
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiological interactions of ferromagnetic iron oxide–carbon nanohybrids with alveolar epithelial cells†
Silvia
Vercellino
 a,
Ida
Kokalari
bc,
Mayra
Liz Cantoral
ab,
Vanya
Petseva
a,
Lorenzo
Cursi
a,
Francesca
Casoli
d,
Valentina
Castagnola
*aef,
Luca
Boselli
*ag and
Ivana
Fenoglio
*b
a,
Ida
Kokalari
bc,
Mayra
Liz Cantoral
ab,
Vanya
Petseva
a,
Lorenzo
Cursi
a,
Francesca
Casoli
d,
Valentina
Castagnola
*aef,
Luca
Boselli
*ag and
Ivana
Fenoglio
*b
aCentre for BioNano Interactions, School of Chemistry, University College Dublin, Belfield, Dublin 4, Ireland. E-mail: valentina.castagnola@iit.it; luca.boselli@iit.it
bDept. of Chemistry, Università di Torino, via P. Giuria 7, 10125 Torino, Italy. E-mail: ivana.fenoglio@unito.it
cDelft University of Technology, Dept. of Chemical Engineering, Van der Maasweg 9, 2629 HZ DELFT, The Netherlands
dInstitute of Materials for Electronics and Magnetism (IMEM), National Research Council (CNR), Parco Area delle Scienze 37/A, Parma 43124, Italy
eCenter for Synaptic Neuroscience and Technology, Istituto Italiano di Tecnologia, Largo Rosanna Benzi 10, 16132, Genova, Italy
fIRCCS Ospedale Policlinico San Martino, Largo Rosanna Benzi 10, 16132, Genova, Italy
gNanobiointeractions and Nanodiagnostics, Istituto Italiano di Tecnologia, Genova, Italy
First published on 17th May 2022
Abstract
Iron oxide nanoparticles (IONPs) have been largely investigated in a plethora of biological fields for their interesting physical–chemical properties, which make them suitable for application in cancer therapy, neuroscience, and imaging. Several encouraging results have been reported in these contexts. However, the possible toxic effects of some IONP formulations can limit their applicability. In this work, IONPs were synthesized with a carbon shell (IONP@C), providing enhanced stability both as colloidal dispersion and in the biological environment. We conducted a careful multiparametric evaluation of IONP@C biological interactions in vitro, providing them with an in vivo-like biological identity. Our hybrid nanoformulation showed no cytotoxic effects on a widely employed model of alveolar epithelial cells for a variety of concentrations and exposure times. The IONP@C were efficiently internalized and TEM analysis allowed the protective role of the carbon shell against intracellular degradation to be assessed. Intracellular redistribution of the IONP@C from the lysosomes to the lamellar bodies was also observed after 72 hours.
Introduction
Iron oxide nanoparticles (IONPs), with their magnetic properties, have been considered for a range of biological applications spanning magnetic resonance imaging (MRI),1,2 neuronal modulation,3 cancer therapy,4 drug delivery,5–8 and magnetic sorting.9–12 Thanks to their magnetic properties that allow for thermotherapy (magnetic hyperthermia)13 and their capability to overcome biological barriers,14 IONPs have also been used to treat glioblastoma, up to clinical trials.15,16In 2009, the Food and Drug Administration (FDA) approved the iron oxide colloidal formulation “ferumoxytol” to treat iron-deficiency anemia in adults with chronic kidney disease.17,18 Ferumoxytol, extensively employed in both preclinical and clinical studies, is composed of a superparamagnetic iron oxide core and a hydrophilic carboxymethyl-dextran shell and has shown potential in several biomedical applications, including immunomodulation.19
Starting from the encouraging results, it is of paramount importance to understand the IONP interactions with the body machinery to be able to engineer IONP formulations that fully exploit their biological potential. Indeed, the cytotoxicity of these NPs towards different biological systems is still controversial.20,21 It has been shown that IONPs can be degraded within the lysosomal compartment releasing iron ions, which, depending on the concentrations and kinetics of release, might induce reactive oxygen species (ROS)-mediated cytotoxicity.22–24 This phenomenon is counterbalanced by the role of intracellular protein cages (such as ferritin) implicated in iron homeostasis. The iron ions, products of IONP degradation, are intracellularly transferred to ferritin that can cage and store them in the acidic physiological environment of the lysosomes, as demonstrated in vitro and in vivo.25
The particle size of IONPs intended for magnetic hyperthermia is generally below the superparamagnetic critical size.26 However, some studies suggest that ferromagnetic IONPs with particles of size around 40 nm exhibit a better heat efficiency, thus allowing to use low-frequency field, producing a magnetic field that minimizes possible damage to healthy tissues.27,28 On the other hand, ferromagnetic particles present lower colloidal stability if compared to superparamagnetic particles due to particle–particle magnetic attraction.
The use of a shell increases the stability of IONPs in the biological environment, also improving their biocompatibility by preventing rapid degradation.29,30 Different coating materials have been investigated, such as silica, inorganic metals (or metal oxides), and, more recently, carbon which can enhance biocompatibility, provide new optical properties and allow for anchoring of molecules through covalent bonding.31,32
Furthermore, it is well-known that the NPs surface chemistry affects their interactions with the airways mucus layer:33 the NPs shell functionalization with hydrophilic moieties, such as PEG,34,35 minimizes the interactions of NPs and mucus, consequently preventing the NPs mucus entrapment and enhancing mucus penetration, that eventually results in an improved cellular uptake.36
Inorganic carbon shell coated IONPs (IONP@C) have been previously synthesized by solvothermal carbonization of ferrocene, leading to superparamagnetic magnetite nanoparticles embedded in a carbonaceous matrix, and used as a carrier for doxorubicin.37–39 IONP@C core–shell nanohybrids are promising materials for medical applications, as they combine the magnetic properties of the core with the optical properties of the carbon shell, but the investigation of their biological interactions has been so far overlooked. In this work, ferromagnetic hybrid core–shell nanoparticles (IONP@C) were synthesized, and their colloidal stability was measured in water and cell media. The bio-nano interactions of IONP@C were investigated on a lung alveolar cellular model. In this kind of study, the role of protein corona40,41 is of paramount importance, as it can be responsible for the mismatch between in vitro and in vivo results.42–45 Indeed, the layer of proteins adsorbed onto the surface of nanomaterials regulates their interactions with the cell membrane,46–50 their intracellular pathway12,51–53 and eventually their final fate in the body.44 In this optics, classic biological milieu supplemented with 10% serum might not be sufficient to give a complete picture of the interactions occurring in vivo, and higher protein concentration should be adopted.46,48,54 For this reason, in our work, we paid particular attention to the use of an appropriate biological environment to analyze IONP@C interactions with cells. From our result, IONP@C showed excellent biocompatibility towards a widely used in vitro model and good stability in the biological environment (including intracellular compartments), making them suitable for promising applications in nanomedicine.
Results and discussion
Synthesis and physical chemical properties of the nanohybrids
For the synthesis of the nanohybrids, a solvothermal method using ferrocene as a precursor was used starting from the protocol described by Wang et al.39 As previously reported, besides temperature and reaction time, the concentration of hydrogen peroxide is a key parameter that determines the size of the nanohybrids.37 The synthesis (see Experimental section) was performed firstly by using 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hydrogen peroxide/ferrocene ratio. Particles of diameter higher than 100 nm were obtained (Fig. S1†). By reducing the ratio to 4:1, particles having a diameter lower than 100 nm were obtained (IONP@C). Also the core/shall sizes ratio (shell thickness) is affected by this parameter.
:1 hydrogen peroxide/ferrocene ratio. Particles of diameter higher than 100 nm were obtained (Fig. S1†). By reducing the ratio to 4:1, particles having a diameter lower than 100 nm were obtained (IONP@C). Also the core/shall sizes ratio (shell thickness) is affected by this parameter.
The morphology of the particles was investigated by transmission electron microscopy (TEM). A representative micrograph is shown in Fig. 1A where the iron oxide core surrounded by a continuous carbon shell is clearly visible. The mean geometrical diameter as measured by statistical TEM analysis was around 50 nm (Fig. 1B). The mean thickness of the shell was 12 nm, while the mean diameter of the iron oxide core resulted to be about 35 nm (Fig. 1B). This value well agrees with the mean hydrodynamic diameter measured by DLS in water (60 nm). The colloidal suspension in water was very stable, and the particles showed a monomodal size distribution (Fig. 1C). A comparison of the dH distributions of different batches from different syntheses is reported in the ESI (Fig. S2A†). The populations were almost identical, demonstrating the good reproducibility of the synthesis.
