
pH-Responsive hyaluronic acid-cloaked polycation/gold nanohybrids for tumor-targeted synergistic photothermal/gene therapy†
Yanjun
Liu‡
ab,
Xiaoguang
Dai‡
a,
Bingran
Yu
 a,
Meiwan
Chen
c,
Nana
Zhao
*a and
Fu-Jian
Xu
*a
a,
Meiwan
Chen
c,
Nana
Zhao
*a and
Fu-Jian
Xu
*a
aState Key Laboratory of Chemical Resource Engineering, Key Laboratory of Biomedical Materials of Natural Macromolecules (Beijing University of Chemical Technology), Beijing Laboratory of Biomedical Materials, College of Materials Sciences and Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: xufj@mail.buct.edu.cn; zhaonn@mail.buct.edu.cn
bDepartment of Materials Engineering, Taiyuan Institute of Technology, Taiyuan 030008, China
cState Key Laboratory of Quality Research in Chinese Medicine, Institute of Chinese Medical Sciences, University of Macau, Macau 999078, China
First published on 12th April 2022
Abstract
The combination of photothermal therapy (PTT) and gene therapy (GT) has attracted intense interest in cancer treatment. However, the lack of long circulation and active tumor targeting reduces the therapeutic efficacy of complementary PTT/GT. In this work, hyaluronic acid (HA)–cloaked gold nanorods–PGED (prepared by ring-opening of polyglycidyl methacrylate (PGMA) with ethylenediamine (ED))/pDNA (AP/pDNA–HA) complexes were prepared to achieve long circulation and tumor targeting for photoacoustic imaging (PAI)-guided synergistic PTT/GT. Gold nanorods endow the complexes with photothermal effect and PAI function. Benefiting from the HA cloak, the AP/pDNA–HA complexes exhibit excellent stability, biocompatibility, long circulation behavior and active targeting. In addition, the pH-responsive characteristic of the Schiff base bonds helps the AP/pDNA–HA complexes to effectively escape from the endosome/lysosome. The antioncogene p53 was employed to investigate the gene transfection efficiency of the delivery system both in vitro and in vivo. The superiority of synergistic PTT/GT is established in a mouse 4T1 breast tumor model. The current study provides a facile strategy for constructing multifunctional gene delivery systems with long circulation and tumor targeting features, which can achieve effective imaging-guided synergistic tumor treatment.
1. Introduction
Gene carriers with photothermal properties could combine photothermal therapy (PTT) and gene therapy (GT) to realize synergistic therapeutic effects.1,2 The photothermal effect that converts light energy into heat can help trigger gene release,3–5 enhance cellular uptake,3,6 and facilitate endosomal/lysosomal escape to promote gene delivery.7,8 Due to the superiority in photothermal performance, photoacoustic imaging (PAI), and facile surface modification, a variety of gold nanorod (Au NR)-based gene carriers with positive charges have been applied in complementary PTT/GT.9–11 However, due to non-specific adsorption and lack of active tumor targeting, positively charged gene carriers are easily cleared from circulation.12,13 Polyethylene glycol (PEG)-modified carriers are thought to extend the circulation time and reduce carrier-associated cytotoxicity.13–15 However, as a hydrophilic shielding material, PEG usually hinders the cellular uptake of carriers and lacks tumor targeting ability, resulting in reduced transfection efficiency.13,16 The development of shielding materials with targeting function to achieve accumulation in the tumor region for effective gene transfection still remains a great challenge. Therefore, it will be desirable to propose a simple and efficient strategy to fabricate Au NR-based gene carriers with prolonged circulation time and tumor-targeted accumulation to realize PAI-guided synergistic PTT/GT.The natural polysaccharide hyaluronic acid (HA) exhibits good biocompatibility, biodegradability, and biological functions.17 HA can bind to the CD44 receptor, which is overexpressed on the tumor cell surface.17 Therefore, HA-modified nanoparticles have been employed to enhance the delivery of chemotherapeutic drugs,18–20 proteins,21,22 or genes23–30 by locating the CD44 receptor. For most cases of gene delivery, cationic polyethyleneimine (PEI) was employed to construct HA-cloaked gene carriers.24,25,28–30 However, high cytotoxicity limits their application in vivo.31 In addition, for the HA-cloaked nanoplatforms used for complementary PTT/GT, the colloidal stability in serum is usually poor due to weak electrostatic interaction between HA and nanoparticles, resulting in protein instability during circulation.25 Moreover, whether PTT/GT could produce synergistic therapeutic effect in vivo remains to be further explored. If HA could be applied to shield Au NR-based gene carriers via chemical bonds to produce stable and biocompatible nanohybrids, an advanced gene delivery system with long circulation, tumor-specific accumulation, and photothermal-enhanced GT could be achieved.
Endosomal escape is a key step in gene delivery while most non-viral gene carriers achieve this step through the “proton sponge effect”.12,32 The shielding layer of gene carriers might interfere with endosomal escape and inhibit transfection efficiency due to steric hindrance.12 To overcome this obstacle, a reversible shielding layer that can detach from the gene carriers in response to the tumor microenvironment would be an ideal choice. Schiff base bonds that are cleavable in the acidic tumor microenvironment have been used for responsive drug or gene delivery.14,33–35 The combination of HA and Au NR-based gene carriers through Schiff base bonding could promote endosomal escape and facilitate synergistic PTT/GT.
Herein we propose the construction of HA-cloaked polycation/gold gene carriers with long circulation and active tumor targeting ability to achieve PAI-guided PTT/GT. Au NRs endow the carriers with photothermal effect and PAI functions. A cationic polymer PGED (prepared by ring-opening of polyglycidyl methacrylate (PGMA) with ethylenediamine (ED))36 was conjugated onto the surface of Au NRs to prepare Au–PGED (AP) for gene delivery.9 HA was then introduced to cloak AP/plasmid DNA (pDNA) complexes through Schiff base bonds, which could realize reversible shielding with pH responsiveness. Through the rational design, prolonged circulation time, active tumor targeting, and enhanced endosomal escape could be achieved for efficient gene transfection (Scheme 1). Meanwhile, under near-infrared (NIR) laser irradiation, synergistic PTT/GT could be realized. The feasibility of the HA-cloaked multifunctional system for PAI-guided PTT/GT was investigated in detail both in vitro and in vivo.
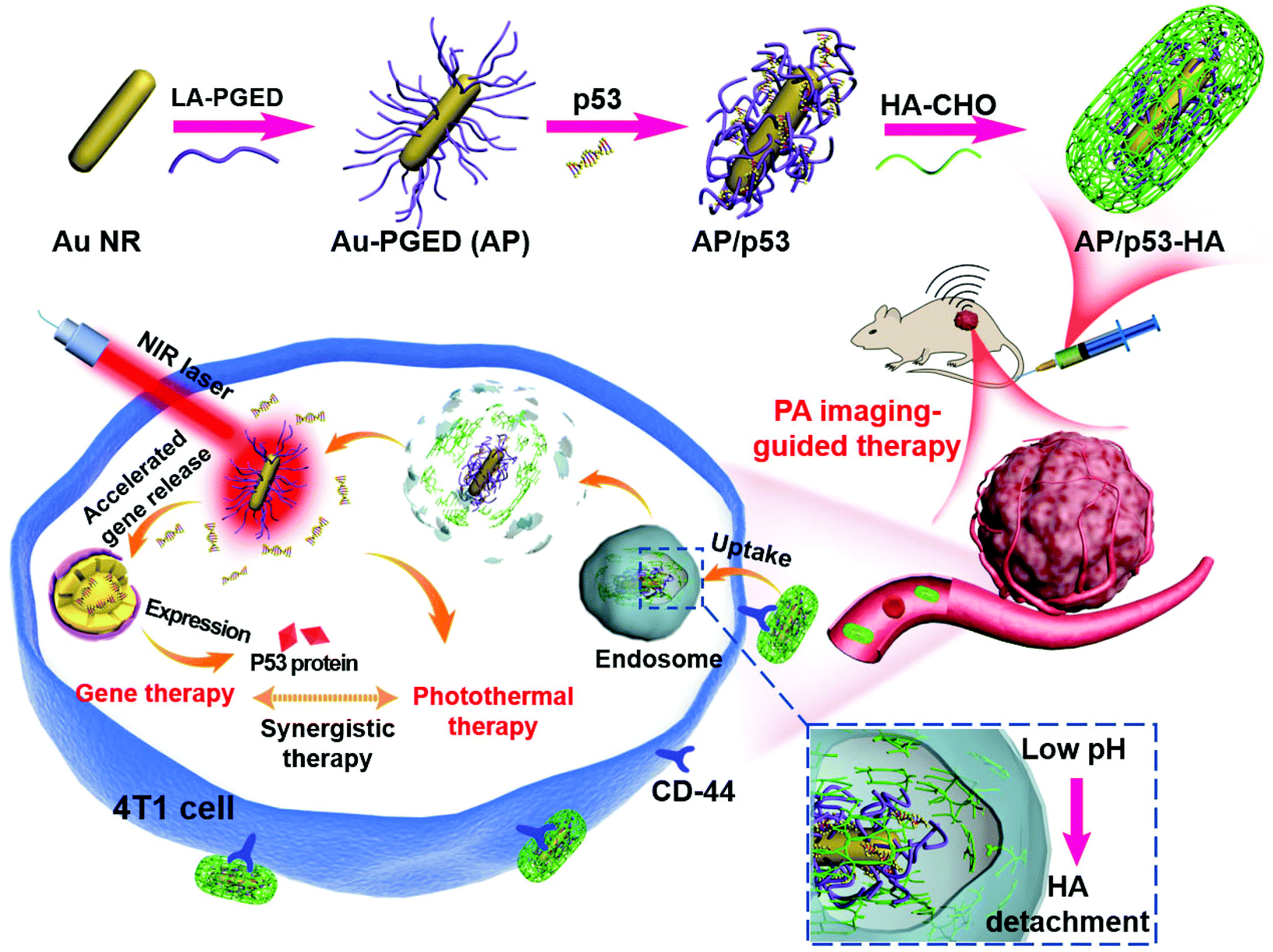 | ||
| Scheme 1 Schematic presentation of the design of AP/p53–HA complexes and their application for tumor-targeted complementary PTT/GT. | ||
2. Experimental
2.1. Preparation of AP nanohybrids
AP nanohybrids were prepared by ligand exchange between gold nanorods and PGED–LA (lipoic acid linked with PGED) through the “grafting-onto” method. The synthesis of gold nanorods was through the seed-mediated approach.37 To prepare PGED–LA, PGMA and PGED were synthetized as previously described.9 PGED–LA was synthesized by conjugating LA with PGED. The detailed information is shown in the ESI.†2.2. Preparation of AP/pDNA–HA complexes
AP/pDNA complexes were firstly prepared at a fixed N/P ratio of 15 and incubated for 30 min. Then, different amounts of HA–CHO (aldehyde groups-bearing hyaluronic acid) were added to the solution to produce AP/pDNA–HA complexes. Weight ratios of HA/PGED in AP/pDNA–HA complexes were 25%, 50%, 75% and 100%, respectively. The detailed procedures can be found in the ESI.†2.3. In vitro gene transfection, cytotoxicity and cellular internalization
Gene transfection of AP/pDNA–HA complexes were performed in HEK293 and 4T1 cell lines as previously described.38 The cell cytotoxicity of AP/pDNA–HA complexes was evaluated through the MTT (3-(4,5-dimethylthiazol-2yl)-2,5-diphenyl tetrazolium bromide) assay.38 Cellular internalization of AP/pDNA–HA was investigated by fluorescence imaging, confocal laser scanning microscopy (CLSM) and flow cytometry. The detailed procedures are displayed in the ESI.†2.4. Complementary PTT/GT in vitro
Complementary antitumor effects of PTT/GT in vitro were evaluated through the MTT and FDA–PI (fluorescein diacetate–propidium iodide) dual staining assays. The 4T1 cells were treated with NIR light irradiation, AP/p53–HA, AP–HA + NIR light irradiation and AP/p53–HA + NIR light irradiation, respectively, as described in the ESI.†2.5. Western blot
The expression of P53 protein in 4T1 cells was determined by the western blot assay. For in vivo testing, the P53 protein was extracted from the tumor tissue. Detailed procedures can be found in the ESI.†2.6. In vivo PAI
AP–HA or AP nanohybrids were injected into 4T1 tumor-bearing mice intravenously. Thereafter, PA images were captured and the PA intensity was recorded as shown in the ESI.†2.7. Complementary PTT/GT in vivo
For tumor therapy, 4T1 tumor-bearing mice were randomly divided into four groups, which were intravenously injected with PBS (control group), AP/p53–HA (GT group), AP–HA + NIR (PTT group), and AP/p53–HA + NIR (PTT/GT group), respectively. Detailed information is given in the ESI.†3. Results and discussion
3.1. Preparation and characterization of the HA-cloaked complexes
The synthetic procedures of the HA-cloaked gene delivery system (AP/pDNA–HA) involve the preparation of AP nanohybrids and the subsequent modification of AP with HA–CHO (Scheme 1). Firstly, AP nanohybrids were prepared by conjugating PGED–LA onto the surface of Au NRs through ligand exchange.9 As shown in the transmission electron microscopy (TEM) image (Fig. 1a), Au NRs (∼40 nm in length and ∼10 nm in diameter) were obtained by a seed-mediated growth strategy. Then, the cationic polymer PGMA (Mn = 1.01 × 104, PDI = 1.31) was synthesized by atom transfer radical polymerization and then reacted with excess ethylenediamine through ring-opening reaction to prepare PGED. Then, PGED–LA was successfully prepared by the conjugation of lipoic acid and PGED. Typical structures of PGMA, PGED and PGED–LA were characterized by 1H NMR spectra, as shown in Fig. S1 of the ESI.† Finally, AP nanohybrids were prepared by conjugating PGED–LA onto the surface of Au NRs. The dynamic light scattering results show that compared with Au NRs, the size of AP nanohybrids increased evidently (from ∼59 to ∼190 nm, Fig. S2 of the ESI†). In addition, a slight redshift of ∼12 nm was observed for the localized surface plasmon resonance (SPR) peak of AP (Fig. 1b), which may be attributed to refractive index change caused by the polymer attached on Au NRs.39 These findings are consistent with our previous work, indicating that PGED–LA was successfully grafted onto the surface of Au NRs.9,40 Thermal gravimetric analysis also verified the conjugation of the polymer in the AP nanohybrids (Fig. S3, ESI†). | ||
| Fig. 1 (a) TEM image of Au NRs. (b) UV-vis spectra of Au NRs and AP. (c) Luciferase gene transfection efficiency of AP/pDNA/HA′ and AP/pDNA–HA complexes (N/P = 15) with different HA/PGED weight ratios in 4T1 cells. Luciferase gene transfection efficiency of AP/pDNA and AP/pDNA–HA complexes with different N/P ratios in 4T1 (d) and HEK293 (e) cell lines. (f) Cytotoxicity of AP/pDNA and AP/pDNA–HA in 4T1 cells. (g) Representative fluorescence images of pEGFP expression in 4T1 cells mediated by PEI/pDNA, PGED/pDNA, AP/pDNA, and AP/pDNA–HA at their optimal N/P ratios, respectively. | ||
Aldehyde-functionalized HA (HA–CHO) was readily synthesized through the oxidation reaction.25 The absorption peak at 1730 cm−1 in the Fourier transform infrared (FTIR) spectra (Fig. S4, ESI†)41 confirms the successful preparation of HA–CHO. The degree of oxidation of the resultant HA–CHO was determined to be ∼50% by the hydroxylamine hydrochloride titration method.42 The reporter plasmid DNA (encoding Renilla luciferase) was then adopted to construct AP/pDNA complexes. The AP/pDNA complexes were prepared at the fixed N/P ratio (calculated from the molar ratio of nitrogen (N) in PGED to phosphate (P) in pDNA) of 15. HA-cloaked complexes AP/pDNA–HA were obtained after the introduction of HA–CHO to AP/pDNA. The electrostatic interactions and Schiff base bonds between HA–CHO and AP/pDNA were supposed to be responsible for the formation of the AP/pDNA–HA complexes. When different amounts of HA–CHO were applied, the weight ratios of HA/PGED in the complexes were 25%, 50%, 75%, and 100%, respectively. For comparison, AP/pDNA/HA′ complexes were also prepared by the electrostatic interaction between AP/pDNA and HA. The particle sizes and zeta potentials of AP/pDNA–HA and AP/pDNA/HA′ complexes investigated by dynamic light scattering confirmed the successful formation of the complexes (Fig. S5a and S5b, ESI†). Due to the dual interactions in the AP/pDNA–HA complexes, the particle size of the AP/pDNA–HA complexes was significantly smaller than that of AP/pDNA/HA′ when the amount of HA was comparable. In addition, the zeta potentials of AP/pDNA–HA and AP/pDNA/HA′ decreased to ∼8 mV at the HA/PGED weight ratio of 50% due to the shielding effect of HA–CHO or HA on positively charged AP/pDNA complexes (∼33 mV). Electrophoretic mobility retardation assay revealed that both AP/pDNA–HA and AP/pDNA/HA′ complexes could condense pDNA effectively at the HA/PGED weight ratio from 25% to 100% (Fig. S5c, ESI†).
3.2. Gene transfection and targeting ability of AP/pDNA–HA complexes
The gene transfection efficiency of the two complexes was evaluated employing luciferase as the reporter gene. As shown in Fig. 1c, gene transfection efficiency of AP/pDNA–HA and AP/pDNA/HA′ complexes in 4T1 cells first increased and then decreased with the increase of the HA/PGED weight ratio. Meanwhile, the AP/pDNA–HA complexes exhibited higher gene transfection efficiencies than AP/pDNA/HA′ at the same HA/PGED weight ratio. The optimal gene transfection efficiency appeared at the HA/PGED weight ratio of 50%, which was probably due to the combined effects of suitable particle size, zeta potential, active targeting, and stability.12 To verify the feasibility of applying the complexes in vivo, the serum stability of AP/pDNA–HA, AP/pDNA/HA′, and AP/pDNA was investigated in cell medium supplemented with 10% fetal bovine serum (FBS). The particle size changes after incubation for different periods are exhibited in Fig. S5d of the ESI.† AP/pDNA displayed much larger particle size in serum compared with the particle size in water, which was due to the strong adsorption of proteins. AP/pDNA/HA′ demonstrated similar behavior, probably resulting from the unstable electrostatic interaction between AP, pDNA, HA, and FBS. In contrast, the AP/pDNA–HA complexes exhibited stable particle size within 200 nm after the incubation in FBS for 96 h. Dual interactions (Schiff base bonding and electrostatic attraction) in the complexes and the resistance of HA to protein adhesion were supposed to contribute to the excellent stability of AP/pDNA–HA.25,43 These results suggest the great potential of AP/pDNA–HA for long circulation in vivo. At the HA/PGED weight ratio of 50%, AP/pDNA–HA complexes exhibited significantly higher gene transfection efficiency than that of AP/pDNA/HA′ or AP/pDNA (Fig. 1c), which could be attributed to the much higher stability. In addition, it is speculated that compared with AP/pDNA, the active targeting of AP/pDNA–HA introduced by HA could compensate for the loss of the positive surface charge on gene transfection efficiency. Therefore, AP/pDNA–HA with an optimal HA/PGED weight ratio of 50% was selected for further studies. To verify the pH-responsive separation of the HA cloak in AP/pDNA–HA complexes, the zeta potential of AP/pDNA–HA over time was evaluated in buffer solutions of pH 5.0 and 7.4 (Fig. S6, ESI†). The apparent increase in zeta potential (from ∼8 to ∼34 mV) in pH 5.0 buffer solution compared to the negligible change in pH 7.4 buffer solution indicates successful detachment of the shielding HA layer.To verify the targeting ability of AP/pDNA–HA for gene delivery, gene transfection mediated by AP/pDNA and HA-cloaked Ap/pDNA (AP/pDNA–HA with the HA/PGED weight ratio of 50%) were evaluated in both CD44-positive 4T1 cells and CD44-negative HEK293 cells. As shown in Fig. 1d, in 4T1 cells, the gene transfection efficiency mediated by AP/pDNA–HA was significantly higher than that mediated by AP/pDNA under various N/P ratios. In contrast, AP/pDNA–HA and AP/pDNA-mediated gene transfection showed no obvious difference in HEK293 cells (Fig. 1e). The enhanced gene transfection efficiency in CD44-positive 4T1 cells was believed to confirm the tumor targeting ability of the AP/pDNA–HA complexes. In addition, the AP/pDNA–HA complexes exhibited obviously higher gene transfection efficiency at the optimal N/P ratio of 15 compared with PEI (25 kDa) and PGED at their optimal N/P ratios of 10 and 15, respectively.44 The HA cloak was also found to evidently decrease the cytotoxicity of AP/pDNA–HA complexes compared with AP/pDNA in both 4T1 cells and HEK293 cells (Fig. 1f and Fig. S7, ESI†). This phenomenon could be attributed to the shielding effect of HA on the positively charged AP/pDNA, indicating the great potential of AP/pDNA–HA in biomedical applications.
The gene expression of enhanced green fluorescent protein (EGFP) was also visualized in 4T1 cells (Fig. 1g), while the percentage of EGFP-positive cells was quantified by the flow cytometry assay (Fig. S8, ESI†). The AP/pDNA–HA complexes exhibited much higher EGFP expression level (∼30.2%) compared with AP/pDNA (∼19.5%), PGED/pDNA (∼11.4%), and PEI/pDNA (∼13.3%) at their corresponding optimal N/P ratios. Therefore, AP/pDNA–HA complexes with the N/P ratio of 15 and HA/PGED weight ratio of 50% could be promising in gene delivery with the superiority of low cytotoxicity, active tumor targeting, and high gene transfection efficiency.
3.3. Cellular internalization
To reveal the mechanism involved in the enhanced gene delivery of AP/pDNA–HA complexes, cellular internalization of PGED/pDNA, PEI/pDNA, AP/pDNA and AP/pDNA–HA complexes was studied. As displayed in Fig. S9 of the ESI,† pDNA was labeled with green fluorescent YOYO-1 to visualize the complexes while nuclei of 4T1 cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) in blue. Much brighter green signals were observed in the AP/pDNA–HA group than other complexes. Furthermore, quantitative analysis by flow cytometry revealed the internalization percentages of 85.7%, 73.9%, 43.2%, and 36.5% for AP/pDNA–HA, AP/pDNA, PEI/pDNA, and PGED/pDNA (Fig. 2a), confirming the enhanced internalization of the AP/pDNA–HA complexes. CLSM was used to monitor the real-time intracellular distribution of AP/pDNA and AP/pDNA–HA. As shown in Fig. 2b, at 0.5 h of incubation, both complexes appeared at the cell membrane. After 4 h, the complexes entered the cells and distributed around the nuclei. Stronger green signals were observed in the cytoplasm of 4T1 cells treated with AP/pDNA–HA complexes, verifying the active tumor targeting ability of HA-cloaked complexes. After 6 h of incubation, the green signals were overlapped with the blue nuclei, indicating the release of pDNA from complexes. Higher fluorescence signals appeared in cells treated with the AP/pDNA–HA complexes. These findings confirm that the enhanced gene transfection of AP/pDNA–HA was due to the increased cellular internalization in 4T1 cells.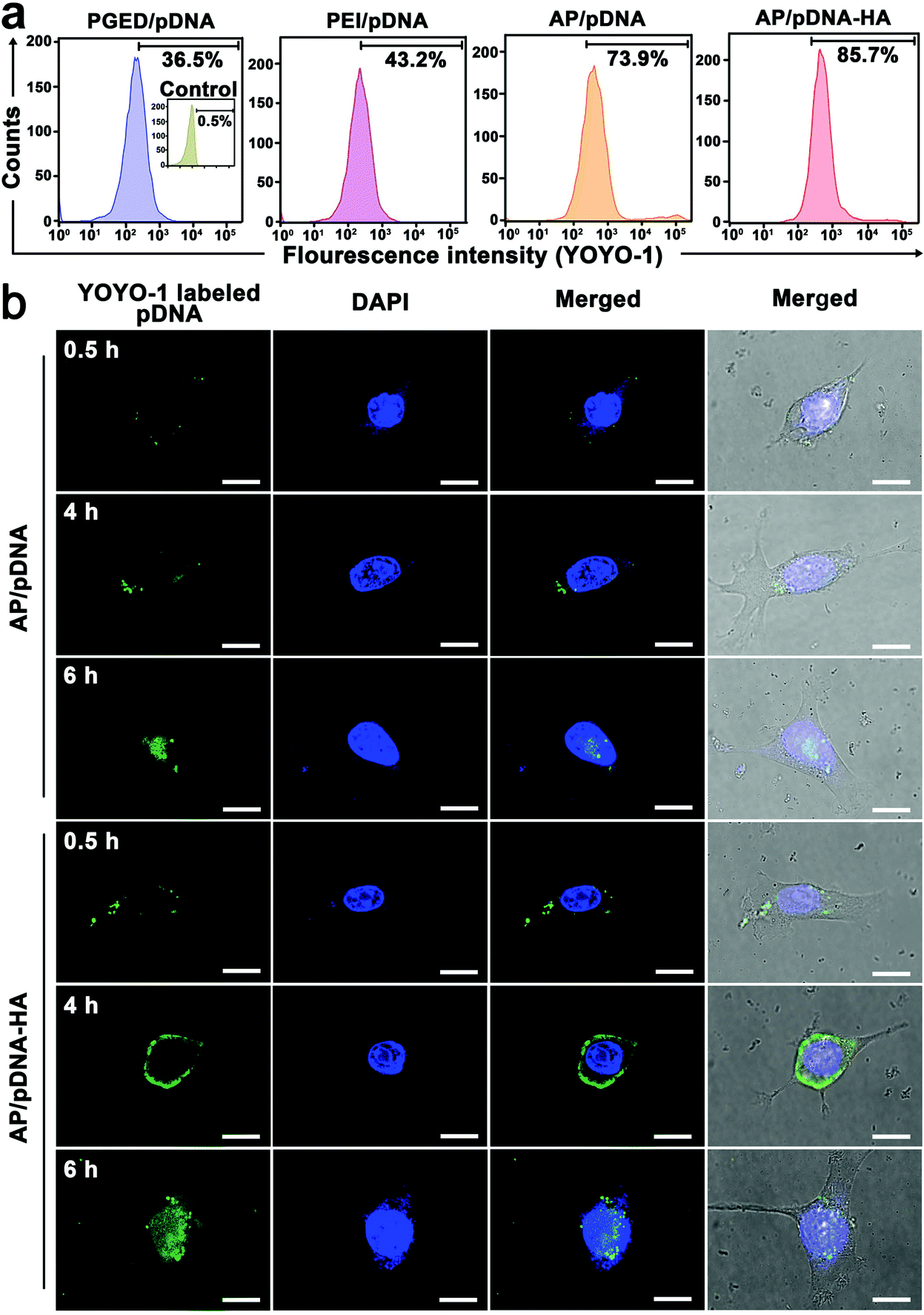 | ||
| Fig. 2 (a) Flow cytometric results of 4T1 cells treated with PGED/pDNA, PEI/pDNA, AP/pDNA and AP/pDNA–HA. (b) CLSM images of 4T1 cells treated with AP/pDNA and AP/pDNA–HA for 0.5, 4, and 6 h, respectively. Scale bar: 10 μm. | ||
3.4. Combined PTT/GT in vitro
The stability of pDNA after the complexes were irradiated with NIR light laser was evaluated before the study of PTT/GT in vitro. As shown in Fig. S10 of the ESI,† pDNA could be successfully released from the AP/pDNA–HA complexes under NIR light irradiation without damage.45 NIR irradiation was supposed to reduce the attraction between complexes and pDNA to promote DNA release.46 Encouraged by the superior gene transfection performance of AP/pDNA–HA in vitro, the antioncogene p53 was used to assess the antitumor effect of the AP/p53–HA complexes (N/P = 15, HA/PGED weight ratio = 50%). 4T1 cells were treated with PGED/p53, AP/p53, and AP/p53–HA complexes, respectively. After gene transfection for 48 h, the percentages of apoptotic 4T1 cancer cells were assessed by flow cytometry. As shown in Fig. 3a, the percentages of apoptotic cells induced by PGED/p53, AP/p53, and AP/p53–HA were 15.79%, 21.45%, and 30.97%, respectively while negligible apoptotic cells in the control group were observed. The most severe apoptosis of 4T1 cells induced by the AP/p53–HA complexes verified the successful delivery of p53 and superior gene transfection performance of HA-cloaked complexes. Western blot assay confirmed the enhanced expression of P53 protein mediated by the HA-cloaked complexes (Fig. 3b). Quantitative analysis revealed that the expression level of P53 protein in the AP/p53–HA group was significantly higher than that in the AP/p53 group (Fig. S11, ESI†).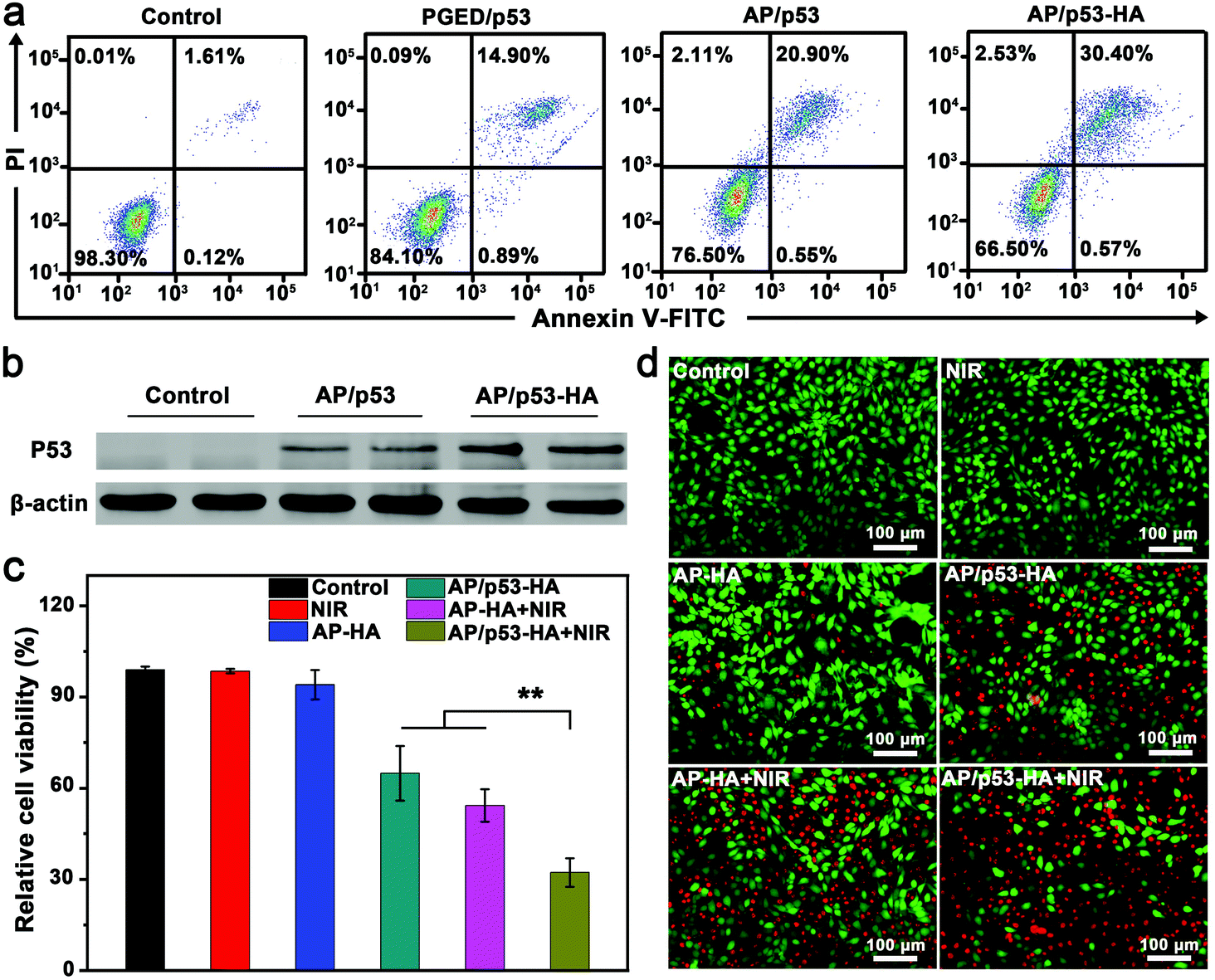 | ||
| Fig. 3 (a) Flow cytometric analysis of apoptotic 4T1 cells after different treatments. (b) Western blot of P53 protein expression in 4T1 cells after 48 h transfection. (c) MTT assay and (d) fluorescence images of FDA–PI-stained 4T1 cells treated with NIR, AP–HA, AP/p53–HA, AP–HA + NIR, and AP/p53–HA + NIR, respectively. | ||
To investigate the complementary PTT/GT in vitro, photothermal effects of AP–HA were first evaluated. The SPR peak of AP–HA was observed at ∼815 nm (Fig. S12a, ESI†) while concentration- and time-dependent temperature variation was recorded under an 808 nm laser at the power density of 1 W cm−2 (Fig. S12b, ESI†). In addition, the excellent photothermal stability of AP/pDNA–HA was confirmed by the cycling test (Fig. S12c, ESI†). The efficacy of combined PTT/GT was investigated by MTT and FDA–PI double staining (Fig. 3c and d). 4T1 cells were treated with NIR light irradiation, AP/p53–HA, AP–HA, AP–HA + NIR light irradiation, and AP/p53–HA + NIR light irradiation, respectively. The viability of 4T1 cells treated with AP/p53–HA and AP–HA + NIR was ∼65% and ∼54%, respectively. In contrast, 4T1 cells treated with NIR light irradiation or AP–HA exhibited negligible cell death. The severe 4T1 cell death mediated by small amounts of AP–HA was supposed to be caused by the accumulation of AP–HA in cells after internalization. These findings confirm that the photothermal effect of AP–HA and p53 delivery can effectively kill tumor cells. Notably, 4T1 cells treated with AP/p53–HA + NIR light exhibited significantly lower cell viability (∼32%) compared with the GT and PTT groups. Accordingly, the severest 4T1 cell death was induced in the AP/p53–HA + NIR group (Fig. 3d), verifying the superiority of complementary PTT/GT. In addition, the excellent biocompatibility of the AP/p53 and AP/p53–HA complexes was confirmed by hemolysis assay (Fig. S13, ESI†).
3.5. In vivo PAI-guided therapy
The superior performance of AP/p53–HA complexes in vitro inspired us to explore the antitumor effect of the complexes in vivo. Owing to the excellent photothermal effect (Fig. S12a, ESI†), AP–HA nanohybrids were expected to achieve PAI in vivo,47 which could monitor the accumulation of the nanohybrids in the tumor.48–50 After intravenous injection of AP–HA or AP in the 4T1 tumor-bearing mice, PAI was explored. As shown in Fig. 4a and Fig. S14 of the ESI,† gradually increasing PA signals were observed at the tumor site until 10 h after intravenous injection, implying the time-dependent accumulation of the nanohybrids. Remarkably, stronger PA signals were observed in the AP–HA-treated tumors compared with AP-administered tumors. The PA intensity of the AP–HA group reached the maximum value at 10 h post-injection (Fig. S15, ESI†), which was 1.8-fold higher than that of the AP group. It is speculated that the HA cloak facilitate the active targeting and long circulation of the AP–HA nanohybrids in vivo. After 10 h, the PA intensity of both AP and AP–HA groups decreased over time, implying that the nanoparticles could be gradually metabolized. In addition, the highest PA intensity at 10 h post-injection indicates a maximum accumulation of the nanohybrids in the tumor, which could be regarded as the optimal time point for PTT. As shown in Fig. 4b, obvious temperature elevation and color change in the infrared thermal images were observed in the AP–HA-treated tumor with NIR light irradiation at 10 h post-injection, which could effectively ablate the tumor cells. In contrast, the temperature rise of the tumor in the control group was negligible. In this regard, AP–HA nanohybrids could be effective for PTT in vivo while imaging-guided tumor treatment could be achieved. | ||
| Fig. 4 (a) PA images and intensity of tumor the site at different time points after the intravenous injection of AP–HA or AP, respectively. Scale bars: 2 mm. (b) Infrared thermal images and temperature plots of tumor-bearing mice intravenously injected with AP–HA or PBS (control) under 808 nm laser irradiation at 10 h post-injection (1 W cm−2, 5 min). (c) Relative growth curves, (d) tumor weights and (e) photos of the excised tumors in different groups. (f) H&E-stained images of the tumor tissue after different treatments. Scale bars: 50 μm. | ||
Complementary PPT/GT in vivo was then performed under the guidance of PAI. The 4T1 tumor-bearing mice were divided into four groups and intravenously administered with PBS (control group), AP/p53–HA (GT group), AP–HA with NIR light irradiation (PTT group), and AP/p53–HA with NIR light irradiation (PPT/GT group), respectively. Treatments were administered every other day for 10 days. For PTT and PPT/GT groups, tumors were irradiated with an 808 nm laser (1 W cm−2, 5 min) only once at 10 h after the first injection, which was regarded as the optimal time point (Fig. 4a). The relative tumor volume of the different groups was recorded every other day (Fig. 4c). The in vivo study was stopped on day 10 as tumors in the PPT/GT group had almost disappeared. After 10 days of treatment, the tumor growth in the PTT/GT group was completely suppressed, while the tumor weight was significantly lower than that of the other groups. In contrast, tumors in the PTT and GT groups still grew rapidly to a certain extent, suggesting the superior antitumor effects of complementary PTT/GT. It is to be noted that the combination of GT and PTT exhibited a synergistic effect (degree of synergy > 0),51 confirming PTT-enhanced GT. The weight and typical photographs of the tumors after different treatments confirmed the satisfactory therapeutic effect of the PTT/GT group (Fig. 4d and e). Taken together, AP/p53–HA complexes are promising to realize PAI-guided synergistic PTT/GT for tumor treatment.
To further assess the antitumor effect of different groups, tumor tissues were analyzed by hematoxylin and eosin (H&E) staining (Fig. 4f). Tumor cells in the control group were normal, while cell apoptosis were observed in the GT group. Tumor cell necrosis including nuclei fragmentation occurred in the PTT group. Remarkably, the dual-modal complementary PTT/GT therapy resulted in the most severe cell death, suggesting the superiority of the synergistic therapeutic effect of the AP/p53–HA complexes.
The expression level of P53 protein was verified by western blot assay. As shown in Fig. 5a, the P53 protein expression was observed in the GT and PTT/GT groups. In addition, quantitative analysis revealed that the P53 expression level in the PTT/GT group was significantly higher than that in the GT group (Fig. 5b). Immunohistochemical staining also confirmed more obvious P53-positive area in the PTT/GT group, compared with the GT group (Fig. 5c). The enhanced gene transfection was speculated to be caused by the photothermal effect-enhanced cellular uptake, endosomal escape, and p53 release from AP/p53–HA complexes,5,6 which also explains why PTT and GT produced a synergistic therapeutic effect.
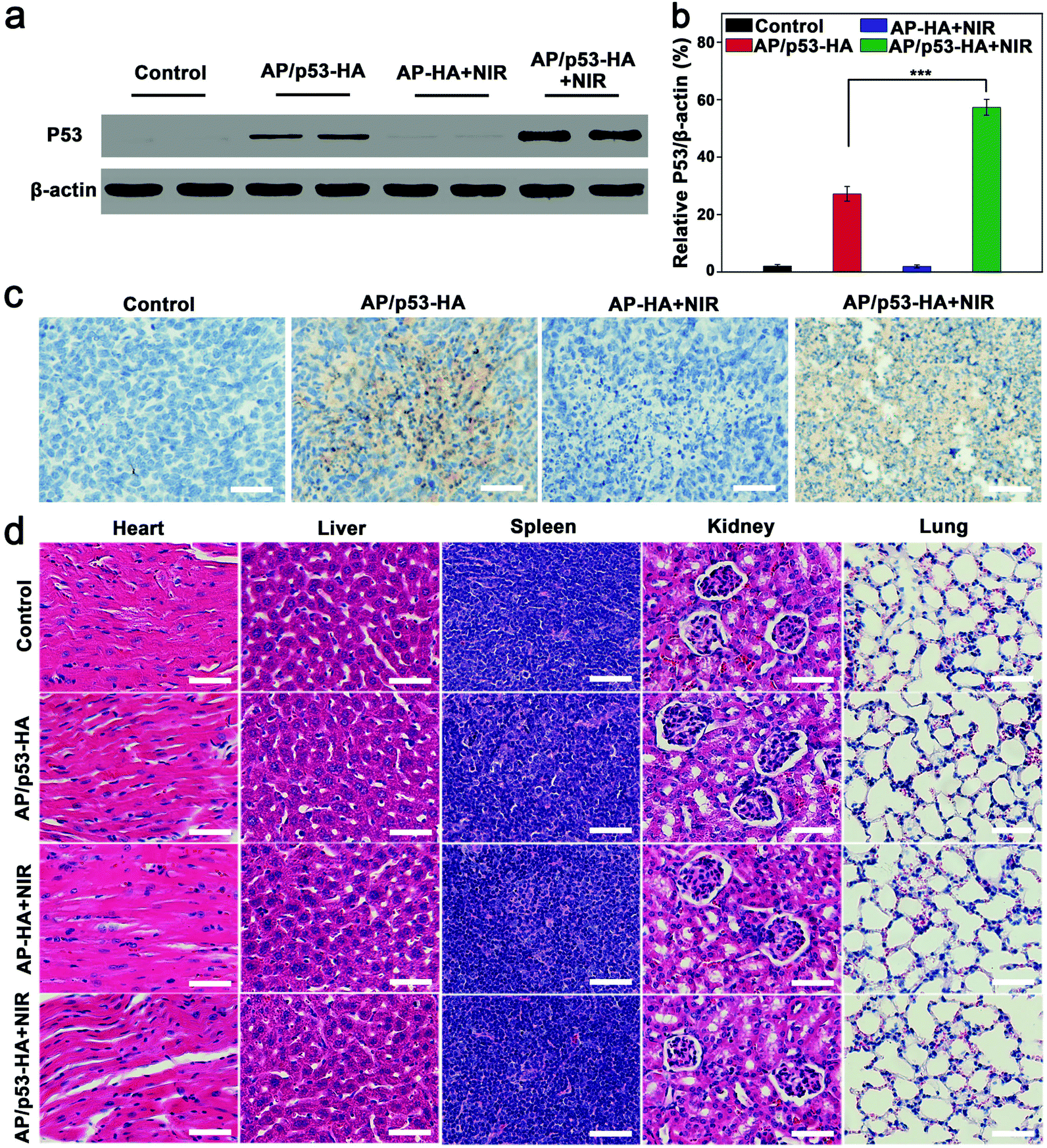 | ||
| Fig. 5 (a) Western blot of P53 protein expression and (b) quantitative analysis of P53 protein in tumor tissues after different treatments. (c) Immunohistochemical analysis of the tumors after different treatments. (d) Histology analysis of major mouse organs after different treatments. Scale bars: 50 μm. | ||
During the treatment, the body weight of the mice was found to increase slightly over time (Fig. S16, ESI†). Furthermore, the organs (heart, liver, spleen, kidneys and lungs) of the mice were resected and analyzed by H&E staining (Fig. 5d). The histological analysis of major organs in different treatment groups did not exhibit obvious abnormality. Taken together, AP/p53–HA complexes possess excellent biocompatibility for PAI-guided synergistic therapy.
4. Conclusions
In summary, HA-cloaked polycation/gold gene carriers with long circulation and active tumor targeting ability were successfully fabricated for PAI-guided synergistic PTT/GT. A cationic polymer PGED was conjugated onto the surface of Au NRs through Au–S bonds to produce AP nanohybrids for pDNA condensation. Then an appropriate amount of HA was readily introduced through Schiff bonds and electrostatic interaction to obtain AP/pDNA–HA complexes. The reversible shielding with pH responsiveness guaranteed the endosomal escape of the complexes for effective gene delivery with compromised cytotoxicity. Taking advantage of the excellent stability and active targeting properties conferred by HA, the HA-cloaked complexes exhibited significantly higher gene transfection efficiency than that of AP/pDNA through enhanced cellular internalization in CD44-positive tumor cells. Furthermore, Au NRs impart photothermal properties to the complexes and complementary PTT/GT was achieved through the combination with antioncogene p53. The feasibility of PAI-guided therapy in vivo was also demonstrated. PAI confirmed that the optimal time point to perform PTT was 10 h after intravenous injection, when a maximum accumulation in the tumor was observed. Remarkably, a synergistic effect was achieved when AP/p53–HA complexes were applied for PTT/GT. Satisfactory antitumor effects resulting from PTT-enhanced GT make the HA-cloaked complexes ideal candidates for PAI-guided synergistic therapy. The current work provides a facile strategy to construct multifunctional gene delivery systems with long circulation and tumor-targeting characteristics for effective tumor treatment.Ethical statement
Animal studies were approved by the Ethical Committee of Chinese Academy of Medical Sciences (CAMS) and Peking Union Medical College and performed under legal protocols.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 52173271, 51922022, and 51773013), the Beijing Outstanding Young Scientist Program (No. BJJWZYJH01201910010024), and the Fundamental Research Funds for the Central Universities (No. BHYC1705A and XK1802-2).References
- W. Fan, B. Yung, P. Huang and X. Chen, Chem. Rev., 2017, 117, 13566–13638 CrossRef CAS PubMed.
- Z. Xie, T. Fan, J. An, W. Choi, Y. Duo, Y. Ge, B. Zhang, G. Nie, N. Xie, T. Zheng, Y. Chen, H. Zhang and J. S. Kim, Chem. Soc. Rev., 2020, 49, 8065–8087 RSC.
- J. Kim, J. Kim, C. Jeong and W. J. Kim, Adv. Drug Delivery Rev., 2016, 98, 99–112 CrossRef CAS PubMed.
- N. Zhao, L. Yan, X. Zhao, X. Chen, A. Li, D. Zheng, X. Zhou, X. Dai and F. J. Xu, Chem. Rev., 2019, 119, 1666–1762 CrossRef CAS PubMed.
- Y. Meng, S. Wang, C. Li, M. Qian, X. Yan, S. Yao, X. Peng, Y. Wang and R. Huang, Biomaterials, 2016, 100, 134–142 CrossRef CAS PubMed.
- Y. Liu, B. Yu, X. Dai, N. Zhao and F. J. Xu, Biomaterials, 2021, 274, 120885 CrossRef CAS PubMed.
- H. Kim and W. J. Kim, Small, 2014, 10, 117–126 CrossRef CAS PubMed.
- F. Ding, X. Gao, X. Huang, H. Ge, M. Xie, J. Qian, J. Song, Y. Li, X. Zhu and C. Zhang, Biomaterials, 2020, 245, 119976 CrossRef CAS PubMed.
- N. Zhao, J. Li, Y. Zhou, Y. Hu, R. Wang, Z. Ji, F. Liu and F. J. Xu, Adv. Funct. Mater., 2016, 26, 5848–5861 CrossRef CAS.
- Y. Hu, Y. Zhou, N. Zhao, F. Liu and F. J. Xu, Small, 2016, 12, 2459–2468 CrossRef CAS PubMed.
- J. Shen, H. C. Kim, C. Mu, E. Gentile, J. Mai, J. Wolfram, L. N. Ji, M. Ferrari, Z. W. Mao and H. Shen, Adv. Healthcare Mater., 2014, 3, 1629–1637 CrossRef CAS PubMed.
- M. S. Shim and Y. J. Kwon, Adv. Drug Delivery Rev., 2012, 64, 1046–1059 CrossRef CAS PubMed.
- X. Guo and L. Huang, Acc. Chem. Res., 2012, 45, 971–979 CrossRef CAS PubMed.
- X. Cai, Y. Li, D. Yue, Q. Yi, S. Li, D. Shi and Z. Gu, J. Mater. Chem. B, 2015, 3, 1507–1517 RSC.
- H. Kim, J. Kim, M. Lee, H. C. Choi and W. J. Kim, Adv. Healthcare Mater., 2016, 5, 1918–1930 CrossRef CAS PubMed.
- G. F. Walker, C. Fella, J. Pelisek, J. Fahrmeir, S. Boeckle, M. Ogris and E. Wagner, Mol. Ther., 2005, 11, 418–425 CrossRef CAS PubMed.
- E. J. Oh, K. Park, K. S. Kim, J. Kim, J. A. Yang, J. H. Kong, M. Y. Lee, A. S. Hoffman and S. K. Hahn, J. Controlled Release, 2010, 141, 2–12 CrossRef CAS PubMed.
- W. Sun, Y. Du, X. Liang, C. Yu, J. Fang, W. Lu, X. Guo, J. Tian, Y. Jin and J. Zheng, Biomaterials, 2019, 217, 119264 CrossRef CAS PubMed.
- Q. Sun, H. Bi, Z. Wang, C. Li, X. Wang, J. Xu, H. Zhu, R. Zhao, F. He, S. Gai and P. Yang, Biomaterials, 2019, 223, 119473 CrossRef CAS PubMed.
- F. Dosio, S. Arpicco, B. Stella and E. Fattal, Adv. Drug Delivery Rev., 2016, 97, 204–236 CrossRef CAS PubMed.
- S. Z. F. Phua, G. Yang, W. Q. Lim, A. Verma, H. Chen, T. Thanabalu and Y. Zhao, ACS Nano, 2019, 13, 4742–4751 CrossRef CAS PubMed.
- G. Huang and H. Huang, J. Controlled Release, 2018, 278, 122–126 CrossRef CAS PubMed.
- X. Y. He, X. H. Ren, Y. Peng, J. P. Zhang, S. L. Ai, B. Y. Liu, C. Xu and S. X. Cheng, Adv. Mater., 2020, 32, 2000208 CrossRef CAS PubMed.
- N. N. Parayath, A. Parikh and M. M. Amiji, Nano Lett., 2018, 18, 3571–3579 CrossRef CAS PubMed.
- Y. Zhang, L. Liu, L. Lin, J. Chen, H. Tian, X. Chen and A. Maruyama, Acta Biomater., 2018, 65, 349–362 CrossRef CAS PubMed.
- Q. Zhang, G. Kuang, S. He, H. Lu, Y. Cheng, D. Zhou and Y. Huang, Nano Lett., 2020, 20, 3039–3049 CrossRef CAS PubMed.
- S. Ganesh, A. K. Iyer, D. V. Morrissey and M. M. Amiji, Biomaterials, 2013, 34, 3489–3502 CrossRef CAS PubMed.
- S. Wang, Y. Tian, W. Tian, J. Sun, S. Zhao, Y. Liu, C. Wang, Y. Tang, X. Ma, Z. Teng and G. Lu, ACS Nano, 2016, 10, 8578–8590 CrossRef CAS PubMed.
- M. Y. Lee, S. J. Park, K. Park, K. S. Kim, H. Lee and S. K. Hahn, ACS Nano, 2011, 5, 6138–6147 CrossRef CAS PubMed.
- L. J. Yan, X. H. Guo, W. P. Wang, Y. R. Hu, S. F. Duan, Y. Liu, Z. Sun, S. N. Huang and H. L. Li, Curr. Cancer Drug Targets, 2019, 19, 330–337 CrossRef CAS PubMed.
- J. Shen, D. J. Zhao, W. Li, Q. L. Hu, Q. W. Wang, F. J. Xu and G. P. Tang, Biomaterials, 2013, 34, 4520–4531 CrossRef CAS PubMed.
- S. Li and L. Huang, Gene Ther., 2000, 7, 31–34 CrossRef CAS PubMed.
- Y. Yu, C. K. Chen, W. C. Law, H. Sun, P. N. Prasad and C. Cheng, Polym. Chem., 2015, 6, 953–961 RSC.
- L. Liu, C. Kong, M. Huo, C. Liu, L. Peng, T. Zhao, Y. Wei, F. Qian and J. Yuan, Chem. Commun., 2018, 54, 9190–9193 RSC.
- Y. Liu, N. Zhao and F. J. Xu, ACS Appl. Mater. Interfaces, 2019, 11, 34707–34716 CrossRef CAS PubMed.
- F. J. Xu, Y. Zhu, M. Y. Chai and F. S. Liu, Acta Biomater., 2011, 7, 3131–3140 CrossRef CAS PubMed.
- B. Nikoobakht and M. A. El-Sayed, Chem. Mater., 2003, 15, 1957–1962 CrossRef CAS.
- X. Lin, N. Zhao, P. Yang, H. Hu and F. J. Xu, Acta Biomater., 2015, 11, 381–392 CrossRef CAS PubMed.
- Z. Zhang, L. Wang, J. Wang, X. Jiang, X. Li, Z. Hu, Y. Ji, X. Wu and C. Chen, Adv. Mater., 2012, 24, 1418–1423 CrossRef CAS PubMed.
- P. Yan, R. Wang, N. Zhao, H. Zhao, D. F. Chen and F. J. Xu, Nanoscale, 2015, 7, 5281–5291 RSC.
- T. Hozumi, T. Kageyama, S. Ohta, J. Fukuda and T. Ito, Biomacromolecules, 2018, 19, 288–297 CrossRef CAS PubMed.
- H. Zhao and N. D. Heindel, Pharm. Res., 1991, 8, 400–402 CrossRef CAS PubMed.
- L. E. Dickinson, C. C. Ho, G. M. Wang, K. J. Stebe and S. Gerecht, Biomaterials, 2010, 31, 5472–5478 CrossRef CAS PubMed.
- R. Wu, X. K. Ding, Y. Qi, Q. Zeng, Y. W. Wu, B. Yu and F. J. Xu, Small, 2018, 14, 1800201 CrossRef PubMed.
- H. Zhao, W. Hu, H. Ma, R. Jiang, Y. Tang, Y. Ji, X. Lu, B. Hou, W. Deng, W. Huang and Q. Fan, Adv. Funct. Mater., 2017, 1702592 CrossRef.
- R. Huschka, J. Zuloaga, M. W. Knight, L. V. Brown, P. Nordlander and N. J. Halas, J. Am. Chem. Soc., 2011, 133, 12247–12255 CrossRef CAS PubMed.
- Y. Liu, P. Bhattarai, Z. Dai and X. Chen, Chem. Soc. Rev., 2019, 48, 2053–2108 RSC.
- W. Huang, R. Chen, Y. Peng, F. Duan, Y. Huang, W. Guo, X. Chen and L. Nie, ACS Nano, 2019, 13, 9561–9570 CrossRef CAS PubMed.
- Q. Li, K. Chen, W. Huang, H. Ma, X. Zhao, J. Zhang, Y. Zhang, C. Fang and L. Nie, Cancer Lett., 2021, 496, 169–178 CrossRef CAS PubMed.
- Q. Yu, S. Huang, Z. Wu, J. Zheng, X. Chen and L. Nie, J. Nucl. Med., 2020, 61, 1079–1085 CrossRef CAS PubMed.
- M. Hegreness, N. Shoresh, D. Damian, D. Hartl and R. Kishony, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13977–13981 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and figures showing data including 1H NMR spectra, thermogravimetric analysis, electrophoretic mobility retardation assay, FTIR spectra, fluorescence images, UV-vis spectra, and body weight evolution. See DOI: https://doi.org/10.1039/d2bm00296e |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |