
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSoluplus®-based dissolving microarray patches loaded with colchicine: towards a minimally invasive treatment and management of gout
Qonita Kurnia
Anjani
 ab,
Akmal Hidayat Bin
Sabri
a,
Natalia
Moreno-Castellanos
c,
Emilia
Utomo
a,
Álvaro
Cárcamo-Martínez
a,
Juan
Domínguez-Robles
a,
Luki Ahmadi Hari
Wardoyo
d and
Ryan F.
Donnelly
*a
ab,
Akmal Hidayat Bin
Sabri
a,
Natalia
Moreno-Castellanos
c,
Emilia
Utomo
a,
Álvaro
Cárcamo-Martínez
a,
Juan
Domínguez-Robles
a,
Luki Ahmadi Hari
Wardoyo
d and
Ryan F.
Donnelly
*a
aSchool of Pharmacy, Queen's University Belfast, Medical Biology Centre, 97 Lisburn Road, Belfast BT9 7BL, UK. E-mail: r.donnelly@qub.ac.uk
bFakultas Farmasi, Universitas Megarezky, Jl. Antang Raya No. 43, Makassar 90234, Indonesia
cBasic Science Department, Faculty of Health, Universidad Industrial de Santander, Bucaramanga 680001, Colombia
dFakultas Seni Rupa dan Desain, Institut Teknologi Bandung, Jl. Ganesa No.10, Bandung 40132, Indonesia
First published on 16th August 2022
Abstract
Considered as one of the most common inflammatory arthritis, gout is characterised by a sudden onset of severe joint pain. As the first-line drug of choice used in treating acute gout, colchicine (CLC) is hindered by poor gastrointestinal permeability as well as unfavourable gastrointestinal side effects. Herein, we present, for the first time, the preparation of microarray array patches (MAPs) made of a polymeric solubiliser, Soluplus®, loaded with CLC for its systemic delivery. The fabricated MAPs displayed acceptable mechanical properties and were capable of being inserted into the skin to a depth of ≈500 μm in full thickness ex vivo neonatal porcine skin, as evidenced by optical coherence tomography. In vitro dermatokinetic studies utilising full thickness neonatal porcine skin demonstrated that the CLC-loaded MAPs delivered CLC across all skin strata, resulting in a delivery efficiency of 73% after 24 hours. Furthermore, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) and cell proliferation assays along with LIVE/DEAD™ staining on the 3T3-L1 cell line showed that the MAP formulation displayed minimal toxicity, with acceptable biocompatibility. Lastly, the anti-inflammatory properties of the formulation were evaluated using a THP-1 macrophage cell line. It was shown that treatment of THP-1 macrophages that are exposed to lipopolysaccharide (LPS) with CLC-loaded MAPs caused a significant (p < 0.05) reduction of TNF-α production, a pro-inflammatory cytokine typically associated with the early onset of acute gout. Accordingly, CLC-loaded MAPs could represent a new minimally-invasive alternative strategy for management of acute gout.
1. Introduction
Gout is considered as one of the most common inflammatory arthritis. Some of the cardinal features of the disease include the sudden onset of severe joint pain which can lead to swelling and redness of the affected area. This form of arthritis typically occurs as a result of hyperuricaemia (serum urate >6.8 mg dL−1), triggering the formation of tophi around the affected joints.1 The disease affects around 4.2% of the UK population, with prevalence rising at a global scale, due to an ever-rising ageing population.2 If left untreated, acute flare-ups can occur and last from hours to weeks, which can impose a significant burden to patients’ quality of life while simultaneously leading to irreversible joint damage.3According to the National Institute of Clinical Excellence (NICE) the current first line pharmacological agents in the management of acute gout are colchicine (CLC), nonsteroidal anti-inflammatory drugs (NSAIDs) and oral corticosteroids.4 Although NSAIDs are efficacious in managing acute gout, this class of drugs, due to the high doses used, may induce adverse side effects, such as gastrointestinal (GI) ulcers in some patients.5–8 On the other hand, treatment with corticosteroids has been shown to provide some level of efficacy in managing acute gout. However, the use of high doses of corticosteroids for extended periods in treating recurrent flare-ups may lead to unwanted side effects in patients in the long run.9 Oral administration of CLC is associated with several drawbacks. Firstly, delivering CLC orally typically results in GI side effects, such as diarrhoea, nausea, vomiting, and abdominal pain which leads to poor treatment compliance.10 Secondly, CLC belong to the class III category based on the biopharmaceutical classification system (BCS), displaying poor gastrointestinal permeability and, hence, significant variability in terms of bioavailability (24–88%) between patients.11
In an attempt to obviate these issues, an alternative formulation strategy could be explored via the use of microarray patches (MAPs). MAPs consist of several micron-size projections that range between 25 and 1000 μm in height, assembled on a supporting baseplate. When MAPs are inserted into the skin, the micro-projections can effectively pierce the stratum corneum, resulting in the formation of transient aqueous channels which can be used to deliver drug molecules to the extensive dermal microcirculation.12 During the early stages of MAP research, most of the needles were produced using inorganic materials, such as metal, glass and silicon. These are known as solid MAPs and are used in tandem with topical formulations for delivering pharmaceuticals into and across the skin. However, microneedle-formed channels have a limited lifetime, reducing the utility of this approach for drug delivery.13–15 This limitation arises due to the intrinsically regenerative nature of the skin, causing the pores to close up quickly within a matter of minutes after application16 up to several hours.17,18 This would not only limit the dose of drugs that can be administered via this strategy but may also lead to considerable variability in the dose delivered. In addition, the application of topical products to MAP-treated skin is not advisable since they are non-sterile and may contain excipients that were never intended to reach the viable skin layers. Owing to this limitation, there has been a paradigm shift to utilise other types of MAPs, such as dissolving MAPs, to administer therapeutics into and across the different layers of the skin. Using the dissolving MAPs approach, the payload is encapsulated within a polymeric matrix that forms the body of the needles. When the needle layers of the MAPs are inserted into the skin, the contact between the matrix with interstitial fluid leads to polymer dissolution which in turn promotes the release of the payload into the skin.
Regarding dissolving MAPs, the selection of biodegradable and biocompatible polymers that can be used to manufacture these types of MAPs are limited.19 To date, the vast majority of dissolving MAPs are manufactured from common pharmaceutical polymers such as carboxy-methylcellulose,20 poly(vinylpyrrolidone) (PVP),21 polyvinyl pyrrolidone-co-vinyl acetate,22 chondroitin sulfate,23 sodium hyaluronate (HA)24 and poly(vinyl alcohol) (PVA).25 In addition, it has been hypothesised that utilising water-soluble polymers typically results in mechanically weaker MAPs relative to non-dissolving inorganic materials, such as silicon or metal.26,27 This is further exacerbated by the encapsulation of a payload, which may further weaken the overall mechanical strength of the MAPs.23 In light of these limitations, there is a need for alternative pharmaceutical-grade polymers which can be used to fabricate MAPs with acceptable mechanical strength whilst enabling effective delivery of the active substance for the treatment of gout.
Soluplus®, polyvinyl caprolactam polyvinyl acetate-polyethylene glycol grafted copolymer, is a novel amphiphilic pharmaceutical polymer which was initially designed to be utilised in the manufacture of amorphous solid dispersions.28 In comparison to classical solubilizers such as Cremophore RH40 and Solutol HS15, Soluplus® can function as a matrix polymer to stabilise formulation, as well as an serving the role of a polymeric solubilizer via micellar solubilisation when present an aqueous milieu.29–32 Owing this characteristic, Soluplus® has been widely used as film former with addition of plasticizer to reduce the brittleness.33,34 Therefore, owing to its mechanical properties, biocompatibility as well as biodegradability, Soluplus® may serve as potentially novel material to fabricate dissolving MAPs.
In the current work, we detail the manufacture of a composite pharmaceutical system consisting of CLC-loaded dissolving MAPs fabricated with the polymeric solubiliser Soluplus®. In order to develop the MAP system, CLC was amorphized via lyophilisation in the presence of varying concentrations of Soluplus®. The properties of CLC post lyophilisation were evaluated and characterised before the drug was loaded into dissolving MAPs. Then, fabricated MAPs were evaluated in terms of mechanical resistance to a compressive force, as well as the insertion profiles of the MAPs into suitable skin models. Furthermore, CLC delivery into and across the skin was assessed in order to understand the drug delivery kinetics. The pharmaceutical system developed in the current work may provide a patient-friendly strategy to administer CLC transdermally in a painless fashion for the treatment of gout.
2. Materials and methods
2.1. Materials
Colchicine (purity, 95%) was procured from Alfa Aesar (Lancashire, UK). Soluplus® was kindly donated by BASF (Ludwigshafen, Germany). PVP 90 kDa (Plasdone™ K-29/32) was obtained by Ashland (Kidderminster, UK). HPLC grade water used in the current work was supplied from a water purification system (Elga PURELAB DV 25, Veolia Water Systems, Dublin, Ireland). All other reagents that were of analytical grade were procured from Sigma-Aldrich (Dorset, UK) or Fisher Scientific (Loughborough, UK). The full-thickness neonatal porcine skins used in the current work were sourced from stillborn piglets within 24 h post-mortem and frozen at −20 °C prior to experimentation.2.2. Preparation of CLC-Soluplus® powder
A serial dilution of Soluplus® solution were prepared at concentrations of 0.25, 0.5, 1.0 and 2.0% w/v. Next, 3 mL of each Soluplus® solution were added to 120 mg of CLC and blended using a SpeedMixer™ DAC 150.1 FVZ-K (GermanEngineering, Hauschild & Co. KG, Hamm, Germany) at 3500 rpm for 3 min. The drug–polymer blend was placed into a temperature-controlled freezer at −80 °C for 3 h prior to lyophilisation. Lyophilisation was carried out using a freeze drier (Virtis™ Advantage XL-70, SP Scientific, Warminster, PA, USA) over the course of 24 h, utilising a primary drying cycle of 13 h, with the shelf temperature that commences at −40 °C. Subsequently, the secondary drying commenced for the remaining 11 h at 25 °C under a vacuum of 50 mTorr (Fig. 1).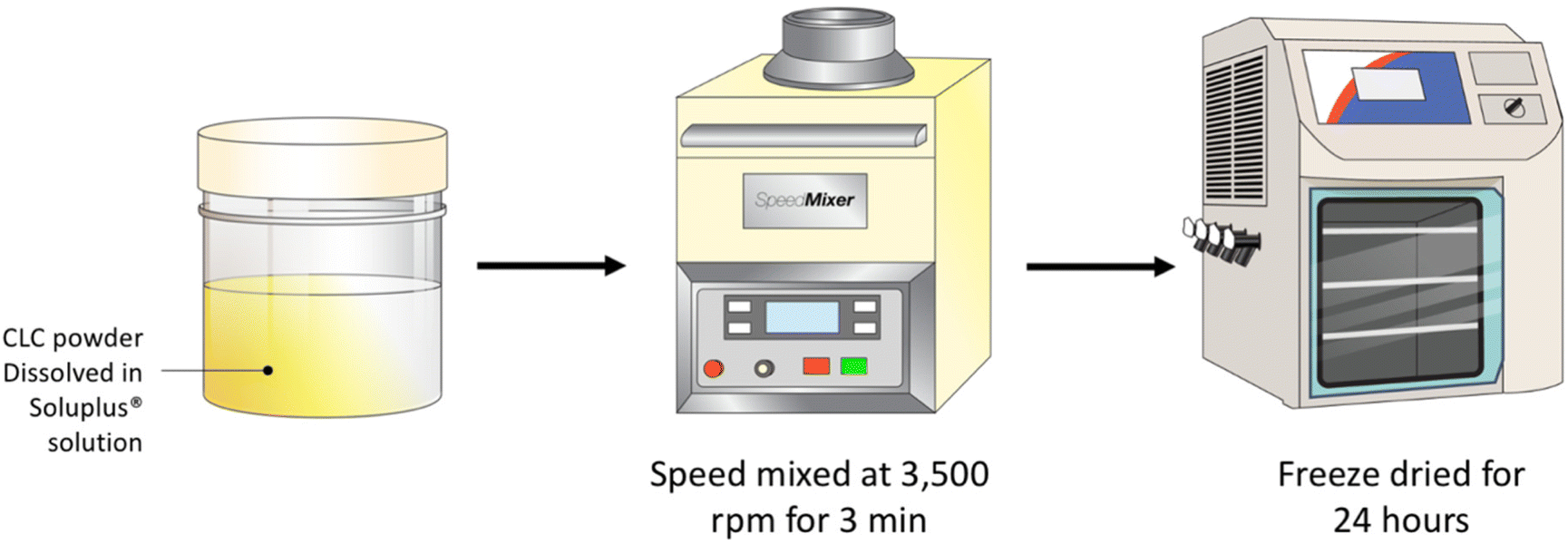 | ||
| Fig. 1 Schematic illustration of the preparation of CLC-Soluplus® powders via lyophilisation. | ||
2.3. Characterisation of CLC-Soluplus® powder
Quasi Elastic Light Scattering (NanoBrook Omni® analyser, Brookhaven, New York, NJ, USA) was utilised in order to evaluate size distribution of the CLC-Soluplus® particles formed. An aliquot (10 μL) of CLC-Soluplus® aqueous dispersion was diluted in 2 ml of deionised water and vortexed at 2500 rpm for 1 min. Next, the diluted mixture was transferred into a disposable cuvette for analysis. The analysis was conducted at a temperature 25 °C while implementing an equilibration time of 3 min prior to sample analysis. All samples were analysed in triplicates. CLC-Soluplus® powder was also visualised using scanning electron microscopy (SEM) analysis with a TM3030 microscope (Hitachi, Krefeld, Germany). Prior to analysis, the samples were left to dry for 24 h under ambient conditions. The chemical interactions between CLC and Soluplus® were investigated using a Fourier transform infrared (FTIR) spectrometer (Accutrac FT/IR-4100™ Series, PerkinElmer, USA). The crystallinity of pure CLC, pure Soluplus®, physical mixture (PM) and CLC-Soluplus® powder were determined using a differential scanning calorimeter DSC Q20 (TA Instruments, Elstree, Hertfordshire, UK) and an X-ray diffractometer (Rigaku Corporation, Kent, England).2.4 Fabrication of dissolving MAPs
A double casting strategy was used to prepare CLC-Soluplus® powder-loaded dissolving MAP, as detailed in our previous work with some adjustment to the protocol.21 Briefly CLC-Soluplus® powder and deionized water (33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :67 w/w) were mixed and 50 mg of the mixture was casted into a silicone mould (16 × 16 pyramidal needle density, 850 μm height, 300 μm width at base, 300 μm interspacing and 0.36 cm2 patch area) as illustrated in Fig. 2 in order to form the needle layer of the formulation. The composition of this layer is outlined in Table 1. The moulds were placed in an enclosed chamber which exert a positive pressure of 4 bar for 5 min. Next, any remaining formulation that was still present above the moulds was meticulously removed using a spatula, before leaving the moulds to air dry for 30 min under the same positive pressure. Subsequently, PDMS rings (external diameter 23 mm, internal diameter 18 mm, thickness 3 mm) was attached to the moulds using 40% of w/w PVA (9–10 kDa). Once the ring has been secured and dried, 850 μL of baseplate solution was added on top of the needle layer. The baseplate solution consisted of 30% w/w of PVP (90 kDa) with the addition of 1.5% of w/w glycerol (as a plasticiser. Upon demoulding, excess sidewalls formed were meticulously removed before the MAPs were left to dry in a thermostaticall-controlled oven set to 37 °C for an additional 12 h.
:67 w/w) were mixed and 50 mg of the mixture was casted into a silicone mould (16 × 16 pyramidal needle density, 850 μm height, 300 μm width at base, 300 μm interspacing and 0.36 cm2 patch area) as illustrated in Fig. 2 in order to form the needle layer of the formulation. The composition of this layer is outlined in Table 1. The moulds were placed in an enclosed chamber which exert a positive pressure of 4 bar for 5 min. Next, any remaining formulation that was still present above the moulds was meticulously removed using a spatula, before leaving the moulds to air dry for 30 min under the same positive pressure. Subsequently, PDMS rings (external diameter 23 mm, internal diameter 18 mm, thickness 3 mm) was attached to the moulds using 40% of w/w PVA (9–10 kDa). Once the ring has been secured and dried, 850 μL of baseplate solution was added on top of the needle layer. The baseplate solution consisted of 30% w/w of PVP (90 kDa) with the addition of 1.5% of w/w glycerol (as a plasticiser. Upon demoulding, excess sidewalls formed were meticulously removed before the MAPs were left to dry in a thermostaticall-controlled oven set to 37 °C for an additional 12 h.
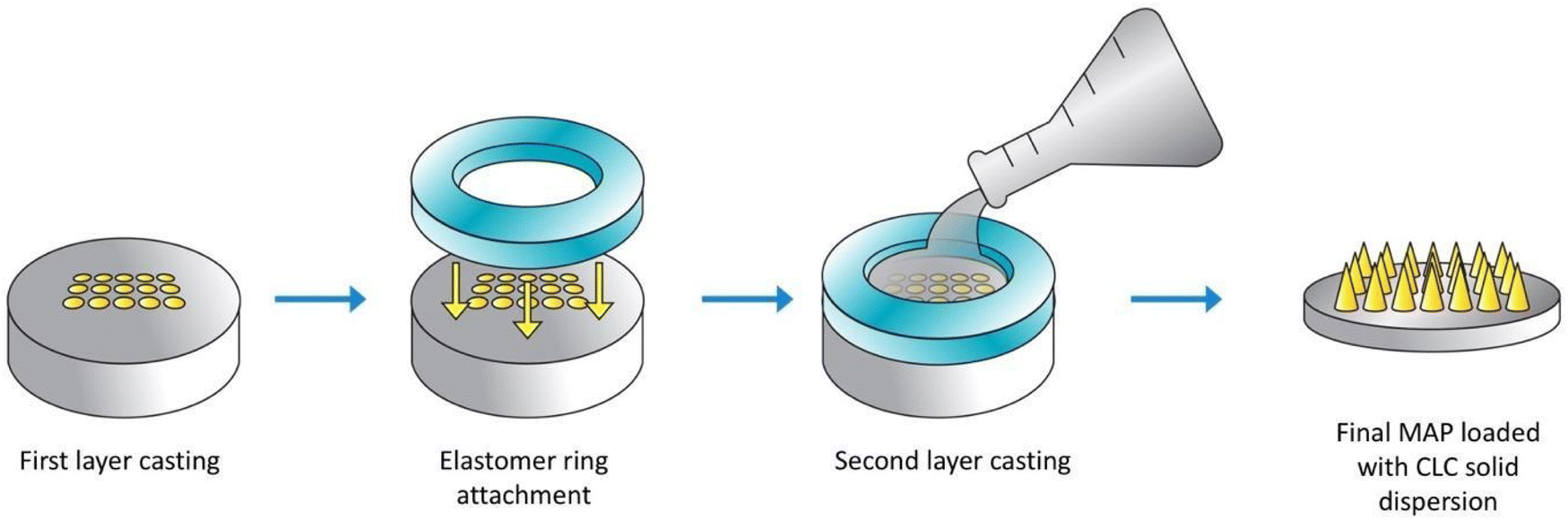 | ||
| Fig. 2 Schematic illustration of the preparation of dissolving MAPs loaded with CLC. | ||
| MAP formulation code | Drug:Soluplus® ratio |
|---|---|
| F1 | 8:1 |
| F2 | 4:1 |
| F3 | 2:1 |
| F4 | 1:1 |
2.5 Mechanical resistance and ex vivo skin insertion study
The architecture and appearance of the MAPs were observed using an optical microscope (Leica EZ4 D, Leica Microsystems, Milton Keynes, UK). Differential scanning calorimetry (DSC) was conducted on the neat drug and respective formulations. This was done using DSC Q100 (TA Instruments, Elstree, Hertfordshire, UK). The resistance of the needles under compression was ascertained using a TA-TX2 Texture Analyser (TA) (Stable Microsystems, Haslemere, UK) using the same parameters which have been previously reported.35,36 Upon exposure to a compressive force of 32 N, changes in needle height was estimated using eqn (1).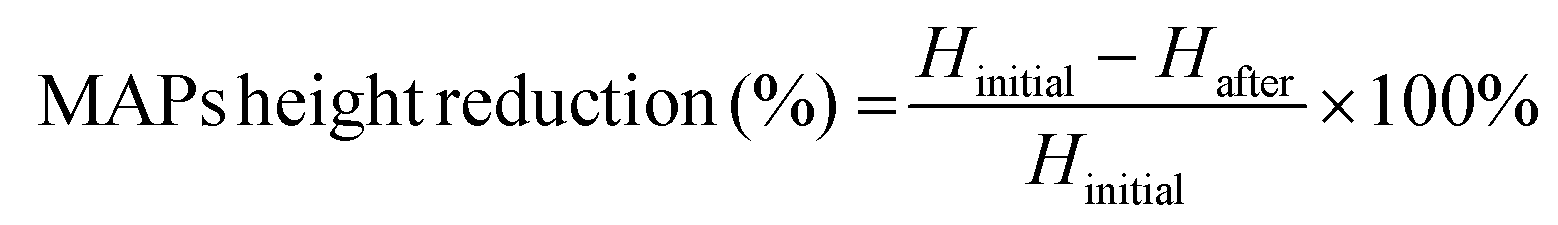 | (1) |
The insertion profile of the MAPs into Parafilm® M and ex vivo neonatal porcine skin (with a thickness of approximately 600 μm) was investigated using an EX-101 optical coherence tomography (OCT) microscope (Michelson Diagnostics Ltd, Kent, UK), as reported previously.37 Upon acquiring the OCT images, the insertion depth of the needles was visualised and calculated using ImageJ® software (National Institutes of Health, Bethesda, MD, USA).
2.6 Evaluation of drug loading within MAPs
The drug loading within MAPs was evaluated by first dissolving the formulation in 4 mL. The dissolution process was promoted by subjecting the patch to a sonication cycle of 30 minutes. The mixture was diluted by a factor of ×2 using acetonitrile and sonicated for an additional 30 min. The final mixture was subjected to a centrifugation cycle of 14500 rpm for 15 min before the sample was analysed via HPLC.
2.7 MAP skin dissolution studies
In order to investigate the time needed for polymeric needles to dissolve within the skin, an in situ ex vivo skin dissolution study was conducted. Excised full-thickness neonatal porcine skin was first immersed in PBS (pH 7.4) for 30 min. This is done to enable the tissue to equilibrate and acclimatise to ambient temperature prior to MAP insertion. CLC-loaded MAPs from F1–F4 were inserted into the ex vivo skin using manual thumb pressure for 30 s. In order to reduce the likelihood of the patch being dislodged from the skin, stainless-steel cylindrical weights (15.0 g) were secured on top of the MAPs.21 The samples were then placed in a thermostatically controlled oven at 37 °C to mimic physiological conditions. At predetermined time points (30 min, 1-hour and 3-hour), MAPs were carefully removed from the skin and their morphology as well as the skin were visualised and captured using a digital microscope.2.8 Dermatokinetic studies
Dermatokinetic studies using excised full thickness neonatal porcine skin with a thickness of approximately 600 μm was performed in order to study the delivery of CLC from MAPs. In this work, vertical Franz diffusion cells (Permergear, Hellertown PA, USA) was used. Using cyanoacrylate adhesive (Stick it® super glue, PLDZ Pattison House, Dublin, ROI), the excised skin was attached to the donor component of the apparatus. PBS (pH 7.4) at 37 °C which has been degassed was used as the receiver fluid. Using a thermostatically regulated water bath, the experimental set up was maintained at 37 ± 1 °C. In addition, the receiver compartment contained 12 mL of PBS (pH 7.4), which was stirred at a constant rate of 600 rpm. The prototype MAPs loaded with CLC were applied to the excised skin under manual pressure for 0.5 min, before being clamped to the receiver compartment. Once inserted, the MAP is secured in place by placing a stainless-steel cylindrical weight (diameter 1.4 cm, height 1.0 cm, weight 15.0 g). In addition, CLC- Soluplus® film that contained a similar drug loading to MAPs (2.8 mg) as well as a CLC solution were used as control. In this case, 20 μL of CLC solution were placed on the skin surface in the donor compartment. In order to reduce the risk of the receiver fluid evaporating throughout the duration of the experiment, the sampling arm and the donor component was meticulously sealed using Parafilm®M. At pre-determined time points, the Franz cells were disassembled and 200 μL of the receiver solution was sampled, filtered and analysed via HPLC. The porcine skins were heat treated at 60 °C for 5 min to facilitate the separation of the epidermis and dermis.38 Upon separating, the epidermal layer was homogenised in 2 mL of acetonitrile using a Vortex™ mixer (Fisons Scientific Equipment, Leicestershire, UK) for 60 seconds and then sonicated for 0.5 hours. On the other hand, the dermal tissue was homogenised in 0.5 ml of deionised water using a Tissue Lyser LT (Qiagen Ltd, Manchester, UK). The homogenisation step was carried out at 50 Hz for 15 min. Upon homogenising, 1 mL of acetonitrile was added to the samples followed by another cycle of homogenisation for 15 minutes. The skin samples were subjected to a centrifugation cycle of 14000 rpm for a period of 15 min. The supernatant was collected and filtered through a 0.45 μm nylon membrane prior to HPLC analysis.
2.9 Biocompatibility study
The biocompatibility of fibroblast cells (3T3-L1 ATCC CL-173) and macrophages cells (THP-1) with CLC and Soluplus® were evaluated through a series of biocompatibility assays. Fibroblast cells were then plated onto 24-well plate and cultured with Dulbecco's modified Eagle's medium (DMEM) growth media (Sigma-Aldrich, St Louis, MS, USA). On the other hand, Human THP-1 monocytes cell lines were grown in suspension using RPMI 1640 media (Sigma-Aldrich, St Louis, MS, USA). THP-1 monocytes were differentiated into THP-1 macrophages by culturing the monocyte at a cell density of 1 × 106 cells per ml followed by a treatment with Phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich, St Louis, MS, USA) at a concentration of 20 ng ml−1, as described previously.39 The monocytes were incubated for 48 h to induce their differentiation. Sterilised MAPs were dispersed with 0.25% (w/v) trypsin-0.53 mM ethylenediaminetetraacetic acid (EDTA) solution before being added to the cell lines. Throughout the study, fibroblast and THP-1 cell lines were supplemented with 10% fetal bovine serum (v/v) (Sigma-Aldrich, St Louis, MS, USA), 1% sodium pyruvate (v/v) (Sigma-Aldrich, St Louis, MS, USA) and 1% antibiotic–antimycotic solution (v/v) containing penicillin, streptomycin, and Amphotericin B (Sigma-Aldrich, St Louis, MS, USA). Cells were incubated with the formulation for 72 h at 37 °C with 5% CO2 under 60% humidified atmosphere.For the MTT (3-(4.5-dimethylthiazol-2-yl)-2.5-diphenyltetrazolium bromide) assay, the growth media of cell lines was replaced with MTT solution (0.5 mg mL−1) for 5 h. Subsequently, the supernatant was removed, followed by the addition of 200 μL dimethylsulfoxide (DMSO) (Sigma-Aldrich, St Louis, MS, USA). Cells were gently shaken for 15 min to enable the dissolution of formazan crystals. The optical absorbance of formazan was measured using a Synergy H1 microplate reader (Agilent Technologies, St Clara, CA, USA) at 570 nm. To confirm the cell viability, LIVE/DEAD™ staining was also conducted. After culturing the cells for 72 h in the presence of CLC-loaded MAP or Soluplus® blank MAPs, the cells were then stained for 5 min at room temperature using 5 μg mL−1 calcein AM and 5 μg mL−1 ethidium homodimer-1 (EthD-1) (Molecular probes, Eugene, Oregon).
Cell proliferation was evaluated on the fibroblast cell line via DNA content assay. This was done using Quant-iT™ PicoGreen® dsDNA Reagent and Kits (Invitrogen, Thermo Scientific, Waltham, MA, USA). Briefly, the cells were carefully washed with PBS (pH 7.0) (Sigma-Aldrich, St Louis, MS, USA) three times before being placed in 1 mL of lysis buffer that consisted of 10 mM Tris (pH 8), 1 mM EDTA, and 0.2% (v/v) Triton X-100. DNA release was done by agitating the samples for 10 s every 5 min for over a period of 0.5 hours. This procedure was done on ice to mitigate sample deterioration due to any excess heat generated. The samples were thawed on ice, and homogenized for a further 10–15 min. Next, samples were treated with 100 μL of DNA-binding fluorescent dye solution, and the fluorescence was recorded using Synergy H1 microplate reader at an excitation wavelength of 480 nm and an emission wavelength of 520 nm. Lambda DNA was used for the standard curve to calculate the amount of DNA that has been released. Samples were prepared in triplicate.
After obtaining the optical density of each sample for cell viability (MTT assay) and mg ml−1 for proliferation (pico-green assay), sample cell viability were expressed in percentage following the eqn (2):
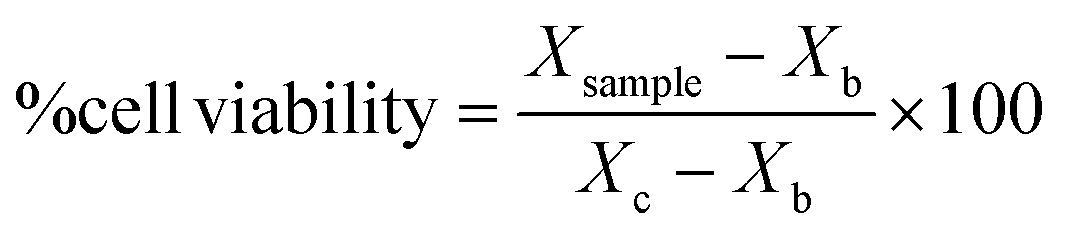 | (2) |
2.10 In vitro anti-inflammation activity
Enzyme-linked immunosorbent assay (ELISA) was used to evaluate TNF-α concentration in the supernatant of THP-1 macrophages when the cells are exposed to lipopolysaccharide (LPS, Sigma-Aldrich, St Louis, MS, USA). This assay was performed on cells that have been treated with either CLC-loaded MAPs or blank Soluplus® MAPs to evaluate if the formulations affected the TNF-α secretion from the THP-1 macrophages during a pro-inflammatory state. In brief, THP-1 cells treated with MAP formulations were incubated for 6 h in the presence of LPS. After this period, cells were centrifuged (1800g, 5 min, 16 °C) and the supernatants were collected and assayed using a TNF-α-specific sandwich quantitative ELISA (Invitrogen, Carlsbad, CA, USA). Total protein concentration was evaluated using the Bradford Protein Assay (Bio-Rad, Hercules, CA, USA) and used to normalise ELISA data. In addition, a control experiment was performed to evaluate if exposure to either CLC-loaded MAPs or blank Soluplus® MAPs would affect TNF-α in the absence of LPS.2.11 High performance liquid chromatography (HPLC) analysis
The CLC content was evaluated by high performance liquid chromatography (HPLC) (Agilent Technologies 1220 Infinity UK Ltd, Stockport, UK). Sample analysis was conducted using Phenomenex® Luna C18 (ODS1) column (150 mm × 4.6 mm i.d. with 5 μm particle size). The mobile phase consisted of acetonitrile and water (50:50 v/v) which was eluted at a flow rate of 1 mL min−1. The detection of CLC was conducted at a detection wavelength, λ of 247 nm under room temperature and pressure. In the current work, sample analysis was conducted using an injection volume of 20 μl. This HPLC method was validated in accordance with the International Council on Harmonisation (ICH) 2005.
2.12 Statistical analysis
Data analysis and presentation was done using GraphPad Prism® version 8.0 (GraphPad Software, San Diego, California, USA). Unless otherwise noted, results are presented as mean ± standard deviation (SD). An unpaired t-test was used to evaluate the statistical difference between two cohorts. In contrast, one-way analysis of variance (ANOVA) was conducted when several treatment cohorts were evaluated for statistical significance. Statistical significances, p < 0.05, were represented based on the following established convention 0.033 (*), 0.002 (**), <0.001 (***) and <0.0001 (****).3. Results and discussion
3.1 Characterisation of CLC-Soluplus® powder
CLC is classified as class III according to the BCS, as this drug displays high aqueous solubility, but poor permeability. Therefore, we developed a composite formulation consisting of MAPs loaded with CLC-Soluplus® powder in an attempt to enhance the transdermal administration of CLC into systemic circulation.Lyophilisation was employed in preparing CLC-Soluplus® powders. Such an approach was utilised, as it offered the highest yield with the potential to be scaled up for commercial manufacture in an industrial setting.40Fig. 3A and B highlights the particle size and polydispersity of CLC-Soluplus® powder prior to, and post lyophilisation. Poly(vinyl caprolactam–polyvinyl acetate–polyethylene glycol) graft copolymer (Soluplus®) was utilised as a polymeric carrier in preparing CLC-Soluplus® powder. It can be seen from Fig. 3A that almost immediately after mixing the drug and Soluplus®, regardless of the concentration of the Soluplus® used, drug particles of 60–70 nm and highly monodisperse (PDI ≈ 0.1) were observed. However, upon lyophilisation, the formulation containing 1.0–2.0% of Soluplus® exhibited an increase in PDI. Nevertheless, these formulations exhibited acceptable PDI, as all values were still below 0.3.41 The increase in PDI and particle size for CLC-Soluplus® powder containing 1.0 and 2.0% Soluplus® may indicate that formulations containing a high polymeric carrier could be more susceptible to the stresses imparted during the lyophilisation process-namely freezing, first and secondary drying. This stress might trigger some degree of aggregation and agglomeration within the particles. However, such an effect was not seen at 0.25% of Soluplus®, which low polymer concentrations.42
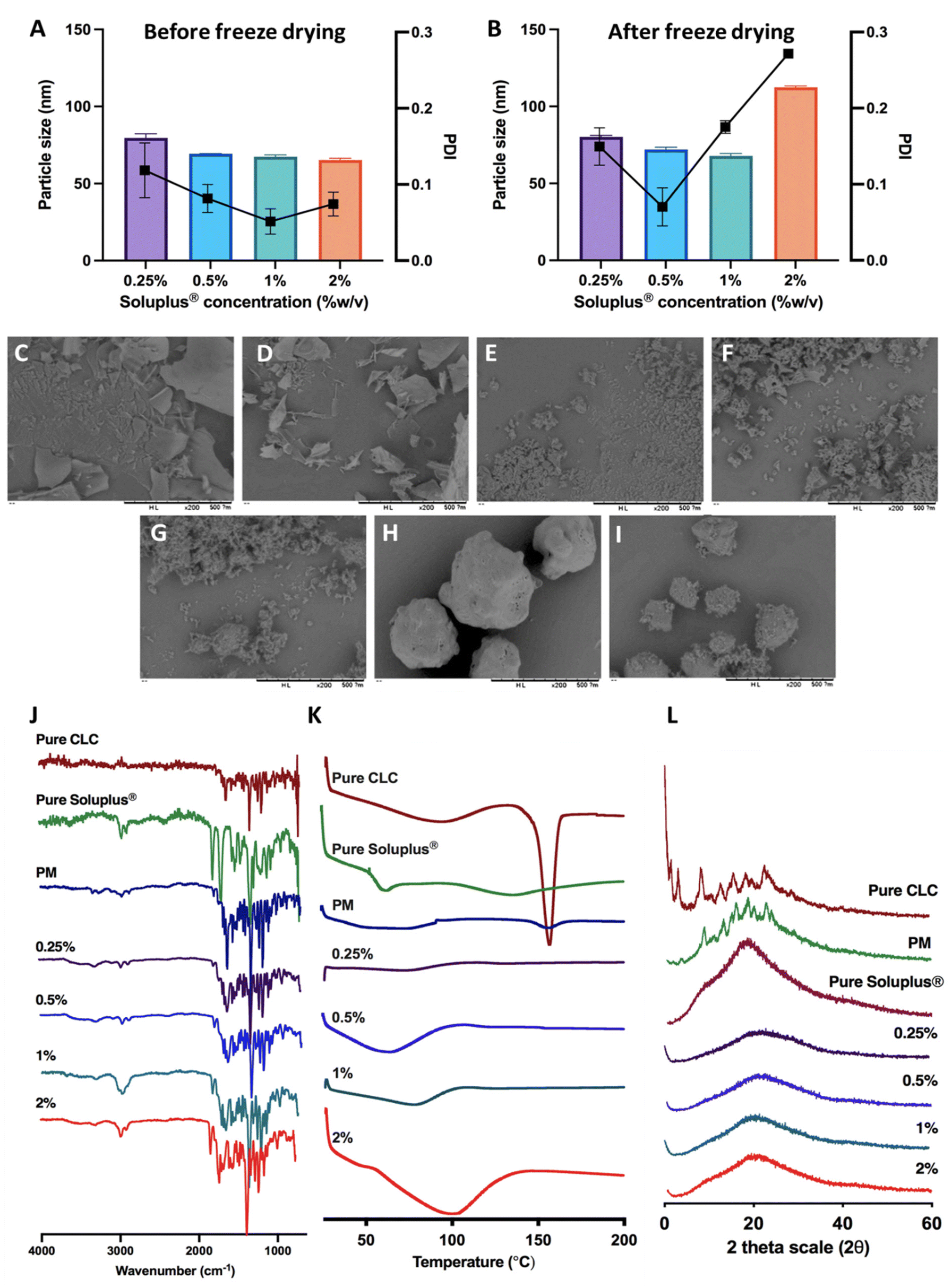 | ||
| Fig. 3 (A) Particle size and polydispersity of CLC-Soluplus® powder prior and (B) post lyophilisation (means ± SD, n = 3). SEM images of CLC-Soluplus® powder at different concentration of Soluplus®, (C) 0.25%, (D) 0.5%, (E) 1%, (F) 2% compared to (G) pure CLC, (H) pure Soluplus® and (I) physical mixture. (J) FT-IR spectra, (K) DSC thermogram and (L) XRD diffractogram of pure CLC, Soluplus®, physical mixture and CLC-Soluplus® powder. | ||
The reduction in CLC particles is intended to promote drug dissolution when the MAPs are inserted into the skin. In addition, there are several publication which have shown that reducing the size of CLC into the nanometer range (<250 nm) helped enhancing the efficacy of the drug. For instance, the recent work by Wang et al.43 has shown that reformulating CLC into nanoparticles with a size of 240 nm helped enhancing the anti-inflammatory effect of the drug in attenuating acute myocardial infarction injury. This finding is also corroborated by the work of Aboumanei & Fayez44 who showed that reformulating CLC into a nanoemulsion with a particle size of ≈100 nm, which is similar to the CLC particle size obtained in our current work, enhanced the pharmacodynamics effect of the drug when the formulation is administered intra-articularly. Collectively, it can be seen that in addition to enhancing drug solubility, reducing the particle size of CLC has been to enhance the therapeutic effect of the drug. Such enhanced efficacy has been attributed to several mechanisms, one being the preferential delivery of the drugs to inflammatory sites because of the enhanced vascular permeability in inflamed tissues, like the case of acute gout.45 Secondly, this may be attributed to the difference in drug uptake when it is formulated as a nanoparticulate system. When the drug is formulated as a nanoparticle, it would be preferentially taken up by immune cells by endocytosis instead of simple diffusion, enabling a higher dose of the drug to act on the immune cells resulting in enhanced anti-inflammatory effect.46
SEM images for pure CLC, pure Soluplus® and CLC-Soluplus® powder are presented in Fig. 3C–I. It was found that the presence of Soluplus® in the formulation influenced the shape of the obtained product. The surface morphology studies revealed that the pure CLC appeared as fine particles, whilst pure Soluplus® appeared as spheres. SEM visualisation of the powder mixtures showed a modification, where it was no longer possible to distinguish between the pure CLC and Soluplus® particles. The particles appeared flakier in comparison to the pure drug when the CLC-Soluplus® powder was prepared with blend containing 0.25–0.50% of Soluplus®. However, when the concentration of Soluplus® used in the blend was 1.0–2.0%, the morphology of the CLC-Soluplus® powder appeared quite similar to the pure drug. Compared to the physical mixture (Fig. 3I), both pure CLC and pure Soluplus® could still be distinguished.
FTIR analysis of CLC and formulations in Fig. 3J shows C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1653 cm−1 while the band at 1551 cm−1 is indicative for N–H bending for CLC.47 Soluplus®, on the other hand, displayed characteristic peaks at 1477 cm−1, correlating to C–O–C stretching, while the band at 1635 cm−1 arises due to the tertiary amide CO group. Lastly, the 1734 cm−1 is attributed to CO ester group.48 It can be seen that the FTIR spectra for CLC before and after being encapsulated into CLC-Soluplus® mixture are similar, indicating that the lyophilisation process did not result in any noticeable changes to the structure and chemical composition of the drug.
O stretching at 1653 cm−1 while the band at 1551 cm−1 is indicative for N–H bending for CLC.47 Soluplus®, on the other hand, displayed characteristic peaks at 1477 cm−1, correlating to C–O–C stretching, while the band at 1635 cm−1 arises due to the tertiary amide CO group. Lastly, the 1734 cm−1 is attributed to CO ester group.48 It can be seen that the FTIR spectra for CLC before and after being encapsulated into CLC-Soluplus® mixture are similar, indicating that the lyophilisation process did not result in any noticeable changes to the structure and chemical composition of the drug.
The DSC thermogram of CLC in Fig. 3K shows a sharp endotherm at around 150 °C, correlating with the melting temperature of CLC.47 Soluplus® showed a broad endotherm at ≈73 °C, which corresponds to the Tg of the polymer.48 It can be also seen that the endotherm became shallower in the physical mixture of the drug with Soluplus® and when CLC was encapsulated into CLC-Soluplus® powder via lyophilisation, the endotherm at 150 °C was absent. Thus, it is postulated that CLC was present in a non-crystalline state post lyophilisation. XRD analysis was done in order to further ascertain the solid state of CLC within the CLC-Soluplus® powder. The XRD diffractogram Fig. 3L showed that CLC-Soluplus® powder were void of any sharp diffractogram peaks, which corroborated the DSC analysis in indicating that CLC is present in an amorphous state. Besides that, it can also be seen that the DSC for pure Soluplus® exhibits an endotherm at 70 °C which corresponds to the Tg of the graft polymer.49 However, when the concentration of the polymer increases from 0.25% to 2% while preparing CLC-Soluplus® mixture we observed a shift and broadening for this Tg. Such observation with increasing Soluplus® concentration within drug–polymer mixtures has also been reported by Altamimi and Neau.50 This may be attributed to the hygroscopic nature of the graft polymer, which entraps more moisture with increasing Soluplus® concentration, resulting in an increase in the hydration of CLC–Soluplus® mixture.51 Such increase in water-soluble polymer hydration has been recently reported to cause a shift and broadening in the Tg of the polymer.52
3.2 Characterisation of dissolving MAPs
After manufacturing and characterising CLC-Soluplus® powder, the system was then used to fabricate the microneedle layer of MAPs. Fig. 4A shows the CLC-loaded dissolving MAPs, with different Soluplus® concentrations. As seen from the microscopy images in Fig. 4A along with the SEM images in Fig. 4B, all manufactured MAPs showed sharp and clear pyramidal projections on a clean and flat baseplate. In addition, all MAPs displayed a tip diameter of ≈7.3 μm (Fig. 4C). Based on findings reported by other researchers, this needle tip diameter is of sufficient sharpness to enable the effective insertion of MAPs into the skin.53,54 | ||
| Fig. 4 (A) Optical microscopy images of CLC-Soluplus® microarray patches (MAPS) with different Soluplus® concentration fabricated via double casting method. (B) SEM images of dissolving MAPs fabricated from Soluplus® which are loaded CLC. (C) Microneedle tip diameter of CLC-loaded MAPs, ≈7.3 μm (n = 5). | ||
Based on Fig. 5A, we can see that the drug loading was ≈ 1.6–1.8 mg per patch for formulations F1, F3 and F4. In contrast, MAPs fabricated with F2 displayed a drug loading of 2.8 mg. As shown in Fig. 5A, F2 showed a higher drug loading than F1. Which is followed by a decrease in drug loading with F3 and F4. This observation may be attributed to the solubilising capacity of Soluplus®.55 In F3 and F4, the ratio of CLC:Soluplus® are 2:1 and 1:1, where it is hypothesised that the solubility capacity of Soluplus® has not yet been fully reached and so enable more drug to be loaded into MAPs. When the ratio of CLC:Soluplus® is 4:1, as depicted in F2, we observed that this drug:polymer ratio enables the highest CLC solubilisation out of the four different drug: polymer ratio investigated and so, the highest drug loading into MAPs formulation. However, when the drug:polymer ratio is further increased to 8:1, as depicted in F1, the solubilising capacity of Soluplus® has been exceeded, and causes some drug to crash out before it could be loaded into the needle tip leading to a decrease in the drug loading observed. In this instance, the concentration of the drug exceeded the solubilising limit of Soluplus® which may result in the phenomenon of known as polymeric micellar saturation which prevent any further drug from being solubilised.32
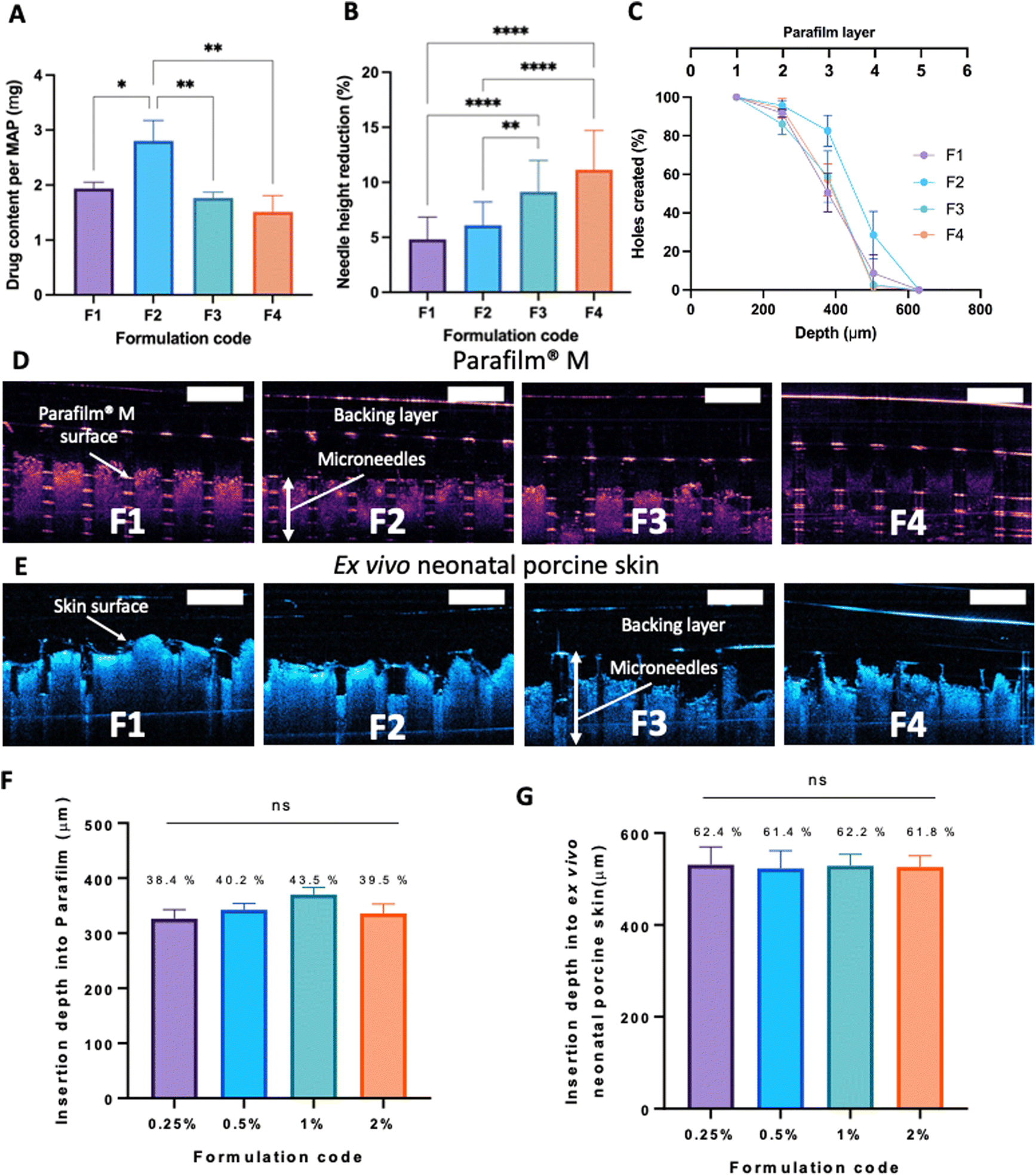 | ||
| Fig. 5 (A) Drug content of CLC-loaded dissolving MAPs (means ± SD, n = 3). (B) MAPs height reduction for needles loaded with CLC-Soluplus® following the application of a 32 N compressive force (means + SD, n = 20). (C) Percentage of channels formed per Parafilm® M layer upon the application of CLC loaded dissolving MAPs (means ± SD, n = 3). Insertion of CLC loaded dissolving MAPs into (D) Parafilm® M and (E) full thickness ex vivo neonatal porcine skin monitored in situ using optical coherence tomography (OCT). Insertion depth of CLC loaded dissolving MAP into (F) Parafilm® M (G) full thickness ex vivo neonatal porcine skin (means + SD, n = 20). Values above each bar represent the percentage of needle successfully inserted into the skin. | ||
After MAP fabrication, their mechanical properties and insertion capabilities were evaluated. The result detailed in Fig. 5B, showed that all the MAPs exhibited a reduction in needle height of ≈10% or less when compressed with a force of 32 N, which is similar to that typically exerted by humans when applying manual thumb pressure, as previously shown.56 Also, it is worth noting that, by increasing the Soluplus®:CLC ratio from 1:8 to 1:1, an increase in the percentage height reduction of the MAPs was seen. It is postulated that this observation may be due to the intrinsically brittle nature of the polymer.57 It has been reported that Soluplus® exhibits a glass transition temperature (Tg) of 73 °C.58 Since the MAPs were subjected to a compressive force under ambient conditions, the polymer is below its Tg and is present in a glassy and brittle state. This predisposes the needles to fracture during compression, especially with an increasing Soluplus® concentration in the first layer formulation. In addition, it has been reported that film formulations prepared from pure Soluplus® without any plasticizer, were found to be brittle and difficult to remove intact from the mould following solvent casting method.34,59 This result indicates that CLC may act as plasticiser for Soluplus®, which similar to the result reported by previous work on Eudragit RS.60 The presence of CLC within a polymeric system could be attributed to a decrease in the attractive force between the molecules, resulting in an increased movement of molecules and hence, decreasing the Tg of the polymer.34,60 Nevertheless, the percentage height reduction for all MAP formulations manufactured were still below 15%, which is similar to the values reported by other work on dissolving MAPs.61,62 Hence, it is expected that MAPs produced would display the mechanical robustness needed to endure the compressive force encountered during MAP application.
MAP penetration into eight layers of Parafilm® M, serving the role as a skin simulant, was performed in order to evaluate the insertion profile of the needles. As shown in Fig. 5C & D, all the formulations breached the Parafilm® stack into the third layers, as visualised in situ using OCT, resulting in an insertion depth of ≈300 μm (Fig. 5F). Furthermore, all the CLC-loaded MAPs investigated breached the first layer of Parafilm® with a 100% efficiency as evidence absence of any error bars in the first layer. Subsequently, an insertion study utilising full thickness ex vivo neonatal porcine skin was performed in order to see if MAPs were able to puncture the skin tissue. Ex vivo neonatal porcine skin was used as a proxy to human skin due to histological and mechanical similarities of the skin tissues between the two species.63 Similar to MAP insertion into the Parafilm®M layer, OCT was employed to visualise needle penetration into the skin, as shown in Fig. 5E. It can be seen that all four formulations resulted in a relatively similar skin insertion depth of ≈520 μm.
It has been widely reported that the elasticity of the skin may play an integral factor in influencing the insertion profile of MAPs into the skin, as previously reported by several researchers.64,65 Such difference in elasticity between the skin relative to the skin simulant, Parafilm®M, may account for the difference in penetration rate observed. It is worth noting that such difference in insertion depth of the MAP into ex vivo skin relative to Parafilm® M has also been reported by us in our previous work.21 This effect can be attributed to the presence of water in the ex vivo skin, which confers a level of lubrication. Hence, aiding the insertion of needles into the skin to a deeper extent when compared to Parafilm® layers.
After conducting an in situ ex vivo insertion study, we also investigated the dissolution profile of the MAPs following skin administration. As shown in Fig. 6, we observed that all four CLC-loaded MAP formulations (F1–F4) displayed an almost similar dissolution profile. After 30 minutes of insertion, we observed that the majority of the needle lengths have dissolved. However, after 1-hour we observed that all needle shafts have completely dissolved for F1, F3 and F4, while some remaining needle lengths still being seen at the base of the patch for F2. This may be attributed to the higher drug loading for F2. However, after 3 hours, we observed that all needles have fully dissolved as evidenced from the flat base plate shown in Fig. 6. The dissolution of needles from the MAPs results in the formation of microchannels on the skin surface, as evidenced by stereoscopic microscope images of the skin (Fig. 6). The fast dissolution profile exhibited by all four CLC-loaded MAP formulations may be attributed to the hydrophilic nature of the needles, given the high aqueous solubility of CLC (45 mg mL−1)66 as well as the presence of Soluplus® along the needle length and its solubilising effect when exposed to dermal interstitial fluid.67
 | ||
| Fig. 6 Skin dissolution studies of MAP loaded with CLC following 30 min, 1-hour and 3-hour skin application. | ||
3.3 Dermatokinetic studies
Upon characterising the respective MAP formulations for their mechanical properties and insertion profiles, the MAP fabricated from CLC and Soluplus® with a ratio 4:1 (F2) was selected for the skin deposition study. This was because this MAP formulation offered the highest drug loading, while displaying a similar mechanical and insertion profile in comparison to the other MAP formulations. When performing the dermatokinetic study, a CLC-Soluplus® film that contained a similar drug loading to MAPs was used as control (2.8 mg). It can be seen from Fig. 7 that no percutaneous permeation of CLC from the film took place. This could be ascribed to the high aqueous solubility of CLC (7 mg mL−1)68 which also exhibits a logP (1.10),69 thus preventing the drug from diffusing across the highly lipophilic stratum corneum.70 In contrast, when CLC was formulated into MAPs, the microprojections on the MAP breached the stratum corneum, which resulted in the administration of the drug across different strata of the skin, as shown in Fig. 7A–D. In addition, we also included an additional control where we evaluated the permeation profile of a solution of CLC. This was done in order to decipher if there is any drug passive diffusion taking place.
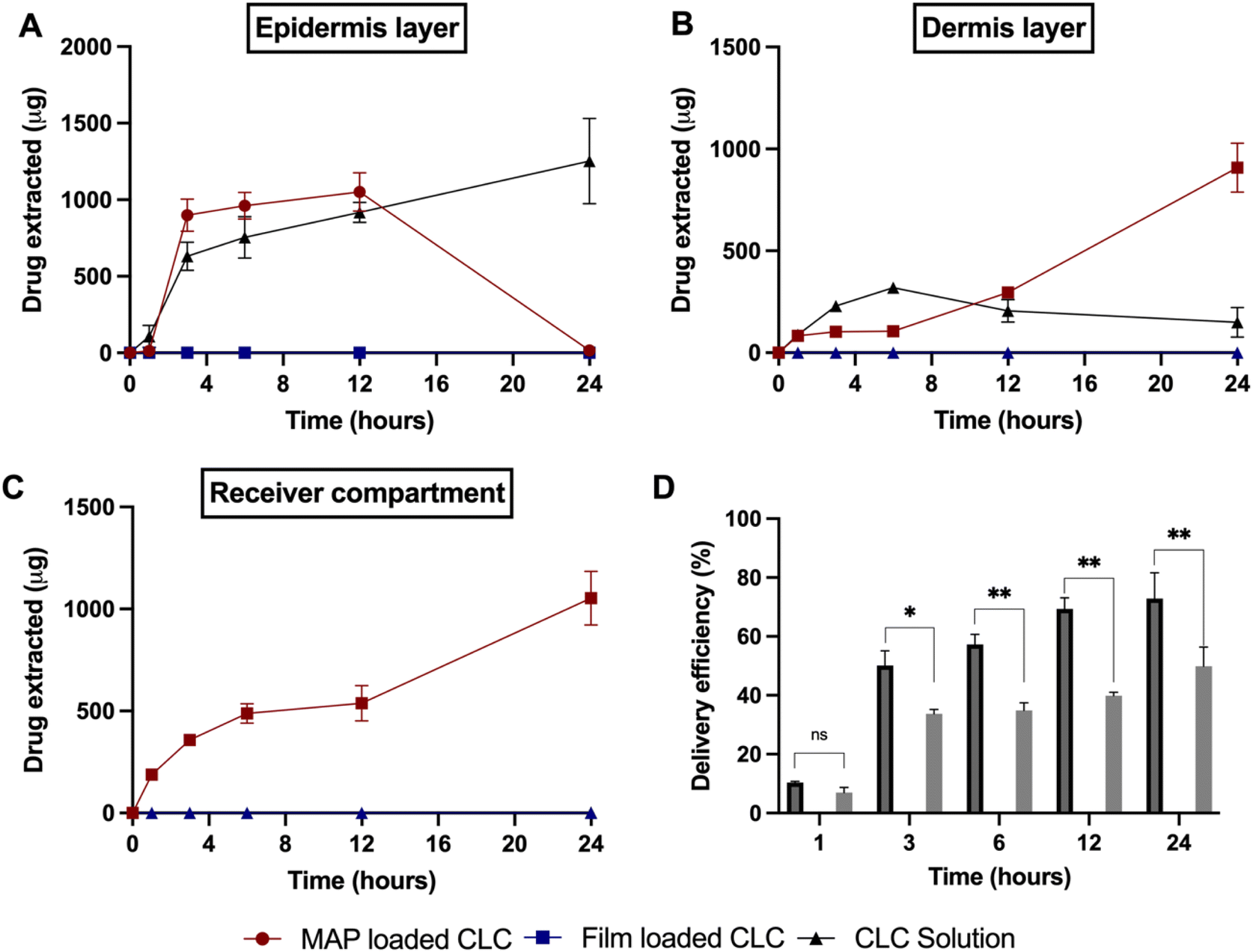 | ||
| Fig. 7 Amounts of colchicine (CLC) extracted from different skin layers: (A) epidermis and (B) dermis from excised neonatal porcine skin at different time points following in vitro Franz cell diffusion study. (C) Amounts of CLC delivered transdermally into the receiver compartment of Franz cells. (D) The delivery efficiency of CLC from dissolving MAPs, film and drug solution over the course of 24 h (values represent means ± SD, n = 4). | ||
As shown in Fig. 7A, the cumulative CLC administered into the epidermis progressively increased from 1 to 3 h, before it plateaued at ≈0.9 mg within 3 to 12 h. After that period, the amount in the epidermis dropped to ≈0 mg. On the other hand, the amount delivered into the dermis and receptor compartment displayed a different profile. It can be seen that there is a rise in the amount of CLC being delivered into both the dermis and receptor compartment as shown in Fig. 7B and C. In addition, when the cumulative CLC delivered (as delivery efficiency (%)) is considered, it can be seen that CLC-Soluplus® loaded MAPs resulted in an increase in delivery efficiency with application time. More importantly, the formulation was capable of achieving a 73% delivery efficiency which equal to ≈1.98 mg of CLC delivered after 24 h of study. With respect to the control consisting of a CLC-solution, we observed that this only led to CLC delivery into the epidermis and dermis. However, there was no measurable transdermal delivery of CLC into the receptor compartment as shown in Fig. 7C. The ability of the drug solution to promote the delivery of the CLC into the skin, particularly the epidermis, may be attributed to the hydration of the stratum corneum upon solution application. The hydration of the stratum corneum has been shown to reduce the barrier function of this layer and promote the diffusion of small molecules, particularly hydrophilic compounds (e.g. CLC) into the epidermis.71,72 The diffusion of CLC into the epidermis continues to increase with time, as shown in Fig. 7A. This suggests that the drug accumulates within this skin strata resulting in the formation of a secondary reservoir which slowly releases the drug into the deeper layers of the skin (dermis) over time.73 However, this passive delivery of CLC via stratum corneum hydration was insufficient to promote the delivery of the drug across the skin, which is the intended target for systemic delivery. The lack of transdermal delivery from the CLC-solution ultimately resulted in the solution displaying a significantly (p < 0.05) lower delivery efficiency than the CLC MAPs, as shown in Fig. 7D.
The enhancement in delivery efficiency achieved using dissolving MAPs fabricated from Soluplus® may be attributed to the combination of the drug being present in an amorphous state in tandem with solubilising properties of Soluplus®. As evidenced from Fig. 3K and L, the presence of CLC in an amorphous state will cause the drug to be at higher Gibbs free energy level. This will accelerate the formation of inter molecular hydrogen bonds between CLC with the water molecules present within the dermal ISF. The formation of this intermolecular hydrogen bonds will occur at a faster rate for the amorphous form of CLC relative to when the drug is present in crystalline state resulting in enhanced drug dissolution.74 In addition, this process is further enhanced due to the high aqueous solubility exhibited by CLC, 7 mg mL−1. In addition, the dissolution of CLC is further promoted by the presence of Soluplus® within the needle matrix. The presence of Soluplus® in a drug containing matrix has been shown to cause the Gibbs free energies of transfer, 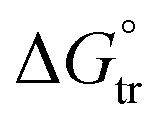 to become more negative.67 This correlates with the drug solubilisation to become more spontaneous in the presence of the polymer. This is ascribed to presence of hydroxyl (–OH) on the polymer which confer Soluplus® with surface active properties which promote drug dissolution via micellar solubilisation.67
to become more negative.67 This correlates with the drug solubilisation to become more spontaneous in the presence of the polymer. This is ascribed to presence of hydroxyl (–OH) on the polymer which confer Soluplus® with surface active properties which promote drug dissolution via micellar solubilisation.67
Regarding drug delivery systems for gout, several researchers have explored the utility of the transdermal route for the administration of anti-inflammatory agents. For instance, Zhang et al.75 developed ethosomes, which are lipid nanocarriers composed of a relatively high concentration of ethanol, for delivering CLC transdermally across the skin. The group was able to demonstrate that ethosome formulations were successful in delivering up to 1.1 mg cm−2 of CLC across the skin.75 From a clinical point a view, the application of ethosomes for the delivery of CLC might not be as practical as MAPs application as ethosomes are typically formulated as a colloid. This could result in unfavourable cosmetic sensation as well as raising the possibility of staining the patients’ clothes.76 All the above could lead to poor patient compliance during clinical translation. Furthermore, the possibility for colloid to spread away and off the skin may result in dosing inaccuracies and inconsistencies. This may raise safety issues, as CLC has a narrow therapeutic index and, hence, accurate dosing will be extremely crucial in managing the disease, whilst mitigating the possibility of CLC-induced toxicity. With respect to MAP formulation, these limitations are not as prominent as the patches contain a defined amount of drug per unit area.
The utilisation of dissolving MAPs as strategy to promote the administration of anti-gout therapeutics across the skin has also been previously documented in literature. For instance, Chen et al.9 formulated and delivered polydatin–cyclodextrin inclusion complexes via dissolving MAPs. Polydatin is an anti-inflammatory molecule isolated from the root of a traditional Chinese herb, Polygonum cuspidatum Sieb. et Zucc, which has shown promising pharmacological properties against gout. In their work, Chen and colleagues managed to load up 2.4 mg of polydatin per MAP, resulting in a delivery of 100 μg after 8 h.9 However, in the current work, using amorphous CLC-Soluplus® loaded dissolving MAPs, it was seen that a total dose of 2.1 mg of CLC within 6 h of application was delivered. In comparison to CLC, which has a logP of 1.1, polydatin has been reported to exhibit a logP of 0.4 indicating that the drug is more water soluble than CLC. Although Chen and co-workers did not evaluate the solid state of polydatin upon loading the compound into dissolving MAP, it could be suggested that the drug may still be present to some degree in a monocrystalline state, resulting in a lower drug delivery profile.5 In the current work, however, it has been demonstrated that despite CLC having a higher logP, the amorphisation of CLC upon loading into dissolving MAPs, may account to a higher and quicker release of payload upon skin application.
More recently, Chen et al.77 have developed dissolving MAP for the delivery of allopurinol, a xanthine oxidase inhibitor, used in the management of gout.77 In their work, Chen and colleagues were successful in loading up to 3.5 mg of allopurinol per patch and demonstrated that their formulations were able to deliver up to 90% of the dose, using a Franz cell set up, within 5 h of application. The higher dose delivered in this work may be attributed to several factors. Firstly, from a physicochemical perspective, allopurinol is a highly water-soluble drug exhibiting a logP of −0.55 which is much lower than CLC, and could result in a more rapid release of the drug upon intradermal deposition.78 Secondly, the higher delivery profile observed may be attributed to the difference in Franz cell set up used by Chen et al.80 compared to the present study. In their work, Chen and co-workers opted to use mouse skin, which is far thinner than the porcine skin.79 Therefore, this may lead to deeper MAPs insertion into the skin model used, and potentially come in direct contact with the receiver fluid, resulting in a higher in the delivery efficiency observed. Moreover, even if the formulation does exhibit a 90% of delivery efficiency, the clinical dose of allopurinol is approximately 200 mg daily while in the case of CLC, the clinical daily dose of the drug is approximately 2 mg a day.80 This would suggest that the patch size needed to deliver CLC would be approximately 0.36 cm2, similar to the one used in the current study, which is far smaller to that would be needed to deliver allopurinol.
With regards to the use of Soluplus® as an excipient in the design and fabrication of MAP-based formulations, Uddin et al.,81 included the graft copolymer as an excipient to aid on the coating of solid MAPs fabricated from stainless steel. Although this approach showed that the polymer was useful for coating microneedle surfaces, the overall approach was limited by the drug loading that could be achieved (50–150 μg).81 Therefore, the use of coated MAPs was deemed ineffective in delivering CLC as the dose needed to achieve therapeutic efficacy is 2000 μg and so, either a thicker coating or a wider MAP array would be needed. However, this may result in lower needle insertions, compromising the delivery efficiency. However, with our current approach, the use of dissolving MAPs by which the drug is loaded into the whole needle length, helped enhancing drug loading into a single MAP patch with an area of 0.36 cm2, thus skipping the need for a wider array size. The use of a MAP with a smaller array area may be more favourable, as it can easily be applied by patients via single thumb pressure.
3.4 Biocompatibility study
Fibroblast cell viability following exposure to blank Soluplus® MAPs and CLC-loaded dissolving MAPs were evaluated using MTT assay and LIVE/DEAD™ staining. The results presented in Fig. 8A revealed that, compared to the control, exposure to both the CLC-loaded MAPs and blank Soluplus® did not compromise the viability of fibroblast cells. This was further confirmed by LIVE/DEAD™ staining (Fig. 8C), which demonstrated that Soluplus® and CLC did not affect fibroblast cell viability due to the absence of any noticeable red fluorescence from the treated cells.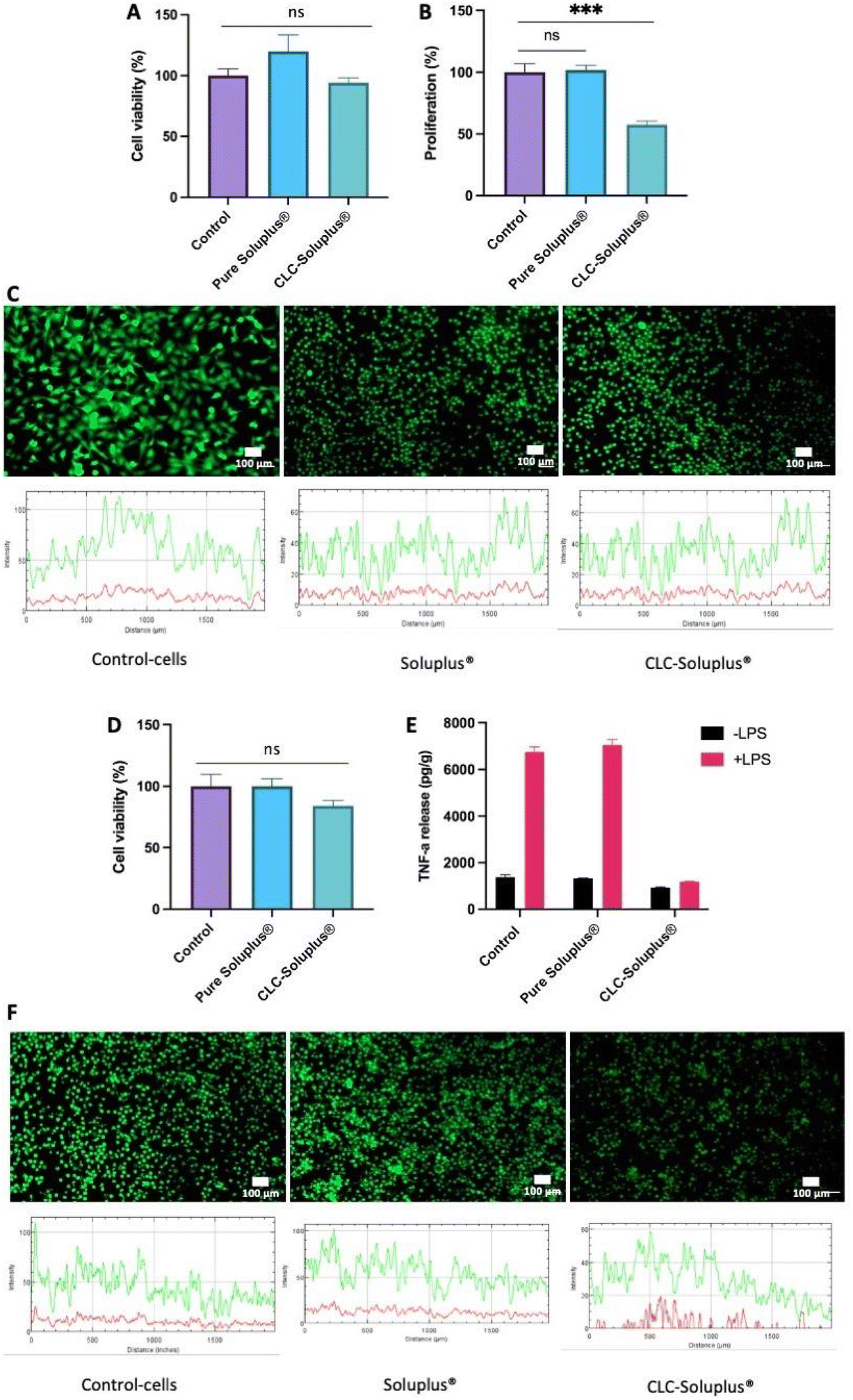 | ||
| Fig. 8 Fibroblast cell viability and proliferation upon exposure to CLC and Soluplus®. (A) MTT assay data showing the percentage of viable cells after a culture period of 72 h. (B) LIVE/DEAD™ staining on control fibroblastic cells, cells treated with Soluplus® and CLC. (Green = FDA (live); red = PI (dead)); scale bar = 100 μm. (C) Cell proliferation evaluated via PicoGreen assay, which evaluated the total DNA content for cells treated with Soluplus® and CLC sample relative to control (post 72 h of culture period). A standard curve of known dsDNA (ng mL−1) was used to calculate the DNA content from the samples. “***” denotes a statistically significant difference (p < 0.001) in cell number with respect to control (plate cells culture). Results are presented as means ± SD (n = 3). Macrophages inflammatory response. MTT assay was also utilised to ascertain the viability of macrophages cell. (D) MTT results show the percentage of viable cells after a culture period of 72 h. (E) TNF-α release to the media by macrophages cultured or not cultured with Soluplus® or CLC and with (pink bars) or without LPS (black bars) (10 μg mL−1) for 6 h by ELISA. Results are shown as concentration normalised to total protein levels (pg per g protein). Data points represent means ± SE (n = 3). (F) Live/dead staining of macrophages cells on control (plate cells culture), Soluplus® and CLC samples. (Green = FDA (live); red = PI (dead)); scale bar = 100 μm. | ||
Additionally, PicoGreen® assay was used to evaluate the impact of CLC-loaded and blank MAPs on cell proliferation (Fig. 8B). It can be seen that exposure to blank Soluplus® MAPs did not result in any significant change in fibroblast proliferation with respect to the control. However, CLC treatment induced a significantly lower degree of fibroblast proliferation in comparison to the control (p < 0.05) and Soluplus® (p < 0.05). The results showed that blank Soluplus® and CLC-loaded MAPs had good cytocompatibility and low cytotoxicity when cultured with fibroblasts. However, it was observed that the fibroblast proliferation rate was significantly lower (p < 0.05) after treatment with CLC-loaded MAP. Previous investigations have reported that CLC alone inhibits fibroblast migration and proliferation, which explains the reduction in cellular proliferation observed in the present work.82–85 Overall, the reduction in fibroblast proliferation without the presence of any noticeable cytotoxicity suggest that the formulation possesses anti-fibrotic and anti-inflammatory properties that maybe highly favourable in the management of acute gout.86
3.5 In vitro anti-inflammation activity
THP1-macrophage viability after exposure to both the CLC loaded MAPs and blank Soluplus® MAPs was evaluated by MTT assay and LIVE/DEAD™ staining. It can be seen in Fig. 8D that there were no observable differences between macrophages cell viability with respect to the control (p > 0.05) following an incubation period of three days with CLC-loaded MAPs. Nevertheless, this level of cell viability is considered as low toxicity in accordance with ISO 10993-5: 2009 standard.87 In contrast, the viability of THP-1 macrophages after Soluplus® treatment remained unaffected, having similar cell viability with respect to the control. In addition, the absence of any red fluorescence following LIVE/DEAD™ staining, as shown in Fig. 8F, supports the findings of the MTT assay, suggesting that the formulations are biocompatible with the THP-1-macrophage cell line.In addition, TNF-α release profile from the cells was assessed in an attempt to elucidate the inflammatory response of macrophages after exposure to blank Soluplus® MAPs and CLC-loaded MAPs. As shown in Fig. 8E, compared to the group control, macrophages cultured in the presence of LPS release higher levels of TNF-α (p < 0.05), supporting the notion of macrophage mediated inflammatory response towards LPS.88 On the other hand, treating the THP-1-macrophages with Soluplus® did not mitigate TNF-α release upon exposure to LPS. This suggests that exposure to the polymeric excipient Soluplus® alone did not result in any anti-inflammatory effect. In contrast, without the presence of LPS, TNF-α release was significantly reduced upon exposure to CLC-loaded MAPs compare to the control (p < 0.05) and Soluplus® treated group (p < 0.05). This shows that, even in the absence of LPS, CLC has a direct effect on THP-1 macrophages under basal conditions.
In the presence of LPS, CLC-loaded MAPs significantly reduced TNF-α release when compared to both control (p < 0.05) and Soluplus® treated groups (p < 0.05). Such a finding suggests that CLC exerts an anti-inflammatory effect on THP-1 macrophages in the presence of a pro-inflammatory mediator. These results show a significant anti-inflammatory effect of CLC-loaded MAPs on macrophages, evidenced by a marked reduction of TNF-α release from such cells. These observations can be corroborated by previously reported mechanistic studies that highlight the inhibitory effect of CLC on inflammatory cytokines, such as TNF-α.89 This anti-inflammatory effect is achieved mainly via the destabilisation of the microtubule network that inhibits the signal transduction of the TNF-α–NF-κB pathway,91 which translates into the reduction of TNF-α post treatment with CLC.
In this sense, it is known that a down-regulation of an acute inflammatory response begins with the suppression of macrophages-mediated inflammatory release of TNF-α.90 As shown in the current work, exposure of THP-1 macrophages to CLC-loaded MAPs resulted in a low TNF-α secretion even in the presence of a pro-inflammatory mediator such as LPS. This reduction in TNF-α secretion can lead to a reduction in apoptosis, necroptosis, pyroptosis91 as well as suppressing further immune responses. Thus, it is postulated that, by inhibiting TNF-α release at an early stage in the TNF-α–NF-κB pathway, a reduction of inflammatory cell infiltration in subjects with gout during an acute inflammatory state could be achieved.89,91 This suggests that CLC-loaded MAPs could potentially be an efficacious treatment for acute gout.
It is worth noting that currently the only marketed dosage form used to administer CLC in patients with gout are oral tablets which cost approximately £4.00 for a packet of 100 tablets.80 Indeed, it would seem that delivering the drug orally may be a more financially viable strategy relative to MAPs. This is because MAPs are considered to be an emerging pharmaceutical technology, which have yet to receive regulatory approval for delivering drug molecules for the management and treatment of medical conditions. It is anticipated that once approved, the initial market price of MAPs would be much higher than the price of generic CLC oral tablets. This price disparity stems from the need to recoup the cost of developing and bringing new pharmaceutical technologies to market which necessitate specialised GMP manufacturing facilities. Nevertheless, it should be noted that most patients with gout are typically those within the geriatric population which tend to suffer dysphagia (swallowing difficulties).92,93 Therefore, the use of oral CLC tablets may not be a viable strategy for gout patients who suffer comorbidities such as dysphagia. This would incur additional nonmedical cost (e.g. hospital visit to administer drug via alternative treatment routes) as well as imposing unwanted intangible costs (e.g. pain and discomfort) associated with alternative treatment procedures resulting in a deterioration in the overall quality adjusted life-year (QALY) for the patients. Therefore, the use of dissolving MAPs as an alternative to oral tablets may help obviate these issues especially in geriatric gout patients who suffer dysphagia. On the other hand, dissolving MAPs may also offer an alternative formulation approach for patients who typically suffer gastrointestinal side effects when taking oral CLC tablets.
4. Conclusion
In conclusion, the current work highlights the fabrication and evaluation of tip-loaded CLC-Soluplus® MAPs for the treatment of acute gout. The fabricated MAPs were capable of bypassing the stratum corneum, resulting in intradermal insertion and release of CLC across the skin with an efficiency of 73% after a 24-hour permeation study. The prototype MAPs also exhibited anti-inflammatory properties as demonstrated by the significant reduction of TNF-α production in THP-1 macrophages treated with CLC-loaded MAPs in the presence of lipopolysaccharide (LPS). Despite the promising result in the current work, further investigations to evaluate the efficacy, stability and safety prior to the clinical application will be paramount in order for this system to have a true clinical impact. Should this system be translated into a healthcare setting, these MAPs may be of clinical benefits particular in gout patients who suffer dysphagia and are unable to take CLC tablets, which is the only dosage form marketed for acute gout. In addition, due to the ever-growing evidence on the anti-inflammatory properties of CLC, this formulation may also be of great clinical value in managing other inflammatory-related diseases, such as familial Mediterranean fever (FMF), Behçet's disease, and pericarditis. Also worth noting, further studies will also be required to evaluate if gout patients would have sufficient manual dexterity to apply these MAPs by themselves or would require the use of MAP-applicator to assist the application of the patch in a reproducible and consistent fashion. Such human factor study would also aid the clinical translation of these prototype MAPs from bench to bedside. Nevertheless, the overall work shown in this study highlights that indeed the transdermal route, via the use of MAPs, is a viable and alternative formulation strategy to administer anti-gout medication.Conflicts of interest
There are no conflicts to declare.References
- T. Neogi, J. Gastroenterol. Hepatol., 2015, 30, 13–26 CrossRef.
- M. Dehlin, L. Jacobsson and E. Roddy, Nat. Rev. Rheumatol., 2020, 16, 380–390 CrossRef PubMed.
- S. K. Rai, L. C. Burns, M. A. De Vera, A. Haji, D. Giustini and H. K. Choi, Semin. Arthritis Rheum., 2015, 45, 75–80 CrossRef PubMed.
- NICE, Gout | Treatment summary | BNF content published by NICE, https://bnf.nice.org.uk/treatment-summary/gout.html, (accessed July 9, 2021).
- M. Drini, Aust. Prescr., 2017, 40, 91 CrossRef PubMed.
- K. H. Katsanos, D. E. Sigounas, D. K. Christodoulou and E. v. Tsianos, Ann. Gastroenterol., 2011, 24, 320 Search PubMed.
- R. I. Russell, Postgrad. Med. J., 2001, 77, 82–88 CrossRef CAS PubMed.
- A. H. Soll, W. M. Weinstein, J. Kurata and D. McCarthy, Ann. Intern. Med., 1991, 114, 307–319 CrossRef CAS PubMed.
- Z. Chen, B. Han, L. Liao, X. Hu, Q. Hu, Y. Gao and Y. Qiu, J. Drug Delivery Sci. Technol., 2020, 55, 101487 CrossRef CAS.
- S. Stewart, K. C. K. Yang, K. Atkins, N. Dalbeth and P. C. Robinson, Arthritis Res. Ther., 2020, 22, 28 CrossRef CAS PubMed.
- G. Cocco, D. C. C. Chu and S. Pandol, Eur. J. Intern. Med., 2010, 21, 503–508 CrossRef CAS PubMed.
- M. Kirkby, A. R. J. Hutton and R. F. Donnelly, Pharm. Res., 2020, 37, 1–18 CrossRef PubMed.
- A. T. Ogunjimi, J. Carr, C. Lawson, N. Ferguson and N. K. Brogden, Sci. Rep., 2020, 10, 1–14 CrossRef PubMed.
- M. Milewski, N. K. Brogden and A. L. Stinchcomb, Expert Opin. Drug Delivery, 2010, 7, 617 CrossRef CAS PubMed.
- Y. C. Kim, J. H. Park and M. R. Prausnitz, Adv. Drug Delivery Rev., 2012, 64, 1547 CrossRef CAS PubMed.
- S. Bal, A. C. Kruithof, H. Liebl, M. Tomerius, J. Bouwstra, J. Lademann and M. Meinke, LaPhL, 2010, 7, 246 Search PubMed.
- J. Gupta, H. S. Gill, S. N. Andrews and M. R. Prausnitz, J. Controlled Release, 2011, 154, 148–155 CrossRef CAS PubMed.
- H. Kalluri and A. K. Banga, Pharm. Res., 2011, 28, 82–94 CrossRef CAS PubMed.
- L. K. Vora, A. J. Courtenay, I. A. Tekko, E. Larrañeta and R. F. Donnelly, Int. J. Biol. Macromol., 2020, 146, 290–298 CrossRef CAS PubMed.
- J. W. Lee, S. O. Choi, E. I. Felner and M. R. Prausnitz, Small, 2011, 7, 531–539 CrossRef CAS PubMed.
- Q. K. Anjani, A. H. B. Sabri, E. Utomo, J. Domínguez-Robles and R. F. Donnelly, Mol. Pharm., 2022, 19(4), 1191–1208 CrossRef CAS PubMed.
- A. H. Sabri, Z. Cater, P. Gurnani, J. Ogilvie, J. Segal, D. J. Scurr and M. Marlow, Int. J. Pharm., 2020, 589, 119808 CrossRef CAS PubMed.
- Y. Ito, M. Hirono, K. Fukushima, N. Sugioka and K. Takada, Int. J. Pharm., 2012, 436, 387–393 CrossRef CAS PubMed.
- P. González-Vázquez, E. Larrañeta, M. T. C. McCrudden, C. Jarrahian, A. Rein-Weston, M. Quintanar-Solares, D. Zehrung, H. McCarthy, A. J. Courtenay and R. F. Donnelly, J. Controlled Release, 2017, 265, 30–40 CrossRef PubMed.
- S. Abdelghany, I. A. Tekko, L. Vora, E. Larrañeta, A. D. Permana and R. F. Donnelly, Pharmaceutics, 2019, 11, 308 CrossRef CAS PubMed.
- K. Ahmed Saeed AL-Japairai, S. Mahmood, S. Hamed Almurisi, J. Reddy Venugopal, A. Rebhi Hilles, M. Azmana and S. Raman, Int. J. Pharm., 2020, 587, 119673 CrossRef CAS PubMed.
- G. Bonfante, H. Lee, L. Bao, J. Park, N. Takama and B. Kim, Micro Nano Syst. Lett., 2020, 8, 1–13 CrossRef.
- U. Paaver, I. Tamm, I. Laidmäe, A. Lust, K. Kirsimäe, P. Veski, K. Kogermann and J. Heinämäki, BioMed Res. Int., 2014, 2014, 789765 Search PubMed.
- J. Fang, Z. Chen, J. Song, J. Li, Y. Han, W. Hou, W. Wang and B. H. Ruan, RSC Chem. Biol., 2021, 2, 1669–1681 RSC.
- Y. Wang, Y. Ding, Y. Xu, C. Wang, Y. Ding, M. Gao, C. Ma, X. Ma and L. Li, Pharm. Dev. Technol., 2020, 25, 865–873 CrossRef CAS PubMed.
- R. S. Bhuptani, A. S. Jain, D. Tikam Makhija, A. Ganpat Jagtap, P. Abdul, R. Hassan, S. Nagarsenker and M. S. Nagarsenker, Indian J. Pharm. Educ. Res., 2016, 50, 277–286 CAS.
- R. Pignatello, R. Corsaro, A. Bonaccorso, E. Zingale, C. Carbone and T. Musumeci, Drug Delivery Transl. Res., 2022, 12, 1991–2006 CrossRef CAS PubMed.
- A. Salawi and S. Nazzal, Int. J. Pharm., 2018, 546, 255–262 CrossRef CAS PubMed.
- H. Lim and S. W. Hoag, AAPS PharmSciTech, 2013, 14, 903–910 CrossRef CAS PubMed.
- R. F. Donnelly, M. T. C. McCrudden, A. Z. Alkilani, E. Larrañeta, E. McAlister, A. J. Courtenay, M. C. Kearney, T. R. Raj Singh, H. O. McCarthy, V. L. Kett, E. Caffarel-Salvador, S. Al-Zahrani and A. D. Woolfson, PLoS One, 2014, 9, e111547 CrossRef PubMed.
- R. F. Donnelly, T. R. R. Singh, M. J. Garland, K. Migalska, R. Majithiya, C. M. McCrudden, P. L. Kole, T. M. T. Mahmood, H. O. McCarthy and A. D. Woolfson, Adv. Funct. Mater., 2012, 22, 4879–4890 CrossRef CAS PubMed.
- Q. K. Anjani, A. D. Permana, Á. Cárcamo-Martínez, J. Domínguez-Robles, I. A. Tekko, E. Larrañeta, L. K. Vora, D. Ramadon and R. F. Donnelly, Eur. J. Pharm. Biopharm., 2021, 158, 294–312 CrossRef CAS PubMed.
- Á. Cárcamo-Martínez, B. Mallon, Q. K. Anjani, J. Domínguez-Robles, E. Utomo, L. K. Vora, I. A. Tekko, E. Larrañeta and R. F. Donnelly, Int. J. Pharm., 2021, 593, 120152 CrossRef PubMed.
- E. W. Baxter, A. E. Graham, N. A. Re, I. M. Carr, J. I. Robinson, S. L. Mackie and A. W. Morgan, J. Immunol. Methods, 2020, 478, 112721 CrossRef CAS PubMed.
- K. Nagapudi and J. Jona, DOI:10.2174/157340708786847852.
- M. Danaei, M. Dehghankhold, S. Ataei, F. H. Davarani, R. Javanmard, A. Dokhani, S. Khorasani and M. R. M. Id, Pharmaceutics, 2018, 10, 1–17 CrossRef PubMed.
- B. Vanbillemont, J. F. Carpenter, C. Probst and T. De Beer, J. Pharm. Sci., 2020, 109, 3308–3318 CrossRef CAS PubMed.
- L. Wang, Y. Peng, L. Song, D. Xia, C. Li, Z. Li, Q. Li, A. Yu, C. Lu and Y. Wang, Cardiovasc. Drugs Ther., 2021, 26, 1–15 CAS.
- M. H. Aboumanei and H. Fayez, Drug Dev. Ind. Pharm., 2021, 47, 770–777 CrossRef CAS PubMed.
- J. A. Nagy, L. Benjamin, H. Zeng, A. M. Dvorak and H. F. Dvorak, Angiogenesis, 2008, 11, 109–119 CrossRef CAS PubMed.
- J. H. Park, D. Dehaini, J. Zhou, M. Holay, R. H. Fang and L. Zhang, Nanoscale Horiz., 2019, 5, 25–42 RSC.
- M. Nasr, H. Younes and R. AbdelRashid, Drug Delivery Transl. Res., 2020, 10, 1302–1313 CrossRef CAS PubMed.
- S. Y. Lin, H. L. Lin, Y. T. Chi, Y. T. Huang, C. Y. Kao and W. H. Hsieh, Int. J. Pharm., 2015, 496, 457–465 CrossRef CAS PubMed.
- R. N. Shamma and M. Basha, Powder Technol., 2013, 237, 406–414 CrossRef CAS.
- M. A. Altamimi and S. H. Neau, Saudi Pharm. J., 2017, 25, 419–439 CrossRef PubMed.
- P. Nowak, A. Krupa, K. Kubat, A. Węgrzyn, H. Harańczyk, A. Ciułkowska and R. Jachowicz, Powder Technol., 2019, 346, 373–384 CrossRef CAS.
- C. Qiao, X. Ma, X. Wang and J. Yao, Polym. Cryst., 2019, 2, e10092 CAS.
- A. M. Römgens, D. L. Bader, J. A. Bouwstra, F. P. T. Baaijens and C. W. J. Oomens, J. Mech. Behav. Biomed. Mater., 2014, 40, 397–405 CrossRef PubMed.
- A. R. Johnson, C. L. Caudill, J. R. Tumbleston, C. J. Bloomquist, K. A. Moga, A. Ermoshkin, D. Shirvanyants, S. J. Mecham, J. C. Luft and J. M. de Simone, PLoS One, 2016, 11, e0162518 CrossRef PubMed.
- J. F. Alopaeus, E. Hagesæther and I. Tho, Pharmaceuticals, 2019, 12, 1–23 CrossRef PubMed.
- E. Larrañeta, J. Moore, E. M. Vicente-Pérez, P. González-Vázquez, R. Lutton, A. D. Woolfson and R. F. Donnelly, Int. J. Pharm., 2014, 472, 65–73 CrossRef PubMed.
- N. Gottschalk, M. Bogdahn, M. Harms and J. Quodbach, Int. J. Pharm., 2021, 597, 120216 CrossRef CAS PubMed.
- S. M. Alshahrani, W. Lu, J. Park, J. T. Morott, B. B. Alsulays, S. Majumdar, N. Langley, K. Kolter, A. Gryczke and M. A. Repka, AAPS PharmSciTech, 2015, 16, 824–834 CrossRef CAS PubMed.
- A. Salawi and S. Nazzal, Int. J. Pharm., 2018, 552, 378–387 CrossRef CAS PubMed.
- F. Siepmann, V. le Brun and J. Siepmann, J. Controlled Release, 2006, 115, 298–306 CrossRef CAS PubMed.
- I. A. Tekko, L. K. Vora, F. Volpe-zanutto, K. Moffatt, C. Jarrahian, H. O. Mccarthy and R. F. Donnelly, Adv. Funct. Mater., 2022, 32, 2106999 CrossRef CAS.
- A. D. Permana, A. J. Paredes, F. Volpe-Zanutto, Q. K. Anjani, E. Utomo and R. F. Donnelly, Eur. J. Pharm. Biopharm., 2020, 154, 50–61 CrossRef CAS PubMed.
- F. Benech-Kieffer, P. Wegrich, R. Schwarzenbach, G. Klecak, T. Weber, J. Leclaire and H. Schaefer, Skin Pharmacol. Physiol., 2000, 13, 324–335 CrossRef CAS PubMed.
- P. Makvandi, M. Kirkby, A. R. J. Hutton, M. Shabani, C. K. Y. Yiu, Z. Baghbantaraghdari, R. Jamaledin, M. Carlotti, B. Mazzolai, V. Mattoli and R. F. Donnelly, Nano-Micro Lett., 2021, 13, 1–41 CrossRef PubMed.
- A. H. Sabri, Z. Cater, J. Ogilvie, D. J. Scurr, M. Marlow and J. Segal, J. Drug Delivery Sci. Technol., 2020, 58, 101766 CrossRef CAS.
- J. W. Cook and J. D. Loudon, Alkaloids: Chem. Physiol., 1952, 2, 261–329 Search PubMed.
- S. Patnaik, L. A. A. Chunduri, M. S. Akilesh, S. S. Bhagavatham and V. Kamisetti, J. Exp. Nanosci., 2016, 11, 916–929, DOI:10.1080/17458080.2016.1178402.
- A. L. Mohamed, H. Elmotasem and A. A. A. Salama, Int. J. Biol. Macromol., 2020, 164, 1149–1163 CrossRef CAS PubMed.
- D. Czerwonka, S. Sobczak, E. Maj, J. Wietrzyk, A. Katrusiak and A. Huczynski, Molecules, 2020, 25, 1–12 CrossRef PubMed.
- N. M. Mahfouz and M. A. Hassan, J. Pharm. Pharmacol., 2001, 841–848 CAS.
- H. Trommer and R. H. H. Neubert, Skin Pharmacol. Physiol., 2006, 19, 106–121 CrossRef CAS PubMed.
- M. I. Foreman, Drug Dev. Ind. Pharm., 2008, 12, 461–463, DOI:10.3109/03639048609026625.
- M. S. Roberts, J. Bouwstra, F. Pirot and F. Falson, Dermatologic, Cosmeceutic, and Cosmetic Development: Therapeutic and Novel Approaches, 2007, pp. 115–124 Search PubMed.
- S. Gurunath, S. Pradeep Kumar, N. K. Basavaraj and P. A. Patil, J. Pharm. Res., 2013, 6, 476–480 CAS.
- Y. Zhang, N. Zhang, H. Song, H. Li, J. Wen, X. Tan and W. Zheng, Drug Delivery, 2019, 26, 70–77 CrossRef CAS PubMed.
- R. Brummer and S. Godersky, Colloids Surf., A, 1999, 152, 89–94 CrossRef CAS.
- J. Chen, X. Liu, S. Liu, Z. He, S. Yu, Z. Ruan and N. Jin, Drug Dev. Ind. Pharm., 2022, 1–9 Search PubMed.
- H. Bundgaard, E. Falch, S. B. Pedersen and G. H. Nielsen, Int. J. Pharm., 1985, 27, 71–80 CrossRef CAS.
- J. C. J. Wei, G. A. Edwards, D. J. Martin, H. Huang, M. L. Crichton and M. A. F. Kendall, Sci. Rep., 2017, 1–17 Search PubMed.
- Joint Formulary Committee, BNF 82 (British National Formulary) September 2021, Pharmaceutical Press, 2021 Search PubMed.
- M. J. Uddin, N. Scoutaris, P. Klepetsanis, B. Chowdhry, M. R. Prausnitz and D. Douroumis, Int. J. Pharm., 2015, 494, 593–602 CrossRef CAS PubMed.
- Y. Liu, B. Xu and X. L. Xu, Chin. J. Exp. Ophthalmol., 2013, 31, 630–635 CAS.
- Q. Wu, H. Liu, J. Liao, N. Zhao, G. Tse, B. Han, L. Chen, Z. Huang and Y. Du, Biomed. Pharmacother., 2020, 129, 110384 CrossRef CAS PubMed.
- N. Sandbo, C. Ngam, E. Torr, S. Kregel, J. Kach and N. Dulin, J. Biol. Chem., 2013, 288, 15466–15473 CrossRef CAS PubMed.
- M. Lemor, S. de Bustros and B. M. Glaser, Arch. Ophthalmol., 1986, 104, 1223–1225 CrossRef CAS PubMed.
- N. Dalbeth and D. O. Haskard, Rheumatology, 2005, 44, 1090–1096 CrossRef CAS PubMed.
- ISO - ISO 10993–5:2009 - Biological evaluation of medical devices—Part 5: Tests for in vitro cytotoxicity, https://www.iso.org/standard/36406.html, (accessed May 31, 2022).
- J. C. Wataha, S. Ratanasathien, C. T. Hanks and Z. Sun, Dent. Mater., 1996, 12, 322–327 CrossRef CAS PubMed.
- F. Zhang, Q. He, C. Helena Qin, P. J. Little, J. Weng and S. Xu, DOI:10.1038/s41401-021-00835-w.
- N. Dalbeth, T. J. Lauterio and H. R. Wolfe, Clin. Ther., 2014, 36, 1465–1479 CrossRef CAS PubMed.
- J. Zhao, K. Wei, P. Jiang, C. Chang, L. Xu, L. Xu, Y. Shi, S. Guo, Y. Xue and D. He, Front. Immunol., 2022, 1566 Search PubMed.
- S. Westmark, D. Melgaard, L. O. Rethmeier and L. H. Ehlers, Clinicoecon Outcomes Res., 2018, 10, 321–326 CrossRef PubMed.
- L. X. Chen, Geriatr. Rheumatol., 2011, 209–212 CAS.
| This journal is © The Royal Society of Chemistry 2022 |