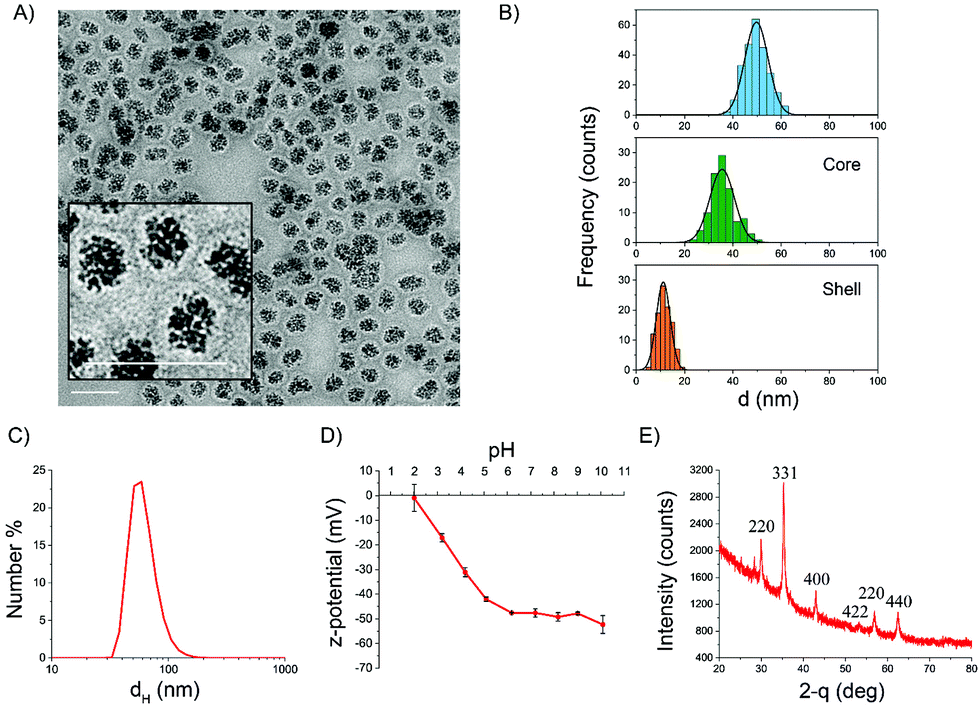 | ||
| Fig. 1 Characterization of IONP@C NPs. (A) TEM micrograph (scale bars are 100 nm); (B) statistical size distribution from TEM imaging for the whole NPs, the iron oxide core, and the carbon shell; (C) size distribution evaluated by DLS (PDI = 0.12); (D) ζ-potential vs. pH evaluated by ELS; (E) XRD diffractogram. | ||
Fig. 1D reports the ζ-potential values as a function of pH range from 2–10, measured by electrophoretic light scattering (ELS). At physiological pH, the ζ-potential was lower than −40 mV, suggesting good stability of the suspension. Indeed, colloids having zeta potential from ±30 to ±60 mV are very stable due to high electrostatic repulsion between particles, which prevents agglomeration.55 A negative zeta potential, which decreased with the pH, suggests the presence of acidic groups on the surface of the carbon shell. The absence of positive values at low pH might be an indication that a small percentage or no iron oxide core is accessible, being mostly embedded by the carbon shell.
XRD analysis was performed in order to identify the crystal phase of the core (Fig. 1E). The XRD pattern indicated that the iron oxide core is composed of magnetite or maghemite phases.56 The nanoparticles showed an evident magnetic response at room temperature, with a specific magnetization value of 15 A m2 kg−1 at 1.8 T, as evaluated considering the total mass of the sample (Fig. 2A). After renormalizing magnetization only to the weight fraction of iron oxide, we could estimate a specific magnetization value in the range of 31–40 A m2 kg−1 at 1.8 T. The magnetization curve measured up to 0.5 T evidenced a ferromagnetic behavior with reduced values of the coercive field and magnetic remanence (HC = 7 mT, Mr/MS = 0.17, inset of Fig. 2A). This result is consistent with the dimensional analysis.57 The susceptibility of the particles to applied magnetic fields was obvious (Fig. S2B†). However, the particles formed a stable colloidal suspension again once the magnetic field was removed.
 | ||
| Fig. 2 Magnetic properties, surface reactivity, and degradation. (A) Magnetization curves of IONP@C measured with AGFM magnetometry at room temperature up to fields of 1.8 T and 0.5 T (the inset shows a zoom of the measurement up to 0.5 T); (B) surface reactivity of IONP@C evaluated by EPR spectroscopy. EPR signals recorded in solutions containing: FeSO4 [1.4 mM], DMPO [51 mM], H2O2 [27 mM] (a) in absence of IONP@C (the four-line EPR signal corresponds to the DMPO/HO˙ adduct) in PBS; (b) in the presence of IONP@C [1 mg mL−1] in PBS; (c) in the presence of IONP@C [1 mg mL−1] in cMEM supplemented with 50% FBS; (d) in the presence of IONP@C [1 mg mL−1], and absence of FeSO4 in PBS. (C) Appearance of the suspensions of IONP@C incubated in NaClO or H2O2, 0.2 or 2 mM. (D) Absorbance at 450 nm of the suspensions of IONP@C incubated in NaClO or H2O2, 0.2 or 2 mM. | ||
Surface reactivity
As previously reported in several studies, carbon nanostructures might have antioxidant properties due to their ability to scavenge ROS.58–61 Oppositely, IONPs have been shown to induce oxidative stress mediated by iron overload.62 The surface reactivity of IONP@C was investigated here to get preliminary information on possible adverse effects elicited in vivo.To investigate the antioxidant properties of IONP@C, hydroxyl radicals were generated by the Fenton reaction as described in the Experimental section and their concentration was evaluated by EPR spectroscopy/spin trapping technique (Fig. 2B).
The high number of radicals generated in this condition is proved by the intense (1:2:2:1) four-line EPR signal recorded, corresponding to the DMPO/HO˙ adduct (aN = aH = 14.6 G) (Fig. 2B, spectrum a).
In the presence of the IONP@C, the EPR signal was suppressed entirely (Fig. 2B, spectrum b), suggesting a high scavenging capability. The same reactivity was observed when the nanoparticles were suspended in the cell culture medium (Fig. 2B, spectrum c). When the particles were incubated with hydrogen peroxide (Fig. 2A, spectrum d), no signals corresponding to hydroxyl or superoxide radicals were detected.
These data further indicate that the carbonaceous shell is able to inhibit or balance the surface reactivity of the core since exposure to the solvent should result in a Fenton-like surface reactivity as previously reported.61,63
Degradability of the carbon shell
Iron oxide nanoparticles have been shown to be degraded by macrophages in spleen and liver with kinetics that depends upon the kind of particles coating.25,64 In the present case, the carbon shell is expected to inhibit the degradation of the IONP@C, potentially leading to accumulation in tissues.58 On the other hand, the carbon shell prevent the rapid release of iron ions in the intracellular compartments that may induce oxidative stress.62 Carbon nanomaterials are known to be degraded by myeloperoxidase,65,66 the enzyme expressed mainly in neutrophils, which generates the strong oxidant hypochlorous acid, and by hydrogen peroxide in macrophages.67,68 To investigate the possible degradation of the carbon shell by these oxidants, the sample has been incubated in NaClO or H2O2 at two different concentrations (0.2 and 2 mM). After 14 days, the suspensions in NaClO turned from brown to a pale red color, suggesting the degradation of the carbon shell that leaves the core exposed to the solvent (Fig. 2C and S3†). Similarly, a clear change of color was observed with H2O2 treatment, but only for the highest concentration. In parallel, a decrease of the absorption at λ = 450 nm was observed (Fig. 2D). An obvious shift of the color from brown to red was also visible in the pellet (Fig. S3†).These preliminary results suggest a possible degradation of the carbon shell in activated neutrophils, while degradation by hydrogen peroxide in the lysosomal milieu of other cell types appears unlikely. Further investigations will be necessary to confirm the relevance of the present findings in cells.
Biological identity of IONP@C in cell media
Protein corona formation is a fundamental aspect to consider when investigating the bio-nano interaction, as it governs the cell receptor-nanoparticles recognition that takes place on the plasma membrane, on which the cellular uptake and the intracellular trafficking largely depend.46,48,69 It also might play a pivotal role in NPs stabilization and toxicity mitigation.47,70The protein corona formation was evaluated by 1D SDS-PAGE electrophoresis. IONP@C were incubated with Fetal Bovine Serum (FBS) at increasing concentrations to mimic both in vitro and in vivo conditions.45 A washing protocol was applied to remove proteins weakly bound to the surface and to analyze the hard protein corona exclusively.71
The SDS page reported in Fig. S4† revealed the formation of complex protein coronas, with some visible changes in the protein profile for an increasing percentage of FBS, indicating that classical in vitro conditions might not be a good proxy for the in vivo bio-nano interactions scenario.
NPs designed for systemic delivery need to be monodispersed and stable in the biological medium used in the toxicological in vitro tests, similar to what is expected in vivo. Therefore NPs stability must be assessed, for the used conditions, before proceeding to the in vitro investigation.
Differential centrifugal sedimentation (DCS) represents an ideal tool to assess the possible occurrence of aggregation/agglomeration processes. IONP@C suspended in MEM supplemented with 10% v/v FBS underwent aggregation. Therefore, we increased the FBS supplement to 50%; this condition resulted in very good stability of the IONP@C stability over time (see Fig. S5†) and was therefore adopted for the following in vitro experiments. Of note, the DCS curve showed a shift towards larger sizes, compatible with the formation of the protein corona layer. However, some parts of the NPs surface might still be exposed to the solvent. In fact, IONP@C suspended in 50% FBS-supplemented MEM to form the protein corona still exhibited antioxidant properties (Fig. 2B).
An alternative strategy leading to similar stability results consisted of forming a pre-corona around the particles before resuspending them in the culture medium.
Biological interactions of IONP@C
In view of a potential application of our nanoparticles against lung tumors, we investigated the interaction of IONP@C with the human lung carcinoma cell line A549, which is also a widely applied in vitro model for cytotoxicity assessment in nanomedicine. A549 cells were exposed to increasing concentrations of IONP@C, up to 200 μg mL−1, at different times. It is important to highlight that, according to the performed protein corona and stability characterization (Fig. S4 and S5†), the culture medium was supplemented with 50% v/v of FBS to mimic a more realistic in vivo-like physiological scenario and ensure long-term stability of the dispersions.MTS assay, which evaluates the mitochondrial reductase enzymatic activity, and is a well-established test to assess cell viability, was employed at first. The cells viability was not compromised by the presence of IONP@C in any of the conditions tested (even though the condition 200 μg mL−1 IONP@C incubated for 72 h exhibited a trend), confirming that the cells are metabolically active (Fig. 3A).
 | ||
| Fig. 3 Cytotoxicity assessment towards A549 cells. (A) MTS viability assay for A549 cells treated with increasing concentrations of NPs suspended in MEM supplemented with 50% FBS for 24, 48, and 72 hours (mean ± SD, n = 3). (B) Cell membrane permeability assay of A549 cells with increasing concentrations of NPs suspended in MEM supplemented with 50% FBS for 24, 48, and 72 hours (mean ± SD, n = 3). (C) HCA cell count based on the Hoechst counterstain in the field imaged. (D) Mitochondrial membrane potential assessment for A549 cells treated with increasing concentrations of NPs suspended in MEM supplemented with 50% FBS for 24, 48, and 72 hours (mean ± SD, n = 3). Statistical analysis: **** p < 0.0001, *** p < 0.001, ** p < 0.01, * p < 0.05 one-way ANOVA/Tukey's tests. | ||
However, when investigating the biocompatibility of a new nanomaterial, a multiparametric study should be performed since the health of the cells can be affected in many ways.72
For this reason, in addition to MTS, a series of other parameters were considered. Cell membrane permeability, a phenomenon typically involved in late-stage apoptosis, was assessed.73 TOPRO-3 is a cell impermeable dye, and, as such, it stains exclusively the apoptotic cells, which have a compromised plasma membrane, intercalating the cell DNA. The cells were counterstained with both Hoechst to count the total number of cells, and TOPRO-3, to count the number of the apoptotic cells. The results shown in Fig. 3B are expressed as a percentage of cells negative for the TOPRO-3 staining over the total cell population. The cells treated with IONP@C showed no significant membrane permeabilization in any of the conditions tested. Again, a trend can be observed for the condition 200 μg mL−1 IONP@C incubated for 72 h.
Similarly, no toxicity was observed in cells treated with IONP@C bearing a pre-corona before dispersion in cell culture medium (Fig. S6†).
High content analysis (HCA) was employed to complement the multiparametric cytotoxicity investigation and ensure that the cells were proliferating during the exposure to IONP@C. The cells were treated as described and then stained with Hoechst, a cell-permeable DNA intercalating dye that allows for counterstain in live cells. The number of cells was calculated using the HCA Cellomics© software, and the total number of cells counted per well is reported in Fig. 3C. As expected, the cell number increased exponentially with increasing times, showing a physiological growth of the cells during the exposure time, meaning that the IONP@C did not interfere with the regular cell division.
Furthermore, to examine the mitochondria state, the cells were then stained with tetramethylrhodamine methyl ester (TMRM), which fluorescence emission is proportional to the mitochondrial membrane potential. As such, a decrease in the dye emission corresponds to a loss of potential, which, triggering the caspases cascade, is a widely recognized marker for early stages of apoptosis.74 The mitochondria can also receive several pro-apoptotic signals from most cell organelles, including lysosomes, peroxisomes, ER, and nucleus.75 Therefore, assessing their health is traditionally considered a robust way to detect toxicity caused by nanomaterials in several cell compartments. In good agreement with the MTS assay, the screening results showed that the mitochondria of NPs-treated cells were healthy, and the metabolic homeostasis was not altered by the treatment (Fig. 3D). However, a significant decrease in mitochondria potential was observed for cells treated for 72 hours with 200 μg mL−1 of IONP@C, confirming a trend already observed with the previous assays.
Similar results were observed in cells treated with IONP@C bearing a pre-corona (Fig. S7†).
To exclude a possible role of the excess of proteins used, viability test were also repeated using 10% FBS-supplemented MEM and IONP@C bearing a precorona to ensure stability (see Fig. S5†). Also in this case, similar results were obtained as reported in Fig. S8.†
It is well documented that IONPs can induce toxicity by affecting the cell homeostasis at different levels, and therefore causing oxidative stress in many cell lines, including A549.22,76–78 Therefore, HCA was also used to assess intracellular ROS production, using a fluorogenic ROS probe, during the incubation with increasing concentrations of IONP@C up to 72 hours. The results reported in Fig. S9† indicated that the carbon shell embedding the iron oxide might act as a shield during the internalization and trafficking processes.
After excluding acute toxicity effects on A549 cells for the conditions tested, we moved into investigating IONP@C internalization. In order to have a statistically relevant quantification of the uptake rate, NP-treated cells were analyzed by flow cytometry. The side scatter (SSC-A) signal is a well-recognized proxy for the label-free evaluation of uptake of some nanomaterials, as it reflects an increased cellular granularity due to nanomaterial accumulation in intracellular organelles and vesicles.46,78–81 Details on the analysis are reported in Fig. S10.† An uptake kinetic was performed following the internalization rate of different concentrations of IONP@C up to 72 hours. From the results depicted in Fig. 4A, we observed a slow uptake kinetic, both concentration- and time-dependent.
 | ||
| Fig. 4 IONP@C uptake and intracellular trafficking. (A) IONP@C uptake in A549 cells as measured by SSC-A by flow cytometry. The NPs were suspended in MEM supplemented with 50% FBS at increasing concentrations, and the cells were treated for 24, 48, and 72 hours (mean ± SD, n = 3). (B) Overtime evaluation of lysosomal pH (mean ± SD, n = 3). Lysotracker™ deep red was used to stain the cells, and its fluorescence emission increase is proportional to the lysosome acidification. Statistical analysis: **** p < 0.0001, *** p < 0.001, ** p < 0.01, * p < 0.05 one-way ANOVA/Tukey's tests. (C) Representative images of A549 cells treated with 200 μg mL−1 of IONP@C or 24 (first lane), 48 (middle lane), and 72 hours (bottom lane). The cells showed a healthy morphology across the conditions tested. Pseudo-colors: blue: nuclei, red: mitochondria, green: lysosomes. Scale bar: 20 μm. | ||
HCA was exploited again to investigate the intracellular trafficking pathways. Cells were stained with Lysotracker™ deep red and imaged with an ArrayScan HCA in live-cell mode. The fluorescent emission of the dye proportionally increases with the decrease of the lysosomal pH; as such, it is a valuable tool to study the endo-lysosomal dynamics. Cells usually react to the nanomaterials accumulation with increased lysosomal activity; therefore, it is commonly expected to observe a lysosomes acidification following NPs uptake.82 As expected, a mild decrease of pH is observed after the 24 hours of incubation, as the NPs are internalized and enclosed in the vesicles, while the acidification reached a peak after 48 hours of exposure (Fig. 4B). The dye signal increased with the dose of NPs used, probably reflecting the higher uptake rate (Fig. 4A). However, IONP@C did not cause long-term acidification as the fluorescence was not higher than the untreated control after 72 hours of continuous exposure (Fig. 4B). This is in good agreement with the previously demonstrated biocompatibility. Note that, in the case of the positive control, the drastic decrease of the Lysotracker signal is merely due to cell death.
A similar trend was observed for the cells treated using protein pre-coated IONP@C, as reported in Fig. S11.†
Previous studies correlated the lysosomal pH increase to the prolonged presence of non-biodegradable nanomaterials (such as gold or carbon) in the endo-lysosomal compartments.83,84 In most cases, the phenomenon also caused a reduction of the proteasomal activity and interference with autophagy,85 followed by apoptosis that, however, was not observed in this study.
We hypothesized that whilst the nanohybrids are not cytotoxic, the large volume of NPs internalized within 48 hours could evoke stress in the NPs containing vesicles and challenge the cell degradative mechanism causing a partial lysosome-related perturbation. However, the cells appear to cope with the induced stress and adapt after 72 hours. In addition, we need to consider the effect of the lysosomal load dilution occurring as a consequence of cell proliferation (Fig. 4C), as previously reported.86 However, the total uptake amount per cell does not decrease at 72 hours as shown in Fig. 4A, leading to think that some intracellular redistribution of the load might occur.
