
Antimicrobial mechanisms of biomaterials: from macro to nano
Shounak
Roy†
 a,
Sanchita
Sarkhel†
a,
Deepali
Bisht
a,
Samerender Nagam
Hanumantharao
b,
Smitha
Rao
b and
Amit
Jaiswal
*a
a,
Sanchita
Sarkhel†
a,
Deepali
Bisht
a,
Samerender Nagam
Hanumantharao
b,
Smitha
Rao
b and
Amit
Jaiswal
*a
aSchool of Basic Sciences, Indian Institute of Technology Mandi, Kamand, Mandi, Himachal Pradesh Pincode: 175075, India. E-mail: j.amit@iitmandi.ac.in
bDepartment of Biomedical Engineering, Michigan Technological University, Houghton, MI 49931, USA
First published on 3rd June 2022
Abstract
Overcoming the global concern of antibiotic resistance is one of the biggest challenges faced by scientists today, and the key to tackling this issue of emerging infectious diseases is the development of next-generation antimicrobials. The rapid emergence of multi-drug resistant microbes, superbugs and mutated strains of viruses has fuelled the search for new and alternative antimicrobial agents with broad-spectrum biocidal activity. Biomaterials, ranging from macroscopic polymers, proteins, and peptides to nanoscale materials such as nanoparticles, nanotubes and nanosheets have emerged as effective antimicrobials. An extensive body of research has established the antibacterial and antiviral efficiencies of different types of biomaterials. What make these materials unique are the different modes through which they interact and exert their antimicrobial activity. This review provides a comprehensive and detailed overview of the diverse modes of interaction between biomaterials and bacteria and viruses, and sheds light on how different biomaterials influence and modulate antimicrobial mechanisms to achieve a high degree of therapeutic efficacy without resistance generation.
 Shounak Roy | Shounak Roy received a Master of Science degree in genetic engineering from the West Bengal University of Technology, India, in 2013. Following this, he received a Master of Technology degree in Biotechnology from the Birla Institute of Technology Mesra, Ranchi, India, in 2015. He received the INSPIRE fellowship from the Department of Science and Technology, Government of India in 2017 to pursue a Ph.D. Currently, he is a Ph.D. candidate at the Indian Institute of Technology, Mandi in the School of Basic Sciences under the supervision of Dr Amit Jaiswal. His main research interests include the synthesis of novel 2D material-based nanocomposites for the development of cancer therapeutics and nano-antimicrobials. |
 Sanchita Sarkhel | Sanchita Sarkhel did her Bachelor's degree in Microbiology at the University of Calcutta. After that, she completed her Master's degree in Microbiology from St. Xavier's College, Kolkata. Currently, she is pursuing her Ph.D. at the Indian Institute of Technology, Mandi, under the guidance of Dr Amit Jaiswal. Her primary research interest lies in biomaterials with antimicrobial and wound healing properties. |
 Deepali Bisht | Deepali Bisht received her Bachelor of Technology degree in Biotechnology from IMS Engineering College, UP, Ghaziabad, India, in 2019. In 2020, she qualified with GATE Biotechnology, a MHRD-funded scholarship in India and is currently pursuing her Master of Technology degree from the Indian Institute of Technology, Mandi, in the School of Basic Sciences. She is working under the supervision of Dr Amit Jaiswal for her Master's thesis, and her research interest includes 2D nanomaterials and their antimicrobial action mechanism. |
 Samerender Nagam Hanumantharao | Dr Hanumantharao is currently a Research Scientist at Neurocarrus Inc., Lincoln, Nebraska, USA. He received his M.S. and Ph.D. in Biomedical Engineering from Michigan Technological University under the supervision of Dr Smitha Rao. He finished his postdoctoral training in the Biomedical Microdevices Lab at Michigan Technological University. His research interests include the synthesis of nanomaterials, drug delivery, tissue engineering and cancer biology. |
 Smitha Rao | Dr Smitha Rao is an Assistant Professor in the Department of Biomedical Engineering at Michigan Technological University. Dr Rao received her M.S. and Ph.D. in Electrical Engineering from UT Arlington, Texas, USA. Dr Rao is a senior member of IEEE, and her research interests include micro- and nano-devices and structures, wound healing, and cancer growth and migration. |
 Amit Jaiswal | Dr Amit Jaiswal is presently an Associate Professor in the School of Basic Sciences at the Indian Institute of Technology, Mandi, India. Dr Jaiswal is also an associate of the Indian Academy of Sciences, Bangalore, India. He completed his Ph.D. in Nanotechnology at the Indian Institute of Technology, Guwahati, India, and post-doctoral research at Washington University in St. Louis, USA, and Technion – Israel Institute of Technology, Haifa, Israel. His research interest is in the synthesis of nanomaterials for sensing, catalysis, drug delivery and diagnostic applications. |
1. Introduction
Recently, the increasing numbers of fatalities related to infectious diseases1–4 have been higher than those attributable to terrorism, weapons of mass destruction and wars combined. This has led to global concern and a shift in focus towards identification, prevention, and treatment of the diseases,5,6 particularly air-borne respiratory diseases, antibiotic-resistant bacterial infections (ARBI) and what are known as super bugs.7 Traditional therapies used against the ARBI have combined different antibiotics, while vaccinations are used to prevent viral infections.8–10 However, the effectiveness of a combination of antibiotics differs, with possible side effects, and hence cannot be optimized. Currently available antiviral targets are very specific to the virus and have differential results based on clinical experience.11 Thus, the widespread emergence of ARBI and novel strains of virus such as SARS-CoV-2 has demonstrated the need for novel techniques for prevention, diagnosis, and therapies.Biomaterials, from macromolecules such as polymers and proteins to nanoparticles belonging to different dimensions, have emerged as promising antimicrobial alternatives to the existing therapeutics. Even before the emergence of antibiotics, antimicrobial peptides (AMP) played a big role in host defense against infection. And for the same reason they are also referred to as host defense peptides. AMPs are low molecular weight peptides of 8–100 amino acids, mainly cationic and amphipathic in nature, and show broad-spectrum activity. They are present almost ubiquitously from prokaryotes to eukaryotes.12 The discovery of antibiotics13 overshadowed the importance of AMPs to an extent, but the rapid emergence of antibiotic resistance among pathogens has brought back attention to AMPs again as a potential agent for antimicrobial activity. Because of some drawbacks like stability, hemolytic activity etc., naturally occurring AMPs have limited applications. To overcome this problem, various synthetic analogs of AMPs like cationic peptides and peptide mimics, and macromolecules like polymers and peptidopolysaccharides have come into play. Some naturally occurring biomolecules like polysaccharides have intrinsic antibacterial activity, whereas others have been developed through chemical modifications of the natural polymeric backbone.14 Cationic peptides and synthetic peptide mimics, which basically mimic the backbone of naturally occurring AMPs but have additional functional groups attached to them, also make for very potent antibacterial biomaterials.15 Furthermore, peptidopolysaccharides, which are combinations of peptides and polysaccharides, possess the ability to mimic the bacterial peptidoglycan layer and show antibacterial activity by interfering with bacterial cell wall synthesis.16
Similarly, there have been tremendous developments in the field of nanoscience in the past decade in the development of next-generation antimicrobials. The definition of nanomaterials is different based on the legislating organizations. The general definition classifies materials of the scale of 1–100 nm in at least one dimension and exhibiting properties distinct from their bulk counterpart as nanomaterials. The nanomaterials can have different structures: spheres, rods, wires, ribbons, tubes, scaffolds, fibers, beads or sheets.17 Further, their origin can be carbon based, inorganic, organic, or composite based. The size-dependent properties and vastly different interactions with their environment promotes the manipulation and use of nanomaterials as anti-microbials.18
In addition to bacteria, viruses are another major cause of global health concern.19 Viruses are protein-coated particles with DNA or RNA as their genetic material and have both living and non-living characteristics. To behave like a living entity, the virus needs to enter its host cell and replicate. Viral replication has six major steps starting from viral adsorption on the host cell surface to new virion release. The traditional antiviral therapies which include antiviral drugs like Acyclovir and Remdesivir20 specifically target viral enzymes involved in viral DNA or RNA synthesis or inhibit proteases, but the problem with such therapies is that viruses quickly mutate and generate resistance against these agents. To address this difficulty, biomaterials have again emerged as an immensely good alternative. Especially, the polyvalency of peptides or polymers gives them diverse antiviral functionalities, which poses difficulty for viruses to develop resistance against them easily.21,22 In parallel, nanoscience also has a promising perspective when it comes to antiviral agents. Different shapes, sizes and chemistry of nanoparticles attack different steps of the viral replication and infection machinery, thereby assaulting viruses from multiple fronts.
Several studies have been carried out so far that have established the broad-spectrum antibacterial and antiviral properties of polymers, peptides and nanoscale biomaterials against a wide variety of bacteria and viruses. These studies have provided readers with detailed insights into the nature of interactions of the wide varieties of biomaterials with different classes of bacteria and viruses, both at structural and molecular level, and how such interactions contribute towards the observed antimicrobial activity. However, it is interesting to note that biomaterials, depending on their class, follow different modes of antimicrobial activity. Several mechanisms of action of these biomaterials have been proposed in the past by independent studies, each shedding light on a different aspect of a particular material's interaction with microbes. Direct physical contact-mediated membrane disruption through pore-forming or non-pore-forming pathways, and oxidative stress causing membrane damage and subsequent cellular oxidation of biomolecules have been proposed as mechanisms of antimicrobial activity.23
The key focus of this review is to provide comprehensive and up-to-date knowledge about the current understanding on how biomaterials ranging from macroscale to nanoscale exert their antibacterial and antiviral actions, and specifically present the readers with an organized overview of their different modes of action. However, discussions, comparisons and evaluations of antibacterial and antiviral efficiencies of different biomaterials in terms of their therapeutic dosage are beyond the scope of this review.
2. Antimicrobial biomaterials
2.1 Macroscale biomaterials
AMPs are internalized through a non-receptor-mediated pathway. Broadly, they can be classified into naturally occurring and chemically synthesized AMPs, but both of them follow more or less the same mechanism of action. The first ever naturally occurring AMP which was reported in humans was lysozyme from nasal mucous. Presently, more than 2500 AMPs have been reported in the Antimicrobial Peptide Database.12 Magainin II and β-defensins are some of the most common AMPs that are synthesized as host defense peptides when cells undergo microbial infection.26,27 Beside immune system-stimulated AMPs, constitutively expressed AMPs are also present in cells and they can be secreted from various sites where microbial infection is possible, such as the oral cavity (Defensins and Cathelicidin),28 gastrointestinal tract (α-Defensins HD-5 and HD-6),29 skin, eye, respiratory tract, and reproductive tract. However, commercial use of naturally occurring AMPs is very limited because of their instability, expensive extraction from host cells and haemolytic activity. To overcome these shortcomings, biological and chemical syntheses of AMPs have been attempted. Biological synthesis of AMPs is much more difficult because it is a time-consuming, tedious and expensive purification process with low yield. Hence, chemical synthesis methods like solid phase synthesis, liquid phase peptide synthesis, and α-amino acid N-carboxyanhydrides ring-opening polymerization (NCA-ROP) peptide synthesis are choices of interest. The first amphipathic polypeptide which was synthesized using the NCA process was P(K12.5F12.5), in which the hydrophilicity was rendered by the cationic lysine residues, whereas the hydrophobicity was provided by L-phenylalanine, L-alanine and L-leucine residues.30 Another synthetic AMP with better cytocompatibility is peptide-g-polymer 6.31 The less positive charge of this peptide makes it less cytotoxic and less haemolytic towards human cells. A series of poly(4-vinyl-N-alkylpyridinium) with different linear alkyl chains ranging from propyl to hexadecyl was developed by Tiller and co-workers, and showed significant bactericidal activity against Staphylococcus aureus.32
 | ||
| Fig. 1 Schematic showing different classes of antimicrobial biomaterials. | ||
2.2 Nanoscale biomaterials or nanomaterials
Gleiter et al. classified nanostructures based on their crystallinity and microstructural features, introducing grain boundary engineering without factoring in the dimensionality.42 The classification was further developed by Pokropivny et al. by using dimensionality of the nanostructures.43 Pokropivny and Skorohod's method of classification can be used to classify most nanostructures except a few which demonstrate the properties of more than one class. Based on the number of dimensions that lie outside the nanoscale range, nanomaterials can be classified as 0-D, 1-D, 2-D, and 3-D material (Fig. 1).43 When all the dimensions of a material are within the nanoscale range (≤100 nm), such a material falls in the category of 0-D materials. Isotropic nanoparticles like spherical nanoparticles and quantum dots, nanocubes, decahedrons, octahedrons, and icosahedrons belong to this category. Materials in which two dimensions are within nanoscale range and one dimension falls outside the nanoscale are termed 1-D materials. Structures with highly anisotropic morphologies such as nanotubes, nanorods, and nanowires fall under this class. In the case of 2-D materials, one of the dimensions is within the nanoscale, and the other two are outside the nanoscale range. Such structures usually take the form of nanosheets, nanoplates, nanofilms etc., which are formed in the kinetically driven regime, where the growth is allowed on two axes while restricted along the third axis. In addition to these three typical morphologies of nanomaterials, there is a fourth class known as 3-D nanomaterials, where the assembly of either one or more types of previously mentioned nanostructures happens to form a complex nanostructure. Such 3-D nanostructures display properties similar to those of their 1-D or 2-D components.3. Antimicrobial mechanisms of macroscale biomaterials
3.1 Mechanism of antibacterial action of AMPs
Various studies from the last few decades have shown that the mode of action of antimicrobial peptides can be broadly classified into three major sub-types, which include pore-forming mechanisms of membrane damage, non-pore-forming mechanisms of membrane damage, and mechanisms that target intracellular processes.(a) Barrel stave model. When an AMP comes into contact with a phospholipid bilayer it forms an amphiphilic secondary structure of α-helixes and causes augmentation in the interfacial region of the membrane. This results in the creation of a void in the hydrocarbon tail region, which leads to generation of a positive curvature strain and membrane thinning on the opposite side.61 Following this, the AMP vertically inserts itself into the phospholipid bilayer, creating a closely compact pore in which the hydrophobic residues of the peptide remain in close contact with the hydrophobic interior of the bilayer (Fig. 2a).62 In 1991, Sansom et al. reported that Peptaibols, peptides with aminoisobutyric acid residues with C-terminal alcohol, showed antibacterial properties following the barrel stave model.63
 | ||
| Fig. 2 Schematic of different models showing "pore-forming" mechanisms of membrane damage by antimicrobial macromolecules. (a) Barrel-stave model, (b) Torroidal pore model, (c) Dirodered Toroidal pore model, (d) Carpet model, (e) Detergent like model, (f) Interfacial model and (g) Electroporation model. | ||
(b) Toroidal model. Unlike the barrel stave model, in the toroidal model the pore lining is formed between the polar residues of the AMPs and the polar head groups of phospholipids (Fig. 2b). In view of this fact, not only can small molecules and ions pass through these pores but also phospholipid itself can pass through these pores, and can even flip-flop at high speed.64 It has also been observed that a part of the peptide itself can translocate inside the phospholipid bilayer. In 1996, this model was first established during the study of magainin-induced membrane pores,65 where at high magainin concentrations water-filled cavities were formed which looked like ‘worm-holes’. Hence, this model is also known as the Worm-hole model.
(c) Disordered toroidal pore model. Molecular dynamics simulations have shown that while interacting with dipalmitoylphosphatidylcholine (DPPC) membranes, Magainin H2 causes more random pore formation at multiple places through the inward twisted conformation of membrane phospholipids. Such pores are referred to as disordered toroidal pores, where one or two peptides can be present in the pore interior, while the rest of the AMPs remain in the pore lining66 (Fig. 2c).
(d) Carpet model. At high peptide-to-lipid ratio and in the presence of negatively charged phospholipids, some AMPs tend to accumulate together and adsorb at high concentrations on the phospholipid bilayer, resembling a carpet.61 As a result of this typical arrangement, the electrostatic repulsive forces existing between positively charged peptides get reduced or diminished to a large extent, which ultimately leads to lysis of the membrane (Fig. 2d).67 This model was first proposed by Shai in the year of 1996 while explaining the antibacterial action of MOA of mammalian cecropin P1 on model membranes.68
(e) Detergent-like model. This model is very much applicable to explain the activities of amphiphilic peptides on membranes. Similar to detergents, amphiphilic peptides form micelles with the lipid bilayer at concentrations above their critical micelle concentration (CMC) and form aggregates (Fig. 2e). This property greatly depends on the detergent/AMP-to-lipid ratio. For example, in the presence of very little detergent there is no negative effect on the membrane; rather, it stabilizes it. However, at intermediate concentrations it starts to form small transient pores, which at high concentrations of the detergent results in disintegration of the lipid bilayer. AMPs can exist both as monomers and oligomers. When present as oligomers, AMPs may act like detergent and form micelles with the lipid bilayer, causing disintegration, loss of membrane barrier, dissolution of the electrostatic gradient across the membrane, interference in energy metabolism of living cells and finally loss of cytoplasm and its constituents.67 Cecropin B is a very good example of an AMP showing bactericidal activity through this detergent-like model.69
(f) Interfacial activity model. The core of the lipid bilayer membrane is one of the most hydrophobic microenvironments present in nature. It acts as a permeability barrier for polar or charged solutes, but itself is surrounded by two bilayer interfacial zones named “zones of tumultuous chemical heterogeneity”.70 When a peptide molecule disturbs the hydrophobic core region of a phospholipid bilayer by interfering in the interfacial region of the bilayer, it causes local rearrangements in vertical lipid packing. As a result of this, separation of the interfacial groups takes place, and the hydrocarbon core region is lost (Fig. 2f). This can be described as a peptide's interfacial activity.25 This model is mainly dependent on the amino acid sequence of the AMP rather than its peptide structure. It requires AMPs to have “imperfect amphipathicity”, where instead of a large hydrophobic segment, such segments are present in the AMP structure which are large enough to traverse the phospholipid bilayer but are interrupted by at least polar residues like arginine or lysine. Such types of AMP translocate through the bilayer along with lipid molecules even at low peptide concentrations, and at higher concentration this causes membrane leakage.25 AMPs like cyclic AMP Rhesus theta Defensin, helical AMP Xenopus Magainin 2, globular peptide Human α-defensin and Human β-defensin, etc. show membrane disruption through the interfacial activity model, independent of their structures but based on their amino acid sequences.
(g) Electroporation model. It is observed that when highly charged molecules bind with the phospholipid bilayer, electrostatic potential is developed on the membrane. If the electrostatic potential is at least 0.2 V across the phospholipid bilayer, then it can create pores across the membrane without changing its conformation (Fig. 2g). Through these pores small molecules and even the peptide itself can pass, as these pores have sizes of 2–4 nm in diameter.71 Miteva et al. reported such a mechanism of action in a single highly charged α-helical segment of NK-lysin.72
(a) Membrane thinning model. When peptides are aligned on the surface of the outer bilayer, they interact with the polar head groups of the phospholipid bilayer and expand the local area. To keep the volume constant, the length of the acyl chains decreases, hence causing membrane thinning (Fig. 3a). The thinning of the membrane depends on the elastic constants of the bilayer, which can extend over a range of approximately 40 Å.73 At a sufficiently high peptide concentration, the deformations created by each peptide will overlap and will create a uniform thinning of the membrane. Therefore, the overall thinning of the membrane is proportional to the peptide-to-lipid ratio. Peptides which are not able to form pores and insert into the bilayer, such as Mag2, GS, and BP100, follow this mechanism for their antimicrobial activity.74
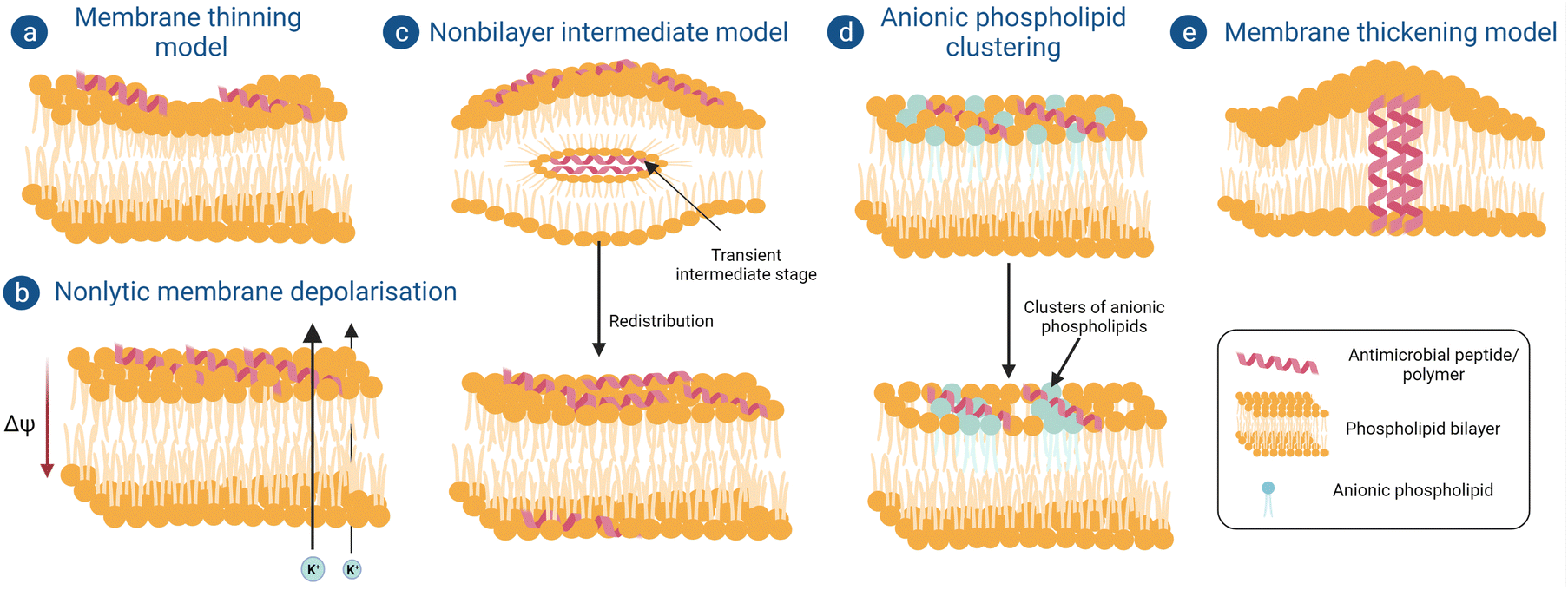 | ||
| Fig. 3 Schematic of different models showing “non-pore-forming” mechanisms of membrane damage by antimicrobial macromolecules. (a) Membrane thinning model, (b) Nonlytic membrane depolarization, (c) Nonbilayer intermediate model, (d) Anionic phospholipid clustering and (e) Membrane thickening model. | ||
(b) Non-lytic membrane depolarization. There are certain peptides which can cause bacterial membrane damage by depolarization without forming actual ion channels or pores. Huang et al. showed that when a cyclic lipopeptide daptomycin binds with the bacterial phospholipid bilayer it inserts its acyl fatty acid chain into the bacterial membrane in a calcium-dependent manner, thereby causing oligomerization of membrane proteins to form ion channels or pores, but this phase is very transient and recovers back quickly.75 Through these pores potassium ions leak, which causes a reduction in membrane potential from −165 mV to −100 mV; as a result of this, the electrochemical gradient-induced proton motif force, which is a prerequisite for adenosine triphosphate synthesis, gets disrupted (Fig. 3b). Adenosine triphosphate plays an important role in many intracellular processes like active transport of nutrients, synthesis of peptidoglycan precursors and many more cell signaling pathways.76
(c) Non-bilayer intermediate model. A few peptides follow another non-pore-forming pathway whereby they become internalized inside the cell without disrupting the membrane integrity (Fig. 3c).77 Powers et al. reported that an antimicrobial peptide, polyphemusin, translocates the lipid bilayer without breaking the integrity of it. Firstly, it interacts with the membrane mainly with the negatively charged polar head groups and partially enters the membrane. After insertion of a sufficient number of peptides, they aggregate and produce a negative curvature stress which forms a non-bilayer intermediate with the remaining peptides inside the hydrophobic core. Finally, this non intermediate core disrupts and the peptides in it redistribute themselves in the outer and inner leaflets of the phospholipid bilayer.78
(d) Anionic phospholipid clustering model. Cationic peptides can attach with the negatively charged polar head groups in the bacterial membrane and form clusters of anionic phospholipids (Fig. 3d). This charge clustering sometimes leads to the formation of transient pores in the membrane and causes infringement of the membrane barrier, leading to leakage of cytoplasmic contents and membrane depolarization. Such mechanism of antimicrobial action was reported for C12K-7α8 peptide.79
(e) Membrane thickening model. While studying the antimicrobial action of an α-helical antimicrobial peptide, peptidyl-glycyl-leucine-carboxyamide (PGLa)80 on a phosphatidyl glycerol model membrane, it was observed that initially they were not able to insert themselves completely inside the membrane. Rather, to match the hydrophobic part of the peptide, hydrophobic tails of the phospholipids shifted towards it, resulting in a local thickening of the membrane and causing bacterial membrane deformation (Fig. 3e). But after crossing a certain peptide concentration, these peptides start to insert themselves into the membrane vertically or at an oblique angle, eventually forming pores like the barrel stave model or toroidal model and causing bacteriolysis.
(a) Cell wall synthesis. There are antimicrobial peptides which target various intracellular pathways of the microorganism. Lantibiotics are a group of lanthionines with antimicrobial peptides against Gram-positive bacteria. Among these Mersacidin is the smallest lantibiotic with antimicrobial properties. Heike et al. reported that the antimicrobial activity shown by this peptide does not involve pore formation; rather, it interferes with peptidoglycan synthesis. In the presence of Mersacidin, glucose uptake is significantly reduced thereby leading to a reduction in the synthesis of cell wall-specific D-amino acids (Fig. 4). However, this does not have any effect on DNA, RNA or protein synthesis. The study suggested that Mersacidin reduced peptidoglycan width from 30–34 nm to 17–20 nm. This caused bacterial cells to rupture because of osmotic pressure. Further, it was also shown that this lantibiotic can reduce overall teichoic acid content in the treated cell.81
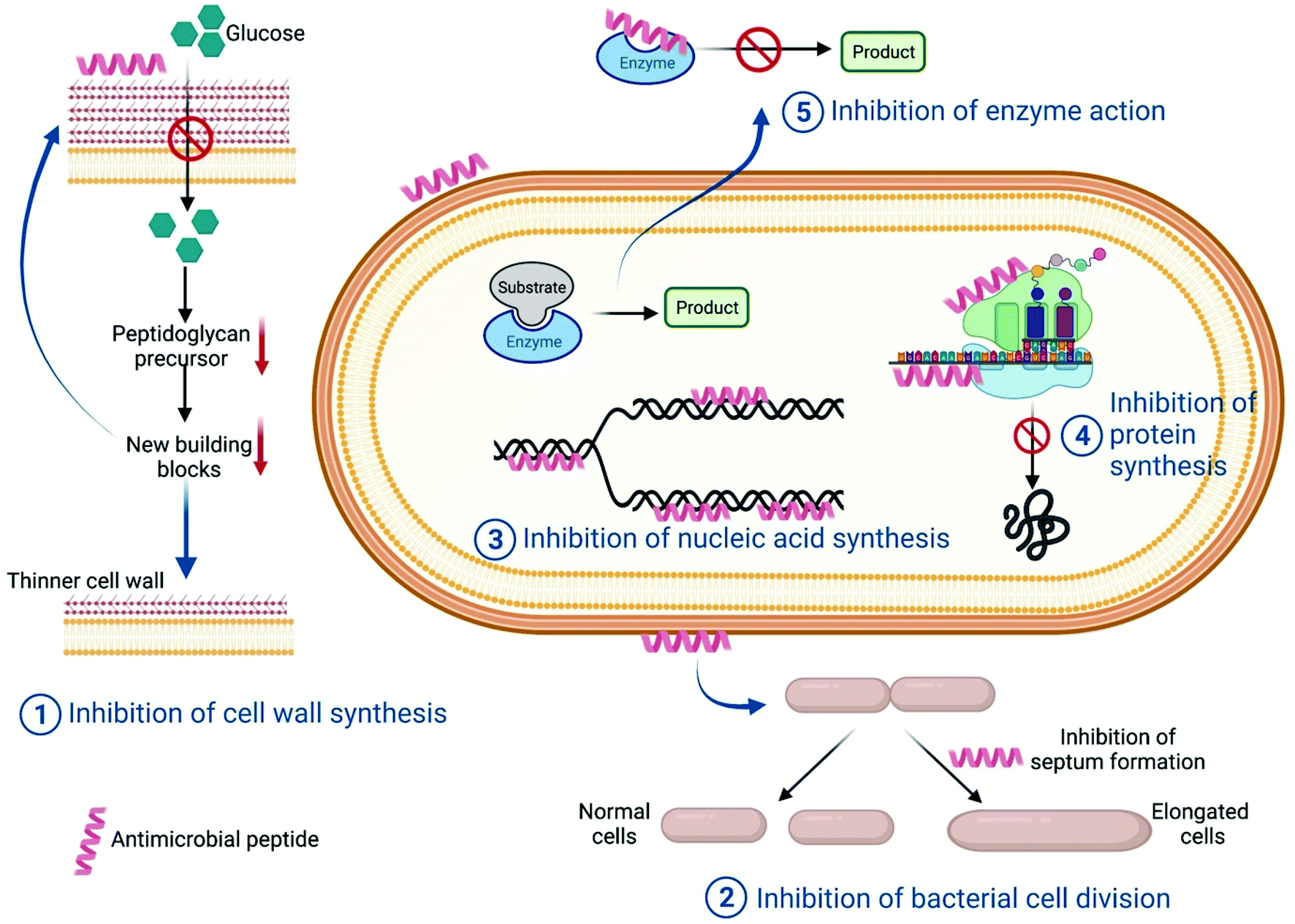 | ||
| Fig. 4 Schematic showing different mechanisms of intracellular targeting pathways by antimicrobial peptides. | ||
(b) Cell division. A few antimicrobial peptides result in the generation of filamentous bacterial cells, which can be a direct consequence of inhibition of various intracellular pathways like improper chromosome segregation, inhibition of septum formation, inhibition of DNA replication or SOS induction. Salomón et al. reported that Microcin J25, a 20 amino acid peptide, results in the formation of filamentous bacterial cells through blocking the septation process of cell division (Fig. 4). In the presence of the antimicrobial peptide, microscopically it was observed that E. coli cells increased in length until they took on a long aseptate filamentous structure. In parallel, cell mass was also increasing which was a clear indication of the increase in bacterial cell length because of inhibition of cell division. It was also found that even in the presence of prophage induction, there was a relatively low phage titer in AMP-treated cells, which suggested it was a non-SOS dependent pathway and hence showed only a bacteriostatic effect.82 A similar effect was also observed for the peptide diptericin in E. coli cells.83
(c) DNA/RNA synthesis. Nucleic acids are a major component of any cell. Both DNA replication and RNA transcription are complex and multistep processes which involve different enzymes and proteins. Hence each step, including the components, can be a potential target for antimicrobial peptides. There are a few AMPs which have structural similarities with proteins that have DNA or RNA-binding domains. Such AMPs bind with the respective nucleic acids and inhibit their function. They can also inhibit any enzymes involved in replication, transcription or post transcriptional pathways (Fig. 4). Buforin II is an antimicrobial peptide which has structural homology with the N-terminal fragment of the DNA-binding protein histone H2A, and binds with the DNA fragment to interrupt DNA or RNA metabolism.84,85
(d) Protein synthesis. After DNA replication and mRNA transcription, translation of mRNA takes place via the 70S ribosome in the case of prokaryotic cells, whereas in eukaryotic cells 80S ribosomes are required for protein translation. Hence this is a very good potential target for antimicrobial activity. Besides, inhibition of any intermediate step can halt protein synthesis (Fig. 4). Bac-7, a 60-residue peptide, can be internalized by inner membrane protein SbmA of E. coli and shows antimicrobial action by targeting the bacterial ribosomes.86 Inhibition of protein synthesis can be targeted by many other antimicrobial peptides.87,88
(e) Enzymatic action inhibition. For a living cell, cellular metabolism is an important process which includes various overlapping and independent biochemical reactions. The total system is like an orchestra, so inhibition of any one reaction can cause the whole cellular system to diminish. Therefore, cellular biochemical reactions are a very common target for antimicrobial peptides. UDP-N-acetylglucosamine acyltransferase enzyme is the enzyme of the first step of lipid A synthesis, which is one of the main components of Gram-negative bacteria's outer membrane. A penta-decapeptide (Peptide 920) targets this enzyme and has a very high affinity towards the active site of this enzyme, thus preventing its catalysis of lipid A. As a result, an incomplete membrane structure will be formed, which is lethal for Gram-negative bacteria89 (Fig. 4).
3.2 Mechanism of antiviral action of antiviral peptides
In today's scenario, viral outbreak and resistance are the biggest global health and economic concerns. This situation acts as a positive stimulus for the search for new antivirals. Antivirals do not always have the property of directly killing or destroying the viruses; rather, in most cases they act by disrupting the machinery of viral infection.90 The process of viral infection involves (i) attachment of virus to the host cell membrane through specific ligand–receptor interactions between the virus and its target cell, which is then followed by (ii) penetration or internalization of the virus inside the host cell. Once inside the host, (iii) the virus hijacks the genetic machinery of the host to replicate its genetic material, followed by its translation into viral proteins. Once the viral genome has been copied and necessary structural components have been synthesized, (iv) packaging of the viral genome into protein structures takes place to form virions, which finally fuse with the host cell membrane to bud-off from the cell surface and get released as progeny viruses. Each stage of this replication machinery can be a potential target for any antiviral biomaterial.i Naturally occurring antiviral peptides: naturally occurring peptides with cationic residues show a similar effect but their amphipathic α-helical conformation and β-sheet structures also play important roles. On the basis of their mode of action, they can be classified as follows:
1. Inhibition of viral attachment on the host cell: lactoferricin, an α-helical cationic peptide containing 21 amino acids, shows antiviral activity by preventing viral attachment to host cells.94,95 It was suggested that lactoferricin binds with the heparan sulfate proteoglycans (HSPGs) present on the mammalian cell surface and therefore blocks viral attachment to the host cell.96
2. Inhibition of viral entry inside the host cell: cationic peptide T22, which has β-sheet conformation, shows antiviral activity against HIV-1, but rather than the cationic moiety, β-sheets play a more important role here by interrupting binding of CXCR4, a T-cell receptor which is utilized by the viral particle for entry into the host cell.97,98
3. Inhibition of viral cell fusion with the host cell: another cationic antiviral peptide obtained from bee venom is Melittin. It was reported that this peptide is able to inhibit Herpes simplex viral infection and Junin virus infection by inhibiting viral particle fusion with the host cell membrane.99
4. Inhibition of viral replication: it was reported by Wachinger et al. that cationic antiviral peptide Cecropin and also Melittin were able to inhibit HIV-1 infection by suppressing activity of the HIV Long terminal repeat.100
ii. Synthetic peptides: Krepstakies et al. reported that 20 amino acid-long peptides containing lysine and arginine residues can successfully inhibit a broad spectrum of viral infections including Human Immunodeficiency Virus (HIV) type1, Herpes Simplex virus (HSV) type 1 and type 2, Hepatitis B virus (HBV) and Hepatitis C virus. Their study showed that the antiviral action of these peptides is observed when used prior to viral infection. The mechanism of action exhibited by these peptides thus mainly involves inhibition of viral attachment to the host cells before membrane fusion.101 Similar results have been shown in studies with 12 amino acid-containing cationic peptide G2 against HSV-2 viral infection. This peptide was able to inhibit not only viral attachment but also viral fusion with the host cells.102
3.3 Mechanism of antibacterial action of polymeric biomaterials
A moderately different mechanism has also been found in the case of another cationic polysaccharide, namely quaternized pullulan.106 Pullulan is a fungal exopolysaccharide secreted by the black fungus Aureobasidium pullulans. Pullulan itself does not have any antibacterial activity; however, cationization of this polysaccharide through the introduction of quaternary ammonium groups into the polysaccharide backbone results in the creation of a highly potent bactericidal agent. Quaternized pullulan has been observed to exert its bactericidal action against both Gram-negative and Gram-positive bacteria through a typical non-pore-forming pathway, which does not involve the formation of classical “pores” for disrupting bacterial membranes.106 Such a mechanism of action is more common for antimicrobial peptides than for antimicrobial polysaccharides. Both atomistic MD simulations and in vitro experiments showed that interaction of the cationic pullulan with the exposed negatively charged polar head groups of the bacterial cell membrane occurs via electrostatic interactions, which results in the polymers getting tightly adsorbed on the outer membrane surface of the bacteria. Subsequently, this leads to the clustering of anionic phospholipids into domains in the bacterial membrane (Fig. 5). These negatively charged clusters or domains cause a difference in the packing of the hydrophobic tails of phospholipids, which ultimately causes a difference in fluidity on one leaflet of the membrane. As a result, the properties of the other leaflet are also affected, a phenomenon known as interleaflet coupling. This phenomenon has the ability to create transient pores in the membrane through which water molecules and ions can pass, thereby resulting in depolarization of the membrane. A combined effect of anionic lipid clustering and membrane depolarization makes the bilayer weak over time, which ultimately results in physical disruption of bilayer integrity and cell death.
