsequently, the hydrogel was placed in a glass graduate (10 mL) containing 5 mL of deionized (DI) water at the respective measuring temperature. Then excessive water was removed and collected in a graduated cylinder (5 mL) to precisely reach the scale of 5 mL on the glass graduate. The relative size of the evaluated hydrogel was determined by aligning to the original mold size (0.3 mL) for hydrogel fabrication.Thermogravimetric analysis (TGA)
TGA was performed towards 4.0–5.0 mg samples by thermogravimetry (TG-209 F1 Iris, Netzsch, Germany) in N2 and air at a heating rate of 1 °C min−1 from 21 °C to 80 °C.Bacterial culture
Staphylococcus aureus ATCC 6538 and Pseudomonas aeruginosa ATCC 43390 were applied for the antibacterial evaluation of the OCT-loaded hydrogels. S. aureus ATCC 6538, P. aeruginosa ATCC 43390, E. coli DSMZ 22312, and S. epidermidis ATCC 49461 were exploited to evaluate their adhesion to abiotic surfaces.Bacterial colonies from an agar plate were incubated in 10 mL of LB in 10 mL TPP tubes at 160 rpm and 37 °C overnight. 100 μL of overnight cultures were subsequently transferred into 10 mL of fresh LB and cultivated for around 2 h until the bacterial exponential growth phase. The bacterial cultures of S. aureus and P. aeruginosa were then diluted with fresh sterile PBS according to previously reported43 to roughly 106 colony forming units (CFUs) per mL.
BioAFM measurements
Surface viscoelasticity analysis of hydrogel 5:5 with and without OCT was carried out using a Flex BioAFM (Nanosurf, Switzerland) in 0.2 mm-filtered PBS (pH 7.4). The spring constant of cantilevers was determined according to its resonance frequency in PBS (pH 7.4). Furthermore, the cantilever was programmed to contact the surface at a speed of 1 μm s−1 until reaching a force of 10 nN. The contact time between the AFM probe and the surface was set as 5 s. Force curve measurements were conducted through a gold-coated cantilever PointProbe® Plus non-contact/soft tapping mode (PPP-NCST-Au, Nanosensor, Switzerland). Young's modulus and surface adhesion force towards the cantilever were then analyzed as reported.3
Single-molecule force spectroscopy (video 1†) was performed after adapting the reported method.38 AFM probes and reference substrates were initially functionalized. Gold-coated AFM cantilevers (PPP-NCST-Au, Nanosensor, Switzerland) and gold substrates (50 nm gold-coated silicon chips, 10 × 10 mm, 10-AU8119, Micro to Nano) were rinsed twice in chloroform for 10 min, then exposed to air plasma (Plasma Cleaner PD-32G, Harrick Plasma, USA) for 10 min, and finally rinsed three times with chloroform, each for 10 min. After the cleaning process, the AFM probes and gold substrates were both immediately immersed into a 1 mM solution containing SH-PEG-NH2 (Mw = 3400 g mol−1, PBL-8063, Creative PEGWorks, Winston Salem, USA):mPEG (Mw = 2000 g mol−1, PLS-605, Creative PEGWorks, USA) of 1:200 (mol:mol) in chloroform for 12–18 hours at room temperature. After the PEG-coating, all the surfaces were cleaned sequentially three times and every time for 3 minutes by the following solvents: chloroform, absolute ethanol, and deionized water. PEG-coated surfaces were immediately activated by immersing the coated surfaces into a 2-(n-morpholino)ethanesulfonic acid buffer at pH 6 containing 10 g L−1n-hydroxysulfosuccinimide and 40 g L−1 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide for 30 minutes at room temperature. The activated PEG-coated gold surfaces (AFM probes and reference substrates) were rinsed three times with PBS buffer at pH 7.4 and subsequently incubated with 0.2 g L−1 human serum albumin in PBS buffer (pH 7.4) for two hours at room temperature. The albumin functionalized gold AFM probes and the reference substrates were rinsed three times with PBS buffer (pH 7.4) containing 0.1% TWEEN20 and stored at 4 °C in PBS buffer (pH 7.4) for up to 72 h. The immobilization of albumin was confirmed by XPS (data not shown).
Force spectroscopy experiments between the human serum albumin functionalized AFM probes and mPEG coated substrates, PDMS 5:1, PDMS 40:1, and hydrogel 5:5 with and without OCT loading were performed using Flex BioAFM similarly to surface viscoelasticity analysis.
Single cell (videos 2 and 3†) and bacterial adhesion (videos 4 and 5†) force with surfaces were analyzed following the previously reported method.44–46 Flex Bio-AFM and a digital pressure controller (Cytosurge, Switzerland) were used, combined with an AxioObserver Z1 inverted microscope (Carl Zeiss, Germany) to ensure precise control throughout the analysis. Pyramidal hollow cantilevers (Nanopipette, Cytosurge, Switzerland) with a nominal spring constant of 2 N m−1 and a 300 nm aperture at their distal end and tipless hollow cantilevers (Micropipette, Cytosurge AG, Switzerland) with a nominal spring constant of 2 N m−1 and an 8 μm aperture at their distal end were used for the measurement of bacterial and mammalian cell adhesion force towards different surfaces. All the cantilevers were treated with air plasma for 30 s. AFM probes were subsequently stored in a desiccator containing 1 mL of Sigmacote siliconizing reagent for 12 hours and finally dried at 100 °C for 1 hour. Moreover, the spring constant of all the cantilevers was determined according to its resonance frequency in PBS buffer (pH 7.4). The microchannel inside the cantilever was filled with filtered deionized water after applying pressure by the digital pressure controller. The spring constant and sensitivity of the cantilever were recalibrated every time after cell remobilization on cantilevers.
Single cell/bacterial force spectroscopy was performed at room temperature in PBS (pH 7.4) with normal human dermal fibroblasts (nHDFs, PromoCell, C-12352), P. aeruginosa ATCC 43390, E. coli DSMZ 22312, S. aureus ATCC 6538, and S. epidermidis 49461, which were dispersed onto a confined region of a glass dish. Cantilevers were brought into contact with the selected cell (micropipette) or bacterium (nanopipette) with a force-setpoint of 10 nN. Upon contact, a negative pressure of 800 mbar was immediately applied to immobilize a cell/bacterium reversibly on the cantilever aperture. The cantilever immobilized with a cell/bacterium was immediately moved to a new glass dish containing a sample and kept in PBS buffer (pH 7.4). The cell/bacterium-probe approached the surface to be tested at a speed of 1 μm s−1 until reaching a force of 10 nN. To ensure a reproducible interaction between cell/bacterium and surface, this force was maintained for 5 s. The cell/bacterium-probe was then retracted at a piezo velocity of 1 μm s−1, and the emerging forces were simultaneously recorded. Fifteen measurements were performed for every surface sample, and at least five different bacterial cells of every strain were applied for every tested surface sample. The adhesion forces were analyzed as reported44 with the SPIP software (Image Metrology A/S, Denmark).
Bacterial adhesion
The bacterial adhesion on abiotic surfaces was performed as reported.3 Gold substrates coated by mPEG, PDMS 40:1, and hydrogel 5:5 with and without loading of OCT were assessed in 12 well plates (TPP, Switzerland). All samples were sterilized with 2 mL 70% ethanol for 20 minutes and then immersed in 2 mL of PBS for 2 hours. 1.5 mL bacterial suspension was added to every well and subsequently incubated at 37 °C for 2 hours without shaking. The bacterial suspension was then removed, and all samples were gently washed twice with fresh PBS (pH 7.4). All samples were subsequently treated with bacterial fixation solution (4% paraformaldehyde and 2.5% glutaraldehyde) for 30 minutes. Later the fixation solution was replaced by surface passivation solution (0.1% bovine serum albumin) for incubation of 5 minutes. The adherent bacteria on sample surfaces were analyzed using an inverted microscope Eclipse Ti2E (Nikon, Japan) with a 40× objective lens.
Antibacterial assay
Antibacterial activity of hydrogels (with and without OCT loading) was first assessed qualitatively. Briefly, 50 μL of bacterial suspension was pipetted on the surface of the hydrogels, and incubated at 30 °C or 37 °C for 2 hours. The hydrogels were then brought to the surface of the BHI agar and held for 2 seconds. The agar plate was afterward incubated at 37 °C for 12 hours. Scan® 300 (interscience, France) was applied to take photos of the agar plates. Every measurement was performed independently three times and every time with 3 replicates.Antibacterial efficacy was quantitatively analyzed by incubating hydrogels (with and without OCT loading) in 45 mL of bacterial suspension (106 CFU ml−1) at 30 °C and 37 °C for 2 hours. 100 μL of bacterial suspension after a serial dilution was plated on a BHI agar plate with three replicates through an automatic plater (easySpiral, interscience, France). Bacterial colonies were subsequently quantified through a colony counter (Scan® 300, interscience, France) after incubation at 37 °C for 12 hours.
Antibacterial efficacy was further analyzed as reported47 using ex vivo human skin samples to mimic the clinical situation. Human skins obtained from Cantonal Hospital St Gallen, Switzerland, were pretreated by removing the fat part and subsequently burned by a stamp (∅ 10 mm, 200 °C) for 5 seconds and then punched into a disc (∅ 20 mm) using a puncher tool. These artificial burn wound samples were washed sequentially once with octenidine dihydrochloride (octenisept® farblos/incolore, Schülke & Mayr GmbH, Norderstedt, Germany) for 5 minutes and three times with Dulbecco's PBS (10 minutes every time). The cleaned wounds were placed inside TPP 6-well plates with 600 μL DMEM (Dulbecco's Modified Eagle Medium) containing 10% fetal calf serum (FCS), and 20 μL of bacterial suspension was then loaded on every wound. After incubating the bacteria-loaded wounds for 24 hours, hydrogels (with and without OCT loading) were placed on the wound surfaces for 2 hours at both 30 °C and 37 °C. The wound samples treated with hydrogels were then transferred to 15 mL TPP tubes and ultrasonicated in 5 mL of PBS for 1 minute. After a serial dilution of the obtained suspensions, 100 μL of the suspension was plated on a BHI agar plate with three replicates through an automatic plater (easySpiral, interscience, France). Bacterial colonies were counted through a colony counter (Scan® 300, interscience, France) after incubation at 37 °C for 12 hours.
Cytotoxicity assay
Cytotoxicity of the hydrogel samples was studied by applying normal human dermal fibroblasts (nHDFs, PromoCell, C-12352) as reported.4 Extracts were prepared with hydrogel 5:5 with and without OCT and negative control (wells without any sample) in DMEM (Dulbecco's Modified Eagle's Medium) containing 1% penicillin/streptomycin (PS) with an extraction ratio of 0.1 g mL−1 (sample mass per extraction volume). The extraction process was subsequently performed in an incubator (30 °C or 37 °C, 100% humidity and 5% CO2) for 24 hours. nHDFs were pre-seeded at a concentration of 10000 cells per well (TPP, Switzerland) in 100 μL of DMEM with 10% FCS for 24 hours and then incubated with 100 μL of 95% extracts diluted with FCS for another 24 hours. The cell viability of the negative control was set as 100%, and the positive control was the cell viability after an incubation with 1% Triton X-100 in DMEM containing 5% FCS. Cell viability was quantified by the absorbance at 490 nm through MTS [(3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium)] assay.48
Histological analysis
Mechanical wounds of 5 mm diameter were generated on ex vivo human skin of 13 mm diameter by punching. The samples were then disinfected with octenisept (Schülke & Mayr GmbH, Norderstedt, Germany) for 5 minutes. The samples were subsequently rinsed in fresh PBS three times and every time for 10 minutes. The rinsed samples were relocated into 12-well plates (TPP Techno Plastic Products AG, Trasadingen, Switzerland), and every well containing 400 μL of DMEM with 10% FCS. 20 μL S. aureus (OD600 0.08) suspension was then added to every wound. Subsequently, the samples were incubated at 37 °C containing 5% CO2 for three days. Afterward, the infected samples were treated with hydrogels at both 30 °C and 37 °C. Two hours later, the wound samples were rinsed with PBS buffer and immersed into fresh 4% formalin at 4 °C for 24 hours. The bacteria on the mechanical wounds were furthermore analyzed through histological staining.Histological staining49,50 was then applied to observe bacteria on these wounds. The wound samples were placed into another fresh 12-well plate and cleaned with deionized water for 45 minutes. Subsequently, the samples were dehydrated by incubating various ethanol of increasing concentrations, rinsed in xylene, and placed in paraffin blocks. 5 μm thick sections were afterward vertically sliced, subsequently deparaffinization in xylene and rehydration in various ethanol of decreasing concentrations. These samples were stained with hematoxylin (HistoLab, Askim, Sweden) and eosin, and rinsed with distilled water in-between.
Statistics
Statistical difference between every sample was assessed by applying unpaired and two-tailed Student's t-test to compare two groups.Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.Author contributions
Fei Pan: conceptualization, methodology, software, validation, formal analysis, investigation, visualization, supervision, project administration, writing – original draft, writing – review and editing. Sixuan Zhang: methodology, investigation, validation. Stefanie Altenried: methodology, validation, investigation. Flavia Zuber: methodology, validation, investigation. Qian Chen: conceptualization, methodology, formal analysis, validation, writing – review and editing. Qun Ren: conceptualization, methodology, validation, investigation, supervision, project administration, writing – review and editingConflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors would like to thank Jörg Grünert and Zhihao Li from the Cantonal Hospital St Gallen, Switzerland, for ex vivo human skin samples, Maximilian Mittelviefhaus for his valuable suggestions on BioAFM settings, Nico Strohmeyer and Daniel Müller for their suggestions on BioAFM, Orane Guillaume-Gentil for her kind review of BioAFM methods and data analysis, and Ruben Pianegonda and Irene Rodriguez for their support in lab work. FP thanks Berna Neidhart for the valuable scientific discussions.References
- J. Hurlow, K. Couch, K. Laforet, L. Bolton, D. Metcalf and P. Bowler, Clinical biofilms: a challenging frontier in wound care, Adv. Wound Care, 2015, 4(5), 295–301 CrossRef.
- S. T. Elkins-Williams, W. A. Marston and C. S. Hultman, Management of the chronic burn wound, Clin. Plast. Surg., 2017, 44(3), 679–687 CrossRef.
- F. Pan, S. Altenried, M. Liu, D. Hegemann, E. Bülbül, J. Moeller, W. W. Schmahl, K. Maniura-Weber and Q. Ren, A nanolayer coating on polydimethylsiloxane surfaces enables a mechanistic study of bacterial adhesion influenced by material surface physicochemistry, Mater. Horiz., 2020, 7(1), 93–103 RSC.
- F. Pan, A. Amarjargal, S. Altenried, M. Liu, F. Zuber, Z. Zeng, R. M. Rossi, K. Maniura-Weber and Q. Ren, Bioresponsive hybrid nanofibers enable controlled drug delivery through glass transition switching at physiological temperature, ACS Appl. Bio Mater., 2021, 4(5), 4271–4279 CrossRef.
- F. Werdin, M. Tenenhaus and H.-O. Rennekampff, Chronic wound care, Lancet, 2008, 372(9653), 1860–1862 CrossRef.
- World Health Organization, Global action plan on antimicrobial resistance, World Health Organization, Geneva, 2015 Search PubMed.
- World Health Organization, Global antimicrobial resistance and use surveillance system (GLASS) report: 2021, ( 2021).
- J. Zhu, Q. Jin, H. Zhao, W. Zhu, Z. Liu and Q. Chen, Reactive Oxygen Species Scavenging Sutures for Enhanced Wound Sealing and Repair, Small Struct., 2021, 2(7), 2100002 CrossRef.
- J. Hu, T. Wei, H. Zhao, M. Chen, Y. Tan, Z. Ji, Q. Jin, J. Shen, Y. Han, N. Yang, L. Chen, Z. Xiao, H. Zhang, Z. Liu and Q. Chen, Mechanically active adhesive and immune regulative dressings for wound closure, Matter, 2021, 4(9), 2985–3000 CrossRef.
- H. Zhao, J. Huang, Y. Li, X. Lv, H. Zhou, H. Wang, Y. Xu, C. Wang, J. Wang and Z. Liu, ROS-scavenging hydrogel to promote healing of bacteria infected diabetic wounds, Biomaterials, 2020, 258, 120286 CrossRef CAS.
- N. Zhao and W. Yuan, Highly adhesive and dual-crosslinking hydrogel via one-pot self-initiated polymerization for efficient antibacterial, antifouling and full-thickness wound healing, Composites, Part B, 2022, 230, 109525 CrossRef CAS.
- A. Chanmugam, D. Langemo, K. Thomason, J. Haan, E. A. Altenburger, A. Tippett, L. Henderson and T. A. Zortman, Relative temperature maximum in wound infection and inflammation as compared with a control subject using long-wave infrared thermography, Adv. Skin Wound Care, 2017, 30(9), 406–414 CrossRef PubMed.
- X. Xu, Y. Liu, W. Fu, M. Yao, Z. Ding, J. Xuan, D. Li, S. Wang, Y. Xia and M. Cao, Poly (N-isopropylacrylamide)-based thermoresponsive composite hydrogels for biomedical applications, Polymers, 2020, 12(3), 580 CrossRef CAS.
- D. L. Huber, R. P. Manginell, M. A. Samara, B.-I. Kim and B. C. Bunker, Programmed adsorption and release of proteins in a microfluidic device, Science, 2003, 301(5631), 352–354 CrossRef CAS PubMed.
- B. Wang, Q. Xu, Z. Ye, H. Liu, Q. Lin, K. Nan, Y. Li, Y. Wang, L. Qi and H. Chen, Copolymer brushes with temperature-triggered, reversibly switchable bactericidal and antifouling properties for biomaterial surfaces, ACS Appl. Mater. Interfaces, 2016, 8(40), 27207–27217 CrossRef CAS.
- M. Oak, R. Mandke and J. Singh, Smart polymers for peptide and protein parenteral sustained delivery, Drug Discovery Today: Technol., 2012, 9(2), e131–e140 CrossRef CAS.
- Y. Zhao, C. Shi, X. Yang, B. Shen, Y. Sun, Y. Chen, X. Xu, H. Sun, K. Yu and B. Yang, pH-and temperature-sensitive hydrogel nanoparticles with dual photoluminescence for bioprobes, ACS Nano, 2016, 10(6), 5856–5863 CrossRef CAS PubMed.
- S. Ziane, S. Schlaubitz, S. Miraux, A. Patwa, C. Lalande, I. Bilem, S. Lepreux, B. Rousseau, J.-F. Le Meins and L. Latxague, A thermosensitive low molecular weight hydrogel as scaffold for tissue engineering, Eur. Cells Mater., 2012, 23, 147–160 CrossRef CAS.
- Y. Tang, X. Cai, Y. Xiang, Y. Zhao, X. Zhang and Z. Wu, Cross-linked antifouling polysaccharide hydrogel coating as extracellular matrix mimics for wound healing, J. Mater. Chem. B, 2017, 5(16), 2989–2999 RSC.
- T. Shimizu, T. Goda, N. Minoura, M. Takai and K. Ishihara, Super-hydrophilic silicone hydrogels with interpenetrating poly (2-methacryloyloxyethyl phosphorylcholine) networks, Biomaterials, 2010, 31(12), 3274–3280 CrossRef CAS PubMed.
- K. Ishihara, K. Fukumoto, Y. Iwasaki and N. Nakabayashi, Modification of polysulfone with phospholipid polymer for improvement of the blood compatibility. Part 2. Protein adsorption and platelet adhesion, Biomaterials, 1999, 20(17), 1553–1559 CrossRef CAS.
- T. Konno and K. Ishihara, Temporal and spatially controllable cell encapsulation using a water-soluble phospholipid polymer with phenylboronic acid moiety, Biomaterials, 2007, 28(10), 1770–1777 CrossRef CAS.
- S. H. Ye, J. Watanabe, M. Takai, Y. Iwasaki and K. Ishihara, High functional hollow fiber membrane modified with phospholipid polymers for a liver assist bioreactor, Biomaterials, 2006, 27(9), 1955–1962 CrossRef CAS.
- T. Goda, T. Konno, M. Takai, T. Moro and K. Ishihara, Biomimetic phosphorylcholine polymer grafting from polydimethylsiloxane surface using photo-induced polymerization, Biomaterials, 2006, 27(30), 5151–5160 CrossRef CAS.
- G. D. Nicodemus and S. J. Bryant, Cell encapsulation in biodegradable hydrogels for tissue engineering applications, Tissue Eng., Part B, 2008, 14(2), 149–165 CrossRef CAS PubMed.
- J. S. Boateng, K. H. Matthews, H. N. Stevens and G. M. Eccleston, Wound healing dressings and drug delivery systems: a review, J. Pharm. Sci., 2008, 97(8), 2892–2923 CrossRef CAS.
- A. Kumar and M. Jaiswal, Design and in vitro investigation of nanocomposite hydrogel based in situ spray dressing for chronic wounds and synthesis of silver nanoparticles using green chemistry, J. Appl. Polym. Sci., 2016, 133(14), 43260 CrossRef.
- S. Tavakoli and A. S. Klar, Advanced Hydrogels as Wound Dressings, Biomolecules, 2020, 10(8), 1169 CrossRef CAS PubMed.
- S. Van Vlierberghe, P. Dubruel and E. Schacht, Biopolymer-based hydrogels as scaffolds for tissue engineering applications: a review, Biomacromolecules, 2011, 12(5), 1387–1408 CrossRef CAS.
- C. Bilici, V. Can, U. Nöchel, M. Behl, A. Lendlein and O. Okay, Melt-processable shape-memory hydrogels with self-healing ability of high mechanical strength, Macromolecules, 2016, 49(19), 7442–7449 CrossRef CAS.
- P. Gupta, K. Vermani and S. Garg, Hydrogels: from controlled release to pH-responsive drug delivery, Drug Discovery Today, 2002, 7(10), 569–579 CrossRef CAS.
- J. Koehler, F. P. Brandl and A. M. Goepferich, Hydrogel wound dressings for bioactive treatment of acute and chronic wounds, Eur. Polym. J., 2018, 100, 1–11 CrossRef CAS.
- L. Du, L. Tong, Y. Jin, J. Jia, Y. Liu, C. Su, S. Yu and X. Li, A multifunctional in situ–forming hydrogel for wound healing, Wound Repair Regen., 2012, 20(6), 904–910 CrossRef.
- P. Webb, Temperatures of skin, subcutaneous tissue, muscle and core in resting men in cold, comfortable and hot conditions, Eur. J. Appl. Physiol. Occup. Physiol., 1992, 64(5), 471–476 CrossRef CAS PubMed.
- T. Koburger, N.-O. Hübner, M. Braun, J. Siebert and A. Kramer, Standardized comparison of antiseptic efficacy of triclosan, PVP–iodine, octenidine dihydrochloride, polyhexanide and chlorhexidine digluconate, J. Antimicrob. Chemother., 2010, 65(8), 1712–1719 CrossRef CAS.
- N.-O. Hübner, J. Siebert and A. Kramer, Octenidine dihydrochloride, a modern antiseptic for skin, mucous membranes and wounds, Skin Pharmacol. Physiol., 2010, 23(5), 244–258 CrossRef.
- L. Zhu, J. Qiu and E. Sakai, A high modulus hydrogel obtained from hydrogen bond reconstruction and its application in vibration damper, RSC Adv., 2017, 7(69), 43755–43763 RSC.
- E. Martines, J. Zhong, J. Muzard, A. Lee, B. Akhremitchev, D. Suter and G. Lee, Single-molecule force spectroscopy of the Aplysia cell adhesion molecule reveals two homophilic bonds, Biophys. J., 2012, 103(4), 649–657 CrossRef CAS.
- R. Singh and M. K. Purkait, Evaluation of mPEG effect on the hydrophilicity and antifouling nature of the PVDF-co-HFP flat sheet polymeric membranes for humic acid removal, J. Water Process Eng., 2016, 14, 9–18 CrossRef.
- C. Kojima, R. Katayama, T. L. Nguyen, Y. Oki, A. Tsujimoto, S.-i. Yusa, K. Shiraishi and A. Matsumoto, Different antifouling effects of random and block copolymers comprising 2-methacryloyloxyethyl phosphorylcholine and dodecyl methacrylate, Eur. Polym. J., 2020, 136, 109932 CrossRef.
- A. V. Fuchs, S. Ritz, S. Pütz, V. Mailänder, K. Landfester and U. Ziener, Bioinspired phosphorylcholine containing polymer films with silver nanoparticles combining antifouling and antibacterial properties, Biomater. Sci., 2013, 1(5), 470–477 RSC.
- D. Church, S. Elsayed, O. Reid, B. Winston and R. Lindsay, Burn wound infections, Clin. Microbiol. Rev., 2006, 19(2), 403–434 CrossRef PubMed.
- F. Pan, S. Altenried, F. Zuber, R. S. Wagner, Y.-H. Su, M. Rottmar, K. Maniura-Weber and Q. Ren, Photo-activated titanium surface confers time dependent bactericidal activity towards Gram positive and negative bacteria, Colloids Surf., B, 2021, 206, 111940 CrossRef PubMed.
- M. Mittelviefhaus, D. B. Müller, T. Zambelli and J. A. Vorholt, A modular atomic force microscopy approach reveals a large range of hydrophobic adhesion forces among bacterial members of the leaf microbiota, ISME J., 2019, 13(7), 1878–1882 CrossRef.
- E. Potthoff, D. Ossola, T. Zambelli and J. A. Vorholt, Bacterial adhesion force quantification by fluidic force microscopy, Nanoscale, 2015, 7(9), 4070–4079 RSC.
- F. Pan, M. Liu, S. Altenried, M. Lei, J. Yang, H. Straub, W. W. Schmahl, K. Maniura-Weber, O. Guillaume-Gentil and Q. Ren, Uncoupling bacterial attachment on and detachment from polydimethylsiloxane surfaces through empirical and simulation studies, J. Colloid Interface Sci., 2022, 622, 419–430 CrossRef PubMed.
- F. Pan, G. Giovannini, S. Zhang, S. Altenried, F. Zuber, Q. Chen, L. F. Boesel and Q. Ren, pH-responsive silica nanoparticles for triggered treatment of skin wound infections, Acta Biomater., 2022, 145, 172–184 CrossRef.
- G. Jin, M. P. Prabhakaran, D. Kai, S. K. Annamalai, K. D. Arunachalam and S. Ramakrishna, Tissue engineered plant extracts as nanofibrous wound dressing, Biomaterials, 2013, 34(3), 724–734 CrossRef.
- M. A. Andersson, L. B. Madsen, A. Schmidtchen and M. Puthia, Development of an Experimental Ex Vivo Wound Model to Evaluate Antimicrobial Efficacy of Topical Formulations, Int. J. Mol. Sci., 2021, 22(9), 5045 CrossRef PubMed.
- S. C. Becerra, D. C. Roy, C. J. Sanchez, R. J. Christy and D. M. Burmeister, An optimized staining technique for the detection of Gram positive and Gram negative bacteria within tissue, BMC Res. Notes, 2016, 9, 216–216 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm01244h |
| ‡ Present address: Department of Chemistry, University of Basel, Mattenstrasse 24a, BPR 1096, 4058 Basel, Switzerland, Email: Fei.Pan@unibas.ch |
| This journal is © The Royal Society of Chemistry 2022 |