
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA defined heat pretreatment of gelatin enables control of hydrolytic stability, stiffness, and microstructural architecture of fibrin–gelatin hydrogel blends†
Mattis
Wachendörfer
a,
Philipp
Schräder
a,
Eva Miriam
Buhl
b,
Alena L.
Palkowitz
a,
Ghazi
Ben Messaoud
cd,
Walter
Richtering
 cd and
Horst
Fischer
*a
cd and
Horst
Fischer
*a
aDepartment of Dental Materials and Biomaterials Research, RWTH Aachen University Hospital, Pauwelsstrasse 30, 52074 Aachen, Germany. E-mail: hfischer@ukaachen.de
bElectron Microscopy Facility, Institute of Pathology, RWTH Aachen University Hospital, Pauwelsstrasse 30, 52074 Aachen, Germany
cInstitute of Physical Chemistry, RWTH Aachen University, Landoltweg 2, 52074 Aachen, Germany
dDWI–Leibniz Institute for Interactive Materials, Forckenbeckstr. 50, 52074 Aachen, Germany
First published on 15th August 2022
Abstract
Fibrin–gelatin hydrogel blends exhibit high potential for tissue engineering in vitro applications. However, the means to tailor these blends in order to control their properties, thus opening up a broad range of new target applications, have been insufficiently explored. We hypothesized that a controlled heat treatment of gelatin prior to blend synthesis enables control of hydrolytic swelling and shrinking, stiffness, and microstructural architecture of fibrin–gelatin based hydrogel blends while providing tremendous long-term stability. We investigated these hydrogel blends’ compressive strength, in vitro degradation stability, and microstructure in order to test this hypothesis. In addition, we examined the gel's ability to support endothelial cell proliferation and stretching of encapsulated smooth muscle cells. This research showed that a controlled heat pretreatment of the gelatin component strongly influenced the stiffness, swelling, shrinking, and microstructural architecture of the final blends regardless of identical gelatin mass fractions. All blends offered high long-term hydrolytic stability. In conclusion, the results of this study open the possibility to use this technique in order to tune low-concentrated, open-porous fibrin-based hydrogels, even in long-term tissue engineering in vitro experiments.
1. Introduction
The cell's microenvironment is known to exert an influence on the responsive cellular behavior and plays a key role in regulating cell functions for both in vitro models and in vivo.1–3 Native extracellular matrix (ECM) is often replaced with hydrogel blends to engineer biomimetic ECM substitutes for tissue engineering and in vitro modeling due to their cell-supportive properties that mimic native ECM.4,5 The ECM substitute's stiffness plays an important role in the control, influence, and tailoring of cellular behavior such as cell migration, cell differentiation and cell morphology due to cell–substrate anchoring and interaction.6–9 The hydrogel microstructure influences not only the mechanical strength of the ECM substitute but greatly impacts cellular processes such as cell spreading and migration and also should enhance the diffusion of nutrients or growth factors and the removal of waste products from the hydrogel.4,10 Besides stiffness and a suitable microstructure, the long-term hydrolytic stability of the hydrogels during incubation at physiological conditions is also essential for long-time cell culture experiments,11,12 as in vitro models are only functioning after a specific period of maturation, during which embedded cells are able to proliferate, differentiate, form cell-to-cell interactions, and express key markers.13 As most hydrogels either swell or compact when infused with cell growth medium, the degree of swelling or compaction should remain as low as possible. In organ-on-a-chip models, the hydrogel is degraded by embedded cells such as fibroblasts, smooth muscle cells or especially endothelial cells, which line the inside of every native vessel lumen and play a key role in the vascularization of in vitro models.14 Importantly, advancing degradation during long-term incubation must be controlled and must not be accompanied by the release of cytotoxic byproducts and adverse chemical reactions that affect cell viability and functionality.15,16 In addition, the optical clarity of hydrogel blends is often mentioned as a helpful asset as it enhances live cell imaging of manufactured in vitro models and enables better observation of cellular processes in situ.11Fibrin is a biomaterial frequently used for ECM substitutes known for its key role in the clotting cascade and diverse properties for tissue engineering applications, such as cell binding sites or controlled release of growth factors.17–19 Depending on calcium and thrombin concentration and pH, fibrin appears either opaque or transparent based on the fiber lateral size and fiber self-assembly, which affects the mechanical properties as well.20,21 The fast degradation and compaction of low-concentrated fibrin-based hydrogels is a critical issue and restricts their use in long-term 3D cell culture.22,23
Gelatin, a cost-effective derivative of collagen, is frequently used for biofabrication processes such as extrusion and as part of hydrogel blends used as ECM substitutes.24–26 During gelatin synthesis from collagen, the pH of the medium will affect the isoelectric point while processing temperature and duration are known to affect the molecular weight of the proteins.27,28 Various processing parameters of gelatin are used throughout the scientific community (e.g. synthesis at 40 °C, 65 °C, 70 °C or 90 °C for various durations).29–33 As gelatin is soluble under physiological conditions, covalent crosslinking is needed to stabilize the gelatin for long-term incubation at physiological temperatures. A cytocompatible crosslinking strategy is the use of microbial transglutaminase to form covalent bonds between gelatin and fibrinogen.25,34–36 The enzyme transglutaminase catalyzes the formation of covalent bonds between γ-carbonyl and ε-amino groups of glutamine and lysine substrates, respectively.37The activity of microbial transglutaminase is calcium-independent as opposed to mammalian transglutaminase such as factor XIII in the clotting cascade, which stabilizes the formed fibrin clot in a calcium-dependent reaction.38–40 The enzymatic crosslinking of gelatin and fibrinogen with transglutaminase is dependent on the reaction temperature, pH and duration. After subsequent thrombin crosslinking, the reactions can result in transparent hydrogels with high potential for combining biomimetic organ-on-a-chip models with in situ live imaging techniques or facilitated confocal imaging.31 Both fibrinogen and gelatin provide the tripeptide arginine–glycine–aspartate sequences (RGD motifs) that facilitate cell adhesion.41
The reproduction of cell–cell and cell–ECM interactions remains challenging in the fields of tissue engineering and biofabrication. Furthermore, every tissue targeted to be mimicked possesses various requirements for realizing a functional in vitro model. Hence, new methods for tailoring hydrogel blends combining the aforementioned requirements are being continually explored and novel materials developed in order to improve in vitro models such as organ-on-a-chip models. A broad range of scientific studies, however, focuses only on the composition of the hydrogel blends without taking into account the mechanisms that occur during synthesis of each hydrogel component and the importance of control over the physiochemical mechanisms during hydrogel blend preparation.2,42–44However, both the interactions of the reaction conditions such as temperature and pH and the properties of the reaction partner strongly influence its final ECM substitutes’ microstructure and physio-mechanical properties, especially in fibrin-based hydrogel blends.45 We hypothesized that a controlled heat pre-treatment of gelatin blended with fibrin can significantly enhance control over swelling and shrinking, stiffness and microstructural architecture of fibrin-based hydrogel blends whilst providing excellent long-term stability. For all of the investigations, we precisely controlled the physiochemical mechanisms (pH, temperature) during synthesis. With our study, we demonstrate that thermal pretreatment of gelatin is a powerful tool to tailor hydrogel blends functioning as ECM substitutes and proved the potential of the novel gel blends in cell culture experiments exemplarily on endothelial cells. We reveal new insights for enhanced, tailorable fibrin-based hydrogels blends that can be used even in long-term tissue engineering in vitro applications.
2. Materials and methods
2.1 Hydrogel stock solution preparation
Gelatin from porcine skin (G2500, gel strength 300, Type A, Sigma-Aldrich, Darmstadt, Germany) was dissolved in phosphate-buffered saline (PBS, Gibco 18912014, Fisher Scientific, Schwerte, Germany) with 150 mg ml−1 at 90 °C for either 2 h or 12 h. After dissolving, the pH of the gelatin was adjusted to 7.4 with 1 M NaOH. The gelatin stock solutions were sterile filtered (0.2 μm sterile Syringe Filter, Filtropur S, Sarstedt, Nürnbrecht, Germany), aliqouted and subsequently frozen at −20 °C. Fibrinogen from bovine plasma (F8630, Sigma-Aldrich, Darmstadt, Germany) was dissolved by stirring with 50 mg ml−1 in PBS at 37 °C for 24 h. The stock solution was then sterile filtered, aliquoted, and frozen at −20 °C. Thrombin from bovine plasma (SRP6556, Sigma-Aldrich, Darmstadt Germany) was diluted in PBS to 100 U ml−1, aliquoted, and frozen at −20 °C. Microbial transglutaminase (SKU: 5060341114533, Special Ingredients, Chesterfield, United Kingdom) was dissolved in PBS with 60 mg ml−1 for 6 h at 37 °C and subsequently sterile filtered, aliqouted, and frozen at −20 °C. Calcium chloride (CaCl2, CN93.2, Carl Roth, Karlsruhe, Germany) was dissolved in Millipore®-water with 250 mmol ml−1, sterile filtered, aliqouted, and frozen at −20 °C.2.2 SDS-page of gelatin solutions
Sodium dodecyl sulfate-polyacrylaminde gel electrophoresis (SDS-page) was performed to assess the molecular weight distribution of gelatin solutions synthetized for 2 h and 12 h at 90 °C, respectively. Materials were from Bio-Rad Laboratories, Inc., Feldkirchen, Germany. Briefly, 1% gelatin samples were prepared and 15 μl of sample were mixed with 5 μl of Laemmli Sample Buffer (1610737) mixed with β-mercaptoethanol with a ratio of 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Samples were mixed for 5 min at 95 °C on a shaker and the Criterion Cell (1656001) was filled with running buffer 10× Tris/Glycine/SDS Buffer (1610732). Precision Plus Protein Blotting Standards (1610376) were filled into the outer columns of the 4–20% Criterion TGX Precast Protein Gel (5671094) while samples were filled in the center columns. Electrophoresis was performed at 40 mA for 15 min using PowerPac Basic (1645050) and subsequently at 80 mA for 30 min until proteins were separated. The gel was first microwaved for 2 min at 400 W and subsequently washed with dH2O on a shaker twice for 5 min. The gels was stained using Coomassie Brilliant Blue R-250 (1610436) for 1 h and destained using Coomassie Brilliant Blue R-250 Destaining Solution (1610438) for 3 h before imaging.