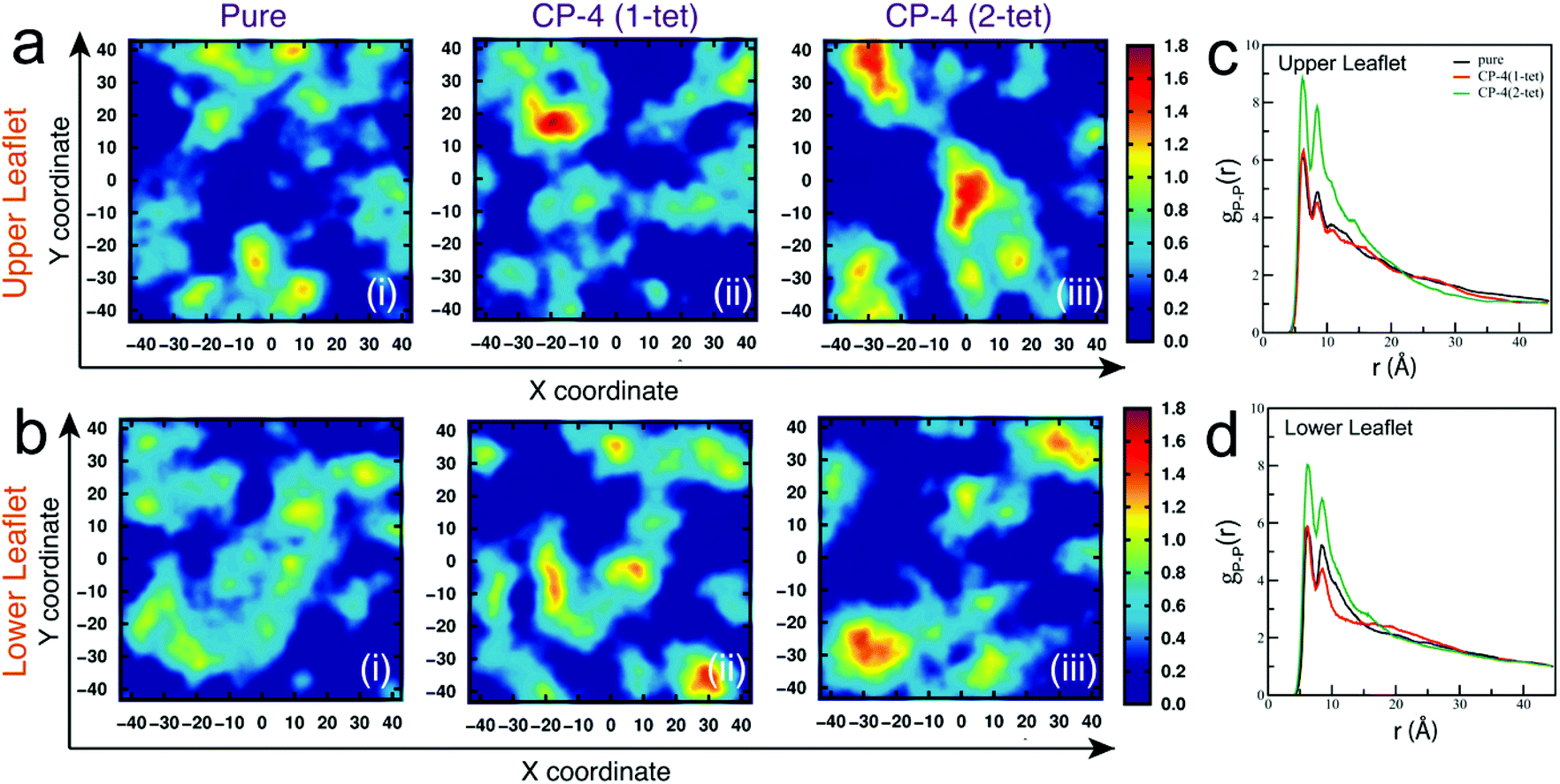 | ||
| Fig. 5 Anionic lipid clustering induced by quaternized pullulan. Two-dimensional number density plots for DOPG lipids in xy-plane orthogonal to membrane normal with subsequent addition of CP-4 molecules (a) upper leaflet and (b) lower leaflet. The units of x and y coordinates and number density are Å and Å-2, respectively. Radial distribution function for P–P atomic pairs of DOPG lipid head groups for (c) upper leaflet (d) lower leaflet. Reproduced with permission.106 Copyright 2022, Royal Society of Chemistry. | ||
i. Biguanide polymers: polyhexamethylene biguanide chloride (PHMB) is the first antimicrobial polymer whose interaction was studied against Gram-negative bacteria E. coli and a model phospholipid membrane. The study suggested that after interaction of the polymer with the cell membrane a domain of acidic phospholipids is created that compromises the integrity of the outer membrane in the case of Gram-negative bacteria. First, it gets adsorbed on the outer membrane and compromises its integrity so that it can internalize further and interact with the inner membrane. This makes the membrane more porous, which results in leakage of K+ ions. At this stage, the polymer mainly inhibits bacterial growth. However, complete loss of membrane function ultimately leads to cell lysis.14,107 4-(2-Hydroxyethyl)aniline hydrochloride(II), 4-(2-hydroxyethyl) phenyl dicyandiamide(III), Nl-4-(2-acryloyl oxyethyl) phenyl-A5-4-chlorophenyl biguanide hydrochloride(VI) etc. are chemically synthesized biguanide polymers which also show antimicrobial activity against both Gram-positive and Gram-negative bacteria.108
ii. Quaternary ammonium or phosphonium polymers: in the case of quaternary ammonium or phosphonium polymers, the basic mechanism of action is similar. Because of the presence of cationic charge, the polymers get absorbed on the outer envelope and damage the outer membrane mainly through non-pore-forming pathways of membrane destruction (Fig. 3). In the majority of cases, the polymers exert their membrane-damaging action without themselves penetrating the membranes.109,110 A copolymer of 2-choroethylvinyl ether and vinylbenzylchloride with immobilized phosphonium ions showed a similar bactericidal effect on both Gram-positive Staphylococcus aureus and Gram-negative E. coli.111 Another copolymer (PEB-b-PDMAEMA), which is copolymerised with octyl bromide, was synthesized by Lenoir et al. by quaternization of amino groups of poly(ethylene-co-butylene)-b-poly[2-(dimethylamino)ethyl-methacrylate]. This copolymer also showed antibacterial activity against E. coli.112 A highly hydrophilic and biocompatible monomer hydroxythylmethacrylate (HEMA) and polyethylene glycol methyl ether methacrylate (PEGMA) were incorporated in quaternized poly(vinylpyridine) (PVP) via copolymerization with 4-vinyl pyridine. This copolymer has shown much better antibacterial activity against E. coli than normal PVP.113 A series of tributyl(4-vinylbenzyl) phosphonium salts with different counter anions and corresponding polymers have shown antibacterial activity against S. aureus where the antibacterial activity emerged from the counter anions.114 Those counter anions which were tightly bound with the phosphonium ions showed relatively less antibacterial activity than the counter anions which completely dissociated into free ions.
iii. N-Halamine polymers: this is a group of bactericidal polymers where N-halamine precursors are covalently bonded with nitrogen atoms of the targeted polymer. Upon halogenation they are converted to N-halamine structures which provide stability to the polymeric structure and allow slow release of free halogens in the environment to show bactericidal effect. N-Halamine polymers have broad-spectrum antibacterial property. Here, not the overall polymer but the free oxidative halogen along with its bound compound (thiol groups or amino groups) comes into direct contact with bacterial cells and results in cellular inactivation.115,116 A recent study shows that N-halamine acrylamide monomer, a hydantoin acrylamide (HA), is produced by forming a hydantoin ring from the ketone moiety of a secondary amide monomer, N-(1,1-dimethyl-3-oxobutyl) acrylamide (DA). HA was copolymerized with a siloxane monomer (SL) using different feed ratios, and this copolymer can be used as a antimicrobial coating on fabric.117,118 Kocer et al. reported another series of water-dispersible biocidal polymers which can be used as antimicrobial paints. These were synthesized by copolymerization of the hydantoin acrylamide and sodium salt of 2-acrylamido-2-methylpropane sulfonic acid.119
3.4 Mechanism of antiviral action of polymeric biomaterials
Polymers, because of their high molecular weight and repetitious structure, have emerged as a promising class of antiviral biomaterial. Polymeric biomaterials can be divided into anionic and cationic polymers, depending on the type of charge they carry on their surface.(a) Sulfated polymers. Sulfated polymers can be both polysaccharides and non-glycosylated polymers.
i. Sulfated polysaccharides: depending on the polymeric subunit and virus particle, different antiviral mechanisms have been observed.122,123
Virucidal activity: some polysaccharides because of their negative charge can directly show a virucidal effect on some envelope viruses by altering the envelope proteins of the viruses so that they are no longer available for interaction with the host cells, hence reducing their virulence. Carrageenan is a sulfated polysaccharide whose λ-type can firmly attach to the Herpes Simplex virus (HSV) and cause structural changes in the viral glycoprotein gB and gC, thereby inactivating the virus for further interaction with host cells.124,125 Chitosan and its oligosaccharides can also have direct virucidal activity against some human enteric viral surrogates.126,127
Inhibition at attachment stage: this is the first stage in viral infection, when virus particles interact with the host cells through their membrane proteins and bind with specific receptors present on the host cell surface. At first this interaction is reversible, but slowly irreversible interaction develops. Various antiviral polymers are available which target the viral membrane proteins so that they are not able to bind with the host cell receptor, or polymers can also bind with the host cell receptors thereby making these receptors no longer available for the viral proteins. Dextran sulfate and heparin were found to interfere with the interaction between HIV glycoprotein gp120 and CD4+ antigen receptors present on the T lymphocytes.128–130 There are many more polymers like sulfated galactan from red algae,131 chitosan derivatives,132 and fucoidan from brown algae133–136 which show antiviral activity using this mechanism.
Inhibition at internalization and uncoating: viral internalization and uncoating occurs after the adsorption step. Internalization and uncoating usually occurs in a stepwise manner though in an allosteric way, but they can also take place simultaneously in some viruses.137 Viral internalization mainly occurs through a vesicular endocytic pathway where viral particles are transported to endosomes or any targeted subcellular organelles through cytoplasm inside a vesicle.138 Some antiviral polymers can block any of the above steps. Kim et al. showed that p-KG03, a sulfated polysaccharide obtained from Gyrodinium impudium, can inhibit Influenza A viral infection if administered during or within 6 h of viral infection. This suggests that the polymer interferes primarily at the adsorption stage and also in the internalization of the virus inside the host cell.139 There are many other polysaccharides, like Carrageenan, which also follow this mechanism against HPV140 and DENV141,142 viruses.
Inhibition of viral replication and transcription: the molecular weight of the polysaccharides plays an important role in inhibiting viral replication and transcription. Large molecular weight polysaccharides which are not able to enter the host cell show their antiviral activity at the adsorption stage; however, the low molecular weight version of the same polysaccharide which can get internalized exhibits its antiviral activity by inhibiting viral genome replication or transcription. It can also interfere in the viral protein translation process. It has been demonstrated by Wang et al. that the antiviral activity of Carrageenan oligosaccharide CO-1 against IAV is not at the adsorption stage. The oligosaccharide cannot even bind to the surface of host MDCK cells directly. Thus, after internalization it inhibits IAV mRNA transcription and protein synthesis.143 Another such example is that of sulfated fucan, which can inhibit HIV by inhibiting its reverse transcriptase enzyme required for viral genome replication.144,145
Indirect antiviral effect: when a cell is infected with virus, it induces the type I interferon system, which is a host's innate antiviral mechanism and prevents viral multiplication inside the host cell. There are a few polymers which induce this host antiviral mechanism, hence indirectly showing antiviral property. A sulfated polysaccharide (SPPMG) obtained from brown algae has shown an indirect antiviral effect on HIV and HBV infection by inducing cellular and humoral immunity of the host cell.146,147 There are various other polymers like λ-Carrageenan148–150 which show a similar antiviral effect.
(a) Sialylated polymers. Sialylated polymers can be defined as polymers having sialic acid components in their structure. This type of polymer especially works against influenza virus. The antiviral mode of action followed by these polymers is inhibition of viral attachment to the host cells.156 The resistance rate of influenza virus is considerably high because the virus particle interacts with the host cell through its surface glycoprotein hemagglutinin with the sialylated glycans present on the host cells which share different properties, hence sulfonated or sulfated polymers are not highly efficient against influenza virus. The antiviral efficiency of these polymers depends on the degree of substitution, and it also increases with substitution.157,158
(b) Phosphonothioate polymers. Since late 1970, nucleotide and nucleoside analogs have been established as antiviral agents but viruses easily adapt to their antiviral mechanism and become resistant.159,160 Synthetic oligonucleotides are promising antivirals, but to increase the in vivo half-life and resist nuclease attack, phosphonothioation of the phosphodiester linkages is done which provides the polymers with anionic as well as amphipathic properties, making them a potential contender for antiviral therapy. As they mimic natural nucleotides, such polymers are also known as Nucleic Acid Polymers (NAPs),161 such as Adenosine 3,5′-cyclic phosphorothioate, Adenosine 5′-phosphorothiolate,162 and REP2139, a phosphorothiolate polymer used against Hepatitis B virus.163 The antiviral efficiency of a NAP is independent of its sequence but very much dependent on the amphipathicity and length. NAPs show a broad spectrum of antiviral activity both under in vitro and in vivo conditions.164–166 The mechanism of action of these polymers is similar to that of sulfated polymers, i.e. it can prevent viral attachment to the host cell surface, and can inhibit later stages like viral assembly.166
(a) Amine-functionalized polymers. The primary mode of action of these polymers against viruses is inhibition of viral attachment to the host cell membrane. Eudragit E100, an ammonium-functionalized polymer which is a copolymer of methyl methacrylate, N,N (dimethylamino) ethyl methacrylate and methyacrylate in 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 composition, shows antiviral activity against a wide variety of viruses such as HSV-2, VSV, bovine viral diarrhea virus, and Measles virus by preventing the viruses from attaching to the host cell membrane.167,168 Molecular dynamics simulations suggest that cationic oligomeric-conjugated polyelectrolytes (OPEs) made of synthetic poly(phenylene ethynylene) show antiviral action against MS2 and T4 bacteriophages by strongly binding with the viral capsid via electrostatic interactions and van der Waals forces, which causes viral capsid destruction.169,170
:2:1 composition, shows antiviral activity against a wide variety of viruses such as HSV-2, VSV, bovine viral diarrhea virus, and Measles virus by preventing the viruses from attaching to the host cell membrane.167,168 Molecular dynamics simulations suggest that cationic oligomeric-conjugated polyelectrolytes (OPEs) made of synthetic poly(phenylene ethynylene) show antiviral action against MS2 and T4 bacteriophages by strongly binding with the viral capsid via electrostatic interactions and van der Waals forces, which causes viral capsid destruction.169,170
(b) Guanidine-functionalized polymers. It has been shown that the guanidine moiety exhibits antiviral activity against both enveloped and non-enveloped viruses. So, polymers functionalized with the guanidine moiety show a broad spectrum of antiviral activity against Equine herpes virus type 1, Rhinotracheitis infectious bovine and Equine infectious anemia virus.171 Such polymers showed antiviral properties only when administered prior to infection, thus suggesting that the mode of action of these polymers was again inhibition of viral attachment to the host cells by itself binding to the phospholipids of the host cell membrane.171
4. Antimicrobial mechanisms of nanoscale biomaterials
In this section, we present a comprehensive and general outline of the possible mechanisms involved in the antibacterial and antiviral action of nanostructures of different dimensionalities such as nanoparticles, nanotubes and nanosheets, and try to provide an overview of the possible sequence of events that might take place when a bacterial cell or a virus interacts with a specific class of nanostructure, ultimately leading to its death. Different factors such as diameter, length (short vs. long), degree of oxidation, surface chemistry, electronic structure (metallic vs. semiconductor), as well as microbial strain and morphology have been taken into consideration in understanding the mechanism of action of these nanostructures. We have restricted our discussion to the inherent antibacterial and antiviral effects of pristine nanostructures of different dimensions without any antimicrobial surface functional agents such as surfactants, polymers or loaded with any other biocidal agents.4.1 Mechanism of antibacterial action of 0D nanoparticles
0D nanoparticles have been widely used as antibacterial agents. Among the different types of nanoparticle explored as antibacterial agents, metal (Ag, Au, Cu) and metal oxide (Cu2O, CuO, ZnO, MgO, TiO2, Al2O3etc.) nanoparticles have been studied the most. Carbon dots too have emerged as promising antibacterial agents. In the following section, we discuss how these 0D nanoparticles interact with bacteria and exert their antibacterial action.In addition to metal ions, the nanoparticles themselves can also penetrate the cell by overcoming the membrane barrier. This generally occurs because of the membrane damage caused by the nanoparticles which results in the formation of pores in the membrane, through which the nanoparticles enter the cells. Inside the cells, the nanoparticles along with their ionic counterparts carry out structural and functional damage to the bacterial cells through oxidative and non-oxidative mechanisms. Inactivation of the phosphomannose isomerase enzyme by direct interaction with Ag NPs has also been observed in bacteria, which caused inhibition of sugar metabolism.186 Similarly, denaturation of DNA and mutation of key DNA repair genes (mutY, mutS, mutM, mutT and nth) have been observed to take place in bacteria via direct interactions between Ag NPs and DNA molecules.187 Ag NPs, due to their ability to penetrate the cells, directly interact with respiratory dehydrogenases involved in the bacterial electron transport system and cause their inhibition or inactivation,188 thus thwarting the bacterial respiration process. Additionally, Ag NPs inhibit protein synthesis via direct interaction with ribosomes and cause its denaturation.183,189,190 However, not all nanoparticles need to enter the cells to exert their antibacterial action. For example, MgO NPs were found to exhibit excellent antibacterial activities through membrane damage without entering the cells.191
The anti-oxidant glutathione (GSH), which acts as a redox regulator in bacterial cells and protects the cells from oxidative damage by maintaining a pro-oxidant–antioxidant equilibrium, has been found to get oxidized to its disulfide form GSSH, leading to its inactivation.198 Nanoparticles of silver, copper, ZnO and TiO2 have been found to cause oxidative damage of intracellular bacterial proteins by depleting the GSH levels in cells.199,200 The ROS-induced oxidative damage to bacterial cells also includes DNA damage, which severely compromises the genetic machinery of bacterial cells and thus inhibits DNA replication and synthesis.200 DNA damage through single and double-strand breaks, formation of base–sugar adducts and complexation of DNA with other molecules resulting in inhibition of replication are known to be caused by free radicals such as hydroxyl radicals and singlet oxygen.
Photocatalysis is another mechanism by which a number of metal oxide nanoparticles like ZnO and TiO2 generate ROS.201 When these nanoparticles are excited with light of a particular energy that is either equal to or greater than their band gap, electrons get excited from the valence band to the conduction band. This process creates highly reactive intermediates called electron–hole pairs, which react with oxygen and water to generate ROS and attack cells and cause damage. In addition to metal oxide nanoparticles, carbon dots have also been shown to exhibit excellent antibacterial activities through the process of photocatalytic ROS production.202 Upon excitation with light ranging from UV to near-IR, C-dots have been found to generate ROS such as singlet oxygen and hydroxyl radicals which act as the main mediators for their observed antibacterial action.
In addition to modulation of signaling pathways, nanoparticles also play a role in regulating bacterial gene expression to a large extent. Exposure of bacterial cells to metal and metal oxide nanoparticles such as Ag, TiO2, MgO, CeO2 has been found to cause differential expression of a variety of genes associated with bacterial membrane structure and function, cellular transport, electron transfer and ATP production, DNA replication and repair, metabolism and stress response (oxidative and non-oxidative). Ag NPs-181 and TiO2 NPs205-induced membrane damage and membrane stress were found to upregulate the expression of genes regulating membrane structure (bolA) and envelope proteins such as outer membrane proteins (OmpA, OmpC, OmpF), periplasmic oligopeptide binding protein A (OppA), and D-methionine binding lipoprotein (MetQ).206 Genes regulating electron transfer (sdhC) and bacterial respiration (cydA and cydB) were found to be differentially expressed in bacteria upon treatment with Ag and CeO2 NPs, indicating the role of these nanoparticles in disrupting the bacterial electron transport system and respiratory chain.206,207 Upregulation of genes conferring protection against ROS-mediated oxidative stress is one of the key aspects of gene regulation observed in bacteria upon treatment with nanoparticles, which again establishes the strong role of nanoparticle-induced oxidative stress in killing of bacteria. Genes such as ahpC, aphF, katE, katG, oxyR and sod genes (A, B, C), associated with regulating redox reactions and peroxide metabolism, were differentially regulated in bacteria by Ag and TiO2 NPs.205,206 Similarly, Ag and TiO2 NPs also regulate the expression of genes related to DNA replication, synthesis, and repair. Downregulation of genes like dnaX, holB, guaC, pyrC, gyrA that are involved in DNA replication and synthesis has been observed in bacterial cells upon treatment with TiO2 NPs.205 Along with this, upregulation of genes involved with DNA repair such as recN, uvrA, uvrD, umuD, ybfE, yebG, ssb, sbmc, and nfo also takes place as a result of treatment with Ag and TiO2 NPs, thereby showing that the cell upon nanoparticle treatment is subjected to stress that prevents the cell from synthesizing new DNA and also prepares it to repair its damaged DNA for survival.205,206 Nanoparticles can also regulate different metabolic pathways in bacteria by acting on target proteins. MgO, CuO and TiO2 nanoparticles have been found to modulate pathways related to metabolism of sugar, nitrogen, amino acids etc. through differential regulation of a variety of genes that either upregulated or downregulated a set of proteins essential for cell survival and growth.191,205,206
Thus, nanoparticles exert their antibacterial action by directly interacting with the bacterial cell wall and membrane where direct physical contact-mediated as well as ROS-mediated pathways cause damage and disruption of membrane permeability and integrity. This results in leakage of cellular components like sugars, proteins and ATP from the membrane-compromised cells, rendering the cells metabolically inactive. Toxic ions leaching out from nanoparticles further contribute towards the ongoing antibacterial action through direct interaction with cellular proteins, nucleic acids and membranes, causing their denaturation and disintegration. Furthermore, excessive generation of intracellular oxidative stress upon cellular penetration of nanoparticles and their toxic ions leads to oxidative damage to cellular structures and components, which completely disrupts the functional integrity of cells through differential regulation of metabolism and different signal transduction pathways (Fig. 6).
 | ||
| Fig. 6 Schematic showing the mechanism of antibacterial action of 0D nanoparticles. | ||
4.2 Mechanism of antiviral action of 0D nanoparticles
As discussed previously, each step in the process of viral infection is a potential target for antiviral agents to act upon for inhibiting viral infection, either by directly inactivating the virus through physical or chemical damage, or by preventing the virus from entering the host cells. These are the two key steps by which majority of nanoparticles (Ag, Au, CuO, SiO2, C-dots) act to exert their antiviral action. Ag NPs have been found to inhibit HIV-1 infection by preventing the binding of HIV-1 virus's surface protein gp120 with its receptor CD4 on host cells.208 This was found to occur because of direct interaction of Ag NPs with the gp120 protein on the virus surface, leading to its structural and functional modification. This inhibited HIV-1 virus infection at the very early stage of virus binding and fusion, thus reducing the infectivity of the virus. In addition to this, the Ag NPs were also found to inhibit HIV-1 infection in both cell-free and cell-associated systems, showing the virucidal property of these nanoparticles to cause direct inactivation of viruses.208 CuO-NPs have been observed to interfere with attachment and entry of hepatitis C virus (HCV) infectious virions in hepatic host cells.209 Carbon dots (C-dots) have also been found to exert antiviral activity against a wide variety of viruses by inhibiting the first step of virus–host cell interaction. Inhibition of binding of norovirus virus-like particle (VLP) to histo-blood group antigen (HBGA) receptors on human cells210 and blocking of infection by flaviviruses (Japanese encephalitis, Zika, and dengue viruses) and non-enveloped viruses (porcine parvovirus and adenovirus-associated virus)211 have been found to take place upon treatment of these viruses with C-dots. A recent study has demonstrated that carbon quantum dots (CQDs) can block the Receptor Binding Domain (RBD) of the SARS-CoV-2 spike protein by increasing the extent of the interaction through formation of hydrogen bonds between functional groups on the C-dot surface and spike amino acid residues.212 In a recent study, molecular docking revealed the anti-viral effect of iron oxides NPs (IONPs) against SARS-CoV-2. It was observed that the strong binding affinity of IONPs to the S1-RBD of viral spike glycoprotein could induce conformational changes in the spike, resulting in virus instability.213 Nanoparticle coatings made of Ag, CuO and ZnO have been reported to exhibit potent anti-SARS-CoV-2 activity. It has been anticipated that these nanoparticles exhibit antiviral property against SARS-CoV-2 through the release of Ag+, Cu2+ and Zn2+ ions, which could lyse the membrane by directly adsorbing on the viral envelope and ROS generation.214 Titanium dioxide (TiO2) nanoparticles inactivated the influenza virus H3N2 by destroying the virus envelope, which resulted in the destruction and disintegration of the virions entirely.215The antiviral action can also be exerted at later stages of the viral infection cycle when the viruses have already infected the cells. In such cases, inhibition of viral DNA/RNA replication, damage to the viral genome and induction of a pro-inflammatory phenotype in host cells are the major ways of preventing further infection and killing the virus. Some nanoparticles like Ag, ZnO, CuO, C-dots and Ag2S nanoclusters have been found to act at this stage.216,217 Ag2S nanoclusters prevented porcine epidemic diarrhea virus (PEDV) infection in host cells by inhibiting the synthesis of viral negative-strand RNA and also activated the production of IFN-stimulating genes (ISGs) and the expression of proinflammatory cytokines by the host cells, which resulted in generating an anti-viral innate immune response in the host cells, helping to inhibit the PEDV infection.218 In addition to preventing viral attachment and binding to the host cell, C-dots also exert antiviral action by activating the type-I interferon response in host cells.219
Lastly, preventing the virions from budding off the host cell surface is another mechanism through which the viral infection can also be restricted. Ag2S nanoclusters were found to inhibit the process of budding off of PEDV virions from Vero cells.218 The nanoparticles might be able to inhibit the cleavage of hemagglutinin–sialic acid receptor interaction between the virions and the host cell membrane by directly binding to neuraminidase proteins and thereby inactivating it in the process.220 Hence, inhibiting the virus from entering the host cells seems to be the most common pathway followed by nanoparticles in implementing their antiviral action (Fig. 7).
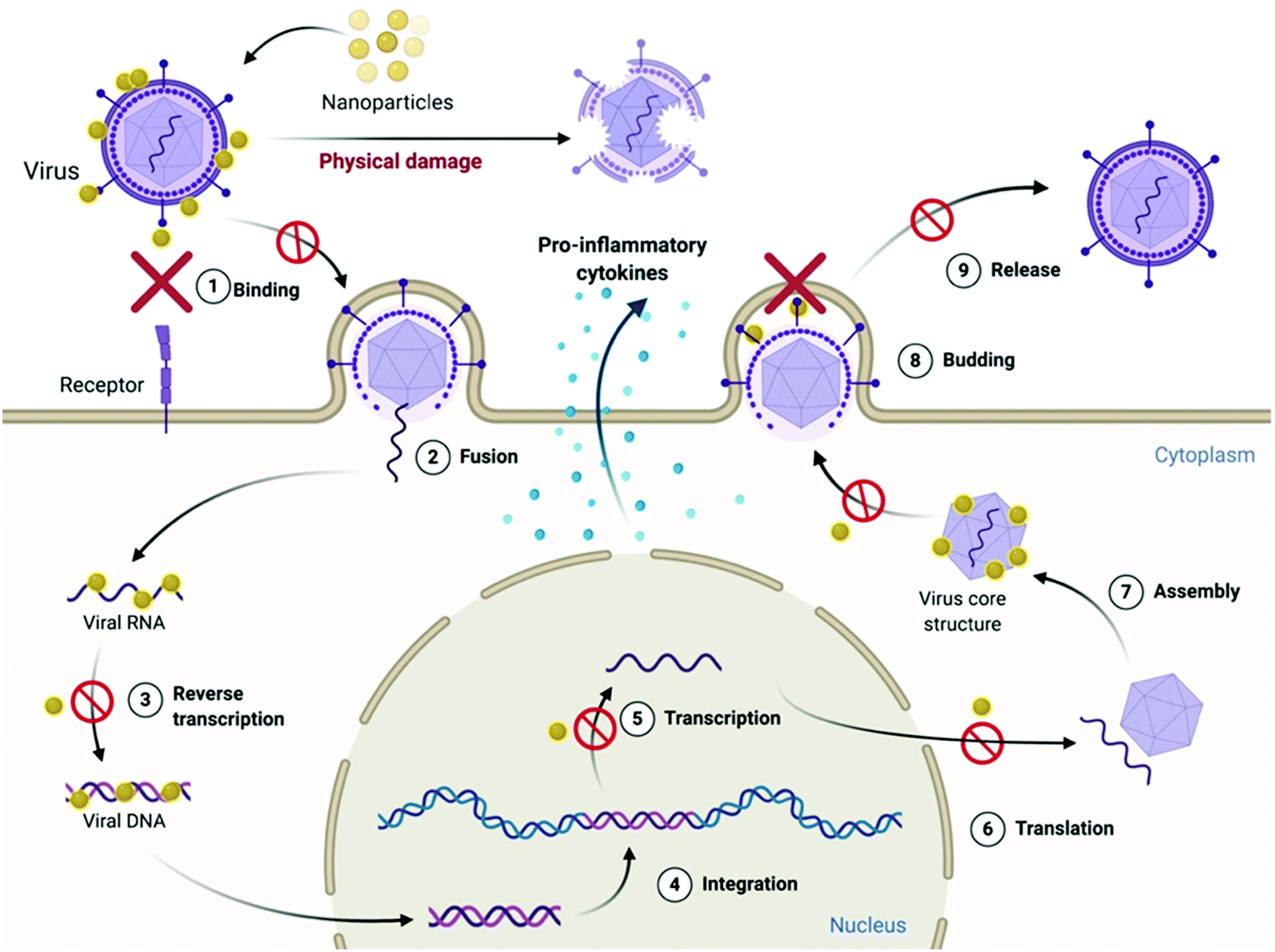 | ||
| Fig. 7 Schematic showing the mechanism of antiviral action of 0D nanoparticles. | ||
4.3 Mechanism of antibacterial action of 1-D nanotubes
 | ||
| Fig. 8 Mechanism of antibacterial action of 1-D nanotubes. (A) SEM images of Salmonella cells (i) without SWCNTs, and the aggregates of cells SWCNTs of (ii) <1 μm, (iii) 1–5 μm, and (iv) ∼5 μm. Reproduced with permission.221 Copyright 2007, American Chemical Society. (B) SEM images contrasting the heights of the high aspect ratio nanotubes (i) 1 μm and (ii) 30 μm VACNTs. False color SEM images of (iii) S. aureus and (iv) P. aeruginosa attached onto VACNT surfaces, revealing the bending of the CNTs and deformation of the bacterial cell membrane. Reproduced with permission.222 Copyright 2018, American Chemical Society. (C) Oxidative stress (ROS)-mediated bacterial cell death. Reproduced with permission.225 Copyright 2014, American Chemical Society. (D) Protection of bacterial cells against oxidative stress by an antioxidant. Reproduced with permission.225 Copyright 2014, American Chemical Society. (E) Representative SEM images of E. coli deposited on SWNT filters. E. coli were deposited on the SWNT filter, incubated for 45 min in isotonic saline, and fixed with glutaraldehyde and osmium tetroxide prior to SEM imaging: (i and ii) <5% metallic, (iii and iv) 30% metallic, and (v and vi) >95% metallic. Note the differences in cell membrane hydration, structure, and roughness between the three samples. Reproduced with permission.226 Copyright 2010, American Chemical Society. | ||
(i) Direct puncturing or piercing of the bacterial membrane: the tubular morphology of the nanotubes makes them behave as “nano-darts” in solution that are capable of directly penetrating through the bacterial lipid bilayer, resulting in loss of membrane permeability and membrane disruption.221,222 This is mainly due to the “non-specific toxicity” of the CNT arising from its hydrophobicity, which allows it to penetrate or partition itself into the hydrophobic lipid bilayer very efficiently, consequently causing membrane stress.226 However, in the case of nanostructured bactericidal surfaces with flexible and high aspect ratio nanotube arrays, the process of generation of membrane stress and its subsequent disruption is governed by the flexibility and elasticity of the vertically aligned nanotubes.222 Owing to this flexible nature, these high aspect ratio nanotubes store a significant amount of elastic energy within them. Upon contact with a bacterial cell, these flexible nanotubes undergo a process of sequential bending and retraction, which subjects the attached cells to progressive stretching and tearing of their adsorbed membranes, ultimately leading to complete membrane deformation and rupture (Fig. 8B).222 The elastic energy previously stored in these nanostructures is released during this process of sequential bending and retraction of the nanotubes, which acts as the main driving force for inducing the membrane stress. The diameter of nanotubes plays a very crucial role at this stage in determining the toxicity of the nanotubes towards bacteria.227 SWCNTs with smaller diameter exert more toxicity through a “diameter-dependent piercing” mechanism in comparison with MWCNTs having a larger diameter.224,227 The presence of a larger surface area in SWCNTs contributes towards more contact and interaction with the bacterial outer membrane, leading to more membrane perturbation and stress. As a result of this induced membrane stress, a number of genes like Sigma factors σS and σE, high-pressure stress-related genes, Tol/Pal and PhoPQ two-component system genes associated with maintaining and preserving the integrity of bacterial outer membrane and envelope get up-regulated.227
(ii) Oxidative stress induced membrane damage: CNTs upon stimulation with light can produce ROS like singlet oxygen species, superoxide anions and hydroxyl radicals, which can directly act upon the bacterial membrane and cause lipid peroxidation that induces conformational changes in membrane proteins, and alters membrane fluidity and integrity, thereby leading to membrane damage and loss in permeability (Fig. 8C).225 This is mainly due to the “specific or reactive toxicity” of the CNT that arises from its electrophilicity and results in the generation of oxidative stress through the disruption/oxidation of key cellular components like lipids, proteins and nucleic acids.226 The electronic structure of the nanotubes determines their reactive nature, with metallic CNTs showing more reactive (oxidative) toxicity towards bacteria as compared with semiconductor CNTs (Fig. 8E).226 Experiments with antioxidant-functionalized CNTs and bare CNTs in the absence of light stimulation have shown inhibition of the antibacterial activity of the CNTs (Fig. 8D). This confirms the pivotal role of ROS-dependent oxidative stress in causing membrane damage, and also establishes that direct physical contact-dependent membrane damage might not be the only process through which the nanotubes exert their antibacterial action.225
There are no significant reports demonstrating the antiviral activity of pristine nanotubes. On the contrary, reports suggest that SWCNTs significantly increase the infectivity of influenza H1N1 virus towards lung epithelial cells by inhibiting expression of anti-viral molecules and pro-inflammatory cytokines by the epithelial cells, increasing the expression of viral attachment receptors and through impairment of mitochondrial function.228,229 Thus, a targeted impairment of anti-viral signaling networks that are vital to immune defense mechanisms in lung cells was induced by the nanotubes.
4.4 Mechanism of antibacterial action of 2-D nanosheets
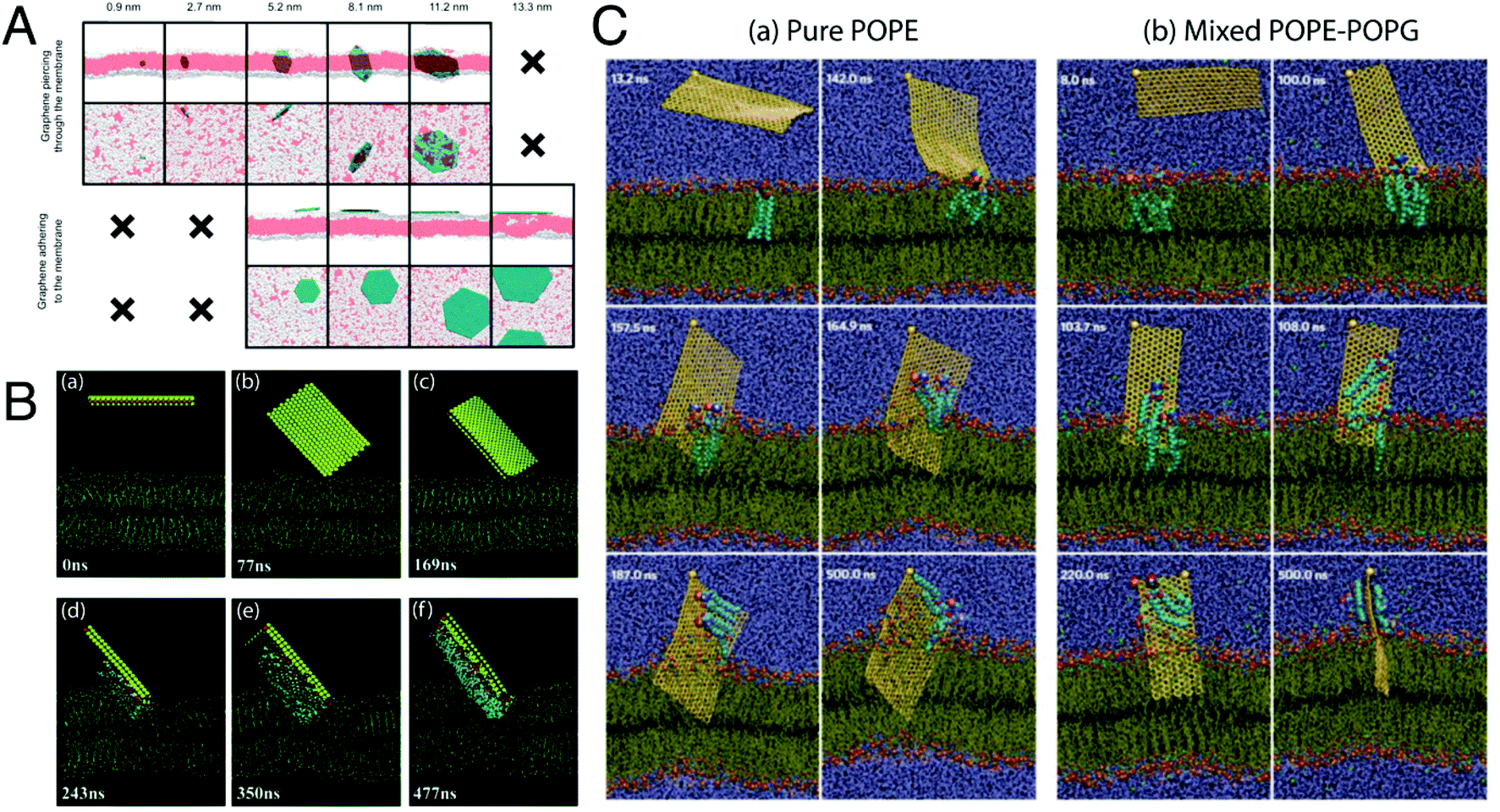 | ||
| Fig. 9 Mechanism of antibacterial action of 2-D nanosheets. (A) Illustrative snapshots, at the end of the simulations, of six graphene nanosheets of increasing size. From left to right, sizes of 0.9, 2.7, 5.2, 8.1, 11.2, and 13.3 nm. White: hydrophilic heads of the phospholipids; red: hydrophobic phospholipid tails; petroleum blue: graphenes. For clarity, water is not shown. The top two rows are different perspectives of the six sheets, as are the bottom two rows. Only the five smaller sheets pierce through the membrane. The four larger sheets adhere to the membrane. Situations not observed in the simulations are indicated by “×”. Reproduced with permission.238 Copyright 2015, American Chemical Society. (B) Representative simulated trajectory of the restrictive simulation. (snapshot time is shown in the lower left corner of each picture.) The MoS2 nanosheet is shown as a yellow rectangular sandwich structure with S atoms in yellow and Mo atoms in pink. The fixed S atom in the corner of the nanosheet is denoted in red. The phospholipids are represented in blue lines with P atoms as khaki spheres. Extracted phospholipid atoms (within 5 Å of the MoS2 nanosheet) are shown as coloured spheres (oxygen, red; nitrogen, navy blue; hydrogen, white; carbon, aqua; and phosphorus, khaki). The molecules of the physiological saline solution are omitted here for expediently displaying the phenomenon of phospholipid extraction. Reproduced with permission.234 Copyright 2018, Royal Society of Chemistry. (C) Graphene nanosheet insertion and lipid extraction. (a and b) Representative simulated trajectories of graphene nanosheet insertion and lipid extraction in the outer membrane (pure POPE) and inner membrane (3:1 mixed POPE–POPG) of E. coli (the snapshot times are shown in the top left corners). Water is shown in violet and the phospholipids in tan lines with hydrophilic charged atoms as coloured spheres (hydrogen, white; oxygen, red; nitrogen, dark blue; carbon, cyan; phosphorus, orange). The graphene sheet is shown as a yellow-bonded sheet with a large sphere marked at one corner as the restrained atom in simulations. Extracted phospholipids are shown as larger spheres. Reproduced with permission.240 Copyright 2013, Nature Nanotechnology. | ||
Sheets having a larger size tend to interact with the bacterial membrane through surface-area mediated physical interaction, using their basal planes to come into contact with the membrane surface and tend to arrange themselves flat on the membrane surface.235 Such kind of interaction generally leads to the “wrapping/trapping” of the cells by the larger-sized sheets (micron sized), which can efficiently wrap around the entire cells and trap them in an isolated environment.231,235,236 In such events, membrane stress in the form of membrane damage or disruption is not that evident in the bacterial cells; however, studies have shown that it can lead to the formation of patches of upturned phospholipids at the sites of nanosheet attachment, thereby causing membrane perturbations in place of disruptions.237,238 As shown in studies with GO nanosheets in suspension, bacterial cells were completely wrapped by the nanosheets having larger lateral dimensions, resulting in bacterial inactivation.235 This observed inactivation actually occurred through the inhibition of proliferation of cells trapped within the sheets. As a result of trapping by the nanosheets the bacterial cells were isolated from their surrounding environment, which prevented the passage and entry of nutrients into the cells. However, such a process of bacterial inactivation through the wrapping/trapping pathway was found to be reversible in nature, as the trapped bacterial cells upon sonication could come out from the traps and regain their viability, and thus this process does not lead to complete bacterial inactivation.236 On the other hand, sheets with smaller size possess sharp edges and corners, which make them behave as “nano-knives” and as a result interact with the bacterial membranes through an edge-mediated physical interaction process that involves direct puncturing or piercing of the lipid bilayer, leading to penetration of the nanosheets into the cytoplasmic region of the cells by creating pores in the bacterial membrane.238 Such an interaction is also observed with nanosheets that have been arranged in the form of a coating or placed on a substrate in a specific orientation.233
The orientation of the nanosheets plays a key role in determining their mode of interaction with the bacterial membrane. Smaller sheets are found to mostly interact with the membrane through a perpendicular orientation, allowing them to penetrate more efficiently through the membrane using their edges, as evidenced in molecular dynamics (MD) simulation studies.238 However, the antibacterial efficiency of nanosheets is not always dictated by a perpendicular orientation. Tilted nanosheets arranged at a particular angle on a substrate are also found to be extremely potent in displaying antibacterial activity.239 This emphasizes the importance of availability of sharp edges and their density in achieving strong antibacterial action. The vertical alignment of the nanosheets on a substrate233 or their tilted arrangement in a particular orientation239 results in providing a surface with a high density of sharp edges, which interact with bacterial cells through the edge-mediated physical interaction process.
243 to MXenes247 and black phosphorous248 nanosheets, provide a picture of this membrane damage process which seems to be quite uniform and shared among these different classes of nanosheets, thereby helping us in proposing a general mechanism of membrane-directed antibacterial action of these nanosheets by taking into consideration the “nanosheets” as a family.
