
Destructive fibrotic teamwork: how both microenvironment stiffness and profibrotic interleukin 13 impair alveolar macrophage phenotype and function†

Abstract
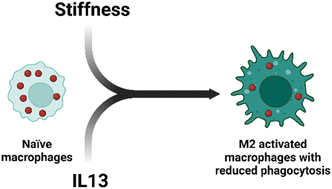
The pulmonary fibrotic microenvironment is characterized by increased stiffness of lung tissue and enhanced secretion of profibrotic soluble cues contributing to a feedback loop that leads to dysregulated wound healing and lung failure. Pinpointing the individual and tandem effects of profibrotic stimuli in impairing immune cell response remains difficult and is needed for improved therapeutic strategies. We utilized a statistical design of experiment (DOE) to investigate how microenvironment stiffness and interleukin 13 (IL13), a profibrotic soluble factor linked with disease severity, contribute to the impaired macrophage response commonly observed in pulmonary fibrosis. We used engineered bioinspired hydrogels of different stiffness, ranging from healthy to fibrotic lung tissue, and cultured murine alveolar macrophages (MH-S cells) with or without IL13 to quantify cell response and analyze independent and synergistic effects. We found that, while both stiffness and IL13 independently influence macrophage morphology, phenotype, phagocytosis and efferocytosis, these factors work synergistically to exacerbate impaired macrophage phenotype and efferocytosis. These unique findings provide insights into how macrophages in fibrotic conditions are not as effective in clearing debris, contributing to fibrosis initiation/progression, and more broadly inform how underlying drivers of fibrosis modulate immune cell response to facilitate therapeutic strategies.
- This article is part of the themed collection: Biomaterials Science Emerging Investigator Series
 Please wait while we load your content...
Please wait while we load your content...