:1. Samples were mixed for 5 min at 95 °C on a shaker and the Criterion Cell (1656001) was filled with running buffer 10× Tris/Glycine/SDS Buffer (1610732). Precision Plus Protein Blotting Standards (1610376) were filled into the outer columns of the 4–20% Criterion TGX Precast Protein Gel (5671094) while samples were filled in the center columns. Electrophoresis was performed at 40 mA for 15 min using PowerPac Basic (1645050) and subsequently at 80 mA for 30 min until proteins were separated. The gel was first microwaved for 2 min at 400 W and subsequently washed with dH2O on a shaker twice for 5 min. The gels was stained using Coomassie Brilliant Blue R-250 (1610436) for 1 h and destained using Coomassie Brilliant Blue R-250 Destaining Solution (1610438) for 3 h before imaging.
2.3 Rheology of gelatin solutions
Rheological characterization of gelatin was used to investigate the effects on the gelatin's properties of the heat treatment temperature and duration and the accompanying changes in molecular weight. The data was measured using a rheometer (Kinexus pro + KNX2500, Netzsch-Gerätebau, Selb, Germany) and the manufacturer's software rSpace. A temperature sweep from 38 °C to 14 °C with a ramp rate of 1.5 K min−1 was performed using a 4°, 40 mm diameter cone and plate geometry with a constant frequency of 1 Hz and constant shear stress of 0.5 Pa, which had been checked to be within the linear viscoelastic region. The gelation point was defined at the temperature of the crossover point of the storage modulus (G′) over the loss modulus (G′′) with a phase angle (δ) lower than 45°. The plateau modulus was determined from the value of G′ at minimum and stable G′′ and δ. Besides the abovementioned gelatins, gelatin with 150 mg ml−1 dissolved for 6 h at 90 °C and gelatin dissolved for 2 h, 6 h and 12 h at 70 °C were also tested. Three samples of each gelatin were measured. To assess the effect of the filtering procedure on the mass fraction of the gelatin, the gelation point of unfiltered and filtered gelatin dissolved for 2 or 12 h at 90 °C was assessed as described above. All other experiments described before and afterwards were performed with filtered and thus sterile gelatin.2.4 Preparation of fibrin–gelatin hydrogel blends
Fibrin–gelatin blends were prepared with final gelatin concentrations of 2.5, 5, 7.5 and 10% (w/v), each using gelatin thermally processed for either 2 h or 12 h at 90 °C, resulting in eight hydrogel blends. During hydrogel synthesis, all solutions were kept and prepared at 37 °C. First, PBS, CaCl2 (250 mmol l−1) and transglutaminase were mixed before the respective amounts of gelatin were added. Tranexamic acid (100 mg ml−1, Carinopharm, Elze, Germany) was added with a final concentration of 0.017% (w/v). Fibrinogen was then added and the pH of the mixture adjusted to between 7.4 and 7.45 using 0.7 M NaOH. Subsequently, the solution was pre-incubated (37 °C, 5% CO2) for 1 h to allow the enzymatic crosslinking of transglutaminase between gelatin and fibrinogen. Finally, thrombin was diluted in PBS to 25 U ml−1 and gently mixed into the hydrogel blend to a final concentration of 1 U ml−1. The blends were processed within 2 min before the start of gelation following thrombin addition, and samples were left at rest at 37 °C for 3 h to ensure complete gelation prior to experimental analysis. The final concentrations of CaCl2, transglutaminase, and fibrinogen were 2.5 mmol ml−1, 0.4% (w/v) and 1% (w/v), respectively.2.5 Mechanical testing of hydrogel blends
Unconfined compression tests were performed to study the compressive strength of the hydrogel blends. The hydrogels were prepared in plastic tubes (13 ml tubes 95 × 16.8 mm, 23160, Sarstedt, Nürnbrecht, Germany) with 12 ml per hydrogel blend. After the incubation, the tubes were cut in sections of approx. 12 mm in height, resulting in five cylindrical hydrogel samples with a diameter d of 14.5 mm. The samples were weighed (m0) and the height was measured. Each sample was then tested using a universal testing machine (Z2.5, zwickiLine, ZwickRoell GmbH & Co. KG, Ulm, Germany) and compressed with 4 mm min−1, a preload force of 0.01 N, and maximum force of 7 N. Data was recorded by the manufacturer's software testXpert II. After the compression, samples were weighed again (m1). Subsequently, the weight loss due to compression was calculated as: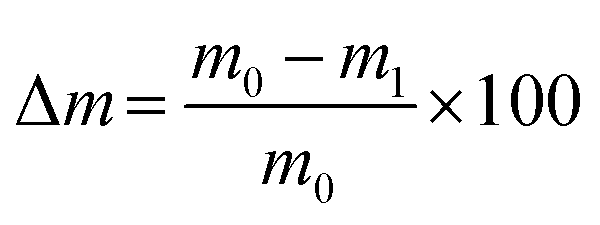 | (1) |
The stress σ was calculated by dividing the force F through the circular cross sectional area. The strain was calculated by dividing the height difference of the initial sample height (H0) and the height H through H0. The Young's moduli for the strain regions of 0–5, 5–10, 10–15, and 15–20% strain were calculated by dividing the stress by the strain. Samples were both tested on the day of synthesis and after 21 days of in vitro degradation (for details see 2.9).
2.6 Investigation of hydrogel microstructure
The microstructure of the hydrogel blends was investigated using scanning electron microscopy (SEM). Hydrogels were prepared in duplicates and with each 100 μl in 96-well plates. After crosslinking, the hydrogels were fixed in 3% (v/v) glutaraldehyde in 0.1 M Sorensen's phosphate buffer. Samples were dehydrated in ethanol (30, 50, 70, 90 and 100%) and critical point dried (E-300 Critical Point Dryer, Polaron Equipment, London, UK). Hydrogels were then sputtered with gold/palladium and imaged using a scanning electron microscope (ESEM XL 30 FEG, FEI, Philips, Eindhoven, Netherlands) under a high vacuum at an acceleration voltage of 10 kV. Pictures were analyzed using ImageJ (National Institutes of Health, Bethesda, MD, USA). To calculate the hydrogels’ porosity, pictures with a magnification of 10000× were chosen, split into six domains of 2 × 2 μm2 and then transformed into binary pictures. All black parts with a diameter of more than 0.005 μm2 were accounted as pores and thereby the average pore size and porosity of the hydrogel was calculated.
2.7 Cell culture
Human umbilical vein endothelial cells (HUVECs) and human umbilical artery smooth muscle cells (HUASMCs) were isolated from the endothelium of the veins or arteries of human umbilical cords after informed consent approved by the Ethics Committee of the Faculty of Medicine, RWTH Aachen University (EK 424/19). HUVECs were cultivated in T75 cell culture flasks previously coated with 2% (w/v) gelatin, whereas HUASMCs were cultured in uncoated flasks. As growth medium, endothelial cell basal medium (C-22111, PromoCell, Heidelberg, Germany) for HUVECs and 1× DMEM (21885, ThermoFisher Scientific, Waltham, MA, USA) for HUASMCs were used and cells were used for experiments between passage two and five.2.8 Cell proliferation study
For the cell proliferation studies, antibiotic antimycotic solution (100×) (A5955, Sigma-Aldrich, Darmstadt, Germany) was added to the hydrogel blends prior to thrombin crosslinking with a final concentration of 1% (v/v). The respective amount was subtracted from the PBS volume. Cell proliferation on hydrogels was investigated over the course of 7 days using the Cell Counting Kit-8 (CCK8-assay, CK04, Dojindo EU, Munich, Germany). Briefly, a water-soluble tetrazolium salt, WST-8, is reduced by the cells’ dehydrogenase activities to create a yellow-colored formazan dye. The amount of dye generated is directly proportional to the number of living cells. The wells of 12-well plates were loaded with 280 μl of the hydrogel blends. After incubation for complete crosslinking, HUVECs were trypsinized and counted, and 1 × 104 HUVECs were seeded onto the gels and covered with 686 μl of growth medium. On day 1, 3, 5, and 7 after seeding, the CCK8-assay was performed. The old medium was removed and the gels were thoroughly washed with PBS to remove any medium residues. The CCK8-solution was mixed with HUVEC medium at a ratio of 1:11. Every well was covered with 370 μl of the CCK8-media-mixture and the well plates were incubated for 135 min. Next, 100 μl of the mixture was given into 96-well plates, whereas three blanks with solution not exposed to cells were also loaded. The optical density (OD) of the samples was measured at 450 nm using a microplate reader (Spectramax M2, Molecular Devices, LLC., San Jose, CA, USA) and data was obtained by the manufacturer's software, Softmax Pro 6. The remaining CCK-8-media-mixture was removed, and the gels again washed with PBS and covered with fresh growth media. The metabolic activity and number of living cells was normalized to the number of living cells on day 1 (D1) in order to calculate the cell multiplication on the respective days as follows: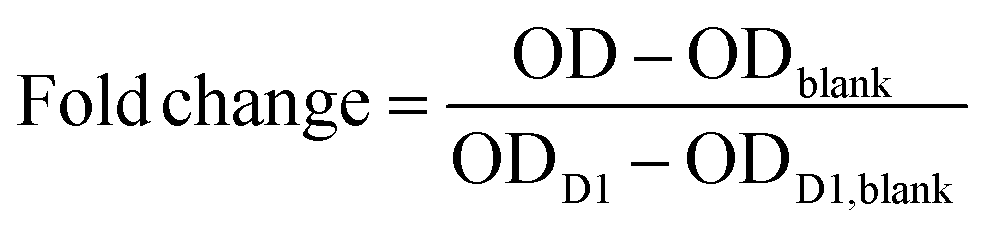 | (2) |
For imaging, the wells of 2-well slides (μ-Slide 2 Well Glass Bottom, Ibidi, Gräfeling, Germany) were each loaded with 360 μl of hydrogel blend. After incubation, 1.45 × 104 HUVECs were seeded onto the hydrogels and covered with 1 ml of growth medium to keep the cell seeding density and media volume constant compared to the 12-well plates. HUVECs were stained using Calcein-AM (425201, BioLegend, Koblenz, Germany) on day 1, 3, 5, and 7. Briefly, Calcein-AM was diluted with dimethyl sulfoxide (DMSO) for a stock solution of 1 mM. For a working solution of 1 μM, the stock solution was further diluted with PBS. The staining solution was added to the wells, followed by incubation of 20 min. The staining solution was removed, samples were washed with PBS, and images were taken on 10 random fields using a fluorescence microscope (Axio Imager·M2m, Carl Zeiss Microscopy Deutschland, Oberkochen, Germany). Experiments were performed with three different donors and 3 samples per donor.
2.9 Analysis of compaction, swelling and degradation rate
To study the compaction, swelling and degradation rate of the hydrogels, five samples per hydrogel blend were prepared with 500 μl each inside a 24-well plate. After sufficient incubation time for complete crosslinking, the hydrogels were removed and weighed (m0). Subsequently, the samples were immersed in 1 ml sterile PBS and incubated. On day 1, 3, 5, 7, 14, 21, and 28 after immersion, the hydrogels were weighed (m) and the pH of the PBS was noted. The PBS was changed three times a week. The weight change was compared to the initial weight and calculated as: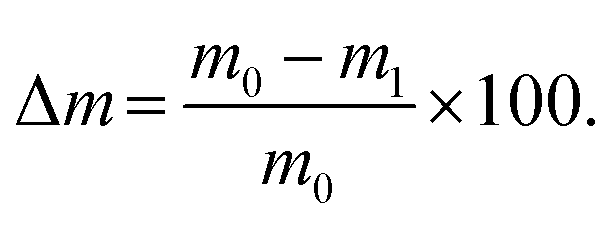 | (3) |
To study the degradation rate of the hydrogels during cell experiments, the hydrogels used for imaging on the respective days were fixed in 4% formaldehyde (28908, ThermoFisher Scientific, Waltham, MA, USA). The hydrogels were then lyophilized (Lyoquest, Telstar, Leeds, UK) and the weight change calculated as described above.
2.10 Statistical analysis
Statistical analysis was performed using GraphPad PRISM 9 (GraphPad Software, San Diego, CA, USA). ANOVA tests were performed in order to study significant differences within groups and comparison between groups with Tukey's post-hoc test. Significance levels of p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***) were defined. Data is presented using box and tukey whiskers.3. Results
3.1 Influence of the heat treatment on molecular weight of gelatin and its rheological properties
The influence of the heat treatment on the molecular weight distribution of gelatin solutions was determined by SDS-page. The resulting rheological properties of the gelatin solutions were characterized by measurements of gelation temperature and plateau modulus. The molecular weight distribution of gelatin shifts to lower values for increased processing time (Fig. 1B). Gelatin processed for 2 h at 90 °C accumulated between 100 and 250 kDa, while gelatin processed for 12 h at 90 °C showed a much broader distribution from 15 to 250 kDa. For gelatin thermally processed at 70 °C, both gelation temperature and plateau modulus remained constant independent of the annealing duration at ca. 24 °C and 8.5 kPa (Fig. 1A). The plateau modulus was stable between 4.5 and 4.9 kPa independent of the preheating duration. The gelation temperature of gelatin decreased with increasing thermal processing time at 90 °C from 25.84 ± 0.25 °C for 2 h of thermal treatment, over 22.31 ± 0.05 °C for 6 h, to 19.56 ± 0.05 °C for 12 h of heat treatment. Similarly, the plateau modulus of the gelatins decreased from 4.86 ± 0.30 kPa over 2.85 ± 0.03 kPa to 1.16 ± 0.03 kPa with increasing preheating duration. No statistical differences were found between the gelation points and plateau moduli of unfiltered and filtered gelatin (Fig. S10†), indicating no shift in gelatin mass fraction due to the filtering procedure.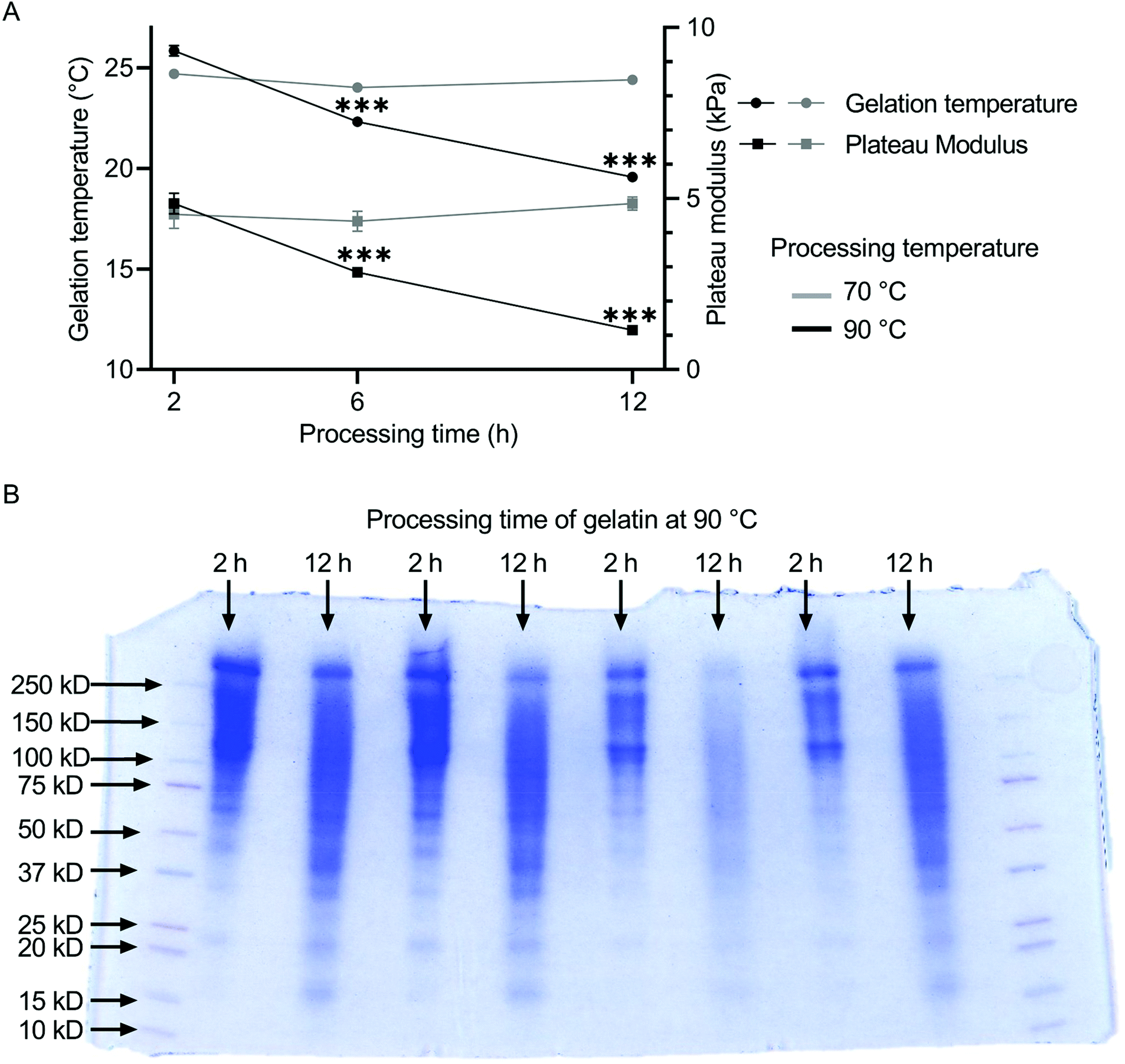 | ||
| Fig. 1 (A) Gelation temperature and plateau modulus decrease significantly when gelatin is processed for 6 h or 12 h compared to 2 h at 90 °C. For the heat treatment at 70 °C, gelation temperature and plateau modulus remain constant. (B) SDS-page of gelatin processed for either 2 or 12 h at 90 °C. The molecular weight distribution shifts to lower values for increased processing time. | ||
3.2 Gel transparency and compressive strength
All fibrin–gelatin blends composed of 2 h processed gelatin appeared transparent (Fig. 2). The turbidity of fibrin–gelatin blends composed of 12 h processed gelatin increased with gelatin concentration. Quantification of optical transparency can be found in Fig. S1.†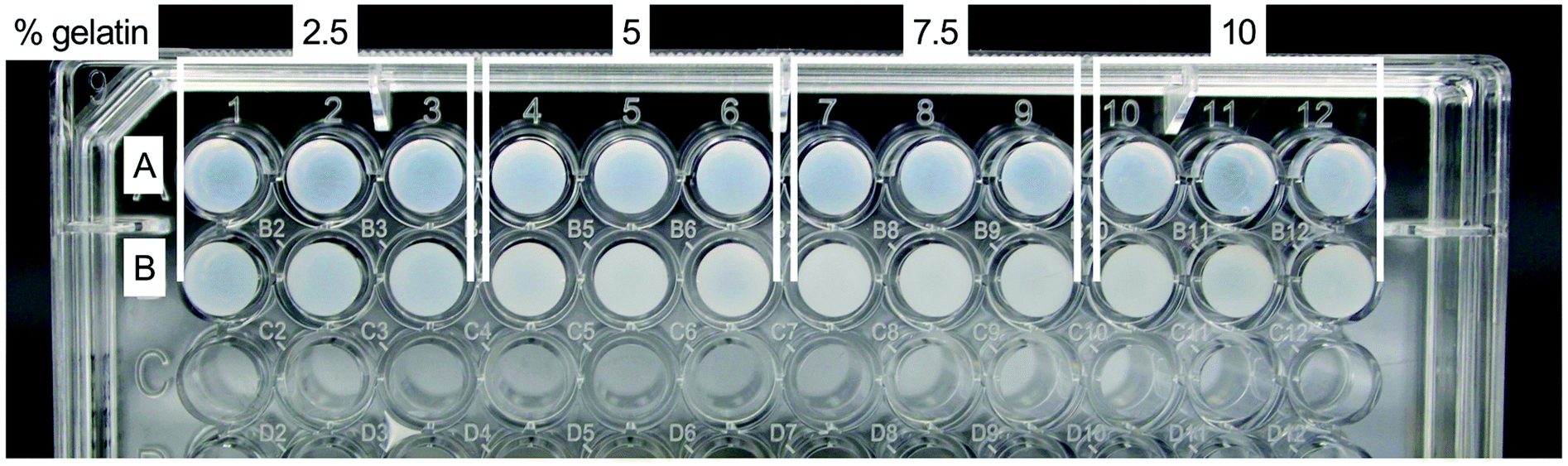 | ||
| Fig. 2 Optical appearance of fibrin–gelatin blends. (A) Fibrin–gelatin blends composed of 1% fibrin and 2 h processed gelatin, blends appear transparent. (B) Fibrin–gelatin blends composed of 1% fibrin and 12 h processed gelatin, blends appear increasingly turbid with gelatin concentration. | ||
For fibrin–gelatin blends with gelatin processed for 2 h, the stiffness of the gels increased with gelatin fraction (Fig. 3A). At 0–5% strain, the 2.5% blends provided a compressive strength of 3.1 ± 0.4 kPa, significantly lower than 5.0 ± 0.7 kPa for 5% blends, 7.5 ± 1.4 kPa for 7.5% blends, and 8.1 ± 1.2 kPa for 10% blends. The gelatin blend with 2.5% provided significantly lower compressive strength at all strain regions. The 5% blend was significantly less stiff compared to the blends with the highest gelatin fraction at 0–5% and 5–10% strain. Conversely, for the fibrin–gelatin blends containing gelatin processed for 12 h, the compressive strength of the gels was inversely proportional to the gelatin fraction (Fig. 3B). At the strain region of 0–5%, the fibrin–gelatin blend with 2.5% provided a compressive strength of 3.9 ± 0.8 kPa, significantly higher than 2.6 ± 0.5 kPa at 5%, 2.6 ± 0.4 kPa at 7.5%, and 1.9 ± 0.3 kPa at 10% gelatin fraction. The relations between the hydrogel blends remained at higher strain regions and the low-concentrated fibrin–gelatin blend remained at the highest compressive strength, while with gelatin fraction the stiffness decreased.
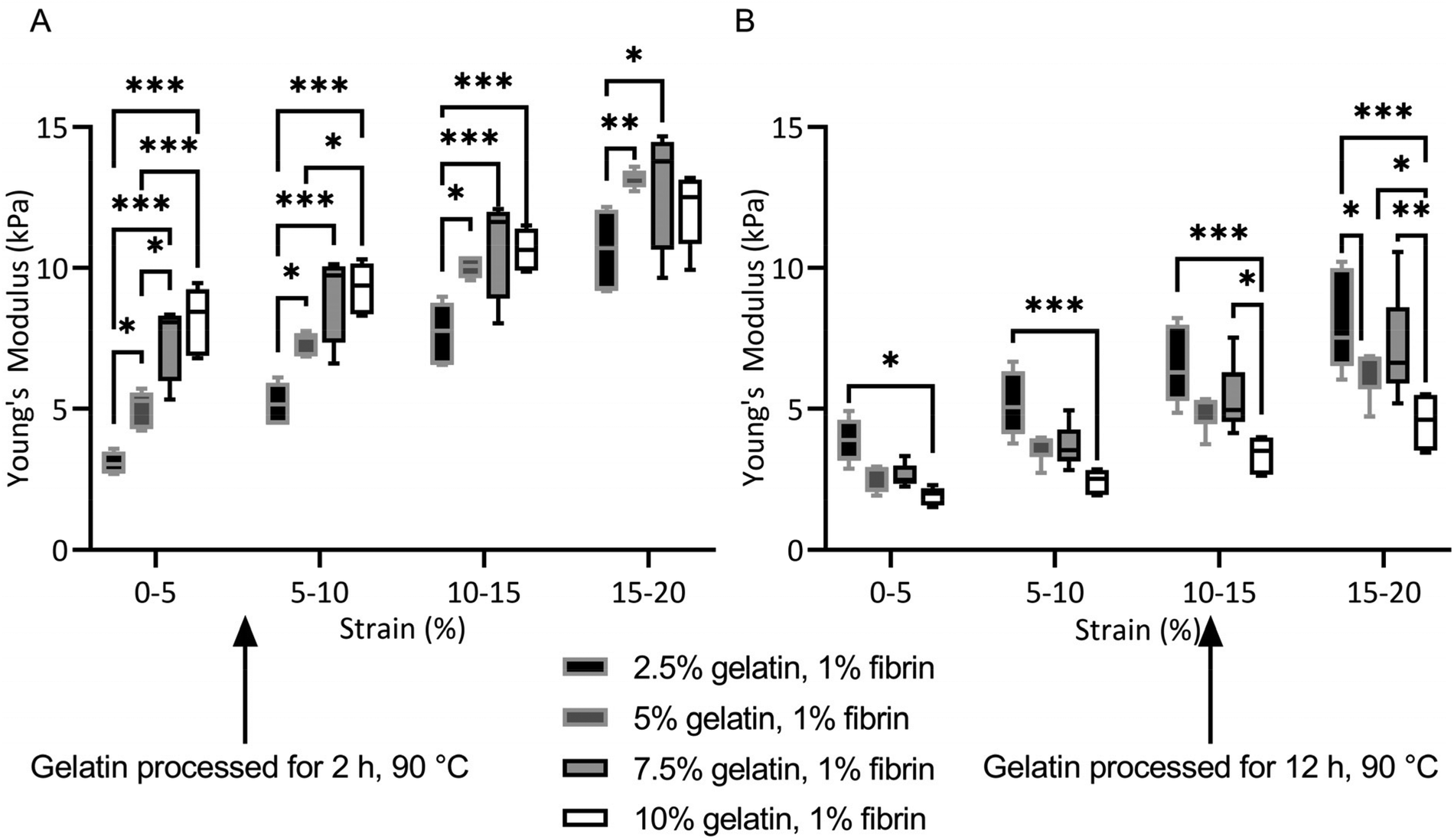 | ||
| Fig. 3 Compressive strength of fibrin-based hydrogel blends. (A) Fibrin–gelatin blends with gelatin preheated for 2 h at 90 °C, Young's modulus increases with gelatin fraction. (B) Fibrin–gelatin blends with gelatin preheated for 12 h at 90 °C, Young's modulus decreases with gelatin fraction. | ||
The blends composed of gelatin preheated for 12 h lost significantly more weight than their identical gelation fraction counterparts (Fig. S2†). After 21 days of in vitro degradation, the blends maintained between 50–100% of their initial Young's modulus (Fig. S3†).
3.3 Hydrogel microstructure
All fibrin–gelatin blends provided an extremely finely pored microstructure with pore sizes less than 1 μm (Fig. 4). The microstructure of fibrin–gelatin blends with 2 h processed gelatin was found to be more uniform and smooth compared to fibrin–gelatin blends containing 12 h processed gelatin. The depicted surface of the latter blends appears more textured and open-porous with more prominent access to adjacent pores. Especially the hydrogel blends with 2.5% and 5% gelatin provide larger pore sizes when the gelatin was thermally processed for 12 h instead of 2 h. Pore size analysis demonstrates that both 2.5% and 5% fibrin–gelatin blends with 12 h processed gelatin show significantly larger pores and higher porosity compared to the other fibrin–gelatin blends (Fig. S5†). | ||
| Fig. 4 SEM pictures of fibrin–gelatin blends containing 1% fibrin. Blends composed of gelatin processed for longer times show a more open-porous, fragmented microstructure. Scale bar identical for all micrographs. | ||
3.4 Endothelial cell proliferation on hydrogel blends
HUVECs proliferated at different rates on the hydrogel blends. However, ultimately a monolayer was formed on all ECM substitutes. Fluorescence images of HUVECs stained with calcein-AM on the specific gels and days can be found in Fig. S7† and Fig. S8.†The two lowest concentrate (2.5%, 5%) fibrin–gelatin blends with gelatin preheated for 2 h provided significantly higher metabolic activity compared to the blends with the highest gelatin proportion (10%), displaying a final fold change on day 7 of 7.09 ± 0.44 and 6.46 ± 0.80 compared to 5.82 ± 0.89 and 5.66 ± 0.60, respectively (Fig. 5A). On fibrin–gelatin blends composed of gelatin preheated for 12 h, significantly higher HUVEC activity was measured for the 5% blend (2.42 ± 0.36) on day 3 after seeding (Fig. 5B). On day 7, both blends 2.5% and 5% provided significantly higher metabolic activity at a fold change of 5.58 ± 0.42 and 5.85 ± 0.92 compared to the blends the highest gelatin amount, with fold changes of 4.53 ± 0.77 and 4.26 ± 0.94, respectively.
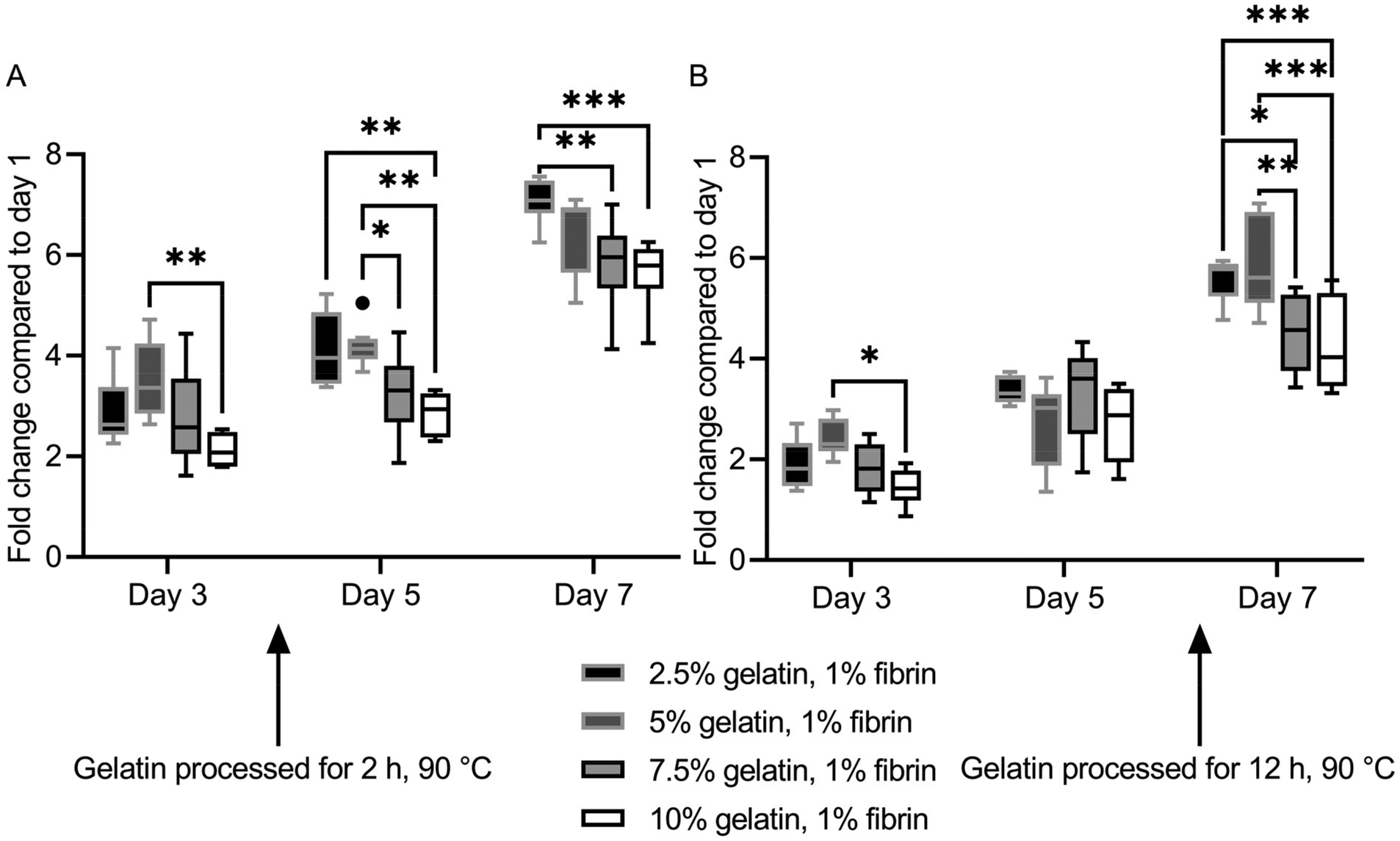 | ||
| Fig. 5 CCK-8 assay of fibrin-based hydrogel blends. (A) Fibrin–gelatin blends with gelatin preheated for 2 h at 90 °C. HUVECs proliferate faster on less stiff blends. (B) Fibrin–gelatin blends with gelatin preheated for 12 h at 90 °C. | ||
3.5 In vitro swelling and degradation of non-cell-laden and cell-laden hydrogels
After immersion in PBS, the fibrin–gelatin blend containing 2.5% of 2 h processed gelatin lost 13 ± 12.1% of the initial weight within the first 3 days, after which the weight was constant until day 21 of incubation (Fig. 6A). After 21 days of the experiment, one of six samples had dissolved, and the remaining samples dissolved within the following 7 days. The 5% blend absorbed fluid (2.7 ± 2.6%) within the first 24 h and provided a constant weight until day 28 with no dissolved samples. The highest concentrated fibrin–gelatin blends containing 2 h processed gelatin initially had a weight gain of 31.5 ± 14.6% and 90.1 ± 8.4% within the first 24 h. The weight decreased over the following days and the gels provided a constant weight between day until day 21. The weight of the 10% blend was continuously significantly higher compared to the initial weight on day 0. Half the samples of the 7.5% blend and 4 samples out of 6 of the 10% blend dissolved within the following 7 days.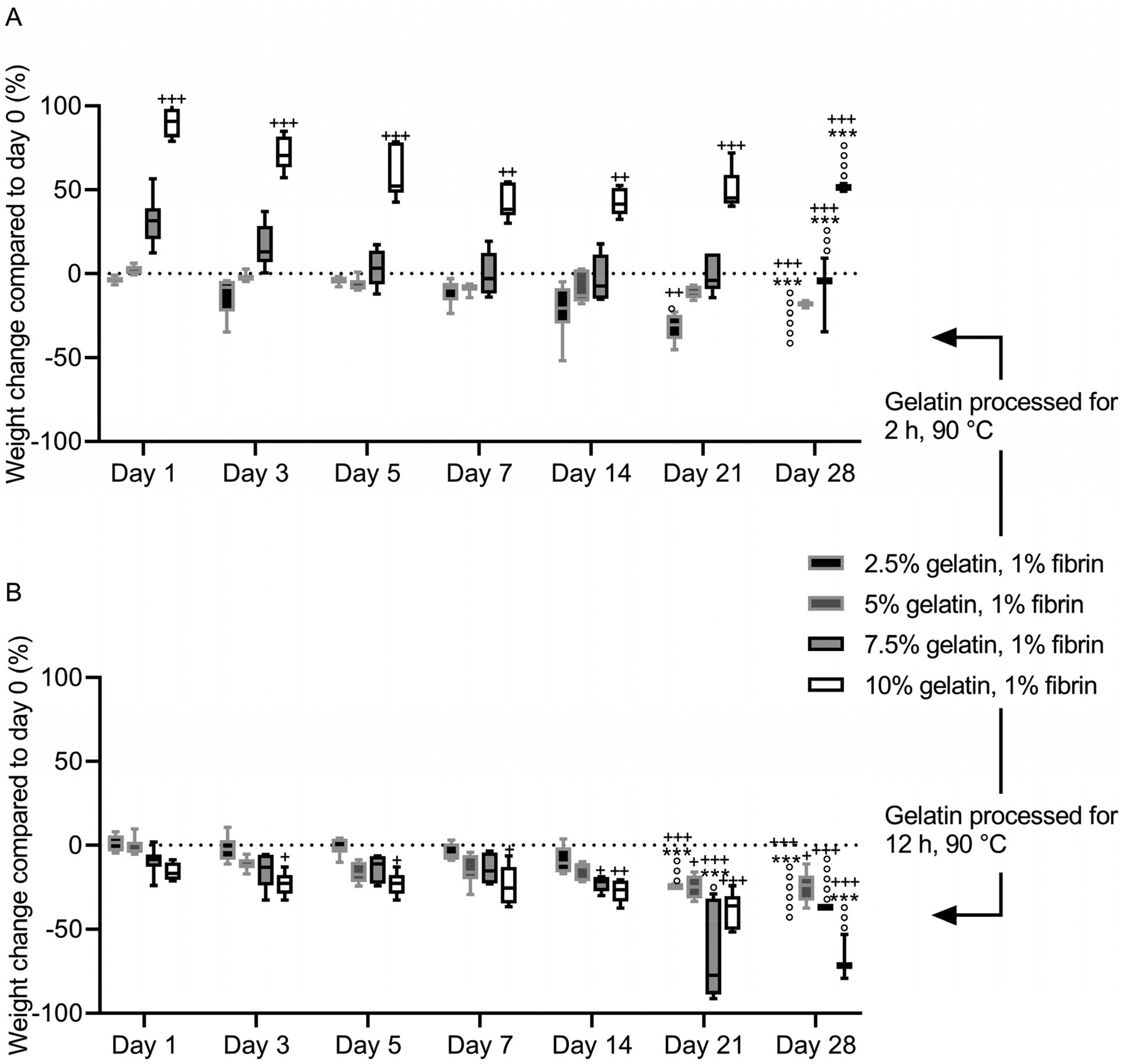 | ||
| Fig. 6 In vitro degradation of fibrin-based hydrogel blends. Significant weight changes compared to day 0 are marked with +/++/+++, while significant weight change to the previous time point is marked with ***. Dissolved samples are marked with °. (A) Fibrin–gelatin blends with gelatin preheated for 2 h at 90 °C. Initial swelling increases with gelatin fraction. (B) Fibrin–gelatin blends with gelatin preheated for 12 h at 90 °C. Initial compaction increases with gelatin fraction. | ||
The weight of fibrin–gelatin blends containing 2.5%, 5% and 7.5% of 12 h processed gelatin were constant, whereas the weight of the 10% blends decreased significantly within the first 3 days (Fig. 6B). The 2.5% blends were stable until day 21 and the next half of the six samples dissolved within the next 7 days. The 5% blends were stable from until day 28 and none of the samples completely dissolved, however the weight on day 28 was significantly lower compared to the initial weight on day 0. The 7.5% and 10% blends were stable until day 21 and day 28, respectively, whereas after 28 days all except one sample of the former blend and half of the latter blend dissolved. The pH of the PBS was between 6.8 and 7.3 in the first weeks. After 28 days, the pH of the PBS increased in all samples (Fig. S4†).
All cell-laden hydrogels were stable during HUVEC proliferation. The lyophilized weight of the blend did not decrease significantly over the term of 7 days (Fig. S6†). Hence, the hydrogels were not degraded by the cells or other medium-activated mechanisms.
4. Discussion
Fibrin-based hydrogels exhibit excellent cell-supporting properties, however high compaction and diminished long-term stability often decrease the scope of potential applications. Previous studies show that this can be reduced by tuning the delicate equilibrium of pH and calcium and thrombin concentration, respectively, or by addition of protease inhibitiors such as aprotinin, aminocaproic acid and tranexamic acid, which enhance the stability of the fibrin clot.46–51 In combination with gelatin, stable hydrogel blends can be achieved, however with high swelling ratios at high gelatin concentrations. This study shows improved processing of low concentrated and open-porous fibrin-based hydrogels with great long-term stability, which can be of great advantage for tissue engineering applications. As the hydrogel components fibrinogen and gelatin are greatly influenced by pH and temperature, precise control of these parameters in this work revealed new insights for the processing of enhanced, tailorable fibrin-based hydrogel blends. We showed that thermal processing of gelatin is a powerful tool to tailor fibrin–gelatin blends.It is known that the molecular weight of gelatin decreases with both increased synthesis temperature and time.30,31 However, the molecular weight of gelatin is usually not discrete but widely distributed as the polydispersity index of gelatin follows a Gaussian curve, showing a broad molar mass distribution.27,30 Both intact and partially hydrolyzed chains are present in gelatin solution and a large variety of supramolecular structures, from globular polymer-like chains to triple-stranded helical fibrils, can form, resulting in different molecular weights.30 Due to long heat exposure, molecules are fragmented into lower molar mass protein derivatives due to hydrolysis, affecting the resulting amino acid composition, whereas primarily the β- and y-chains contribute to the gelation properties.52,53 In our research, this mechanism was highlighted by the SDS-PAGE of the gelatin solutions (Fig. 1B). We could observe that the broad distribution of gelatin molecular weight shifts towards lower molecular weights when the gelatin is processed for longer. The effects of the shift in molecular weight distribution were showed by the rheological investigation of the gelatin sol–gel transition (Fig. 1A). Both gelation the temperature and plateau modulus of gelatin decreased distinctly when pretreated for longer periods at 90 °C, reflecting the reduced strength and gelation behavior of lower molecular weight gelatins observed in previous studies.28,54 It must be noted that the annealing of gelatin with 12 h at 90 °C represents an extensive and extreme gelatin heat treatment, yet as demonstrated from rheological characterization the preheated protein maintained its gelation ability. We further showed that our filtering procedure does not affect gelatin mass fraction (Fig. S10†). Previous studies found that gel the strength and the turbidity of gelatin solution decreases and increases, respectively, with increased annealing time.53 We could also observe this as gelatin solutions pretreated for 12 h at 90 °C provided higher turbidity of the resulting fibrin–gelatin blends increased with gelatin mass fraction (Fig. 2, Fig. S1†).
The effect of molecular weight and gel strength are distinctly reflected in the different compressive strength of the fibrin–gelatin blends. For blends containing gelatin processed for 2 h, the Young's modulus increased with gelatin concentration as expected and observed by others in hydrogels when crosslinker concentration or mass fraction were increased.55 Conversely, the Young's modulus of fibrin–gelatin blends containing the longer-processed gelatin decreased with increasing gelatin mass fraction, indicating that due to the lower molecular weight of the gelatin and its decreased gel strength, increasing gelatin concentration does not strengthen but rather weakens the interpenetrating network. During compression, liquid is squeezed out between the gel fibrils as they rotate in the direction of stretch.56 A high degree of water loss should be avoided as it can lead to detachment of hydrogel blends from the bioreactor surface or stability loss of the hydrogel's structure under the load.29 The increased fluid loss due to compression could indicate insufficiently transglutaminase-mediated crosslinking of gelatin and fibrinogen during pre-incubation (Fig. S2†). The efficiency of crosslinking and formation of isopeptide bonds is dependent on accessibility to the transglutaminase of the target amino acid groups in gelatin and fibrinogen. Due to the extended heat treatment of the gelatin, the accessibility of the gelatin target amino groups may decrease, resulting in a lower degree of crosslinking. Hence, excess transglutaminase and isolated gelatin could leak out during compression. The crosslinking degree increases with gelatin mass fraction, therefore the fluid loss due to compression decreases.
All fibrin–gelatin blends containing 2 h processed gelatin provide a dense, homogenous microstructure with pore sizes of 200–250 nm and porosities of approx. 20%. For fibrin–gelatin blends composed of 12 h processed gelatin, a more open-porous microstructure was found. For 2.5% and 5% gelatin blends, larger pore sizes can be observed along with a rougher surface structure. These observations are substantiated by the microstructure analysis (Fig. S5†), where significantly larger pores are found for the two lower concentrated blends. The porosity of these blends composed of 12 h processed gelatin is likewise higher than its counterparts. When the gelatin concentration is increased to 7.5% and 10%, similar pore sizes are found when comparing blends containing 12 h to 2 h processed gelatin, however the microstructure seems to be rougher. Increasing the gelatin concentration also increases the resulting number of polymers per volume, which leads to a higher density of reactive groups and therefore denser hydrogel for a constant and sufficient transglutaminase concentration. This also increases the degree of crosslinking, resulting in a denser microstructure with smaller pores and overall decreased porosity. The mechanical properties mainly depend on the density of branching points, as reduced distance of crosslink points results in a tighter network with smaller pore sizes, which increases mechanical strength.20,57,58 The molecular weight of the reaction partners affects the gel's final microstructure and porosity,4 which we also observed due to the reduced molecular weight of the longer-processed gelatin. Although at high gelatin concentrations the crosslinking density and density of branching points is similar within the interpenetrating networks, the mechanical strength of the blends containing longer-processed gelatin is comparatively lower as described above. This is likely due to the decreased molecular weight of the gelatin, resulting in both a reduced gel strength of the gelatin itself and a reduced bond strength with the enzymatically crosslinked fibrinogen. We would like to emphasize that especially the lower concentrated fibrin–gelatin blends could be of interest for further research, since they are identical in chemical composition, provide similar mechanical strength at low strain regions and yet provide distinct microstructures with porosities at different levels of magnitude. The decoupling of hydrogel composition and strength from the microstructure allows the investigation of one factor independently of the other and is a feature often targeted in interpenetrating networks but only yet inadequately explored and investigated.10
The molecular weight of polymers prior to gelation affects not only the final gel microstructure and porosity accompanied by changes in mechanical properties, but also the overall degradation behavior.4,16 Initial swelling of immersed hydrogels is often observed at high molecular weights, whereas the swelling ratio decreases for hydrogels with lower molecular weight.52 In the in vitro degradation test of the fibrin–gelatin blends, we observed similar behavior, as the fibrin–gelatin blends composed of high concentrations of shorter-processed gelatin initially swelled up to 90%. In contrast, the counterparts composed of longer-processed gelatin, hence with lower molecular weight, initially shrank to a low degree. In addition, the fibrin–gelatin blends composed of shorter-processed gelatin provided slightly higher stability with fewer samples dissolved after 28 days. The increased molecular weight results in longer kinetic chains accompanied by a higher degree of polymerization and increased stability, consistent with previous studies.16 However, all blends showed excellent degradation stability, also shown by compression test after 21 days of degradation, whereas most of the intial Young's modulus was preserved. Moreover, even after 21 and 28 days of degradation, samples were easy to handle and perform experiments with. As the in vitro degradation was performed with non-cell-laden gels, the underlying degradation mechanism is likely to be hydrolysis due to the fact that hydrolytically unstable bonds are dissolved over time. The pH of the PBS was constant when the gels were stable, indicating a stable degree of ionization of functional groups. The pH rose when the weight of the gels began decreasing after ca. 21 days of incubation, also observed elsewhere.11,59 Fibrin is sensitive to proteolytic degradation by metalloproteinases or plasmin, and plasminogen is present in most of the fibrin preparations, which might also have initially accelerated the gels’ degradation.46 We added tranexamic acid as a fibrinolysis inhibitor in the initial gel mixture, but it is probable that it was flushed out within the first days. For longer degradation stability, fibrinolysis inhibitors could be added directly into the surrounding medium, however 21 days of stability are usually sufficient for a broad range of in vitro applications.
As mentioned above, the compaction of fibrin-based hydrogels during immersion is a well-known issue. Swelling and shrinking of hydrogels is often measured optically, however such methods are flawed since gels are often only recorded two-dimensionally and the degree of swelling or shrinking is then reconstituted by the degree of area extension or reduction.42,60,61 Here, we measured the weight of the immersed gels at specific time points. Because the wet weight of the hydrogel is dominated by the weight of the retained water and not the comparatively low weight of the fibrous polymer network, we deduced that the hydrogels’ wet weight provides immediate information about gel volume, as the density of PBS is similar to pure water. Hence, when the gel compacts its volume is reduced, decreasing the weight of retained fluid. Reversely, when the gel swells, its volume increases, increasing the weight of retained fluid. The swelling/shrinking degree and long-term stability are dependent on the immersion medium (e.g., PBS with monovalent, most cell culture media with both monovalent and divalent ions), and its salinity and resulting osmotic pressure.4,25,57
As expected, all fibrin-based blends showed their well-known cell-active properties, as HUVECs proliferated on the gels and ultimately formed monolayers within 7 days. The blends were stable during cell proliferation and were not degraded by the cells, in contrast to the rapid hydrogel degradation by HUVECs reported elsewhere.13,62 This emphasizes the enhanced and outstanding stability of the blends and confirms the suitability of our synthesis approach for long-term, cell-laden applications. Encapsulating smooth muscle cells or fibroblasts in the ECM substitutes could be a challenge, however these cells showed high viability and stretching within similar or more dense networks (collagen-agarose, fibrin, pure collagen).63,64 Moreover, in preliminary experiments encapsulated smooth muscle cells showed high viability (>85%) and characteristic stretching after 7 days and no bulk degradation of the hydrogels was observed over 3 weeks of cultivation (Fig. S9†). Optically transparent gels provided penetration depths of 1–1.5 mm using confocal microscopy. Encapsulation of other cells in combination with other cell medium could change the observed degradation dynamics. Bulk degradation of fibrin can be modified by the addition of aprotinin, aminocaproic acid or tranexamic acid to the medium as mentioned beforehand. Partial degradation, however, favors the development of pericellular and extracellular matrices, cell anchoring and penetration, and can be beneficial for smooth muscle cell proliferation and vascularization.52,65,66 The fibrin-based gels can be locally degraded by cells expressing metalloproteinase, making them generally suitable for cell migration or angiogenesis studies.67,68 As mentioned above, perfusion is required for vascularization in dense or highly concentrated and dense fibrin networks.69 For cell migration, the space between pores must be large enough or must be created by degradation of ECM.70,71 While the fixation of hydrogel samples with glutaraldehyde introduces small changes in pore size and shape, the following dehydration (critical point drying) and the high vacuum during SEM is expected to result in more considerable changes to the hydrogel microstructure due to shrinkage.72 Hence, the pore sizes of the hydrated gels are expected to be even larger than depicted in SEM images.
5. Conclusion
We have shown that a defined preheatment of gelatin accompanied by changes in molecular weight enables control of the mechanical behavior, microstructure, and in vitro degradation kinetics of fibrin–gelatin hydrogels while maintaining the support of endothelial cell proliferation. We emphasize the importance of understanding and controlling the physiochemical parameters during hydrogel synthesis in order to design improved, tailorable fibrin-based hydrogel blends for tissue engineering applications. Our approach allowed the decoupling of the dependency of the resulting microstructure from the chemical composition and stiffness for low-concentrated fibrin–gelatin blends.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support of the European Regional Development Fund (Interreg Euregio Maas-Rhein) of the European Union (grant EMR116). This work was supported by the Confocal Microscopy Facility, a Core Facility of the Interdisciplinary Center for Clinical Research (IZKF) Aachen within the Faculty of Medicine at RWTH Aachen University.References
- K. Bott, Z. Upton, K. Schrobback, M. Ehrbar, J. A. Hubbell, M. P. Lutolf and S. C. Rizzi, The effect of matrix characteristics on fibroblast proliferation in 3D gels, Biomaterials, 2010, 31, 8454–8464 CrossRef CAS PubMed.
- R. DeVolder and H.-J. Kong, Hydrogels for in vivo-like three-dimensional cellular studies, Wiley Interdiscip. Rev.: Syst. Biol. Med., 2012, 4, 351–365 CAS.
- G. Charras and E. Sahai, Physical influences of the extracellular environment on cell migration, Nat. Rev. Mol. Cell Biol., 2014, 15, 813–824 CrossRef CAS PubMed.
- M. Tenje, F. Cantoni, A. M. Porras Hernández, S. S. Searle, S. Johansson, L. Barbe, M. Antfolk and H. Pohlit, A practical guide to microfabrication and patterning of hydrogels for biomimetic cell culture scaffolds, Organs-on-a-Chip, 2020, 2, 100003 CrossRef.
- A. D. Doyle, N. Carvajal, A. Jin, K. Matsumoto and K. M. Yamada, Local 3D matrix microenvironment regulates cell migration through spatiotemporal dynamics of contractility-dependent adhesions, Nat. Commun., 2015, 6, 8720 CrossRef CAS PubMed.
- T. G. Vladkova, Surface Engineered Polymeric Biomaterials with Improved Biocontact Properties, Int. J. Polym. Sci., 2010, 2010, 1–22 CrossRef.
- A. Leal-Egaña, A. Díaz-Cuenca and A. R. Boccaccini, Tuning of cell-biomaterial anchorage for tissue regeneration, Adv. Mater., 2013, 25, 4049–4057 CrossRef PubMed.
- M. Guvendiren and J. A. Burdick, Stiffening hydrogels to probe short- and long-term cellular responses to dynamic mechanics, Nat. Commun., 2012, 3, 792 CrossRef PubMed.
- X. Tong and F. Yang, Engineering interpenetrating network hydrogels as biomimetic cell niche with independently tunable biochemical and mechanical properties, Biomaterials, 2014, 35, 1807–1815 CrossRef CAS.
- J. Yang, Y. Li, Y. Liu, D. Li, L. Zhang, Q. Wang, Y. Xiao and X. Zhang, Influence of hydrogel network microstructures on mesenchymal stem cell chondrogenesis in vitro and in vivo, Acta Biomater., 2019, 91, 159–172 CrossRef CAS PubMed.
- S. R. Caliari and J. A. Burdick, A practical guide to hydrogels for cell culture, Nat. Methods, 2016, 13, 405–414 CrossRef CAS PubMed.
- F. Akther, P. Little, Z. Li, N.-T. Nguyen and H. T. Ta, Hydrogels as artificial matrices for cell seeding in microfluidic devices, RSC Adv., 2020, 10, 43682–43703 RSC.
- Y. Liu, S. Rayatpisheh, S. Y. Chew and M. B. Chan-Park, Impact of endothelial cells on 3D cultured smooth muscle cells in a biomimetic hydrogel, ACS Appl. Mater. Interfaces, 2012, 4, 1378–1387 CrossRef CAS.
- H. Liu, Y. Wang, K. Cui, Y. Guo, X. Zhang and J. Qin, Advances in Hydrogels in Organoids and Organs-on-a-Chip, Adv. Mater., 2019, 31, e1902042 CrossRef.
- V. Dhote, S. Skaalure, U. Akalp, J. Roberts, S. J. Bryant and F. J. Vernerey, On the role of hydrogel structure and degradation in controlling the transport of cell-secreted matrix molecules for engineered cartilage, J. Mech. Behav. Biomed. Mater., 2013, 19, 61–74 CrossRef CAS PubMed.
- G. D. Nicodemus and S. J. Bryant, Cell encapsulation in biodegradable hydrogels for tissue engineering applications, Tissue Eng., Part B, 2008, 14, 149–165 CrossRef CAS PubMed.
- T. A. E. Ahmed, E. V. Dare and M. Hincke, Fibrin: a versatile scaffold for tissue engineering applications, Tissue Eng., Part B, 2008, 14, 199–215 CrossRef CAS PubMed.
- S. H. Bhang, O. Jeon, C. Y. Choi, Y. H. K. Kwon and B.-S. Kim, Controlled release of nerve growth factor from fibrin gel, J. Biomed. Mater. Res., 2007, 80, 998–1002 CrossRef PubMed.
- B. Bujoli, J.-C. Scimeca and E. Verron, Fibrin as a Multipurpose Physiological Platform for Bone Tissue Engineering and Targeted Delivery of Bioactive Compounds, Pharmaceutics, 2019, 11, 556 CrossRef CAS.
- J. Wedgwood, A. J. Freemont and N. Tirelli, Rheological and Turbidity Study of Fibrin Hydrogels, Macromol. Symp., 2013, 334, 117–125 CrossRef CAS.
- A. C. Brown and T. H. Barker, Fibrin-based biomaterials: modulation of macroscopic properties through rational design at the molecular level, Acta Biomater., 2014, 10, 1502–1514 CrossRef CAS.
- Y. Li, H. Meng, Y. Liu and B. P. Lee, Fibrin gel as an injectable biodegradable scaffold and cell carrier for tissue engineering, Sci. World J., 2015, 2015, 685690 Search PubMed.
- R. A. Shirwaiker, M. F. Purser and R. A. Wysk, in Rapid Prototyping of Biomaterials, Elsevier, 2014, pp. 176–200 Search PubMed.
- L. Gasperini, J. F. Mano and R. L. Reis, Natural polymers for the microencapsulation of cells, J. R. Soc., Interface, 2014, 11, 20140817 CrossRef PubMed.
- X. Zhao, S. A. Irvine, A. Agrawal, Y. Cao, P. Q. Lim, S. Y. Tan and S. S. Venkatraman, 3D patterned substrates for bioartificial blood vessels - The effect of hydrogels on aligned cells on a biomaterial surface, Acta Biomater., 2015, 26, 159–168 CrossRef CAS PubMed.
- A. P. Golden and J. Tien, Fabrication of microfluidic hydrogels using molded gelatin as a sacrificial element, Lab Chip, 2007, 7, 720–725 RSC.
- M. Meyer and B. Morgenstern, Characterization of gelatine and acid soluble collagen by size exclusion chromatography coupled with multi angle light scattering (SEC-MALS), Biomacromolecules, 2003, 4, 1727–1732 CrossRef CAS PubMed.
- C. Joly-Duhamel, D. Hellio and M. Djabourov, All Gelatin Networks: 1. Biodiversity and Physical Chemistry, Langmuir, 2002, 18, 7208–7217 CrossRef CAS.
- J. Schöneberg, F. de Lorenzi, B. Theek, A. Blaeser, D. Rommel, A. J. C. Kuehne, F. Kießling and H. Fischer, Engineering biofunctional in vitro vessel models using a multilayer bioprinting technique, Sci. Rep., 2018, 8, 10430 CrossRef PubMed.
- B. Crawshaw, D. Z. Herrick, W. Gao, E. P. Maziarz and X. M. Liu, in Chemicals and Materials from Renewable Resources, ed. J. J. Bozell, American Chemical Society, Washington, DC, 2001, pp. 51–74 Search PubMed.
- D. B. Kolesky, K. A. Homan, M. A. Skylar-Scott and J. A. Lewis, Three-dimensional bioprinting of thick vascularized tissues, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3179–3184 CrossRef CAS PubMed.
- A. Bettadapur, G. C. Suh, N. A. Geisse, E. R. Wang, C. Hua, H. A. Huber, A. A. Viscio, J. Y. Kim, J. B. Strickland and M. L. McCain, Prolonged Culture of Aligned Skeletal Myotubes on Micromolded Gelatin Hydrogels, Sci. Rep., 2016, 6, 28855 CrossRef CAS PubMed.
- A. B. Bello, D. Kim, D. Kim, H. Park and S.-H. Lee, Engineering and Functionalization of Gelatin Biomaterials: From Cell Culture to Medical Applications, Tissue Eng., Part B, 2020, 26, 164–180 CrossRef CAS.
- Y. Liu, R. Weng, W. Wang, X. Wei, J. Li, X. Chen, Y. Liu, F. Lu and Y. Li, Tunable physical and mechanical properties of gelatin hydrogel after transglutaminase crosslinking on two gelatin types, Int. J. Biol. Macromol., 2020, 162, 405–413 CrossRef CAS.
- G. Yang, Z. Xiao, X. Ren, H. Long, H. Qian, K. Ma and Y. Guo, Enzymatically crosslinked gelatin hydrogel promotes the proliferation of adipose tissue-derived stromal cells, PeerJ, 2016, 4, e2497 CrossRef PubMed.
- M. Savoca, E. Tonoli, A. Atobatele and E. Verderio, Biocatalysis by Transglutaminases: A Review of Biotechnological Applications, Micromachines, 2018, 9, 562 CrossRef PubMed.
- K. Yokoyama, N. Nio and Y. Kikuchi, Properties and applications of microbial transglutaminase, Appl. Microbiol. Biotechnol., 2004, 64, 447–454 CrossRef CAS PubMed.
- R. I. Litvinov and J. W. Weisel, Fibrin mechanical properties and their structural origins, Matrix Biol., 2017, 60–61, 110–123 CrossRef CAS.
- E. P. Sproul, R. T. Hannan and A. C. Brown, in Biomaterials for Tissue Engineering, ed. K. Chawla, Springer New York, New York, NY, 2018, pp. 85–99 Search PubMed.
- A. Trengove, S. Duchi, C. Onofrillo, C. D. O'Connell, C. Di Bella and A. J. O'Connor, Microbial Transglutaminase Improves ex vivo Adhesion of Gelatin Methacryloyl Hydrogels to Human Cartilage, Front. Med. Technol., 2021, 3, 773673 CrossRef.
- M. Gomez-Florit, A. Pardo, R. M. A. Domingues, A. L. Graça, P. S. Babo, R. L. Reis and M. E. Gomes, Natural-Based Hydrogels for Tissue Engineering Applications, Molecules, 2020, 25, 5858 CrossRef CAS.
- S. L. Rowe, S. Lee and J. P. Stegemann, Influence of thrombin concentration on the mechanical and morphological properties of cell-seeded fibrin hydrogels, Acta Biomater., 2007, 3, 59–67 CrossRef CAS PubMed.
- C. M. A. P. Schuh, A. G. E. Day, H. Redl and J. Phillips, An Optimized Collagen-Fibrin Blend Engineered Neural Tissue Promotes Peripheral Nerve Repair, Tissue Eng., Part A, 2018, 24, 1332–1340 CrossRef CAS PubMed.
- V. K. Lai, S. P. Lake, C. R. Frey, R. T. Tranquillo and V. H. Barocas, Mechanical behavior of collagen-fibrin co-gels reflects transition from series to parallel interactions with increasing collagen content, J. Biomech. Eng., 2012, 134, 11004 CrossRef PubMed.
- A. Maleki, A.-L. Kjøniksen and B. Nyström, Characterization of the chemical degradation of hyaluronic acid during chemical gelation in the presence of different cross-linker agents, Carbohydr. Res., 2007, 342, 2776–2792 CrossRef CAS.
- K. M. Lorentz, S. Kontos, P. Frey and J. A. Hubbell, Engineered aprotinin for improved stability of fibrin biomaterials, Biomaterials, 2011, 32, 430–438 CrossRef CAS PubMed.
- R. Picetti, H. Shakur-Still, R. L. Medcalf, J. F. Standing and I. Roberts, What concentration of tranexamic acid is needed to inhibit fibrinolysis? A systematic review of pharmacodynamics studies, Blood Coagulation Fibrinolysis, 2019, 30, 1–10 CrossRef CAS PubMed.
- S. Jockenhoevel and T. C. Flanagan, in Tissue Engineering for Tissue and Organ Regeneration, ed. D. Eberli, InTech, 2011 Search PubMed.
- D. K. Jarrell, E. J. Vanderslice, M. L. Lennon, A. C. Lyons, M. C. VeDepo and J. G. Jacot, Increasing salinity of fibrinogen solvent generates stable fibrin hydrogels for cell delivery or tissue engineering, PLoS One, 2021, 16, e0239242 CrossRef CAS.
- E. Potier, J. Noailly, C. M. Sprecher and K. Ito, Influencing biophysical properties of fibrin with buffer solutions, J. Mater. Sci., 2010, 45, 2494–2503 CrossRef CAS.
- D. Eyrich, F. Brandl, B. Appel, H. Wiese, G. Maier, M. Wenzel, R. Staudenmaier, A. Goepferich and T. Blunk, Long-term stable fibrin gels for cartilage engineering, Biomaterials, 2007, 28, 55–65 CrossRef CAS PubMed.
- B. Sarker, R. Singh, R. Silva, J. A. Roether, J. Kaschta, R. Detsch, D. W. Schubert, I. Cicha and A. R. Boccaccini, Evaluation of fibroblasts adhesion and proliferation on alginate-gelatin crosslinked hydrogel, PLoS One, 2014, 9, e107952 CrossRef PubMed.
- S. Chuaychan, S. Benjakul and H. Kishimura, Characteristics and Gelling Property of Gelatin from Scale of Spotted Golden Goatfish (Parupeneus heptacanthus), J. Food Process. Preserv., 2017, 41, e13139 CrossRef.
- J. Eysturskarð, I. J. Haug, N. Elharfaoui, M. Djabourov and K. I. Draget, Structural and mechanical properties of fish gelatin as a function of extraction conditions, Food Hydrocolloids, 2009, 23, 1702–1711 CrossRef.
- M. L. Oyen, Mechanical characterisation of hydrogel materials, Int. Mater. Rev., 2014, 59, 44–59 CrossRef CAS.
- T. T. Tower, M. R. Neidert and R. T. Tranquillo, Fiber alignment imaging during mechanical testing of soft tissues, Ann. Biomed. Eng., 2002, 30, 1221–1233 CrossRef PubMed.
- T. Q. Bui, V. D. Cao, N. B. D. Do, T. E. Christoffersen, W. Wang and A.-L. Kjøniksen, Salinity Gradient Energy from Expansion and Contraction of Poly(allylamine hydrochloride) Hydrogels, ACS Appl. Mater. Interfaces, 2018, 10, 22218–22225 CrossRef CAS PubMed.
- P. A. Janmey, J. P. Winer and J. W. Weisel, Fibrin gels and their clinical and bioengineering applications, J. R. Soc., Interface, 2009, 6, 1–10 CrossRef CAS PubMed.
- S. Eliyahu, A. Galitsky, E. Ritov and H. Bianco-Peled, Hybrid Acrylated Chitosan and Thiolated Pectin Cross-Linked Hydrogels with Tunable Properties, Polymers, 2021, 13 DOI:10.3390/polym13020266.
- N. J. Kaiser, R. J. Kant, A. J. Minor and K. L. K. Coulombe, Optimizing Blended Collagen-Fibrin Hydrogels for Cardiac Tissue Engineering with Human iPSC-derived Cardiomyocytes, ACS Biomater. Sci. Eng., 2019, 5, 887–899 CrossRef CAS PubMed.
- G.-Z. Jin and H.-W. Kim, Effects of Type I Collagen Concentration in Hydrogel on the Growth and Phenotypic Expression of Rat Chondrocytes, Tissue Eng. Regener. Med., 2017, 14, 383–391 CrossRef CAS PubMed.
- A. Krüger-Genge, S. Hauser, A. T. Neffe, Y. Liu, A. Lendlein, J. Pietzsch and F. Jung, Response of Endothelial Cells to Gelatin-Based Hydrogels, ACS Biomater. Sci. Eng., 2021, 7, 527–540 CrossRef PubMed.
- H. Liu, M. Wu, Y. Jia, L. Niu, G. Huang and F. Xu, Control of fibroblast shape in sequentially formed 3D hybrid hydrogels regulates cellular responses to microenvironmental cues, NPG Asia Mater., 2020, 12 DOI:10.1038/s41427-020-0226-7.
- D. Lam, H. A. Enright, S. K. G. Peters, M. L. Moya, D. A. Soscia, J. Cadena, J. A. Alvarado, K. S. Kulp, E. K. Wheeler and N. O. Fischer, Optimizing cell encapsulation condition in ECM-Collagen I hydrogels to support 3D neuronal cultures, J. Neurosci. Methods, 2020, 329, 108460 CrossRef PubMed.
- K. A. Ahmann, J. S. Weinbaum, S. L. Johnson and R. T. Tranquillo, Fibrin degradation enhances vascular smooth muscle cell proliferation and matrix deposition in fibrin-based tissue constructs fabricated in vitro, Tissue Eng., Part A, 2010, 16, 3261–3270 CrossRef CAS PubMed.
- B. Trappmann, B. M. Baker, W. J. Polacheck, C. K. Choi, J. A. Burdick and C. S. Chen, Matrix degradability controls multicellularity of 3D cell migration, Nat. Commun., 2017, 8, 371 CrossRef PubMed.
- A. Collen, R. Hanemaaijer, F. Lupu, P. H. A. Quax, N. van Lent, J. Grimbergen, E. Peters, P. Koolwijk and V. W. M. van Hinsbergh, Membrane-type matrix metalloproteinase-mediated angiogenesis in a fibrin-collagen matrix, Blood, 2003, 101, 1810–1817 CrossRef CAS PubMed.
- L. T. Vu, G. Jain, B. D. Veres and P. Rajagopalan, Cell migration on planar and three-dimensional matrices: a hydrogel-based perspective, Tissue Eng., Part B, 2015, 21, 67–74 CrossRef CAS PubMed.
- S. Zippusch, F. Helms, S. Lau, M. Klingenberg, C. Schrimpf, A. Haverich, M. Wilhelmi and U. Böer, Perfusion promotes endothelialized pore formation in high concentration fibrin gels otherwise unsuitable for tube development, Int. J. Artif. Organs, 2021, 44, 130–138 CrossRef CAS PubMed.
- X. Li, Y. Dai, T. Shen and C. Gao, Induced migration of endothelial cells into 3D scaffolds by chemoattractants secreted by pro-inflammatory macrophages in situ, Regener. Biomater., 2017, 4, 139–148 CrossRef CAS PubMed.
- A. Bertz, S. Wöhl-Bruhn, S. Miethe, B. Tiersch, J. Koetz, M. Hust, H. Bunjes and H. Menzel, Encapsulation of proteins in hydrogel carrier systems for controlled drug delivery: Influence of network structure and drug size on release rate, J. Biotechnol., 2013, 163, 243–249 CrossRef CAS PubMed.
- H. Muri, L. Hoang and D. Hjelme, Mapping Nanoparticles in Hydrogels: A Comparison of Preparation Methods for Electron Microscopy, Appl. Sci., 2018, 8, 2446 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm00214k |
| This journal is © The Royal Society of Chemistry 2022 |