
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced human T cell expansion with inverse opal hydrogels†
Fabião
Santos
 ab,
Julia
Valderas-Gutiérrez
a,
Eduardo
Pérez del Río
ab,
Miquel
Castellote-Borrell
ac,
Xavier Rodriguez
Rodriguez
ac,
Jaume
Veciana
ab,
Imma
Ratera
ab and
Judith
Guasch
*abc
ab,
Julia
Valderas-Gutiérrez
a,
Eduardo
Pérez del Río
ab,
Miquel
Castellote-Borrell
ac,
Xavier Rodriguez
Rodriguez
ac,
Jaume
Veciana
ab,
Imma
Ratera
ab and
Judith
Guasch
*abc
aInstitute of Materials Science of Barcelona (ICMAB-CSIC), Campus UAB, Bellaterra, 08193, Spain. E-mail: jguasch@icmab.es
bNetworking Research Center on Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN), Campus UAB, Bellaterra, 08193, Spain
cDynamic Biomimetics for Cancer Immunotherapy, Max Planck Partner Group, ICMAB-CSIC, Campus UAB, Bellaterra, 08193, Spain
First published on 6th June 2022
Abstract
Advanced personalized immunotherapies still have to overcome several biomedical and technical limitations before they become a routine cancer treatment in spite of recent achievements. In adoptive cell therapy (ACT), the capacity to obtain adequate numbers of therapeutic T cells in the patients following ex vivo treatment should be improved. Moreover, the time and costs to produce these T cells should be reduced. In this work, inverse opal (IOPAL) 3D hydrogels consisting of poly(ethylene) glycol (PEG) covalently combined with heparin were engineered to resemble the environment of lymph nodes, where T cells get activated and proliferate. The introduction of an IOPAL strategy allowed a precise control on the porosity of the hydrogels, providing an increase in the proliferation of primary human CD4+ T cells, when compared with state-of-the-art expansion systems. Additionally, the IOPAL hydrogels also showed a superior expansion compared to hydrogels with the same composition, but without the predetermined pore structure. In summary, we have shown the beneficial effect of having an IOPAL architecture in our 3D hydrogels to help achieving large numbers of cells, while maintaining the desired selected phenotypes required for ACT.
1. Introduction
Adoptive cell therapy (ACT) is an immunotherapy in which T cells are usually harvested from the blood or tumor tissue of patients, genetically modified and/or selected in the laboratory, expanded ex vivo in large numbers, and reintroduced into the same patients.1 These cells are modified ex vivo to improve the targeting and killing of cancer cells, through T cell receptor (TCR) engineering or the use of chimeric antigen receptor (CAR) T cells.2 Alternatively, tumor-specific T cells can be selected among the rest, such as in endogenous T cell (ETC) therapy or in ACT performed with tumor-infiltrating lymphocytes (TILs).3,4 In any case, the resulting T cells are then capable of specifically recognizing and eliminating malignant cells. In some cases, these cells hold the potential of maintaining their healing properties for years through memory subsets, thus helping the patient immune system to fight cancer recurrence.5–9T cells divide in a few subtypes, which play specific roles in ACT. CD8+ T cells can directly recognize cancer cells and display the main cytotoxic function, while CD4+ T cells were known to help directing the immune response and activating effector immune cells, mainly cytotoxic CD8+ T cells.10 Interestingly, recent evidence suggests that CD4+ T cells can also kill cancer cells,11–13 and that specific mixtures of CD4+ and CD8+ T cells can provide the highest antitumoral activities due to synergistic effects between subsets.14–16
Even though the potential of ACT is undoubtful, there are several limitations to overcome before it becomes a first line anti-cancer therapy.17 For example, a successful T cell ex vivo activation, proliferation, and differentiation that result in an optimal product in vivo to achieve long-term efficacy is needed.18–20In vivo T cells are primarily activated by antigen presenting cells (APCs) through the immunological synapse (IS).21 In the IS, there is a main interaction between the TCR of T cells and the antigen-loaded major histocompatibility complex (MHC) of APCs. Additionally, there are costimulatory and adhesion signals that ensure a proper T cell activation and proliferation, such as the CD28/B7 and LFA-1/ICAM-1 pairs, respectively.22 Not surprisingly, the ex vivo T cell activation and expansion methods have been focused on mimicking the IS,19,23 including materials that allow spatial control at the micro- and nanoscale.24–30 The current gold standard method to activate and expand T cells relies on artificial APCs consisting of polymeric magnetic beads (such as Dynabeads; ThermoFisher Scientific) that are coated with stimulating antibodies (usually, anti-CD3 and anti-CD28).31 Although these beads are easy to manipulate and remove by magnetic separation, they are not able to provide environmental signals naturally present in the native secondary lymphoid organs (SLOs), which upon antigen recognition, provide an optimal environment for T cell proliferation.32
SLOs and particularly the lymph nodes (LNs) have a well interconnected structure with compartmentalized microdomains, which separate almost exclusively T or B cells. The T cell zones contain CD4+ and CD8+ T cells as well as subsets of dendritic cells anchored to a network of fibroblastic reticular cells and reticular fibers. The stromal structures are known to be critical for immune cell migration, activation, and survival.33
Two-dimensional (2D) culture methods are therefore not suitable to mimic the natural three-dimensional (3D) environment of cells in vivo. To tackle this limitation, natural or synthetic 3D hydrogels can be used to better mimic the native ECM of the LNs.34,35 Recently, we have described hydrogels consisting of poly(ethylene glycol) (PEG) covalently combined with heparin, which were successfully used to culture T cells.36 PEG is a polymer that confers adequate structural and mechanical properties to the hydrogel.37 Heparin is a sulfated glycosaminoglycan naturally present in the ECM, with a high number of negative charges provided by carboxylate and sulfate groups.38 Although this molecule is commonly known as an anticoagulant, it can be used as an anchoring point for cationic molecules and basic peptides through electrostatic interactions.39
Furthermore, PEG hydrogels with enlarged and homogeneous pore structures using the inverse opal (IOPAL) or inverted colloidal crystal technique, as a porogen method,40 were shown to increase T cell migration41 and be useful for drug delivery of soluble T cell activators.42 This method is reported to provide a porosity above 70%, being the reproducibility of the synthesis process one of its main advantages.43 Moreover, the uniform interconnectivity achieved leads to a more homogeneous distribution of macromolecules and cells inside the matrix.40,43
Here we describe a family of IOPAL 3D hydrogels consisting of PEG and heparin. These IOPAL 3D PEG–Hep hydrogels were synthesized and characterized to imitate the conditions of the ECM of the LNs, with the objective of increasing primary human (CD4+) T cell proliferation and tuning differentiation for ACT. CD4+ T cell proliferation was found to be enhanced when using the IOPAL 3D PEG–Hep hydrogels in combination with the artificial APCs Dynabeads. These hydrogels not only improved the performance of the state-of-the-art suspension systems, but also that of the hydrogel in its bulk form. Moreover, we demonstrated the capacity of such hydrogels to influence the phenotype obtained, naïve (TN), central memory (TCM), and effector memory (TEM), which is closely related to the clinical outcomes.44,45
2. Materials and methods
2.1 Materials
A 10% w/v aqueous suspension of poly(methyl methacrylate) (PMMA) beads with a diameter of 78.3 ± 1.7 μm was purchased from microParticles GmbH (Germany). Heparin and penicillin/streptomycin (P/S) was acquired from Fisher Scientific (Spain). Fetal bovine serum (FBS), the CellTrace carboxyfluorescein diacetate succinimidyl ester (CFSE) cell proliferation kit, and Dynabeads were supplied by Thermo Fisher Scientific (USA). The CD4+ T cell isolation kit was obtained from Miltenyi Biotec GmbH (Germany). The anti-human CD3 FITC, CD4 PE, CD45RO FITC antibodies, and their controls used for flow cytometry were bought from Immunotools GmbH (Germany), whereas CD62L PE and its control were purchased from BioLegend (USA). Lymphoprep was acquired from Stemcell Technologies (Canada). 4-arm thiolated PEG (Mn 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), N-(2-aminoethyl)maleimide trifluoroacetate salt (AEM), 1-hydroxybenzotriazole hydrate (HOBT), N-(3-dimethylamino-propyl)-N-ethylcarbodiimide hydrochloride (EDC·HCl), 2-(N-morpholino)ethanesulfonic acid (MES), Dulbecco's phosphate buffered saline (PBS), Roswell Park Memorial Institute (RPMI)-1640 cell culture media, propidium iodide (PI), and the rest of the products not otherwise specified were obtained from Merck (USA).