Upon establishment of primary physical contact with the phospholipid bilayer, the nanosheets begin to interact with the lipid head groups of the membrane through a wide variety of interactive forces such as electrostatic, van der Waals's and hydrophobic interactions, depending on the chemical nature of the nanosheets. Graphene sheets interact primarily through hydrophobic interactions,240 while the oxygen-containing functional groups of GO and rGO,238 and Mo and S atoms of MoS2 nanosheets234 interact through electrostatic forces with the phospholipid heads (Fig. 9B & C). As a result, the nanosheets start to embed themselves into the phospholipid membranes, creating dents or troughs on the membrane surface.234 These membrane perturbations lead to the generation of patches of upturned phospholipids on the surface, as discussed before, and mark the initiation of membrane disruption.237,238 The embedding of the nanosheets into the lipid bilayer intensifies the ongoing interactions between the lipid molecules and the nanosheet surface, as a result of which deepening of dents is observed which in turn exposes the hydrophobic lipid tails buried in the membrane core. Strong interactions take place between the nanosheet surface and the hydrophobic lipid tails resulting in destructive extraction of phospholipid molecules from the membrane onto the surface of the nanosheets. MD simulation studies using both graphene240 and MoS2234 nanosheets clearly show the process of phospholipid extraction by these nanosheets. MoS2 nanosheets were found to mediate this process through the formation of dents on the membrane surface (Fig. 9B), whereas graphene nanosheets caused direct penetration of the lipid bilayer along with lipid extraction (Fig. 9C). This event of phospholipid extraction disturbs the membrane integrity to a large extent, causing rapid depolarization of the membranes.244 The membranes lose their permeability barrier and become more vulnerable to further damage. However, it is to be noted that at this stage, the membrane is still intact and not fully disintegrated. For complete disintegration of the membrane to take place, the already embedded nanosheets now need to further penetrate into the membrane and puncture through the lipid bilayer. At this point, the edge-mediated physical interaction starts to dominate. The process of phospholipid extraction weakens the bilayer structure, as a result of which it becomes easier for the embedded nanosheets to penetrate further into the membrane, ultimately causing the membrane to rupture and disintegrate. This results in leakage of the cytoplasmic contents of the cells, such as DNA, RNA and proteins.
• ROS-dependent oxidative stress: Nanosheets are capable of generating ROS or free radicals by interacting with molecular oxygen available in the surrounding environment. The oxygen molecules adsorb on the defects and edges of the nanosheets and get reduced into free radicals such as superoxide anion, hydrogen peroxide, singlet oxygen and hydroxyl radical.231,249 The degree of oxidation and the presence of surface defects on the nanosheets are key factors in determining the ROS-producing abilities of nanosheets. Studies on GO and rGO have shown GO sheets to exert more antibacterial activity as compared with rGO, through generation of more intracellular ROS.250,251 The greater oxidation capacity of GO nanosheets can be attributed to the presence of numerous oxygen-containing functional groups and increased defect density on their surface in comparison with rGO.251–253 In addition to these factors, the size of nanosheets also influences their oxidation capacity and thus antibacterial action. In studies involving GO,231 MoS2244 and MXene247 nanosheets, a size-dependent antibacterial action was observed to take place, with smaller nanosheets showing stronger antibacterial action as compared with their larger counterparts. This is again due to the higher oxidative nature of smaller nanosheets because with decrease in size, the density of surface defects increases, and the higher the defect density, the greater is the ROS-generating ability of the nanosheets. The membrane-directed antibacterial action of nanosheets also results in the formation of oxidative stress inside the bacterial cells. This, coupled with the nanosheets’ intrinsic material property to generate abiotic ROS, overpowers the bacterial antioxidant mechanism and results in disturbing the redox equilibrium of the cells through GSH oxidation. The free radicals cause further damage to the cells through oxidation of key bacterial components like DNA, RNA, proteins, lipids etc., thereby rendering them non-functional.254 Along with these, the oxidative stress generated can also damage the bacterial membrane through peroxidation of the membrane phospholipids, which in turn generates more reactive oxygen species like lipid peroxide radicals that participate in propagating the on-going oxidative damage.255
• Charge (electron) transfer mediated ROS-independent oxidative stress: Apart from ROS-mediated oxidative stress, nanosheets can also exert oxidative stress on bacteria through a mechanism arising from the conductive nature of nanosheets without the involvement of free radical generation. Studies with GO,233 MoS2,242,243,256 WS2,243 and MoSe2243 nanosheets, which are all known to have good electrical conductivity, have shown their involvement in directly disrupting or inhibiting a key biochemical process or structure of bacterial cells through the process of electron transfer. The nanosheets, upon interacting with the bacterial membrane, form a conductive layer on the surface of the membrane. This conductive layer then behaves as an electron pump and transfers electrons from bacterial membrane or from intracellular components to the outside environment, thereby oxidizing the bacterial components in the process.241,257 Several studies have reported the direct GSH oxidizing properties of different nanosheets, which have helped in establishing the fact that nanosheets are also capable of directly disturbing the redox equilibrium of bacterial cells and create oxidative stress in a ROS-independent manner. The higher GSH oxidizing property of rGO observed in comparison with GO sheets is also attributed to the better conductivity of rGO than GO.241 Such a process of nanosheet-assisted electron transfer from bacteria is widely considered to be the primary mechanism of antibacterial action of nanosheets, especially when such nanosheets are employed in the form of coatings and substrates.
The above discussions provide a clear picture that the antibacterial action of nanosheets is not just a result of physical damage to the bacterial cells caused by the sharp edges/corners of these nanosheets, but also due to the chemical nature of the different nanosheets that gives rise to their oxidative mode of action. The consequences of membrane-directed mechano-bactericidal action of nanosheets coupled with ROS-dependent and independent pathways of oxidative stress generation make the bacterial cells completely lose their structural and functional integrity, finally leading to cell death (Fig. 10).
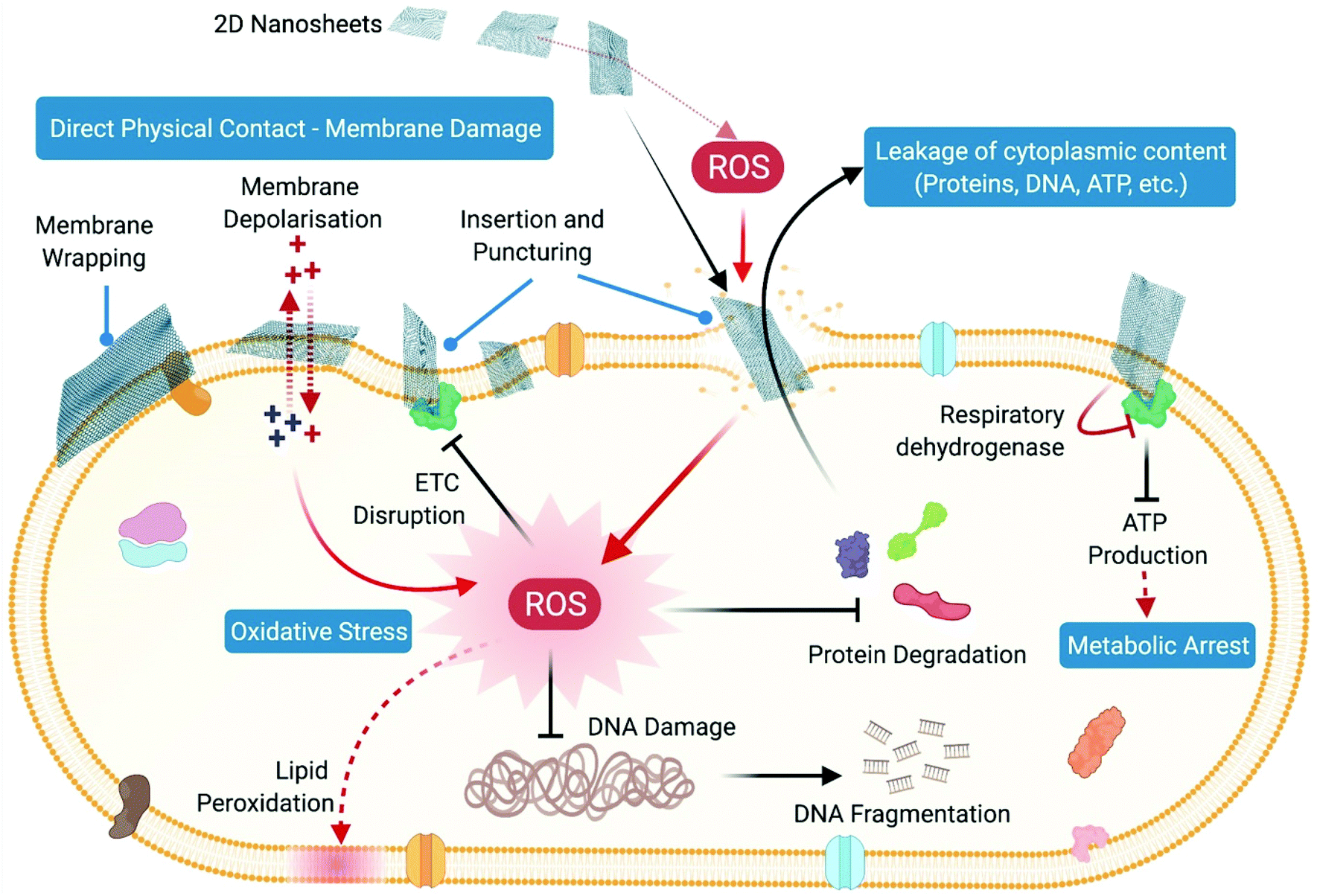 | ||
| Fig. 10 Schematic showing the mechanism of antibacterial action of 2D nanosheets. | ||
4.5 Mechanism of antiviral action of 2D nanosheets
The antiviral action of nanosheets has been attributed to their negative surface charge and the presence of a layered nanoscale morphology with sharp edges and corners.258 The primary mechanism of antiviral action observed in studies involving GO and rGO nanosheets against both enveloped and non-enveloped viruses is the direct interaction of the nanosheets with the virus surface through strong electrostatic interactions, which ultimately results in inducing physical damage to the viral structure through a sharp edge-mediated effect, as also observed in case of bacteria.258The importance of a monolayer structure of nanosheets in determining their antiviral activity was evaluated by comparing the antiviral properties of graphite, graphite oxide, graphene oxide and reduced graphene oxide,258 and it was observed that only GO and rGO with a monolayer structure and nanoscale size could demonstrate significant antiviral activity, whereas graphite and graphite oxide, which act as precursors to these nanostructures, showed very weak or complete absence of antiviral activity due to their multi-layered morphology and larger lateral size.258
In addition to morphology and size, surface charge is another key factor influencing the antiviral action of nanosheets. Studies indicated that it is the charge density and not charge identity that actually determines the antiviral activity of these nanosheets.259 GO and rGO both possess similar net negative charge but differ in the type of functional groups present on their surfaces. The antiviral activity of GO and rGO was found to be similar, demonstrating that the presence of different functional groups did not affect the antiviral action.258 This has been further supported by comparing the antiviral properties of GO and sulfonated rGO nanosheets.259 It was observed that the introduction of additional negatively charged sulfonate groups in the GO structure did not improve its antiviral activity, as both GO and sulfonated rGO had the same negative charge density.
Detailed studies on understanding the exact nature of viral inactivation by GO and MoS2 nanosheets have shown that these nanosheets are not able to inhibit viral infection in cell lines if the virus has already attached to the cells or the cells have been pre-incubated with nanosheets before virus addition.258,260 The nanosheets were found to be effective only when they were pre-incubated with the virus particles prior to infection. Thus, the main mode of action of these nanosheets is by preventing the entry of viruses into cells by capturing them prior to cellular attachment.258,259
Once the nanosheets get attached to the surface of the virus particles, the presence of sharp edges and corners of nanosheets can cause physical damage to the virus structure as evidenced through transmission electron microscopy in the case of both enveloped and non-enveloped viruses. It has been observed that GO nanosheets are quite capable of completely disrupting the virus morphology by damaging the viral envelope, capsid, and viral proteins such as spike proteins which are present on the surface of the virus. Interaction of GO nanosheets with pseudorabies virus (PRV) and PEDV was found to destroy the viral envelope and spike proteins of these viruses258 (Fig. 11A–D). Similar results were also observed with enterovirus 71 (EV71) whose regular hexahedron morphology was completely broken by GO, and with influenza A virus subtype H9N2 where the spherical/filamentous virus particles were found to be collapsed upon interaction with GO nanosheets and their spike proteins were drastically destroyed261 (Fig. 11E). Similarly, physical disruption of herpes simplex virus-1 (HSV-1) by MoS2 nanosheets has also been observed. The sharp edges of the nanosheets were found to disrupt the viral envelope, leading to leakage of internal viral material.260
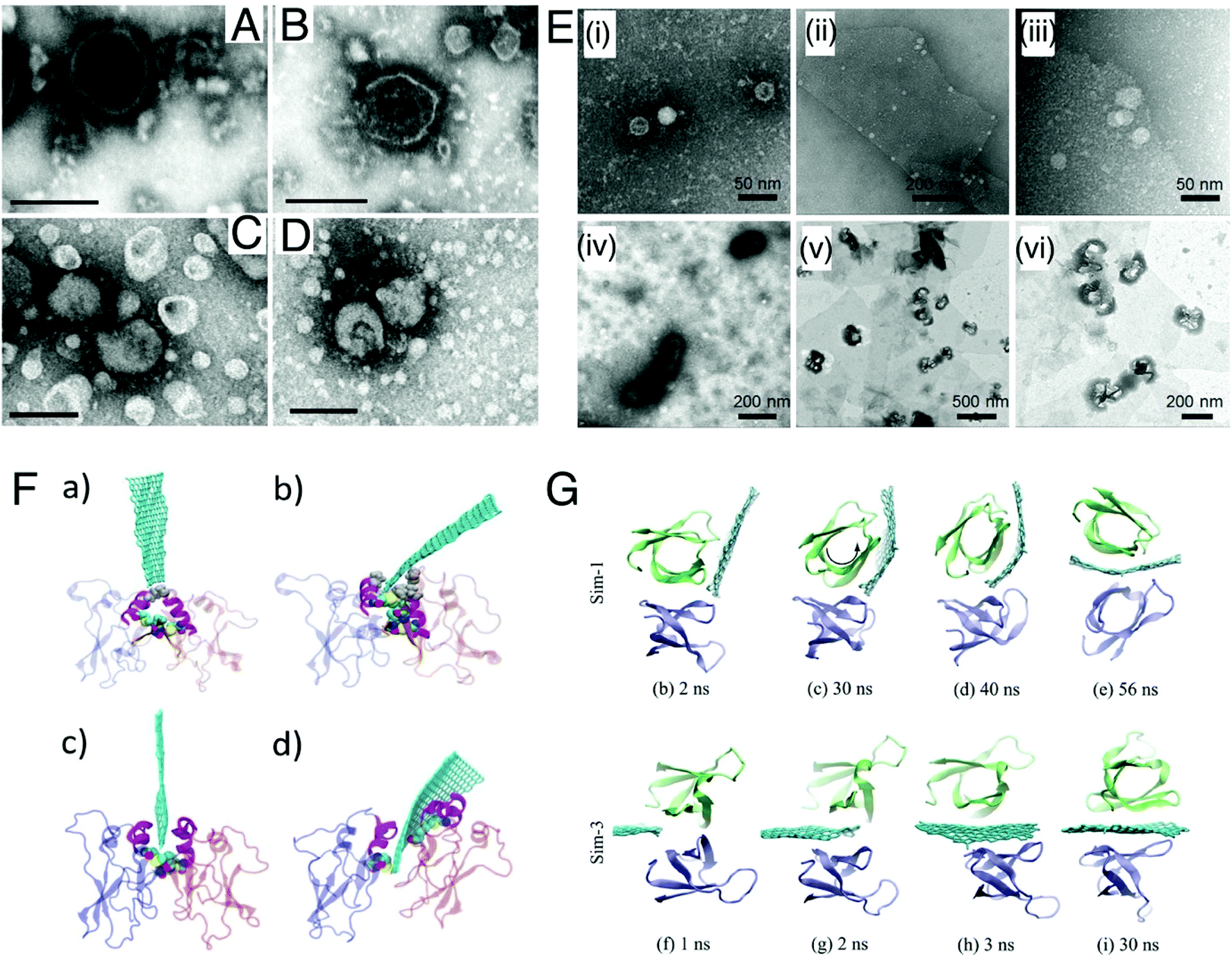 | ||
| Fig. 11 Mechanism of antiviral action of 2D nanosheets. Transmission electron microscopic images of GO-treated viruses. (A) PRV control; (B) PRV treated with GO for 1 h; (C) PEDV control; and (D) PEDV incubated with GO for 1 h. Scale bars: 200 nm (A and B) and 100 nm (C and D). Reproduced with permission.258 Copyright 2015, American Chemical Society. (E) TEM images of negative stained native EV71 (i) and GO captured EV71 complexes at (ii) low and (iii) high magnification; TEM images of negative stained (iv) H9N2 and GO captured H9N2 complexes at (v) low and (vi) high magnification with thermal heating treatment. Reproduced with permission.261 Copyright 2015, Wiley. (F) MD snapshots of the graphene insertion through the hexamer CTD–CTD interface. (a and b) Initial breaking of A229/P234 and (c and d) graphene disrupting the M241-I307 pair and inserting farther into the interface. Reproduced with permission.262 Copyright 2017, Elsevier. (G) Snapshots of the insertion process of a graphene sheet into the dimer. Reproduced with permission.263 Copyright 2015, American Chemical Society. | ||
This disruption of the viral structure and viral proteins by nanosheets can also arise from the chemical properties of nanosheets, where the nanosheets can mediate their antiviral action through disruption of key protein–protein interactions that play crucial roles in maintaining virus structural integrity and infectivity. MD simulation showed that graphene nanosheets can interact very strongly with Ebola virus VP40 oligomers through hydrophobic interactions and break apart these oligomers by penetrating through them (Fig. 11F). VP40 proteins are very crucial in forming the Ebola virus matrix.262 VP40 hexamers undergo oligomerization through their C-terminal domains with adjacent hexamers, forming VP40 filaments which make up the entire Ebola virus matrix. Thus, disrupting the formation of these filaments can directly inactivate the virus and act as a mode of antiviral action. Graphene sheets, owing to their strong hydrophobic nature, insert themselves through the hydrophobic CTD–CTD interactions of the hexamers and separate the hexameric domains from each other. Similar observations have also been reported for HIV-1 integrase proteins, where the dimeric conformation of the integrase protein is destroyed by graphene nanosheets263 (Fig. 11G).
From molecular docking analyses it has been recently revealed that GO binds with strong affinity to the SARS-CoV-2 spike glycoprotein and its mutated form (Fig. 12A & B).264 The GO nanosheets were also capable of interacting with the host receptor ACE2 and its complex with the spike protein spike–ACE2 complex. Upon binding, GO nanosheets acted as a hydrogen donor and acceptor at major positions on the surface of the spike protein and interacted with the proteins via non-covalent interactions. Among such interactions, GO interaction at the RBD region of SARS-CoV-2 via electrostatic and hydrogen bonding is of great significance for being an antiviral agent.264 Another study provided detailed insight about deactivation of the SARS-CoV-2 protease Mpro by disabling of the active site and reduced expression upon direct interaction with graphene materials (pristine graphene, GO and defective graphene) (Fig. 12C).265 Molecular dynamics simulations showed that Mpro adsorbs onto the surface of graphene through hydrophobic interaction, while with its derivatives the protein interacted with its hydrophilic amino acids. The molecular structure of Mpro and its active site is stabilized by several hydrogen bonds. The effective adsorption of the active site on graphene derivative caused hydrogen bond breakage, which induced instability in the posture and conformation of the active site resulting in loss of function (viral replication and transcription) of Mpro. By damaging the active site of Mpro, graphene has emerged as an efficient inhibitor of SARS-CoV-2 infection.265 In another recent study, Shahbazi et al. studied the effect of multiple 2D nanomaterials (bismuthene, graphene, phosphorene, P-doped graphene, and functionalized P-doped graphene) on SARS-CoV-2 and claimed that the surface chemistry and architecture of these nanomaterials has a strong influence on the inhibition of SARS-CoV-2 infectivity. Nearly all 2D nanomaterials reduced the hydrophobic interaction and hydrogen bonding between spike and ACE2 by distorting the stable conformation of the spike structure. Along with this, the graphene and functionalized P-doped graphene nanosheets directly contacted with lipid, hydrophobic components, and ion channels of the envelope membrane of the virion and penetrated inside the membrane, causing viral membrane disruption. Inactivation of Mpro by functionalized P-doped graphene nanosheet makes it significant inhibitor to combat SARS-CoV-2 infection completely.266 GO has proven to be an excellent adsorbent nanomaterial due to the availability of negatively charged oxygen functional groups on its dual sides. The positive patches of amino acid residues present on the spike glycoprotein and nucleocapsid protein of SARS-CoV-2 subsequently adsorb via electrostatic interaction onto the negative surface of GO nanosheet. This direct contact of GO–viral protein causes destruction of structural integrity of both viral proteins, thereby leading to loss of function followed by viral deactivation. The presence of the oxygen functional group plays significant role in exhibiting antiviral activity against the SARS-CoV-2 as compared with reduced GO nanosheets due to its ability to facilitate effective electrostatic interaction with virions (Fig. 12D).267 Bidimensional graphene nanosheets are flexible in nature too, which along with polyglycerol sulfate (PGS) and aliphatic chain functionalization provides a platform for containment of the Feline coronavirus (FCoV) via electrostatic interactions with alkyl chains of graphene nanosheets and rupturing via hydrophobic interactions directly with graphene nanosheets. In the virustatic and virucidal mode of action, the functionalized graphene nanosheets prohibit the entry of virions by exhibiting electrostatic interaction between negatively charged Polyglycerol Sulfate present in graphene derivative and positively charged amino acid residues of the spike glycoprotein of FCoV and inactivation by causing envelope disintegration upon interacting with lipids of the viral envelope.268 This has led to the utilization of graphene and its derivatives as an antiviral material for developing personal protective equipment such as face masks.269,270
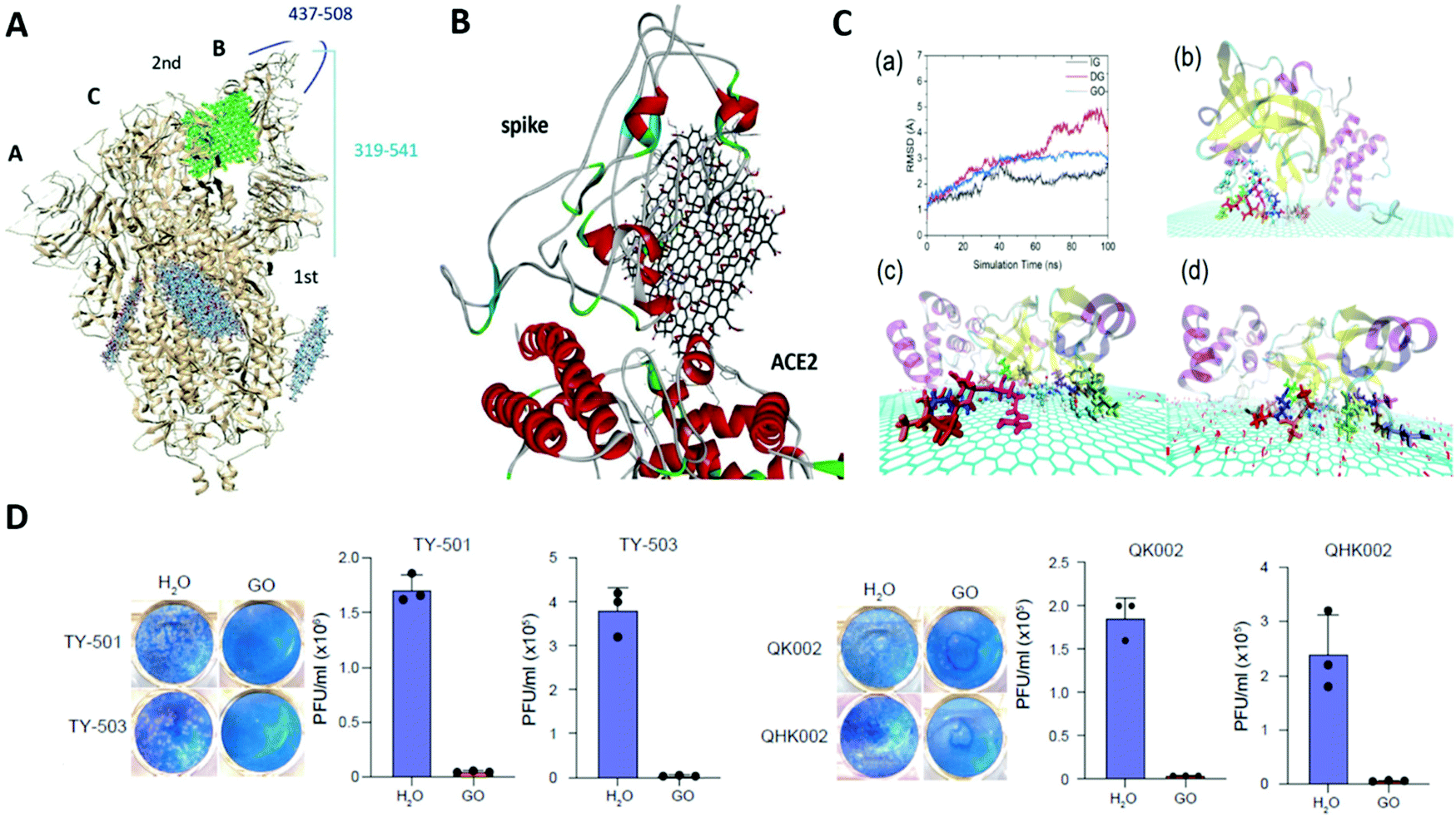 | ||
| Fig. 12 Anti-SARS-CoV-2 activity of 2D nanosheets. (A) Binding of GO at different sites of open state of spike protein (6VYB). (B) Binding of GO with spike-ACE2 complex (6M0J) with binding affinity −9.1 kcal mol−1. Reproduced with permission. Copyright 2021, Wiley.264 (C) Deactivation of the SARS-CoV-2 protease Mpro by GO. (a) RMSD of the active pocket of Mpro for all systems during 100 ns MD simulations. Representative snapshots of Mpro adsorbed onto (b) IG, (c) DG, and (d) GO. Reproduced with permission.265 Copyright 2021, American Chemical Society. (D) Anti-SARS-CoV-2 activity against Brazilian strains (TY7-501 and TY7-503) and U.K. strains (QK002 and QHN001) in the presence and absence of GO (100 μg mL−1) using plaque assay. Reproduced with permission.267 Copyright 2021, American Chemical Society. | ||
Thus, nanosheets with a net negative surface charge and a monolayer morphology exert their antiviral activity by strongly attaching themselves to the surface of positively charged virus particles through electrostatic interactions, thereby capturing them on their surface. This in turn inhibits the attachment of these viruses to the cell surface and thus prevents viral entry into the cells. This is coupled with nanosheet edge-mediated physical destruction of the viruses, which finally leads to their inactivation.
5. Conclusion and future perspective
In summary, the mechanism of action of antibacterial and antiviral agents ranging from macroscale polymers, proteins and peptides to nanoscale particles, tubes and sheets is provided. The mechanism of antibacterial action of macroscale biomaterials, such as antimicrobial peptides and polymers, mainly occurs by targeting the bacterial membrane. These biomaterials damage the cell membrane either through “pore-forming” pathways or via “non-pore-forming” pathways of membrane disruption ultimately leading to cellular lysis. In addition to membrane-targeting mechanisms, antimicrobial peptides also exert their bactericidal activities by inhibiting any of the key intracellular processes like nucleic acids synthesis, cell wall synthesis, protein synthesis and so on. Polymers, both natural and synthetic, can have intrinsic antimicrobial activities, or they can be functionalized with different chemical groups to mimic the antimicrobial peptides. Polymers exert their antimicrobial activity mainly by targeting the cell membrane through pathways similar to that of antimicrobial peptides. Peptido-polysaccharides, which are a combination of peptides and polysaccharides, act as bacterial cell wall mimics and thus interfere with cell wall synthesis and induce osmotic instability.The antibacterial action of nanoparticles, nanotubes and nanosheets is mainly dictated by two main pathways: (i) membrane damage, and (ii) oxidative stress. All kinds of nanostructures, irrespective of their size, morphology and surface chemistry, initiate the process of antibacterial activity by establishing direct physical contact with the bacterial membrane; this then proceeds towards the induction of membrane stress followed by membrane damage. This process of generation of membrane stress differs among the different classes of nanostructures. Nanoparticles mostly adhere to the membrane surface and cause local perturbations leading to changes in membrane permeability and integrity, without directly penetrating or puncturing the lipid bilayer. On the other hand, nanotubes and nanosheets directly puncture and pierce the bacterial membranes using their sharp edges, corners and narrow tips, creating pores in the lipid bilayer. The generation of oxidative stress in the form of ROS is observed for all types of nanostructures, and leads to oxidative damage of lipids, proteins, nucleic acids and membrane components. Nanoparticles are found to exert this oxidative stress mainly through a ROS-dependent pathway, whereas nanotubes and nanosheets contribute to the overall oxidative stress-mediated cell death involving both ROS-dependent and ROS-independent pathways. The intrinsic material properties of nanotubes and nanosheets participate in direct electron transfer to intracellular components such as proteins and nucleic acids, leading to their oxidation without the involvement of free radicals. On the other hand, cellular uptake of nanoparticles and their dissolved/leached out ions delivers direct damage to proteins and nucleic acids through dephosphorylation, intercalation, condensation and disintegration.
Focusing on the virucidal properties of biomaterials, it can be stated that polymers and peptides act by making each step of the viral infection and replication a potential target. However, nanoparticles can exert their antiviral action at multiple stages of virus infection, such as directly damaging the viral structure, preventing the virus from entering the host cells by binding to receptors, inhibition of viral DNA/RNA replication, damage to the viral genome, induction of a pro-inflammatory phenotype in host cells and by blocking the budding off and release of viral progenies from the host cell surface. Nanosheets are found to act upon the viruses by directly destroying their structure and membrane/envelope proteins. Nanotubes, on the other hand, are not found to exert antiviral activity.
Overall, this review provides a comprehensive understanding about how different classes of biomaterials interact with bacteria and viruses to exert their antimicrobial action. It is very important to understand the nature and specific outcome of such interactions to develop better antimicrobial biomaterials. By knowing the detailed mechanism of action of the biomaterials, researchers will be able to develop novel antimicrobial agents against which bacteria or viruses cannot easily become resistant. Furthermore, understanding these diverse mechanisms will enable researchers to choose the most appropriate biomaterials for specific applications, such as for biofilm eradication, development of coating materials for biomedical implants, wound healing materials, tissue engineering scaffolds etc. It will also assist in developing better strategies for designing next-generation antimicrobial PPEs and antiviral agents. Combining macroscale and nanoscale biomaterials into a single antimicrobial platform may provide biomaterials with better therapeutic efficacy through the combined effects of the individual properties of each component.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors acknowledge the support from the BioX Centre and Advanced Materials Research Centre (AMRC), the Indian Institute of Technology Mandi for Research and Infrastructure facility. Shounak Roy would like to acknowledge DST INSPIRE Fellowship [IF160513] for providing doctoral fellowship. Fig. 1–4, 6, 7 and 10 and the graphical abstract was created with BioRender.com and the authors gratefully acknowledge BioRender.References
- M. L. Cohen, Nature, 2000, 406, 762–767 CrossRef CAS PubMed.
- D. E. Bloom and D. Cadarette, Front. Immunol., 2019, 10, 549 CrossRef PubMed.
- H. H. Khachfe, M. Chahrour, J. Sammouri, H. Salhab, B. E. Makki and M. Fares, Cureus, 2020, 12(3), e7313 Search PubMed.
- A. Findlater and I. I. Bogoch, Trends Parasitol., 2018, 34, 772–783 CrossRef PubMed.
- J. P. Koplan, T. C. Bond, M. H. Merson, K. S. Reddy, M. H. Rodriguez, N. K. Sewankambo and J. N. Wasserheit, Lancet, 2009, 373, 1993–1995 CrossRef.
- N. I. Nii-Trebi, BioMed Res. Int., 2017, 2017, 5245021 Search PubMed.
- W. Hall, Superbugs: An arms race against bacteria, Harvard University Press, 2018 Search PubMed.
- B. Aslam, W. Wang, M. I. Arshad, M. Khurshid, S. Muzammil, M. H. Rasool, M. A. Nisar, R. F. Alvi, M. A. Aslam and M. U. Qamar, Infect. Drug Resist., 2018, 11, 1645 CrossRef CAS PubMed.
- M. Frieri, K. Kumar and A. Boutin, J. Infect. Public Health, 2017, 10, 369–378 CrossRef PubMed.
- I. Yelin and R. Kishony, Cell, 2018, 172, 1136–1136 CrossRef CAS PubMed.
- H.-P. Shih, X. Zhang and A. M. Aronov, Nat. Rev. Drug Discovery, 2018, 17, 19–33 CrossRef CAS PubMed.
- L.-j. Zhang and R. L. Gallo, Curr. Biol., 2016, 26, R14–R19 CrossRef CAS PubMed.
- S. Y. Tan and Y. Tatsumura, Singapore Med. J., 2015, 56, 366 CrossRef PubMed.
- T. Franklin and G. Snow, in Biochemistry and molecular biology of antimicrobial drug action, Springer, 1998, pp. 43–59 Search PubMed.
- K. Matsuzaki, O. Murase, N. Fujii and K. Miyajima, Biochemistry, 1996, 35, 11361–11368 CrossRef CAS PubMed.
- P. Li, C. Zhou, S. Rayatpisheh, K. Ye, Y. F. Poon, P. T. Hammond, H. Duan and M. B. Chan-Park, Adv. Mater., 2012, 24, 4130–4137 CrossRef CAS PubMed.
- N. Baig, I. Kammakakam and W. Falath, Mater. Adv., 2021, 2, 1821–1871 RSC.
- E. O. Ogunsona, R. Muthuraj, E. Ojogbo, O. Valerio and T. H. Mekonnen, Appl. Mater. Today, 2020, 18, 100473 CrossRef.
- G. G. Luo and S. J. Gao, J. Med. Virol., 2020, 92, 399 CrossRef CAS PubMed.
- C. S. Adamson, K. Chibale, R. J. Goss, M. Jaspars, D. J. Newman and R. A. Dorrington, Chem. Soc. Rev., 2021, 50, 3647–3655 RSC.
- M. Ito, M. Baba, A. Sato, R. Pauwels, E. De Clercq and S. Shigeta, Antiviral Res., 1987, 7, 361–367 CrossRef CAS PubMed.
- A. Grassauer, R. Weinmuellner, C. Meier, A. Pretsch, E. Prieschl-Grassauer and H. Unger, Virol. J., 2008, 5, 1–13 CrossRef PubMed.
- L. Yu, K. Li, J. Zhang, H. Jin, A. Saleem, Q. Song, Q. Jia and P. Li, ACS Appl. Bio Mater., 2022, 5(2), 366–393 CrossRef CAS PubMed.
- M. A. Abdalla, J. Nat. Med., 2016, 70, 708–720 CrossRef CAS PubMed.
- W. C. Wimley, ACS Chem. Biol., 2010, 5, 905–917 CrossRef CAS PubMed.
- E. F. Palermo, I. Sovadinova and K. Kuroda, Biomacromolecules, 2009, 10, 3098–3107 CrossRef CAS PubMed.
- E. R. Cobo and K. Chadee, Pathogens, 2013, 2, 177–192 CrossRef PubMed.
- M. Zasloff, Lancet, 2002, 360, 1116–1117 CrossRef.
- H. Tanabe, T. Sato, J. Watari, A. Maemoto, M. Fujiya, T. Kono, T. Ashida, T. Ayabe and Y. Kohgo, Helicobacter, 2008, 13, 370–379 CrossRef CAS PubMed.
- C. Zhou, X. Qi, P. Li, W. N. Chen, L. Mouad, M. W. Chang, S. S. Leong and M. B. Chan-Park, Biomacromolecules, 2010, 11, 60–67 CrossRef CAS PubMed.
- J. Gao, M. Wang, F. Wang and J. Du, Biomacromolecules, 2016, 17, 2080–2086 CrossRef CAS PubMed.
- J. C. Tiller, C. J. Liao, K. Lewis and A. M. Klibanov, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5981–5985 CrossRef CAS PubMed.
- Y. Hu, Y. Du, J. Yang, Y. Tang, J. Li and X. Wang, Polymer, 2007, 48, 3098–3106 CrossRef CAS.
- M. Kong, X. G. Chen, C. S. Liu, C. G. Liu and X. H. Meng, Colloids Surf., B, 2008, 65, 197–202 CrossRef CAS PubMed.
- Y. Zhang, J. Jiang and Y. Chen, Polymer, 1999, 40, 6189–6198 CrossRef CAS.
- H. Tan, R. Ma, C. Lin, Z. Liu and T. Tang, Int. J. Mol. Sci., 2013, 14, 1854–1869 CrossRef CAS PubMed.
- Y. Chen, S. Worley, J. Kim, C.-I. Wei, T.-Y. Chen, J. Santiago, J. Williams and G. Sun, Ind. Eng. Chem. Res., 2003, 42, 280–284 CrossRef CAS.
- T. J. Cuthbert, B. Hisey, T. D. Harrison, J. F. Trant, E. R. Gillies and P. J. Ragogna, Angew. Chem., Int. Ed., 2018, 57, 12707–12710 CrossRef CAS PubMed.
- M. N. Stadtmueller, S. Bhatia, P. Kiran, M. Hilsch, V. Reiter-Scherer, L. Adam, B. Parshad, M. Budt, S. Klenk, K. Sellrie, D. Lauster, P. H. Seeberger, C. P. R. Hackenberger, A. Herrmann, R. Haag and T. Wolff, J. Med. Chem., 2021, 64, 12774–12789 CrossRef CAS PubMed.
- B. P. Mowery, S. E. Lee, D. A. Kissounko, R. F. Epand, R. M. Epand, B. Weisblum, S. S. Stahl and S. H. Gellman, J. Am. Chem. Soc., 2007, 129, 15474–15476 CrossRef CAS PubMed.
- S. Saidjalolov, Z. Edoo, M. Fonvielle, L. Mayer, L. Iannazzo, M. Arthur, M. Etheve-Quelquejeu and E. Braud, Chemistry, 2021, 27, 3542–3551 CrossRef CAS PubMed.
- H. Gleiter, Acta Mater., 2000, 48, 1–29 CrossRef CAS.
- V. Pokropivny and V. Skorokhod, Phys. E, 2008, 40, 2521–2525 CrossRef CAS.
- W. Niu, L. Zhang and G. Xu, Nanoscale, 2013, 5, 3172–3181 RSC.
- P. Innocenzi and L. Stagi, Chem. Sci., 2020, 11, 6606–6622 RSC.
- H. Sun, L. Wu, W. Wei and X. Qu, Mater. Today, 2013, 16, 433–442 CrossRef CAS.
- A. Al-Jumaili, S. Alancherry, K. Bazaka and M. V. Jacob, Materials, 2017, 10, 1066 CrossRef PubMed.
- A. Jaiswal, S. S. Ghosh and A. Chattopadhyay, Chem. Commun., 2012, 48, 407–409 RSC.
- R. Hirlekar, M. Yamagar, H. Garse, M. Vij and V. Kadam, Asian J. Pharm. Clin. Res., 2009, 2, 17–27 CAS.
- T. Mocan, C. T. Matea, T. Pop, O. Mosteanu, A. D. Buzoianu, S. Suciu, C. Puia, C. Zdrehus, C. Iancu and L. Mocan, Cell. Mol. Life Sci., 2017, 74, 3467–3479 CrossRef CAS PubMed.
- S. Lal, J. H. Hafner, N. J. Halas, S. Link and P. Nordlander, Acc. Chem. Res., 2012, 45, 1887–1895 CrossRef CAS PubMed.
- J. Lu, H. Liu, X. Zhang and C. H. Sow, Nanoscale, 2018, 10, 17456–17476 RSC.
- B. Mandl, J. Stangl, E. Hilner, A. A. Zakharov, K. Hillerich, A. W. Dey, L. Samuelson, G. Bauer, K. Deppert and A. Mikkelsen, Nano Lett., 2010, 10, 4443–4449 CrossRef CAS PubMed.
- G. Milano, S. Porro, I. Valov and C. Ricciardi, Adv. Electron. Mater., 2019, 5, 1800909 CrossRef.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- G. R. Bhimanapati, Z. Lin, V. Meunier, Y. Jung, J. Cha, S. Das, D. Xiao, Y. Son, M. S. Strano and V. R. Cooper, ACS Nano, 2015, 9, 11509–11539 CrossRef CAS PubMed.
- R. Kurapati, K. Kostarelos, M. Prato and A. Bianco, Adv. Mater., 2016, 28, 6052–6074 CrossRef CAS PubMed.
- C. Tan, X. Cao, X.-J. Wu, Q. He, J. Yang, X. Zhang, J. Chen, W. Zhao, S. Han and G.-H. Nam, Chem. Rev., 2017, 117, 6225–6331 CrossRef CAS PubMed.
- C. Gokce, C. Gurcan, O. Besbinar, M. A. Unal and A. Yilmazer, Nanoscale, 2021, 14, 239–249 RSC.
- V. Yadav, S. Roy, P. Singh, Z. Khan and A. Jaiswal, Small, 2019, 15, 1803706 CrossRef PubMed.
- K. Matsuzaki, Adv. Exp. Med. Biol., 2019, 1117, 9–16 CrossRef CAS PubMed.
- G. Ehrenstein and H. Lecar, Q. Rev. Biophys., 1977, 10, 1–34 CrossRef CAS PubMed.
- M. S. Sansom, Prog. Biophys. Mol. Biol., 1991, 55, 139–235 CrossRef CAS PubMed.
- K. Matsuzaki, O. Murase, N. Fujii and K. Miyajima, Biochemistry, 1996, 35, 11361–11368 CrossRef CAS PubMed.
- S. J. Ludtke, K. He, W. T. Heller, T. A. Harroun, L. Yang and H. W. Huang, Biochemistry, 1996, 35, 13723–13728 CrossRef CAS PubMed.
- D. Sengupta, H. Leontiadou, A. E. Mark and S. J. Marrink, Biochim. Biophys. Acta, 2008, 1778, 2308–2317 CrossRef CAS PubMed.
- B. Bechinger and K. Lohner, Biochim. Biophys. Acta, 2006, 1758, 1529–1539 CrossRef CAS PubMed.
- E. Gazit, I. R. Miller, P. C. Biggin, M. S. Sansom and Y. Shai, J. Mol. Biol., 1996, 258, 860–870 CrossRef CAS PubMed.
- M. Vaara and T. Vaara, Antimicrob. Agents Chemother., 1994, 38, 2498–2501 CrossRef CAS PubMed.
- M. C. Wiener and S. H. White, Biophys. J., 1992, 61, 434–447 CrossRef CAS PubMed.
- A. Wiese, M. Münstermann, T. Gutsmann, B. Lindner, K. Kawahara, U. Zähringer and U. Seydel, J. Membr. Biol., 1998, 162, 127–138 CrossRef CAS PubMed.
- M. Miteva, M. Andersson, A. Karshikoff and G. Otting, FEBS Lett., 1999, 462, 155–158 CrossRef CAS PubMed.
- S. Ludtke, K. He and H. Huang, Biochemistry, 1995, 34, 16764–16769 CrossRef CAS PubMed.
- S. L. Grage, S. Afonin, S. Kara, G. Buth and A. S. Ulrich, Front. Cell Dev. Biol., 2016, 4, 65 Search PubMed.
- H. W. Huang, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183395 CrossRef CAS PubMed.
- L. Jeu and H. B. Fung, Clin. Ther., 2004, 26, 1728–1757 CrossRef CAS PubMed.
- E. F. Haney, S. Nathoo, H. J. Vogel and E. J. Prenner, Chem. Phys. Lipids, 2010, 163, 82–93 CrossRef CAS PubMed.
- J. P. Powers, A. Tan, A. Ramamoorthy and R. E. Hancock, Biochemistry, 2005, 44, 15504–15513 CrossRef CAS PubMed.
- R. M. Epand and R. F. Epand, J. Pept. Sci., 2011, 17, 298–305 CrossRef CAS PubMed.
- G. Pabst, S. L. Grage, S. Danner-Pongratz, W. Jing, A. S. Ulrich, A. Watts, K. Lohner and A. Hickel, Biophys. J., 2008, 95, 5779–5788 CrossRef CAS PubMed.
- H. Brötz, G. Bierbaum, A. Markus, E. Molitor and H. G. Sahl, Antimicrob. Agents Chemother., 1995, 39, 714–719 CrossRef PubMed.
- R. A. Salomón and R. N. Farías, J. Bacteriol., 1992, 174, 7428–7435 CrossRef PubMed.
- M. Ishikawa, T. Kubo and S. Natori, Biochem. J., 1992, 287(Pt 2), 573–578 CrossRef CAS PubMed.
- C. B. Park, H. S. Kim and S. C. Kim, Biochem. Biophys. Res. Commun., 1998, 244, 253–257 CrossRef CAS PubMed.
- G. A. Birkemo, T. Lüders, Ø. Andersen, I. F. Nes and J. Nissen-Meyer, Biochim. Biophys. Acta, 2003, 1646, 207–215 CrossRef CAS.
- M. Mardirossian, R. Grzela, C. Giglione, T. Meinnel, R. Gennaro, P. Mergaert and M. Scocchi, Chem. Biol., 2014, 21, 1639–1647 CrossRef CAS PubMed.
- A. Patrzykat, C. L. Friedrich, L. Zhang, V. Mendoza and R. E. Hancock, Antimicrob. Agents Chemother., 2002, 46, 605–614 CrossRef CAS PubMed.
- C. L. Friedrich, A. Rozek, A. Patrzykat and R. E. Hancock, J. Biol. Chem., 2001, 276, 24015–24022 CrossRef CAS PubMed.
- M. Dangkulwanich, C. R. H. Raetz and A. H. Williams, Sci. Rep., 2019, 9, 3947 CrossRef PubMed.
- R. H. Bianculli, J. D. Mase and M. D. Schulz, Macromolecules, 2020, 53, 9158–9186 CrossRef CAS.
- S. Sarrazin, W. C. Lamanna and J. D. Esko, Cold Spring Harbor Perspect. Biol., 2011, 3, a004952 Search PubMed.
- V. Cagno, E. D. Tseligka, S. T. Jones and C. Tapparel, Viruses, 2019, 11(7), 596 CrossRef CAS PubMed.
- C. Herrscher, P. Roingeard and E. Blanchard, Cells, 2020, 9(6), 1486 CrossRef CAS PubMed.
- H. Jenssen, J. H. Andersen, D. Mantzilas and T. J. Gutteberg, Antiviral Res., 2004, 64, 119–126 CrossRef CAS PubMed.
- E. M. Jones, A. Smart, G. Bloomberg, L. Burgess and M. R. Millar, J. Appl. Bacteriol., 1994, 77, 208–214 CrossRef CAS PubMed.
- J. H. Andersen, H. Jenssen and T. J. Gutteberg, Antiviral Res., 2003, 58, 209–215 CrossRef CAS PubMed.
- T. Murakami, T. Nakajima, Y. Koyanagi, K. Tachibana, N. Fujii, H. Tamamura, N. Yoshida, M. Waki, A. Matsumoto, O. Yoshie, T. Kishimoto, N. Yamamoto and T. Nagasawa, J. Exp. Med., 1997, 186, 1389–1393 CrossRef CAS PubMed.
- H. Tamamura, T. Murakami, M. Masuda, A. Otaka, W. Takada, T. Ibuka, H. Nakashima, M. Waki, A. Matsumoto and N. Yamamoto, et al. , Biochem. Biophys. Res. Commun., 1994, 205, 1729–1735 CrossRef CAS PubMed.
- V. C. A. Matanic and V. Castilla, Int. J. Antimicrob. Agents, 2004, 23, 382–389 CrossRef PubMed.
- M. Wachinger, A. Kleinschmidt, D. Winder, N. von Pechmann, A. Ludvigsen, M. Neumann, R. Holle, B. Salmons, V. Erfle and R. Brack-Werner, J. Gen. Virol., 1998, 79, 731–740 CrossRef CAS PubMed.
- M. Krepstakies, J. Lucifora, C. H. Nagel, M. B. Zeisel, B. Holstermann, H. Hohenberg, I. Kowalski, T. Gutsmann, T. F. Baumert, K. Brandenburg, J. Hauber and U. Protzer, J. Infect. Dis., 2012, 205, 1654–1664 CrossRef CAS PubMed.
- M. M. Ali, G. A. Karasneh, M. J. Jarding, V. Tiwari and D. Shukla, J. Virol., 2012, 86, 6434–6443 CrossRef CAS PubMed.
- A. V. Domashevskiy and D. J. Goss, Toxins, 2015, 7, 274–298 CrossRef CAS PubMed.
- N. Yahi, J. Fantini, K. Mabrouk, C. Tamalet, P. de Micco, J. van Rietschoten, H. Rochat and J. M. Sabatier, J. Virol., 1994, 68, 5714–5720 CrossRef CAS PubMed.
- M. A. Matica, F. L. Aachmann, A. Tøndervik, H. Sletta and V. Ostafe, Int. J. Mol. Sci., 2019, 20, 5889 CrossRef CAS PubMed.
- S. Roy, M. Kumari, P. Haloi, S. Chawla, V. B. Konkimalla, A. Kumar, H. K. Kashyap and A. Jaiswal, Biomater. Sci., 2022, 10, 581–601 RSC.
- T. J. Franklin and G. A. Snow, Biochemistry and molecular biology of antimicrobial drug action, Springer Science & Business Media, 2005 Search PubMed.
- T. Ikeda, H. Yamaguchi and S. Tazuke, Antimicrob. Agents Chemother., 1984, 26, 139–144 CrossRef CAS PubMed.
- T. Tashiro, Macromol. Mater. Eng., 2001, 286, 63–87 CrossRef CAS.
- T. Ikeda, H. Hirayama, H. Yamaguchi, S. Tazuke and M. Watanabe, Antimicrob. Agents Chemother., 1986, 30, 132–136 CrossRef CAS PubMed.
- E. R. Kenawy and Y. A. G. Mahmoud, Macromol. Biosci., 2003, 3, 107–116 CrossRef CAS.
- S. Lenoir, C. Pagnoulle, C. Detrembleur, M. Galleni and R. Jérôme, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1214–1224 CrossRef CAS.
- P. H. Sellenet, B. Allison, B. M. Applegate and J. P. Youngblood, Biomacromolecules, 2007, 8, 19–23 CrossRef CAS PubMed.
- A. Kanazawa, T. Ikeda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 1441–1447 CrossRef CAS.
- E.-R. Kenawy, S. Worley and R. Broughton, Biomacromolecules, 2007, 8, 1359–1384 CrossRef CAS PubMed.
- L. Kou, J. Liang, X. Ren, H. Kocer, S. Worley, Y.-M. Tzou and T. Huang, Ind. Eng. Chem. Res., 2009, 48, 6521–6526 CrossRef CAS.
- B. Demir, R. M. Broughton, M. Qiao, T.-S. Huang and S. Worley, Molecules, 2017, 22, 1582 CrossRef PubMed.
- I. Cerkez, H. B. Kocer, S. Worley, R. Broughton and T. Huang, Langmuir, 2011, 27, 4091–4097 CrossRef CAS PubMed.
- H. B. Kocer, Prog. Org. Coat., 2012, 74, 100–105 CrossRef CAS.
- P. Li, C. Zhou, S. Rayatpisheh, K. Ye, Y. F. Poon, P. T. Hammond, H. Duan and M. B. Chan-Park, Adv. Mater., 2012, 24, 4130–4137 CrossRef CAS PubMed.
- Y. Chen, L. Yu, B. Zhang, W. Feng, M. Xu, L. Gao, N. Liu, Q. Wang, X. Huang, P. Li and W. Huang, Biomacromolecules, 2019, 20, 2230–2240 CrossRef CAS PubMed.
- M. J. Carlucci, L. A. Scolaro, M. D. Noseda, A. S. Cerezo and E. B. Damonte, Antiviral Res., 2004, 64, 137–141 CrossRef CAS PubMed.
- E. B. Damonte, M. C. Matulewicz and A. S. Cerezo, Curr. Med. Chem., 2004, 11, 2399–2419 CrossRef CAS PubMed.
- M. J. Carlucci, M. Ciancia, M. C. Matulewicz, A. S. Cerezo and E. B. Damonte, Antiviral Res., 1999, 43, 93–102 CrossRef CAS PubMed.
- M. J. Carlucci, L. A. Scolaro and E. B. Damonte, J. Med. Virol., 2002, 68, 92–98 CrossRef CAS PubMed.
- R. Davis, S. Zivanovic, D. H. D'Souza and P. M. Davidson, Food Microbiol., 2012, 32, 57–62 CrossRef CAS PubMed.
- X. Su, S. Zivanovic and D. H. D'Souza, J. Food Prot., 2009, 72, 2623–2628 CrossRef CAS PubMed.
- M. Ito, M. Baba, A. Sato, R. Pauwels, E. De Clercq and S. Shigeta, Antiviral Res., 1987, 7, 361–367 CrossRef CAS PubMed.
- M. Baba, R. Pauwels, J. Balzarini, J. Arnout, J. Desmyter and E. De Clercq, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 6132–6136 CrossRef CAS PubMed.
- H. Mitsuya, D. J. Looney, S. Kuno, R. Ueno, F. Wong-Staal and S. Broder, Science, 1988, 240, 646–649 CrossRef CAS PubMed.
- S. Mazumder, P. K. Ghosal, C. A. Pujol, M. J. Carlucci, E. B. Damonte and B. Ray, Int. J. Biol. Macromol., 2002, 31, 87–95 CrossRef CAS PubMed.
- M. A. Sosa, F. Fazely, J. A. Koch, S. V. Vercellotti and R. M. Ruprecht, Biochem. Biophys. Res. Commun., 1991, 174, 489–496 CrossRef CAS PubMed.
- J. Trinchero, N. M. Ponce, O. L. Córdoba, M. L. Flores, S. Pampuro, C. A. Stortz, H. Salomón and G. Turk, Phytother. Res., 2009, 23, 707–712 CrossRef CAS PubMed.
- S. C. Feldman, S. Reynaldi, C. A. Stortz, A. S. Cerezo and E. B. Damont, Phytomedicine, 1999, 6, 335–340 CrossRef CAS PubMed.
- N. M. Ponce, C. A. Pujol, E. B. Damonte, M. L. Flores and C. A. Stortz, Carbohydr. Res., 2003, 338, 153–165 CrossRef CAS PubMed.
- M. Baba, R. Snoeck, R. Pauwels and E. de Clercq, Antimicrob. Agents Chemother., 1988, 32, 1742–1745 CrossRef CAS PubMed.
- A. Grassauer, R. Weinmuellner, C. Meier, A. Pretsch, E. Prieschl-Grassauer and H. Unger, Virol. J., 2008, 5, 107 CrossRef PubMed.
- J. Mercer, M. Schelhaas and A. Helenius, Annu. Rev. Biochem., 2010, 79, 803–833 CrossRef CAS PubMed.
- M. Kim, J. H. Yim, S. Y. Kim, H. S. Kim, W. G. Lee, S. J. Kim, P. S. Kang and C. K. Lee, Antiviral Res., 2012, 93, 253–259 CrossRef CAS PubMed.
- C. B. Buck, C. D. Thompson, J. N. Roberts, M. Müller, D. R. Lowy and J. T. Schiller, PLoS Pathog., 2006, 2, e69 CrossRef PubMed.
- L. B. Talarico and E. B. Damonte, Virology, 2007, 363, 473–485 CrossRef CAS PubMed.
- L. B. Talarico, C. A. Pujol, R. G. Zibetti, P. C. Faría, M. D. Noseda, M. E. Duarte and E. B. Damonte, Antiviral Res., 2005, 66, 103–110 CrossRef CAS PubMed.
- W. Wang, P. Zhang, C. Hao, X. E. Zhang, Z. Q. Cui and H. S. Guan, Antiviral Res., 2011, 92, 237–246 CrossRef CAS PubMed.
- X. Xianliang, G. Meiyu, G. Huashi and L. Zelin, Chin. J. Mar. Drugs, 2000, 19, 15–18 Search PubMed.
- K. C. Queiroz, V. P. Medeiros, L. S. Queiroz, L. R. Abreu, H. A. Rocha, C. V. Ferreira, M. B. Jucá, H. Aoyama and E. L. Leite, Biomed. Pharmacother., 2008, 62, 303–307 CrossRef CAS PubMed.
- X. Xianliang, D. Hua, G. Meiyu, L. Pingfang, L. Yingxia and G. Huashi, Chin. J. Mar. Drugs, 2000, 19, 4–8 Search PubMed.
- B.-f. Jiang, X.-f. Xu, L. Li and W. Yuan, Mod. Prev. Med., 2003, 30, 517–518 Search PubMed.
- E. V. Turner and G. Sonnenfeld, Infect. Immun., 1979, 25, 467–469 CrossRef CAS PubMed.
- G. Zhou, Y. Sun, H. Xin, Y. Zhang, Z. Li and Z. Xu, Pharmacol. Res., 2004, 50, 47–53 CrossRef CAS PubMed.
- H. Yuan, J. Song, X. Li, N. Li and J. Dai, Cancer Lett., 2006, 243, 228–234 CrossRef CAS PubMed.
- M. Lüscher-Mattli, Antiviral Chem. Chemother., 2000, 11, 249–259 CrossRef PubMed.
- D. Huskens, K. Vermeire, A. T. Profy and D. Schols, Antiviral Res., 2009, 84, 38–47 CrossRef CAS PubMed.
- M. J. Keller, B. Zerhouni-Layachi, N. Cheshenko, M. John, K. Hogarty, A. Kasowitz, C. L. Goldberg, S. Wallenstein, A. T. Profy, M. E. Klotman and B. C. Herold, J. Infect. Dis., 2006, 193, 27–35 CrossRef CAS PubMed.
- R. Rupp, S. L. Rosenthal and L. R. Stanberry, Int. J. Nanomed., 2007, 2, 561–566 CAS.
- C. S. Dezzutti, V. N. James, A. Ramos, S. T. Sullivan, A. Siddig, T. J. Bush, L. A. Grohskopf, L. Paxton, S. Subbarao and C. E. Hart, Antimicrob. Agents Chemother., 2004, 48, 3834–3844 CrossRef CAS PubMed.
- J. Chung, Y. Jung, C. Hong, S. Kim, S. Moon, E. A. Kwak, B. J. Hwang, S. H. Park, B. L. Seong, D. H. Kweon and W. J. Chung, J. Colloid Interface Sci., 2021, 583, 267–278 CrossRef CAS PubMed.
- T. Tanaka, K. Nakashima, S. Tsuji, X. Han, J. Zhao, Y. Honda, K. Sakakibara, Y. Kurebayashi, T. Takahashi and T. Suzuki, Polym. Chem., 2019, 10, 5124–5130 RSC.
- S. Tsuji, Y. Aso, H. Ohara and T. Tanaka, J. Polym. Sci., 2020, 58, 548–556 CrossRef CAS.
- E. D. Clercq, Nat. Rev. Drug Discovery, 2007, 6, 941–941 CrossRef.
- L. Menéndez-Arias, Virus Res., 2008, 134, 124–146 CrossRef PubMed.
- S. Agrawal, J. Tang and D. Brown, J. Chromatogr. A, 1990, 509, 396–399 CrossRef CAS PubMed.
- F. Eckstein, J. Am. Chem. Soc., 1970, 92, 4718–4723 CrossRef CAS PubMed.
- A. Vaillant, ACS Infect. Dis., 2018, 5, 675–687 CrossRef PubMed.
- R. D. Cardin, F. J. Bravo, A. P. Sewell, J. Cummins, L. Flamand, J. M. Juteau, D. I. Bernstein and A. Vaillant, Virol. J., 2009, 6, 214 CrossRef PubMed.
- D. I. Bernstein, N. Goyette, R. Cardin, E. R. Kern, G. Boivin, J. Ireland, J. M. Juteau and A. Vaillant, Antimicrob. Agents Chemother., 2008, 52, 2727–2733 CrossRef CAS PubMed.
- M. Blanchet, V. Sinnathamby, A. Vaillant and P. Labonté, Antiviral Res., 2019, 164, 97–105 CrossRef CAS PubMed.
- M. L. Guzmán, R. H. Manzo and M. E. Olivera, Mol. Pharm., 2012, 9, 2424–2433 CrossRef PubMed.
- R. V. Alasino, I. D. Bianco, M. S. Vitali, J. A. Zarzur and D. M. Beltramo, Macromol. Biosci., 2007, 7, 1132–1138 CrossRef CAS PubMed.
- Y. Wang, T. D. Canady, Z. Zhou, Y. Tang, D. N. Price, D. G. Bear, E. Y. Chi, K. S. Schanze and D. G. Whitten, ACS Appl. Mater. Interfaces, 2011, 3, 2209–2214 CrossRef CAS PubMed.
- T. D. Martin, E. H. Hill, D. G. Whitten, E. Y. Chi and D. G. Evans, Langmuir, 2016, 32, 12542–12551 CrossRef CAS PubMed.
- A. Kuroki, J. Tay, G. H. Lee and Y. Y. Yang, Adv. Healthcare Mater., 2021, 10, 2101113 CrossRef CAS PubMed.
- A. Abbaszadegan, Y. Ghahramani, A. Gholami, B. Hemmateenejad, S. Dorostkar, M. Nabavizadeh and H. Sharghi, J. Nanomater., 2015, 2015, 720654 Search PubMed.
- W. Schreurs and H. Rosenberg, J. Bacteriol., 1982, 152, 7–13 CrossRef CAS PubMed.
- M. Ansari, H. Khan, A. Khan, S. S. Cameotra, Q. Saquib and J. Musarrat, J. Appl. Microbiol., 2014, 116, 772–783 CrossRef CAS PubMed.
- A. R. Nalwade and A. Jadhav, Arch. Appl. Sci. Res., 2013, 5, 45–49 CAS.
- I. Sondi and B. Salopek-Sondi, J. Colloid Interface Sci., 2004, 275, 177–182 CrossRef CAS PubMed.
- S. Pal, Y. K. Tak and J. M. Song, Appl. Environ. Microbiol., 2007, 73, 1712–1720 CrossRef CAS PubMed.
- J. Yu, W. Zhang, Y. Li, G. Wang, L. Yang, J. Jin, Q. Chen and M. Huang, Biomed. Mater., 2014, 10, 015001 CrossRef PubMed.
- A. Lesniak, A. Salvati, M. J. Santos-Martinez, M. W. Radomski, K. A. Dawson and C. Åberg, J. Am. Chem. Soc., 2013, 135, 1438–1444 CrossRef CAS PubMed.
- A. Kędziora, M. Speruda, E. Krzyżewska, J. Rybka, A. Łukowiak and G. Bugla-Płoskońska, Int. J. Mol. Sci., 2018, 19, 444 CrossRef PubMed.
- C.-N. Lok, C.-M. Ho, R. Chen, Q.-Y. He, W.-Y. Yu, H. Sun, P. K.-H. Tam, J.-F. Chiu and C.-M. Che, J. Proteome Res., 2006, 5, 916–924 CrossRef CAS PubMed.
- U. Klueh, V. Wagner, S. Kelly, A. Johnson and J. Bryers, J. Biomed. Mater. Res., 2000, 53, 621–631 CrossRef CAS PubMed.
- J. R. Morones, J. L. Elechiguerra, A. Camacho, K. Holt, J. B. Kouri, J. T. Ramírez and M. J. Yacaman, Nanotechnology, 2005, 16, 2346 CrossRef CAS PubMed.
- Q. L. Feng, J. Wu, G. Q. Chen, F. Cui, T. Kim and J. Kim, J. Biomed. Mater. Res., 2000, 52, 662–668 CrossRef CAS PubMed.
- A. K. Chatterjee, R. Chakraborty and T. Basu, Nanotechnology, 2014, 25, 135101 CrossRef PubMed.
- R. Bhattacharya and P. Mukherjee, Adv. Drug Delivery Rev., 2008, 60, 1289–1306 CrossRef CAS PubMed.
- M. Radzig, V. Nadtochenko, O. Koksharova, J. Kiwi, V. Lipasova and I. Khmel, Colloids Surf., B, 2013, 102, 300–306 CrossRef CAS PubMed.
- T. Parandhaman, A. Das, B. Ramalingam, D. Samanta, T. Sastry, A. B. Mandal and S. K. Das, J. Hazard. Mater., 2015, 290, 117–126 CrossRef CAS PubMed.
- W. K. Jung, H. C. Koo, K. W. Kim, S. Shin, S. H. Kim and Y. H. Park, Appl. Environ. Microbiol., 2008, 74, 2171–2178 CrossRef CAS PubMed.
- M. K. Rai, S. Deshmukh, A. Ingle and A. Gade, J. Appl. Microbiol., 2012, 112, 841–852 CrossRef CAS PubMed.
- Y. H. Leung, A. M. Ng, X. Xu, Z. Shen, L. A. Gethings, M. T. Wong, C. M. Chan, M. Y. Guo, Y. H. Ng and A. B. Djurišić, Small, 2014, 10, 1171–1183 CrossRef CAS PubMed.
- Y. Li, W. Zhang, J. Niu and Y. Chen, ACS Nano, 2012, 6, 5164–5173 CrossRef CAS PubMed.
- E. Cabiscol, J. Tamarit and J. Ros, Int. Microbiol., 2000, 3(1), 3–8 CAS.
- S. R. Sarker, M. Hossain, S. A. Polash, M. Takikawa, R. D. Shubhra, T. Saha, Z. Islam, M. Hossain, M. Hasan and S. Takeoka, Front. Bioeng. Biotechnol., 2019, 7, 239 CrossRef PubMed.
- R. Dutta, B. P. Nenavathu, M. K. Gangishetty and A. Reddy, Colloids Surf., B, 2012, 94, 143–150 CrossRef CAS PubMed.
- Y.-M. Zhang and C. O. Rock, Nat. Rev. Microbiol., 2008, 6, 222–233 CrossRef CAS PubMed.
- D. R. Janero, Free Radicals Biol. Med., 1990, 9, 515–540 CrossRef CAS PubMed.
- C. Gaucher, A. Boudier, J. Bonetti, I. Clarot, P. Leroy and M. Parent, Antioxidants, 2018, 7, 62 CrossRef CAS PubMed.
- O. Choi and Z. Hu, Environ. Sci. Technol., 2008, 42, 4583–4588 CrossRef CAS PubMed.
- A. Kumar, A. K. Pandey, S. S. Singh, R. Shanker and A. Dhawan, Free Radicals Biol. Med., 2011, 51, 1872–1881 CrossRef CAS PubMed.
- R. J. Barnes, R. Molina, J. Xu, P. J. Dobson and I. P. Thompson, J. Nanopart. Res., 2013, 15, 1432 CrossRef.
- X. Dong, W. Liang, M. J. Meziani, Y.-P. Sun and L. Yang, Theranostics, 2020, 10, 671 CrossRef CAS PubMed.
- J. Deutscher and M. H. Saier Jr., J. Mol. Microbiol. Biotechnol., 2005, 9, 125–131 CAS.
- S. Shrivastava, T. Bera, A. Roy, G. Singh, P. Ramachandrarao and D. Dash, Nanotechnology, 2007, 18, 225103 CrossRef.
- B. Sohm, F. Immel, P. Bauda and C. Pagnout, Proteomics, 2015, 15, 98–113 CrossRef CAS PubMed.
- N. Gou, A. Onnis-Hayden and A. Z. Gu, Environ. Sci. Technol., 2010, 44, 5964–5970 CrossRef CAS PubMed.
- D. A. Pelletier, A. K. Suresh, G. A. Holton, C. K. McKeown, W. Wang, B. Gu, N. P. Mortensen, D. P. Allison, D. C. Joy and M. R. Allison, Appl. Environ. Microbiol., 2010, 76, 7981–7989 CrossRef CAS PubMed.
- H. H. Lara, N. V. Ayala-Nuñez, L. Ixtepan-Turrent and C. Rodriguez-Padilla, J. Nanobiotechnol., 2010, 8, 1–10 CrossRef PubMed.
- X. Hang, H. Peng, H. Song, Z. Qi, X. Miao and W. Xu, J. Virol. Methods, 2015, 222, 150–157 CrossRef CAS PubMed.
- X. Dong, M. M. Moyer, F. Yang, Y.-P. Sun and L. Yang, Sci. Rep., 2017, 7, 1–10 CrossRef PubMed.
- S. Huang, J. Gu, J. Ye, B. Fang, S. Wan, C. Wang, U. Ashraf, Q. Li, X. Wang and L. Shao, J. Colloid Interface Sci., 2019, 542, 198–206 CrossRef CAS PubMed.
- Z. Ramezani, M. R. Dayer, S. Noorizadeh and M. Thompson, COVID, 2021, 1, 120–129 CrossRef CAS.
- Y. Abo-Zeid, N. S. Ismail, G. R. McLean and N. M. Hamdy, Eur. J. Pharm. Sci., 2020, 153, 105465 CrossRef CAS PubMed.
- P. Merkl, S. Long, G. M. McInerney and G. A. Sotiriou, Nanomaterials, 2021, 11, 1312 CrossRef CAS PubMed.
- N. Mazurkova, Y. E. Spitsyna, N. Shikina, Z. Ismagilov, S. Zagrebel'nyi and E. Ryabchikova, Nanotechnol. Russ., 2010, 5, 417–420 CrossRef.
- H. Ghaffari, A. Tavakoli, A. Moradi, A. Tabarraei, F. Bokharaei-Salim, M. Zahmatkeshan, M. Farahmand, D. Javanmard, S. J. Kiani and M. Esghaei, J. Biomed. Sci., 2019, 26, 1–10 CrossRef CAS PubMed.
- A. Tavakoli, A. Ataei-Pirkooh, G. M. Sadeghi, F. Bokharaei-Salim, P. Sahrapour, S. J. Kiani, M. Moghoofei, M. Farahmand, D. Javanmard and S. H. Monavari, Nanomedicine, 2018, 13, 2675–2690 CrossRef CAS PubMed.
- T. Du, J. Liang, N. Dong, J. Lu, Y. Fu, L. Fang, S. Xiao and H. Han, ACS Appl. Mater. Interfaces, 2018, 10, 4369–4378 CrossRef CAS PubMed.
- T. Du, J. Liang, N. Dong, L. Liu, L. Fang, S. Xiao and H. Han, Carbon, 2016, 110, 278–285 CrossRef CAS.
- S. Gurunathan, M. Qasim, Y. Choi, J. T. Do, C. Park, K. Hong, J.-H. Kim and H. Song, Nanomaterials, 2020, 10, 1645 CrossRef CAS PubMed.
- S. Kang, M. Pinault, L. D. Pfefferle and M. Elimelech, Langmuir, 2007, 23, 8670–8673 CrossRef CAS PubMed.
- D. P. Linklater, M. De Volder, V. A. Baulin, M. Werner, S. Jessl, M. Golozar, L. Maggini, S. Rubanov, E. Hanssen and S. Juodkazis, ACS Nano, 2018, 12, 6657–6667 CrossRef CAS PubMed.
- C. Yang, J. Mamouni, Y. Tang and L. Yang, Langmuir, 2010, 26, 16013–16019 CrossRef CAS PubMed.
- H. Chen, B. Wang, D. Gao, M. Guan, L. Zheng, H. Ouyang, Z. Chai, Y. Zhao and W. Feng, Small, 2013, 9, 2735–2746 CrossRef CAS PubMed.
- K. Rajavel, R. Gomathi, S. Manian and R. T. Rajendra Kumar, Langmuir, 2014, 30, 592–601 CrossRef CAS PubMed.
- C. D. Vecitis, K. R. Zodrow, S. Kang and M. Elimelech, ACS Nano, 2010, 4, 5471–5479 CrossRef CAS PubMed.
- S. Kang, M. Herzberg, D. F. Rodrigues and M. Elimelech, Langmuir, 2008, 24, 6409–6413 CrossRef CAS PubMed.
- H. Chen, S. T. Humes, S. E. Robinson, J. C. Loeb, I. V. Sabaraya, N. B. Saleh, R. B. Khattri, M. E. Merritt, C. J. Martyniuk and J. A. Lednicky, Nanotoxicology, 2019, 13, 1176–1196 CrossRef CAS PubMed.
- P. Sanpui, X. Zheng, J. C. Loeb, J. H. Bisesi Jr., I. A. Khan, A. N. Afrooz, K. Liu, A. R. Badireddy, M. R. Wiesner and P. L. Ferguson, Part. Fibre Toxicol., 2014, 11, 66 CrossRef PubMed.
- X. Zou, L. Zhang, Z. Wang and Y. Luo, J. Am. Chem. Soc., 2016, 138, 2064–2077 CrossRef CAS PubMed.
- F. Perreault, A. F. De Faria, S. Nejati and M. Elimelech, ACS Nano, 2015, 9, 7226–7236 CrossRef CAS PubMed.
- I. Zucker, J. R. Werber, Z. S. Fishman, S. M. Hashmi, U. R. Gabinet, X. Lu, C. O. Osuji, L. D. Pfefferle and M. Elimelech, Environ. Sci. Technol. Lett., 2017, 4, 404–409 CrossRef CAS.
- X. Lu, X. Feng, J. R. Werber, C. Chu, I. Zucker, J.-H. Kim, C. O. Osuji and M. Elimelech, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E9793–E9801 CAS.
- R. Wu, X. Ou, R. Tian, J. Zhang, H. Jin, M. Dong, J. Li and L. Liu, Nanoscale, 2018, 10, 20162–20170 RSC.
- S. Liu, M. Hu, T. H. Zeng, R. Wu, R. Jiang, J. Wei, L. Wang, J. Kong and Y. Chen, Langmuir, 2012, 28, 12364–12372 CrossRef CAS PubMed.
- O. Akhavan, E. Ghaderi and A. Esfandiar, J. Phys. Chem. B, 2011, 115, 6279–6288 CrossRef CAS PubMed.
- J. Chen, H. Peng, X. Wang, F. Shao, Z. Yuan and H. Han, Nanoscale, 2014, 6, 1879–1889 RSC.
- M. Dallavalle, M. Calvaresi, A. Bottoni, M. Melle-Franco and F. Zerbetto, ACS Appl. Mater. Interfaces, 2015, 7, 4406–4414 CrossRef CAS PubMed.
- V. T. Pham, V. K. Truong, M. D. Quinn, S. M. Notley, Y. Guo, V. A. Baulin, M. Al Kobaisi, R. J. Crawford and E. P. Ivanova, ACS Nano, 2015, 9, 8458–8467 CrossRef CAS PubMed.
- Y. Tu, M. Lv, P. Xiu, T. Huynh, M. Zhang, M. Castelli, Z. Liu, Q. Huang, C. Fan and H. Fang, Nat. Nanotechnol., 2013, 8, 594 CrossRef CAS PubMed.
- S. Liu, T. H. Zeng, M. Hofmann, E. Burcombe, J. Wei, R. Jiang, J. Kong and Y. Chen, ACS Nano, 2011, 5, 6971–6980 CrossRef CAS PubMed.
- X. Yang, J. Li, T. Liang, C. Ma, Y. Zhang, H. Chen, N. Hanagata, H. Su and M. Xu, Nanoscale, 2014, 6, 10126–10133 RSC.
- T. I. Kim, J. Kim, I.-J. Park, K.-O. Cho and S.-Y. Choi, 2D Mater., 2019, 6, 025025 CrossRef CAS.
- S. Roy, A. Mondal, V. Yadav, A. Sarkar, R. Banerjee, P. Sanpui and A. Jaiswal, ACS Appl. Bio Mater., 2019, 2, 2738–2755 CrossRef CAS PubMed.
- X. Liu, G. Duan, W. Li, Z. Zhou and R. Zhou, RSC Adv., 2017, 7, 37873–37880 RSC.
- G. R. Navale, C. S. Rout, K. N. Gohil, M. S. Dharne, D. J. Late and S. S. Shinde, RSC Adv., 2015, 5, 74726–74733 RSC.
- A. Arabi Shamsabadi, M. Sharifian Gh, B. Anasori and M. Soroush, ACS Sustainable Chem. Eng., 2018, 6, 16586–16596 CrossRef CAS.
- Z. Xiong, X. Zhang, S. Zhang, L. Lei, W. Ma, D. Li, W. Wang, Q. Zhao and B. Xing, Ecotoxicol. Environ. Saf., 2018, 161, 507–514 CrossRef CAS PubMed.
- X. Liu, S. Sen, J. Liu, I. Kulaots, D. Geohegan, A. Kane, A. A. Puretzky, C. M. Rouleau, K. L. More and G. T. R. Palmore, Small, 2011, 7, 2775–2785 CrossRef CAS PubMed.
- S. Gurunathan, J. W. Han, A. A. Dayem, V. Eppakayala and J.-H. Kim, Int. J. Nanomed., 2012, 7, 5901 CrossRef CAS PubMed.
- S. Gurunathan, J. W. Han, A. A. Dayem, V. Eppakayala, M.-R. Park, D.-N. Kwon and J.-H. Kim, J. Ind. Eng. Chem., 2013, 19, 1280–1288 CrossRef CAS.
- J. Chen, X. Wang and H. Han, J. Nanopart. Res., 2013, 15, 1658 CrossRef.
- Y. L. F. Musico, C. M. Santos, M. L. P. Dalida and D. F. Rodrigues, ACS Sustainable Chem. Eng., 2014, 2, 1559–1565 CrossRef CAS.
- J. D. West and L. J. Marnett, Chem. Res. Toxicol., 2006, 19, 173–194 Search PubMed.
- K. Krishnamoorthy, M. Veerapandian, L.-H. Zhang, K. Yun and S. J. Kim, J. Phys. Chem. C, 2012, 116, 17280–17287 CrossRef CAS.
- P. Kumar, S. Roy, A. Sarkar and A. Jaiswal, ACS Appl. Mater. Interfaces, 2021, 13, 12912–12927 CrossRef CAS PubMed.
- J. Li, G. Wang, H. Zhu, M. Zhang, X. Zheng, Z. Di, X. Liu and X. Wang, Sci. Rep., 2014, 4, 1–8 CrossRef PubMed.
- S. Ye, K. Shao, Z. Li, N. Guo, Y. Zuo, Q. Li, Z. Lu, L. Chen, Q. He and H. Han, ACS Appl. Mater. Interfaces, 2015, 7, 21571–21579 CrossRef CAS PubMed.
- M. Sametband, I. Kalt, A. Gedanken and R. Sarid, ACS Appl. Mater. Interfaces, 2014, 6, 1228–1235 CrossRef CAS PubMed.
- M. Singh, C. Zannella, L. Altucci, F. Bajardi, A. Chianese, A. Damasco, M. R. Del Sorbo, R. di Girolamo, V. Foliero and G. Franci, Front. Bioeng. Biotechnol., 2020, 8, 1056 Search PubMed.
- Z. Song, X. Wang, G. Zhu, Q. Nian, H. Zhou, D. Yang, C. Qin and R. Tang, Small, 2015, 11, 1171–1176 CrossRef CAS PubMed.
- R. Pokhrel, N. Bhattarai, K. A. Johnson, B. S. Gerstman, R. V. Stahelin and P. P. Chapagain, Biochem. Biophys. Res. Commun., 2017, 493, 176–181 CrossRef PubMed.
- B. Luan, T. Huynh, L. Zhao and R. Zhou, ACS Nano, 2015, 9, 663–669 CrossRef CAS PubMed.
- M. A. Unal, F. Bayrakdar, H. Nazir, O. Besbinar, C. Gurcan, N. Lozano, L. M. Arellano, S. Yalcin, O. Panatli and D. Celik, Small, 2021, 17, 2101483 CrossRef CAS PubMed.
- J. Wang, Y. Yu, T. Leng, Y. Li and S.-T. Lee, ACS Appl. Mater. Interfaces, 2021, 14(1), 191–200 CrossRef PubMed.
- M. Khedri, R. Maleki, M. Dahri, M. M. Sadeghi, S. Rezvantalab, H. A. Santos and M.-A. Shahbazi, Drug Delivery Transl. Res., 2021, 1–15 Search PubMed.
- M. Fukuda, M. S. Islam, R. Shimizu, H. Nassar, N. N. Rabin, Y. Takahashi, Y. Sekine, L. F. Lindoy, T. Fukuda and T. Ikeda, ACS Appl. Nano Mater., 2021, 4, 11881–11887 CrossRef CAS.
- I. S. Donskyi, C. Nie, K. Ludwig, J. Trimpert, R. Ahmed, E. Quaas, K. Achazi, J. Radnik, M. Adeli and R. Haag, Small, 2021, 17, 2007091 CrossRef CAS PubMed.
- F. De Maio, V. Palmieri, G. Babini, A. Augello, I. Palucci, G. Perini, A. Salustri, P. Spilman, M. De Spirito and M. Sanguinetti, Iscience, 2021, 24, 102788 CrossRef CAS PubMed.
- Z. Lin, Z. Wang, X. Zhang and D. Diao, Nano Res., 2021, 14, 1110–1115 CrossRef CAS PubMed.
Footnote |
| † Equal contributing authors. |
| This journal is © The Royal Society of Chemistry 2022 |