
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermosensitive hydrogels to deliver reactive species generated by cold atmospheric plasma: a case study with methylcellulose†
Xavi
Solé-Martí
 abc,
Tània
Vilella
ab,
Cédric
Labay
abc,
Francesco
Tampieri
abc,
Maria-Pau
Ginebra
abd and
Cristina
Canal
*abc
abc,
Tània
Vilella
ab,
Cédric
Labay
abc,
Francesco
Tampieri
abc,
Maria-Pau
Ginebra
abd and
Cristina
Canal
*abc
aBiomaterials, Biomechanics and Tissue Engineering Group, Department of Materials Science and Engineering, and Research Centre for Biomedical Engineering (CREB), Universitat Politècnica de Catalunya (UPC), c/Eduard Maristany 14, 08019 Barcelona, Spain. E-mail: cristina.canal@upc.edu; Tel: +34 934 017 810
bBarcelona Research Center in Multiscale Science and Engineering, UPC, 08019 Barcelona, Spain
cInstitut de Recerca Sant Joan de Déu, Santa Rosa 39-57, 08950 Esplugues de Llobregat, Spain
dInstitute for Bioengineering of Catalonia (IBEC), Barcelona Institute of Science and Technology (BIST), c/Baldiri i Reixach 10-12, 08028 Barcelona, Spain
First published on 9th June 2022
Abstract
Hydrogels have been recently proposed as suitable materials to generate reactive oxygen and nitrogen species (RONS) upon gas-plasma treatment, and postulated as promising alternatives to conventional cancer therapies. Acting as delivery vehicles that allow a controlled release of RONS to the diseased site, plasma-treated hydrogels can overcome some of the limitations presented by plasma-treated liquids in in vivo therapies. In this work, we optimized the composition of a methylcellulose (MC) hydrogel to confer it with the ability to form a gel at physiological temperatures while remaining in the liquid phase at room temperature to allow gas-plasma treatment with suitable formation of plasma-generated RONS. MC hydrogels demonstrated the capacity for generation, prolonged storage and release of RONS. This release induced cytotoxic effects on the osteosarcoma cancer cell line MG-63, reducing its cell viability in a dose-response manner. These promising results postulate plasma-treated thermosensitive hydrogels as good candidates to provide local anticancer therapies.
1 Introduction
Plasma-treated liquids (PTLs) are produced when the reactive oxygen and nitrogen species (RONS) generated from a gas-plasma source are transferred to a liquid medium.1,2 Research in this field has shown that PTLs can be used to target malignant tumors and selectively kill cancer cells without damaging surrounding healthy cells/tissues.3–6 For this reason, anticancer therapies based on PTLs have moved forward during the last few years, reporting beneficial effects already in in vivo models.7 Compared to direct treatment with gas plasmas, PTLs represent a promising alternative when treating malignant tumors located in internal organs, which would require open surgery to be treated with a gas-plasma device.8 Therefore, in view of translation of this technology to the clinic, the development of PTLs that can be stored and administered by injection to the tumor would be a very valuable asset. However, the injection of a liquid to the tumor site might result in fast diffusion throughout the whole body due to blood flow, thus reducing the efficiency and efficacy of the PTL treatment. Besides, multiple PTL injections are needed to induce biological effects in vivo.9–12 For these reasons, new strategies with hydrogel-forming polymer solutions are under investigation, with the aim of designing new efficient vehicles that allow a localized release of RONS directly to the tumor site.13–15 In these studies, gelatin and alginate solutions were reported not only to foster the generation of RONS compared to certain saline solutions, but also to be suitable vehicles for the delivery of RONS, inducing cytotoxic effects to cancer cells without affecting nonmalignant cells. The latter show ionic crosslinking of the alginate chains in the presence of divalent cations like Ca2+, but these and other strategies based on adding crosslinkers can have some drawbacks such as wash-out of the RONS generated by the plasma treatment.13 Recently, a thermosensitive synthetic hydrogel based on polylactide and polyethyleneglycol and undergoing in situ gel formation has also been investigated as a RONS delivery vehicle.16 By modulating their sol-gel transition temperature, thermosensitive hydrogels can be conveniently injected with minimal invasive surgery and also fill irregular shaped defects, sometimes created after a tumor resection. Methylcellulose (MC) is a water-soluble cellulose derivative that can form a thermoreversible hydrogel in water upon heating. At low temperature water molecules form a cage-like structure surrounding the methoxy groups and thus solubilizing MC.17–19 However, high temperatures cause the destruction of the cage structure of water and a three-dimensional hydrogel network is formed due to hydrophobic interactions of the methoxy groups of MC. The sol-gel transition temperature of pure MC is ∼60 °C, too high for in vivo purposes.20 However, the gel point can be altered by the addition of salts, based on the Hofmeister series.21In this work, it is our aim to describe for the first time the ability of thermosensitive MC hydrogels to generate, store and release plasma-generated RONS for cancer treatment purposes. To do so, we optimized the composition of a MC hydrogel to confer the ability to form a gel at physiological temperatures while remaining in the liquid phase at room temperature to allow the suitable formation and homogeneous distribution of plasma-generated RONS. The effect of plasma treatment on MC physical–chemical properties are analyzed, together with the in vitro cytotoxic effects induced by the release of plasma-generated RONS to cancer cells.
2 Materials and methods
2.1 Sample preparation
Methylcellulose (Metolose® SM 4000, Shin-Etsu Chemical Co., Japan) with a methoxy content of 29.7% was used for the preparation of the hydrogels. MC solutions were prepared by dispersing the weighted MC powder with hot deionized water (approximately 80 °C) using the SpeedMixer™ (DAC 150.1 FVZ-k, 3500 rpm) for 5 minutes to obtain a homogeneous dispersion. Then, the solution was stirred with a magnetic stirrer inside an ice bath until a clear solution was obtained and stored in the refrigerator. Solutions containing di-sodium hydrogen phosphate (HP) (Na2HPO4·2H2O, purity >99.5%, Fluka) were prepared by mixing the salt with the MC powder prior to dissolution. All solutions were gently stirred for 15 minutes prior to testing. Samples were named xMCyHP, where x and y are the weight percent of MC and HP in the solution, respectively.2.2 Plasma treatment
Plasma treatment of the different MC solutions was performed using a commercial plasma jet kINPen® IND (Neoplas tools GmbH, Germany) operated with Ar gas (Ar 5.0, PRAXAIR, Spain). Gas flow was set to 1 L min−1. The treatments were done on 1000 μL of sample, in a 24-well plate, at 10 mm distance between the nozzle of the jet and the surface of the solution. Different plasma treatments (60, 180 and 300 s) were evaluated. During plasma treatment, MC solutions were magnetically stirred to ensure the homogeneous distribution of the generated reactive species (Fig. S1†).2.3 Quantification of RONS
The generation of reactive species was quantified as reported in the protocol.22 In brief, the determination of species such as NO2−, NO3−, H2O2 and OH radicals in MC solutions and release media (culture medium or phosphate buffered saline (PBS)) was performed as follows.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 in HP, and 50 μL of the Amplex red/HRP reagent were added to 200 μL of the diluted sample in a 96-well plate for incubation at 37 °C for 30 min. Fluorescence measurements were performed with a Synergy™ HTX Multi-Mode Microplate Reader using 560 nm and 590 nm as excitation and emission wavelengths, respectively. Standard solutions for the calibration curves to correlate fluorescence with concentration were prepared by diluting a 30 wt% H2O2 solution (Sigma Aldrich, USA).
:100 in HP, and 50 μL of the Amplex red/HRP reagent were added to 200 μL of the diluted sample in a 96-well plate for incubation at 37 °C for 30 min. Fluorescence measurements were performed with a Synergy™ HTX Multi-Mode Microplate Reader using 560 nm and 590 nm as excitation and emission wavelengths, respectively. Standard solutions for the calibration curves to correlate fluorescence with concentration were prepared by diluting a 30 wt% H2O2 solution (Sigma Aldrich, USA).
Moreover, the stability of these reactive species was also quantified for sample 1MC3.5HP after one gelling cycle: following plasma treatment, samples were gelled at 37 °C for 24 h in the incubator and subsequently cooled down at room temperature to solubilize again MC. Then, RONS were quantified following the procedure described in 2.3. This experiment was possible due to the thermoreversible properties of 1MC3.5HP.26–28
2.4 Characterization of methylcellulose
:10 (thus obtaining 0.1MC) before plasma-treatment and then analyzed as explained.
:10 with phosphate buffer (thus obtaining 0.1MC) before plasma treatment and analyzed subsequently.
where the hydrogel weight was calculated as the weight of the gelled hydrogel after 10 min, 30 min, 1 h, 2 h, 4 h, 6 h, 24 h and 72 h immersion. Samples were kept at 37 °C during the experiment.
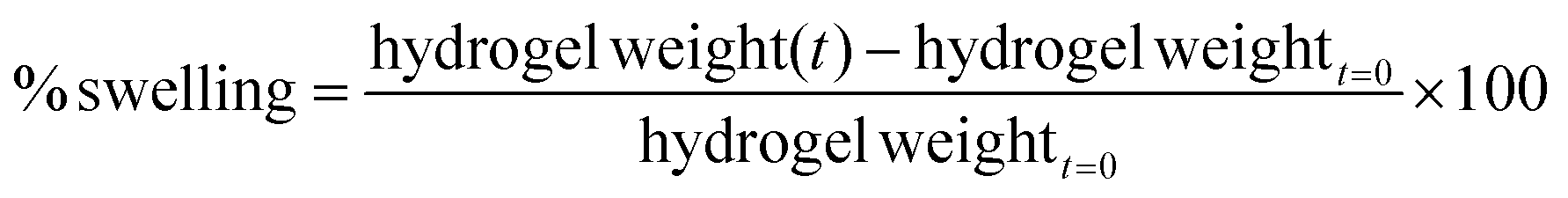
2.5 Cell culture
The osteosarcoma cell line MG-63 (CRL-1427™, ATCC collection) was employed. Cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM) (Gibco™, Carlsbad, CA, USA), supplemented with 10% fetal bovine serum (FBS) (Gibco™), 2 mM L-glutamine (Gibco™), 100 units per mL penicillin (Gibco™) and 100 μg mL−1 streptomycin (Gibco™), designated as “complete cell culture medium”. Cells were maintained and expanded at 37 °C, 95% humidity and 5% CO2. MC powder was sterilized by low-pressure plasma treatment using a low-pressure radio-frequency plasma (Femto plasma surface technologies, Diener Electronics, Germany) and the HP solutions were filtered at 37 °C using a 0.22 μm syringe filter (Millipore, Merck, Germany). All the processes leading to the preparation of 1MC3.5HP were carried out under sterile conditions.2.6 Statistical analysis
Comparison between multiple groups was made using one-way ANOVA. A two-tailed t-test was performed when two values were compared. p-values <0.05 were considered statistically significant.3 Results and discussion
3.1 Gel point determination
The gel point of 1MC solutions with increasing concentrations of HP was evaluated by rheology tests and microDSC (Fig. 1i and ii). In the rheology test, the crossover of storage modulus (G′′) and loss modulus (G′) was used as the indicator of the gel point,19,30 whereas for the microDSC test, the gel point was determined at the endothermic peak of the microDSC curves. As expected, the higher the concentration of HP in the 1MC solution, the lower the gel point. The results obtained for both experiments are comparable (Fig. 1iii). The sample 1MC3.5HP was selected as the optimal composition since its gel point is at T ≈ 35.2 °C – right below the physiological temperature (Fig. 1iv) – and was employed in subsequent experiments.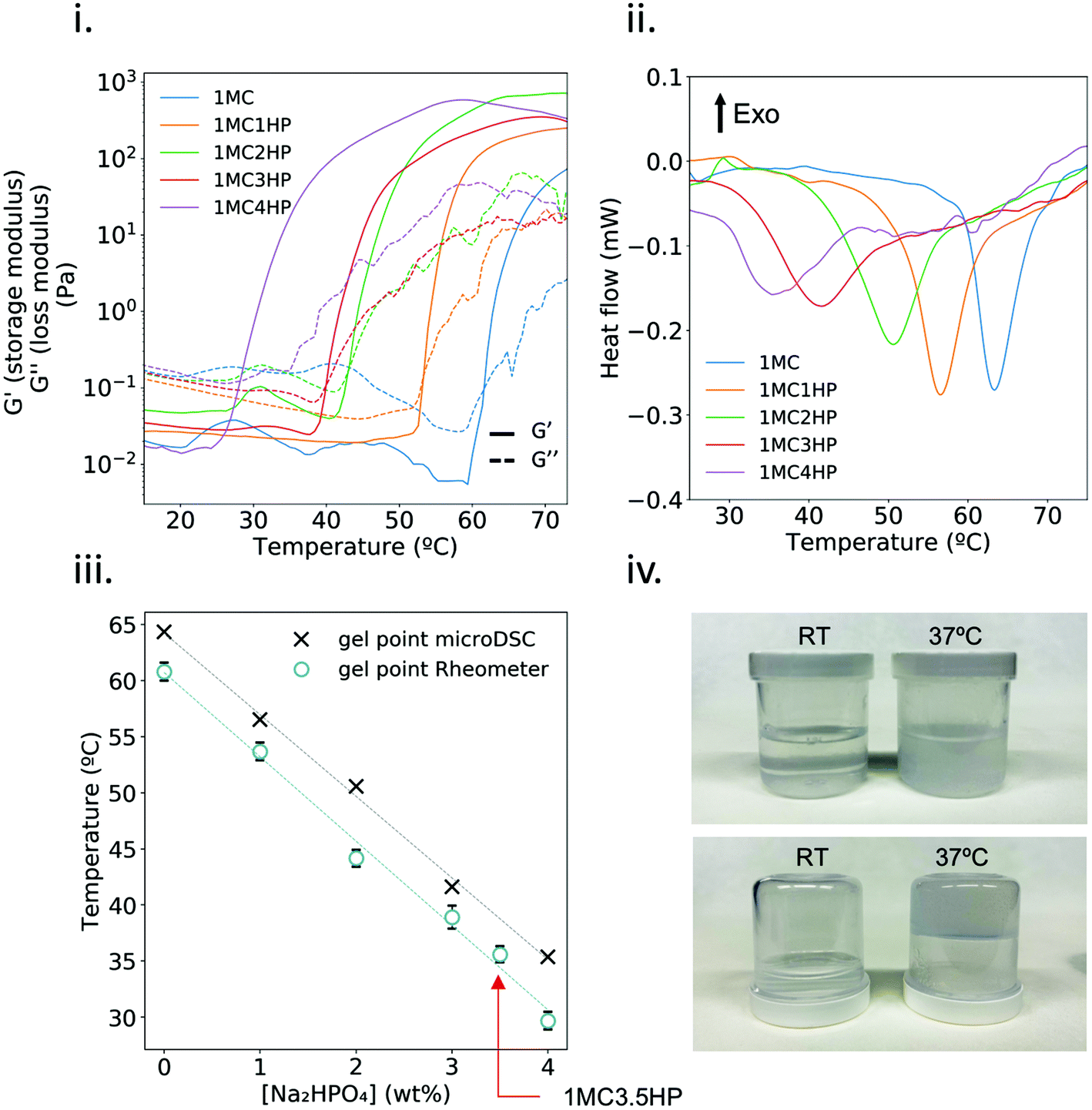 | ||
| Fig. 1 Determination of the gel point for the different HP concentrations in 1MC. (i) Storage (G′) and loss (G′′) modulus as a function of temperature and (ii) microDSC heating curves. In both cases the heating rate was 1 °C min−1. (iii) Comparison of the gel point obtained by microDSC (n = 1) and rheological analysis (n = 2, except 1MC3.5HP; n = 4). Red arrow indicates the gel point of the 1MC3.5HP sample. (iv) Pictures of 1MC3.5HP at room temperature (RT) and after crosslinking at 37 °C. | ||
3.2 Characterization of methylcellulose after plasma treatment
A thermal camera was used to monitor the temperature of MC solutions during plasma treatment. Fig. 2i and ii show that, even though plasma treatment did increase the temperature of the well containing the sample, the temperature of MC solutions always remained below the crosslinking temperature, and thus staying in the liquid state, which was associated with a homogeneous treatment to the whole sample (Fig. S1†).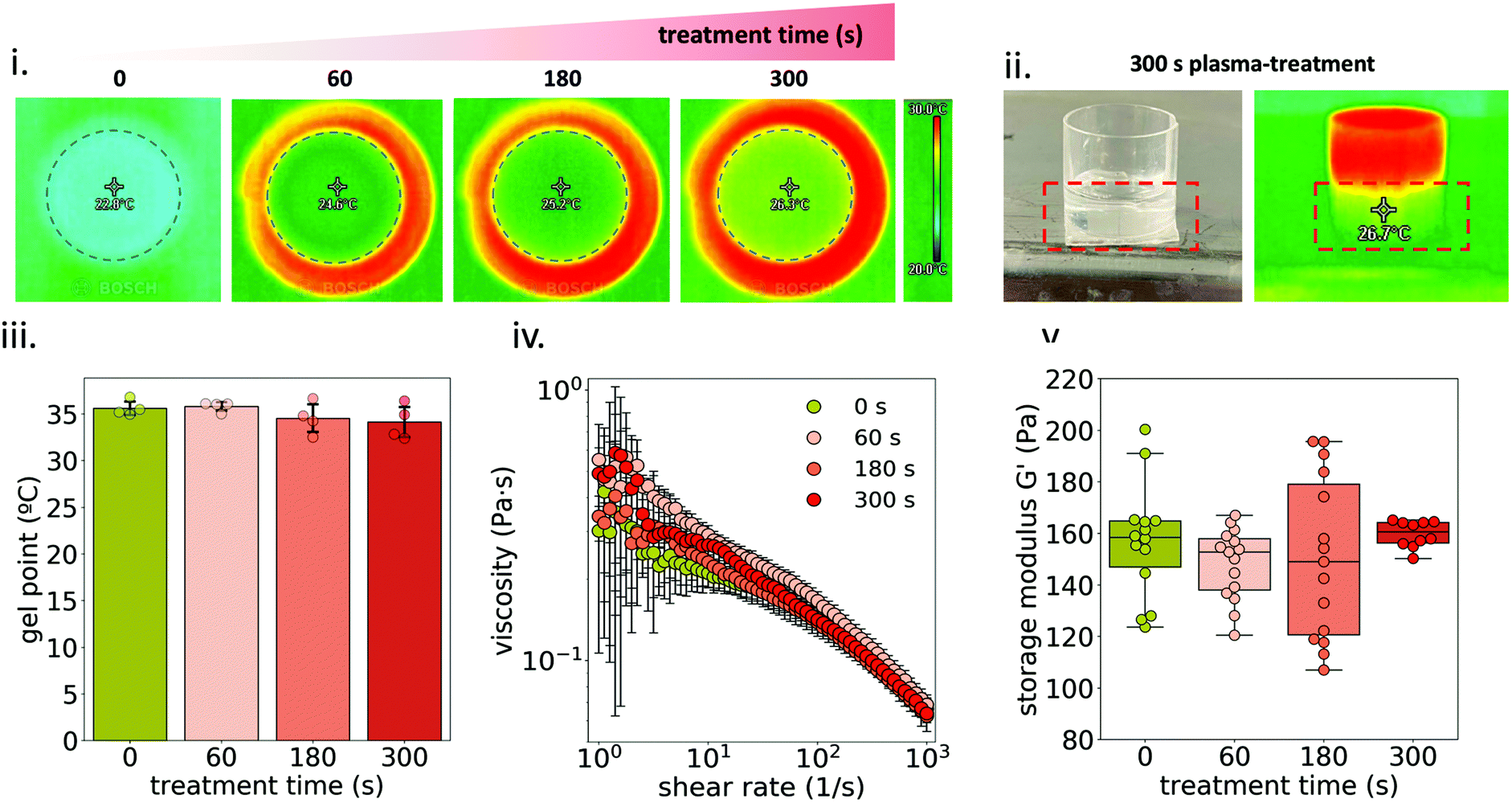 | ||
| Fig. 2 Gel point and rheological analysis of plasma-treated 1MC3.5HP. (i) Thermal camera images of the surface of MC solutions after 60, 180 and 300 s plasma treatment. The temperature value is measured at the center of the well plate, right below the plasma jet plume. (ii) Thermal camera image of the side view of the well after 300 s plasma-treatment. (iii) Gel point comparison between non-treated and plasma-treated hydrogels. (iv) Viscosity curves of 1MC3.5HP at different plasma-treatment times before hydrogel formation. (v) Storage modulus (G′) of 1MC3.5HP at different plasma-treatment times after hydrogel formation. | ||
Temperature ramp measurements were conducted in order to determine the gel point of the plasma-treated hydrogel (Fig. 2iii) with no difference among treated and non-treated 1MC3.5HP. This indicates that plasma-treatment did not affect the crosslinking ability of the hydrogel.
The viscosity of 1MC3.5HP showed a shear-thinning behavior, in accordance with other studies (Fig. 2iv).31,32 Although high variability was recorded at low shear stress, the viscosity of 1MC3.5HP did not significantly change due to the plasma-treatment. Similarly, storage modulus values did not vary either, meaning that the viscoelastic properties of the material remained unchanged after plasma-treatment (Fig. 2v). This indicates that the rheological properties of 1MC3.5HP – both before and after crosslinking – were not affected by plasma.
To further study changes in MC physico-chemical properties, SEC (Fig. 3i) was used to analyze pristine and plasma-treated 1MC diluted 1:10 before the treatment (0.1MC). Two peaks were detected in the chromatograms: peak A (at ∼5.3 min) and peak B (at ∼10.25 min). The former is attributed to the elution of the MC and the latter to the elution of the solvent. At increasing plasma-treatment time, peak A gradually shifted to higher elution times, reflecting a lower molecular weight. This effect was only observed for diluted samples (0.1MC) (Fig. S2†). This indicates that plasma treatment could only break up a small proportion of MC polymeric chains and was only appreciable in 0.1MC samples. Consequently, this minor reduction of molecular weight in MC did not have an influence in the rheological behavior of the material (Fig. 2iv and v). Since peak B is attributed to the lower limit of the column, the elution time at this point did not shift. However, the intensity of the peak increased at a higher plasma treatment time. This effect was attributed to changes in the refractive index of the solvent. A higher plasma-treatment time was related to a higher concentration of RONS in the solvent that led to higher intensity values.
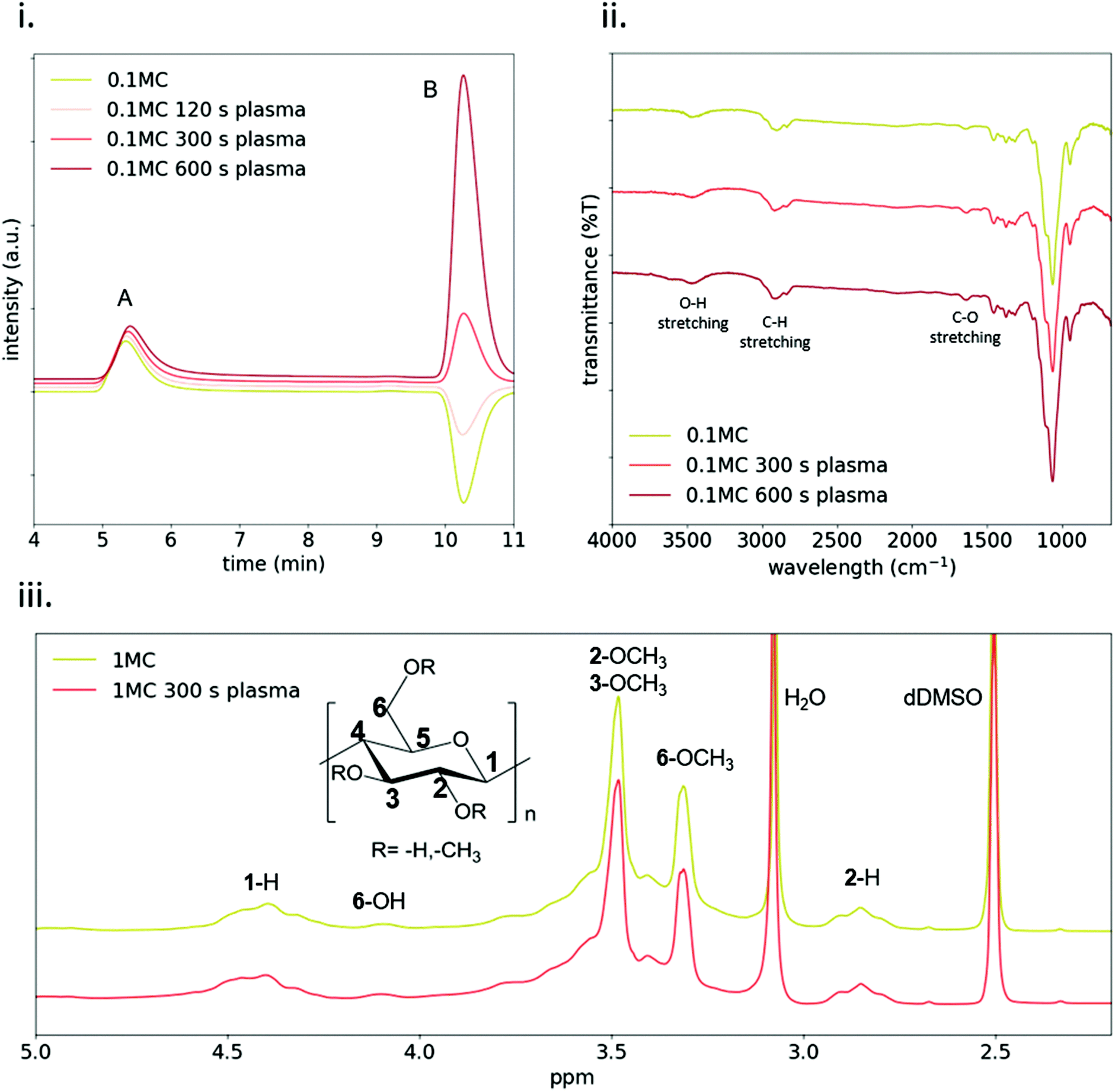 | ||
| Fig. 3 Chemical characterization of plasma-treated MC. (i) Analysis by size exclusion chromatography of diluted (1:10) plasma-treated MC solutions. (ii) ATR-IR spectra of MC films at different plasma treatment times. (iii) 1H NMR spectra of MC at different plasma treatment times (30 g L−1 in dDMSO at 80 °C). | ||
ATR-IR and 1H NMR spectroscopies were used to study any possible functional group modification or the generation of side products due to the interaction of plasma species with MC. Fig. 3ii shows the ATR-IR spectra of both pristine and plasma-treated 0.1MC. The band at 3475 cm−1 is assigned to O–H stretching, and the bands at 2900 cm−1 and 1640 cm−1 to C–H stretching and C–O stretching, respectively.33,34 The region from 1500 cm−1 to 500 cm−1 is considered the ‘fingerprint’ region of cellulose and cellulose derivatives.35,36 No changes in absorbance were detected at any wavelength after plasma treatment. Moreover, no new absorbance bands were recorded, in agreement with previous studies that analyzed plasma-treated hydrogels by infrared spectroscopy.13,16,37 Similar results were obtained with NMR spectroscopy (Fig. 3iii). The identification of the different peaks was performed as described in ref. 38–40. As shown, the spectrum of the pristine MC was the same as that for the plasma-treated sample. All these results strongly suggest that, at the concentrations used during the experiments, plasma treatment did not induce any evident functional group modification on the MC chemical structure.
3.3 Quantification and stability of RONS
The formation of NO2−, NO3− and H2O2 due to plasma treatment was quantified in three different liquids: 3.5HP, 1MC and 1MC3.5HP (Fig. 4i and ii). In all cases, these reactive species showed a linear increase in their concentration in the liquid with longer treatment times. The highest concentration values were obtained with 1MC3.5HP, generating ∼75, ∼165 and ∼340 μM NO2−, NO3− and H2O2, respectively, after 300 s of plasma treatment (Fig. 4i). These concentrations are in the same order of magnitude compared to other plasma-treated polysaccharide solutions (i.e. alginate13), while being several-fold larger than some typical isotonic saline solutions.6,41,42 The shelf stability of these reactive species in 1MC3.5HP was monitored at 4 °C for up to 72 h. Only the concentration of H2O2 and NO3− experienced a slight reduction after 72 h storage at 4 °C (Fig. 4iii). In view of future clinical applications, it is of interest that plasma-treated hydrogels/liquids are stable during storage for a relatively long time. The concentration of H2O2 and NO3− in a plasma-treated water has been shown to be stable even after 21 days after treatment.43 In contrast, it is known that the presence of some amino acids (such as cysteine and methionine) in plasma-treated liquids can drastically reduce the concentration of long-lived RONS during storage.44 In our case, the polysaccharide chains of MC in solution did not have a significant impact on the stability of the reactive species, thus being a good substrate for the generation and stability of RONS for at least 48 h (Fig. 4i and iii).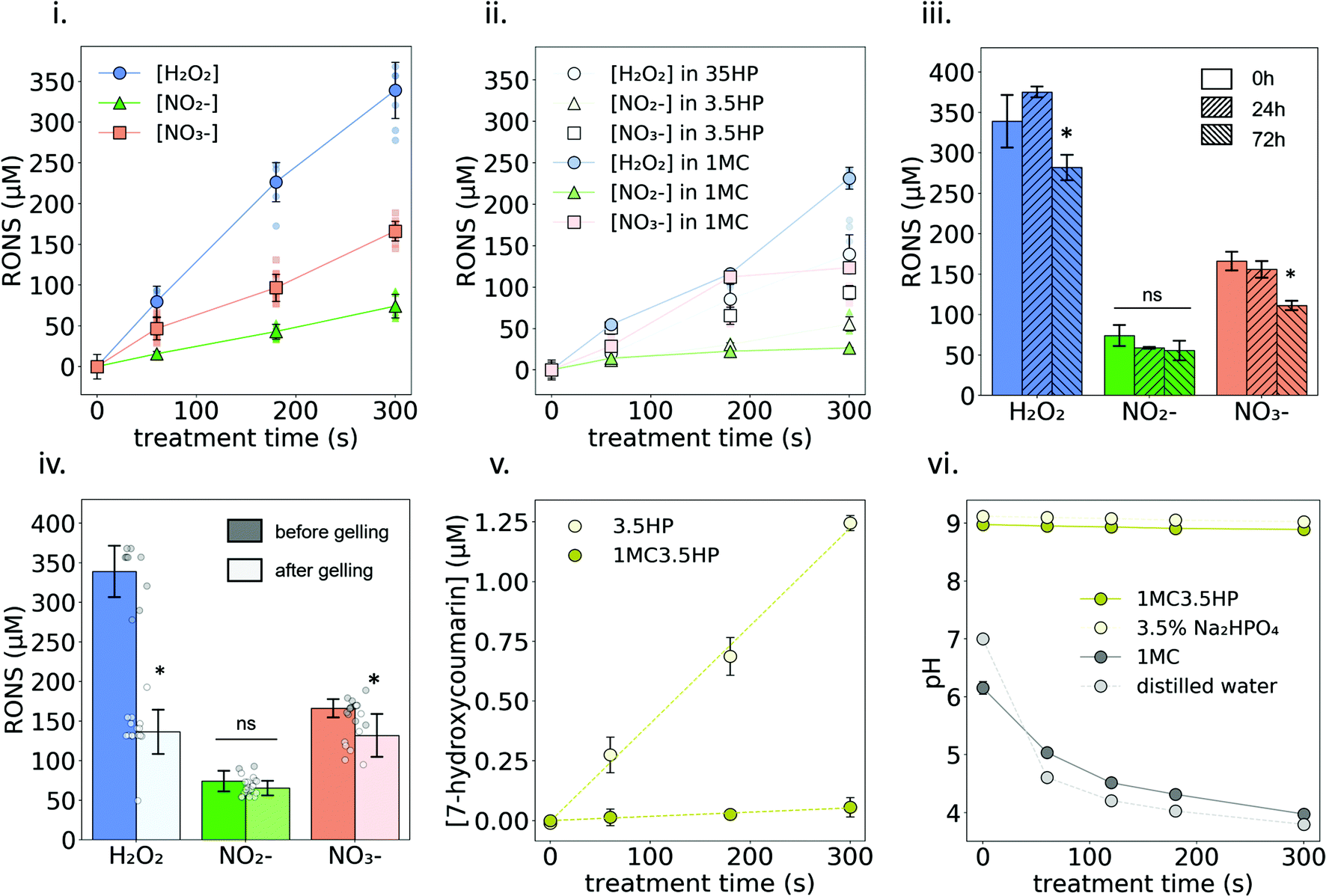 | ||
| Fig. 4 Quantification and stability of plasma-generated RONS in 1MC35HP. (i) Concentration of H2O2, NO2− and NO3− in plasma treated 1MC3.5HP solution. Small markers represent each individual measure while big markers represent the mean value at each time point. (ii) Concentration of H2O2, NO2− and NO3− in plasma treated 1MC and 3.5HP solutions. (iii) Stability of RONS after 24 h and 72 h storage in the fridge. Asterisks denote statistically significant differences with respect to time 0 h (*p < 0.05). (iv) Stability of RONS after one gelling cycle. Asterisks denote statistically significant differences with respect to before gelling (*p < 0.05). (v) Indirect quantification of the production rate of OH radicals. (vi) pH evolution at increasing plasma treatment time of 1MC dissolved either in 3.5HP or in distilled water. | ||
However, when plasma-generated RONS were measured after one gelling cycle, the stability of H2O2 was clearly affected, to approximately half of its final concentration (Fig. 4iv). Incubation of the samples at the gelling temperature (37 °C) did not affect the stability of H2O2 without the presence of MC (Fig. S3†). Therefore, the formation of a hydrogel might be one of the reasons that led to H2O2 decomposition. The concentration of NO3− was slightly reduced to ∼135 μM while NO2− did not experience any significant change in concentration.
Regarding short-lived reactive species, OH radicals were indirectly measured using a COU probe. As shown in Fig. 4v, the concentration of hCOU rises linearly with the increase of the plasma-treatment time. However, a much lower concentration is reached when the treated solution contains MC (see Fig. S4† for results with different MC concentrations). These results suggest that the presence of MC reduced the availability of OH radicals to react with the chemical probe. Nonetheless, the scavenging effect produced by MC on ˙OH did not have an impact on the formation of NO2−, NO3− or H2O2. In fact, having MC in the solution slightly promoted the formation of H2O2 (Fig. 4i and ii). This suggests that H2O2 was not mainly formed by the combination of the dissolved OH radicals45 but may rather come from secondary reactions involving OH radicals and the anhydroglucose units of MC. The differences observed due to the presence of MC in the concentration of NO2− and NO3− were not relevant (Fig. 4i and ii). Other biopolymer solutions13–15 have also shown an increase in the formation of NO2− and/or H2O2 upon plasma-treatment compared with solvent only, indicating an important secondary contribution to their generation that depends on the specific chemistry of the plasma-treated liquid/hydrogel.
The pH of 1MC rapidly decreased upon plasma treatment, leading to an acidic pH (pH ≈ 4) for the longest treatment time (Fig. 4vi). It is well known that plasma-treatment decreases the pH of non-buffered liquids mostly due to the generation of nitrous and nitric acid.25 However, the pH values of 3.5HP and 1MC3.5HP remained constant (pH ≈ 9) due to the buffering effect of HP salt.
3.4 Swelling and release of RONS
The swelling behavior was assessed by immersing 1MC3.5HP in PBS at 37 °C, showing that the hydrogel could quickly take up around ∼70% of its initial weight (Fig. 5i).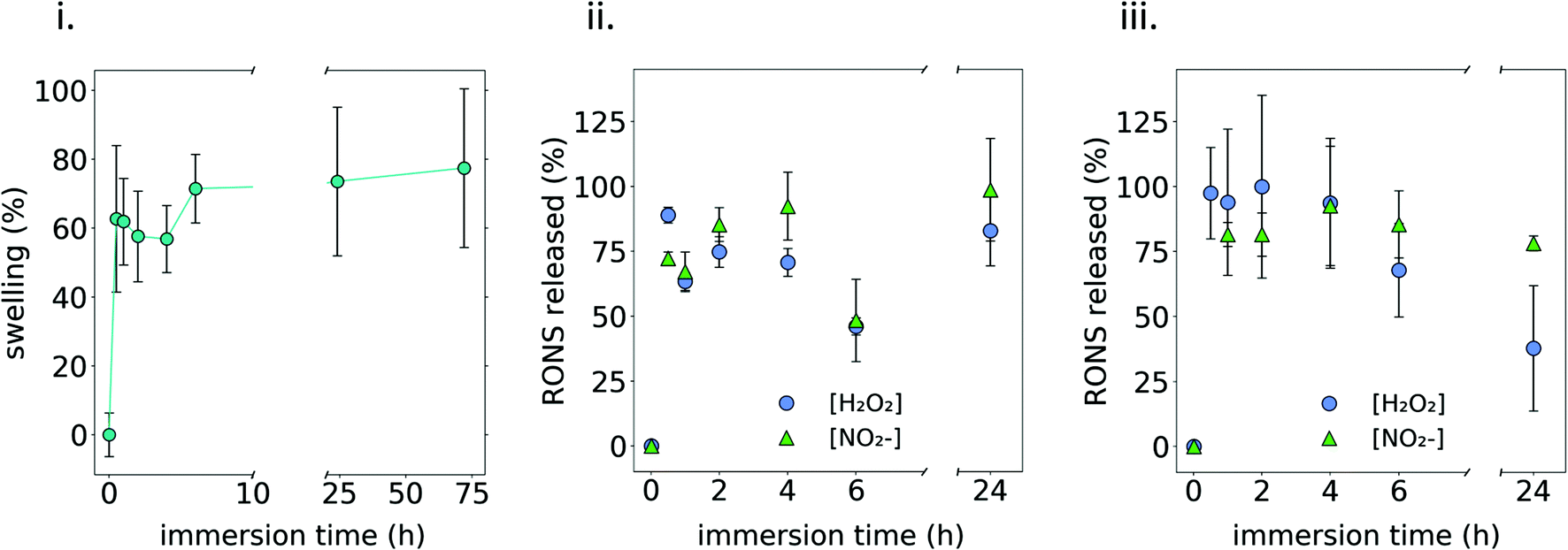 | ||
| Fig. 5 Swelling of the hydrogel and release of plasma generated RONS in 1MC35HP. (i) Swelling of the 1MC3.5HP hydrogel after immersion in PBS at 37 °C. (ii) Release kinetics of plasma generated RONS after immersion of the 1MC3.5HP hydrogel in PBS at 37 °C. (iii) Release kinetics of plasma generated RONS after immersion of the 1MC3.5HP hydrogel in DMEM at 37 °C. | ||
Two different media (PBS and complete culture medium (DMEM)) were used to quantify the release of RONS from the plasma-treated 1MC3.5HP. As observed in Fig. 5ii and iii, an initial burst release took place during the first hour of immersion in PBS, releasing ∼80% of the initial amount of NO2− and H2O2. Then, the amount of these RONS remained constant following 24 h of experiment. This agrees with the release observed from the hydrogel containing NO2− and H2O2 added from standard solutions (Fig. S5†) that are released by diffusion. If compared to the concentrations of RONS quantified in DMEM, an ageing effect was observed when the release took place in this cell culture medium, as the amount of H2O2 decreased considerably after 24 h. As reported in ref. 44, some amino acids in cell culture medium have a clear effect on the stability of RONS. This was also observed in ref. 41, where the addition of FBS to a plasma treated liquid was related to a 30% decrease in the concentration of peroxides. This could be one of the reasons explaining why the concentration of NO2− and H2O2 decreased after 24 h in DMEM as it contains a high concentration of proteins.
It is important to highlight that both the swelling and release of RONS showed a very similar behaviour. Since water uptake kinetics are monitored by swelling, Fig. 5i indicates that water could diffuse into the 3D polymeric network of the MC hydrogel within the first hours of immersion until an equilibrium state was reached. Similarly, RONS could diffuse out to the release media from the 3D polymeric network and an equilibrium state was reached also within the first hours after immersion (Fig. 5ii and iii). Considering the small size of NO2− and H2O2, much below the hydrogel mesh size, the diffusion of these species out of the MC polymeric network was easy and, after 1 h immersion, RONS were completely released. Strategies might have to be considered in future to further slow down this release.
3.5 In vitro anticancer performance
Osteosarcoma cells MG-63 were exposed to plasma-treated 1MC3.5HP and cell viability was evaluated (Fig. 6). As shown in Fig. 6i, the non-treated MC hydrogel can be considered a biocompatible material, since no signs of cell death were observed. In contrast, the plasma-treated hydrogel reduced the metabolic activity of cancer cells in a treatment time-dependent manner, by inducing higher cytotoxicity at a longer plasma-treatment time (Fig. 6i and ii). Short treatment time (60 s) already showed a small reduction in the metabolic activity (∼89%) after 24 h of exposure. These cells were able to recover after 72 h, increasing their metabolic activity up to levels similar to the control. However, at a long treatment time (300 s), a sharp decrease of the metabolic activity after 24 h (∼35%) is observed, which further decreased (∼20%) after 72 h.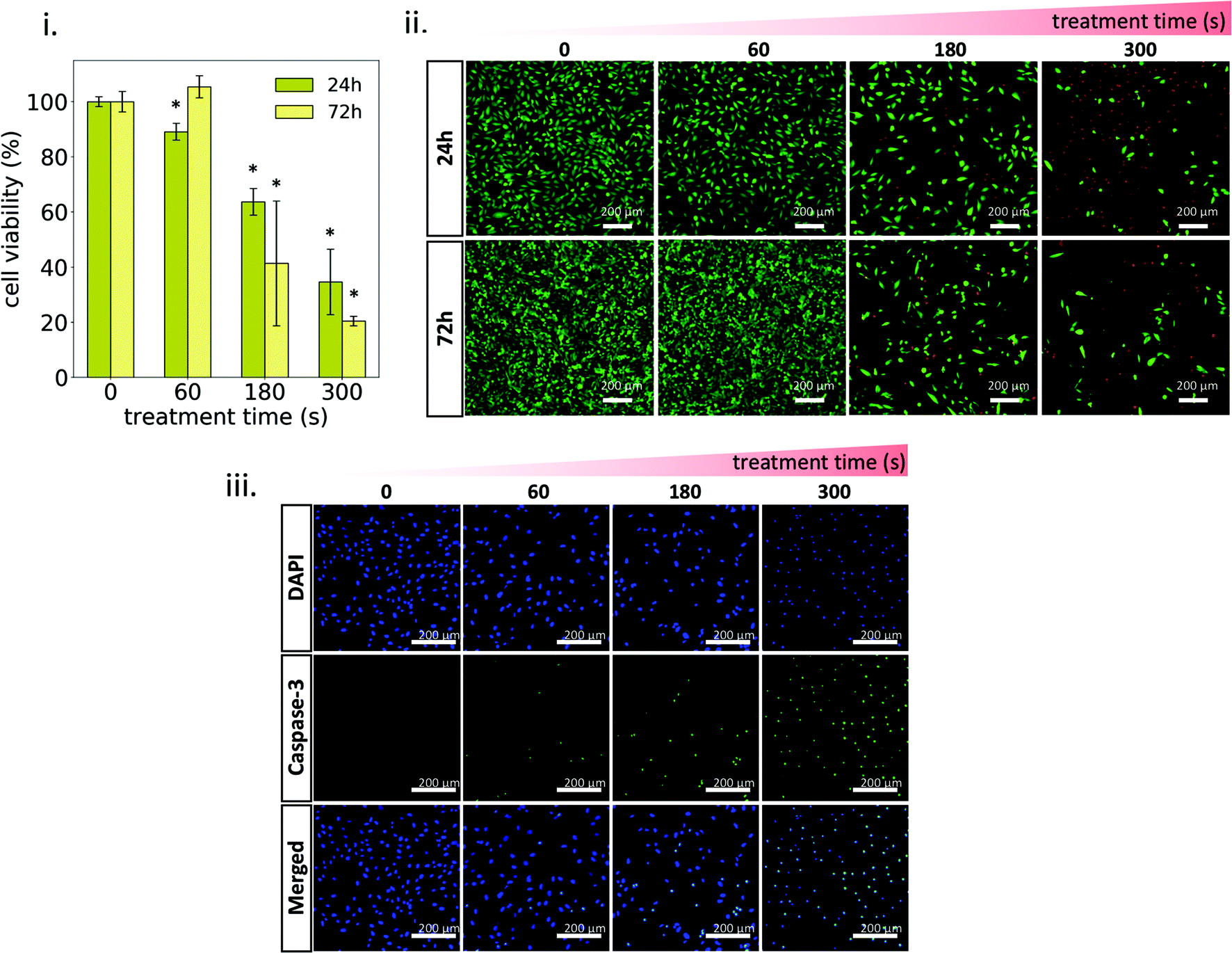 | ||
| Fig. 6 Effects of the plasma-treated hydrogel on MG-63 cells. (i) Cell viability determined at 24 h and 72 h. (ii) Live/Dead assay showing live cells (green) and dead cells (red) after 24 h and 72 h exposure to the plasma-treated hydrogel. (iii) Caspase-3 activity after 24 h exposure to the plasma-treated hydrogel. Asterisks denote statistically significant differences with respect to the control (treatment time = 0 s) (*p < 0.05). | ||
Confocal images of Live/Dead assay were in agreement with cytotoxicity results, where the dose-response effect of plasma-treatment can be clearly observed (Fig. 6ii). This dose-response effect has already been reported by several authors employing different types of plasma-treated liquids as well as plasma-treated hydrogels.45–47 In all cases, the cytotoxic effect of these treatments is based on the delivery of adequate concentrations of plasma-generated RONS to the cancer cell culture.48,49 The oxidative stress generated by the supply of these exogenous RONS has been demonstrated to trigger specific biological pathways in cells that eventually lead to apoptosis.50 In our case, oxidative stress was induced by the release of RONS from the plasma-treated 1MC3.5HP (Fig. 5iii) and higher cytotoxic levels were obtained at a higher concentration of RONS in the culture medium (Fig. 5i and iii). Moreover, the reduced viability observed upon plasma treatment resulted in the activation of the apoptotic marker caspase-3 (Fig. 6iii). This marker showed higher activity at a higher plasma-treatment time, thus confirming the dose-response effect of plasma-treatment. Another sign of apoptotic cell death is chromatin condensation.51,52 This was qualitatively detected for plasma-treated samples, since higher fluorescence intensities and smaller diameters of cell nuclei were observed (Fig. 6iii).
In all, the thermosensitive 1MC3.5HP hydrogel described here has clear advantages regarding the generation and the delivery of RONS to the cell culture. The concentration of RONS delivered by the hydrogel induced similar anticancer effects compared to several types of PTLs.
4 Conclusions
This work presents for the first time a thermosensitive MC hydrogel used as a delivery system of RONS generated by gas-plasma. The polymeric solution designed here (1MC3.5HP) is able to form a hydrogel at physiological temperatures while remaining a liquid at room temperature to allow plasma treatment and the formation of RONS. Although a minor fragmentation of the polymeric chains of MC was observed after plasma treatment, the rheological properties of the biomaterial were not affected, as happened also with its gelling ability. The anticancer effects of plasma-treated MC are time-dependent, showing increased cytotoxicity at longer plasma treatment times related to the higher concentration of RONS generated and released from the MC hydrogel.Author contributions
Conceptualization: Xavi Solé-Martí, Cristina Canal. Data curation: Xavi Solé-Martí, Tània Vilella. Formal analysis: Xavi Solé-Martí, Tània Vilella, Cédric Labay, Francesco Tampieri. Investigation: Xavi Solé-Martí, Tània Vilella, Cédric Labay, Francesco Tampieri. Writing – original draft: Xavi Solé-Martí. Writing – review and editing: Cédric Labay, Francesco Tampieri, Maria-Pau Ginebra, Cristina Canal. Supervision: Cristina Canal. Funding acquisition: Maria-Pau Ginebra, Cristina Canal.Conflicts of interest
The authors declare no competing interests. Images created with BioRender.comAcknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no. 714793). The authors acknowledge the financial support of MINECO for the PID2019-103892RB-I00/AEI/10.13039/501100011033 project. The authors belong to the SGR2017-1165. The authors acknowledge Generalitat de Catalunya for the Scholarship of Xavi Solé-Martí (2020 FI-B00999). Support for the research of Maria-Pau Ginebra and Cristina Canal was received through the ICREA Academia Award for excellence in research, funded by the Generalitat de Catalunya.References
- P. J. Bruggeman, M. J. Kushner, B. R. Locke, J. G. E. Gardeniers, W. G. Graham, D. B. Graves, R. C. H. M. Hofman-Caris, D. Maric, J. P. Reid, E. Ceriani, D. Fernandez Rivas, J. E. Foster, S. C. Garrick, Y. Gorbanev, S. Hamaguchi, F. Iza, H. Jablonowski, E. Klimova, J. Kolb, F. Krcma, P. Lukes, Z. Machala, I. Marinov, D. Mariotti, S. Mededovic Thagard, D. Minakata, E. C. Neyts, J. Pawlat, Z. L. Petrovic, R. Pflieger, S. Reuter, D. C. Schram, S. Schröter, M. Shiraiwa, B. Tarabová, P. A. Tsai, J. R. R. Verlet, T. Von Woedtke, K. R. Wilson, K. Yasui and G. Zvereva, Plasma Sources Sci. Technol., 2016, 25, 53002 CrossRef.
- P. Bruggeman and C. Leys, J. Phys. D: Appl. Phys., 2009, 42, 53001 CrossRef.
- Y. Li, T. Tang, H. J. Lee and K. Song, Int. J. Mol. Sci., 2021, 22, 3956 CrossRef CAS PubMed.
- E. Biscop, A. Lin, W. Van Boxem, J. Van Loenhout, J. De Backer, C. Deben, S. Dewilde, E. Smits and A. Bogaerts, Cancers, 2019, 11, 1287 CrossRef CAS PubMed.
- R. Silva-Teixeira, M. Laranjo, B. Lopes, C. Almeida-Ferreira, A. C. Gonçalves, T. Rodrigues, P. Matafome, A. B. Sarmento-Ribeiro, F. Caramelo and M. F. Botelho, Free Radicals Biol. Med., 2021, 171, 302–313 CrossRef CAS PubMed.
- M. Mateu-Sanz, J. Tornín, B. Brulin, A. Khlyustova, M. P. Ginebra, P. Layrolle and C. Canal, Cancers, 2020, 12, 227 CrossRef CAS PubMed.
- X. Solé-Martí, A. Espona-Noguera, M. P. Ginebra and C. Canal, Cancers, 2021, 13, 1–17 CrossRef PubMed.
- Z. Chen, H. Simonyan, X. Cheng, E. Gjika, L. Lin, J. Canady, J. H. Sherman, C. Young and M. Keidar, Cancers, 2017, 9, 1–16 CrossRef CAS PubMed.
- F. Saadati, H. Mahdikia, H. A. Abbaszadeh, M. A. Abdollahifar, M. S. Khoramgah and B. Shokri, Sci. Rep., 2018, 8, 7689 CrossRef PubMed.
- F. Utsumi, H. Kajiyama, K. Nakamura, H. Tanaka, M. Mizuno, K. Ishikawa, H. Kondo, H. Kano, M. Hori and F. Kikkawa, PLoS One, 2013, 8, 81576 CrossRef PubMed.
- K. R. Liedtke, S. Bekeschus, A. Kaeding, C. Hackbarth, J. P. Kuehn, C. D. Heidecke, W. Von Bernstorff, T. Von Woedtke and L. I. Partecke, Sci. Rep., 2017, 7, 8319 CrossRef PubMed.
- K. R. Liedtke, E. Freund, C. Hackbarth, C. D. Heidecke, L. I. Partecke and S. Bekeschus, Clin. Plasma Med., 2018, 11, 10–17 CrossRef.
- C. Labay, I. Hamouda, F. Tampieri, M. P. Ginebra and C. Canal, Sci. Rep., 2019, 9, 16160 CrossRef PubMed.
- C. Labay, M. Roldán, F. Tampieri, A. Stancampiano, P. E. Bocanegra, M. P. Ginebra and C. Canal, ACS Appl. Mater. Interfaces, 2020, 12, 47256–47269 CrossRef CAS PubMed.
- X. Solé-Martí, C. Labay, Y. Raymond, J. Franch, R. Benitez, M.-P. Ginebra and C. Canal, Submitted for Publication.
- H. Zhang, S. Xu, J. Zhang, Z. Wang, D. Liu, L. Guo, C. Cheng, Y. Cheng, D. Xu, M. G. Kong, M. Rong and P. K. Chu, Biomaterials, 2021, 276, 121057 CrossRef CAS PubMed.
- M. H. Kim, B. S. Kim, H. Park, J. Lee and W. H. Park, Int. J. Biol. Macromol., 2018, 109, 57–64 CrossRef CAS PubMed.
- M. C. Tate, D. A. Shear, S. W. Hoffman, D. G. Stein and M. C. LaPlaca, Biomaterials, 2001, 22, 1113–1123 CrossRef CAS PubMed.
- M. K. Bain, B. Bhowmick, D. Maity, D. Mondal, M. M. R. Mollick, D. Rana and D. Chattopadhyay, Int. J. Biol. Macromol., 2012, 51, 831–836 CrossRef CAS PubMed.
- N. Li, E. Liu and C. H. Lim, J. Phys. Chem. B, 2007, 111, 6410–6416 CrossRef PubMed.
- S. Q. Liu, S. C. Joshi and Y. C. Lam, J. Appl. Polym. Sci., 2008, 109, 363–372 CrossRef CAS.
- J. Tornin, C. Labay, F. Tampieri, M. P. Ginebra and C. Canal, Nat. Protoc., 2021, 16, 2826–2850 CrossRef CAS PubMed.
- I. Guevara, J. Iwanejko, A. Dembińska-Kieć, J. Pankiewicz, A. Wanat, P. Anna, I. Goł
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) bek, S. Bartuś, M. Malczewska-Malec and A. Szczudlik, Clin. Chim. Acta, 1998, 274, 177–188 CrossRef CAS.
bek, S. Bartuś, M. Malczewska-Malec and A. Szczudlik, Clin. Chim. Acta, 1998, 274, 177–188 CrossRef CAS. - M. J. Moorcroft, J. Davis and R. G. Compton, Talanta, 2001, 54, 785–803 CrossRef CAS PubMed.
- A. Khlyustova, C. Labay, Z. Machala, M. P. Ginebra and C. Canal, Front. Chem. Sci. Eng., 2019, 13, 238–252 CrossRef CAS.
- M. Takahashi, M. Shimazaki and J. Yamamoto, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 91–100 CrossRef CAS.
- S. Morozova, M. L. Coughlin, J. T. Early, S. P. Ertem, T. M. Reineke, F. S. Bates and T. P. Lodge, Macromolecules, 2019, 52, 7740–7748 CrossRef CAS.
- S. A. Arvidson, J. R. Lott, J. W. McAllister, J. Zhang, F. S. Bates, T. P. Lodge, R. L. Sammler, Y. Li and M. Brackhagen, Macromolecules, 2013, 46, 300–309 CrossRef CAS.
- V. Veronico, P. Favia, F. Fracassi, R. Gristina and E. Sardella, Plasma Processes Polym., 2021, 18, 2100062 CrossRef CAS.
- L. Li and Y. Aoki, Macromolecules, 1997, 30, 7835–7841 CrossRef CAS.
- L. Deng, Y. Liu, L. Yang, J. Z. Yi, F. Deng and L. M. Zhang, Colloids Surf., B, 2020, 194, 111159 CrossRef CAS PubMed.
- K. K. Tha Goh, M. S. Mei Wee and Y. Hemar, Food Funct., 2013, 4, 627–634 RSC.
- G. R. Filho, R. M. N. de Assunção, J. G. Vieira, C. da S. Meireles, D. A. Cerqueira, H. da Silva Barud, S. J. L. Ribeiro and Y. Messaddeq, Polym. Degrad. Stab., 2007, 92, 205–210 CrossRef.
- M. Baiardo, G. Frisoni, M. Scandola and A. Licciardello, J. Appl. Polym. Sci., 2002, 83, 38–45 CrossRef CAS.
- R. G. Zhbankov, Infrared Spectra of Cellulose and its Derivatives, Springer US, Boston, MA, 1995 Search PubMed.
- H. Akinosho, S. Hawkins and L. Wicker, Carbohydr. Polym., 2013, 98, 276–281 CrossRef CAS PubMed.
- Z. Liu, Y. Zheng, J. Dang, J. Zhang, F. Dong, K. Wang and J. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 22941–22949 CrossRef CAS PubMed.
- P. L. Nasatto, F. Pignon, J. L. M. Silveira, M. E. R. Duarte, M. D. Noseda and M. Rinaudo, Polymers, 2015, 7, 777–803 CrossRef CAS.
- Y. Sekiguchi, C. Sawatari and T. Kondo, Polym. Bull., 2002, 47, 547–554 CrossRef CAS.
- M. Karakawa, Y. Mikawa, H. Kamitakahara and F. Nakatsubo, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 4167–4179 CrossRef CAS.
- J. Tornín, A. Villasante, X. Solé-Martí, M. P. Ginebra and C. Canal, Free Radicals Biol. Med., 2021, 164, 107–118 CrossRef PubMed.
- K. R. Liedtke, E. Freund, M. Hermes, S. Oswald, C. D. Heidecke, L. I. Partecke and S. Bekeschus, Cancers, 2020, 12, 123 CrossRef CAS PubMed.
- I. E. Vlad and S. D. Anghel, J. Electrost., 2017, 87, 284–292 CrossRef CAS.
- D. Yan, N. Nourmohammadi, K. Bian, F. Murad, J. H. Sherman and M. Keidar, Sci. Rep., 2016, 6, 26016 CrossRef CAS PubMed.
- N. K. Kaushik, B. Ghimire, Y. Li, M. Adhikari, M. Veerana, N. Kaushik, N. Jha, B. Adhikari, S. J. Lee, K. Masur, T. Von Woedtke, K. D. Weltmann and E. H. Choi, Biol. Chem., 2018, 400, 39–62 Search PubMed.
- Y. Sato, S. Yamada, S. Takeda, N. Hattori, K. Nakamura, H. Tanaka, M. Mizuno, M. Hori and Y. Kodera, Ann. Surg. Oncol., 2018, 25, 299–307 CrossRef PubMed.
- D. Yan, J. H. Sherman and M. Keidar, Anticancer Agents Med. Chem., 2017, 18, 769–775 CrossRef PubMed.
- D. Yan, A. Talbot, N. Nourmohammadi, X. Cheng, J. Canady, J. Sherman and M. Keidar, Sci. Rep., 2015, 5, 1–17 Search PubMed.
- G. Bauer, Redox Biol., 2019, 26, 101291 CrossRef CAS PubMed.
- M. Mateu-Sanz, J. Tornín, M. P. Ginebra and C. Canal, J. Clin. Med., 2021, 10, 1–29 Search PubMed.
- D. Gümbel, S. Bekeschus, N. Gelbrich, M. Napp, A. Ekkernkamp, A. Kramer and M. B. Stope, Int. J. Mol. Sci., 2017, 18, 2004 CrossRef PubMed.
- G. Majno and I. Joris, Am. J. Pathol., 1995, 146, 3–15 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm00308b |
| This journal is © The Royal Society of Chemistry 2022 |