000 g mol−1), N-(2-aminoethyl)maleimide trifluoroacetate salt (AEM), 1-hydroxybenzotriazole hydrate (HOBT), N-(3-dimethylamino-propyl)-N-ethylcarbodiimide hydrochloride (EDC·HCl), 2-(N-morpholino)ethanesulfonic acid (MES), Dulbecco's phosphate buffered saline (PBS), Roswell Park Memorial Institute (RPMI)-1640 cell culture media, propidium iodide (PI), and the rest of the products not otherwise specified were obtained from Merck (USA).
2.2 Synthesis of bulk and IOPAL PEG–Hep hydrogels
The bulk 3D PEG–Hep hydrogels were produced based on a recent publication of our group.36 In brief, heparin was first functionalized with maleimide groups yielding a Mal-Hep derivative. Then, PEG–Hep hydrogels were formed through a Michael reaction between the Mal-Hep derivative and a 4-arm thiolated PEG in a ratio of 1.5:1 in PBS. The volume of each bulk hydrogel was of 30 μL, which were added to a home-made Teflon template with 5 mm-diameter wells and left at least for 2 h in the incubator (37 ºC). This reaction resulted in a covalent crosslink and the consequent hydrogel formation with 3 wt% of PEG. For the IOPAL 3D PEG–Hep hydrogel formation, we adapted a protocol previously described.46 In this case, 100 μL of a 10% w/v aqueous suspension of non-crosslinked PMMA beads (78.3 ± 1.7 μm diameter) were added in the same home-made well plate template, and left for 24 h until the solvent evaporated. Thus, the opal was formed. Afterwards, a PEG–Hep hydrogel mixture analogous to the one described above for the bulk PEG–Hep hydrogels (3 wt% PEG) was prepared by mixing solutions of 4-arm thiolated PEG and Mal-Hep in PBS. 30 μL of the hydrogel mixture were added on top of the PMMA opal in the template and left to infiltrate and solidify for at least 48 h in the incubator (37 °C). Opal & PEG–Hep hydrogel hybrids were removed from the template after hydrogel formation. Then, the PMMA opal was dissolved by introducing the hybrids in glacial acetic acid (AcOH) for 72 h at 40 °C with agitation (150 rpm; orbital shaker). After this period, the hydrogels became completely transparent, indicating the successful removal of the PMMA beads. Finally, the resulting IOPAL PEG–Hep hydrogels were washed three times with PBS to remove the acid. After 1 h of UV sterilization, all hydrogels were rinsed with the cell culture media RPMI with 10% FBS and 1% P/S, and incubated until seeding at 37 °C.
2.3 Environmental scanning electron microscopy (ESEM)
Hydrogels were stored in the incubator at 37 °C under aqueous conditions until analyzed (Nbulkhydrogels = 3; NIOPALhydrogels = 3). To image the hydrated bulk and IOPAL hydrogels, the pressure and temperature of the vacuum chamber of a FEI Quanta 650F environmental scanning electron microscope (Thermo Fisher Scientific, USA) were slowly decreased until the structures could be observed.
2.4 X-ray microtomography
For these measurements, large IOPAL hydrogels were formed (2 mm in height and 1 cm in diameter), following the same protocol above mentioned (NIOPALhydrogels = 2). In this case though, the amount of aqueous suspension of PMMA beads and hydrogel precursor mixtures were 333 μL and 100 μL, respectively. The samples immersed in PBS were frozen with liquid nitrogen and lyophilized before the analysis. The instrument used was a Skyscan 1272 high-resolution micro computed tomography (Bruker, USA). The scanning time was 3 h with a minimum resolution of 5 μm, without any filter and with a peak voltage of 40–50 kV.
2.5 Rheometry
The equipment used was a Rheometer HAAKE RheoStress RS600 (Thermo Electron Corporation, USA) with a 10 mm diameter rotor. The larger IOPAL hydrogels described above were also employed for these measurements (NIOPALhydrogels = 2), which were all done at 37 °C. Strain and frequency sweeps were performed in the linear-viscoelastic (LVE) regime. The strain sweeps were executed at a constant frequency of 1.0 Hz and a pressure of 1–50 Pa, whereas in the frequency sweeps, we maintained a constant pressure of 50 Pa and varied the frequency from 0.01 Hz to 1.0 Hz.
2.6 CD4+ T cell culture in bulk and IOPAL PEG–Hep hydrogels
Buffy coats from healthy adult donors were supplied by “Banc de Sang i Teixits” (Barcelona, Spain) after the approval of the research project by the “Ethics Committee on Animal and Human Experimentation” of the Autonomous University of Barcelona (No. 5099). Primary human CD4+ T cells were obtained following an established protocol.28,29,35 In summary, it consists of a density gradient centrifugation with Ficoll to isolate peripheral blood mononuclear cells, and then separating the primary human CD4+ T cells with a commercial CD4+ T cell isolation kit. Cell purity was measured by flow cytometry with the antibodies anti-human CD3 FITC, anti-human CD4 PE, and the respective negative controls. Only samples that were >90% (usually >95%) positive for both CD3+ and CD4+ were employed. On the same day of cell isolation, the CD4+ T cells were seeded on the bulk and IOPAL hydrogels, which were placed in 96-well plates (105 cells per well). CD4+ T cells were seeded at a concentration of 106 cells per ml and a 1:1 ratio of Dynabeads (1 bead per cell) in the supplemented RPMI medium. The activating beads were kept in culture until measurements, when they were extracted from the cell suspension with a magnet, as suggested by the manufacturer. The pore size and interconnectivity of the hydrogels ensure proper cell and Dynabead infiltration through the structure.
2.7 CD4+ T cell viability, proliferation, and differentiation in bulk and IOPAL PEG–Hep hydrogels
In all experiments, CD4+ T cells were firstly harvested from the hydrogels through vigorous pipetting. For cell viability experiments, PI was used to stain CD4+ T cells on day 5 at a concentration of 1 mg ml−1 during 3 min at room temperature before flow cytometry measurements (Ndonors = 4). For proliferation analysis, CD4+ T cells were stained with a CFSE cell proliferation kit following the instructions of the manufacturer before seeding. On day 6, cells were analyzed by flow cytometry after recovering them from the hydrogels through vigorous pipetting (Ndonors = 7). To minimize the variability caused by the different donors, the results obtained were normalized to the positive control of each donor, which was assigned a value of 1. For the differentiation analysis, CD4+ T cells were analyzed on day 5 by flow cytometry, after removing them from the hydrogels, as explained above (Ndonors = 6). Anti-human CD45 RO FITC, CD62L PE, and the corresponding negative controls were used to stain the CD4+ T cells for 30 min at 0 °C before measurements.2.8 Flow cytometry
Flow cytometry experiments were performed with a CytoFLEX LX (Beckman Coulter, USA) and a BD FACSCanto (BD Bioscience, USA). For all measurements, cells were resuspended in PBS with 0.1% FBS.2.9 Confocal microscopy
3D images of a volume of 1.5 mm × 1.5 mm × 0.4 mm were obtained with a Leica TCS SP5 confocal microscope (Leica, Germany) equipped with a 10× objective on day 5. They show CFSE-stained CD4+ T cells inside an IOPAL PEG–Hep hydrogel on day 5 of culture (NIOPALhydrogels = 2). In the ESI,† Dynabeads can also be observed (black dots) in a video obtained in brightfield mode.
