
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Current progress in bionanomaterials to modulate the epigenome
Anna D. Y.
Rhodes†
a,
Jose Antonio
Duran-Mota†
ab and
Nuria
Oliva
 *a
*a
aDepartment of Bioengineering, Imperial College London, London W12 0BZ, UK. E-mail: n.oliva-jorge@imperial.ac.uk
bMaterials Engineering Group (GEMAT), IQS Barcelona, Barcelona 08017, Spain
First published on 21st July 2022
Abstract
Recent advances in genomics during the 1990s have made it possible to study and identify genetic and epigenetic responses of cells and tissues to various drugs and environmental factors. This has accelerated the number of targets available to treat a range of diseases from cancer to wound healing disorders. Equally interesting is the understanding of how bio- and nanomaterials alter gene expression through epigenetic mechanisms, and whether they have the potential to elicit a positive therapeutic response without requiring additional biomolecule delivery. In fact, from a cell's perspective, a biomaterial is nothing more than an environmental factor, and so it has the power to epigenetically modulate gene expression of cells in contact with it. Understanding these epigenetic interactions between biomaterials and cells will open new avenues in the development of technologies that can not only provide biological signals (i.e. drugs, growth factors) necessary for therapy and regeneration, but also intimately interact with cells to promote the expression of genes of interest. This review article aims to summarise the current state-of-the-art and progress on the development of bio- and nanomaterials to modulate the epigenome.
Introduction
Biomaterials possess the ability to modulate gene expression and hence, regenerate tissues at a molecular level.1 For example, biomaterials may be used as depots for the delivery of biological molecules such as growth factors that promote cell growth and differentiation.2–4 However, the power of biomaterials extends far beyond their ability to simply deliver drugs or other biomolecules. Mechanical properties, physicochemical cues, and biological stimuli that cells experience when in contact with a biomaterial all contribute to a cell's epigenetic state and ultimately cell function.5 The biomaterials community has exploited this phenomenon to explore how these cues, particularly topography,6 surface chemistry,7 matrix elasticity,8,9 and other mechanical stimuli, influence primarily stem cell differentiation but also other outputs, including cell reprogramming, proliferation, migration, and nuclear deformation.10 These changes in cell function are mediated by biophysical forces that are mechanotransduced from the extracellular space to the nucleus, resulting in the regulation of epigenetic modifier activity and chromatin or nuclear lamina remodelling.5 However, these epigenetic phenomena that govern observed changes in expression have remained relatively underexplored,11 especially in differentiated cell types that establish pathological milieux. Therefore, further elucidation of the nature of epigenetic biomaterial-tissue interactions will enable the development of a new class of therapeutic biomaterials that can reprogram expression of diseased tissues to healthy ones. In this review, we discuss key findings that illuminate the interface between biomaterials and epigenetics (Fig. 1) with implications in the areas of cancer modelling and therapy, tissue engineering, and regenerative medicine.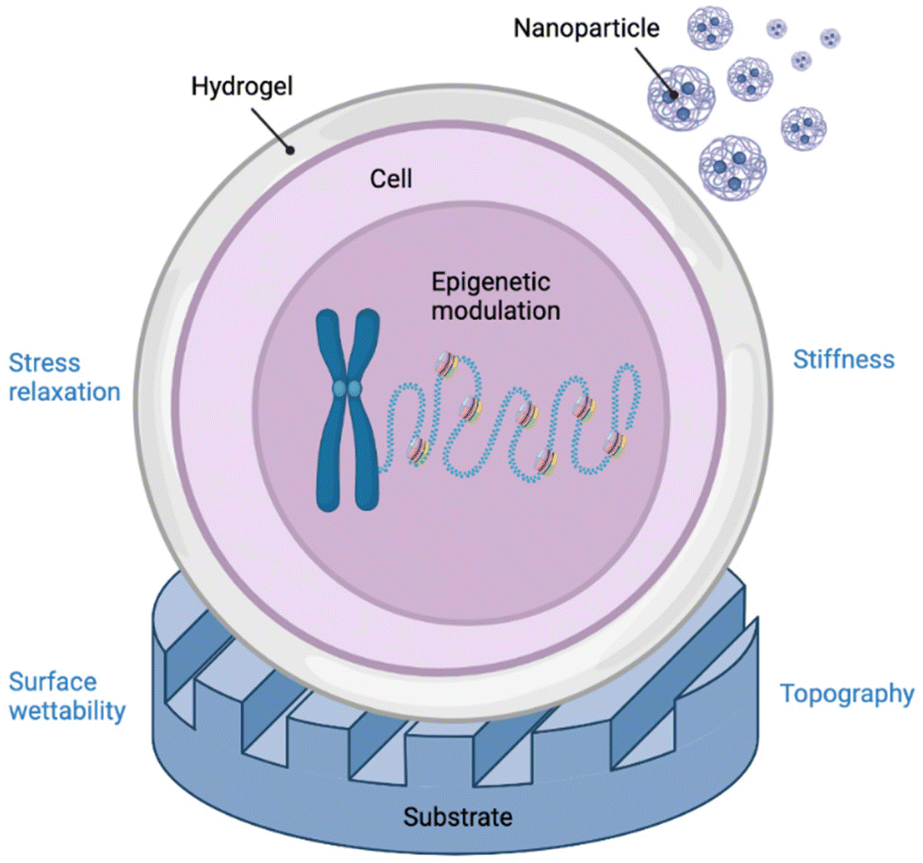 | ||
| Fig. 1 Graphical abstract depicting the various interactions of bio- and nanomaterials with DNA at an epigenetic level. Created with BioRender.com. | ||
Epigenetic regulation
While the basic mechanisms by which biological systems activate genes of interest have been well characterised, a complete understanding of how biomaterials may modulate the expression of genes is still being unravelled. Regulation of the epigenome is required for normal gene expression patterns and development, and for the prevention of inappropriate activation/inhibition of signalling pathways and disease progression.12 For example, various cancers have been associated with aberrant epigenetic signatures. However, unlike genetic mutations, epigenetic marks are generally reversible. For example, DNA methylation or demethylation can be reversed using DNA methyltransferase (DNMT) enzymes and their inhibitors, respectively. Similarly, histone acetyltransferases (HATs) and histone deacetylases (HDACs) can reversibly add or remove acetyl groups from histone proteins, respectively. Moreover, specific microRNAs (miRNAs) can upregulate or downregulate gene expression of different genes by binding to mRNA transcripts. Hence, this versatility and reversibility render epigenetic marks attractive therapeutic targets.12 For more details describing epigenetic molecular mechanisms, we refer the reader to Allis & Jenuwein.13 In this review, we restrict our discussion to the ways in which bionanomaterials’ properties can trigger epigenetic cues and signals (DNA methylation, histone modifications, and ncRNAs) to regulate gene expression at the pre- and post-transcriptional levels.Bionanomaterials and epigenetics in stem cells
The defining hallmarks of stem cells (perpetual self-renewal and the ability to differentiate into at least one specialised cell type) make them invaluable resources in tissue engineering and regenerative medicine. However, their biology must be fully understood and characterised before their complete therapeutic potential can be realised.14 As a result, researchers have focused their efforts on how various biochemical, biophysical, and material cues influence stem cell behaviour. Arguably, studies have focused primarily on how differentiation is affected, but these cues can also regulate cell migration, morphology, proliferation, and reprogramming.10,15 For example, many groups have demonstrated that substrate stiffness can determine stem cell lineage specification via mimicking native tissue stiffness. A seminal publication by Engler et al.8 showed that human mesenchymal stem cells (hMSCs) cultured on softer substrates (elastic modulus, E = 0.1–1 kPa) preferentially differentiated into cells which exhibited neuronal morphology, whereas hMSCs cultured on intermediate (E = 8–17 kPa) and stiff substrates (E = 25–40 kPa) tended to express myogenic and osteogenic markers, respectively.8 At present, researchers are focusing on decoding the mechanisms that result in these phenomena, including characterising the interplay between material properties and epigenetic regulation. Recent work has demonstrated that the biophysical features of biomaterials, such as stiffness, surface roughness, patterning, and wetting characteristics,16 may alter a cell's epigenetic state through several epigenetic regulatory mechanisms, including chromatin remodelling and DNA methylation. Table 1 summarises the genes, proteins, and pathways found to have been affected by biomaterials via epigenetic modulation.| Material description | Epigenetic modulation | Downstream gene expression modulation | Ref. |
|---|---|---|---|
| Monolayer of graphene on glass substrate | H3K4me3 enrichment in pluripotency regulator promoters (OCT4, NANOG) in mouse fibroblasts | Upregulation in pluripotency regulator (OCT4, NANOG, SOX2) expression and epithelial (E-CAD, EP-CAM, CLDN3) markers; downregulation in mesenchymal (N-CAD, SLUG, ZEB1) markers | 18 |
| Microgrooved PDMS substrate | AcH3 enrichment in pluripotency regulator promoters (OCT4, NANOG, SOX2) caused by HDAC2 downregulation; H3K4me2 and H3K4me3 enrichment in mouse fibroblasts | Upregulation in pluripotency (OCT4, NANOG, SOX2) and epithelial markers (E-CAD, EP-CAM, KRT8, OCLN, CLDN3); downregulation in mesenchymal markers (TGFBR1, SNAI1, SNAI2, VIM, ITGB1) | 21 |
| PDMS substrate stiffness | Softer substrates (E = 1.5, 15 kPa) induced increased euchromatin and decreased heterochromatin contents in human mesenchymal stem cells | Upregulation in pluripotency markers (OCT3/4, SOX2, NANOG) | 23 |
| Microgrooved collagen-coated PDMS membranes | Microgrooves elicited histone hyperacetylation and AcH3 enrichment in human cardiac progenitor cells | Upregulation of mature cardiac marker (sarcomeric MyHC, ACTC1, NPPA) expression | 28 |
| Microgrooved Matrigel-coated PDMS substrates | Microgrooves elicited AcH3 enrichment in mouse fibroblasts | Upregulation of mature cardiac transcription factor (GATA4, MEF2C, TBX5, NKX 2.5, HAND2) expression, mature cardiac marker (α-SMA, cTnT, TNNT2, ACTN2) expression, and MKL1 expression | 31 |
| PDMS substrate stiffness | Stiff substrates (E = 140 kPa) induced downregulation of H3K9-methylating genes and inhibited nuclear peripheral organization of H3K9me3-marked chromatin in cardiomyocytes | Downregulation of cardiac transcription factor (GATA4, HAND2, MEF2C, NKX2-5), structural protein (ATP2A2, MYH6, SCN5A, TNNI3), and histone methyltransferase (KDM6A, EED, EHMT1, EHMT2, SETDB1) expression | 34 |
| Polyacrylamide hydrogel stiffness | Stiff substrates (E = 50 kPa) induced downregulated miR-6740-5p expression in HUVECs | Suppressed miR-6740-5p led to increased ET-1 production | 38 |
| Nanogrooved Matrigel-coated PUA substrates | Nanogrooves induced enrichment of H3K4me3 marks in mouse embryonic fibroblasts | Upregulation of dopaminergic (TH, DAT) and epithelial (E-CAD, CLDN3, EP-CAM) expression; no trend observed with mesenchymal (ZEB1, N-CAD, SLUG) expression | 43 |
| Gelatin, gelatin-hydroxyapatite, gelatin-hydroxyapatite-pig brain substrates | Gelatin biomaterials induced upregulation of HDAC3 expression in mouse embryonic fibroblasts | Upregulation of astrocytic transcription factor (NFIA, NFIB, SOX9) expression | 44 |
| Free nano-hydroxyapatite | Osteogenic (ALP) expression in bone marrow stromal cells | Downregulation in ALP expression | 50 |
| TSA-laden PLLA aligned fiber scaffold | Increased AcH3 and AcH4 enrichment and downregulation of HDAC1 activity | Enhanced tenogenic (SCX, MKX, EYA1, HOXA11, DCN) expression | 54 |
| G1, G3, and G5 polyamidoamine dendrimer surfaces | Distinct differences in H3K9me3, H3K27me3, and H3K9ac levels on G1, G3, and G5 surfaces | Differentiation to smooth, skeletal, and cardiac muscle lineages | 59 |
| Silver (AgNP), gold (AuNP), and superparamagnetic iron oxide nanoparticles (SPIONs) | No differences in DNA methylation as a result of treatment with nanoparticles in HepG2 hepatocarcinoma cells | Significant changes in nine pro- and anti-tumour miRNAs expression | 63 |
| Collagen-alginate interpenetrating network (IPN)-based matrices to soften the ECM | Soft matrices (E = 0.5–1 kPa) induced recruitment of DNA methylation inhibitors TET2, GRHL2, and KMT2A in gastric cancer cells | Decrease of YAP overexpression; YAP overexpression is linked to aggravation of cell migration, invasion and promotion of a malignant phenotype | 65 |
| Alginate IPN hydrogel stiffness | Stiff matrices (E = 2 kPa) led to increased HDAC3 and HDAC8 activity compared with soft matrices (E = 100 Pa) in breast cancer epithelial cells | Increased accessibility of transcription factor Sp1 target genes | 66 |
| Collagen-Matrigel gel stiffness | Softer substrates (E = 0.25 kPa) added activating histone marks to CD133 promoter and repressing histone marks to THBS2 promoter in liver cancer stem cells | Upregulation of CD133 expression and downregulation of THBS2 expression | 70 |
| Fibrin gel stiffness | Stiff gels induced increased Tet2 enzyme activity in murine melanoma cells | Upregulation of genes that inhibit cell cycle progression | 72 |
| PDMS substrate stiffness | No significant differences in DNA methylation in human mesenchymal stem cells | No significant differences in senescence associated β-galactosidase | 76 |
| PEG-crosslinked collagen hydrogel stiffness | Stiff hydrogels (E = 5–6 kPa) decreased accessibility of genes in bivalent chromatin domains in hair follicle stem cells | Softer hydrogels (E = 1–3 kPa) restored expression of key HFSC self-renewal (CCND2, CCNE1, CDC34, CDKL1, HMGA2) and differentiation (LEF1, RSPO3, WNT2B, WNT7A) genes | 78 |
| Carbon dots of different surface charge | mRNA and miRNA expression and DNA methylation of human embryonic lung fibroblasts after exposure to carbon dots | Positive dots influenced pathways related to dysregulation of immune response processes, tumorigenesis, and cell cycle regulation; negative dots induced changes in cell proliferation, apoptosis, oxidative stress, and cycle regulation | 80 |
| TiO2 nanoparticles | Global DNA methylation and hydroxymethylation alteration in mice lungs following nasal inhalation of TiO2 nanoparticles | Alteration of methylation in promoter of TNF-α and Thy-1, key in the inflammatory response and fibration | 81 |
| PEG hydrogel | Long-term culture on stiff (E = 4.7 kPa) hydrogels induces global chromatin condensation in fibroblasts | Chromatin condensation is permanent, and contributes to persistent fibroblast activation | 82 |
Reprogramming
Although reprogramming technology has advanced significantly since 2006, induced pluripotent stem cell (iPSC) generation still relies heavily on viral-induced transcription factor (TF) overexpression, which introduces safety risks such as unexpected genomic integration and insertional mutagenesis.16 In addition, the reprogramming process is naturally inefficient due to epigenetic barriers that stabilise gene expression in terminally differentiated cells.17 Therefore, several studies have investigated the addition of factors such as silencing miRNAs, mRNAs, gene-editing machinery such as CRISPR-Cas9, and epigenetic ‘signals’ in an effort to identify an alternative strategy that could replace viral methods and increase efficiency in the future.16 We focus our discussion on how material properties can induce epigenetic modifications that lead to enhanced reprogramming.Yoo et al.18 investigated the capability of a graphene monolayer to induce reprogramming of mouse fibroblasts to an embryonic-like state. Interestingly, they found that pluripotency master regulators, such as OCT4, NANOG, and SOX2, were markedly upregulated in graphene-coated substrate cultures compared with uncoated cultures. They hypothesised that this observed difference was due to the graphene substrate enhancing induction of the mesenchymal-to-epithelial transition (MET), an essential process for reprogramming.19–21 To investigate this theory, they measured enrichment of histone modifications at various transcription start sites (TSSs) to determine the extent of chromatin remodelling. Indeed, they found that the TSSs of OCT4 and NANOG were enriched for H3K4me3, a histone mark associated with gene activation and MET.22 In addition, cells cultured on graphene substrates exhibited significantly increased expression of epithelial markers, including E-CAD. These results demonstrate the MET-promoting potential of graphene and its ability to increase reprogramming efficiency through an epigenetic mechanism.18
Another study conducted by Downing et al.21 explored the effects of microgrooved (10, 20, and 40 μm spacing) polydimethylsiloxane (PDMS) substrates on cell reprogramming. The authors initially observed that microgrooves elicited significant upregulation in fibroblast-derived NANOG+ iPSC colonies compared with flat surfaces, and subsequently hypothesised that the topography increased the prevalence of histone H3 acetylation (AcH3) marks, which are known to be involved with reprogramming. Surprisingly, they found that the microgrooves increased global AcH3 marks within the promoter regions of OCT4, SOX2, and NANOG in mouse fibroblasts even in the absence of reprogramming TFs, which was subsequently found to be due to downregulation of HDAC2. In addition, there was increased methylation of H3K4me2 and H3K4me3, marks that are also critical for reprogramming. Most notably, the microgrooves elicited analogous histone modifications compared to the sole introduction of OCT4, SOX2, KLF4, and MYC (OSKM) or small-molecule epigenetic modifiers, such as valproic acid (VPA) and tranylcypromine hydrochloride (TCP).21 Therefore, material cues may have the potential to entirely replace conventional virus-induced TF overexpression-based methods, circumventing safety concerns. However, expansion of the iPSC-like colonies was limited, suggesting that microgrooved surfaces only activate the early phase of reprogramming. In concert, the activity of several genes associated with both epithelial and mesenchymal phenotypes were monitored. All investigated epithelial markers including E-CAD were upregulated, whereas all mesenchymal markers including TGFBR1 were downregulated. There was also an increase in H3K4me2 in the E-CAD promoter, again emphasising the role of MET.21
Downing et al.21 additionally sought to elucidate how important cytoskeletal reorganization was in causing the epigenetic changes associated with cell reprogramming. As expected, cells cultured on the microgrooves exhibited greater alignment and nuclear elongation. To investigate the role of alignment, they used nanofibers in both aligned and random orientations. Indeed, aligned nanofibers globally increased AcH3, H3K4me2, and H3K4me3 marks, and significantly upregulated NANOG expression compared with random fibres. To determine the importance of elongated cell morphology, they used micropatterning techniques to closely control cell shape on flat surfaces. They found that elongated cells displayed significantly higher levels of nuclear AcH3, H3K4me2, and H3K4me3, compared with circular cells. Together, these results illustrate the key roles of cell alignment and morphology, parameters that can be controlled using biomaterials, in overcoming epigenetic barriers, i.e. chromatin remodelling, to achieve cell reprogramming.21
Several groups have also investigated the effect of substrate stiffness on reprogramming efficiency. Gerardo et al.23 investigated this phenomenon when reprogramming hMSCs to iPSCs on PDMS substrates of varying stiffness. Previous studies have identified a negative correlation between ‘stemness’ and nuclear stiffness – multipotent and terminally differentiated cells possess a higher cellular stiffness compared with pluripotent stem cells.24 Considering this, they hypothesised that culturing hMSCs on soft substrates would induce reprogramming to iPSCs via promoting nuclear relaxation and softening the cell as a whole. They found that the hMSCs cultured on softer substrates (E = 1.5 and 15 kPa) exhibited increased euchromatin, i.e. enriched H4K16ac (transcriptionally permissive, relaxed chromatin) and decreased heterochromatin (transcriptionally restrictive, condensed chromatin) contents compared with hMSCs cultured on stiff GPa-range substrates. Pluripotent cells are characterised by high levels of euchromatin, while gradual enrichment of heterochromatic regions ensues during differentiation.25 This dynamic epigenomic structure blocks transcriptional machinery and TFs from physically accessing irrelevant developmental programmes that are not necessary once a cell has ‘chosen’ its fate.23 These results show that by providing a mechanical environment that facilitates decreased cellular stiffness, reprogramming to a pluripotent state may be enhanced.
Differentiation
Multi- or unipotent stem cells are present in almost all adult tissues. They serve to facilitate repair and rejuvenation of damaged, diseased, or aged tissues via differentiation to replace the specialised cells that were lost.26 For example, following injury to the skin, basal epidermal stem cells differentiate and migrate towards the surface to close the wound.27 However, some organs and tissues have limited intrinsic regenerative potential28 due to a paucity of tissue-resident stem cells.Although induced cardiomyocytes (iCMs) have been successfully generated both in vitro and in vivo from cardiac fibroblasts via direct reprogramming TFs (GATA4, MEF2C, and TBX5), the efficiency remains extremely low (10–15%). Another limitation is that iCMs do not reach complete maturation in vitro due to the lack of an in vivo-like structural organisation.30 A study by Morez et al.28 sought to increase the efficiency of de novo cardiomyocyte generation from CPCs by using topographical cues that offer enhanced recapitulation of the 3D cardiac environment and circumvent epigenetic barriers associated with direct lineage reprogramming.28 For example, a high degree of chromatin condensation or histone deacetylation, typical of differentiated cells, prevents transcriptional regulators from accessing DNA binding/recognition sites.17 They found that microgrooves elicited histone hyperacetylation in a baseline-dependent manner, unwinding the chromatin. They also identified a significant positive correlation between nuclear elongation and AcH3 levels. Furthermore, they concluded that histone acetylation directly resulted in increased reprogramming efficiency. This finding suggests that material-induced histone acetylation not only improves reprogramming cells to iPSCs but also enhances direct conversion of cells to highly differentiated states. In addition, the combination of their established cardiac differentiation protocol and the microgrooves proved to be synergistic, resulting in a two-fold increase in cells that were positive for sarcomeric MyHC, ACTC1, and NPPA compared with flat surfaces,28 likely owing to the relaxed chromatin structure.
Sia et al.31 also questioned the augmenting effect of microtopography as well as substrate stiffness on direct conversion efficiency of mouse fibroblasts to CMs.31 Cells cultured on microgrooved substrates (3 and 5 μm spacing) yielded a higher proportion of α-SMA- and cTnT-positive cells (both of which are markers of contractility)32,33 compared to flat substrates. They concluded that the microgrooves increased conversion efficiency through increased AcH3 marks. Interestingly, there were no significant differences in the yield of α-SMA-positive cells comparing substrates which had stiffnesses of 1, 21 and 62 kPa.31
In contrast, the work of Seelbinder et al.34 suggested that there is a correlation between stiffness and chromatin structure, which is important for cardiac development and tissue maintenance. It is known that perturbations to the mechanical properties of the heart, due to genetic disorders and chronic conditions, lead to onset of disease states as a result of diminished CM gene expression and consequent attenuated cardiac function.35 Therefore, they sought to further elucidate the role of environmental mechanics in CM de-differentiation. They observed that culturing CMs on stiff substrates (E = 140 kPa) elicited downregulation of histone proteins that are replication-dependent, suggesting a reduction in cell proliferation. There was also downregulation of H1, H2a, and H3 histone variants, which are involved in cell differentiation, implying considerable de-differentiation. Overall, these results show that stiff environments result in acquisition of an epigenetic profile that impairs CM function. In contrast, culturing CMs on a substrate that mimicked tissue stiffness (E = 13 kPa) supported normal spatial chromatin organisation and gene expression characteristic of healthy CMs.34
Stiffness has also been shown to be an important biophysical cue for endothelial cells, which line the inner wall of blood vessels.36 Vascular stiffening has been linked to endothelial dysfunction, which is an early indicator of cardiovascular disease.37 To further probe how stiffness regulates endothelial function, Song et al.38 cultured human umbilical vein endothelial cells (HUVECs) on polyacrylamide hydrogels with several stiffnesses (E = 4, 25, and 50 kPa), and performed a miRNA array analysis to identify mechanosensitive miRNAs. Normal stiffness of the basement membrane has been reported to be in the range of E = 2.5–8 kPa.39 They found that miR-6740-5p was significantly downregulated on the stiffest substrate (E = 50 kPa), compared with the softest substrate (E = 4 kPa). Using a bioinformatics approach, they identified EDN1 as a target of miR-6740-5p. EDN1 mRNA codes for ET-1, which is a powerful vasoconstrictor – its upregulation has been associated with high blood pressure and the development of atherosclerotic plaques. Ultimately, this result shows that increased stiffness leads to the establishment of a vascular environment that favours hypertension through an epigenetic mechanism.38
Kantawong et al.44 explored the potential of gelatin-hydroxyapatite-pig brain biomaterials in generating induced neural stem cells (iNSCs) from MEFs. iNSCs are not only capable of differentiating into neurons, but also into glial cells including astrocytes and oligodendrocytes.45 They found that cells cultured on the gelatin biomaterials had increased HDAC3 expression. HDACs have been shown to repress miR-124,46 whose target is PTBP1 – this TF has been implicated in suppressing neuronal differentiation whilst activating astrocytic differentiation.47 They postulated that decreased miR-124 expression could have caused increased PTBP1 expression, leading to a preference to differentiate into the astrocytic lineage. Indeed, culture on the gelatin materials upregulated astrocytic TF (NFIA, NFIB, SOX9) expression even without viral TF infection.44 Currently, astrocyte generation is slow in vitro,48 which is a considerable limiting factor for studying neurodegenerative disease. Future studies could focus on further optimising gelatin-based substrates to shorten the differentiation period.
Tendon is another tissue with poor regenerative capacity that often requires clinical intervention.51 The conventional treatments for tendon injuries present detrimental side effects including donor site morbidity and risk of disease transmission,52 as well as being relatively short-term solutions.53 An alternative approach that holds promise is tendon tissue engineering, which uses human tendon stem/progenitor cells (hTSPCs) seeded in scaffolds that replicate the physiological array of parallel collagen fibres in the tendon microenvironment to direct tenogenesis.54 Zhang et al.54 used the synergistic combination of electrospun poly(L-lactic acid) (PLLA) fibres and an HDAC inhibitor (TSA) to promote tendon differentiation. The aligned PLLA fibres resulted in elongated cell morphology, provided remarkable mechanical properties, as well as sustained drug release, which was due to a higher degree of polymer crystallinity because of TSA incorporation. SCX, a marker of tendon differentiation was approximately 2-fold higher on parallel aligned TSA fibres compared with just aligned fibres with no active HDAC inhibitor and randomly aligned TSA fibres. They confirmed that the increased differentiation efficiency was in fact due to chromatin remodelling and increased AcH3 and AcH4 marks, as well as a decrease in HDAC1.54
hMSCs are another cell type of interest to both clinicians and researchers for their potential to differentiate into many different tissues, including cartilage, bone, fat, muscle, and skin.55 Endogenous hMSCs are a key component enabling both tissue development and long-term maintenance, and as such, the depletion of the hMSC reservoirs in tissues results in degenerative or age-related disease onset.56 As well as their ability to directly differentiate, hMSCs have the potential to regenerate tissues by promoting normal tissue remodelling in a paracrine manner. To date, a number of studies have investigated the effect of polyamidoamine (PAMAM) dendrimer generation number on cell fate within the mesenchymal lineage.57,58 Ayuningtyas et al.59 explored the mechanism governing muscle lineage switching of hMSCs through migration and cellular and nuclear deformation on dendrimer surfaces. Cells cultured on G3 and G5 surfaces exhibited more dynamic temporal stretching and contraction, which is representative of active migration, compared with cells grown on a G1 surface. Interestingly, on G5 surfaces, the cells developed rounder shapes and spreading was inhibited across the surface, leading to formation of spherical aggregates. Accompanying these migratory behaviours were substantial conformational changes of the cytoskeleton and nuclear lamina deformation, which in turn led to chromatin reorganisation. Overall, they found H3K9me3 levels were consistently decreased on the G3 and G5 surfaces, whereas both H3K27me3 and H3K9ac levels were decreased in the short-term but increased in the long-term. In contrast, nuclear lamina deformation was comparatively lower in G1 cells and was correlated with the upregulation of H3K9ac in the short-term, and upregulation of H3K9me3 in the long-term, suppressing hMSC differentiation. It is thought that initial downregulation of H3K27me3 in the short-term causes unwinding of chromatin and increased gene transcription, initiating the differentiation process,60 whereas at later stages, the increase in H3K27me3 suppresses genetic programs of alternative specific cell fate programs not within the muscle lineages. Importantly, this material-induced spectrum of histone modification resulted in diverse differentiation patterns; differentiation for G1, G3, and G5 cells tended towards smooth muscle, skeletal muscle and cardiac muscle lineages, respectively.59
Bionanomaterials and epigenetics in cancer cells
As stated above, various cancers have been associated with aberrant epigenetic signatures. Hence, it is not surprising that the scientific community has placed a large effort on delivering drugs and other therapeutics to modulate epigenetics and overall gene expression in cancer cells (beyond the scope of this review).61,62 For example, nanoparticles and nano-carriers (NCs) have been used for decades to address the challenges associated with delivery of drugs with poor stability or bioavailability. However, only recently has it been suggested that the ability of nanoparticles to modify cells’ epigenetics extends beyond simply delivering therapeutics. The rapid advance of sequencing technologies has recently revealed the effects of nanocarrier size, surface charge, shape, and the administration dose on epigenetic regulation. Brzóska et al.63 studied the change of miRNA expression and DNA methylation status of different genes related to inflammatory processes and apoptosis in HepG2 hepatocarcinoma cells after exposure of mildly cytotoxic doses of silver, gold, and iron oxide nanoparticles. No significant difference was shown in DNA methylation, but the expression of nine miRNAs was significantly altered by treatment using these nanoparticles. Specifically, silver nanoparticles induced more changes in miRNAs expression than gold, followed by iron oxide nanoparticles, thus showing a dependence on the type of material.63Beyond material type, its mechanical properties are also defining traits to modulate cancer cell epigenetics. Generally, a stiffer ECM is a characteristic of malignant tumours – this environment leads to aggravation of cell migration and invasion, as well as promoting a malignant phenotype in surrounding cells.64 Jang et al.65 showed that softening a stiff ECM (from E = 7 to 0.5–1 kPa) in a gastric cancer model could epigenetically reverse elevated YAP protein expression, through recruitment of DNA methylation inhibitors, namely TET2, GRHL2, and, KMT2A. YAP is a mechano-sensitive transcription factor that is associated with metastasis and malignant behaviour. Stowers et al.66 explored this phenomenon in the context of breast cancer; they showed that stiff interpenetrating networks (IPNs) comprised of alginate and reconstituted basement membrane matrix (E = 2 kPa) promoted the development of an invasive phenotype, whereas IPNs with stiffness comparable to native mammary tissue (E = 100 Pa) promoted healthy, acinar morphology. Stiff matrices led to increased HDAC3 and HDAC8 activity and increased accessibility of Sp1 target genes; Sp1 is a transcription factor that is upregulated in breast cancer.67
Interestingly, there is evidence that stiffness is actually non-uniform within a tumour,68 which has important implications for cancer cell invasiveness and drug resistance. In contrast to the notion of increased bulk stiffness promoting malignancy, some research groups have shown that soft matrices promote the plasticity of cancer stem cell (CSC) populations – CSCs are thought to be the main drivers behind tumorigenesis.69 In this sense, Ng et al.70 investigated the role of matrix stiffness in regulating the ‘stemness’ of CD133+ liver CSCs with THBS2 deficiency in hepatocellular carcinoma. They observed that the CSCs grown on softer collagen-Matrigel gels (E = 0.25 kPa) had more ‘aggressive’ phenotypes – there was increased CD133 expression and reduced THBS2 production, compared with stiffer gels (E = 15 kPa). CD133 is a marker of self-renewal potential,71 and THBS2 is known to interfere with tumour growth and angiogenesis by suppressing matrix metalloproteinase (MMP) activity.70 To further examine the mechanism underlying the changes in protein expression, they looked for differential histone H3 modifications. They discovered that upon culture on softer supports, transcriptionally activating H3K4me3 and H3K9ac marks were enriched within the CD133 promoter, whereas repressing H3K9me3 and H3K27me3 marks were present in the THBS2 promoter. Interestingly, they determined that there was a positive feed-forward loop where induced THBS2 downregulation led to further accumulation of CD133-high CSCs, which enabled the maintenance of localised ‘soft’ regions in the ECM. This in turn, ultimately confers enhanced metastasis due to decreased ECM presence.70 Furthermore, Liu et al.72 showed that a stiff mechanical environment can induce dormancy in tumorigenic cells via an epigenetic mechanism. They cultured murine B16 melanoma tumour-repopulating cells (TRCs) in several fibrin gel formulations (E = 90, 450, 1050, and 2000 Pa); they found that the stiffer gels induced the nuclear translocation of Cdc42, which resulted in activation of a hydroxymethylating enzyme, Tet2. This activation then led to upregulation of genes that inhibit cell cycle progression, including p21 and p27.72 In the future, designing bespoke biomaterial scaffolds with heterogeneous stiffness will be important to mimic the diverse mechanical landscape of tumours, and ultimately to conserve the functional phenotypes of distinct cell populations within a tumour.
Bionanomaterials and epigenetics in primary cells
Ageing
All primary cells enter a senescent state after reaching approximately 50 cell divisions, also known as the Hayflick Limit.73 Hallmarks of cellular senescence include flattened morphology and proliferative cessation despite an optimal growth environment with sufficient mitogenic stimuli; the aggregation of senescent cells has been confirmed to provoke cellular aging and dysfunction.74 Acquisition of a senescent phenotype has been linked to epigenetic changes; Schellenberg et al.75 demonstrated that senescent hMSCs had distinct DNA methylation profiles that corresponded with repressive histone marks. They found that senescent hMSCs had significantly differentially methylated CpG sites in genes associated with development, including DLX5, CDKN2B, and HOXD10. This differential methylation was correlated to absence of activating-H3K4me3 and presence of repressing-H3K9me3 marks.75 Schellenberg et al.76 then asked whether substrate stiffness could affect senescence onset. Interestingly, they showed that hMSCs cultured until senescence on elastic substrates did not display the typical flattened morphology characteristic of a senescent cell and remained relatively condensed. Despite this, there was no significant difference in the activity of senescence associated β-galactosidase (SA-β-GAL) or DNA methylation as a function of matrix stiffness.76Another characteristic of cellular aging is decline in stem cell number, resulting in diminished regenerative capacity of tissues.77 Koester et al.78 sought to investigate if restoration of youthful niche mechanics could rescue aged stem cells using the hair follicle as a model. Initially, they characterised both young and aged hair follicle stem cells (HFSCs) and they discovered that the aged HFSCs showed large-scale reduced chromatin accessibility compared to the young HFSCs, particularly in promoter regions of genes that regulate tissue development and differentiation (Fig. 2A and B). One category of regions with decreased accessibility in the aged HFSCs were promoters with antagonising epigenetic marks, namely activating-H3K4me3 and repressing-H3K27me3 (Fig. 2C). These regions are known as bivalent chromatin domains. Gene ontology enrichment analyses demonstrated that these bivalent genes were involved with HFSC differentiation and self-renewal, which compromises adequate cell cycle entry and tissue turnover in aged HFSCs. Interestingly, placing the aged HFSCs in a young niche activated previously repressed bivalent genes, leading to a return of self-renewal capacity, equivalent to that of young HFSCs. They later discovered that upregulation of various collagens and laminins that comprise the extracellular matrix (ECM) and the basement membrane (BM) was associated with the aged HFSC phenotype. These alterations led to thickening and stiffening of the BM (Fig. 2D and E), so they hypothesised that matrix stiffness, as a single parameter, was the dominating regulator of HFSC potency. Indeed, culturing both young and old HFSCs on a soft matrix (E = 1–3 kPa) produced similar activation of bivalent regions, whereas culturing young HFSCs on a stiff hydrogel matrix (E = 5–6 kPa) that recapitulated the aged BM was adequate to induce impaired stem cell self-renewal and deactivation of bivalent regions (Fig. 2F).78 This finding emphasises the advantage in circumventing intrinsic manipulation of cells by exploiting the power of relatively simple external cues to modulate cell behaviour in a non-invasive manner.
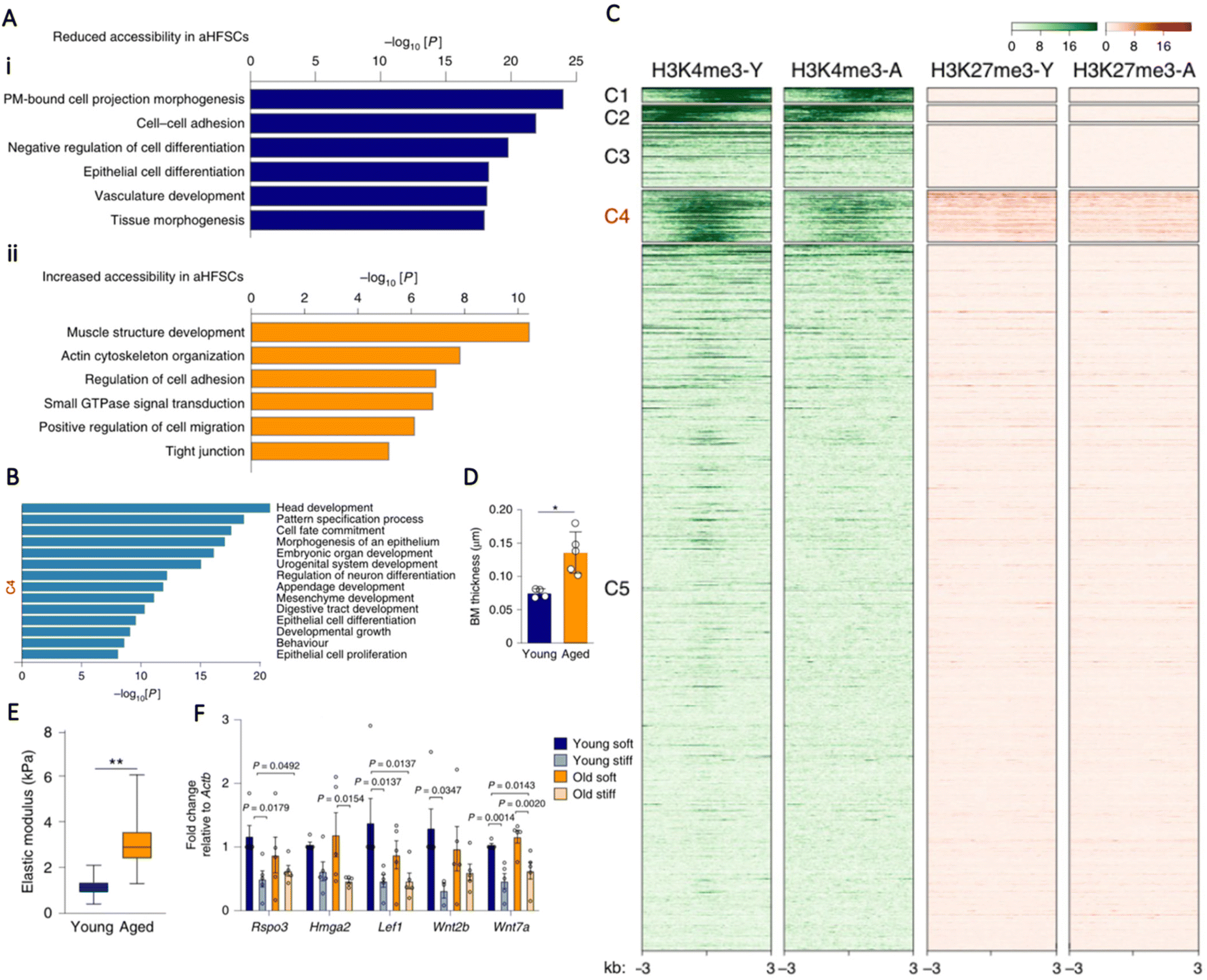 | ||
| Fig. 2 (A) Gene ontology (GO) term analyses of peaks that have increased accessibility in young (i) and aged (ii) HFSCs demonstrate that developmental and cell differentiation processes have decreased accessibility, whereas genes that are involved in actin cytoskeleton organisation and the regulation of cell adhesion have increased accessibility in aged HFSCs. (B) GO term analyses of the genes identified in cluster 4 (C4) are related to developmental processes, cell fate, and differentiation. (C) k-means clustering of ATAC-seq peaks (not shown) with diminished accessibility in aged HFSCs. Peak cluster 4 (C4) shows differential epigenetic modification with both H3K4me3 and H3K27me3 in young HFSCs, and decreased H3K4me3 at these sites in aged HFSCs. Colour scales represent reads per genomic content. (D) Aged HFSCs exhibit increased basement membrane (BM) thickness compared to young HFSCs. (E) Aged HFSCs exhibit increased stiffness compared to young HFSCs. (F) RT-qPCR analysis of bivalent genes related to HFSC activation and self-renewal in young and aged HFSCs cultured in soft and stiff hydrogels. Note the downregulated expression in stiff hydrogels. n = 5 cultures per group. Statistical analysis was performed using ANOVA and Fisher's Least Significant Difference test. P values are indicated on the graph. Data are mean ± s.e.m.78 Reproduced from ref. 78 with permission from Springer Nature, copyright 2021. | ||
Inflammation
Bionanomaterials’ properties have recently been shown to elicit epigenetic changes, including DNA and/or RNA methylation, hydroxymethylation, histone modification, and changes in miRNA expression, linked to oxidative stress due to ROS overproduction, MAPK signalling cascade dysregulation, and inflammation.20 Indeed, nanoparticle composition, size and shape, as well as biomaterial surface charge and stiffness can control both immunostimulation and immunosuppression.79 For example, Sima et al.80 studied the mRNA and miRNA expression of the full genome of human embryonic lung fibroblasts (HEL 12469) as well as DNA methylation after exposure to positive or negative carbon dots. Generally, negative dots showed more differences in expression than positive dots, with only a few of them overlapping in both groups. After a pathway analysis, it was shown that positive dots influenced pathways related to dysregulation of immune response processes, tumorigenesis, and cell cycle regulation, whereas the negative dots induced changes in cell proliferation, apoptosis, oxidative stress, and cycle regulation. Similar trends were shown when studying miRNAs. Interestingly, within the same group, the number of dysregulated mRNAs increased when increasing dose and exposure time.80 Taken together, these observations demonstrate that nanoparticle charge can induce epigenetic changes. Interestingly, characteristics of the host organism also play a critical role in epigenetic response to bionanomaterials. Ma et al.81 treated two groups of mice, comprised of young and adult populations, with TiO2 nanoparticles via nasal inhalation. Global methylation and hydroxymethylation in lungs were measured. As a result of the treatment, pulmonary inflammation and fibrosis were induced more severely in young mice due to a decrease in global methylation and hydroxymethylation only in this group. The altered methylation affected the TNF-α and Thy-1 promoters. Overall, the young mice presented 197 genes that were upregulated compared to the adult group.81 Deciphering nanoparticle-tissue interactions and underlying mechanisms will allow for a more comprehensive and personalised use of materials in biomedicine and avoid possible toxicity and safety concerns.Biomaterials have also been explored as useful tools to probe the biology underlying fibrosis, which is a consequence of dysregulated wound healing. Walker et al.82 used photo-softening PEG hydrogels to investigate aberrant fibroblast activation, which is known to contribute to fibrotic tissue development. They showed that activated fibroblasts, also known as myofibroblasts, cultured on stiff hydrogels (storage modulus, G′ = 4.5 kPa) for 1–3 days were capable of ‘deactivating’ upon hydrogel softening (G′ = 1.7 kPa), whereas cells that were cultured for 1–5 days were only partially deactivated. Interestingly, a period for more than 7 days rendered the cells unable to deactivate, even after the pathological ‘stiff’ stimulus was withdrawn. They subsequently identified that the persistently activated myofibroblasts possessed globally condensed chromatin, compared with transiently activated fibroblasts, which was correlated with increased HDAC activity.
Conclusions and future directions
Advances in chemistry, physics, and biology, integrated with medicine, have enlightened the path to a better understanding of biomaterials and their interactions with cells and tissues. The biomaterials field has focused on stiffness as the predominant material property that affects cell behaviour. Whilst stiffness has been proven to be a potent stimulus for epigenetic remodelling particularly in cancer, other material parameters whose epigenetic effects in cells remain under-characterised include topography and stress relaxation. Highly uniform topography (2D micro/nanogrooves) has been shown to alter cell shape and thus promote epigenetic marks that support cell reprogramming, which will be useful for optimised iPSC/cardiomyocyte/neuron generation. Further elucidation of the effects that 3D hydrogel- or scaffold-based culture has on cell shape and subsequent epigenetic consequences warrants further investigation, especially in organ modelling/tissue engineering applications. Furthermore, viscoelastic hydrogels that mimic the natural viscoelasticity of biological tissues have been shown to enhance several cell outputs, as opposed to purely elastic hydrogels. To our knowledge, there have been no studies as of yet that have explored the effects of viscoelasticity/stress relaxation on epigenetic remodelling to guide cell fate or phenotype. In the future, these parameters should be considered equally as significant as stiffness to generate highly representative tissue/organ models. Equally interesting is the understanding of how bio- and nanomaterials alter gene expression, and whether they have the potential to overcome the limited drugability of some targets, like tumour suppressor genes.Despite the significant leaps taken, there is inevitably a wealth of bionanomaterial-cell interactions that remain un-characterised. The increasing availability of highly informative molecular techniques will enable a better understanding of this relatively young interface, leading to bioactive materials capable of modulating cell epigenetics in a therapeutic manner.
Author contributions
A. D. Y. R. and J. A. D. M. wrote the original draft manuscript. A. D. Y. R., J. A. D. M. and N. O. reviewed and edited the manuscript. N. O. conceptualised the ideas and acquired the funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. O. acknowledges an Imperial College Research Fellowship.References
- C. Ning, L. Zhou and G. Tan, Mater. Today, 2016, 19, 2–3 CrossRef.
- Y. Meng, X. Li, Z. Li, C. Liu, J. Zhao, J. Wang, Y. Liu, X. Yuan, Z. Cui and X. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 5783–5793 CrossRef CAS PubMed.
- Z. Geng, X. Wang, J. Zhao, Z. Li, L. Ma, S. Zhu, Y. Liang, Z. Cui, H. He and X. Yang, Biomater. Sci., 2018, 6, 2694–2703 RSC.
- W. Song, C. Yang, D. Q. Svend Le, Y. Zhang and J. Kjems, ACS Appl. Mater. Interfaces, 2018, 10, 7756–7764 CrossRef CAS PubMed.
- S. W. Crowder, V. Leonardo, T. Whittaker, P. Papathanasiou and M. M. Stevens, Cell Stem Cell, 2016, 18, 39–52 CrossRef CAS PubMed.
- G. Abagnale, M. Steger, V. H. Nguyen, N. Hersch, A. Sechi, S. Joussen, B. Denecke, R. Merkel, B. Hoffmann, A. Dreser, U. Schnakenberg, A. Gillner and W. Wagner, Biomaterials, 2015, 61, 316–326 CrossRef CAS PubMed.
- W. Li, K. Li, W. Wei and S. Ding, Cell Stem Cell, 2013, 13, 270–283 CrossRef CAS PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- D. E. Discher, P. Janmey and Y.-L. Wang, Science, 2005, 310, 1139–1143 CrossRef CAS PubMed.
- L. Lv, Y. Tang, P. Zhang, Y. Liu, X. Bai and Y. Zhou, Tissue Eng., Part B, 2018, 24, 112–132 CrossRef PubMed.
- S. Nemec and K. A. Kilian, Nat. Rev. Mater., 2021, 6, 69–83 CrossRef CAS.
- S. Sharma, T. K. Kelly and P. A. Jones, Carcinogenesis, 2010, 31, 27–36 CrossRef CAS PubMed.
- C. D. Allis and T. Jenuwein, Nat. Rev. Genet., 2016, 17, 487–500 CrossRef CAS PubMed.
- J. K. Biehl and B. Russell, J. Cardiovasc. Nurs., 2009, 24, 98–105 CrossRef PubMed.
- J. Li, Y. Liu, Y. Zhang, B. Yao, Enhejirigala, Z. Li, W. Song, Y. Wang, X. Duan, X. Yuan, X. Fu and S. Huang, Front. Cell Dev. Biol., 2021, 9, 397 Search PubMed.
- A. K. Gaharwar, I. Singh and A. Khademhosseini, Nat. Rev. Mater., 2020, 5, 686–705 CrossRef CAS.
- V. Pasque, J. Jullien, K. Miyamoto, R. P. Halley-Stott and J. B. Gurdon, Trends Genet., 2011, 27, 516–525 CrossRef CAS PubMed.
- J. Yoo, J. Kim, S. Baek, Y. Park, H. Im and J. Kim, Biomaterials, 2014, 35, 8321–8329 CrossRef CAS PubMed.
- J. Chen, Q. Han and D. Pei, J. Mol. Cell Biol., 2012, 4, 66–69 CrossRef CAS PubMed.
- M. A. Esteban, X. Bao, Q. Zhuang, T. Zhou, B. Qin and D. Pei, Curr. Opin. Genet. Dev., 2012, 22, 423–428 CrossRef CAS PubMed.
- T. L. Downing, J. Soto, C. Morez, T. Houssin, A. Fritz, F. Yuan, J. Chu, S. Patel, D. V. Schaffer and S. Li, Nat. Mater., 2013, 12, 1154–1162 CrossRef CAS PubMed.
- X. Liu, C. Wang, W. Liu, J. Li, C. Li, X. Kou, J. Chen, Y. Zhao, H. Gao, H. Wang, Y. Zhang, Y. Gao and S. Gao, Nature, 2016, 537, 558–562 CrossRef CAS PubMed.
- H. Gerardo, A. Lima, J. Carvalho, J. R. D. Ramos, S. Couceiro, R. D. M. Travasso, R. P. das Neves and M. Grãos, Sci. Rep., 2019, 9, 9086 CrossRef PubMed.
- J. D. Pajerowski, K. N. Dahl, F. L. Zhong, P. J. Sammak and D. E. Discher, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 15619–15624 CrossRef CAS PubMed.
- E. Meshorer, D. Yellajoshula, E. George, P. J. Scambler, D. T. Brown and T. Misteli, Dev. Cell, 2006, 10, 105–116 CrossRef CAS PubMed.
- N. S. Hwang, C. Zhang, Y.-S. Hwang and S. Varghese, Wiley Interdiscip. Rev.: Syst. Biol. Med., 2009, 1, 97–106 CAS.
- M. Takeo, W. Lee and M. Ito, Cold Spring Harbor Perspect. Med., 2015, 5(1), a023267 CrossRef PubMed.
- C. Morez, M. Noseda, M. A. Paiva, E. Belian, M. D. Schneider and M. M. Stevens, Biomaterials, 2015, 70, 94–104 CrossRef CAS PubMed.
- Z. Lin and W. T. Pu, Sci. Transl. Med., 2014, 6, 239rv1 Search PubMed.
- L. Qian, Y. Huang, C. I. Spencer, A. Foley, V. Vedantham, L. Liu, S. J. Conway, J. Fu and D. Srivastava, Nature, 2012, 485, 593–598 CrossRef CAS PubMed.
- J. Sia, P. Yu, D. Srivastava and S. Li, Biomaterials, 2016, 103, 1–11 CrossRef CAS PubMed.
- S. P. Potta, H. Liang, J. Winkler, M. X. Doss, S. Chen, V. Wagh, K. Pfannkuche, J. Hescheler and A. Sachinidis, Cell. Physiol. Biochem., 2010, 25, 595–604 CrossRef CAS PubMed.
- S. Sharma, P. G. Jackson and J. Makan, J. Clin. Pathol., 2004, 57, 1025–1026 CrossRef CAS PubMed.
- B. Seelbinder, S. Ghosh, S. E. Schneider, A. K. Scott, A. G. Berman, C. J. Goergen, K. B. Margulies, K. C. Bedi, E. Casas, A. R. Swearingen, J. Brumbaugh, S. Calve and C. P. Neu, Nat. Biomed. Eng., 2021, 5, 1500–1516 CrossRef CAS PubMed.
- B. Thienpont, J. M. Aronsen, E. L. Robinson, H. Okkenhaug, E. Loche, A. Ferrini, P. Brien, K. Alkass, A. Tomasso, A. Agrawal, O. Bergmann, I. Sjaastad, W. Reik and H. L. Roderick, J. Clin. Invest., 2017, 127, 335–348 CrossRef PubMed.
- F. Ataollahi, S. Pramanik, A. Moradi, A. Dalilottojari, B. Pingguan-Murphy, W. A. B. Wan Abas and N. A. Abu Osman, J. Biomed. Mater. Res., Part A, 2015, 103, 2203–2213 CrossRef CAS PubMed.
- S. M. L. Wallace, Yasmin, C. M. McEniery, K. M. Mäki-Petäjä, A. D. Booth, J. R. Cockcroft and I. B. Wilkinson, Hypertension, 2007, 50, 228–233 CrossRef CAS PubMed.
- X. Song, Z. Sun, G. Chen, P. Shang, G. You, J. Zhao, S. Liu, D. Han and H. Zhou, Acta Biomater., 2019, 100, 52–60 CrossRef CAS PubMed.
- K. M. Stroka, I. Levitan and H. Aranda-Espinoza, J. Biomech., 2012, 45, 1828–1834 CrossRef PubMed.
- B. N. Dugger and D. W. Dickson, Cold Spring Harbor Perspect. Biol., 2017, 9, a028035 CrossRef PubMed.
- P. G. Nagappan, H. Chen and D.-Y. Wang, Mil. Med. Res., 2020, 7, 30 Search PubMed.
- T. Vierbuchen, A. Ostermeier, Z. P. Pang, Y. Kokubu, T. C. Südhof and M. Wernig, Nature, 2010, 463, 1035–1041 CrossRef CAS PubMed.
- J. Yoo, M. Noh, H. Kim, N. L. Jeon, B.-S. Kim and J. Kim, Biomaterials, 2015, 45, 36–45 CrossRef CAS PubMed.
- F. Kantawong, C. Saksiriwisitkul, C. Riyapa, S. Limpakdee, P. Wanachantararak and T. Kuboki, BioImpacts, 2018, 8, 129–138 CrossRef CAS PubMed.
- G.-H. Liu, F. Yi, K. Suzuki, J. Qu and J. C. I. Belmonte, Cell Res., 2012, 22, 1087–1091 CrossRef CAS PubMed.
- D. Wang, H. Zhang, M. Li, M. G. Frid, A. R. Flockton, B. A. McKeon, M. E. Yeager, M. A. Fini, N. W. Morrell, S. S. Pullamsetti, S. Velegala, W. Seeger, T. A. McKinsey, C. C. Sucharov and K. R. Stenmark, Circ. Res., 2014, 114, 67–78 CrossRef CAS PubMed.
- W. H. Neo, K. Yap, S. H. Lee, L. S. Looi, P. Khandelia, S. X. Neo, E. V. Makeyev and I. Su, J. Biol. Chem., 2014, 289, 20788–20801 CrossRef CAS PubMed.
- J. Tcw, M. Wang, A. A. Pimenova, K. R. Bowles, B. J. Hartley, E. Lacin, S. I. Machlovi, R. Abdelaal, C. M. Karch, H. Phatnani, P. A. Slesinger, B. Zhang, A. M. Goate and K. J. Brennand, Stem Cell Rep., 2017, 9, 600–614 CrossRef CAS PubMed.
- R. Dimitriou, E. Jones, D. McGonagle and P. V. Giannoudis, BMC Med., 2011, 9, 66 CrossRef PubMed.
- S.-W. Ha, H. L. Jang, K. T. Nam and G. R. J. Beck, Biomaterials, 2015, 65, 32–42 CrossRef CAS PubMed.
- Z. Yin, X. Chen, J. L. Chen, W. L. Shen, T. M. Hieu Nguyen, L. Gao and H. W. Ouyang, Biomaterials, 2010, 31, 2163–2175 CrossRef CAS PubMed.
- G. Vunjak-Novakovic, G. Altman, R. Horan and D. L. Kaplan, Annu. Rev. Biomed. Eng., 2004, 6, 131–156 CrossRef CAS PubMed.
- J. C.-H. Goh, H.-W. Ouyang, S.-H. Teoh, C. K. C. Chan and E.-H. Lee, Tissue Eng., 2003, 9(Suppl 1), S31–S44 CrossRef CAS PubMed.
- C. Zhang, X. Wang, E. Zhang, L. Yang, H. Yuan, W. Tu, H. Zhang, Z. Yin, W. Shen, X. Chen, Y. Zhang and H. Ouyang, Acta Biomater., 2018, 66, 141–156 CrossRef CAS PubMed.
- A. J. Rosenbaum, D. A. Grande and J. S. Dines, Organogenesis, 2008, 4, 23–27 CrossRef PubMed.
- F. Barry and M. Murphy, Nat. Rev. Rheumatol., 2013, 9, 584–594 CrossRef CAS PubMed.
- M.-H. Kim, M. Kino-oka, A. Saito, Y. Sawa and M. Taya, J. Biosci. Bioeng., 2010, 109, 55–61 CrossRef CAS PubMed.
- Y. Ogawa, M.-H. Kim and M. Kino-oka, J. Biosci. Bioeng., 2016, 122, 627–632 CrossRef CAS PubMed.
- F. D. Ayuningtyas, M.-H. Kim and M. Kino-Oka, Acta Biomater., 2020, 106, 170–180 CrossRef CAS PubMed.
- J. Wang, S. T. Jia and S. Jia, Trends Genet., 2016, 32, 284–294 CrossRef CAS PubMed.
- Y. Cheng, C. He, M. Wang, X. Ma, F. Mo, S. Yang, J. Han and X. Wei, Signal Transduction Targeted Ther., 2019, 4, 62 CrossRef PubMed.
- S. Ghasemi, Pharmacogenomics J., 2020, 20, 367–379 CrossRef CAS PubMed.
- K. Brzóska, I. Grądzka and M. Kruszewski, Materials, 2019, 12, 1038 CrossRef PubMed.
- R. Stowers and O. Chaudhuri, Nat. Biomed. Eng., 2021, 5, 8–10 CrossRef CAS PubMed.
- M. Jang, J. An, S. W. Oh, J. Y. Lim, J. Kim, J. K. Choi, J. H. Cheong and P. Kim, Nat. Biomed. Eng., 2021, 5, 114–123 CrossRef CAS PubMed.
- R. S. Stowers, A. Shcherbina, J. Israeli, J. J. Gruber, J. Chang, S. Nam, A. Rabiee, M. N. Teruel, M. P. Snyder, A. Kundaje and O. Chaudhuri, Nat. Biomed. Eng., 2019, 3, 1009–1019 CrossRef PubMed.
- L. Li, P. Gao, Y. Li, Y. Shen, J. Xie, D. Sun, A. Xue, Z. Zhao, Z. Xu, M. Zhang, B. Li and J. Jiang, Breast Cancer Res. Treat., 2014, 147, 487–500 CrossRef CAS PubMed.
- M. Plodinec, M. Loparic, C. A. Monnier, E. C. Obermann, R. Zanetti-Dallenbach, P. Oertle, J. T. Hyotyla, U. Aebi, M. Bentires-Alj, R. Y. H. Lim and C.-A. Schoenenberger, Nat. Nanotechnol., 2012, 7, 757–765 CrossRef CAS PubMed.
- E. Batlle and H. Clevers, Nat. Med., 2017, 23, 1124–1134 CrossRef CAS PubMed.
- K.-Y. Ng, Q. T. Shea, T.-L. Wong, S. T. Luk, M. Tong, C.-M. Lo, K. Man, J.-P. Yun, X.-Y. Guan, T. K. Lee, Y.-P. Zheng and S. Ma, Adv. Sci., 2021, 8, 2002483 CrossRef CAS PubMed.
- Z. Li, Exp. Hematol. Oncol., 2013, 2, 17 CrossRef CAS PubMed.
- Y. Liu, J. Lv, X. Liang, X. Yin, L. Zhang, D. Chen, X. Jin, R. Fiskesund, K. Tang, J. Ma, H. Zhang, W. Dong, S. Mo, T. Zhang, F. Cheng, Y. Zhou, J. Xie, N. Wang and B. Huang, Cancer Res., 2018, 78, 3926–3937 CrossRef CAS PubMed.
- G. Watts, Lancet, 2011, 377, 2075 CrossRef.
- R. Di Micco, V. Krizhanovsky, D. Baker and F. d'Adda di Fagagna, Nat. Rev. Mol. Cell Biol., 2021, 22, 75–95 CrossRef CAS PubMed.
- A. Schellenberg, Q. Lin, H. Schüler, C. M. Koch, S. Joussen, B. Denecke, G. Walenda, N. Pallua, C. V. Suschek, M. Zenke and W. Wagner, Aging, 2011, 3, 873–888 CrossRef CAS PubMed.
- A. Schellenberg, S. Joussen, K. Moser, N. Hampe, N. Hersch, H. Hemeda, J. Schnitker, B. Denecke, Q. Lin, N. Pallua, M. Zenke, R. Merkel, B. Hoffmann and W. Wagner, Biomaterials, 2014, 35, 6351–6358 CrossRef CAS PubMed.
- C. López-Otín, M. A. Blasco, L. Partridge, M. Serrano and G. Kroemer, Cell, 2013, 153, 1194–1217 CrossRef PubMed.
- J. Koester, Y. A. Miroshnikova, S. Ghatak, C. A. Chacón-Martínez, J. Morgner, X. Li, I. Atanassov, J. Altmüller, D. E. Birk, M. Koch, W. Bloch, M. Bartusel, C. M. Niessen, A. Rada-Iglesias and S. A. Wickström, Nat. Cell Biol., 2021, 23, 771–781 CrossRef CAS PubMed.
- Q. Wei, Y. Su, H. Xin, L. Zhang, J. Ding and X. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 56719–56724 CrossRef CAS PubMed.
- M. Sima, K. Vrbova, T. Zavodna, K. Honkova, I. Chvojkova, A. Ambroz, J. Klema, A. Rossnerova, K. Polakova, T. Malina, J. Belza, J. Topinka and P. Rossner Jr., Int. J. Mol. Sci., 2020, 21, 4763 CrossRef CAS PubMed.
- Y. Ma, Y. Guo, H. Ye, K. Huang, Z. Lv and Y. Ke, Ecotoxicol. Environ. Saf., 2019, 176, 1–10 CrossRef CAS PubMed.
- C. J. Walker, C. Crocini, D. Ramirez, A. R. Killaars, J. C. Grim, B. A. Aguado, K. Clark, M. A. Allen, R. D. Dowell, L. A. Leinwand and K. S. Anseth, Nat. Biomed. Eng., 2021, 5, 1485–1499 CrossRef CAS PubMed.
Footnote |
| † Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |