
Inflammatory serum factors from aortic valve stenosis patients modulate sex differences in valvular myofibroblast activation and osteoblast-like differentiation†
Brandon J.
Vogt
 abc,
Douglas K.
Peters
d,
Kristi S.
Anseth
cd and
Brian A.
Aguado
*ab
abc,
Douglas K.
Peters
d,
Kristi S.
Anseth
cd and
Brian A.
Aguado
*ab
aDepartment of Bioengineering, University of California San Diego, La Jolla, CA 92093, USA. E-mail: baguado@eng.ucsd.edu
bSanford Consortium for Regenerative Medicine, La Jolla, CA 92037, USA
cDepartment of Chemical and Biological Engineering, University of Colorado Boulder, CO 80303, USA
dBioFrontiers Institute, University of Colorado Boulder, CO 80309, USA
First published on 7th October 2022
Abstract
Aortic valve stenosis (AVS) is a sexually dimorphic cardiovascular disease that is driven by fibrosis and calcification of the aortic valve leaflets. Circulating inflammatory factors present in serum from AVS patients contribute to sex differences in valve fibro-calcification by driving the activation of valvular interstitial cells (VICs) to myofibroblasts and/or osteoblast-like cells. However, the molecular mechanisms by which inflammatory factors contribute to sex-specific valve fibro-calcification remain largely unknown. In this study, we identified inflammatory factors present in serum samples from AVS patients that regulate sex-specific myofibroblast activation and osteoblast-like differentiation. After correlating serum proteomic datasets with clinical and in vitro myofibroblast datasets, we identified annexin A2 and cystatin C as candidate inflammatory factors that correlate with both AVS patient severity and myofibroblast activation measurements in vitro. Validation experiments utilizing hydrogel biomaterials as cell culture platforms that mimic the valve extracellular matrix confirmed that annexin A2 and cystatin C promote sex-specific VIC activation to myofibroblasts via p38 MAPK signaling. Additionally, annexin A2 and cystatin C increase osteoblast-like differentiation primarily in male VICs. Our results implicate serum inflammatory factors as potential AVS biomarkers that also contribute to sexually dimorphic AVS progression by driving VIC myofibroblast activation and/or osteoblast-like differentiation. Collectively, the results herein further our overall understanding as to how biological sex may impact inflammation-driven AVS and may lead to the development of sex-specific drug treatment strategies.
Introduction
Aortic valve stenosis (AVS) is a progressive cardiovascular disease that affects nearly 13% of adults over the age of 75 and is characterized by the fibrosis and/or calcification of valve tissue.1–5 Currently, AVS patients are treated with surgical or transcatheter aortic valve replacements, but not all patients are eligible for these procedures. For example, most patients suffering from a severe cardiovascular or pulmonary disease are considered high risk for undergoing valve replacement surgery due to compounding risk factors.6 Furthermore, valve replacement patients are at risk of restenosis, which can cause severe health problems for recovering patients within a year of valve replacement surgery.7 Due to the significant limitations of surgical valve replacement procedures, there is an opportunity to develop or repurpose existing drugs to treat AVS as a suitable alternative to surgical intervention.8Given that AVS progression is heterogeneous between patients as a function of sex, age, and valve phenotype,3,4,9,10 drug treatments may need to be customized for an individual patient or group of patients. Men diagnosed with AVS typically experience increased calcification of aortic valve leaflets relative to women, who instead experience more severe and persistent fibrosis.10 Aortic valve calcification impairs heart function more severely than fibrosis by restricting valve movement and promoting regurgitation, resulting in higher mortality rates in men.11 Thus, biological sex is acknowledged as one key factor for predicting AVS patient outcomes. Sex also plays a significant role in treatment response and disease prevalence, and the sex differences observed are often context-specific.12–14 Notably, AVS is more prevalent in women than men, especially for patients over the age of 75.4 Given that sex is a potent driver of AVS disease manifestation and overall patient outcomes, sex should be considered as a biological variable to understand mechanisms of AVS progression with the opportunity for the development of sex-specific drug treatments.15
Immune cells that secrete pro-inflammatory factors are known to drive sexually dimorphic AVS progression. Prior work has shown that pro-inflammatory factors promote resident fibroblast-like cells in valve tissue, known as valvular interstitial cells (VICs), to adopt diseased myofibroblast and/or osteoblast-like phenotypes.16–18 The arrival of key inflammatory cells to the aortic valve leaflet induces alterations in the VIC microenvironment that drive valve thickening and calcification.15 For example, pro-inflammatory macrophages secrete several cytokines that stimulate VICs to adopt an osteoblast-like phenotype, which promotes calcification.16–18 Secreted inflammatory factors, such as interleukin 1 beta (IL-1β), tumor necrosis factor alpha (TNF-α), and interleukin 6 (IL-6), among others, have long been recognized as drivers of fibro-calcification in the valve,16,19,20 although sex differences in VIC responses to inflammatory cues are less clear. Previous work has found an increased abundance of anti-inflammatory proteins, such as IL-1β and TNF-α, in male AVS patient sera following transcatheter aortic valve replacement (TAVR) relative to female sera,21 which may mediate the increased myofibroblast deactivation and osteoblast-like differentiation observed in male VICs.16 Although anti-inflammatory factors such as IL-1β and TNF-α have been shown to impact VIC phenotypes, the role of additional serum factors in driving sex dimorphisms in myofibroblast activation and osteoblast-like differentiation remains unknown.
In this contribution, we identified inflammatory factors that partially drive sex-specific myofibroblast activation and/or osteoblast-like differentiation, which also correlate to clinical measurements of AVS disease severity. Our approach to identify these factors was to correlate previously published AVS serum proteome data21 to (1) myofibroblast activation levels in fibroblasts cultured with patient sera in vitro and (2) known clinical measures of AVS disease severity. We hypothesized that inflammatory factors would drive increased myofibroblast activation in female VICs and increased osteoblast-like differentiation in male VICs. To test this hypothesis, we seeded male or female VICs on bioengineered hydrogels that recapitulate the extracellular matrix stiffness of healthy aortic valve leaflets to serve as more physiologically relevant culture substrates than standard tissue culture polystyrene (TCPS).22–24 We then exposed VICs to physiologically relevant doses of either annexin A2 or cystatin C and observed sex-specific changes in both myofibroblast activation and osteoblast-like differentiation as measured by alpha-smooth muscle actin (a-SMA) stress fiber formation and runt-related transcription factor 2 (RUNX2) nuclear localization. Furthermore, we identified and implicated p38 MAPK signaling as a pathway that partially regulates sex-specific VIC responses to annexin A2 and cystatin C. Overall, our approach revealed key inflammatory serum proteins that contribute to sexually dimorphic VIC phenotypes during AVS progression.
Methods
Study design and serum protein quantification
The relative abundance of serum sample proteins were determined in a previous publication.21 Serum samples were collected both immediately before and approximately one month after AVS patients underwent a TAVR procedure. Sample collection was approved by the Colorado Multiple Institutional Review Board (CO-MIRB 07-0516) and all patients gave informed consent by submitting an IRB-approved form. All samples were deidentified. Upon review from an interventional cardiologist and a cardiac surgeon, all patients were deemed appropriate candidates for TAVR, which was performed using a Sapien 3 (Edwards Lifesciences) balloon-expandable transcatheter heart valve. Overall, 12 pre-TAVR and 12 post-TAVR (8 male and 4 female) serum samples were obtained and analyzed using a SOMAscan DNA aptamer array (SomaLogic) according to the manufacturer's guidelines. The relative abundance of over 1300 proteins were analyzed using relative fluorescence units (RFU) based on a fluorescence array of the SOMAmer reagents.Computational analysis of AVS patient serum proteome
A code was developed in R that processes large matrices, filters for positive and negative correlations, forms correlation matrices with statistically significant proteins, performs a cluster analysis, outputs correlation matrices based on cluster groups, and performs a KEGG enrichment analysis. This code is now freely available upon request through GitHub. Clinically significant candidate proteins that likely drive myofibroblast activation were identified by filtering for proteins with significant correlations to clinical data and previous in vitro myofibroblast activation data collected from valvular interstitial cells and adult rat ventricular fibroblasts cultured in patient sera.21 Clinically significant proteins had a correlation factor above 0.8 or below −0.8 (corresponding to a p-value of 0.00178) with at least one clinical measure of AVS severity. The correlation threshold was raised to ±0.9 (p-value of 0.00232) when analyzing only male patient data to account for a decrease in sample size from 12 to 8. When correlating serum protein RFU values to previously published in vitro myofibroblast activation data, a correlation factor of ±0.75 (p-value of 0.0321) was used since only one output was being measured compared to the twelve outputs used for patient clinical data.Hydrogel fabrication
Hydrogels were formed on 12 mm circular glass coverslips for immunostaining experiments or 25 mm coverslips for RT-PCR experiments, which were plasma-treated with oxygen and submerged in a solution of toluene (Sigma-Aldrich) with 15% vol/vol mercaptopropyltrimethoxysilane (MPTS, Sigma-Aldrich) and 5% vol/vol 2-butylamine (Sigma-Aldrich) for 2 hours. Coverslips were then removed from the solution, washed with toluene, dried in a 60 °C oven, and sterilized with 70% ethanol. Polyethylene glycol-norbornene (PEG-Nb) was synthesized as previously described.25 Gel precursor solution composed of 4% wt/vol PEG-Nb was prepared by adding 8-arm 40 kDa PEG-Nb at a 0.99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 thiol-to-ene ratio, 2 mmol L−1 CRGDS cell adhesive peptide (Bachem), and a 5 kDa PEG-dithiol crosslinker (JenKem) to phosphate buffered saline (PBS, Sigma-Aldrich). 1.7 mmol L−1 of lithium phenyl-2,4,6-trimethyl-benzoylphosphinate was added to the solution to serve as a photoinitiator. Gels were formed by pipetting 14 μL or 65 μL of gel precursor solution onto a Sigmacote (Sigma) treated glass slide, then covering the solution with a thiolated coverslip (final gel thickness = 130 μm). Gels were then photopolymerized at 4 mW cm−2 for 3 minutes, removed from the glass slide with a razor blade, and sterilized in 5% isopropyl alcohol (Thermo Fisher) in PBS for 30 minutes. After 3 washes with PBS, gels were incubated at 37 °C and 5% carbon dioxide overnight in VIC media composed of Media 199 (Life Technologies), 1% fetal bovine serum (FBS, Life Technologies), 1 μg mL−1 amphotericin B (Thermo Fisher), 50 U mL−1 penicillin (Sigma), and 50 μg mL−1 streptomycin (Sigma).
:1 thiol-to-ene ratio, 2 mmol L−1 CRGDS cell adhesive peptide (Bachem), and a 5 kDa PEG-dithiol crosslinker (JenKem) to phosphate buffered saline (PBS, Sigma-Aldrich). 1.7 mmol L−1 of lithium phenyl-2,4,6-trimethyl-benzoylphosphinate was added to the solution to serve as a photoinitiator. Gels were formed by pipetting 14 μL or 65 μL of gel precursor solution onto a Sigmacote (Sigma) treated glass slide, then covering the solution with a thiolated coverslip (final gel thickness = 130 μm). Gels were then photopolymerized at 4 mW cm−2 for 3 minutes, removed from the glass slide with a razor blade, and sterilized in 5% isopropyl alcohol (Thermo Fisher) in PBS for 30 minutes. After 3 washes with PBS, gels were incubated at 37 °C and 5% carbon dioxide overnight in VIC media composed of Media 199 (Life Technologies), 1% fetal bovine serum (FBS, Life Technologies), 1 μg mL−1 amphotericin B (Thermo Fisher), 50 U mL−1 penicillin (Sigma), and 50 μg mL−1 streptomycin (Sigma).
Rheology
PEG hydrogel precursor solutions (4% wt/vol PEG-Nb) were photopolymerized in situ on a HR3 rheometer (TA Instruments) and characterized using a parallel plate geometry (8 mm diameter). Storage (G′) and loss (G′′) measurements were made using oscillatory shear rheology with an amplitude of 1% and frequency of 1 Hz. The shear modulus values were converted to Young's modulus (E) using the formula E = 2G′(1 + ν) with a Poisson's ratio of ν = 0.5, assuming G′ ⋙ G′′.VIC isolation and culture
VICs were obtained from 5–8-month-old adult pigs (Hormel and Midwest Research Swine) using previously described methods.21 For each isolation, valve leaflets were pooled based on sex with a minimum of three biological replicates per sex. A total of three separate VIC isolations were used for this manuscript, with one isolation used to generate data seen in Fig. 2 and ESI Fig. 4 and 5,† a second isolation used for Fig. 4, and a third isolation used for ESI Fig. 6.† The aortic valve leaflets were washed with Earle's Balanced Salt Solution (EBSS, Sigma-Aldrich) mixed with 50 U mL−1 of penicillin, 1 μg mL−1 amphotericin B, and 50 μg mL−1 streptomycin. The porcine valves were collected in an EBSS solution with 250 units of type II collagenase (Worthington) per mL. Valves were incubated and shaken at 37 °C for 30 minutes, vortexed for 30 seconds, then placed in a fresh collagenase solution and shaken while being incubated at 37 °C for 60 minutes. Porcine valves were then vortexed for 2 minutes and filtered through a 100 μm cell strainer. VICs were centrifuged for 10 minutes at 0.2g and cell pellets were resuspended in VIC growth media composed of Media 199, 15% FBS, 1 μg mL−1 amphotericin B, 50 U mL−1 penicillin, and 50 μg mL−1 streptomycin. Harvested VICs were cultured on tissue culture treated polystyrene (TCPS) at 37 °C and 5% carbon dioxide for expansion. Upon reaching 70–80% confluency, cells were collected using trypsin (Life Technologies), counted by an automated hemocytometer, and seeded on hydrogels at 40000 cells per well for immunostaining experiments or 180000 cells per well for RT-PCR.
Protein validation and p38 MAPK inhibition studies
VICs were seeded on hydrogels (E ≃ 5 kPa, ESI Fig. 1†) and treated with either human recombinant annexin A2 (R&D Systems) or cystatin C (R&D Systems) at various concentrations. For inhibition studies, cells were treated with a known p38 MAPK inhibitor, SB203580 (Selleck Chemicals), at the previously optimized concentration of 20 μM.21 After immunostaining (refer to the following section), gel samples were imaged on a glass-bottom 24 well plate (Cellvis) using either an Operetta High-Content Imaging System (PerkinElmer, Part no. HH12000000) or Nikon Eclipse Ti2-E. Sample images were analyzed by adapting a publicly sourced MATLAB code from GitHub.26 Myofibroblast activation was assessed by quantifying α-SMA gradient mean intensity values and osteoblast-like differentiation was measured by quantifying RUNX2 nuclear intensity normalized to RUNX2 cell mask intensity. Individual cell values were analyzed for at least 5 images per gel, with a minimum of two gels per condition.Immunostaining
After culturing on hydrogels for 48 hours, VICs were fixed using 4% wt/vol paraformaldehyde (PFA, Thermo Fisher) in PBS. After 20 minutes, the PFA solution was removed, and cells were permeabilized using 0.1% Triton-X-100 (Fisher Scientific) in PBS for one hour then blocked with 5% bovine serum albumin (BSA, Sigma-Aldrich) overnight at 4 °C. The following day, VICs were treated with a mouse anti-αSMA primary antibody (Abcam, 1:300 dilution, ab7817) and a rabbit anti-RUNX2 primary antibody (Abcam, 1:250 dilution, ab23981). One hour later, VICs were washed twice with PBS with 0.05% vol/vol Tween20 (Sigma) then treated with a staining solution containing goat anti-mouse Alexa Fluor 488 at a 1:300 dilution (Life Technologies), goat anti-rabbit Alexa Fluor 647 at a 1:300 dilution (Life Technologies), 4′-6-diamidino-2-phenyindole at a 1:500 dilution (Life Technologies), and HCS Cell Mask at a 1:5000 dilution (Life Technologies). After soaking for one hour in the dark, VICs were washed with PBS with 0.05% vol/vol Tween20 and stored in PBS until imaging.
RT-PCR
25 mm hydrogels seeded with VICs were inverted cell-side down into lysis buffer (Qiagen RNEasy Micro Kit) for 2 minutes, then flipped over and rinsed with 70% ethanol (Sigma). RNA was collected from all samples following Qiagen's RNEasy Micro Kit protocol and assessed for quality using a NanoDrop 2000 spectrophotometer (Thermo Fisher). Following the manufacturer's protocol, cDNA was formed using an iScript Synthesis kit (Bio-Rad) and corresponding mRNA expression was measured using iQ SYBR Green Supermix (Bio-Rad) on a CFX384 iCycler (Bio-Rad). All genes were normalized to the RPL30 gene. Primer sequences can be found in ESI Table 1.†Statistical analysis
Unless otherwise stated, statistical significance was determined using one-way ANOVA with Tukey posttests in GraphPad Prism. Due to the large number of data points used in this study (N > 350), the threshold for significance was set at the lowest commonly used cutoff of P < 0.0001. Recognizing that the large sample size impacts the p-value, we also determined significant differences between groups independent of data size. Specifically, we calculated the Cohen's d-value between conditions to measure the effect size for differences between groups that were determined to be statistically significant. Effect size changes were set at commonly used thresholds27–29 and are represented using the following key: *** = d > 0.8, ** = d > 0.5, * = d > 0.2 for differences between groups; ## = d > 0.5, # = d > 0.2 for differences from the male control group; $ = d > 0.2 for differences from the female control group. All data is shown with the mean ± standard deviation as calculated in Prism. The p-values seen in Fig. 1C–F were calculated using a standard t-test in Excel based on Pearson's correlation coefficient (R), which was obtained from the correlation analysis outlined previously.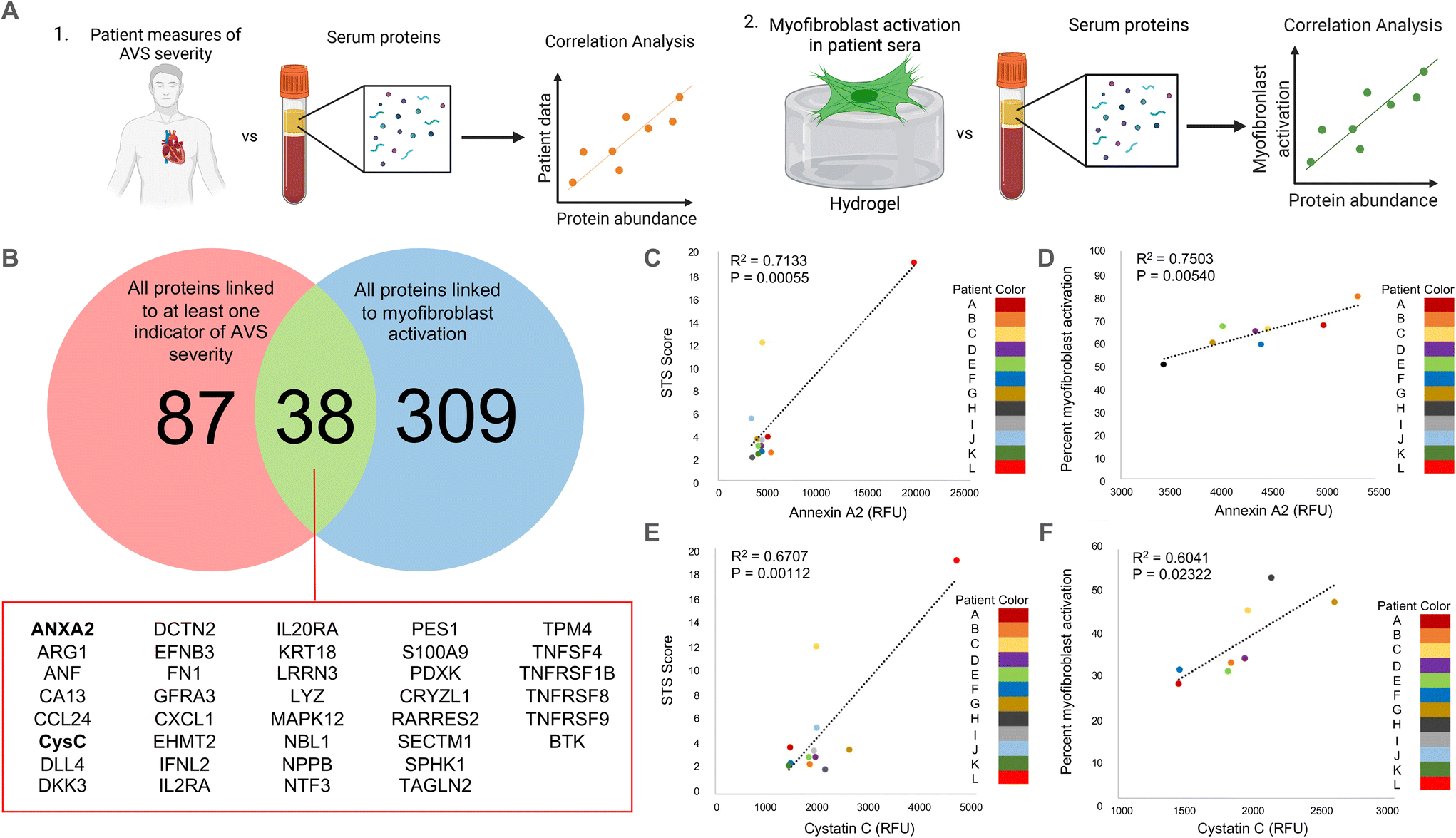 | ||
| Fig. 1 Correlation analysis to determine clinically relevant candidate proteins in patient serum samples. (A) Schematic describing our approach to identify clinically relevant candidate proteins that are significantly correlated with (1) quantified measurements of AVS severity in patients and (2) in vitro myofibroblast activation data. (B) Venn diagram showing the overlap between proteins significantly correlated (R > 0.8) with at least one quantified measure of AVS severity and proteins significantly correlated (R > 0.75) with myofibroblast activation in vitro. Full protein names for abbreviated proteins are shown in ESI Table 2.† (C and D) Correlation plots comparing annexin A2 abundance and (C) patient STS score or (D) percent myofibroblast activation in vitro. (E and F) Correlation plot comparing cystatin C abundance and (E) patient STS score or (F) percent myofibroblast activation in vitro. | ||
Results
Annexin A2 and cystatin C identified as clinically relevant candidate serum factors that drive VIC myofibroblast activation
As a strategy to identify serum proteins that are both clinically significant and likely driving sex-specific myofibroblast activation and/or osteoblast-like differentiation, we re-analyzed previously published AVS patient serum proteome data.21 Specifically, we correlated the relative fluorescence unit (RFU) values that indicate abundances of over 1300 serum proteins in 12 different patient samples with (1) patient echocardiography data and multimorbidity scores that indicate AVS disease severity and (2) our previously published in vitro myofibroblast activation data set21 (Fig. 1A). We identified 125 clinically significant proteins and 347 proteins with significant correlations to myofibroblast activation. Upon comparing the two protein groups, only 38 candidate proteins showed strong correlations to patient measures of AVS severity and myofibroblast activation (Fig. 1B).Next, we selected two factors for in vitro validation based on a literature search to find candidate proteins with strong ties to the inflammatory response, calcification, fibrosis, and cardiovascular disease severity. The first protein, annexin A2, was selected as a candidate protein for validation due to previous work showing that annexin A2 induces human blood macrophages to release pro-inflammatory cytokines.30,31 Additionally, annexin A2 has also been shown to activate lung fibroblasts and promote matrix vesicle calcification in vascular smooth muscle cells.32,33 In our analysis, annexin A2 was significantly positively correlated with both AVS patient Society of Thoracic Surgeons (STS) score (Fig. 1C) and in vitro myofibroblast activation (Fig. 1D). The next factor selected for further validation was cystatin C, as previous work has shown that cystatin C increases cardiovascular disease risk,34 is expressed by inflammatory cells,35 and contributes to lung fibrosis by altering the extracellular matrix.36 Like annexin A2, cystatin C RFU values were significantly and positively correlated with AVS patient STS score (Fig. 1E) and myofibroblast activation (Fig. 1F). We also performed a logarithmic transformation and a linear regression analysis on our data to confirm no strong outliers. Our approach revealed significant correlations (p < 0.05) between annexin A2 or cystatin C abundance and patient STS score (ESI Fig. 2†).
Correlations for this study were performed using both male (n = 8) and female (n = 4) patient samples. However, acknowledging sex dimorphisms in AVS, we also repeated our correlation analyses using only male patient serum to identify candidate proteins specific to male patients. No correlation analysis was performed using only female patient data due to the limited sample size. Using male patient serum, we found 208 clinically significant proteins and 347 proteins significantly correlated with myofibroblast activation, with only 60 proteins present in each group (ESI Fig. 3A†). Interestingly, out of the 38 candidate proteins identified using male and female patient data, only 17 overlapped with the 60 candidate proteins identified using only male patient data, including annexin A2 and cystatin C (ESI Fig. 3B†).
Annexin A2 and cystatin C drive VIC myofibroblast activation and osteoblast-like differentiation on hydrogels
As an initial validation, male and female VICs were isolated from porcine aortic valve leaflets, seeded on TCPS, and cultured for 48 hours with physiologically relevant concentrations of annexin A2 and cystatin C.37–40 We hypothesized that we would observe increased myofibroblast activation in female VICs and increased osteoblast-like differentiation in male VICs in response to annexin A2 and cystatin C. However, aside from an increase in male VIC RUNX2 nuclear localization when cultured with high doses of cystatin C, annexin A2 and cystatin C had a minimal impact on VIC myofibroblast activation and osteoblast-like differentiation (ESI Fig. 4†).VICs are mechanosensitive22–24 and readily activate to myofibroblasts when cultured on platforms with a supraphysiologic stiffness, such as TCPS. We posit that annexin A2 and cystatin C had a minimal effect on VIC phenotype due to VICs inherently activating to myofibroblasts on TCPS that does not effectively recapitulate the stiffness of native valve tissue. To generate a more physiologically relevant microenvironment, we bioengineered hydrogels using thiol–ene click chemistry25 to form a cross-linked polymer network with the peptide CRGDS added as a pendant functional group to promote cell adhesion. By adjusting polymer network concentration, we were able to form hydrogels with a stiffness similar to healthy aortic valve tissue and maintain VIC quiescence.22–24
We then repeated our validation experiments using hydrogels and observed significant changes in VIC myofibroblast activation and RUNX2 nuclear localization (Fig. 2A). Physiologically relevant concentrations of annexin A2 elicited significant increases in α-SMA gradient mean intensity in both male and female VICs on hydrogels (Fig. 2C). However, male VICs had a statistically significant increase in α-SMA intensity across all annexin A2 conditions whereas female VICs only had increases at the 100 and 1000 ng mL−1 conditions. Although, female VICs showed a greater increase in α-SMA intensity than male VICs at those concentrations. In the same experiment, RUNX2 nuclear localization was measured to assess osteoblast-like differentiation (Fig. 2D). Female VICs showed an increase in RUNX2 nuclear localization when exposed to 10 ng mL−1 of annexin A2, but these effects diminished at higher concentrations. In contrast, male VICs showed a decrease in RUNX2 expression with 10 ng mL−1 of annexin A2 but then showed a significant increase at the 1000 ng mL−1 concentration. Collectively, our data show that annexin A2 promotes myofibroblast activation in both male and female VICs, but primarily increases RUNX2 nuclear localization in male VICs.
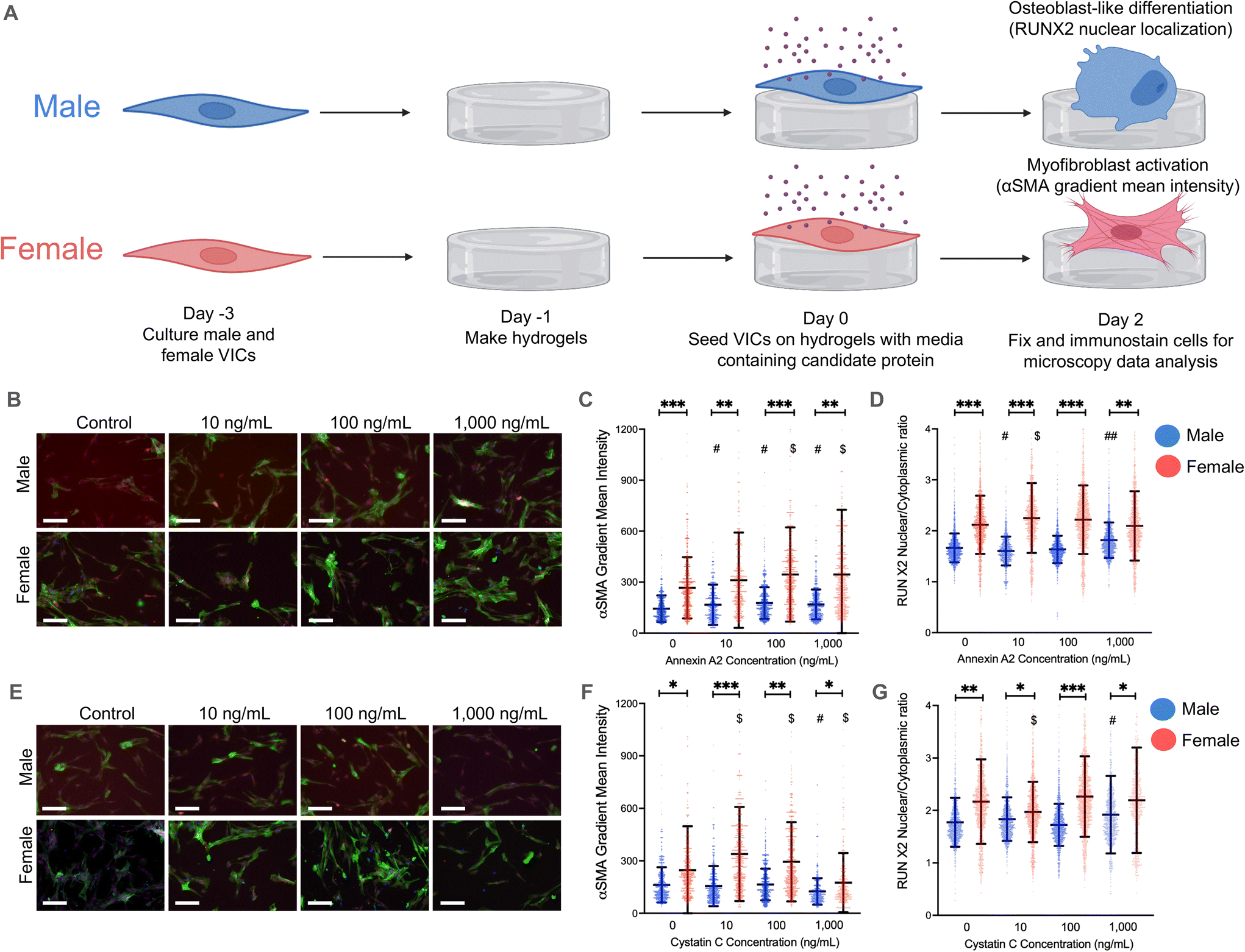 | ||
| Fig. 2 Annexin A2 and cystatin C drive VIC myofibroblast activation and osteoblast-like differentiation. (A) Male and female VICs were cultured for 48 hours on hydrogels with media containing annexin A2 or cystatin C. Myofibroblast activation was measured through α-SMA gradient mean intensity and osteoblast-like differentiation was measured through RUNX2 nuclear localization. (B) Representative images, (C) quantified α-SMA gradient mean intensity, and (D) RUNX2 nuclear localization in VICs treated with annexin A2 and cultured on hydrogels. (E) Representative images, (F) quantified α-SMA gradient mean intensity, and (G) RUNX2 nuclear localization in VICs treated with cystatin C and cultured on hydrogels. For immunofluorescent stains: green = α-SMA; red = RUNX2; blue = DAPI. For all graphs, data is shown as mean ± standard deviation with statistical significance determined by one-way ANOVA with Tukey posttests (P < 0.0001) and effect size measured between statistically significant groups by the Cohen's d-value indicated by *** = d > 0.8, ** = d > 0.5, * = d > 0.2 for sex differences; ## = d > 0.5, # = d > 0.2 for differences relative to the male control group; $ = d > 0.2 for differences relative to the female control group. Scale bar = 100 μm. | ||
On hydrogels, cystatin C had a minimal impact on male VIC α-SMA gradient mean intensity, with only the highest concentration of cystatin C inducing a meaningful change in α-SMA levels (Fig. 2F). At this concentration, male VICs showed a statistically significant decrease in α-SMA gradient intensity. In contrast, female VICs were more responsive to cystatin C, with significant increases in α-SMA intensity observed at both 10 and 100 ng mL−1. Although cystatin C had a more significant impact on female VIC myofibroblast activation, cystatin C had a larger effect on male VIC RUNX2 nuclear localization (Fig. 2G). Male VICs showed a significant increase in RUNX2 nuclear localization at the highest concentration of cystatin C, whereas female VICs only showed a significant decrease in RUNX2 nuclear localization at the lowest concentration of cystatin C. Overall, our data suggest that cystatin C only causes significant increases in myofibroblast activation in female VICs and increases RUNX2 nuclear localization uniquely in male VICs. To supplement our analyses, we also reanalyzed our data by setting an α-SMA gradient mean threshold for myofibroblast activation and quantified the percent activation of male and female VICs. This alternative method of analysis confirmed the observed effects of annexin A2 and cystatin C on male and female VIC myofibroblast activation (ESI Fig. 5†).
We also quantified gene expression of myofibroblast markers [ACTA2 (alpha smooth muscle actin) and COL1A1 (collagen type 1 alpha 1)] and osteoblast markers [OPN (osteopontin) and RUNX2] in VICs to validate key findings (ESI Fig. 6†). Specifically, we found increased ACTA2 and COL1A1 expression in female VICs relative to male VICs (ESI Fig. 6A and B†) and increased COL1A1 expression uniquely in female VICs when cultured with cystatin C (ESI Fig. 6B†). We did not observe significant sex differences in RUNX2 expression, confirming the need to evaluate RUNX2 nuclear localization (ESI Fig. 6C†). We also observed increased OPN expression in female VICs relative to male VICs (ESI Fig. 6D†), corroborating previous work suggesting increased OPN expression may drive reduced osteoblast-like differentiation.41
Cluster and pathway enrichment analyses of candidate proteins identify p38 MAPK signaling as a candidate pathway regulating myofibroblast activation
We next sought to identify potential signaling pathways through which annexin A2 and cystatin C drive sex-specific VIC myofibroblast activation and osteoblast-like differentiation. First, a K-means cluster analysis was performed on our identified clinically significant proteins. Our K-means analysis categorized proteins into three distinct clusters with minimal overlap based on similar correlation values with clinical data (Fig. 3A). Interestingly, annexin A2 and cystatin C clustered closely together in cluster group 2, which was the largest group of proteins. Next, we performed a KEGG pathway enrichment analysis on each cluster to determine candidate signaling pathways for the identified protein clusters. We hypothesized that protein clusters would be upstream of the p38 MAPK pathway since our previous work identified p38 MAPK signaling as a key driver of myofibroblast activation in patient sera.21 KEGG enrichment analysis revealed that clusters 1 and 2 enriched to MAPK signaling, while cluster 3 did not (Fig. 3B–D). Analyzing the correlations with clinical data for each cluster showed that cluster 1 proteins were primarily positively correlated with patient white blood cell count, cluster 2 proteins positively correlated with STS score, and cluster 3 proteins positively correlated with patient stroke volume index (Fig. 3E–G).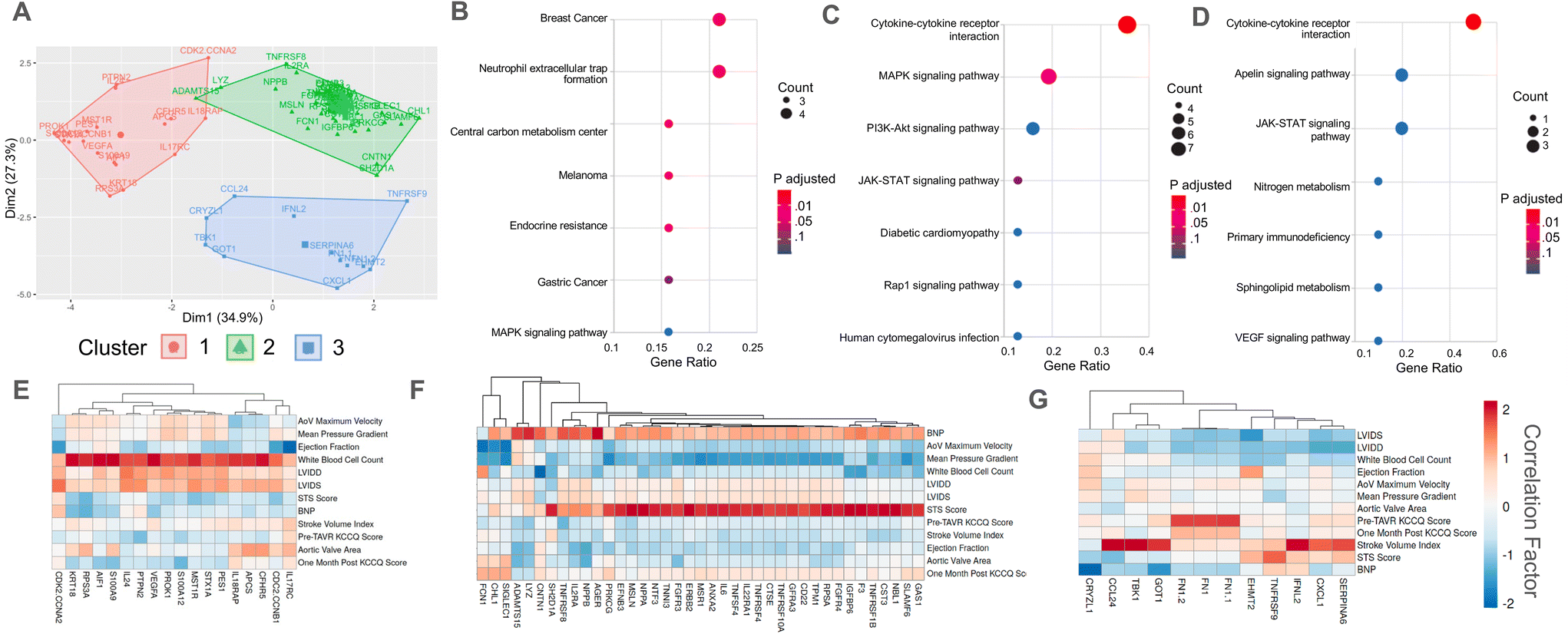 | ||
| Fig. 3 KEGG pathway enrichment analysis implicates the p38 MAPK signaling pathway downstream of annexin A2 and cystatin C. (A) K-means cluster plot grouping serum proteins significantly correlated (R > 0.8) with at least one quantified measurement of AVS disease severity. (B–D) Top seven enriched KEGG signaling pathways for (B) cluster 1, (C) cluster 2, and (D) cluster 3. (E–G) Correlation matrices showing (E) cluster 1, (F) cluster 2, and (G) cluster 3 proteins that significantly correlate (R > 0.8) with at least one measure of AVS severity. | ||
A K-means analysis was also performed using the clinically significant proteins identified using only male patient serum samples (ESI Fig. 3C†). This analysis also organized proteins into three distinct clusters, with individual cluster groups having similar correlations to clinical data (ESI Fig. 3D–F†). Two of the three clusters from the male patient data overlapped with the clusters from the pooled patient data, with one cluster characterized by a positive correlation with stroke volume index and another grouped by a positive correlation with patient STS score. However, in contrast to the third cluster from the pooled patient data, the third cluster group from the male patient data was associated with a negative correlation with stroke volume index.
Annexin A2 and cystatin C drive VIC myofibroblast activation through p38 MAPK signaling
We next validated annexin A2 and cystatin C as potential regulators of VIC phenotypes via p38 MAPK signaling. We treated VICs with low and high doses of annexin A2 or cystatin C and added 20 μM of the p38 MAPK inhibitor SB20358021 (Fig. 4A). We hypothesized that both candidate proteins regulate VIC phenotypic changes through the p38 MAPK pathway, since previous studies have shown that annexin A2 and cystatin C act through MAPK signaling42–44 and our cluster analysis showed that annexin A2 and cystatin C enrich to the MAPK pathway.
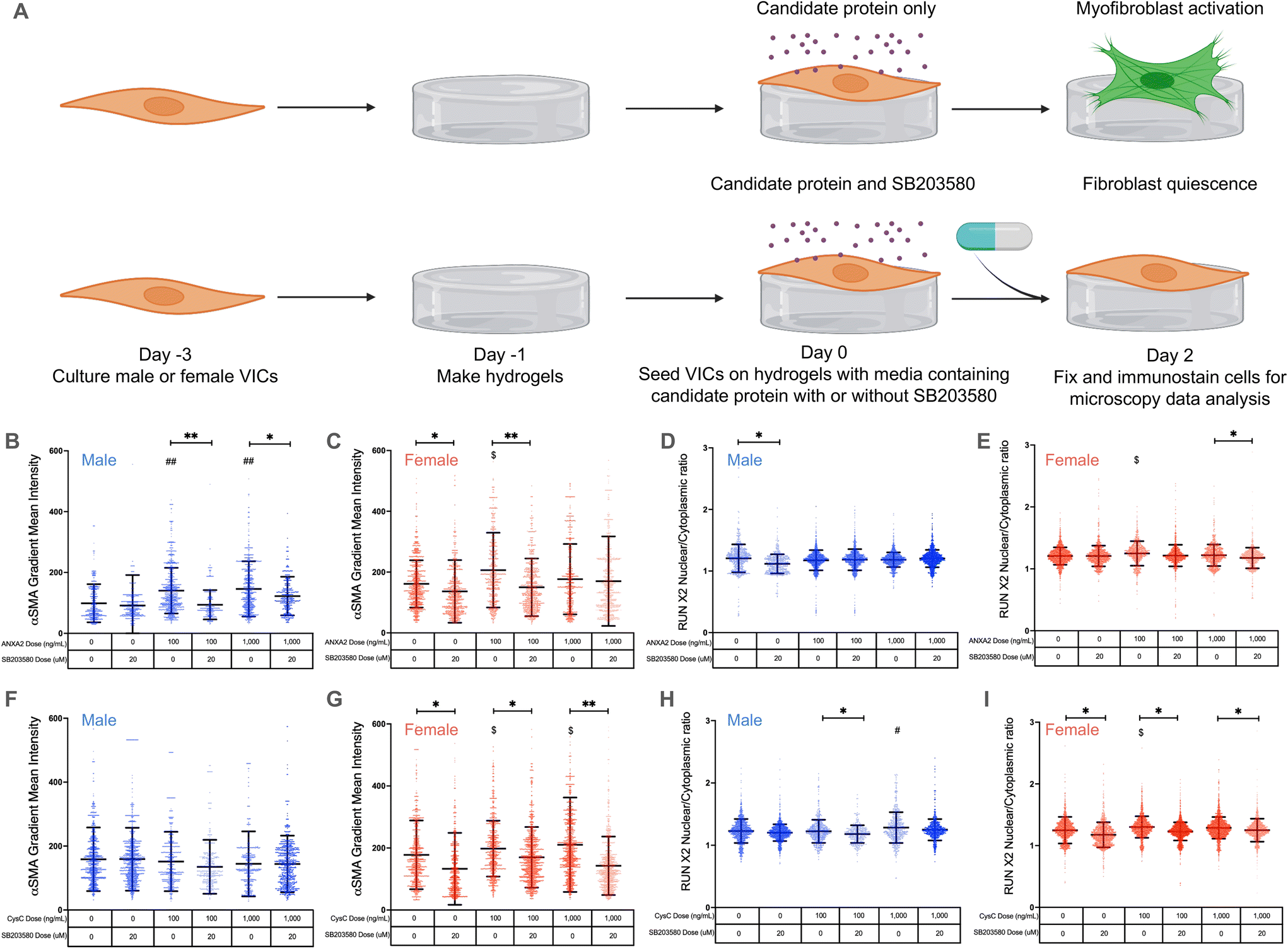 | ||
| Fig. 4 Annexin A2 and cystatin C drive VIC myofibroblast activation via p38 MAPK signaling. (A) Male and female VICs were cultured for 48 hours on hydrogels with media containing either annexin A2 or cystatin C with or without SB203580. Myofibroblast activation was measured through α-SMA gradient mean intensity and osteoblast-like differentiation was measured through RUNX2 nuclear localization. (B and C) α-SMA gradient mean intensity data for (B) male VICs and (C) female VICs treated with annexin A2 and SB203580 on hydrogels. (D and E) RUNX2 nuclear localization data in (D) male VICs and (E) female VICs treated with annexin A2 and SB203580 on hydrogels. (F and G) α-SMA gradient mean intensity data for (F) male VICs and (G) female VICs treated with cystatin C and SB203580 on hydrogels. (H and I) RUNX2 nuclear localization data in (H) male VICs and (I) female VICs treated with cystatin C and SB203580 on hydrogels. For all graphs, data is shown as mean ± standard deviation with statistical significance determined by one-way ANOVA with Tukey posttests (P < 0.0001) and effect size measured between statistically significant groups by the Cohen's d-value indicated by ** = d > 0.5, * = d > 0.2 for differences between groups; ## = d > 0.5, # = d > 0.2 for differences from the male control group; $ = d > 0.2 for differences from the female control group. | ||
Annexin A2 again caused a significant increase in male VIC myofibroblast activation at concentrations of 100 and 1000 ng mL−1 (Fig. 4B). When cultured with SB203580, male VICs showed a significant reduction in α-SMA gradient mean intensity even in the presence of annexin A2. Similar effects were observed in female VICs, with 100 ng mL−1 of annexin A2 causing a significant increase in myofibroblast activation without SB203580; the effect was abrogated with SB203580 (Fig. 4C). Interestingly, female VIC myofibroblast activation was significantly reduced with SB203580 whereas male VICs showed no significant responses to the p38 MAPK inhibitor alone (Fig. 4C and G). No significant differences in RUNX2 nuclear localization were observed in male VICs cultured with annexin A2 and in the presence or absence of SB203580 (Fig. 4D). In contrast, annexin A2 caused a significant increase in female VIC RUNX2 nuclear localization at 100 ng mL−1, and SB203580 caused a decrease in RUNX2 nuclear localization at the highest dose of annexin A2 tested (Fig. 4E). Taken together, these data suggest that annexin A2 promotes myofibroblast activation, but not osteoblast-like differentiation through p38 MAPK signaling in both male and female VICs.
Once again, cystatin C had no impact on male VIC myofibroblast activation as no significant changes in α-SMA gradient mean intensity were observed, regardless of cystatin C concentration or presence of SB203580 (Fig. 4F). On the other hand, cystatin C consistently induced significant increases in female VIC myofibroblast activation (Fig. 4G). When cultured with cystatin C and SB203580, female VICs showed significant reductions in α-SMA gradient mean intensity relative to cells without SB203580. Cystatin C also significantly increased male VIC RUNX2 nuclear localization at the highest concentration tested, and SB203580 reduced RUNX2 nuclear localization at the 100 ng mL−1 concentration (Fig. 4H). Like annexin A2, cystatin C caused a significant increase in female VIC RUNX2 nuclear localization, but this effect was not observed at the highest dose tested (Fig. 4I). Cumulatively, these data suggest that cystatin C activates female VICs through the p38 MAPK pathway but promotes osteoblast-like differentiation in male VICs likely via an alternate pathway.
Our core findings that annexin A2 and cystatin C regulate sexually dimorphic VIC phenotypes via p38 MAPK signaling were also validated using RT-PCR (ESI Fig. 6†). We found COL1A1 expression was uniquely decreased in female VICs treated with SB203580 alone (ESI Fig. 6B†), COL1A1 expression uniquely increased only in female VICs treated with cystatin C, and COL1A1 expression was significantly decreased in female VICs treated with both SB203580 and cystatin C (ESI Fig. 6B†). We also confirmed that osteoblast-like differentiation is not regulated via p38 MAPK signaling as osteoblast-associated gene expression did not decrease significantly in the presence of SB203580 (ESI Fig. 6C and D†).
Discussion
Here, we identified key inflammatory secreted factors from AVS patient serum that are both clinically significant and demonstrate strong links to myofibroblast activation. In vitro validation of two of these factors, annexin A2 and cystatin C, confirmed that these proteins contribute to sexually dimorphic AVS progression by driving sex-specific VIC phenotypes that reflect sex dimorphisms observed during AVS progression. Specifically, we found that annexin A2 increases myofibroblast activation in male and female VICs, whereas cystatin C only impacts female VIC activation. Although, annexin A2 and cystatin C both increase RUNX2 nuclear localization primarily in male VICs. Other researchers have also found that secreted factors contribute to sex dimorphisms in heart disease, such as one study that found that matrix metalloproteinase 12 (MMP12) is more expressed in men with atherosclerosis and removing MMP12 eliminates any sex differences observed in aortic stiffness.45 Another noteworthy study showed that hearts from men and women age differently, with hearts from women exhibiting greater increases in inflammation and pro-inflammatory factors over time.46 An additional study of interest found that men with atherosclerosis have increased concentrations of IL-1β, TNF α, and IL-6.47 Given that IL-1β and TNF-α are anti-fibrotic while IL-6 is pro-calcific, these findings showcase the importance of analyzing secreted proteins to help explain sex differences in AVS progression, such as increased calcification in men. In the future, exploring the combinatorial effects of annexin A2 and cystatin C on fibrosis and calcification in AVS may be fruitful, given the complete proteome in patient sera elicits patient-specific effects on myofibroblast activation.21,48 Additionally, although porcine VICs are known to respond similarly to human VICs when treated with biochemical factors,49 reproducing key experiments using human VICs would further enhance the clinical relevance of our findings.We further showed that annexin A2 and cystatin C modulate sex-specific VIC myofibroblast activation through p38 MAPK signaling. Our K-means cluster analysis sorted our candidate factors into three groups based on correlations to clinical measures of AVS severity. Interestingly, annexin A2 and cystatin C grouped closely together, which explains the similar effects of these factors on VIC phenotype. Our KEGG pathway enrichment analysis broadly identified MAPK signaling as a potential pathway for annexin A2 and cystatin C to promote myofibroblast activation. Our analysis aligns with previous work suggesting that proteins upstream of p38 MAPK increase myofibroblast activation in VICs cultured with AVS patient serum.21 Moreover, other researchers have found that TGF-β1 induces a myofibroblast phenotype in VICs via p38 MAPK signaling.50
Our in vitro experiments demonstrated that annexin A2 and cystatin C had similar effects on female VICs, with both factors increasing α-SMA gradient mean intensity through p38 MAPK signaling while having a minimal impact on RUNX2 nuclear localization. Annexin A2 also increased α-SMA gradient mean intensity through p38 MAPK signaling in male VICs, whereas cystatin C did not affect male VIC myofibroblast activation. We also observed reduced ACTA2 gene expression and α-SMA stress fiber formation only in female VICs treated with SB203580, suggesting that female VICs uniquely activate via p38 MAPK signaling as a mechanosensing pathway. Although RT-PCR corroborated several of our findings, sex-specific myofibroblast and osteoblast-like gene expression was not observed in VICs treated with annexin A2. Our findings underscore the importance of measuring α-SMA stress fiber formation and the nuclear localization of key markers of osteoblast-like differentiation at the protein level over analyzing solely gene expression of key markers. We also observed variations in the sex differences in α-SMA gradient mean intensity and RUNX2 nuclear localization at baseline (Fig. 2 and 4), which is likely due to batch-to-batch variability or activity in other pathways associated with MAPK signaling, including JNK and ERK pathways. However, despite these variations in the absence of annexin A2 and cystatin C, similar trends in phenotype across experiments were still observed. Moreover, significant increases in these measures of VIC phenotype were still observed in response to annexin A2 and cystatin C, showcasing the significance and reproducibility of our findings that annexin A2 and cystatin C promote sexually dimorphic phenotypes in VICs through p38 MAPK signaling.
Although we implicate p38 MAPK signaling in myofibroblast activation, other studies have shown that p38 MAPK signaling is also involved in promoting osteoblast-like phenotypes in VICs.51 VIC signaling networks are complex and predicting their effects on regulating the transitions from a quiescent fibroblast to myofibroblast or osteoblast-like phenotype remains an ongoing area of research.48 Previous work has suggested that pro-inflammatory secreted factors mediate a quiescent VIC to myofibroblast transition, followed by proliferation, then termination in an osteoblast-like phenotype.16 Our data suggest that annexin A2 and cystatin C increase both myofibroblast and osteoblast-like phenotypes. However, experiments evaluating VIC phenotypes at longer time points are needed to determine which sex-specific phenotypes remain dominant over time. Our findings showing sex dimorphisms in p38 MAPK signaling also align with other studies that found similar observations in other diseases.52,53 For example, in an inflammatory disease model of myocardial ischemia, male rats showed increased p38 MAPK signaling relative to female rats after an ischemia/reperfusion injury.52 Future studies probing how annexin A2 and cystatin C utilize p38 MAPK signaling in vivo to promote VIC differentiation are needed and may further corroborate the in vitro cellular sex dimorphisms observed here.
Using hydrogels as precision biomaterials54,55 enabled our assessment of annexin A2 and cystatin C as drivers of sex-specific myofibroblast activation and osteoblast-like differentiation. Several studies have used hydrogels as mimics of the native aortic valve matrix.22–24 In these studies, VICs remained quiescent due to a soft hydrogel matrix comparable to native valve tissue, which deactivates mechanosensitive pathways that promote myofibroblast activation via integrin-mediated interactions with RGD on the synthetic hydrogel matrix.56 Hydrogels have also been shown to exacerbate observed differences in myofibroblast activation and osteoblast-like differentiation, as measured by α-SMA and RUNX2, relative to TCPS controls.21,49 In line with our previous work, the use of hydrogels allowed for a more robust characterization of the sex-specific effects of inflammatory factors on VIC phenotype. Moving forward, improved models that more closely mimic the native valvular cell microenvironment may further exacerbate the observed sex differences in inflammatory cue responses. For example, culturing VICs in three-dimensional hydrogel systems may provide more physiologically relevant VIC responses to biochemical factors.57 Other approaches include using photolithographic patterning to crosslink PEG diacrylate into PEG hydrogels to reproduce the anisotropic structure of native valve tissue and coculturing VICs with valvular endothelial cells.58,59 Nonetheless, the reported findings here showcase the value of using hydrogels that mimic aortic valve tissue to better assess the effects of inflammatory factors on VIC (myo)fibroblast phenotypes and more broadly highlight the utility of using biomaterials as tools to investigate sex dimorphisms.60
We suggest that in addition to contributing to AVS progression, annexin A2 and cystatin C also serve as biomarkers of AVS that are positively correlated with patient STS score. Our findings support previous studies that found that increased serum levels of annexin A2 and cystatin C are directly associated with increased cardiovascular disease severity.40,61–63 Annexin A2 serum levels in healthy patients are typically around 15 ng mL−1 and may not vary by sex.38,64 Serum levels of annexin A2 are elevated in a variety of cardiovascular diseases, including coronary artery disease and heart failure, and can approach 1000 ng mL−1.61,62,65 Similarly, cystatin C serum concentrations typically range from 600 to over 1000 ng mL−1 and are increased in patients at high risk for heart failure and coronary heart disease.39,40,63 Interestingly, healthy male patients have significantly higher levels of cystatin C in serum compared to female patients,66 which offers another possible insight into sex differences in AVS progression as our results show that cystatin C promotes an osteoblast-like phenotype primarily in male VICs. Previous work has also outlined a role for key factors to serve as both biomarkers of and contributors to heart valve disease, with one study implicating apolipoprotein C-III as a contributor to valve calcification beyond serving solely as a biomarker of calcific aortic valve disease.67 Our study was limited by patient sample size (n = 12), so a larger-scale study is required to fully validate annexin A2 and cystatin C as reliable biomarkers. Additionally, separating serum samples by sex and running separate correlation analyses will help identify sex-specific candidate inflammatory factors. Our correlation analyses using only male patient data identified several potential biomarkers unique to men and offer intriguing data suggesting significant heterogeneity in AVS biomarkers based on sex, but further investigation is required. Regardless, the work herein identified two candidate proteins to serve as biomarkers indicative of AVS progression that may also contribute to sex-specific cellular phenotypes during AVS progression.
Overall, this work showcases the importance of accounting for both sex and inflammation to improve the accuracy of in vitro models of AVS.68 Based on our findings, we propose sex-specific mechanisms for the effects of the inflammatory factors annexin A2 and cystatin C on VIC phenotype (Fig. 5A and B). Increased myofibroblast activation observed in female VICs is likely due to mechanosensitive pathway activity, potentially regulated via genes that escape X-chromosome inactivation (XCI)69 or transcription factor activity.70 Indeed, prior work has outlined a role for two XCI escape genes, BMX and STS, in regulating increased myofibroblast activation in female VICs through Rho-associated protein kinase signaling.49 We suggest XCI escape genes that participate in p38 MAPK signaling may partially regulate the increased myofibroblast activation observed uniquely in female VICs in the absence of inflammatory factors. Sex differences in cellular phenotypes observed in our study may also be due to sex-specific epigenetic modifications that persist in cell culture.71,72 For example, gonadal hormones are known to modify patterns of acetylation and methylation before birth and can regulate phenotypes in adult somatic cells.73 Additionally, XCI escape genes that code for histone demethylases such as KDM5C74 could lead to increased demethylation of profibrotic genes in female VICs, promoting a myofibroblast phenotype. Notable sex differences were observed across all data analyzed in this study from the patient proteome to VIC responses to biochemical factors, which is expected given the known sex differences in VIC gene expression.75 Sex dimorphisms in the patient serum proteome have also been observed in other cardiovascular diseases, with studies finding that female atherosclerosis patients have higher serum concentrations of active TGF-β,76 and female coronary artery disease patients have increased IL-6 compared to males.77 Even though we only validated annexin A2 and cystatin C as drivers of sex-specific VIC differentiation, our work identified over 300 inflammatory factors linked to myofibroblast activation. Given that individual inflammatory factors can drive VIC phenotypic changes, the role of inflammation in driving sex-specific fibro-calcification cannot be overlooked.
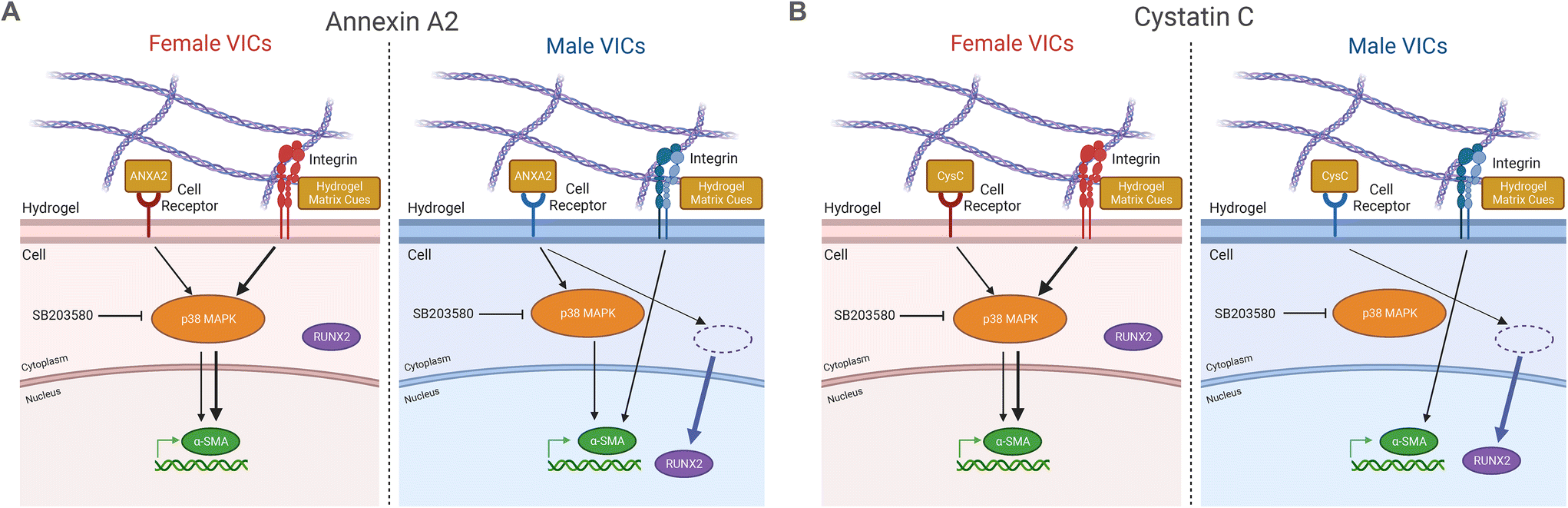 | ||
| Fig. 5 Proposed mechanisms for annexin A2 and cystatin C driving sex-specific VIC myofibroblast and osteoblast-like differentiation. (A) Annexin A2 promotes myofibroblast activation in male and female VICs through p38 MAPK signaling, but osteoblast-like differentiation only in male VICs via an alternate pathway. (B) Cystatin C promotes myofibroblast activation in female VICs through p38 MAPK signaling and osteoblast-like differentiation in male VICs via an alternate pathway. | ||
Conclusion
Using an approach that considered both clinical and in vitro data, we identified clinically relevant AVS serum secreted factors that likely drive VIC myofibroblast activation and osteoblast-like differentiation. We utilized hydrogels that mimic the aortic valve microenvironment to clearly assess the sex-specific effects of physiologically relevant doses of annexin A2 and cystatin C on VIC phenotype. Our findings show a sexually dimorphic response to these factors that was largely driven through p38 MAPK signaling. Further illuminating the sex-specific biological mechanisms of inflammatory proteins and identifying other serum factors that promote AVS progression may lead to the development of more customized and accurate disease models.Conflicts of interest
No conflicts of interest to disclose.Acknowledgements
We acknowledge the help and resources of Dr Eric Griffis at the Nikon Imaging Center at the University of California San Diego and Dr Joe Dragavon at the BioFrontiers Advanced Light Microscopy Core located within the BioFrontiers Institute at the University of Colorado Boulder. B. J. Vogt acknowledges funding from the NIH (R00 HL148542 and R01 HL132353). D. K. Peters acknowledges funding from the NIH (R01 AI151636). K. S. Anseth acknowledges funding from the NIH (R01 HL142935, RO1 HL132353). B. A. Aguado acknowledges funding from the NIH (R00 HL148542 and R25 HL145817), the Burroughs Wellcome Fund Postdoctoral Enrichment Program, and the American Heart Association Career Development Award (942253). Fig. 1A, 2A, 4A, and 5 were created with Biorender.References
- R. L. J. Osnabrugge, D. Mylotte, S. J. Head, N. M. V. Mieghem, V. T. Nkomo, C. M. LeReun, A. J. J. C. Bogers, N. Piazza and A. P. Kappetein, J. Am. Coll. Cardiol., 2013, 62, 1002–1012 CrossRef PubMed.
- N. Bhatia, S. S. Basra, A. H. Skolnick and N. K. Wenger, J. Geriatr. Cardiol., 2016, 13, 941–944 Search PubMed.
- Y. Sritharen, M. Enriquez-Sarano, H. V. Schaff, G. Casaclang-Verzosa and J. D. Miller, Physiology, 2017, 32, 182–196 CrossRef PubMed.
- M. Toyofuku, T. Taniguchi, T. Morimoto, K. Yamaji, Y. Furukawa, K. Takahashi, T. Tamura, H. Shiomi, K. Ando, N. Kanamori, K. Murata, T. Kitai, Y. Kawase, C. Izumi, M. Miyake, H. Mitsuoka, M. Kato, Y. Hirano, S. Matsuda, T. Inada, T. Murakami, Y. Takeuchi, K. Yamane, M. Ishii, E. Minamino-Muta, T. Kato, M. Inoko, T. Ikeda, A. Komasa, K. Ishii, K. Hotta, N. Higashitani, Y. Kato, Y. Inuzuka, C. Maeda, T. Jinnai, Y. Morikami, N. Saito, K. Minatoya, T. Kimura and on behalf of the C. A. R. Investigators, Circ. J., 2017, 81, CJ-16–CJ-1244 CrossRef PubMed.
- E. Aikawa and J. D. Hutcheson, N. Engl. J. Med., 2022, 386, 1372–1374 CrossRef PubMed.
- A. Kumar, K. Sato, J. Narayanswami, K. Banerjee, K. Andress, C. Lokhande, D. Mohananey, A. K. Anumandla, A. R. Khan, A. C. Sawant, V. Menon, A. Krishnaswamy, E. M. Tuzcu, W. A. Jaber, S. Mick, L. G. Svensson and S. R. Kapadia, Circ.: Cardiovasc. Interventions, 2018, 11, e006664 Search PubMed.
- P. J. Goleski, M. Reisman and C. W. Don, Catheter. Cardiovasc. Interv., 2018, 91, 165–168 CrossRef PubMed.
- J. D. Hutcheson, E. Aikawa and W. D. Merryman, Nat. Rev. Cardiol., 2014, 11, 218–231 CrossRef CAS PubMed.
- S. Masjedi, Y. Lei, J. Patel and Z. Ferdous, Heart Vessels, 2017, 32, 217–228 CrossRef PubMed.
- M. Voisine, M. Hervault, M. Shen, A. Boilard, B. Filion, M. Rosa, Y. Bossé, P. Mathieu, N. Côté and M. Clavel, J. Am. Heart Assoc., 2020, 9, e015610 CrossRef CAS PubMed.
- D. Cramariuc, B. P. Rogge, M. T. Lønnebakken, K. Boman, E. Bahlmann, C. Gohlke-Bärwolf, J. B. Chambers, T. R. Pedersen and E. Gerdts, Heart, 2015, 101, 209 CrossRef CAS PubMed.
- K. H. Humphries, S. Toggweiler, J. Rodés-Cabau, L. Nombela-Franco, E. Dumont, D. A. Wood, A. B. Willson, R. K. Binder, M. Freeman, M. K. Lee, M. Gao, M. Izadnegahdar, J. Ye, A. Cheung and J. G. Webb, J. Am. Coll. Cardiol., 2012, 60, 882–886 CrossRef PubMed.
- R. Zusterzeel, N. K. Mishra, H. Beydoun, J. Laschinger, C. Wu, L. M. Dong, J.-A. Lin, D. Marinac-Dabic, D. G. Strauss and D. A. Caños, J. Women's Health, 2018, 27, 808–814 CrossRef PubMed.
- S. Kodali, M. R. Williams, D. Doshi, R. T. Hahn, K. H. Humphries, V. T. Nkomo, D. J. Cohen, P. S. Douglas, M. Mack, K. Xu, L. Svensson, V. H. Thourani, E. M. Tuzcu, N. J. Weissman, M. Leon and A. J. Kirtane, Ann. Intern. Med., 2016, 164, 377 CrossRef PubMed.
- A. Hulin, A. Hego, P. Lancellotti and C. Oury, Front. Cardiovasc. Med., 2018, 5, 21 CrossRef PubMed.
- J. C. Grim, B. A. Aguado, B. J. Vogt, D. Batan, C. L. Andrichik, M. E. Schroeder, A. Gonzalez-Rodriguez, F. M. Yavitt, R. M. Weiss and K. S. Anseth, Arterioscler., Thromb., Vasc. Biol., 2020, 40(11), 296–308 CrossRef PubMed.
- G. Li, W. Qiao, W. Zhang, F. Li, J. Shi and N. Dong, J. Thorac. Cardiovasc. Surg., 2017, 153, 1318–1327 CrossRef PubMed.
- I. Parra-Izquierdo, I. Castaños-Mollor, J. López, C. Gómez, J. A. S. Román, M. S. Crespo and C. García-Rodrígue, Biochim. Biophys. Acta, Mol. Basis Dis., 2019, 1865, 2168–2179 CrossRef PubMed.
- J. J. Kaden, C.-E. Dempfle, R. Grobholz, H.-T. Tran, R. Kılıç, A. Sarıkoç, M. Brueckmann, C. Vahl, S. Hagl, K. K. Haase and M. Borggrefe, Atherosclerosis, 2003, 170, 205–211 CrossRef.
- J. Swierszcz, J. Dubiel and K. Sztefko, J. Heart Valve Dis., 2011, 20, 639–649 Search PubMed.
- B. A. Aguado, K. B. Schuetze, J. C. Grim, C. J. Walker, A. C. Cox, T. L. Ceccato, A.-C. Tan, C. C. Sucharov, L. A. Leinwand, M. R. G. Taylor, T. A. McKinsey and K. S. Anseth, Sci. Transl. Med., 2019, 11, eaav3233 CrossRef CAS PubMed.
- A. M. Kloxin, J. A. Benton and K. S. Anseth, Biomaterials, 2010, 31, 1–8 CrossRef CAS PubMed.
- H. Wang, S. M. Haeger, A. M. Kloxin, L. A. Leinwand and K. S. Anseth, PLoS One, 2012, 7, e39969 CrossRef CAS PubMed.
- H. Wang, M. W. Tibbitt, S. J. Langer, L. A. Leinwand and K. S. Anseth, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 19336–19341 CrossRef CAS PubMed.
- B. D. Fairbanks, M. P. Schwartz, A. E. Halevi, C. R. Nuttelman, C. N. Bowman and K. S. Anseth, Adv. Mater., 2009, 21, 5005–5010 CrossRef CAS PubMed.
- D. Peters, Collaborations-AnsethLab-OperettaImageSortingandAnalysis, Github, 2021 Search PubMed.
- S. Galarza, H. Kim, N. Atay, S. R. Peyton and J. M. Munson, Bioeng. Transl. Med., 2019, 5, e10148 Search PubMed.
- L. D. Hayes, P. Herbert, N. Sculthorpe and F. Grace, Exp. Gerontol., 2020, 140, 111074 CrossRef CAS PubMed.
- J. L. Steel, D. A. Geller, K. H. Kim, L. H. Butterfield, M. Spring, J. Grady, W. Sun, W. Marsh, M. Antoni, M. A. Dew, V. Helgeson, R. Schulz and A. Tsung, Cancer, 2016, 122, 1270–1282 CrossRef PubMed.
- J. F. A. Swisher, N. Burton, S. M. Bacot, S. N. Vogel and G. M. Feldman, Blood, 2010, 115, 549–558 CrossRef CAS PubMed.
- J. F. A. Swisher, U. Khatri and G. M. Feldman, J. Leukocyte Biol., 2007, 82, 1174–1184 CrossRef CAS PubMed.
- M. Schuliga, J. Jaffar, A. Berhan, S. Langenbach, T. Harris, D. Waters, P. V. S. Lee, C. Grainge, G. Westall, D. Knight and A. G. Stewart, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2017, 312, L772–L782 CrossRef PubMed.
- N. X. Chen, K. D. O'Neill, X. Chen and S. M. Moe, J. Bone Miner. Res., 2008, 23, 1798–1805 CrossRef CAS PubMed.
- J. V. Salgado, F. L. Souza and B. J. Salgado, J. Cardiol., 2013, 62, 331–335 CrossRef PubMed.
- P. C. A. Namboodiripad, J. Oral Biol. Craniofac. Res., 2014, 4, 42–46 CrossRef.
- M. Kasabova, A. Joulin-Giet, F. Lecaille, B. F. Gilmore, S. Marchand-Adam, A. Saidi and G. Lalmanach, J. Biol. Chem., 2014, 289, 16239–16251 CrossRef PubMed.
- N. A. Lokman, C. Ricciardelli, A. N. Stephens, T. W. Jobling, P. Hoffmann and M. K. Oehler, Diagnostics, 2021, 11, 69 CrossRef PubMed.
- L. D. Gibbs, K. Mansheim, S. Maji, R. Nandy, C. M. Lewis, J. K. Vishwanatha and P. Chaudhary, Cancers, 2020, 13, 2 CrossRef PubMed.
- P. Villa, M. Jiménez, M.-C. Soriano, J. Manzanares and P. Casasnovas, Crit. Care, 2005, 9, R139 CrossRef PubMed.
- M. J. Sarnak and R. Katz, Ann. Intern. Med., 2005, 142, 497–505 CrossRef CAS PubMed.
- M. E. Schroeder, D. Batan, A. G. Rodriguez, K. F. Speckl, D. K. Peters, B. E. Kirkpatrick, G. K. Hach, C. J. Walker, J. C. Grim, B. A. Aguado, R. M. Weiss and K. S. Anseth, Bioeng. Transl. Med., 2022 DOI:10.1002/btm2.10358.
- F. Li, H. Chung, S. V. Reddy, G. Lu, N. Kurihara, A. Z. Zhao and G. D. Roodman, J. Bone Miner. Res., 2005, 20, 1161–1167 CrossRef CAS PubMed.
- B. Wegiel, T. Jiborn, M. Abrahamson, L. Helczynski, L. Otterbein, J. L. Persson and A. Bjartell, PLoS One, 2009, 4, e7953 CrossRef PubMed.
- Y. Shen, X. Zhang, C. Li, X. Wang, Y. Ye, J. Yuan, H. Gong, Y. Zou and J. Ge, Ann. Transl. Med., 2020, 8, 1514–1514 CrossRef CAS PubMed.
- S. Liu, A. Bajpai, E. A. Hawthorne, Y. Bae, P. Castagnino, J. Monslow, E. Puré, K. L. Spiller and R. K. Assoian, JCI Insight, 2019, 4, e122742 CrossRef PubMed.
- M. L. B. de Arellano, S. Pozdniakova, A. A. Kühl, I. Baczko, Y. Ladilov and V. Regitz-Zagrosek, Aging, 2019, 11, 1918–1933 CrossRef PubMed.
- S. Bernardi, B. Toffoli, F. Tonon, M. Francica, E. Campagnolo, T. Ferretti, S. Comar, F. Giudici, E. Stenner and B. Fabris, Int. J. Mol. Sci., 2020, 21, 3861 CrossRef CAS PubMed.
- J. D. Rogers, B. A. Aguado, K. M. Watts, K. S. Anseth and W. J. Richardson, Proc. Natl. Acad. Sci. U. S. A., 2022, 8, 199 Search PubMed.
- B. A. Aguado, C. J. Walker, J. C. Grim, M. E. Schroeder, D. Batan, B. J. Vogt, A. G. Rodriguez, J. A. Schwisow, K. S. Moulton, R. M. Weiss, D. D. Heistad, L. A. Leinwand and K. S. Anseth, Circulation, 2022, 145, 513–530 CrossRef CAS PubMed.
- J. D. Hutcheson, L. M. Ryzhova, V. Setola and W. D. Merryman, J. Mol. Cell Cardiol., 2012, 53, 707–714 CrossRef CAS PubMed.
- F. Xie, F. Li, R. Li, Z. Liu, J. Shi, C. Zhang and N. Dong, Life Sci., 2020, 257, 118086 CrossRef CAS PubMed.
- M. Wang, L. Baker, B. M. Tsai, K. K. Meldrum and D. R. Meldrum, Am. J. Physiol.: Endocrinol. Metab., 2005, 288, E321–E326 CrossRef CAS PubMed.
- D. N. Krementsov, R. Noubade, J. A. Dragon, K. Otsu, M. Rincon and C. Teuscher, Ann. Neurol., 2013, 75, 50–66 CrossRef PubMed.
- B. A. Aguado, J. C. Grim, A. M. Rosales, J. J. Watson-Capps and K. S. Anseth, Sci. Transl. Med., 2018, 10(424) CAS.
- K. C. Fogg, N.-H. Tseng, S. R. Peyton, P. Holeman, S. T. McLoughlin, J. P. Fisher, A. Sutton, A. Shikanov, J. S. Gnecco, K. M. Knight, E. Slaby, J. D. Weaver, N. Hashemi, Y. Zhang, M. D. House, B. J. Vogt, B. A. Aguado, J. Bradford, J. L. Robinson, P. K. Thomas, A. G. Lau and M. L. Oyen, J. Phys. Mater., 2022 Search PubMed.
- X. Gu and K. S. Masters, J. Biomed. Mater. Res., Part A, 2010, 93, 1620–1630 Search PubMed.
- A. G. Rodriguez, M. E. Schroeder, C. J. Walker and K. S. Anseth, APL Bioeng., 2018, 2, 046104 CrossRef PubMed.
- X. Zhang, B. Xu, D. S. Puperi, A. L. Yonezawa, Y. Wu, H. Tseng, M. L. Cuchiara, J. L. West and K. J. Grande-Allen, Acta Biomater., 2015, 14, 11–21 CrossRef PubMed.
- H. Tseng, L. R. Balaoing, B. Grigoryan, R. M. Raphael, T. C. Killian, G. R. Souza and K. J. Grande-Allen, Acta Biomater., 2014, 10, 173–182 CrossRef PubMed.
- N. E. Félex-Vélez, R. M. Gorashi and B. A. Aguado, J. Mater. Chem. B, 2022, 10, 7089–7098 RSC.
- D. Benevolensky, Y. Belikova, R. Mohammadzadeh, P. Trouvé, F. Marotte, F. Russo-Marie, J.-L. Samuel and D. Charlemagne, Lab. Invest., 2000, 80, 123–133 CrossRef PubMed.
- Z. Lin, D. Liu, Y. Xue, T. Wu, X. Zeng, Z. Guo and S. Cao, J. South. Med. Univ., 2020, 40, 382–387 CAS.
- T. Dent, Atherosclerosis, 2010, 213, 352–362 CrossRef CAS PubMed.
- W. Zhang, C. Gao, S. Zhang and G. Fang, J. Maxillofac. Oral Surg., 2017, 75, 1081–1087 CrossRef PubMed.
- M. K. Shaker, H. I. A. Fattah, G. S. Sabbour, I. F. Montasser, S. M. Abdelhakam, E. E. Hadidy, R. Yousry and A. K. E. Dorry, World J. Hepatol., 2017, 9, 469–476 CrossRef PubMed.
- D. Groesbeck, A. Köttgen, R. Parekh, E. Selvin, G. J. Schwartz, J. Coresh and S. Furth, Clin. J. Am. Soc. Nephrol., 2008, 3, 1777–1785 CrossRef CAS PubMed.
- F. Schlotter, R. C. C. de Freitas, M. A. Rogers, M. C. Blaser, P.-J. Wu, H. Higashi, A. Halu, F. Iqbal, A. B. Andraski, C. N. Rodia, S. Kuraoka, J. R. Wen, M. Creager, T. Pham, J. D. Hutcheson, S. C. Body, A. B. Kohan, F. M. Sacks, M. Aikawa, S. A. Singh and E. Aikawa, J. Biol. Chem., 2021, 296, 100193 CrossRef CAS PubMed.
- A. M. Porras, C. M. McCoy and K. S. Masters, Circ. Res., 2017, 120, 604–606 CrossRef CAS PubMed.
- P. Avner and E. Heard, Nat. Rev. Genet., 2001, 2, 59–67 CrossRef CAS PubMed.
- B. D. James and J. B. Allen, Adv. Healthc. Mater., 2021, 10, 2100735 CrossRef CAS PubMed.
- V. Regitz-Zagrosek, Sex and Gender Differences in Pharmacology, 2012 Search PubMed.
- C. J. Walker, D. Batan, C. T. Bishop, D. Ramirez, B. A. Aguado, M. E. Schroeder, C. Crocini, J. Schwisow, K. Moulton, L. Macdougall, R. M. Weiss, M. A. Allen, R. Dowell, L. A. Leinwand and K. S. Anseth, Bioeng. Transl. Med., 2022, 7(3) CAS.
- T.-Y. Zhang and M. J. Meaney, Annu. Rev. Psychol., 2010, 61, 439–466 CrossRef PubMed.
- J. B. Berletch, W. Ma, F. Yang, J. Shendure, W. S. Noble, C. M. Disteche and X. Deng, PLoS Genet., 2015, 11, e1005079 CrossRef PubMed.
- C. M. McCoy, D. Q. Nicholas and K. S. Masters, PLoS One, 2012, 7, e39980 CrossRef CAS PubMed.
- D. J. Grainger, P. R. Kemp, J. C. Metcalfe, A. C. Liu, R. M. Lawn, N. R. Williams, A. A. Grace, P. M. Schofield and A. Chauhan, Nat. Med., 1995, 1, 74–79 CrossRef PubMed.
- S. Sullivan, M. Hammadah, K. Wilmot, R. Ramadan, B. D. Pearce, A. Shah, B. Kaseer, M. M. Gafeer, B. B. Lima, J. H. Kim, L. Ward, Y. Ko, T. T. Lewis, A. Hankus, L. Elon, L. Li, J. D. Bremner, P. Raggi, A. Quyyumi and V. Vaccarino, J. Am. Heart Assoc., 2018, 7, e010329 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm00844k |
| This journal is © The Royal Society of Chemistry 2022 |