2.10 Data treatment and statistical analyses
Flow cytometry data was analyzed with the FlowJo software (FlowJo LLC, BD, USA). Data processing was performed with OriginPro (OriginLab Corp., USA). In the box plots, the boxes correspond to the interquartile range defined by the 25th and 75th percentiles, the central line is the median, the whiskers show one standard deviation, and □ is the average. In the bar graphs, errors represent one standard deviation. The Mann Whitney U-test was used to evaluate the statistical significance of the cell viability, proliferation, and differentiation assays, as well as for the pore size distributions.3. Results and discussion
3.1 Synthesis of bulk and IOPAL PEG–Hep hydrogels
Bulk 3D PEG–Hep hydrogels were produced through a Michael reaction between a 4-arm thiolated PEG and maleimide-functionalized heparin, based on a recent publication of our group.36 For the IOPAL PEG–Hep hydrogel formation, an opal structure was first created by using non-crosslinked PMMA beads of 78 μm in diameter. Thus, a highly-organized colloidal crystal of PMMA microspheres was formed during gradual solvent evaporation. Afterwards, a PEG–Hep hydrogel mixture analogous to the one described above for the bulk PEG–Hep hydrogels, was added on the PMMA opal and left to infiltrate. After the formation of the hydrogel, the PMMA opal was dissolved in AcOH without visible hydrogel damage, as previously shown in similar systems consisting of PEG or gelatin hydrogels.46 Consequently, completely transparent hydrogels were obtained, indicating the successful removal of the PMMA beads (Fig. 1).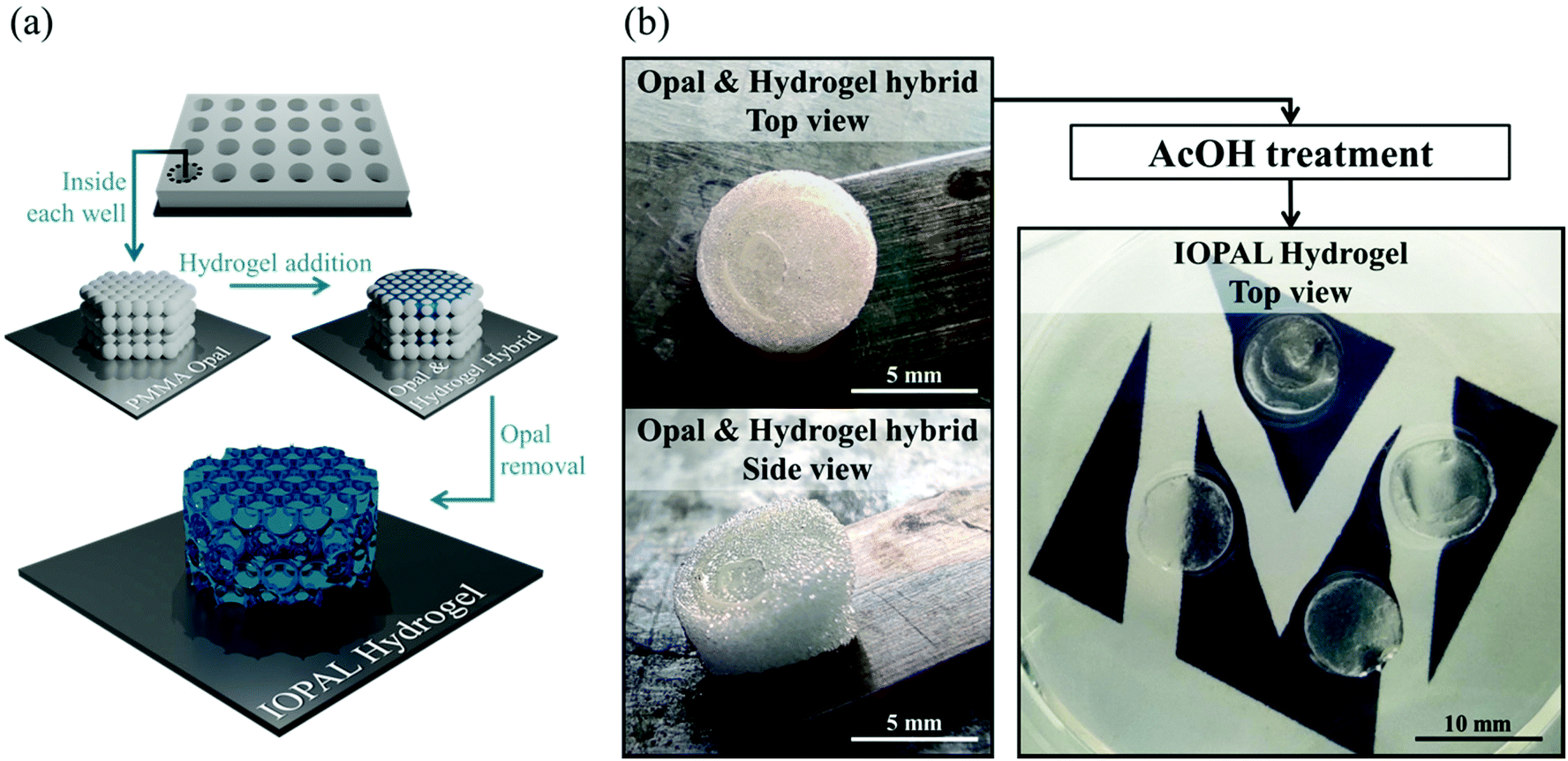 | ||
| Fig. 1 Formation of the IOPAL PEG–Hep hydrogel. (a) Simplified scheme of the formation of an IOPAL PEG–Hep hydrogel. (b) Photographs of the opal & hydrogel hybrid before the AcOH treatment, when they are white and opaque. After AcOH treatment, they result in transparent hydrogels, indicating the complete removal of the PMMA beads. | ||
3.2 Structural properties of the IOPAL PEG–Hep hydrogels
Both bulk and IOPAL hydrogels were studied by environmental scanning electron microscopy (ESEM) and their pore size ranges were measured (Fig. 2a and b). The average pore size of the bulk hydrogels was found to be 47 μm with a range of 9–204 μm, in agreement with the reported values.36 On the other hand, the average pore size present in the IOPAL hydrogels was 77 μm with a range of 24–165 μm, in accordance with the size of the PMMA beads used (78.7 μm ± 1.7 μm). Although the pore size distribution was reduced in comparison with the bulk hydrogels thanks to the effect of the porogen, it was significantly larger than the standard deviation of the PMMA beads. This outcome can be explained by the porous nature of the material, which results from the composition and concentration of its two components, PEG and heparin. In principle, one could narrow this distribution by increasing the percentage of PEG.36 In this case, the pore diameter of ca. 80 μm was chosen, as it is expected to result in an almost unconstrained migration, similar to the effect of collagen fibers in the LNs.41 Furthermore, it is known that T cells aggregate forming clusters during the activation stage with Dynabeads.47 Thus, having a 3D environment that allows such aggregates is expected to be beneficial for T cell proliferation. As this aggregation is rather inhomogeneous, having a few pores that are larger might be beneficial to support its growth. It is also worth mentioning that the pore size dictated by the opal structure could easily be tuned at the microscale by varying the diameter of the PMMA beads, as previously shown in a similar system.41 However, a few parameters should be contemplated. As mentioned above, the intrinsic porosity of the hydrogels should be considered, especially for the smaller pore sizes, whereas potential structure collapses may affect IOPAL hydrogels with larger pore sizes. In both cases, higher concentrations of PEG would help minimize these issues by reducing the inherent porosity of PEG–Hep hydrogels and increasing their stiffness, respectively.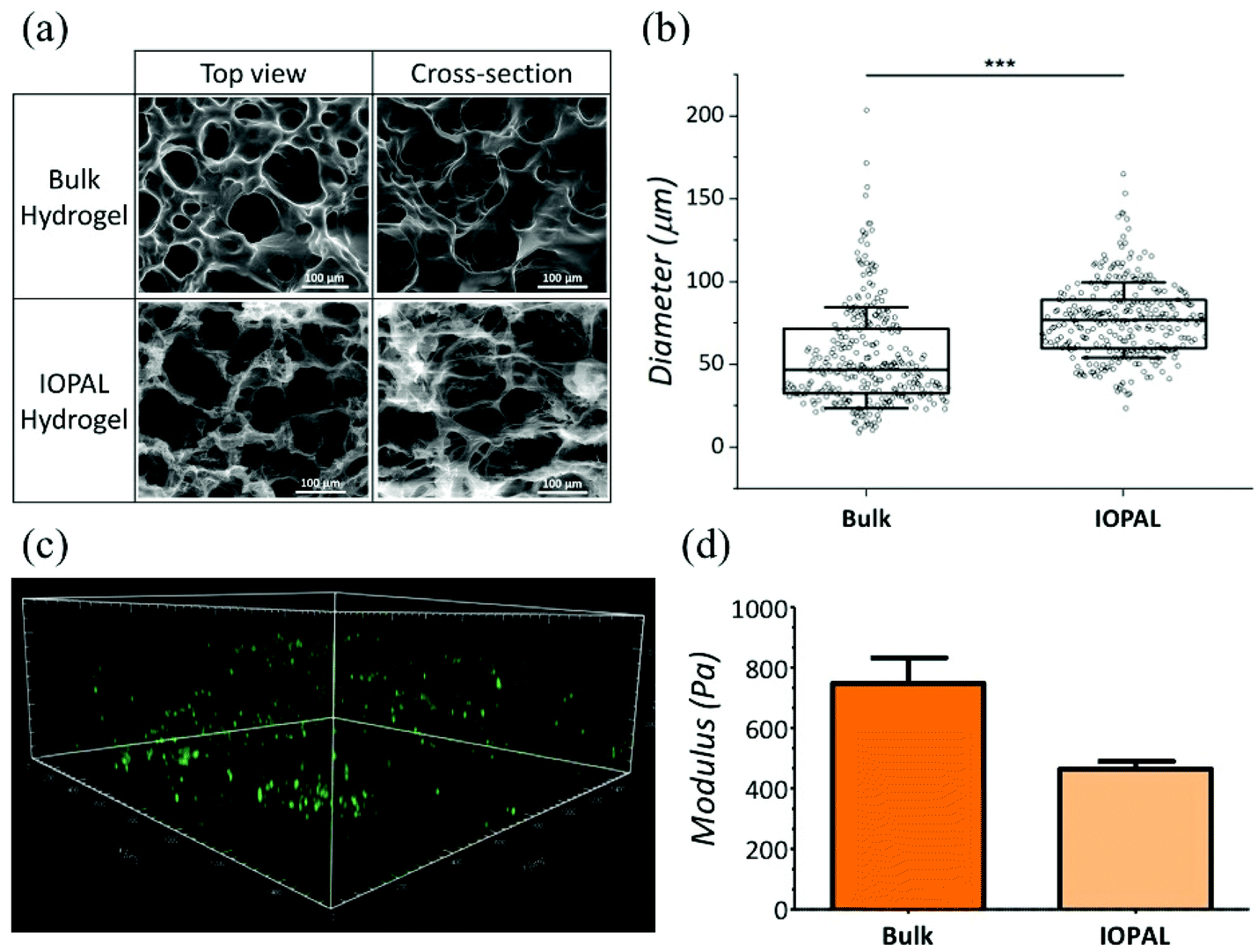 | ||
| Fig. 2 Structural properties of IOPAL PEG–Hep hydrogels. (a) ESEM images and (b) pore size evaluation of bulk and IOPAL PEG–Hep hydrogels. The statistical significance was determined by the Mann–Whitney U test (***p < 0.001). (c) 3D confocal projection showing CFSE-stained primary human CD4+ T cells in a representative IOPAL PEG–Hep hydrogel after 5 days of incubation (area = 1.5 cm × 1.5 cm × 0.4 cm). (d) Storage modulus (G′) of bulk (NHydrogels = 2) and IOPAL (NHydrogels = 2) hydrogels. Bars are mean + standard deviation. | ||
In addition to pore size, the connectivity of the hydrogels is crucial for T cell migration and interaction with Dynabeads in order to improve T cell proliferation. Interestingly, the connectivity density of the IOPAL system was found to be four times superior compared to that of the bulk hydrogel by X-ray microtomography (Fig. S1†). This result is in accordance with the expected nature of the IOPAL structures, which show interconnected pores.40
Additionally, T cell infiltration in the IOPAL hydrogels was confirmed by confocal microscopy. Specifically, primary human CD4+ T cells from adult donors were stained with CFSE, which is an intracellular dye that is only fluorescent in viable cells, and it is widely used for proliferation studies (Fig. 2c). In these hydrogels, we believe that human T cells, with a diameter of ca. 7 μm,35 can infiltrate by the effect of gravity and then move through the interconnected IOPAL structure (pore diameter of 78 μm) with an almost unconstrained migration. In addition, the Dynabeads, which are also infiltrating through the porous structure of the hydrogels, may facilitate the process (see ESI video†).
3.3 Mechanical properties of the IOPAL PEG–Hep hydrogels
The mechanical properties of IOPAL PEG–Hep hydrogels were characterized by small-amplitude oscillatory shear (SAOS) rheology (Fig. 2d and S2†) in the LVE regime.48 The storage modulus (G′) obtained for the IOPAL hydrogels was of 0.46 ± 0.01 kPa, which is slightly lower than the one of the bulk hydrogel (0.75 ± 0.08 kPa),36 as expected. This small difference can be explained by the homogeneously larger and more interconnected pores of the IOPAL structure compared to the bulk. Taking into account the intrinsic variability of the measurements, these values can be considered comparable to previously reported lymphoid tissues (1.5–3.0 kPa),49,50 although higher values were also found for LNs in breast and thyroid cancers.51,52 It is also worth mentioning that cells also contribute to tissue stiffening, which may further reduce differences once the hydrogels are loaded.3.4 Primary human CD4+ T cell culture using IOPAL PEG–Hep hydrogels
To determine the influence of the increased porosity and interconnectivity given by the IOPAL PEG–Hep hydrogels compared to the bulk ones, primary human CD4+ T cell viability, differentiation, and proliferation were measured by flow cytometry at days 5 or 6, as standard time points. Before each flow cytometry measurement, cells were carefully removed from the hydrogels through vigorous pipetting in order to maximize cell recovery. In both cases, T cells were polyclonally stimulated with the artificial APCs Dynabeads (ratio 1:1), which were maintained in culture until measurements. A positive control consisting of T cells with Dynabeads in suspension, i.e. the state-of-the-art T cell expansion methodology, was also included for comparison. It is also worth mentioning that primary human CD4+ T cells from adult donors were used to test the performance of the IOPAL PEG–Hep hydrogels for two main reasons. First, there is a growing body of research that is pointing out the usefulness of CD4+ T cells in the clinics.10–16 Second, CD4+ T cells is an abundant cell population in comparison with other relevant T cell types such as CD8+ T cells.
A PI viability test was performed 5 days after seeding (Fig. 3). As shown by flow cytometry, more viable cells were detected in the IOPAL hydrogels (26.1% PI+) than in the bulk ones (31.8% PI+). In both hydrogels, cells were more viable than in suspension cultures (control+; 39.9% PI+). However, only the IOPAL hydrogels showed non-statistically significant differences with the negative control (cells without Dynabeads; 15.5% PI+). Thus, the IOPAL hydrogels are able to provide a cell viability similar than that of non-activated cells.
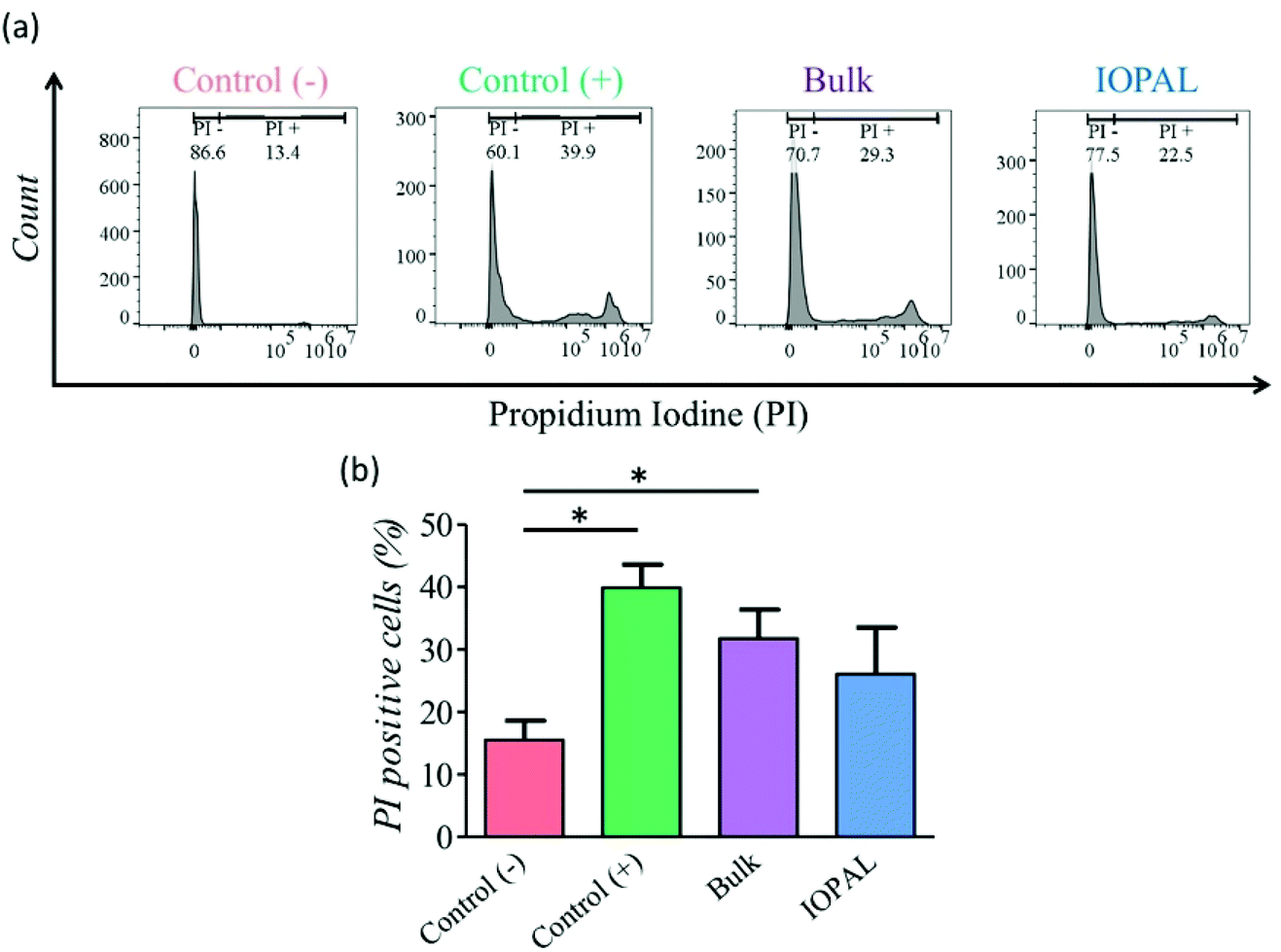 | ||
| Fig. 3 Effect of IOPAL and bulk PEG–Hep hydrogels on primary human CD4+ T cell viability. (a) Representative flow cytometry histograms of the (b) PI viability test performed to CD4+ T cells after 5 days of culture with Dynabeads in IOPAL and bulk PEG–Hep hydrogels, or in suspension (control (+)). Control (−) represents cells in suspension without Dynabeads. Bars are mean + standard deviation (Ndonors = 4). Significance was determined by the Mann–Whitney U test (*p < 0.05). | ||
The proliferation assay was performed using CFSE-stained cells analyzed by flow cytometry after 6 days of culture.53 Specifically, we analyzed the expansion index which gives information about the growth of the whole culture, as a ratio between the final and the starting number of cells. This parameter is therefore indicating the total number of cells achieved after culture, and thus, it is crucial for cell therapies. Additionally, we also evaluated the replication index, defined by the fold-expansion of the culture, but only taking into account the activated cells, and the proliferation index related with the average number of divisions that stimulated cells have undergone. The higher the proliferation index, the higher the cellular proliferative response provided by the culture environment. Consequently, these three parameters provide information about the cell response to the activation and proliferation stimuli (see ESI†).54 Given the donor-to-donor variability, the proliferation results (Fig. 4a–c) were normalized in each experiment to the positive control, consisting of CD4+ T cells with Dynabeads in suspension. For that reason, all the positive controls index values are 1. The non-normalized data can be found in Fig. S3.†
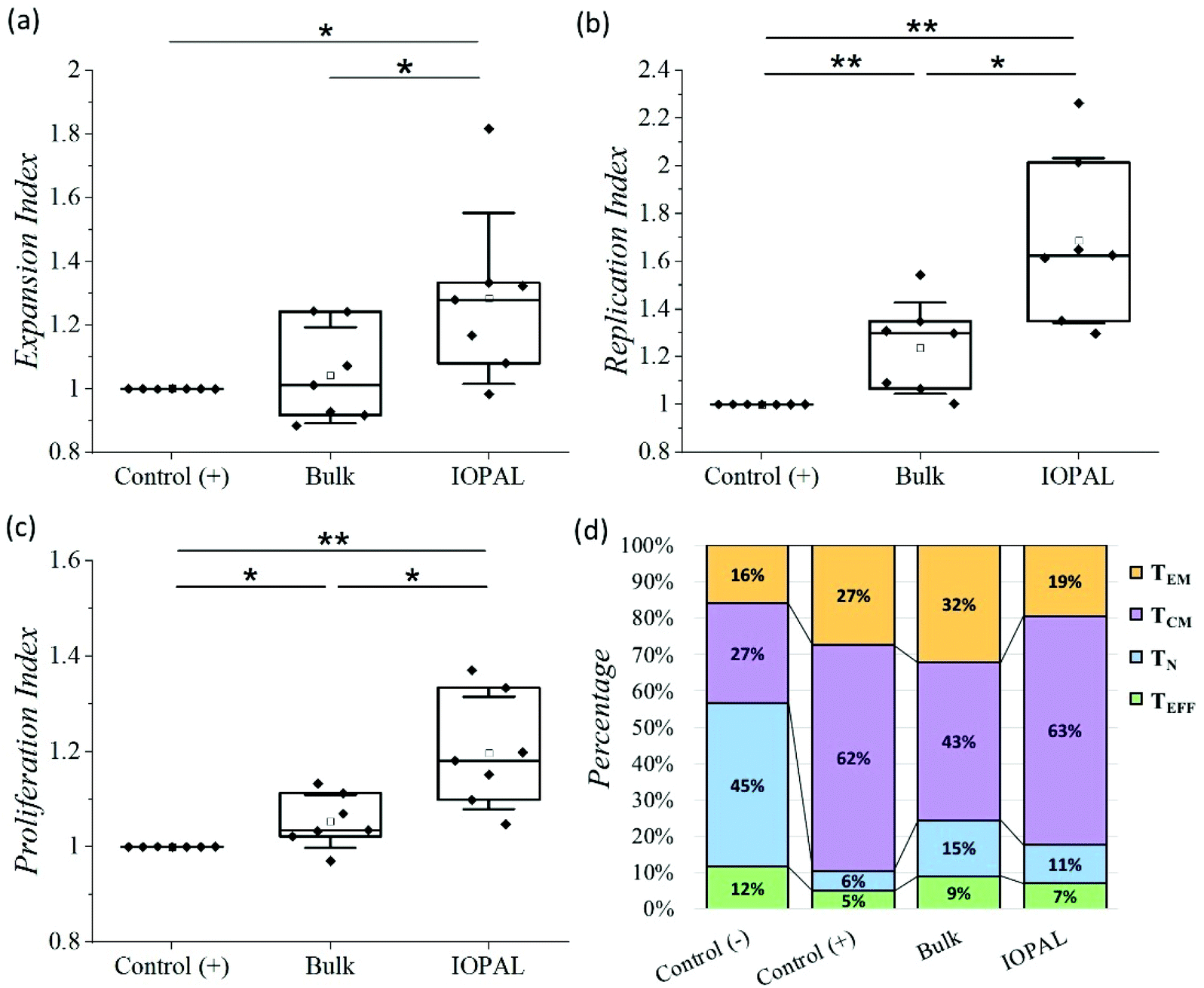 | ||
| Fig. 4 Effect of IOPAL and bulk PEG–Hep hydrogels on primary human CD4+ T cell proliferation. (a) Normalized expansion, (b) replication, and (c) proliferation indexes of CD4+ T cells stimulated with Dynabeads 6 days after seeding on bulk and IOPAL PEG–Hep hydrogels (Ndonors = 7). (d) Percentage of naïve (TN), effector (TEFF), effector memory (TEM), and central memory (TCM) CD4+ T cells on day 5 (Ndonors = 6). The negative control consists of cells seeded in suspension without Dynabeads, whereas in the positive control, cells are seeded in suspension with Dynabeads. Cells seeded in the hydrogels are always stimulated with Dynabeads. Statistical significance was determined by the Mann–Whitney U test (*p < 0.05, **p < 0.01). | ||
The median normalized proliferation, expansion, and replication indexes for the bulk hydrogels were 1.03, 1.01, and 1.29, respectively, in accordance with our previous results.36 The same three normalized indexes were significantly higher for the IOPAL hydrogels, being 1.18, 1.28, and 1.63. Both hydrogels showed statistically significant increases compared to the positive controls, except for the expansion index in the bulk hydrogel, even though a slight increase of 1% was observed. The major improvements were observed for the replication index with the IOPAL hydrogels. Specifically, these hydrogels resulted in an improvement of 63% compared to the cells activated in suspension (positive control) and an increase of 34% in comparison with the bulk hydrogels. This indicates that the responding cells that get activated in the hydrogels proliferate more than the activated cells in suspension. In summary, we can confirm that the increase in pore size, homogeneity, and interconnectivity introduced by the inverse opal strategy, contributed to an even better overall cell growth than the standard bulk hydrogels.
Finally, the phenotypes of the expanded primary human CD4+ T cells were analyzed after 5 days of culture. In particular, the subpopulations of naïve (TN; CD45RO−/CD62L+), effector (TEFF; CD45RO−/CD62L−), effector memory (TEM; CD45RO+/CD62L−), and central memory (TCM; CD45RO+/CD62L+) were quantified by flow cytometry (Fig. 4d, S4 and S5†), given their clinical importance in ACT.45