To further investigate the intracellular trafficking of the IONP@C, and to gain a better insight into the effect of the NPs on the lysosomal compartment, we employed TEM. For this experiment, A549 cells were treated with 100 μg mL−1 of IONP@C, suspended in 50% FBS supplemented media for 24, 48, or 72 hours. More details on the TEM samples preparation are given in the Experimental section. As a first observation, the cells’ morphology appeared healthy (Fig. 5 and Fig. S12†), confirming the observed biocompatibility for the conditions used.
 | ||
| Fig. 5 TEM micrographs showing IONP@C uptake and intracellular trafficking. A549 cells were treated with 100 μg mL−1 of IONP@C for (A) 24, (B) 48, and (C) 72 hours before TEM observation. After 24 and 48 hours, the NPs are enclosed in vesicles, presumably late endosomes and lysosomes. After 72 hours, NPs are mainly observed in multi-layered vesicles, known as lamellar bodies (LB). N = nucleus. | ||
For all the time points, the IONP@C structure appeared intact, with the magnetic core surrounded by the carbon shell, suggesting that the shell is preserved in the intracellular environment and no or very little degradation occurred. This confirms the poor susceptibility of the carbon shell to oxidants (hydrogen peroxide), thus possibly confirming the protective role of the carbon shell inhibiting the reported iron oxide peroxidase-like activity (and thus potential oxidative stress) at the lysosome pH.87
After 24 hours of exposure, NPs IONP@C could be found clustered inside lipidic vesicles (Fig. 5A) spread across the cytoplasm. This observation combined with HCA is coherent with the hypothesis of NPs accumulation in early or late endosomes. In particular, as several particles are contained in each vesicle, it can be hypothesized that the vesicles belong to a late phase of the endo-lysosomal pathway, where many vesicles are fused together to form a late endosome.
After 48 hours, a similar intracellular distribution was observed, with many particles clustered in each vesicle (Fig. 5B). By comparing this observation with the HCA results, it can be hypothesized that NPs are now accumulating in lysosomes. A higher number of clusters was observed due to the higher amount of NPs internalized in 48 hours (Fig. 4A). In rare cases, NPs were found localized in the lamellar bodies (LB) (Fig. S13†). These observations are consistent with the endo-lysosomal pathway, typically followed by NPs in the internalization process.51
The 72 hours sample showed a very different distribution, with most of the NPs embedded in LB (Fig. 5C), suggesting an intracellular redistribution. Lamellar bodies are lysosome-related organelles which formation in A549 cells can be associated with the exporting of NPs.88
The formation of lamellar bodies in A549 cells in the presence of different kinds of nanomaterials, including carbon nanotubes,89 gold,88 and silica,90 was previously reported, suggesting that NPs might have a role in increasing the production and secretion of these vesicles.89,91 Their biogenesis is strictly correlated to the multivesicular bodies generation and the lysosomes health status, that if compromised, can give rise to phospholipidic toxicity phenomena.92 It is known that the physiological LBs role in A549 is secreting surfactant and related mucus, in a primarily important function of alveolar cells,93 and therefore they can also be associated with the exporting of NPs.88 Although the mechanisms involved are not clear yet, the export of NPs is a well-reported phenomenon in multiple cell lines.94,95
This observation seems to be in good agreement with the data obtained by HCA (Fig. 4B). Lysosomes showed increased acidification after 48 hours of exposure to IONP@C, suggesting some swelling to incorporate the materials and some energy devoted to attempted degradation. The new change of lysosomal pH after 72 hours, despite high uptake values (Fig. 4A), coupled with the TEM imaging observation, suggests that the cells are adapting and responding to NPs accumulation by intracellular redistribution of the particles’ load.
Experimental
Synthesis of IONP@C nanohybrids
The solvothermal synthesis was used for the synthesis of IONP@C, modifying a previously reported method.39 IONP@C were synthesized as follows: 100 mg of ferrocene (Fe(C5H5)2, ≥98%, F408 Sigma-Aldrich), were dissolved in 30 mL of acetone and sonicated with a probe sonicator (Bandelin Sonopuls HD 3100) for 30 minutes in dark conditions, placed in an ice bath. Then 2.5 mL or 0.25 mL of hydrogen peroxide 30% w/w (H2O2, 30%, H1009 Sigma-Aldrich) were slowly dropped in the ferrocene solution, under magnetic stirring at room temperature, and then kept under vigorous stirring for 30 minutes.The final solution was transferred in a 100 mL stainless steel Teflon-lined autoclave (Büchi AG) and kept in the oven at 200 °C for 24 hours. Nanoparticles were then purified in order to remove the ferrocene in excess: the suspension was centrifuged at 6000 rpm for 30 min, and the supernatant was discarded; the compact pellet consisting of nanoparticles was resuspended in acetone after the first centrifugation and then in ultrapure water. The washing process was repeated three times until the supernatant appeared colorless instead of yellow.
Dynamic light scattering
The hydrodynamic diameter of IONP@C was evaluated by dynamic light scattering (Nano ZS Malvern Instruments, UK). The measurement was performed immediately after the purification process on samples diluted 50× with ultrapure water, sonicated in an ice bath for 2 minutes at 30% of amplitude with a probe sonicator (Bandelin Sonopuls HD 3100, Berlin, Germany).Electrophoretic light scattering
The ζ-potential was determined in a pH range of 10–12 by electrophoretic light scattering (Nano ZS Malvern Instruments, UK). The samples were suspended in ultrapure water at the concentration of 0.8 mg mL−1, and the pH of the nanoparticle suspensions was adjusted with 0.1 M NaOH, 0.01 M HCl, and 0.001 M HCl (purchased from Sigma-Aldrich).Transmission electron microscopy
Samples were prepared by drop-casting 1 microliter of sample on glow-discharged grids (PELCO® TEM grid Support Films of Formvar Formvar/Carbon TED PELLA INC.) then air-dried. TEM experiments were performed on a FEI Tecnai G2 20 Twin operating at 120 kV, and TEM micrographs analysis was carried out using “ImageJ” software to obtain the statistical size distributions.X-ray diffraction
Powder X-ray diffraction (XRD) analysis was performed using a X'Pert PRO MPD (PANalytical) X-ray diffractometer equipped with Cu Kα radiation, configured in Bragg-Brentano geometry over the 2θ range of 15–80°. Phase identification was performed using the X'Pert High-Score software. The nanoparticles suspension was air-dried at 70 °C before analysis.Magnetometry
Magnetization loops were measured at room temperature by an alternating gradient force magnetometer (AGFM) from Princeton Measurements corp. The measurements were realized first in a magnetic field up 1.8 T and then in a field up to 0.5 T.Scavenging capability toward OH radicals
The scavenging activity of IONP@C towards hydroxyl radicals was evaluated using electronic paramagnetic resonance (EPR)/spin trapping technique (Miniscope MS100, Magnettech, Berlin, Germany). The following parameters were applied: B0 fields = 3344.14 G; sweep = 119.19 G; sweep time = 80 s; smooth = 0.00 s; steps = 4096; no. pass. = 2; modulation = 1000 mG; MW atten. = 7 dB; gain = 2E2/9E2.A volume of 0.2 mL of IONP@C suspension (1 mg mL−1 in phosphate buffer saline (PBS) 0.01 M, pH 7.4) was transferred in a cuvette and stirred in the dark. 0.25 mL of a solution 0.15 M of 5,5-dimethyl-1-pyrroline N-oxide (DMPO) in water, 0.1 mL of PBS 0.01 M, pH 7.4 and 0.08 mL of a solution 13 mM of FeSO4 were added. The reaction was started by adding 0.1 mL of a solution 0.2 M of H2O2. The reaction was also repeated in the absence of IONP@C as a control. EPR spectra were measured on the suspension after 10 minutes of incubation in the dark. In the case of IONP@C, the suspension was filtered after the incubation, and the spectrum was immediately recorded.
The antioxidant activity of the sample was also tested after incubation in the presence of cell culture media: MEM supplemented with 50% FBS. IONP@C suspension was incubated for 24 hours, at 37 °C, in dark conditions. The sample was then subjected to centrifugation at 10000 rpm for 15 minutes. The pellet was suspended in PBS 10 mM, pH 7.4, obtaining a final concentration of 1 mg mL−1. The scavenging capacity of the sample was tested as previously described.
Fenton-like reaction
0.2 mL of IONP@C suspended in water (1 mg mL−1) was added to 0.1 mL of PBS 0.01 M, pH 7.4 and 0.08 mL of ultrapure water and then incubated with H2O2 (0.2 M, 0.1 mL), for 10 minutes, in dark conditions, in the presence of DMPO (0.15 M, 250 μL) as a spin trap. The sample was analyzed by EPR spectroscopy using the instrumental configuration previously described.Degradability by endogenous oxidants
The degradability of IONP@C was investigated by employing a solution of hypochlorite and/or hydrogen peroxide, the two main oxidants present in the phagosome. The final concentration of H2O2 or NaClO was 0.2 mM, or 2 mM. Samples consisted of 1 mg of IONP@C dispersed in PBS, 10 mM. The final volume of the sample tubes (10 mL) was kept constant for all solutions. The pH of the solution was set at 4.5. The samples were incubated for 14 days at 37 °C, and NaClO or H2O2 were added daily to keep a constant concentration of the oxidants. The degradation was evaluated by visual observation and UV-Vis spectroscopy (Uvikon, Kontron Instruments, Inc., Everett, MA).Differential centrifugal sedimentation (DCS)
CPS Disc Centrifuge DC24000 (CPS Instruments) was used. The disc was set in motion at a constant speed, from 18000 rpm to 20000 rpm, and the disc chamber was filled with an 8–24 wt% sucrose density gradient.
Protein corona characterization by gel electrophoresis
Nanoparticles were incubated at 37 °C for 1 hour in FBS in a range of concentrations: 10%, 50%, and 80% v/v in phosphate buffer saline (PBS) 0.01 M. After incubation, the protein-coated nanoparticles were washed three times by centrifugation to remove the unbound free proteins from the solution.The pellet was then resuspended in 20 μl of PBS 0.01 M and 10 μl of denaturation solution prepared with dithiothreitol (DTT) and sodium dodecyl sulfate (SDS) 3× loading buffer in ratio 1:10. The mixture was incubated for 5 min at 100 °C to complete the denaturation process. Conventional 10% SDS-PAGE was performed, and run for about 45 min at 130 V. 15 μl of each sample was loaded in a 10 wells SDS-PAGE gel. Pierce™ silver stain kit was used to visualize the protein bands; the gels were imaged with a G-BOX XT4 (Syngene) equipped with a UV filter and a white light source.
Cell culture
Human carcinoma cell line A549, alveolar basal epithelial cells, was obtained from the American Type Culture Collection (ATCC) and cultured according to standard tissue culture protocols and sterile technique. Cells were maintained in Minimum Essential Media (MEM, Gibco), supplemented with 10% FBS (Gibco), 1% penicillin/streptomycin (Invitrogen), and 1% MEM non-essential amino acids (HyClone), referred to as complete MEM (cMEM). Cells were grown at 37 °C in a humidified atmosphere of 5% CO2 and subcultured after reaching 70–80% confluency. Cells were screened monthly for mycoplasma contaminations using the MycoAlert mycoplasma detection kit.MTS assay
The MTS assay (Promega) was performed following manufacturer instructions. 5000 cells per well were seeded in a 96-wells plate (Cell Star) 24 hours prior to treatment and let adhere to the plate. The cells were then treated with different concentrations of NPs or placed in fresh cMEM (UT control) or incubated for 1 min with 0.1% Triton X-100 (Merck-Sigma) for the positive control for cytotoxicity. Results were then obtained by measuring absorbance at 490 nm using a plate reader (Varioskan Flash, Thermofisher), the UT values were normalized to 100% viability.High content analysis
For the HCA experiments, 5000 cells per well were seeded into 96-wells plate (Cell Star) 24 hours in advance. The cells were treated with the NPs, placed in fresh cMEM (UT control), or treated with commercial amine-modified polystyrene nanoparticles (NP-NH2, L0780, Sigma-Aldrich) as a toxic NPs positive control (+ve crtl) for cytotoxicity.96 The cells were stained with the following dyes (all purchased from ThermoFisher) for 15 minutes, at 37 °C: Lysotracker deep red (1 mM), YO-PRO-3 iodide (1 mM), tetramethylrhodamine methyl ester (TMRM, 100 nM), and Hoechst 33342 (20 mM). The images were acquired in live-cell mode, in a pre-warmed chamber (37 °C in a humidified atmosphere of 5% CO2), using a 20× objective, with the following combination of wavelength filters: Hoechst was excited through a 365 ± 50 nm bandpass filter and fluorescence emission was collected through a 515 ± 20 nm bandpass filter; YOPRO-1 was excited through a 475 ± 40 nm bandpass filter, and fluorescence emission was collected through a 515 ± 20 nm bandpass filter; TMRM was excited through a 549 ± 8 nm bandpass filter and fluorescence emission was collected through a 600 ± 25 nm bandpass filter; Lysotracker deep red was excited through a 655 ± 30 nm bandpass filter, and fluorescence emission was collected through a 730 ± 50 nm bandpass filter. The analysis pipeline was set according to the manufacturer's instructions, and fixed thresholds were used for each parameter to separate background noise from the fluorophores signal.Flow cytometry
The cells for flow cytometry analysis were seeded in a 12-well plate at a cell density of 50000 cells per well 24 hours prior to the NPs exposure. After the nanoparticle treatment, the cells were harvested by trypsinization, washed in PBS, and fixed in 4% paraformaldehyde. The cells were run in a Cytoflex LX (Beckman Coulter) flow cytometer. The side scattering signal per cell was measured for ≥50000 events collected in the single-cell population.
TEM imaging cell preparation
The cells were seeded in a 12-wells plate at a cell density of 50000 cells per well 24 hours prior to the NPs exposure. The cells were treated as previously described, washed, and fixed with a solution composed of 2.5% glutaraldehyde in 0.1 M Sorensen phosphate buffer. The cells were then washed with Sorensen phosphate buffer and fixed for 1 hour in 1% osmium tetroxide in deionized water. Cells were dehydrated with increasing ethanol concentration (from 70% to 100%). Samples were then immersed in an acetone/Epon (1:1 vol/vol) mixture for 1 hour before being transferred to pure Epon and embedded at 37 °C for 2 hours. The polymerization step was carried out at 60 °C for 24 h. The ultrathin sections (80 nm) were mounted on copper grids after sectioning using an ultramicrotome equipped with a diamond knife (Leica U6). The sections were stained with 2% uranyl acetate for 20 minutes, washed carefully, and stained with 3% lead citrate for 5 minutes. The images were acquired with a FEI TECNAI G2 20 Twin TEM operating at an accelerating voltage of 120 kV.
Conclusions
In conclusion, hybrid ferromagnetic IONPs with a carbon coating were synthesized and fully characterized for their size distribution, colloidal stability in water and cellular media, magnetic properties, and degradability. The carbon shell was shown to impart high stability in aqueous media, resistance to degradation, and antioxidant properties. The particles were found to be efficiently internalized by A549 cells in simulated in vivo conditions for the protein corona formation (known to drive the interactions with cell membrane receptors) without eliciting noticeable acute toxic effects. The cytotoxicity of IONP@C was evaluated using a multiparametric approach to cover a broad spectrum of biological readouts following IONP@C interactions with the cells.Intracellular trafficking was also investigated, confirming the protective role of the carbon shell in the intracellular compartments up to 72 hours and suggesting an intracellular redistribution of the NPs cargo from the lysosome to the lamellar bodies at this time point.
The concentration and time window for safe usage of IONP@C was identified as the first step in view of future development for biomedical applications such as cancer therapy.
Author contributions
SV performed most of the experiments and helped with the data analysis and manuscript writing. IK performed the nanoparticles synthesis and characterization. MLC performed part of the biological experiments. VP and LC performed TEM imaging. FC performed the magnetic characterization. VC, LB, and IF conceived and directed the work and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SV and MLC have been the recipient of a grant from the EC Erasmus+ program. VC acknowledges IRCCS Ospedale Policlinico San Martino (Ricerca Corrente and 5 × 1000 grants).The authors thank Dr Matteo Signorile for his assistance in XRD analysis. Access to and use of the UCD Conway Facilities is gratefully acknowledged.
Notes and references
- J.-s. Choi, S. Kim, D. Yoo, T.-H. Shin, H. Kim, M. D. Gomes, S. H. Kim, A. Pines and J. Cheon, Distance-dependent magnetic resonance tuning as a versatile MRI sensing platform for biological targets, Nat. Mater., 2017, 16(5), 537–542 CrossRef CAS PubMed.
- M. Jeon, M. V. Halbert, Z. R. Stephen and M. Zhang, Iron oxide nanoparticles as T1 contrast agents for magnetic resonance imaging: fundamentals, challenges, applications, and prospectives, Adv. Mater., 2021, 33(23), 1906539 CrossRef CAS PubMed.
- T.-H. Shin and J. Cheon, Synergism of nanomaterials with physical stimuli for biology and medicine, Acc. Chem. Res., 2017, 50(3), 567–572 CrossRef CAS PubMed.
- S. M. Dadfar, K. Roemhild, N. I. Drude, S. von Stillfried, R. Knüchel, F. Kiessling and T. Lammers, Iron oxide nanoparticles: Diagnostic, therapeutic and theranostic applications, Adv. Drug Delivery Rev., 2019, 138, 302–325 CrossRef CAS.
- S. Ayyanaar, C. Balachandran, R. C. Bhaskar, M. P. Kesavan, S. Aoki, R. P. Raja, J. Rajesh, T. J. Webster and G. Rajagopal, ROS-responsive chitosan coated magnetic iron oxide nanoparticles as potential vehicles for targeted drug delivery in cancer therapy, Int. J. Nanomed., 2020, 15, 3333 CrossRef CAS PubMed.
- F. Soetaert, P. Korangath, D. Serantes, S. Fiering and R. Ivkov, Cancer therapy with iron oxide nanoparticles: Agents of thermal and immune therapies, Adv. Drug Delivery Rev., 2020, 163, 65–83 CrossRef PubMed.
- C. Ansari, G. A. Tikhomirov, S. H. Hong, R. A. Falconer, P. M. Loadman, J. H. Gill, R. Castaneda, F. K. Hazard, L. Tong and O. D. Lenkov, Development of novel tumor–targeted theranostic nanoparticles activated by membrane–type matrix metalloproteinases for combined cancer magnetic resonance imaging and therapy, Small, 2014, 10(3), 566–575 CrossRef CAS PubMed.
- R. Balzaretti, F. Meder, M. P. Monopoli, L. Boselli, I. Armenia, L. Pollegioni, G. Bernardini and R. Gornati, Synthesis, characterization and programmable toxicity of iron oxide nanoparticles conjugated with D-amino acid oxidase, RSC Adv., 2017, 7(3), 1439–1442 RSC.
- H. Xu, Z. P. Aguilar, L. Yang, M. Kuang, H. Duan, Y. Xiong, H. Wei and A. Wang, Antibody conjugated magnetic iron oxide nanoparticles for cancer cell separation in fresh whole blood, Biomaterials, 2011, 32(36), 9758–9765 CrossRef CAS.
- S. P. Schwaminger, S. A. Blank-Shim, I. Scheifele, V. Pipich, P. Fraga-García and S. Berensmeier, Design of interactions between nanomaterials and proteins: a highly affine peptide tag to bare iron oxide nanoparticles for magnetic protein separation, Biotechnol. J., 2019, 14(3), 1800055 CrossRef.
- J. A. Thomas, F. Schnell, Y. Kaveh-Baghbaderani, S. Berensmeier and S. P. Schwaminger, Immunomagnetic separation of microorganisms with iron oxide nanoparticles, Chemosensors, 2020, 8(1), 17 CrossRef CAS.
- L. Cursi, S. Vercellino, M. M. McCafferty, E. Sheridan, V. Petseva, L. Adumeau and K. A. Dawson, Multifunctional superparamagnetic nanoparticles with a fluorescent silica shell for the in vitro study of bio-nano interactions at the subcellular scale, Nanoscale, 2021, 13(38), 16324–16338 RSC.
- A. Espinosa, R. Di Corato, J. Kolosnjaj-Tabi, P. Flaud, T. Pellegrino and C. Wilhelm, Duality of iron oxide nanoparticles in cancer therapy: amplification of heating efficiency by magnetic hyperthermia and photothermal bimodal treatment, ACS Nano, 2016, 10(2), 2436–2446 CrossRef CAS PubMed.
- A. Ivask, E. H. Pilkington, T. Blin, A. Käkinen, H. Vija, M. Visnapuu, J. F. Quinn, M. R. Whittaker, R. Qiao and T. P. Davis, Uptake and transcytosis of functionalized superparamagnetic iron oxide nanoparticles in an in vitro blood brain barrier model, Biomater. Sci., 2018, 6(2), 314–323 RSC.
- K. Maier-Hauff, R. Rothe, R. Scholz, U. Gneveckow, P. Wust, B. Thiesen, A. Feussner, A. Von Deimling, N. Waldoefner and R. Felix, Intracranial thermotherapy using magnetic nanoparticles combined with external beam radiotherapy: results of a feasibility study on patients with glioblastoma multiforme, J. Neuro-Oncol., 2007, 81(1), 53–60 CrossRef CAS PubMed.
- K. Maier-Hauff, F. Ulrich, D. Nestler, H. Niehoff, P. Wust, B. Thiesen, H. Orawa, V. Budach and A. Jordan, Efficacy and safety of intratumoral thermotherapy using magnetic iron-oxide nanoparticles combined with external beam radiotherapy on patients with recurrent glioblastoma multiforme, J. Neuro-Oncol., 2011, 103(2), 317–324 CrossRef PubMed.
- I. Blumenstein, S. Shanbhag, P. Langguth, P. A. Kalra, H. Zoller and W. Lim, Newer formulations of intravenous iron: a review of their chemistry and key safety aspects–hypersensitivity, hypophosphatemia, and cardiovascular safety, Expert Opin. Drug Saf., 2021, 1–13 Search PubMed.
- Y. Huang, J. C. Hsu, H. Koo and D. P. Cormode, Repurposing ferumoxytol: Diagnostic and therapeutic applications of an FDA-approved nanoparticle, Theranostics, 2022, 12(2), 796–816 CrossRef CAS PubMed.
- S. Zanganeh, G. Hutter, R. Spitler, O. Lenkov, M. Mahmoudi, A. Shaw, J. S. Pajarinen, H. Nejadnik, S. Goodman and M. Moseley, Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues, Nat. Nanotechnol., 2016, 11(11), 986–994 CrossRef CAS.
- S. Z. Imam, S. M. Lantz-McPeak, E. Cuevas, H. Rosas-Hernandez, S. Liachenko, Y. Zhang, S. Sarkar, J. Ramu, B. L. Robinson and Y. Jones, Iron oxide nanoparticles induce dopaminergic damage: in vitro pathways and in vivo imaging reveals mechanism of neuronal damage, Mol. Neurobiol., 2015, 52(2), 913–926 CrossRef CAS.
- Q. Feng, Y. Liu, J. Huang, K. Chen, J. Huang and K. Xiao, Uptake, distribution, clearance, and toxicity of iron oxide nanoparticles with different sizes and coatings, Sci. Rep., 2018, 8(1), 1–13 CAS.
- U. S. Gaharwar, R. Meena and P. Rajamani, Iron oxide nanoparticles induced cytotoxicity, oxidative stress and DNA damage in lymphocytes, J. Appl. Toxicol., 2017, 37(10), 1232–1244 CrossRef CAS PubMed.
- C. Petters, K. Thiel and R. Dringen, Lysosomal iron liberation is responsible for the vulnerability of brain microglial cells to iron oxide nanoparticles: comparison with neurons and astrocytes, Nanotoxicology, 2016, 10(3), 332–342 CrossRef CAS.
- J. Kolosnjaj-Tabi, L. Lartigue, Y. Javed, N. Luciani, T. Pellegrino, C. Wilhelm, D. Alloyeau and F. Gazeau, Biotransformations of magnetic nanoparticles in the body, Nano Today, 2016, 11(3), 280–284 CrossRef CAS.
- J. Volatron, F. Carn, J. Kolosnjaj-Tabi, Y. Javed, Q. L. Vuong, Y. Gossuin, C. Ménager, N. Luciani, G. Charron and M. Hémadi, Ferritin protein regulates the degradation of iron oxide nanoparticles, Small, 2017, 13(2), 1602030 CrossRef PubMed.
- S. M. Dadfar, D. Camozzi, M. Darguzyte, K. Roemhild, P. Varvarà, J. Metselaar, S. Banala, M. Straub, N. Güvener and U. Engelmann, Size-isolation of superparamagnetic iron oxide nanoparticles improves MRI, MPI and hyperthermia performance, J. Nanobiotechnol., 2020, 18(1), 1–13 CrossRef.
- P. Guardia, R. Di Corato, L. Lartigue, C. Wilhelm, A. Espinosa, M. Garcia-Hernandez, F. Gazeau, L. Manna and T. Pellegrino, Water-soluble iron oxide nanocubes with high values of specific absorption rate for cancer cell hyperthermia treatment, ACS Nano, 2012, 6(4), 3080–3091 CrossRef CAS PubMed.
- P. J. Sugumaran, X.-L. Liu, T. S. Herng, E. Peng and J. Ding, GO-functionalized large magnetic iron oxide nanoparticles with enhanced colloidal stability and hyperthermia performance, ACS Appl. Mater. Interfaces, 2019, 11(25), 22703–22713 CrossRef CAS.
- M. A. Malvindi, V. De Matteis, A. Galeone, V. Brunetti, G. C. Anyfantis, A. Athanassiou, R. Cingolani and P. P. Pompa, Toxicity assessment of silica coated iron oxide nanoparticles and biocompatibility improvement by surface engineering, PLoS One, 2014, 9(1), e85835 CrossRef PubMed.
- A. Ruiz, L. Gutiérrez, P. Cáceres-Vélez, D. Santos, S. B. Chaves, M. L. Fascineli, M. P. García, R. Azevedo and M. Morales, Biotransformation of magnetic nanoparticles as a function of coating in a rat model, Nanoscale, 2015, 7(39), 16321–16329 RSC.
- K. Wu, D. Su, J. Liu, R. Saha and J.-P. Wang, Magnetic nanoparticles in nanomedicine: a review of recent advances, Nanotechnology, 2019, 30(50), 502003 CrossRef CAS PubMed.
- Z. Karimi, L. Karimi and H. Shokrollahi, Nano-magnetic particles used in biomedicine: Core and coating materials, Mater. Sci. Eng., C, 2013, 33(5), 2465–2475 CrossRef CAS PubMed.
- J. das Neves, R. S. Arzi and A. Sosnik, Molecular and cellular cues governing nanomaterial–mucosae interactions: From nanomedicine to nanotoxicology, Chem. Soc. Rev., 2020, 49(14), 5058–5100 RSC.
- E. D. Mansfield, R. Victor, R. M. Kowalczyk, I. Grillo, R. Hoogenboom, K. Sillence, P. Hole, A. C. Williams and V. V. Khutoryanskiy, Side chain variations radically alter the diffusion of poly (2-alkyl-2-oxazoline) functionalised nanoparticles through a mucosal barrier, Biomater. Sci., 2016, 4(9), 1318–1327 RSC.
- M. Yang, S. K. Lai, Y. Y. Wang, W. Zhong, C. Happe, M. Zhang, J. Fu and J. Hanes, Biodegradable nanoparticles composed entirely of safe materials that rapidly penetrate human mucus, Angew. Chem., 2011, 123(11), 2645–2648 CrossRef.
- S. Mura, H. Hillaireau, J. Nicolas, S. Kerdine-Römer, B. Le Droumaguet, C. Deloménie, V. r. Nicolas, M. Pallardy, N. Tsapis and E. Fattal, Biodegradable nanoparticles meet the bronchial airway barrier: how surface properties affect their interaction with mucus and epithelial cells, Biomacromolecules, 2011, 12(11), 4136–4143 CrossRef CAS PubMed.
- H. Wang, Q.-W. Chen, Y.-F. Yu, K. Cheng and Y.-B. Sun, Size-and solvent-dependent magnetically responsive optical diffraction of carbon-encapsulated superparamagnetic colloidal photonic crystals, J. Phys. Chem. C, 2011, 115(23), 11427–11434 CrossRef CAS.
- H. Wang, Q.-W. Chen, L.-X. Sun, H.-p. Qi, X. Yang, S. Zhou and J. Xiong, Magnetic-field-induced formation of one-dimensional magnetite nanochains, Langmuir, 2009, 25(12), 7135–7139 CrossRef CAS PubMed.
- H. Wang, J. Shen, Y. Li, Z. Wei, G. Cao, Z. Gai, K. Hong, P. Banerjee and S. Zhou, Magnetic iron oxide–fluorescent carbon dots integrated nanoparticles for dual-modal imaging, near-infrared light-responsive drug carrier and photothermal therapy, Biomater. Sci., 2014, 2(6), 915–923 RSC.
- I. Fenoglio, B. Fubini, E. M. Ghibaudi and F. Turci, Multiple aspects of the interaction of biomacromolecules with inorganic surfaces, Adv. Drug Delivery Rev., 2011, 63(13), 1186–1209 CrossRef CAS PubMed.
- M. P. Monopoli, C. Aberg, A. Salvati and K. A. Dawson, Biomolecular coronas provide the biological identity of nanosized materials, Nat. Nanotechnol., 2012, 7(12), 779–786 CrossRef CAS PubMed.
- G. Stepien, M. Moros, M. Pérez-Hernández, M. Monge, L. Gutiérrez, R. M. Fratila, M. d. las Heras, S. Menao Guillén, J. J. Puente Lanzarote, C. Solans, J. Pardo and J. M. d. la Fuente, Effect of surface chemistry and associated protein corona on the long-term biodegradation of iron oxide nanoparticles in vivo, ACS Appl. Mater. Interfaces, 2018, 10(5), 4548–4560 CrossRef CAS PubMed.
- M. Levy, N. Luciani, D. Alloyeau, D. Elgrabli, V. Deveaux, C. Pechoux, S. Chat, G. Wang, N. Vats and F. Gendron, Long term in vivo biotransformation of iron oxide nanoparticles, Biomaterials, 2011, 32(16), 3988–3999 CrossRef CAS PubMed.
- G. Caracciolo, O. C. Farokhzad and M. Mahmoudi, Biological identity of nanoparticles in vivo: clinical implications of the protein corona, Trends Biotechnol., 2017, 35(3), 257–264 CrossRef CAS PubMed.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. Baldelli Bombelli and K. A. Dawson, Physical−Chemical Aspects of Protein Corona: Relevance to in Vitro and in Vivo Biological Impacts of Nanoparticles, J. Am. Chem. Soc., 2011, 133(8), 2525–2534 CrossRef CAS PubMed.
- F. Alnasser, V. Castagnola, L. Boselli, M. Esquivel-Gaon, E. Efeoglu, J. McIntyre, H. J. Byrne and K. A. Dawson, Graphene Nanoflakes Uptake Mediated by Scavenger Receptors, Nano Lett., 2019, 2, 1260–1268 CrossRef PubMed.
- V. Castagnola, W. Zhao, L. Boselli, M. L. Giudice, F. Meder, E. Polo, K. Paton, C. Backes, J. Coleman and K. Dawson, Biological recognition of graphene nanoflakes, Nat. Commun., 2018, 9(1), 1577 CrossRef CAS PubMed.
- S. Lara, F. Alnasser, E. Polo, D. Garry, M. C. Lo Giudice, D. R. Hristov, L. Rocks, A. Salvati, Y. Yan and K. A. Dawson, Identification of Receptor Binding to the Biomolecular Corona of Nanoparticles, ACS Nano, 2017, 1884–1893 CrossRef CAS.
- L. Boselli, E. Polo, V. Castagnola and K. A. Dawson, Regimes of biomolecular ultrasmall nanoparticle interactions, Angew. Chem., Int. Ed., 2017, 56(15), 4215–4218 CrossRef CAS PubMed.
- F. Muraca, L. Boselli, V. Castagnola and K. A. Dawson, Ultrasmall Gold Nanoparticle Cellular Uptake: Influence of Transient Bionano Interactions, ACS Appl. Bio Mater., 2020, 3(6), 3800–3808 CrossRef CAS PubMed.
- E. Sheridan, S. Vercellino, L. Cursi, L. Adumeau, J. A. Behan and K. A. Dawson, Understanding intracellular nanoparticle trafficking fates through spatiotemporally resolved magnetic nanoparticle recovery, Nanoscale Adv., 2021, 3, 2397–2410 RSC.
- M. Qin, J. Zhang, M. Li, D. Yang, D. Liu, S. Song, J. Fu, H. Zhang, W. Dai and X. Wang, Proteomic analysis of intracellular protein corona of nanoparticles elucidates nano-trafficking network and nano-bio interactions, Theranostics, 2020, 10(3), 1213 CrossRef CAS PubMed.
- Y. T. Ho, R. D. Kamm and J. C. Y. Kah, Influence of protein corona and caveolae-mediated endocytosis on nanoparticle uptake and transcytosis, Nanoscale, 2018, 10(26), 12386–12397 RSC.
- P. E. Costantini, M. Di Giosia, L. Ulfo, A. Petrosino, R. Saporetti, C. Fimognari, P. P. Pompa, A. Danielli, E. Turrini and L. Boselli, Spiky Gold Nanoparticles for the Photothermal Eradication of Colon Cancer Cells, Nanomaterials, 2021, 11(6), 1608 CrossRef CAS PubMed.
- F. Baldassarre, M. Cacciola and G. Ciccarella, A predictive model of iron oxide nanoparticles flocculation tuning Z-potential in aqueous environment for biological application, J. Nanopart. Res., 2015, 17(9), 1–21 CrossRef CAS.
- M. C. Morris , Standard X-ray Diffraction Powder Patterns: Section 15–data for 112 Substances, US Department of Commerce, National Bureau of Standards, 1976.
- S. Tong, C. A. Quinto, L. Zhang, P. Mohindra and G. Bao, Size-dependent heating of magnetic iron oxide nanoparticles, ACS Nano, 2017, 11(7), 6808–6816 CrossRef CAS PubMed.
- E. Carella, M. Ghiazza, M. Alfè, E. Gazzano, D. Ghigo, V. Gargiulo, A. Ciajolo, B. Fubini and I. Fenoglio, Graphenic nanoparticles from combustion sources scavenge hydroxyl radicals depending upon their structure, BioNanoScience, 2013, 3(2), 112–122 CrossRef.
- S. Tsuruoka, H. Matsumoto, K. Koyama, E. Akiba, T. Yanagisawa, F. R. Cassee, N. Saito, Y. Usui, S. Kobayashi and D. W. Porter, Radical scavenging reaction kinetics with multiwalled carbon nanotubes, Carbon, 2015, 83, 232–239 CrossRef CAS PubMed.
- I. Fenoglio, M. Tomatis, D. Lison, J. Muller, A. Fonseca, J. B. Nagy and B. Fubini, Reactivity of carbon nanotubes: free radical generation or scavenging activity?, Free Radicals Biol. Med., 2006, 40(7), 1227–1233 CrossRef CAS PubMed.
- I. Kokalari, R. Gassino, A. M. Giovannozzi, L. Croin, E. Gazzano, E. Bergamaschi, A. M. Rossi, G. Perrone, C. Riganti and J. Ponti, Pro-and anti-oxidant properties of near-infrared (NIR) light responsive carbon nanoparticles, Free Radicals Biol. Med., 2019, 134, 165–176 CrossRef CAS PubMed.
- Y. Wei, M. Zhao, F. Yang, Y. Mao, H. Xie and Q. Zhou, Iron overload by superparamagnetic iron oxide nanoparticles is a high risk factor in cirrhosis by a systems toxicology assessment, Sci. Rep., 2016, 6(1), 1–11 CrossRef PubMed.
- Q. An, C. Sun, D. Li, K. Xu, J. Guo and C. Wang, Peroxidase-like activity of Fe3O4@ carbon nanoparticles enhances ascorbic acid-induced oxidative stress and selective damage to PC-3 prostate cancer cells, ACS Appl. Mater. Interfaces, 2013, 5(24), 13248–13257 CrossRef CAS PubMed.
- N. Feliu, D. Docter, M. Heine, P. Del Pino, S. Ashraf, J. Kolosnjaj-Tabi, P. Macchiarini, P. Nielsen, D. Alloyeau and F. Gazeau, In vivo degeneration and the fate of inorganic nanoparticles, Chem. Soc. Rev., 2016, 45(9), 2440–2457 RSC.
- K. Bhattacharya, S. P. Mukherjee, A. Gallud, S. C. Burkert, S. Bistarelli, S. Bellucci, M. Bottini, A. Star and B. Fadeel, Biological interactions of carbon-based nanomaterials: From coronation to degradation, Nanomedicine, 2016, 12(2), 333–351 CrossRef CAS PubMed.
- I. Kokalari, S. Keshavan, M. Rahman, E. Gazzano, G. Barzan, L. Mandrile, A. Giovannozzi, J. Ponti, G. Antonello and M. Monopoli, Efficacy, biocompatibility and degradability of carbon nanoparticles for photothermal therapy of lung cancer, Nanomedicine, 2021, 16(9), 689–707 CrossRef CAS PubMed.
- V. E. Kagan, A. A. Kapralov, C. M. St. Croix, S. C. Watkins, E. R. Kisin, G. P. Kotchey, K. Balasubramanian, I. I. Vlasova, J. Yu and K. Kim, Lung macrophages “digest” carbon nanotubes using a superoxide/peroxynitrite oxidative pathway, ACS Nano, 2014, 8(6), 5610–5621 CrossRef CAS PubMed.
- W. Xing, G. Lalwani, I. Rusakova and B. Sitharaman, Degradation of graphene by hydrogen peroxide, Part. Part. Syst. Charact., 2014, 31(7), 745–750 CrossRef CAS.
- S. Lara, A. Perez-Potti, L. M. Herda, L. Adumeau, K. A. Dawson and Y. Yan, Differential Recognition of Nanoparticle Protein Corona and Modified Low Density Lipoprotein by Macrophage Receptor with Collagenous Structure, ACS Nano, 2018, 4930–4937 CrossRef CAS PubMed.
- G. Duan, S.-g. Kang, X. Tian, J. A. Garate, L. Zhao, C. Ge and R. Zhou, Protein corona mitigates the cytotoxicity of graphene oxide by reducing its physical interaction with cell membrane, Nanoscale, 2015, 7(37), 15214–15224 RSC.
- M. P. Monopoli, A. S. Pitek, I. Lynch and K. A. Dawson, Formation and characterization of the nanoparticle–protein corona, in Nanomaterial interfaces in biology, Springer, 2013, pp. 137–155 Search PubMed.
- S. J. Soenen, B. Manshian, J. M. Montenegro, F. Amin, B. Meermann, T. Thiron, M. Cornelissen, F. Vanhaecke, S. Doak and W. J. Parak, Cytotoxic effects of gold nanoparticles: a multiparametric study, ACS Nano, 2012, 6(7), 5767–5783 CrossRef CAS PubMed.
- S. Anguissola, D. Garry, A. Salvati, P. J. O'Brien and K. A. Dawson, High content analysis provides mechanistic insights on the pathways of toxicity induced by amine-modified polystyrene nanoparticles, PLoS One, 2014, 9(9), e108025 CrossRef PubMed.
- J. D. Ly, D. R. Grubb and A. Lawen, The mitochondrial membrane potential (Δψ m) in apoptosis; an update, Apoptosis, 2003, 8(2), 115–128 CrossRef CAS.
- M. Xia, Y. Zhang, K. Jin, Z. Lu, Z. Zeng and W. Xiong, Communication between mitochondria and other organelles: a brand-new perspective on mitochondria in cancer, Cell Biosci., 2019, 9(1), 1–19 CrossRef PubMed.
- S. Naqvi, M. Samim, M. Abdin, F. J. Ahmed, A. Maitra, C. Prashant and A. K. Dinda, Concentration-dependent toxicity of iron oxide nanoparticles mediated by increased oxidative stress, Int. J. Nanomed., 2010, 5, 983 CrossRef CAS PubMed.
- M. O. Ansari, N. Parveen, M. F. Ahmad, A. L. Wani, S. Afrin, Y. Rahman, S. Jameel, Y. A. Khan, H. R. Siddique and M. Tabish, Evaluation of DNA interaction, genotoxicity and oxidative stress induced by iron oxide nanoparticles both in vitro and in vivo: Attenuation by thymoquinone, Sci. Rep., 2019, 9(1), 1–14 Search PubMed.
- A. Martin and A. Sarkar, Overview on biological implications of metal oxide nanoparticle exposure to human alveolar A549 cell line, Nanotoxicology, 2017, 11(6), 713–724 CAS.
- A. Jochums, E. Friehs, F. Sambale, A. Lavrentieva, D. Bahnemann and T. Scheper, Revelation of different nanoparticle-uptake behavior in two standard cell lines NIH/3T3 and A549 by flow cytometry and time-lapse imaging, Toxics, 2017, 5(3), 15 CrossRef PubMed.
- M. Esquivel-Gaon, N. H. Nguyen, M. F. Sgroi, D. Pullini, F. Gili, D. Mangherini, A. I. Pruna, P. Rosicka, A. Sevcu and V. Castagnola, In vitro and environmental toxicity of reduced graphene oxide as an additive in automotive lubricants, Nanoscale, 2018, 10(14), 6539–6548 RSC.
- M. Kucki, L. Diener, N. Bohmer, C. Hirsch, H. F. Krug, V. Palermo and P. Wick, Uptake of label-free graphene oxide by Caco-2 cells is dependent on the cell differentiation status, J. Nanobiotechnol., 2017, 15(1), 46 CrossRef PubMed.
- S. Lerch, S. Ritz, K. Bley, C. Messerschmidt, C. K. Weiss, A. Musyanovych, K. Landfester and V. Mailänder, Nanoprobing the acidification process during intracellular uptake and trafficking, Nanomedicine, 2015, 11(6), 1585–1596 CrossRef CAS PubMed.
- B. B. Manshian, S. Pokhrel, L. Mädler and S. J. Soenen, The impact of nanoparticle-driven lysosomal alkalinization on cellular functionality, J. Nanobiotechnol., 2018, 16(1), 1–13 CrossRef.
- M. Wei, Z. Fu, C. Wang, W. Zheng, S. Li and W. Le, Graphene oxide nanocolloids induce autophagy-lysosome dysfunction in mouse embryonic stem cells, J. Biomed. Nanotechnol., 2019, 15(2), 340–351 CrossRef CAS PubMed.
- X. Ma, Y. Wu, S. Jin, Y. Tian, X. Zhang, Y. Zhao, L. Yu and X.-J. Liang, Gold nanoparticles induce autophagosome accumulation through size-dependent nanoparticle uptake and lysosome impairment, ACS Nano, 2011, 5(11), 8629–8639 CrossRef CAS PubMed.
- J. A. Kim, C. Åberg, A. Salvati and K. A. Dawson, Role of cell cycle on the cellular uptake and dilution of nanoparticles in a cell population, Nat. Nanotechnol., 2012, 7(1), 62 CrossRef CAS PubMed.
- Z. Chen, J.-J. Yin, Y.-T. Zhou, Y. Zhang, L. Song, M. Song, S. Hu and N. Gu, Dual enzyme-like activities of iron oxide nanoparticles and their implication for diminishing cytotoxicity, ACS Nano, 2012, 6(5), 4001–4012 CrossRef CAS PubMed.
- M. Wang and N. O. Petersen, Lipid-coated gold nanoparticles promote lamellar body formation in A549 cells, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2013, 1831(6), 1089–1097 CrossRef CAS PubMed.
- M. Davoren, E. Herzog, A. Casey, B. Cottineau, G. Chambers, H. J. Byrne and F. M. Lyng, In vitro toxicity evaluation of single walled carbon nanotubes on human A549 lung cells, Toxicol. In Vitro, 2007, 21(3), 438–448 CrossRef CAS PubMed.
- F. Moret, F. Selvestrel, E. Lubian, M. Mognato, L. Celotti, F. Mancin and E. Reddi, PEGylation of ORMOSIL nanoparticles differently modulates the in vitro toxicity toward human lung cells, Arch. Toxicol., 2015, 89(4), 607–620 CrossRef CAS.
- M. Esquivel-Gaon, S. Anguissola, D. Garry, A. d. C. Gallegos-Melgar, J. M. Saldana, K. A. Dawson, A. De Vizcaya-Ruiz and L. M. Del Razo, Bismuth-based nanoparticles as the environmentally friendly replacement for lead-based piezoelectrics, RSC Adv., 2015, 5(35), 27295–27304 RSC.
- J. A. Shayman and A. Abe, Drug induced phospholipidosis: an acquired lysosomal storage disorder, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2013, 1831(3), 602–611 CrossRef CAS PubMed.
- J. A. Whitsett, S. E. Wert and T. E. Weaver, Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease, Annu. Rev. Med., 2010, 61, 105–119 CrossRef CAS PubMed.
- Z. Chu, Y. Huang, Q. Tao and Q. Li, Cellular uptake, evolution, and excretion of silica nanoparticles in human cells, Nanoscale, 2011, 3(8), 3291–3299 RSC.
- R. E. Serda, A. Mack, A. L. Van De Ven, S. Ferrati, K. Dunner Jr., B. Godin, C. Chiappini, M. Landry, L. Brousseau and X. Liu, Logic–Embedded Vectors for Intracellular Partitioning, Endosomal Escape, and Exocytosis of Nanoparticles, Small, 2010, 6(23), 2691–2700 CrossRef CAS PubMed.
- F. Wang, A. Salvati and P. Boya, Lysosome-dependent cell death and deregulated autophagy induced by amine-modified polystyrene nanoparticles, Open Biol., 2018, 8(4), 170271 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm00220e |
| This journal is © The Royal Society of Chemistry 2022 |