
Multidomain peptide hydrogel adjuvants elicit strong bias towards humoral immunity†
Brett H.
Pogostin
 a,
Marina H.
Yu
a,
Alon R.
Azares
b,
Erin M.
Euliano
a,
Cheuk Sun Edwin
Lai
a,
Gabriel
Saenz
c,
Samuel X.
Wu
a,
Adam C.
Farsheed
a,
Sarah M.
Melhorn
a,
Tyler P.
Graf
a,
Darren G.
Woodside
b,
Jeffrey D.
Hartgerink
ac and
Kevin J.
McHugh
*a
a,
Marina H.
Yu
a,
Alon R.
Azares
b,
Erin M.
Euliano
a,
Cheuk Sun Edwin
Lai
a,
Gabriel
Saenz
c,
Samuel X.
Wu
a,
Adam C.
Farsheed
a,
Sarah M.
Melhorn
a,
Tyler P.
Graf
a,
Darren G.
Woodside
b,
Jeffrey D.
Hartgerink
ac and
Kevin J.
McHugh
*a
aDepartment of Bioengineering, Rice University, Houston, TX 77005, USA. E-mail: kevin.mchugh@rice.edu; Tel: +1 (713) 348-8089
bMolecular Cardiology Research Laboratories, Texas Heart Institute, Houston, TX 77030, USA
cDepartment of Chemistry, Rice University, Houston, TX 77005, USA
First published on 8th September 2022
Abstract
Adjuvants play a critical role in enhancing vaccine efficacy; however, there is a need to develop new immunomodulatory compounds to address emerging pathogens and to expand the use of immunotherapies. Multidomain peptides (MDPs) are materials composed of canonical amino acids that form injectable supramolecular hydrogels under physiological salt and pH conditions. MDP hydrogels are rapidly infiltrated by immune cells in vivo and have previously been shown to influence cytokine production. Therefore, we hypothesized that these immunostimulatory characteristics would allow MDPs to function as vaccine adjuvants. Herein, we demonstrate that loading antigen into MDP hydrogels does not interfere with their rheological properties and that positively charged MDPs can act as antigen depots, as demonstrated by their ability to release ovalbumin (OVA) over a period of 7–9 days in vivo. Mice vaccinated with MDP-adjuvanted antigen generated significantly higher IgG titers than mice treated with the unadjuvanted control, suggesting that these hydrogels potentiate humoral immunity. Interestingly, MDP hydrogels did not elicit a robust cellular immune response, as indicated by the lower production of IgG2c and smaller populations of tetramer-positive CD8+ T splenocytes compared to mice vaccinated alum-adjuvanted OVA. Together, the data suggest that MDP hydrogel adjuvants strongly bias the immune response towards humoral immunity while evoking a very limited cellular immune response. As a result, MDPs may have the potential to serve as adjuvants for applications that benefit exclusively from humoral immunity.
Introduction
Vaccines are a critical component of the global healthcare system and are estimated to have prevented over 37 million deaths between 2000 and 2019.1 Due to their resounding success, there has been growing interest in using vaccines to treat pathologies outside of infectious diseases ranging from cancer to Alzheimer's disease.2,3 Adjuvants are a critical component of modern vaccines that enhance their efficacy by improving immunogenicity, stability, and delivery of antigens.4–6 In addition to serving as immunostimulants, adjuvants that form antigen depots extend vaccine residence time in the body, which has been shown to improve the antigen-specific immune response to vaccines.7,8 Further, adjuvants have demonstrated the ability to direct the phenotype of adaptive immune response generated to a specific antigen.9 The two prominent adaptive effector responses are cellular (Th1) and humoral (Th2) immunity, which are distinct immunological pathways important for preventing and treating different types of pathologies. Th1 immunity is characterized by the activation of antigen-specific cytotoxic CD8+ T cells and is most useful for protection against cancer and intracellular pathogens. Th2 immunity, on the other hand, is characterized by the production of neutralizing antibodies and is effective at preventing and clearing extracellular pathogens, such as multicellular parasites.10 Th2 immunity has also been suggested to protect against amyloidogenic neurodegenerative pathologies like Alzheimer's disease.11,12 The ability of adjuvants to direct the immune response to optimally address a specific pathology is critical for conferring immunological protection.13,14 Despite the importance of adjuvants, only nine vaccine adjuvants have been included in FDA-approved infectious disease vaccines.15,16 As our understanding of the immune system has grown, so has the potential for engineering new classes of adjuvants that favorably direct the type of immune response generated for a particular pathology.15Recently, self-assembling peptides have shown promise as adjuvants.17 Since these materials are composed solely of amino acids, they are inherently biocompatible, and their properties can be readily tuned by altering the amino acid sequence.18 Previous research has established the ability of peptide self-assemblies to act as effective vaccine adjuvants. One of the first self-assembled peptide adjuvant systems developed was the fiber-forming peptide Q11 (Ac-QQKFQFQFEQQ-Am), which, when linked to pathogen-associated peptide epitopes, led to a strong and balanced immune response to a variety of clinically relevant antigens.19–24 Peptide nanoparticles, nanovesicles, dendrimers, and other self-assembly architectures have also shown promise as vaccine adjuvants.17 Many of these materials, however, rely on the use of known short immunological peptide epitopes as antigens to confer immunity. This requires knowledge of the immunogenic epitopes on the protein antigen, which is not always known for unique cancer mutations and emerging pathogens. Furthermore, some epitopes are not efficacious outside their immunity-conferring conformation in the context of the full native protein or multi-protein assembly. Thus, there is a need to develop antigen-agnostic peptide adjuvants that can deliver protein subunit antigens and even whole inactivated viruses.
Peptide-based supramolecular hydrogels are well-suited for adjuvating and delivering protein antigens. These materials are shear-thinning and self-healing, which allows them to be injected through a standard needle as a liquid and then reform as a gel in vivo, as opposed to covalent hydrogels that require surgical implantation or in situ crosslinking. Further, the bulk material properties of peptide hydrogels can be tuned by varying the peptide sequence, the nature/degree of peptide crosslinking, or the peptide concentration.25,26 Their ability to deliver protein payloads in a controlled manner using electrostatic interactions and physical entrapment has been extensively explored.27 The tetrapeptide hydrogel Nap-GFFY is one of the most widely studied peptide hydrogel adjuvants, which elicits both humoral and cellular immune responses to clinically relevant vaccine antigens.28–30 These sequences, however, use noncanonical amino acids and modifications that can complicate the synthetic pathway and introduce potentially bioactive non-standard amino acid degradation products that can have undesirable effects in vivo.31–33
Multidomain peptide (MDP) hydrogels are peptides composed exclusively of canonical amino acid residues that self-assemble into β-sheet rich fibrils that crosslink with divalent ions to form supramolecular hydrogels under physiological salt and pH conditions. Although there are many variants,26,34,35 MDPs generally have the structure Ac-XX(SL)6XX-Am, where Ser (S) and Leu (L) repeats create an amphiphilic core that favors β-sheet formation with “X” being one of the charged canonical amino acids: Lys, Arg, Asp, or Glu (Fig. 1).36 The naming convention for MDP sequences is X2, where “X” is the single letter code for the flanking charged amino acid used in the peptide (Table 1). The most widely investigated MDP hydrogel to date is K2, and this peptide has shown promise for regenerative medicine and drug delivery applications.37–41 Previous investigations have established that the foreign body reaction to MDP hydrogels can be tuned by altering the charged amino acid in the “X” position. It was previously found that inflammation can be increased by including positively charged Lys or Arg residues, while the inclusion of negatively charged Asp and Glu residues leads to minimal inflammation. These materials are infiltrated by immune cells within one day and their chemical functionality controls both the types of immune cells that infiltrate the gel and the local cytokine production profile.36
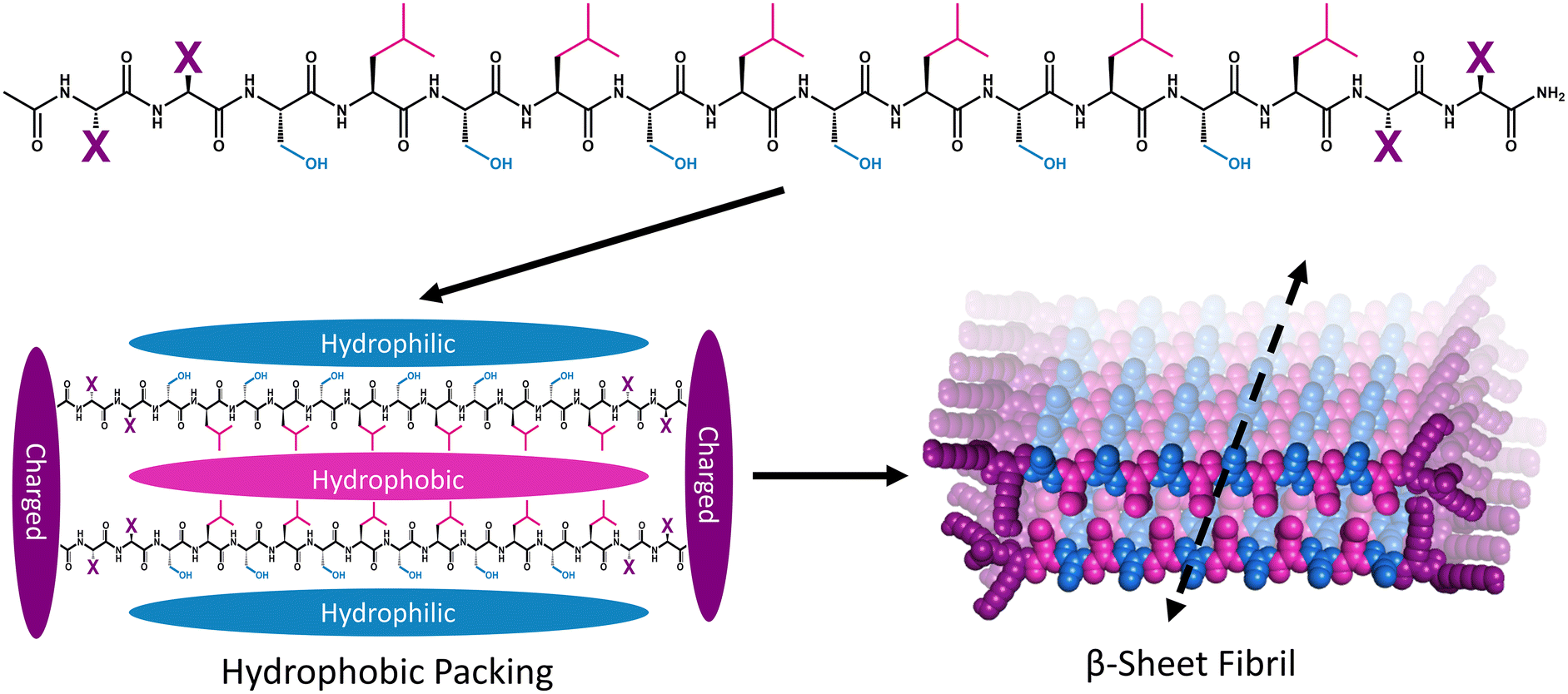 | ||
| Fig. 1 Schematic showing the general primary, secondary, and tertiary structure of MDP highlighting the hydrophilic (blue), hydrophobic (pink), and charged (purple) domains. Peptide monomers assemble via hydrophobic and electrostatic interactions to form beta-sheet-rich fibers that extend along the peptide backbone hydrogen-bonding axis to form fibrils. | ||
| Name | “X” residue | Sequence | Monomer charge |
|---|---|---|---|
| K2 | Lysine (K) | Ac-KK(SL)6KK-Am | +4 |
| R2 | Arginine (R) | Ac-RR(SL)6RR-Am | +4 |
| E2 | Glutamate (E) | Ac-EE(SL)6EE-Am | −4 |
Due to the strong immunostimulatory properties and drug delivery capacities of positively charged MDPs, we hypothesized that these peptides could be formulated as effective adjuvants for subunit vaccines. Using the model antigen ovalbumin (OVA), we demonstrate that positively charged MDP hydrogels can delay the release of antigen to a similar extent as the common clinical adjuvant, alum, and serve as an adjuvant that strongly biases the adaptive immune response towards humoral immunity.
Materials and methods
Solid-phase peptide synthesis (SSPS) and purification
Peptides were synthesized manually using standard Fmoc-based chemistry. Resins and Fmoc-protected amino acids were supplied by Novabiochem (Millipore Sigma, Burlington, MA). Briefly, Fmoc-protected low-loading MBHA rink-amide resin (1 equiv.) was deprotected with two steps of excess 25% v/v piperidine in DMF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMSO 1:1 for 5 min each. All Fmoc deprotection steps were performed identically and checked using the ninhydrin test for primary amines. Coupling was achieved with the addition of Fmoc-protected amino acids (4 equiv.) preactivated for 1 min with HATU (3.95 eq.) and DIEA (6 eq.) in a minimal amount of DMF:DMSO 1:1 that were then added to the resin for 20–30 min at RT. This process was repeated until the peptide was complete. The N-terminus was acetylated with the addition of acetic anhydride (200 eq.) and DIEA (75 eq.) in DCM two times for 45 min. Peptides were cleaved from the resin by reacting it with a cleavage cocktail composed of TFA:Anisole:TIPS:EDT:H2O 90:2.5:2.5:2.5:2.5 for 3 h at RT. TFA was evaporated off with a stream of nitrogen gas to a final volume of 1 mL. The peptide was triturated with cold diethyl ether.
:DMSO 1:1 for 5 min each. All Fmoc deprotection steps were performed identically and checked using the ninhydrin test for primary amines. Coupling was achieved with the addition of Fmoc-protected amino acids (4 equiv.) preactivated for 1 min with HATU (3.95 eq.) and DIEA (6 eq.) in a minimal amount of DMF:DMSO 1:1 that were then added to the resin for 20–30 min at RT. This process was repeated until the peptide was complete. The N-terminus was acetylated with the addition of acetic anhydride (200 eq.) and DIEA (75 eq.) in DCM two times for 45 min. Peptides were cleaved from the resin by reacting it with a cleavage cocktail composed of TFA:Anisole:TIPS:EDT:H2O 90:2.5:2.5:2.5:2.5 for 3 h at RT. TFA was evaporated off with a stream of nitrogen gas to a final volume of 1 mL. The peptide was triturated with cold diethyl ether.
Crude peptides were purified by high-performance liquid chromatography (HPLC) on XBridge Protein BEH C4 OBD Prep column (Waters Corporation, Milford, MA) using two different solvent systems. For positively charged peptides, a solvent system of Milli-Q (MQ) water with 0.05% TFA (solvent A) and acetonitrile with 0.05% TFA (solvent B) was used for purification. Negatively charged peptides were purified using MQ water with 4 mM acetic acid and 5 mM ammonium hydroxide (solvent A) and acetonitrile with the same buffer (solvent B). Peptide solutions (10–15 mg mL−1) were injected on the column in a volume of 1 mL of 80% solvent A and 20% solvent B. Purification was achieved using a 3% solvent B/min gradient from 5% to 50% solvent B while monitoring the absorbance at 220 nm with a TUV detector. Matrix-assisted laser desorption/ionization mass spectrometry (MALDI MS) and ultra-performance liquid chromatography (UPLC) were used to confirm sample identity and purity (Fig. S1–S3†). Pure peptides were checked for endotoxin content using the ToxinSensor gel clot assay (Genscript, Piscataway, NJ). Peptides were only used in vivo if they contained less than 0.25 EU per mg.
Protein fluorophore labeling
OVA (Millipore Sigma, Burlington, MA) was labeled with tetrafluorophenyl (TFP) ester functionalized AZDye 647 (Click Chemistry Tools, Scottsdale, AZ) using the protocol provided by the supplier. In brief, protein (1 eq.) was dissolved in 0.2 M sodium bicarbonate buffer (pH 8.5) at a concentration of 10 mg mL−1. TFP ester functionalized AZDye 647 (5 eq.) was dissolved in MQ water to 10 mg mL−1 and was slowly added to the protein solution. The reaction proceeded at RT in the dark for 1 h. Excess 1.5M hydroxylamine solution was added to quench excess dye for 1 h at RT. Labeled protein was isolated using a size exclusion PD-10 Sephadex™ G-25 M gel filtration column (Cytiva, Marlborough, MA) and the degree of labeling was determined by UV-vis. The labeled OVA was purified by Pierce endotoxin removal beads (ThermoFisher Scientific, Waltham, MA), sterilized by passing through a 0.2 μm PTFE syringe filter (CELLTREAT, Pepperell, MA), and verified to be endotoxin-free (less than 0.25 EU per mg) with the ToxinSensor gel clot assay.Vaccine preparation
Multidomain peptides were prepared by making 20 mg mL−1 peptide stock solutions in 0.4 mg mL−1 endotoxin-free OVA (Worthington Biochemical Corp., Lakewood, NJ) with 102 mg mL−1 low endotoxin sucrose (Millipore Sigma, Burlington, MA) dissolved in ultrapure endotoxin-free water (Cytiva, Marlborough, MA). The resulting OVA concentration was confirmed by UV-vis using the extinction coefficient of 30590 cm−1 M−1. Once the peptide was completely dissolved, the solution was diluted to 10 mg mL−1 peptide with 1X Hank's balanced salt solution (HBSS), which was supplemented with 45 mM MgCl2 for E2. Solutions were then incubated overnight to allow for complete gelation. Alum samples were prepared by performing a 1:1 dilution of 2% Alhydrogel® adjuvant (InvivoGen, San Diego, CA) with stock solutions of 0.4 mg mL−1 endotoxin-free OVA with 102 mg mL−1 low endotoxin sucrose dissolved in ultrapure endotoxin-free water. Antigen-only vaccines were prepared by performing a 1:1 dilution of 1X HBSS with the same stock solutions of OVA with sucrose. The final concentrations of vaccine components were 10 mg mL−1 adjuvant (MDP or alum), 0.2 mg mL−1 OVA, and 51 mg mL−1 sucrose.
Rheology
Oscillatory rheology measurements were collected on an AR-G2 rheometer (TA Instruments, New Castle, DE). Rheology was performed on 75 μL samples at room temperature that were transferred to the instrument with a truncated 200 μL pipet to minimize shear. Samples were equilibrated at 1 rad s−1 and 1% strain for 30 min. The final hydrogel strength was determined by obtaining the storage (G′) and loss (G′′) moduli at the end of this period. After equilibration, a frequency sweep was performed over the range of 0.1–10 rad s−1 at a constant 1% strain. Shear recovery experiments were conducted after the frequency sweep by allowing the gel to equilibrate for 2 min at 1 rad s−1 and 1% strain and then sheared for 1 minute at 200% strain. Recovery was then monitored for 10 min at the starting conditions and the percent recovery was calculated by taking the final storage modulus (G′) values at the end of the 10 min and comparing them to the values at the end of the initial two-minute equilibration.Scanning electron microscopy
Scanning Electron Microscopy (SEM) was used to analyze the nano- and microscale features of gels. Samples were dehydrated in a series of ethanol dilutions (30%, 50%, 60%, 70%, 80%, 90%, 2 × 100%) and then dried in a Leica EM CPD300 Critical Point Dryer (Leica Microsystems, Wetzlar, Germany). Next, samples were transferred to a Denton Desk V Sputter System (Denton Vacuum, Moorestown, NJ) and coated with approximately 5 nm of gold. Finally, samples were imaged with a Helios NanoLab 660 Scanning Electron Microscope (FEI Company, Hillsboro, OR) at a voltage of 1 kV and current of 25 pA.In vitro release assays
Gels were prepared with 10% fluorescent OVA the day before the assay and hydrogelation proceeded at RT in the dark overnight. Gels were then plated (50 μL) into a custom 3D-printed 48-well plate that is described in more depth in the ESI (Fig. S4†). The gels rested for 5 min before supernatant (400 μL) was then added to each well. The supernatant was 1X HBSS (supplemented with 45 mM MgCl2 for E2). Solutions that mimicked 100% release were prepared in the same way as the gels but without peptide. The assay was then monitored on a microplate reader (Tecan, Männedorf, Switzerland) and incubated at 37 °C with orbital shaking (142 rpm) without media replacement. Time points were recorded every 45 min, and the percent release was calculated by dividing the fluorescent intensity of the sample by the intensity of the 100% release solution at each time point. The first measurement taken after preparation was used to determine OVA loading efficiency in the hydrogel (Fig. S5†).In vivo release assays
All animal experiments in this study were carried out in accordance with an IACUC-approved protocol (Rice University protocol 20–065). The day before injection, MDPs, alum, and antigen alone were prepared as described in the vaccine preparation section with OVA and doped with 20% AZDye 647 labeled OVA. Vaccine solutions (50 μL) were loaded into 300 μL insulin syringes (BD Biosciences, Franklin Lakes, NJ) and stored at 4 °C overnight. The day before injections, female C57BL/6J mice (Charles River Laboratories, Wilmington, MA) were shaved, and then a depilatory cream was used to remove all fur from the injection site. Right before injections, the background tissue autofluorescence (Ex/Em 640/700 nm) was acquired for each mouse using a PerkinElmer (Waltham, MA) Spectrum in vivo imaging system (IVIS). Samples were then injected bilaterally subcutaneously and imaged by IVIS at several time points until the fluorescence from the OVA was no longer discernable from the background. The percentage of OVA released was calculated by drawing equally sized rectangular regions of interest around the injection site and dividing the observed total background-subtracted radiant efficiency by the maximum total background-subtracted radiant efficiency observed on the first day of the experiment.Flow cytometry
Equal numbers of male and female C57BL/6J mice were injected with 50 μL of each vaccine and control prepared as previously described. Spleens were harvested 3 weeks after injection, and splenocytes were isolated. In brief, spleens were homogenized in flow buffer (BioLegend, San Diego, CA) and then centrifuged at 250 RCF for 5 min at 4 °C. The supernatant was then aspirated, and the cells were resuspended in 1 mL cold ACK lysis buffer (Quality Biological, Gaithersburg, MD) for 30 seconds and then diluted in 15 mL of flow buffer. Cells were then spun down again and resuspended in flow buffer to make a splenocyte stock solution at a concentration of 2 × 107 cells per mL. An aliquot of the cell stock solution (50 μL) was then diluted with 43 μL flow buffer and 2 μL rat anti-mouse CD16/Cd32 Fc block (BD Biosciences, Franklin Lakes, NJ). SIINFEKL tetramer (0.15 ng) from the NIH tetramer core (Emery University, GA) was added and samples were incubated for 45 min on ice in the dark. Afterward, the samples were stained with antibodies (Table S1†) and incubated for 30 min on ice in the dark (or 20 min at RT for staining chemokine receptors). The samples were then spun at 200 RCF for 7 min and washed twice: once with flow buffer and once with PBS. After the final wash, cells were resuspended in 1 mL PBS and stained with Invitrogen blue live/dead fixable dead cell stain (ThermoFisher Scientific, Waltham, MA) for 30 min on ice in the dark. The cells were then centrifuged and resuspended in 200 μL 1% (v/v) formalin in flow buffer, passed through a 35 μm strainer (Corning Inc., Corning, NY), and stored at 4 °C in the dark until analyzed on a BD SORP FACSAria I (BD Biosciences, Franklin Lakes, NJ). Data were analyzed using FCS Express 5 (De Novo, Los Angeles, CA). For analysis, a cell-only control and fluorescent minus one controls were used to guide manual gating (Fig. S6†).IgG, IgG2c, and IgM titer quantification
Equal numbers of nine-week-old male and female C57BL/6J mice were purchased and bled via the submandibular vein using a 5 mm lancet (Medipoint Inc., Mineola, NY) one week prior to injection. Blood (200 uL) was collected in clotting microvettes (Sarstedt, Nümbrecht, Germany) using the published protocol from the supplier. Blood was stored on ice until serum was separated by centrifugation at 4 °C at 3000 RCF for 20 min. Serum was stored in 20 μL aliquots in LoBind Eppendorf tubes (Eppendorf, Hamburg, Germany) at −20 °C until used. The day before injection, injections were prepared as described above and doped with 20% AZDye 647 labeled low endotoxin OVA. Solutions (50 μL) were loaded into insulin syringes and stored at 4 °C overnight. C57BL/6J mice in an equal male:female ratio for each group were shaved and then injected with OVA vaccine formulations subcutaneously. Blood was collected as previously described every two weeks from one week after injection through the 11-week time point.OVA-specific antibody titers were quantified by using a modified ELISA protocol. Each well in Nunc Maxisorp 96-well plates (ThermoFisher Scientific, Waltham, MA) was coated with 100 μL of 1 μg mL−1 OVA in a 100 mM pH 9.6 carbonate–bicarbonate buffer. Plates were coated overnight on an orbital shaker at 4 °C and were then washed three times with PBST (0.5% Tween 20 in PBS) using a 96-well automatic BioTek Microplate Washer 405 (Agilent, Santa Clara, CA). Wells were then blocked with Blotto non-fat dry milk (Rockland Immunochemicals, Limerick, PA) dissolved at 50 mg mL−1 in PBST for 2 h at RT on an orbital shaker. After an initial 10- to 20-fold dilution, serial 2-fold dilutions of serum were prepared in the Blotto solution. Blocking solution was removed from the plates and 50 μL of each serum dilution was then added and incubated at RT on an orbital shaker for 2 h. Plates were then washed 3 times with PBST and 100 μL anti-mouse IgG, IgG2c, or IgM HRP-conjugated secondary antibodies in blocking solution (Table S2†) was added to each well and incubated in the dark at RT for 2 h on an orbital shaker. To develop the plates, secondary antibody was removed by washing the plates 5 times with PBST then 100 μL of SureBlue TMB substrate solution (SeraCare Life Sciences inc., Milford, MA) was added to each well and incubated for 5 min. The reaction was stopped with 1 M sulfuric acid and then the absorbance at 450 nm was read immediately on a Tecan M1000 microplate reader (Männedorf, Switzerland). Titers were then calculated by determining the lowest serum dilution that was at least 2-fold higher than the background signal from mouse matched naïve serum collected one week pre-injection.
Histology
The day before injection, vaccines were prepared as described above with low endotoxin OVA and doped with 20% AZDye 647 labeled OVA. Solutions (50 μL) were loaded into insulin syringes and stored at 4 °C overnight. Female C57BL/6J mice were shaved and injected subcutaneously bilaterally with different vaccine groups on each flank. After 3 days, the mice were sacrificed, and the hydrogels were harvested and fixed in 4% paraformaldehyde in PBS at 4 °C overnight. The samples were then soaked in 70%, 80%, 95%, and 100% ethanol each for one hr with the first and last conditions being repeated twice. The samples were then soaked in xylenes three times for one hr each before being fixed in 50 °C paraffin wax overnight. Samples were then cooled and cut into 5 μm sections using a Cut 4060 microtome (Olympus, Center Valley, PA). Sections were placed in a warm water bath and then set onto HistoBond + glass slides (VWR, Radnor, PA). Slides were dried and then baked overnight at 45 °C. Samples were then stained with hematoxylin and eosin (H&E) using a standard staining protocol36 and imaged at the MD Anderson Histology Core (Houston, TX). Caspase-3 apoptosis staining and imaging were performed by the MD Anderson Histology Core. Images were processed using ImageJ.42Statistics
Single group comparisons were analyzed by unpaired Student's t-tests. Multiple group comparisons were calculated by ordinary one- or two-way ANOVA with Tukey's multiple comparison test. Calculations were performed in GraphPad Prism 9. Statistical significance in all figures is denoted with asterisks as follows: * = p < 0.05; ** = p < 0.01; *** = p < 0.001; **** = p < 0.0001.Results and discussion
Material characterization
In some cases, the mechanical properties of supramolecular peptide hydrogels can be sensitive to the presence of encapsulated protein.43 To determine if that loading protein antigen into MDP hydrogels would negatively impact their physical properties, MDP gels with 0.2 mg mL−1 OVA were characterized by oscillatory rheology to determine the storage (G′) and loss (G′′) moduli and shear recovery of the hydrogels. For all gels and conditions tested, G′ > G′′ and G′ was largely frequency-independent over the tested range, indicating that the materials behave as viscoelastic hydrogels over the frequency regime of 0.1–10 rad s−1 (Fig. S7†). Statistically significant, but modest, improvements in gel G′ were observed in positively charged K2 and R2 hydrogels upon the inclusion of OVA, but not in E2 (Fig. 2A). Shear recovery was quantified by exposing the gels to a deformation force corresponding to 200% strain for one minute and monitoring the recovery in G′ for 10 min after deformation. The positively charged MDP hydrogels had higher shear recovery in the presence of OVA while no statistically significant difference was observed in E2 (Fig. 2B). These observed changes in G′ and shear recovery are likely due to the protein's net negative charge coordinating with positively charged peptide fibers in K2 and R2 hydrogels. These results are consistent with observed stiffening and improved energy dissipation upon the addition of several negatively charged proteins to a positively charged protein-based hydrogel made from aggregated lysozyme.43 We conclude that including antigen does not significantly impede, and may improve, the ability of MDPs to form a shear-recovering hydrogel under the conditions tested.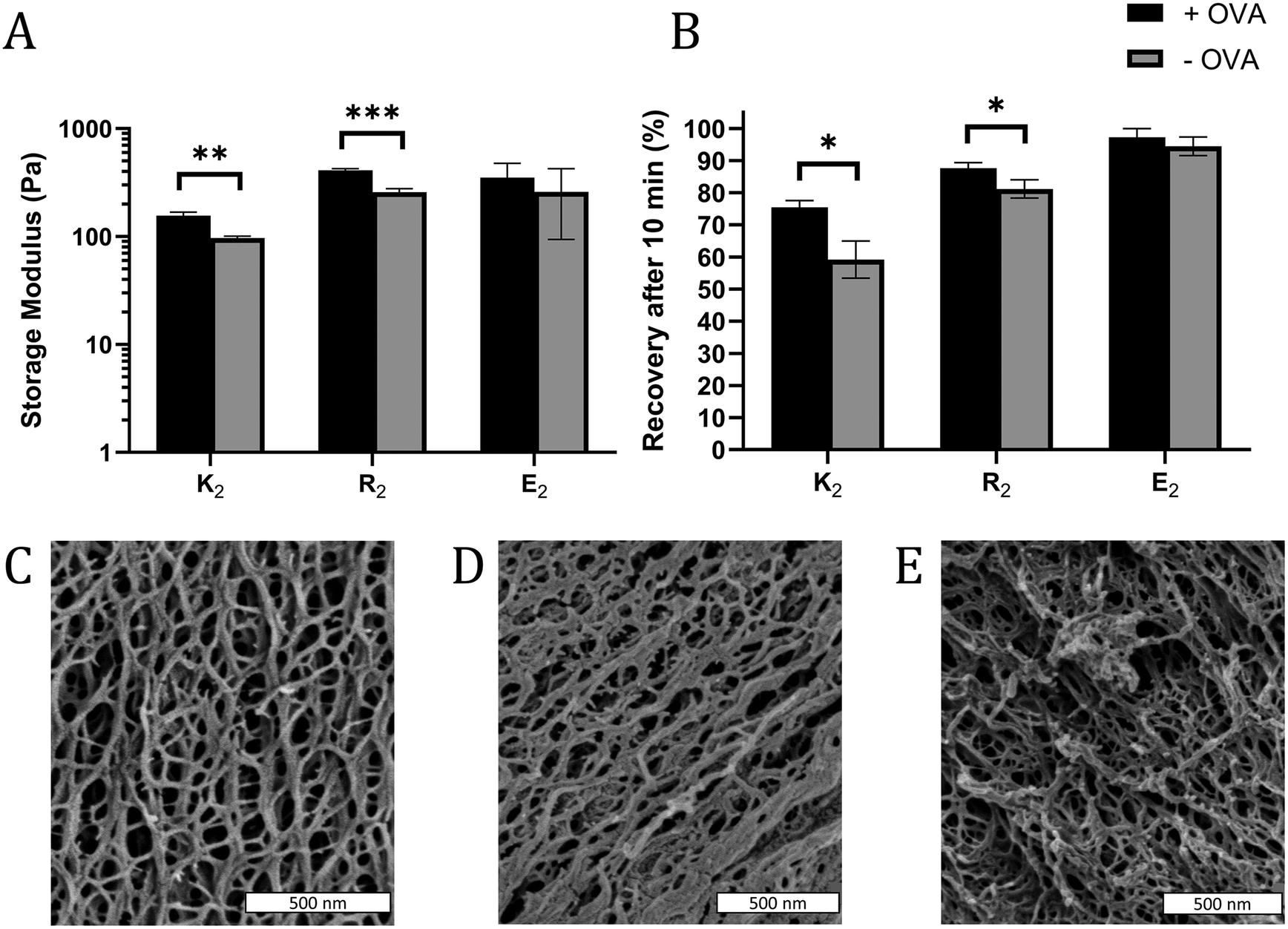 | ||
| Fig. 2 (A) Storage modulus (G′) of hydrogels after 30 min of equilibration time at 1% strain and a frequency of 1 rad s−1, and (B) the percent recovery of G′ 10 min after a one-minute shear event at 200% strain with (black bars) and without (grey bars) OVA (n = 3). SEM images of (C) K2, (D) R2, and (E) and E2 show that all three MDP hydrogels form an extensive nanofiber network. | ||
SEM was used to investigate the nanostructure of the MDP hydrogels. All three MDPs imaged are seen to have a dense and porous nanofiber network (Fig. 2C–E). Although the critical point drying process necessary for SEM preserves some of the porosity of the hydrogels, SEM has some limitations as the sample preparation conditions are relatively harsh for low concentration self-assembling hydrogels and the images are a 2D projection of a 3D network. Thus, the mesh size of the network cannot be measured directly by SEM. These images and previous transmission electron microscopy studies36 suggest that these peptides form long and relatively rigid micrometer length-scale fibers. Mathematical models of similar semiflexible polymer networks show that mesh size should be primarily scale with the polymer concentration.44 Thus, as K2, R2, and E2 peptide monomers have molecular weights within 7% of each other and are dissolved at the same w/v concentration, they should all have similar mesh sizes.
In vitro and in vivo antigen release
The rheology results indicate the presence of attractive electrostatic interactions between the negatively charged OVA and the positively charged MDP hydrogels, K2 and R2. Also, it has been well established that loading a charged material with a payload of the opposite charge will delay release due to attractive electrostatic interactions.45,46 Thus, we hypothesized that these interactions would allow K2 and R2 MDPs to act as antigenic depots and extend the release of negatively charged OVA (isoelectric point = 4.5). To test this hypothesis in vitro, fluorescently labeled OVA was loaded into hydrogels or absorbed onto alum. MDP hydrogels and alum were plated at the bottom of a custom 3D-printed 48-well plate and covered in HBSS. The fluorescent intensity of the supernatant adjacent to the material was monitored with a microplate reader over 24 h without supernatant replacement to measure the equilibrium release of OVA from the hydrogels. All MDP hydrogels were found to have high OVA loading efficiencies of over 95% (Fig. S5†). Since alum is a colloidal suspension of hydrogel microparticles, it did not form a self-supporting hydrogel under the in vitro conditions tested and thus dissipated upon the addition of supernatant, which makes OVA appear to release very quickly. The E2 hydrogels completely released OVA within 12 h while K2 and R2 hydrogels never achieved complete release over the course of 24 hrs (Fig. 3A). Since all three peptides should have similar mesh sizes, the change in release kinetics may be attributed to their differences in chemical functionality and charge. E2 reached equilibrium at >95% OVA released demonstrating the absence of attractive interactions between the negatively charged peptide hydrogel and the negatively charged protein. The positively charged MDPs exhibited much lower release equilibria (less than 10%), suggesting strong attractive interactions between OVA and the peptide fibers. R2 had the lowest percentage of released OVA (4.23 ± 0.53%) by the end of the 24 h, which may be due to the increased hydrogen bonding potential of the arginine sidechain over that of lysine.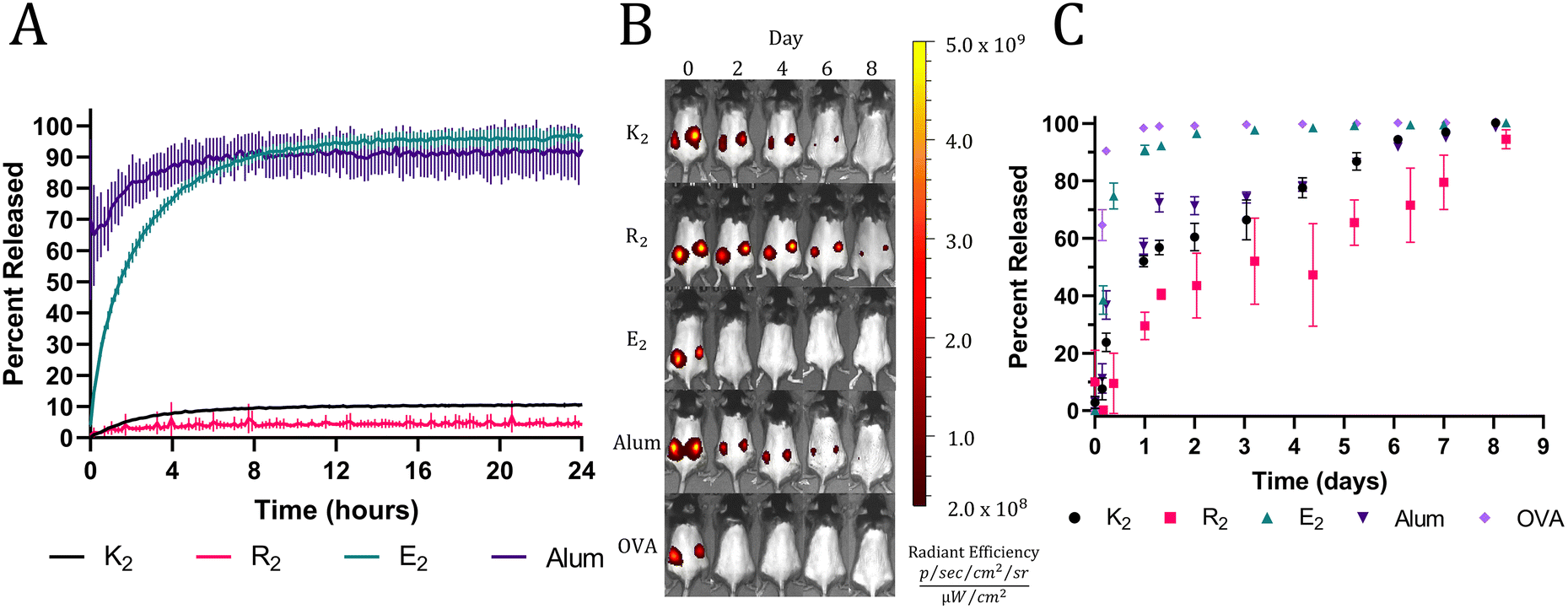 | ||
| Fig. 3 (A) In vitro equilibrium release of fluorescently labeled OVA from MDP hydrogels and alum. (B) Representative IVIS images of C57BL/6J mice injected with vaccines formulated with fluorescently labeled OVA over 8 days. (C) Calculated percent of fluorescently labeled OVA released (n = 5). | ||
In vivo release assays were performed using fluorescently labeled OVA and to track protein release from the injection site by monitoring the loss of fluorescent signal. In vitro trends were recapitulated in vivo with high fidelity, apart from alum likely due to incompatibility with the in vitro release assay, as previously mentioned. A bolus injection of soluble OVA by itself was quickly cleared from the injection site followed by E2, which surpassed a mean of 95% release on days 1 and 2, respectively. All the other groups took significantly longer to release OVA and exceeded a mean of 95% OVA released on day 7 for alum and K2 and day 9 for R2 (Fig. 3B and C). The slightly longer residence time of OVA in R2 is consistent between both in vitro and in vivo assays; however, the differences between R2, K2, and alum are small. These data demonstrate the ability of positively charged MDPs to exhibit a non-inferior antigen depot effect compared to alum, the most widely used clinical adjuvant.15
Adjuvancy of MDP hydrogels
The ability of MDP hydrogels to act as adjuvants and potentiate Th1 and Th2 immunity was assessed by flow cytometry and anti-OVA antibody titer quantification. Mice were monitored for 11 weeks post-vaccination and blood was collected every two weeks and analyzed for anti-OVA IgG antibodies by ELISA. At week 3, alum and the MDPs generated humoral immune responses that were not statistically significantly different; however, by week 11, mice that received alum-adjuvanted vaccines had the highest anti-OVA IgG titer of 221.03 (Fig. 4A). Of the MDPs, K2 generated the highest IgG titer (217.20) that was significantly greater than E2 (213.75) while R2 (215.82) was not significantly different from either of the other MDPs at week 11 (Fig. 4B). The lack of any significant differences between R2 and E2 IgG titers, despite a more than 7-day difference in antigen residence time, suggests that the depot effect plays a marginal role in MDP adjuvancy if any. This conclusion is further supported by the fact that both R2 and E2 IgG titers plateau at the same time, 3 weeks post-vaccination, notwithstanding differences in antigen release rate (Fig. 4A). Thus, the foreign body reaction to the different chemical functionalities of MDP hydrogels is likely the primary factor that dictates the resulting immune response. It should be noted that one mouse in the E2 group did not respond to the vaccine and was excluded from the analysis. It is unclear if a procedural error or natural biological variation is responsible for this lack of a response.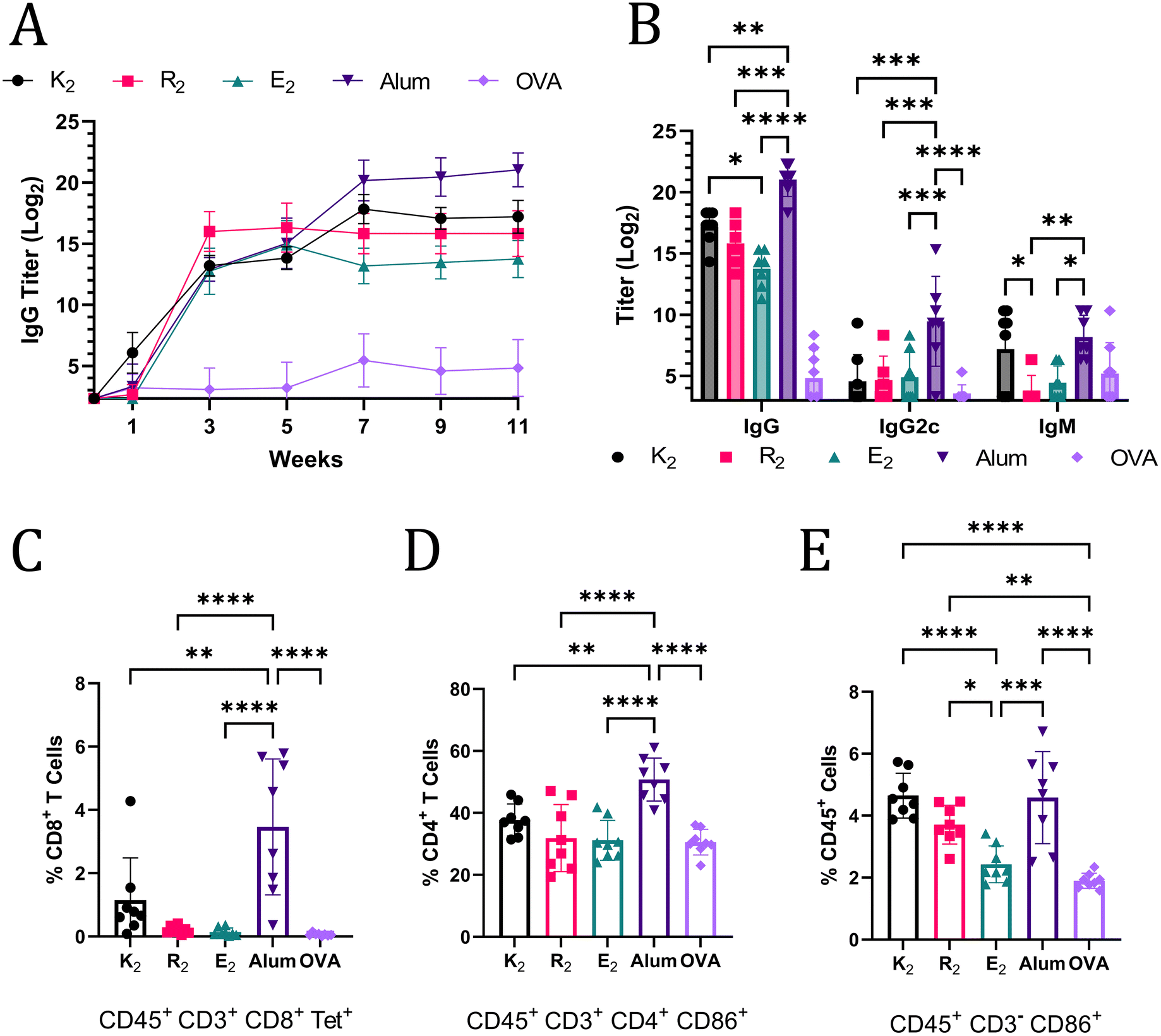 | ||
| Fig. 4 (A) Anti-OVA IgG titers over the course of 11-weeks after OVA vaccination administered with MDPs, alum, or no adjuvant (n = 6–8). (B) Week 11 anti-Ova titers of IgG, IgG2c, and IgM. All MDPs generated statistically significantly higher IgG titers than the OVA-alone injection but are not marked on the graph for the sake of clarity. Panels C–E show data from flow cytometry on splenocytes of vaccinated C57BL/6J mice harvested 3-weeks after inoculation (n = 8). (C) Tetramer+ CD8+ T cells graphed as a percent of CD45+ CD3+ CD8+ cells. (D) Activated CD4+ T cells graphed as a percent of CD45+ CD3+ CD4+ cells that express CD86. (E) Activated non-T lymphocyte immune cells (CD45+ CD3−) graphed as a percent of CD45+ cells that express CD86. | ||
IgG2c and IgM titers were also quantified to provide a more complete picture of the immune response at the terminal time point. IgM titers are present in early immune responses, but can also play an important role in pathogen neutralization.47,48 The only MDP that generated a comparable IgM immune response to alum was K2, further reinforcing the assessment that K2 generates the most robust humoral immunity of the MDP hydrogels tested. IgG2c (an analog of IgG2a) antibody production is associated with Th1 immunity in C57BL/6J mice.49 Alum adjuvancy generated significantly higher titers of anti-OVA IgG2c than all the MDPs, which generated almost no IgG2c (Fig. 4B). Alum is known to generate a very limited Th1 immune response.50 Thus, these data, which show that MDPs generate an even more muted Th1 immune response than alum, suggest that MDPs potentiate nearly exclusive humoral (Th2) immunity.
The absence of a Th1 immune response was confirmed by flow cytometry. Mice vaccinated with MDP and alum adjuvanted vaccines were sacrificed 3 weeks after injection and their splenocytes were labeled with CD45 antibodies to isolate immune cell populations. CD3 antibodies, in combination with CD8 and CD4 antibodies, were used to enable gating of T cell populations. Immune cells displaying an “active” phenotype were identified using CD86,51 which is a critical receptor bridging adaptive and innate immune activation.52–54 A SIINFEKL MHC I tetramer was employed to identify antigen-specific T cell receptors on CD8+ T cells, which would be indicative of a Th1 immune response. Mice vaccinated with alum-adjuvanted OVA had a significantly larger, but still relatively small, proportion of CD8+ Tetramer+ cells compared to all the other groups. These results are congruent with the conclusion that MDP hydrogel adjuvants elicit a more muted Th1 immune response than alum, and, thus, have a stronger bias towards Th2 immunity. The general trend in the percent of activated CD4+ CD86+ T cells also match observed IgG levels with the alum adjuvant eliciting the largest population (Fig. 4D). Alum, K2, and R2 all generated statistically larger populations of activated non-T lymphocyte immune cells (CD45+ CD3− CD86+) as a percent of total immune cells in the spleen when compared to OVA and E2 injections (Fig. 4E). CD86 expression on CD3− immune cells is responsible for activating T cells, stimulating cytokine production, and promoting T cell proliferation.55 Additionally, CD86 expression on immune cells has been shown to be highly correlated to the immune response to vaccines.56 Thus, these data suggest that alum, K2, R2, and alum can activate the innate immune system for extended periods of time and create an environment conducive to prolonged T cell activation in the spleen.
Histological characterization of adjuvants
Tissue was collected from the injection site 3 days after vaccination and stained with H&E to evaluate tissue morphology and cell infiltration into the gels. No evidence of the materials administered was observed in mice injected with soluble OVA only or OVA-loaded E2 (Fig. 5A and B), which is consistent with the observation from the in vivo release assay that the antigen is completely cleared within 24 h. These data also suggest that OVA and E2 lead to minimal inflammation at the site of injection and that the negatively charged MDP is completely degraded by day 3. In contrast, boluses of K2, R2, and alum were found at the injection site with large amounts of cellular infiltration (Fig. 5C and D). Qualitatively, K2 and R2 were more heavily infiltrated by cells than alum at the 3-day time point as seen by the greater number of purple nuclei within the boluses. Higher magnification images of these materials show that cells infiltrate the gels differently depending on the chemical functionality. In the case of alum and K2, this infiltration appears to be well-distributed throughout the gel, in contrast to R2 where cells appear to infiltrate along cracks in the hydrogel (Fig. 5F–H). Heterogeneous cellular infiltration has been previously observed with R2 hydrogels and may be associated with the increased inflammatory nature of this material.36 The impact that this phenomenon might have on the resulting adaptive immune response remains unclear.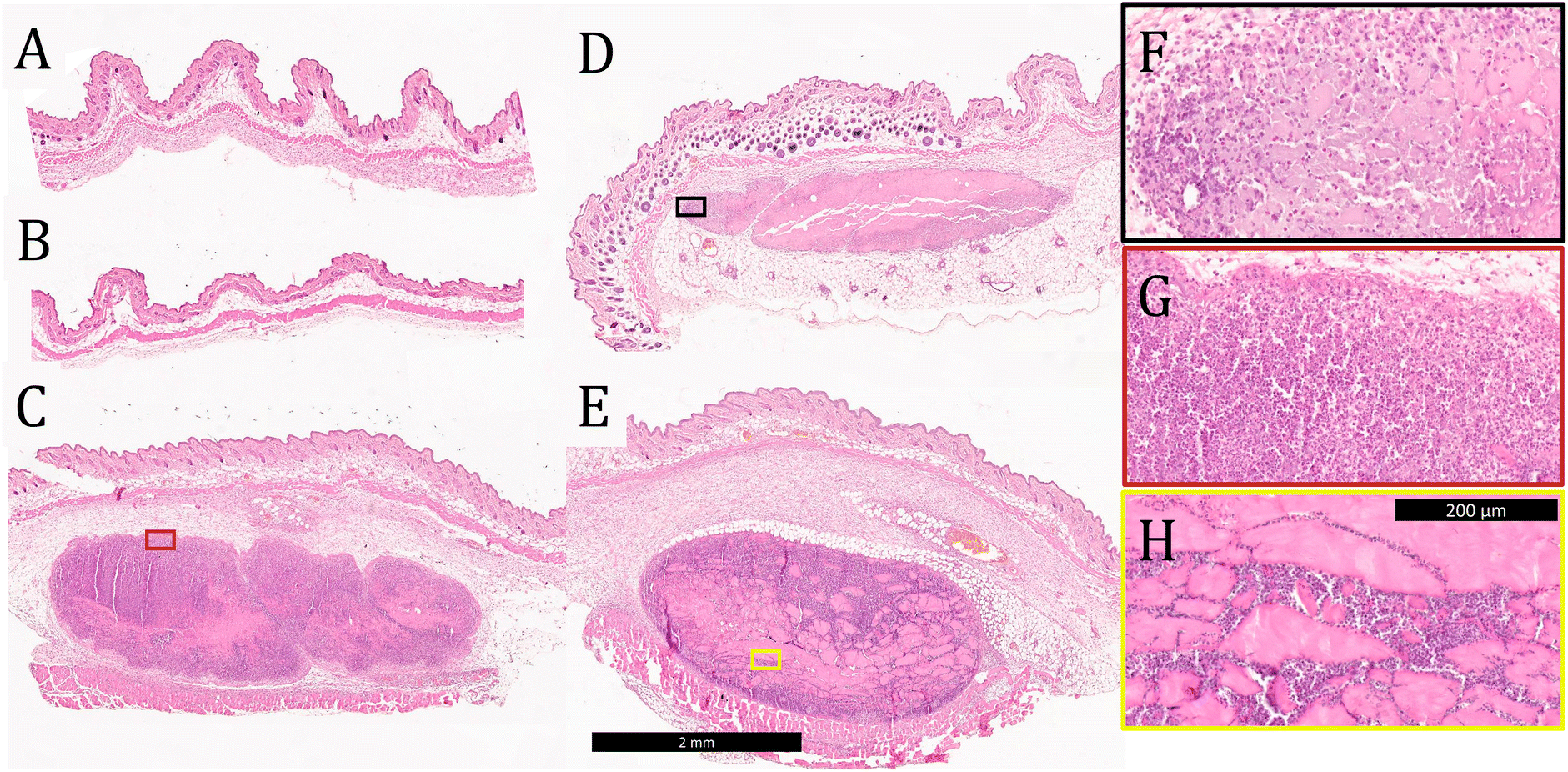 | ||
| Fig. 5 Representative images of H&E-stained C57BL/6J mouse tissue sections 3 days after injection with (A) OVA, (B) OVA + E2, (C) OVA + K2, (D) OVA + alum, and (E) OVA + R2 where the black scale bar represents 2 mm. Higher magnification images of (F) OVA + alum, (G) OVA + K2, and (H) OVA + R2, where the black scale bar represents 200 μm and the colored box outlines show the section of the larger tissue section being focused. | ||
Cleaved caspase-3 apoptosis immunostaining was performed on adjuvants that persisted 3 days after injection to determine biocompatibility. Images of tissue sections from E2 and OVA-only injections that do not have evidence of the injection at day 3 are included in the ESI (Fig. S9†). Qualitatively, K2, R2, and alum are all well tolerated in vivo as demonstrated by similarly low levels of cleaved caspase-3 staining inside and around the boluses (Fig. 6). Furthermore, these images show that K2 and R2 are more highly infiltrated by cells than alum as indicated by the larger number of blue nuclei in the hydrogels, which is consistent with the H&E staining of these materials (Fig. 5C–E). These results suggest that all materials tested cause minimal tissue damage at the site of injection and that the mechanism of adjuvancy of MDP hydrogels is not overt cytotoxicity.
 | ||
| Fig. 6 Representative caspase-3 staining of C57BL/6J mouse tissue collected 3 days after injection. Brown indicates the presence of caspase-3 (apoptosis), and blue is a nuclei counterstain. Images of OVA in (A) K2, (B) R2, and (C) alum where the black scale bar represents 200 μm show minimal apoptosis inside and around the adjuvants. | ||
Innate immune response
Histology and release data demonstrate that alum and positively charged MDP hydrogels have a longer persistence time in the body than E2 or OVA-only injections. This is in agreement with previous investigations that have shown that positively charged gels degrade slower than negatively charged gels in vivo.36 It was also observed that alum, K2, and R2 generated significantly larger populations of activated non-T lymphocyte (CD3−) immune cells in the spleen at the 3-week time point compared to soluble OVA and E2 (Fig. 4E). This elevation in activated CD3− immune cells is likely the result of the inflammatory nature and longer persistence time of these materials. R2 (not loaded with any protein) has been previously shown to be the most pro-inflammatory MDP hydrogel of variants tested.36 Interestingly, however, the increased inflammatory response to this material did not lead to the generation of the best antigen-specific adaptive immune response or the highest observed populations of activated CD3− immune cells. These results suggest that a high magnitude of local non-specific inflammation generated by R2 at the site of injection is not necessarily beneficial for the activation of spleen-resident CD3− immune cells or, as a result, the generation of a robust adaptive immune response.The results in Fig. 4E show that positively charged MDPs have significantly higher populations of activated CD3− immune cells than the negatively charged E2 hydrogel. These results are congruent with previous investigations that found positively charged MDPs to result in greater local inflammation and increased production of inflammatory cytokines compared to negatively charged MDPs in vivo.36 The higher degree of activation in spleen-resident CD3− immune cells from positively charged MDPs is likely due to the proinflammatory nature of strongly cationic materials. Previous investigations have found that modifying lipid nanoparticles and chitosan-based microparticles with positive charges enhanced production of proinflammatory cytokines through activation of the complement system.57,58 It has also been shown that cationic liposomes evoke proinflammatory responses in neutrophils whereas anionic liposomes do not. These effects were attributed to the ability of cationic liposomes to disrupt the negatively charged cell membrane of neutrophils and lead to the production of proinflammatory superoxide compounds.59 We suspect that a combination of cellular stress due to membrane disruption and complement activation is responsible for increased activation of CD3− immune cells, which will be further explored in future investigations.
Humoral and cellular immune responses
In this study, we investigate the potential for self-assembling peptide hydrogels to act as adjuvants for a model antigen. All MDP hydrogels tested generated significantly improved antigen-specific humoral immune responses over the unadjuvanted vaccine control, but the antibody titers generated were not as high as antigen adjuvanted with alum (Fig. 4A). Of the MDP hydrogels, K2 generated the highest antigen-specific IgG and IgM titers while E2 generated the lowest IgG titers at week 11 (Fig. 4B). These differences are likely due to the varying inflammatory nature of these two peptides, which is supported by a previous fundamental study of the innate immune response to MDPs that showed that E2 is minimally inflammatory in vivo.36 Surprisingly, E2 generated statistically non-inferior IgG and IgM responses compared to R2 despite a drastic 7-day difference in antigen residence time and the differing inflammatory nature between the two hydrogels (Fig. 3C and 4B). IgG titers for vaccines adjuvanted with both R2 and E2 plateau around 3 weeks post-injection (Fig. 4A). These data suggest that the depot effect may not be the driving force behind the immune response generated by MDPs.None of the adjuvants generated a particularly robust cellular immune response; however, as indicated by significantly lower IgG2c titers and smaller populations of CD8+ tetramer+ splenocytes than alum, MDP hydrogels potentiated nearly exclusive humoral immunity (Fig. 4B and C). As alum is already known to be a limited Th1 adjuvant, the fact that MDPs generated an even smaller Th1 response indicates that these materials potentiate a strongly biased Th2 immune response. Minimal differences were observed in populations of activated CD86+ CD8+ T cells for all the groups tested. The only statistically significant difference observed was that alum had a higher percentage of activated CD8+ T cells than E2, indicating that it may induce the weakest Th1 activation of the adjuvants tested (Fig. S8A†). To elucidate the mechanism behind this preference for Th2 immunity and determine if any other immune pathways are being activated in response to MDP adjuvants, helper T cell sub-populations in the spleens of vaccinated mice were characterized 3 weeks after inoculation (Fig. S8B†). No significant differences were observed between the adjuvanted vaccines and unadjuvanted control in the populations of Th1, Th2, Th9, follicular helper T cells (TfH), and Th17 cells. These data suggest that the preference for Th2 immunity is not a result of higher populations of B cell activating TfH and Th2 cells or lower populations of Th1 cells in the spleens of mice vaccinated with MDP-adjuvanted OVA. Additionally, there seems to be no generation of Th9 and Th17 helper T cell populations with the alum or MDP hydrogel groups. The only significant differences observed were between alum and E2 in the populations of Th1 and Th9 cells, further supporting the conclusion that E2 is the weakest adjuvant of the three MDP hydrogels tested. Future investigations will be needed to elucidate the mechanism behind the preference for Th2 immunity in vaccines adjuvanted with MDP hydrogels.
The ability to generate exclusive Th2 immunity is of interest for immunomodulation purposes. For example, in Alzheimer's disease vaccine research, it has been hypothesized that the generation of Th1 immunity may hinder vaccine efficacy by leading to side effects such as organ damage and acute meningoencephalitis.60,61 The results of the current study suggest that MDPs may be good adjuvant candidates for these vaccines, and the immunogenicity of MDP hydrogels in older animals should be investigated in future studies. In addition to being potentially useful for Alzheimer's disease, MDP hydrogel adjuvants may be useful in infectious disease applications as well. A balanced Th1/Th2 immune response is favorable for many applications; however, vaccines against extracellular pathogens generally aim to confer protective humoral immunity.10 MDP hydrogel adjuvants may also provide an alternative vaccine adjuvant to alum for individuals who are known to have experienced aluminum-mediated side effects from previous infectious disease vaccines.62
Conclusion
In this study, we have demonstrated that MDP hydrogels potentiate strongly biased Th2 immunity to the model antigen OVA. The positively charged MDP hydrogels, but not the negatively charged E2 MDP, were able to act as antigen depots that released OVA over 7–9 days. Furthermore, these materials generated significantly higher antigen-specific IgG titers than the unadjuvanted vaccine control. These materials could be useful in a broad range of applications for pathologies that would benefit from a biased Th2 immune response such as Alzheimer's disease and certain infectious diseases. Future studies will aim to improve upon the immunostimulatory and immunomodulatory potential of MDP hydrogels to provide full control of the immune phenotype elicited.Author contributions
Conceptualization, B. H. P., J. D. H., and K. J. M.; data curation, B. H. P.; formal analysis, B. H. P., J. D. H., and K. J. M.; funding acquisition, J. D. H., and K. J. M.; investigation, B. H. P., M. H. Y., A. R. A., E. M. E., C. S. E. L., G. S., S. X. W., A. C. F., and S. M. M.; methodology, B. H. P, and A. R. A.; project administration, B. H. P., J. D. H., and K. J. M.; resources, B. H. P., A. R. A., G. S., and T. P. G.; supervision, D. G. W., J. D. H., and K. J. M., validation, B. H. P., visualization, B. H. P., J. D. H., and K. J. M.; writing – original draft, B. H. P., writing – review and editing, B. H. P., E. M. E., C. S. E. L., T. P. G., D. G. W., J. D. H., and K. J. M.Conflicts of interest
K. J. M. is a paid consultant for Particles for Humanity, a public benefit corporation, though the technology described herein is unrelated to that role.Acknowledgements
This work was supported in full (100%) by public funding. B. H. P., M. H. Y., and C. S. E. L. were supported by the National Science Foundation Graduate Research Fellowship Program. This work was supported by the National Institutes of Health Grants R03EB031495, K22AI146215, R01DE021798, and R01DE030140. The following reagent was obtained through the NIH Tetramer Core Facility: SIINFEKL tetramer. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors would like to acknowledge Amy Caivano for her help with flow cytometry and Joseph Swain for helping design Fig. 1. The table of contents figure was created using BioRender.com.References
- X. Li, C. Mukandavire, Z. M. Cucunubá, S. Echeverria Londono, K. Abbas, H. E. Clapham, M. Jit, H. L. Johnson, T. Papadopoulos, E. Vynnycky, M. Brisson, E. D. Carter, A. Clark, M. J. de Villiers, K. Eilertson, M. J. Ferrari, I. Gamkrelidze, K. A. M. Gaythorpe, N. C. Grassly, T. B. Hallett, W. Hinsley, M. L. Jackson, K. Jean, A. Karachaliou, P. Klepac, J. Lessler, X. Li, S. M. Moore, S. Nayagam, D. M. Nguyen, H. Razavi, D. Razavi-Shearer, S. Resch, C. Sanderson, S. Sweet, S. Sy, Y. Tam, H. Tanvir, Q. M. Tran, C. L. Trotter, S. Truelove, K. van Zandvoort, S. Verguet, N. Walker, A. Winter, K. Woodruff, N. M. Ferguson and T. Garske, Estimating the health impact of vaccination against ten pathogens in 98 low-income and middle-income countries from 2000 to 2030: a modelling study, Lancet, 2021, 397, 398–408 CrossRef.
- A. H. Banday, S. Jeelani and V. J. Hruby, Cancer vaccine adjuvants – recent clinical progress and future perspectives, Immunopharmacol. Immunotoxicol., 2015, 37, 1–11 CrossRef CAS.
- D. J. Marciani, A retrospective analysis of the Alzheimer's disease vaccine progress – The critical need for new development strategies, J. Neurochem., 2016, 137, 687–700 CrossRef CAS PubMed.
- D. T. O'Hagan, R. N. Lodaya and G. Lofano, The continued advance of vaccine adjuvants – ‘we can work it out’, Semin. Immunol., 2020, 50, 101426 CrossRef.
- Y. Perrie, E. Kastner, R. Kaur, A. Wilkinson and A. J. Ingham, A case-study investigating the physicochemical characteristics that dictate the function of a liposomal adjuvant, Hum. Vaccines Immunother., 2013, 9, 1374–1381 CrossRef CAS.
- O. S. Kumru, S. B. Joshi, D. E. Smith, C. R. Middaugh, T. Prusik and D. B. Volkin, Vaccine instability in the cold chain: Mechanisms, analysis and formulation strategies, Biologicals, 2014, 42, 237–259 CrossRef CAS PubMed.
- R. Kaur, V. W. Bramwell, D. J. Kirby and Y. Perrie, Pegylation of DDA:TDB liposomal adjuvants reduces the vaccine depot effect and alters the Th1/Th2 immune responses, J. Controlled Release, 2012, 158, 72–77 CrossRef CAS PubMed.
- T. Jansen, M. P. M. Hofmans, M. J. G. Theelen, F. G. A. Manders and V. E. J. C. Schijns, Dose and timing requirements for immunogenicity of viral poultry vaccine antigen: investigations of emulsion-based depot function, Avian Pathol., 2007, 36, 361–365 CrossRef CAS PubMed.
- N. Garcon and M. Friede, in Plotkin's Vaccines, Elesvier, Philadelphia, PA, 7th edn, 2018, pp. 61–74 Search PubMed.
- P. Kidd, Th1/Th2 balance: the hypothesis, its limitations, and implications for health and disease, Altern. Med. Rev., 2003, 8, 223–246 Search PubMed.
- S. Romagnani, T-cell subsets (Th1 versus Th2), Ann. Allergy, Asthma, Immunol., 2000, 85, 9–21 CrossRef CAS.
- C. H. van Dyck, Anti-Amyloid-β Monoclonal Antibodies for Alzheimer's Disease: Pitfalls and Promise, Biol. Psychiatry, 2018, 83, 311–319 CrossRef CAS.
- L. A. R. Froes, M. A. B. Trindade and M. N. Sotto, Immunology of leprosy, Int. Rev. Immunol., 2020, 1–21 Search PubMed.
- E. A. McNeela and K. H. G. Mills, Manipulating the immune system: humoral versus cell-mediated immunity, Adv. Drug Delivery Rev., 2001, 51, 43–54 CrossRef CAS PubMed.
- B. H. Pogostin and K. J. McHugh, Novel Vaccine Adjuvants as Key Tools for Improving Pandemic Preparedness, Bioengineering, 2021, 8, 155 CrossRef CAS PubMed.
- P. Kalita and T. Tripathi, Methodological advances in the design of peptide-based vaccines, Drug Discovery Today, 2022, 27(5), 1367–1380 CrossRef CAS PubMed.
- T. Abudula, K. Bhatt, L. J. Eggermont, N. O’Hare, A. Memic and S. A. Bencherif, Supramolecular Self-Assembled Peptide-Based Vaccines: Current State and Future Perspectives, Front. Chem., 2020, 8 DOI:10.3389/fchem.2020.598160.
- E. D. Santis and M. G. Ryadnov, Peptide self-assembly for nanomaterials: the old new kid on the block, Chem. Soc. Rev., 2015, 44, 8288–8300 RSC.
- J. S. Rudra, Y. F. Tian, J. P. Jung and J. H. Collier, A self-assembling peptide acting as an immune adjuvant, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 622–627 CrossRef CAS PubMed.
- R. R. Pompano, J. Chen, E. A. Verbus, H. Han, A. Fridman, T. McNeely, J. H. Collier and A. S. Chong, Titrating T-Cell Epitopes within Self-Assembled Vaccines Optimizes CD4+ Helper T Cell and Antibody Outputs, Adv. Healthcare Mater., 2014, 3, 1898–1908 CrossRef CAS PubMed.
- G. A. Hudalla, J. A. Modica, Y. F. Tian, J. S. Rudra, A. S. Chong, T. Sun, M. Mrksich and J. H. Collier, A self-adjuvanting supramolecular vaccine carrying a folded protein antigen, Adv. Healthcare Mater., 2013, 2(8), 1114–1119 CrossRef CAS.
- C. Fries, M. Dennis, J. Eudailey, A. Moody, S. Permar, J. Collier and G. Fouda, Multivalent antigen presentation increases the antibody binding breadth and neutralizing potency upon the immunization with a self-assembling HIV env vaccine, J. Int. AIDS Soc., 2021, 24(S1), 45 Search PubMed.
- S. H. Kelly, E. E. Opolot, Y. Wu, B. Cossette, A. K. Varadhan and J. H. Collier, Tabletized Supramolecular Assemblies for Sublingual Peptide Immunization, Adv. Healthcare Mater., 2021, 10, 2001614 CrossRef CAS PubMed.
- Y. Si, Y. Wen, S. H. Kelly, A. S. Chong and J. H. Collier, Intranasal delivery of adjuvant-free peptide nanofibers elicits resident CD8+T cell responses, J. Controlled Release, 2018, 282, 120–130 CrossRef CAS PubMed.
- C. Yan and D. J. Pochan, Rheological properties of peptide -based hydrogels for biomedical and other applications, Chem. Soc. Rev., 2010, 39, 3528–3540 RSC.
- I.-C. Li and J. D. Hartgerink, Covalent Capture of Aligned Self-Assembling Nanofibers, J. Am. Chem. Soc., 2017, 139, 8044–8050 CrossRef CAS PubMed.
- S. E. Miller, Y. Yamada, N. Patel, E. Suárez, C. Andrews, S. Tau, B. T. Luke, R. E. Cachau and J. P. Schneider, Electrostatically Driven Guanidinium Interaction Domains that Control Hydrogel-Mediated Protein Delivery In Vivo, ACS Cent. Sci., 2019, 5, 1750–1759 CrossRef CAS PubMed.
- Y. Tian, H. Wang, Y. Liu, L. Mao, W. Chen, Z. Zhu, W. Liu, W. Zheng, Y. Zhao, D. Kong, Z. Yang, W. Zhang, Y. Shao and X. Jiang, A Peptide-Based Nanofibrous Hydrogel as a Promising DNA Nanovector for Optimizing the Efficacy of HIV Vaccine, Nano Lett., 2014, 14, 1439–1445 CrossRef CAS PubMed.
- H. Wang, Z. Luo, Y. Wang, T. He, C. Yang, C. Ren, L. Ma, C. Gong, X. Li and Z. Yang, Enzyme-Catalyzed Formation of Supramolecular Hydrogels as Promising Vaccine Adjuvants, Adv. Funct. Mater., 2016, 26, 1822–1829 CrossRef CAS.
- H. Jia, Y. Shang, H. Cao, Y. Gao, J. Liu, L. Yang, C. Yang, C. Ren, Z. Wang and J. Liu, A minimalist supramolecular nanovaccine forcefully propels the Tfh cell and GC B cell responses, Chem. Eng. J., 2022, 134782 CrossRef CAS.
- T. J. Bullwinkle, N. M. Reynolds, M. Raina, A. Moghal, E. Matsa, A. Rajkovic, H. Kayadibi, F. Fazlollahi, C. Ryan, N. Howitz, K. F. Faull, B. A. Lazazzera and M. Ibba, Oxidation of cellular amino acid pools leads to cytotoxic mistranslation of the genetic code, eLife, 2014, 3, e02501 CrossRef PubMed.
- K. J. Rodgers and N. Shiozawa, Misincorporation of amino acid analogues into proteins by biosynthesis, Int. J. Biochem. Cell Biol., 2008, 40, 1452–1466 CrossRef CAS PubMed.
- H. Zou, L. Li, T. Zhang, M. Shi, N. Zhang, J. Huang and M. Xian, Biosynthesis and biotechnological application of non-canonical amino acids: Complex and unclear, Biotechnol. Adv., 2018, 36, 1917–1927 CrossRef CAS PubMed.
- T. L. Lopez-Silva, D. G. Leach, I.-C. Li, X. Wang and J. D. Hartgerink, Self-Assembling Multidomain Peptides: Design and Characterization of Neutral Peptide-Based Materials with pH and Ionic Strength Independent Self-Assembly, ACS Biomater. Sci. Eng., 2019, 5, 977–985 CrossRef CAS PubMed.
- V. A. Kumar, S. Shi, B. K. Wang, I.-C. Li, A. A. Jalan, B. Sarkar, N. C. Wickremasinghe and J. D. Hartgerink, Drug-Triggered and Cross-Linked Self-Assembling Nanofibrous Hydrogels, J. Am. Chem. Soc., 2015, 137, 4823–4830 CrossRef CAS.
- T. L. Lopez-Silva, D. G. Leach, A. Azares, I.-C. Li, D. G. Woodside and J. D. Hartgerink, Chemical functionality of multidomain peptide hydrogels governs early host immune response, Biomaterials, 2020, 231, 119667 CrossRef CAS PubMed.
- L. Aulisa, H. Dong and J. D. Hartgerink, Self-Assembly of Multidomain Peptides: Sequence Variation Allows Control over Cross-Linking and Viscoelasticity, Biomacromolecules, 2009, 10, 2694–2698 CrossRef CAS.
- N. C. Carrejo, A. N. Moore, T. L. Lopez Silva, D. G. Leach, I.-C. Li, D. R. Walker and J. D. Hartgerink, Multidomain Peptide Hydrogel Accelerates Healing of Full-Thickness Wounds in Diabetic Mice, ACS Biomater. Sci. Eng., 2018, 4, 1386–1396 CrossRef CAS PubMed.
- A. N. Moore and J. D. Hartgerink, Self-Assembling Multidomain Peptide Nanofibers for Delivery of Bioactive Molecules and Tissue Regeneration, Acc. Chem. Res., 2017, 50, 714–722 CrossRef CAS PubMed.
- N. C. Wickremasinghe, V. A. Kumar and J. D. Hartgerink, Two-Step Self-Assembly of Liposome-Multidomain Peptide Nanofiber Hydrogel for Time-Controlled Release, Biomacromolecules, 2014, 15, 3587–3595 CrossRef CAS PubMed.
- D. G. Leach, N. Dharmaraj, T. L. Lopez-Silva, J. R. Venzor, B. H. Pogostin, A. G. Sikora, J. D. Hartgerink and S. Young, Biomaterial-Facilitated Immunotherapy for Established Oral Cancers, ACS Biomater. Sci. Eng., 2021, 7, 415–421 CrossRef CAS PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, NIH Image to ImageJ: 25 years of image analysis, Nat. Methods, 2012, 9, 671–675 CrossRef CAS.
- M. C. E. van Dalen, J. Vaneyck, S. A. Semerdzhiev, M. Karperien, J. N. Post and M. M. A. E. Claessens, Protein Adsorption Enhances Energy Dissipation in Networks of Lysozyme Amyloid Fibrils, Langmuir, 2021, 37, 7349–7355 CrossRef CAS PubMed.
- C. P. Broedersz and F. C. MacKintosh, Modeling semiflexible polymer networks, Rev. Mod. Phys., 2014, 86, 995–1036 CrossRef CAS.
- D. G. Leach, N. Dharmaraj, S. L. Piotrowski, T. L. Lopez-Silva, Y. L. Lei, A. G. Sikora, S. Young and J. D. Hartgerink, STINGel: Controlled release of a cyclic dinucleotide for enhanced cancer immunotherapy, Biomaterials, 2018, 163, 67–75 CrossRef CAS PubMed.
- D. G. Fatouros, D. A. Lamprou, A. J. Urquhart, S. N. Yannopoulos, I. S. Vizirianakis, S. Zhang and S. Koutsopoulos, Lipid-like Self-Assembling Peptide Nanovesicles for Drug Delivery, ACS Appl. Mater. Interfaces, 2014, 6, 8184–8189 CrossRef CAS PubMed.
- S. Khurana, S. Fuentes, E. M. Coyle, S. Ravichandran, R. T. Davey and J. H. Beigel, Human antibody repertoire after VSV-Ebola vaccination identifies novel targets and virus-neutralizing IgM antibodies, Nat. Med., 2016, 22, 1439–1447 CrossRef CAS PubMed.
- S. Park and M. H. Nahm, Older Adults Have a Low Capacity To Opsonize Pneumococci Due to Low IgM Antibody Response to Pneumococcal Vaccinations, Infect. Immun., 2011, 79(1), 314–320 CrossRef CAS PubMed.
- R. M. Martin, J. L. Brady and A. M. Lew, The need for IgG2c specific antiserum when isotyping antibodies from C57BL/6 and NOD mice, J. Immunol. Methods, 1998, 212, 187–192 CrossRef CAS.
- H. HogenEsch, Mechanisms of stimulation of the immune response by aluminum adjuvants, Vaccine, 2002, 20, S34–S39 CrossRef CAS PubMed.
- K. C. Wolthers, S. A. Otto, S. M. A. Lens, D. N. Kolbach, R. A. W. van Lier, F. Miedema and L. Meyaard, Increased expression of CD80, CD86 and CD70 on T cells from HIV-infected individuals upon activation in vitro: regulation by CD4+ T cells, Eur. J. Immunol., 1996, 26, 1700–1706 CrossRef CAS PubMed.
- S. Axelsson, A. Magnuson, A. Lange, A. Alshamari, E. H. Hörnquist and O. Hultgren, A combination of the activation marker CD86 and the immune checkpoint marker B and T lymphocyte attenuator (BTLA) indicates a putative permissive activation state of B cell subtypes in healthy blood donors independent of age and sex, BMC Immunol., 2020, 21, 14 CrossRef CAS.
- L. Conti, R. Casetti, M. Cardone, B. Varano, A. Martino, F. Belardelli, F. Poccia and S. Gessani, Reciprocal Activating Interaction Between Dendritic Cells and Pamidronate-Stimulated γδ T Cells: Role of CD86 and Inflammatory Cytokines, J. Immunol., 2005, 174, 252–260 CrossRef CAS.
- A. Paine, H. Kirchner, S. Immenschuh, M. Oelke, R. Blasczyk and B. Eiz-Vesper, IL-2 upregulates CD86 expression on human CD4(+) and CD8(+) T cells, J. Immunol., 2012, 188, 1620–1629 CrossRef CAS.
- L. L. Lanier, S. O'Fallon, C. Somoza, J. H. Phillips, P. S. Linsley, K. Okumura, D. Ito and M. Azuma, CD80 (B7) and CD86 (B70) provide similar costimulatory signals for T cell proliferation, cytokine production, and generation of CTL, J. Immunol., 1995, 154, 97–105 CAS.
- D. van Duin, H. G. Allore, S. Mohanty, S. Ginter, F. K. Newman, R. B. Belshe, R. Medzhitov and A. C. Shaw, Prevaccine Determination of the Expression of Costimulatory B7 Molecules in Activated Monocytes Predicts Influenza Vaccine Responses in Young and Older Adults, J. Infect. Dis., 2007, 195, 1590–1597 CrossRef CAS PubMed.
- C. Salvador-Morales, L. Zhang, R. Langer and O. C. Farokhzad, Immunocompatibility properties of lipid–polymer hybrid nanoparticles with heterogeneous surface functional groups, Biomaterials, 2009, 30, 2231–2240 CrossRef CAS PubMed.
- Y. Liu, Y. Yin, L. Wang, W. Zhang, X. Chen, X. Yang, J. Xu and G. Ma, Engineering Biomaterial-Associated Complement Activation to Improve Vaccine Efficacy, Biomacromolecules, 2013, 14, 3321–3328 CrossRef CAS PubMed.
- T.-L. Hwang, C.-Y. Hsu, I. A. Aljuffali, C.-H. Chen, Y.-T. Chang and J.-Y. Fang, Cationic liposomes evoke proinflammatory mediator release and neutrophil extracellular traps (NETs) toward human neutrophils, Colloids Surf., B, 2015, 128, 119–126 CrossRef CAS PubMed.
- D. J. Marciani, Alzheimer's disease vaccine development: A new strategy focusing on immune modulation, J. Neuroimmunol., 2015, 287, 54–63 CrossRef CAS PubMed.
- D. J. Marciani, New Th2 adjuvants for preventive and active immunotherapy of neurodegenerative proteinopathies, Drug Discovery Today, 2014, 19, 912–920 CrossRef CAS PubMed.
- L. Tomljenovic and C. A. Shaw, in Vaccines and Autoimmunity, John Wiley & Sons, Ltd, 2015, pp. 43–56 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm01242a |
| This journal is © The Royal Society of Chemistry 2022 |