As mentioned above, there is a donor-to-donor variability that should be considered. For this reason, we also analyzed the percentages of CD4+ T cells that express CD45RO and CD62L without stimulation with Dynabeads (negative control). The main phenotype obtained in the negative control was TN with a median value of 45%. As expected, this subpopulation was found to be less preponderant when cells were activated. Specifically, the positive control, bulk, and IOPAL hydrogels showed TN percentages of 6, 15, and 11%, respectively. For the TEM phenotype, median values rose to 27% for T cells in suspension, 32% for bulk hydrogels, and 19% for IOPAL. Thus, the IOPAL hydrogels did not promote the TEM phenotype compared to suspension and the bulk hydrogels. In contrast, the TEFF phenotype resulted in lower percentages when cells were activated. A 5, 9, and 7% were obtained for the positive control, bulk, and IOPAL hydrogels, whereas the negative control showed a 12% of the total cell population. Remarkably, the median value for the TCM phenotype obtained in CD4+ T cells cultured in the IOPAL hydrogels was of 63%. This percentage is higher than the 43% obtained with the bulk hydrogels and comparable with the 62% obtained in suspension. It is worth mentioning that the TCM phenotype is of great importance for the clinics due to its capacity to mediate an effective and sustained response.
In conclusion, IOPAL hydrogels provide higher numbers of viable cells, but with less differentiated phenotypes. We suggest that this result is a consequence of the higher mobility of cells and beads inside the interconnected structure of the IOPAL hydrogels, which may favor more but shorter interactions between them. Thus, this system is ideal to achieve the requirements of the clinics, which are the ex vivo production of large numbers of cells with phenotypes able to persist in vivo, such as the central memory T cells.18–20
4. Conclusions
IOPAL 3D PEG–Hep hydrogels were synthesized, characterized, and used for primary human CD4+ T cell culture. Not only they resulted in an improvement in cell viability and proliferation when compared to the state-of-the-art methodologies, but also to its bulk form. This indicates the importance of pore size and interconnectivity on T cell activation, which we suggest that facilitates the interaction with Dynabeads. Moreover, we demonstrated the capacity of such hydrogels to influence the phenotype obtained, which is related to the clinical outcomes. Thus, these hydrogels have the potential to help surpassing the current limitation of ACT of producing large amounts of cells with therapeutic phenotypes. This limitation is especially important in ETC and ACT based on TILs, i.e. in therapies with non-engineered T cells. Additionally, these hydrogels could be explored for implantation, as both their components, PEG and heparin are approved for human use. Heparin is a widely used anticoagulant,55 whereas PEG has also various uses, especially in the cosmetic and pharmaceutical industries.56 Thus, both bulk and IOPAL PEG–Hep hydrogels could potentially be useful implants, e.g. as depots to deliver T cells in vivo after tumor resection to completely eliminate residual cancer cells, or even more interesting, for future in vivo ACT.57 Nevertheless, a complete study should previously be performed, which would include hydrogel degradability, in vivo biocompatibility, and T cell releasing capacity. Moreover, the IOPAL strategy could be applied to other hydrogels used in the growing field of 3D cell cultures for regenerative medicine and organoid engineering.Author contributions
Fabião Santos: conceptualization, methodology, investigation, formal analysis, writing – original draft, data curation, writing – review & editing; Julia Valderas-Gutiérrez: investigation, formal analysis, data curation; Eduardo Pérez del Río: investigation, writing – review & editing; Miquel Castellote-Borrell: investigation, writing – review & editing; Xavier Rodriguez Rodriguez: investigation; Jaume Veciana: funding acquisition, writing – review & editing; Imma Ratera: supervision, funding acquisition, writing – review & editing; Judith Guasch: conceptualization, methodology, validation, writing – original draft, writing – review & editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge D. P. Rosenblatt for proofreading this manuscript and “Servei de Microscòpia” from UAB for assistance in the confocal microscopy. This work has been developed inside the “Materials Science” PhD program of UAB. The authors are grateful for the financial support received from the Spanish Government (PID2020-115296RA-I00, PID2019-105622RBI00, and the “Ramón y Cajal” program (RYC-2017-22614)), the Networking Research Center on Bioengineering, Biomaterials, and Nanomedicine (CIBER-BBN) through the projects “Alycia” (No. BBN18PI01) and “Gels4ACT” (No. BBN20PIV02), as well as CSIC for a JAE Intro fellowship. The work was funded as well by the Max Planck Society through the Max Planck Partner Group “Dynamic Biomimetics for Cancer Immunotherapy” in collaboration with the Max Planck for Medical Research (Heidelberg, Germany). This research was also supported by the European Union's Horizon 2020 research and innovation programme H2020-MSCA-COFUND-2016 (DOC-FAM, grant agreement no. 754397). ICMAB-CSIC acknowledges support from the Severo Ochoa programme for centres of excellence in R&D (FUNFUTURE, CEX2019-000917-S).References
- C. Barbari, T. Fontaine, P. Parajuli, N. Lamichhane, S. Jakubski, P. Lamichhane and R. R. Deshmukh, Int. J. Mol. Sci., 2020, 21, 5009 CrossRef CAS PubMed.
- D. T. Harris and D. M. Kranz, Trends Pharmacol. Sci., 2016, 37, 220–230 CrossRef CAS PubMed.
- M. Saint-Jean, A.-C. Knol, C. Volteau, G. Quéreux, L. Peuvrel, A. Brocard, M.-C. Pandolfino, S. Saiagh, J.-M. Nguyen, C. Bedane, N. Basset-Seguin, A. Khammari and B. Dréno, J. Immunol. Res., 2018, 2018, 1 CrossRef PubMed.
- C. Yee, G. Lizee and A. J. Schueneman, Cancer J., 2015, 21, 492–500 CrossRef CAS PubMed.
- N. V. Frey, S. Gill, E. O. Hexner, S. Schuster, S. Nasta, A. Loren, J. Svoboda, E. Stadtmauer, D. J. Landsburg, A. Mato, B. L. Levine, S. F. Lacey, J. J. Melenhorst, E. Veloso, A. Gaymon, E. Pequignot, X. Shan, W.-T. Hwang, C. H. June and D. L. Porter, J. Clin. Oncol., 2020, 38, 2862–2871 CrossRef CAS PubMed.
- F. L. Locke, A. Ghobadi, C. A. Jacobson, D. B. Miklos, L. J. Lekakis, O. O. Oluwole, Y. Lin, I. Braunschweig, B. T. Hill, J. M. Timmerman, A. Deol, P. M. Reagan, P. Stiff, I. W. Flinn, U. Farooq, A. Goy, P. A. McSweeney, J. Munoz, T. Siddiqi, J. C. Chavez, A. F. Herrera, N. L. Bartlett, J. S. Wiezorek, L. Navale, A. Xue, Y. Jiang, A. Bot, J. M. Rossi, J. J. Kim, W. Y. Go and S. S. Neelapu, Lancet Oncol., 2019, 20, 31–42 CrossRef CAS PubMed.
- R. Andersen, M. Donia, E. Ellebaek, T. H. Borch, P. Kongsted, T. Z. Iversen, L. R. Hölmich, H. W. Hendel, Ö. Met, M. H. Andersen, P. T. Straten and I. M. Svane, Clin. Cancer Res., 2016, 22, 3734–3745 CrossRef CAS PubMed.
- S. A. Rosenberg, J. C. Yang, R. M. Sherry, U. S. Kammula, M. S. Hughes, G. Q. Phan, D. E. Citrin, N. P. Restifo, P. F. Robbins, J. R. Wunderlich, K. E. Morton, C. M. Laurencot, S. M. Steinberg, D. E. White and M. E. Dudley, Clin. Cancer Res., 2011, 17, 4550–4557 CrossRef CAS PubMed.
- T. Lövgren, M. Wolodarski, S. Wickström, U. Edbäck, M. Wallin, E. Martell, K. Markland, P. Blomberg, M. Nyström, A. Lundqvist, H. Jacobsson, G. Ullenhag, P. Ljungman, J. Hansson, G. Masucci, R. Tell, I. Poschke, L. Adamson, J. Mattsson and R. Kiessling, OncoImmunology, 2020, 9, 1792058 CrossRef PubMed.
- R. E. Tay, E. K. Richardson and H. C. Toh, Cancer Gene Ther., 2021, 28, 5–17 CrossRef CAS PubMed.
- D. Y. Oh and L. Fong, Immunity, 2021, 54, 2701–2711 CrossRef CAS PubMed.
- L. Poncette, J. Bluhm and T. Blankenstein, Curr. Opin. Immunol., 2022, 74, 18–24 CrossRef CAS PubMed.
- S. E. Brightman, M. S. Naradikian, A. M. Miller and S. P. Schoenberger, J. Leukocyte Biol., 2020, 107, 625–633 CrossRef CAS PubMed.
- D. Sommermeyer, M. Hudecek, P. L. Kosasih, T. Gogishvili, D. G. Maloney, C. J. Turtle and S. R. Riddell, Leukemia, 2016, 30, 492–500 CrossRef CAS PubMed.
- C. J. Turtle, L.-A. Hanafi, C. Berger, M. Hudecek, B. Pender, E. Robinson, R. Hawkins, C. Chaney, S. Cherian, X. Chen, L. Soma, B. Wood, D. Li, S. Heimfeld, S. R. Riddell and D. G. Maloney, Sci. Transl. Med., 2016, 8, 355ra116 Search PubMed.
- C. J. Turtle, L.-A. Hanafi, C. Berger, T. A. Gooley, S. Cherian, M. Hudecek, D. Sommermeyer, K. Melville, B. Pender, T. M. Budiarto, E. Robinson, N. N. Steevens, C. Chaney, L. Soma, X. Chen, C. Yeung, B. Wood, D. Li, J. Cao, S. Heimfeld, M. C. Jensen, S. R. Riddell and D. G. Maloney, J. Clin. Invest., 2016, 126, 2123–2138 CrossRef PubMed.
- R. K. Iyer, P. A. Bowles, H. Kim and A. Dulgar-Tulloch, Front. Med., 2018, 5, 1–9 CrossRef PubMed.
- A. D. Fesnak, C. H. June and B. L. Levine, Nat. Rev. Cancer, 2016, 16, 566–581 CrossRef CAS PubMed.
- A. Isser, N. K. Livingston and J. P. Schneck, Biomaterials, 2021, 268, 120584 CrossRef CAS PubMed.
- A. M. Krackhardt, B. Anliker, M. Hildebrandt, M. Bachmann, S. B. Eichmüller, D. M. Nettelbeck, M. Renner, L. Uharek, G. Willimsky, M. Schmitt, W. S. Wels and M. Schüssler-Lenz, Cancer Immunol. Immunother., 2018, 67, 513–523 CrossRef CAS PubMed.
- A. Grakoui, S. K. Bromley, C. Sumen, M. M. Davis, A. S. Shaw, P. M. Allen and M. L. Dustin, Science, 1999, 285, 221–227 CrossRef CAS PubMed.
- M. F. Bachmann, K. McKall-Faienza, R. Schmits, D. Bouchard, J. Beach, D. E. Speiser, T. W. Mak and P. S. Ohashi, Immunity, 1997, 7, 549–557 CrossRef CAS PubMed.
- W. K. Wong, B. Yin, A. Rakhmatullina, J. Zhou and S. H. D. Wong, Eng. Regener., 2021, 2, 70–81 Search PubMed.
- H. Cai, J. Muller, D. Depoil, V. Mayya, M. P. Sheetz, M. L. Dustin and S. J. Wind, Nat. Nanotechnol., 2018, 13, 610–617 CrossRef CAS PubMed.
- E. Tabdanov, S. Gondarenko, S. Kumari, A. Liapis, M. L. Dustin, M. P. Sheetz, L. C. Kam and T. Iskratsch, Integr. Biol., 2015, 7, 1272–1284 CrossRef CAS PubMed.
- J. Deeg, M. Axmann, J. Matic, A. Liapis, D. Depoil, J. Afrose, S. Curado, M. L. Dustin and J. P. Spatz, Nano Lett., 2013, 13, 5619–5626 CrossRef CAS PubMed.
- J. Matic, J. Deeg, A. Scheffold, I. Goldstein and J. P. Spatz, Nano Lett., 2013, 13, 5090–5097 CrossRef CAS PubMed.
- J. Guasch, M. Hoffmann, J. Diemer, H. Riahinezhad, S. Neubauer, H. Kessler and J. P. Spatz, Nano Lett., 2018, 18, 5899–5904 CrossRef CAS PubMed.
- J. Guasch, C. A. Muth, J. Diemer, H. Riahinezhad and J. P. Spatz, Nano Lett., 2017, 17, 6110–6116 CrossRef CAS PubMed.
- X. Shou, H. Zhang, D. Wu, L. Zhong, D. Ni, T. Kong, Y. Zhao and Y. Zhao, Small, 2021, 17, 2006955 CrossRef CAS PubMed.
- A. Trickett and Y. L. Kwan, J. Immunol. Methods, 2003, 275, 251–255 CrossRef CAS PubMed.
- T. Cupedo, A. Stroock and M. Coles, Front. Immunol., 2012, 3, 1–6 Search PubMed.
- S. N. Mueller and R. N. Germain, Nat. Rev. Immunol., 2009, 9, 618–629 CrossRef CAS PubMed.
- A. Singh and N. A. Peppas, Adv. Mater., 2014, 26, 6530–6541 CrossRef CAS PubMed.
- E. Pérez del Río, M. Martinez Miguel, J. Veciana, I. Ratera and J. Guasch, ACS Omega, 2018, 3, 5273–5280 CrossRef PubMed.
- E. Pérez del Río, F. Santos, X. R. Rodriguez, M. Martínez-Miguel, R. Roca-Pinilla, A. Arís, E. Garcia-Fruitós, J. Veciana, J. P. Spatz, I. Ratera and J. Guasch, Biomaterials, 2020, 259, 120313 CrossRef PubMed.
- D. Aydin, I. Louban, N. Perschmann, J. Bluemmel, T. Lohmueller, E. A. Cavalcanti-Adam, T. L. Haas, H. Walczak, H. Kessler, R. Fiammengo and J. P. Spatz, Langmuir, 2010, 26, 15472–15480 CrossRef CAS PubMed.
- S. J. Paluck, T. H. Nguyen and H. D. Maynard, Biomacromolecules, 2016, 17, 3417–3440 CrossRef CAS PubMed.
- M. C. Z. Meneghetti, A. J. Hughes, T. R. Rudd, H. B. Nader, A. K. Powell, E. A. Yates and M. A. Lima, J. R. Soc., Interface, 2015, 12, 20150589 CrossRef PubMed.
- Y. S. Zhang, C. Zhu and Y. Xia, Adv. Mater., 2017, 29, 1701115 CrossRef PubMed.
- A. N. Stachowiak and D. J. Irvine, J. Biomed. Mater. Res., Part A, 2008, 85, 815–828 CrossRef PubMed.
- X. Shou, Y. Liu, D. Wu, H. Zhang, Y. Zhao, W. Sun and X. Shen, Chem. Eng. J., 2021, 408, 127349 CrossRef CAS.
- Y. S. Zhang, S.-W. Choi and Y. Xia, Soft Matter, 2013, 9, 9747–9754 RSC.
- S. Krishna, F. J. Lowery, A. R. Copeland, E. Bahadiroglu, R. Mukherjee, L. Jia, J. T. Anibal, A. Sachs, S. O. Adebola, D. Gurusamy, Z. Yu, V. Hill, J. J. Gartner, Y. F. Li, M. Parkhurst, B. Paria, P. Kvistborg, M. C. Kelly, S. L. Goff, G. Altan-Bonnet, P. F. Robbins and S. A. Rosenberg, Science, 2020, 370, 1328–1334 CrossRef CAS PubMed.
- A. L. Garfall, E. K. Dancy, A. D. Cohen, W.-T. Hwang, J. A. Fraietta, M. M. Davis, B. L. Levine, D. L. Siegel, E. A. Stadtmauer, D. T. Vogl, A. Waxman, A. P. Rapoport, M. C. Milone, C. H. June and J. J. Melenhorst, Blood Adv., 2019, 3, 2812–2815 CrossRef CAS PubMed.
- A. N. Stachowiak, A. Bershteyn, E. Tzatzalos and D. J. Irvine, Adv. Mater., 2005, 17, 399–403 CrossRef CAS.
- S. Adutler-Lieber, I. Zaretsky, H. Sabany, E. Kartvelishvily, O. Golani, B. Geiger and N. Friedman, Blood Adv., 2017, 1, 1016–1030 CrossRef CAS PubMed.
- J. M. Zuidema, C. J. Rivet, R. J. Gilbert and F. A. Morrison, J. Biomed. Mater. Res., Part B, 2014, 102, 1063–1073 CrossRef PubMed.
- S. Hirsch, J. Guo, R. Reiter, S. Papazoglou, T. Kroencke, J. Braun and I. Sack, Magn. Reson. Med., 2014, 71, 267–277 CrossRef PubMed.
- F. N. U. Apoorva, Y. F. Tian, T. M. Pierpont, D. M. Bassen, L. Cerchietti, J. T. Butcher, R. S. Weiss and A. Singh, J. Biomed. Mater. Res., Part A, 2017, 105, 1833–1844 CrossRef CAS PubMed.
- S. J. Bae, J. T. Park, A. Y. Park, J. H. Youk, J. W. Lim, H. W. Lee, H. M. Lee, S. G. Ahn, E. J. Son and J. Jeong, J. Breast Cancer, 2018, 21, 190–196 CrossRef PubMed.
- J. You, J. Chen, F. Xiang, Y. Song, S. Khamis, C. Lu, Q. Lv, Y. Zhang and M. Xie, J. Med. Ultrason., 2018, 45, 251–259 CrossRef PubMed.
- A. B. Lyons and C. R. Parish, J. Immunol. Methods, 1994, 171, 131–137 CrossRef CAS PubMed.
- M. Roederer, Cytometry, Part A, 2011, 79A, 95–101 CrossRef PubMed.
- B. Mulloy, J. Hogwood, E. Gray, R. Lever and C. P. Page, Pharmacol. Rev., 2016, 68, 76–141 CrossRef PubMed.
- A. A. D'souza and R. Shegokar, Expert Opin. Drug Delivery, 2016, 13, 1257–1275 CrossRef PubMed.
- H. Ma, C. He and X. Chen, Macromol. Biosci., 2021, 21, 2100039 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm00486k |
| This journal is © The Royal Society of Chemistry 2022 |