
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electroconductive and injectable hydrogels based on gelatin and PEDOT:PSS for a minimally invasive approach in nervous tissue regeneration†
Franco
Furlani
 *a,
Margherita
Montanari
a,
Nicola
Sangiorgi
a,
Emanuela
Saracino
b,
Elisabetta
Campodoni
a,
Alessandra
Sanson
a,
Valentina
Benfenati
b,
Anna
Tampieri
a,
Silvia
Panseri
a and
Monica
Sandri
*a
*a,
Margherita
Montanari
a,
Nicola
Sangiorgi
a,
Emanuela
Saracino
b,
Elisabetta
Campodoni
a,
Alessandra
Sanson
a,
Valentina
Benfenati
b,
Anna
Tampieri
a,
Silvia
Panseri
a and
Monica
Sandri
*a
aNational Research Council of Italy – Institute of Science and Technology for Ceramics, (ISTEC-CNR), Via Granarolo 64, I – 48018, Faenza, RA, Italy. E-mail: franco.furlani@istec.cnr.it; monica.sandri@istec.cnr.it; Tel: +39 0546 699 776 Tel: +39 0546 699 761
bNational Research Council of Italy – Institute of Organic Synthesis and Photoreactivity (ISOF-CNR), via Gobetti, 101, I – 40129, Bologna, Italy
First published on 7th March 2022
Abstract
This work describes the development of electroconductive hydrogels as injectable matrices for neural tissue regeneration by exploiting a biocompatible conductive polymer – poly(3,4-ethylenedioxythiophene)-poly(styrenesulfonate) (PEDOT:PSS) – combined with a biomimetic polymer network made of gelatin. Our approach involved also genipin – a natural cross-linking agent – to promote gelation of gelatin networks embedding PEDOT:PSS. The achieved results suggest that physical–chemical properties of the resulting hydrogels, like impedance, gelation time, mechanical properties, swelling and degradation in physiological conditions, can be finely tuned by the amount of PEDOT:PSS and genipin used in the formulation. Furthermore, the presence of PEDOT:PSS (i) enhances the electrical conductivity, (ii) improves the shear modulus of the resulting hydrogels though (iii) partially impairing their resistance to shear deformation, (iv) reduces gelation time and (v) reduces their swelling ability in physiological medium. Additionally, the resulting electroconductive hydrogels demonstrate enhanced adhesion and growth of primary rat cortical astrocytes. Given the permissive interaction of hydrogels with primary astrocytes, the presented biomimetic, electroconductive and injectable hydrogels display potential applications as minimally invasive systems for neurological therapies and damaged brain tissue repair.
1. Introduction
Hydrogels are highly hydrated networks and are widely used for different applications, spanning from biomedicine to soft electronics.1–3 Some of the most advanced and studied applications of hydrogels deal with the biomedical field, as injectable sensors and scaffolds for regenerative medicine.1,2,4–6 In detail, regenerative medicine approaches require suitable networks able to mimic natural tissue properties, like biochemical milieu, spatial composition, and mechanical performance. Ideally, mimicking the biochemical and biomechanical behavior of the natural extra-cellular matrix (ECM) is a robust strategy for biomaterial development.2,7 Naturally occurring biopolymers are currently exploited to fabricate ECM mimics, since they display most of the abovementioned properties. In this scenario, collagen and its derivatives, e.g. gelatin, represent very attractive biopolymers for this purpose.8–12 Collagen is a highly structured biopolymer present in all human (and animal) tissues, spanning from connective to nervous tissue.13 Unfortunately, collagen is expensive and is difficult to handle since it is insoluble in physiological conditions of pH, temperature and osmolarity. On the other hand, gelatin can be produced by partial degradation of collagen from food waste (e.g. pig skin). Gelatin is a less expensive material which is easy to handle, store and sterilize and can be combined with other functional materials in order to form different structures including hydrogels.Recently, great effort has been devoted to developing the electroconductive biomaterials suitable for the regeneration of electroconductive tissues, like nervous and muscular tissues.14–19 In detail, conductive materials have emerged which are able to enhance cell adhesion, cell growth, differentiation of neural stem cells towards neurons and astrocytes, and formation of neuronal networks.20–23 Indeed, nervous tissue is frequently damaged by trauma and diseases and displays poor self-healing ability.9,24 Thus, different conductive polymers, i.e. polymers able to conduct electrons, among which is poly(3,4-ethylenedioxythiophene) (PEDOT), poly-pyrrole, and poly-aniline, were extensively studied.14,25–29 These polymers can be exploited to fabricate mainly films.30,31 Indeed, these polymers were combined with different materials in order to design hybrid conductive biomaterials for regenerative medicine for electrically conductive tissues.14,18,19,32–41 In this context, the permissive interaction of the biomaterials with glial cells, and in particular with astrocytes, has been recently in the spotlight as a strategy to reduce the inflammatory gliotic reaction induced by brain implants, which critically determine the therapeutic outcome.42,43
Herein we report an original method to form electroconductive biomaterials in the form of hydrogels that can be injected into the lesion with a minimally invasive approach and with a controlled and tunable gelation timing. The hydrogels are based on gelatin and PEDOT conjugated with poly(styrenesulfonate) (PEDOT:PSS) in order to enhance its solubility and stability in water media. Reticulation is promoted by the natural crosslinker genipin without the need of any chemical modification of gelatin, e.g. methacrylation, and of UV-curing. Biocompatibility toward primary rat neocortical astrocytes and performance of this set of materials is reported, confirming the potential that hydrogels could play a role in electroconductive tissue regeneration, especially in neural regeneration.
2. Materials and methods
2.1 Materials
Food-grade gelatin from pig skin, Bloom 280, was purchased from Italgelatine S.p.A. (Italy). Genipin was purchased from Wako Chemicals (U.S.A.). Poly(3,4-ethylenedioxythiophene)-poly(styrenesulfonate) (PEDOT:PSS), 1.1% in H2O, neutral pH, high-conductivity grade, was purchased from Sigma Aldrich (U.S.A.). Sodium azide, ethanol, paraformaldehyde, and Dulbecco's phosphate buffered saline (DPBS) modified, without calcium chloride and magnesium chloride, liquid, sterile-filtered, suitable for cell culture were purchased from Sigma Aldrich (U.S.A.). All reagents and chemicals were of high-purity grade. DAPI (4′,6-diamidino-2-phenylindole, dihydrochloride) reagent was from ThermoFisher Scientific (Waltham, MA, US).2.2 Preparation of gelatin and gelatin/PEDOT:PSS hydrogels
A typical gelatin-based hydrogel was prepared as follows: (i) deionized water was added to gelatin powder and (ii) gelatin was solubilized by heating up to 45 °C by employing a magnetic stirrer equipped with a heating plate and temperature probe; then (iii) genipin solution 10 mg mL−1 in deionized water, sonicated for 30 minutes prior its use, was added and finally (iv) 10 mL of the resulting mixture was transferred in a Petri dish (diameter = 60 mm) and incubated at 37 °C for 24 h (Scheme 1). With the aim of finding a sample able to gel in physiological conditions, different compositions were investigated, in particular with gelatin ranging from 5 to 10% (w/w respect to the total weight of the sample) and genipin from 0.02 to 1% (w/w respect to gelatin) (Table S1†). | ||
| Scheme 1 Schematic representation of gelatin-based hydrogels synthesis. (A) Preparation of genipin crosslinked gelatin hydrogels. (B) Preparation of hydrogels based on gelatin and PEDOT:PSS by using genipin as crosslinking agent. | ||
In the case of electroconductive hydrogels, PEDOT:PSS solution was added to gelatin solution, in suitable amount to achieve ratios ranging from 0 to 0.3% (w/w respect to the total weight of the sample), prior to genipin addition. Furthermore, the amount of deionized water was adjusted in order to fabricate hydrogels with the same concentration (w/w) of gelatin. The final composition of hydrogels was gelatin 10% (w/w respect to the total weight of the sample), genipin 1% (w/w respect to gelatin) and PEDOT:PSS equal to 0, 0.1 and 0.3% (w/w respect to the total weight of the sample). These PEDOT:PSS concentrations were selected since the resulting mixture showed high viscosity and PEDOT:PSS concentrations higher than 0.3% was difficult to handle. Indeed, higher concentrations of PEDOT:PSS entail reduced gelation times. Additionally, PEDOT:PSS concentrations within this range were previously reported to affect cell behavior.18,19
2.3 Rheological characterization
Rheological measurements were performed using a Bohlin C-VOR 120 rotational rheometer equipped with a thermostatic unit (KTB 30). Rheological tests were performed in oscillatory shear conditions on crosslinked hydrogels (after incubation for 24 h at 37 °C). After sample preparation (according to section 2.2), the resulting hydrogel (thickness = 2.7 mm) was then punched with a 20 mm diameter punch before being transferred on to the rheometer plate. The mechanical spectra (frequency sweep, stress (τ) = 5 Pa, well within the linear viscoelasticity range and in the frequency (ν) range 0.01–10 Hz) and the extension of the linear viscoelastic regime (stress sweep tests at ν = 1 Hz, stress range 1 < τ < 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Pa) were investigated. Tests were performed at temperature of 37 °C using a smooth parallel stainless-steel plate apparatus, diameter = 20 mm, as the measuring device and fixing the gap to 2.7 mm.
000 Pa) were investigated. Tests were performed at temperature of 37 °C using a smooth parallel stainless-steel plate apparatus, diameter = 20 mm, as the measuring device and fixing the gap to 2.7 mm.
The experimental settings used to evaluate gelation kinetics are the following: stainless steel plates with 4° cone/plate geometry, diameter = 40 mm, and gap 0.150 mm. Time sweep experiments were performed in strain-controlled conditions, with deformation, γ, of 0.01, kept constant throughout the experiment, frequency (ν) of 3 and 5 Hz and time of 6 h. Upon addition of genipin, samples were mixed under stirring for about 10 s to form uniform samples and poured on the plate. The values of storage G′ (elastic response) and loss G′′ (viscous response) moduli were recorded as a function of time. Time sweep experiments were performed at 37 °C.
In all the cases, except for stress sweep tests, silicone oil (viscosity 50 cSt, purchased from Sigma, USA) was used to seal the interface between the two plates in order to improve thermal control and limit solvent evaporation.
2.4 Dynamic mechanical analysis (DMA)
DMA was performed on the crosslinked hydrogels. After sample preparation (according to section 2.2), the resulting hydrogel (thickness = 5.2 mm) was then punched with a 15 mm diameter punch before being transferred on to the DMA plate. Stress–strain curves were collected using dynamic mechanical analyzer DMA Q800 (TA instruments, Italy) in compressive mode. Tests were performed at 37 °C and in a vapor saturated environment to prevent solvent evaporation.2.5 Electrochemical impedance spectroscopy (EIS)
EIS measurements were performed on the crosslinked hydrogels. After sample preparation (according to section 2.2), the resulting hydrogel (thickness = 1.2 mm) was punched with a 6 mm diameter punch prior before being transferred between two gold flaps of a symmetrical electrochemical cell (Fig. S1†). The electrochemical cell was manufactured by fixing two gold flaps to a plastic bar by exploiting an insulating adhesive tape (Scheme S1†).EIS measurements were performed with an AUTOLAB PGSTAT302N-FRA32 M electrochemical workstation (Metrohm, the Netherlands) controlled by the Nova 2.1 software. EIS measurements were performed in the frequency range between 105 and 10−2 Hz, with a signal amplitude of 10 mV, with an applied potential equal to 0 V and at room temperature. The obtained Nyquist plots were fitted using the Z-View software (Scribner Associates).
This experimental setup was used both for the as-prepared hydrogels (0, 0.1 and 0.3% w/w PEDOT:PSS) and for hydrogels fabricated using PEDOT:PSS sterilized by exploiting different techniques, namely (i) autoclaving and (ii) gamma radiation at 25 kGy of both liquid and freeze-dried (and subsequently re-hydrated with the same amount of water as before hydrogel synthesis) PEDOT:PSS.
The same set of experiments were also performed on hydrogels fabricated using phosphate buffered saline as solvent.
2.6 Swelling and degradation tests
Swelling and degradation tests were performed on the crosslinked hydrogels. After sample preparation (according to section 2.2), the resulting hydrogel was then punched with a 6 and a 20 mm diameter punch. The resulting hydrogels (diameter = 6 mm, thickness = 2.4 mm) were transferred into wells of a 24-well plate containing 2 mL of DPBS supplemented with 0.1% w/v of sodium azide (to avoid mold and bacteria contamination). The resulting multi-well plate was then placed at T = 37 °C under shaking. At selected time points – specifically 1, 3, 7, 14 and 21 days – hydrogels were removed from wells and weighed in order to evaluate hydrogel swelling. Data are reported as % of mass gained with respect to the initial weight of three samples (± standard deviation, SD), calculated as:where W0 is the hydrogels weight at time 0 and Wt is the weight of the same at a selected time.

At the abovementioned selected time points, hydrogels (diameter = 20 mm, thickness = 2.4 mm) were also freeze-dried and weighed in order to investigate hydrogel degradation. In the case of degradation tests, hydrogels were transferred into wells of a 6-well plate containing 6 mL of DPBS supplemented with 0.1% w/v of sodium azide. Data are reported as % of mass lost with respect to the initial weight of three samples (± standard deviation, SD), calculated as:
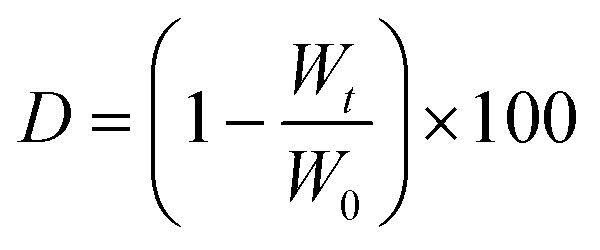
2.7 Fourier transform infra-red spectroscopy (FT-IR)
Fourier transform infrared (FT-IR) spectra of the samples were measured with a Nicolet 5700 spectrometer (Thermo Fisher Scientific Inc., Waltham, MA, USA) in ATR mode using an ATR iD7 accessory. Instrumental resolution was set up to 4 cm−1, and 16 scans were collected per sample in the range spanning from 4000 to 400 cm−1.Freeze-dried hydrogels with different compositions and their singular components (i.e. PEDOT:PSS and gelatin) were considered. The same set of analyses were performed on freeze-dried hydrogels used for the degradation tests (described in section 2.6) and collected at different timeframes after incubation in PBS.
2.8 UV-visible spectroscopy
UV-visible spectrophotometric analyses were performed on supernatants collected during hydrogel degradation tests (described in section 2.6) and collected at different timeframes after incubation in PBS. Samples (2 mL) were transferred in disposable plastic cuvettes and analyzed at 450 nm with a PerkinElmer Lambda 750 double beam spectrophotometer. The supernatant collected from the sample without PEDOT:PSS was used as the blank.2.9 Sample processing, cell culturing and viability testing
All single hydrogel components, i.e. gelatin powder, genipin powder and freeze-dried PEDOT:PSS, were sterilized by gamma irradiation at 25 kGy. Samples were prepared according to section 2.2 in sterile conditions. In this case, after mixing the components 1 mL of the mixture was cast in order to cover the area of each of the 24 wells (diameter = 20 mm, Thermo Fisher multiwell). Electroconductive hydrogels were fabricated with PEDOT:PSS final concentration equal to 0.3% w/w respect to the total weight of the sample. Once cast, the samples were maintained in a thermostable and humified cell incubator at 37 °C for 24 h. Then, the hydrogels were overnight conditioned with 1.5 ml of PBS for each well before cell plating.2.10 Cells staining
Astrocytes plated on hydrogels after 2 days were washed with phosphate-buffered saline for 5 minutes, fixed with paraformaldehyde 4% (w/v) for 2 h and washed with PBS for 5 min. Cells were then incubated in Triton X-100 0.1% (v/v) in PBS for 5 min to promote cell permeabilization. Cells were washed with PBS for 5 min and incubated with DAPI 300 nM in PBS for 5 min. Cell nuclei were visualized by an inverted fluorescence microscope Eclipse TS 100 (Nikon, Japan).2.11 Environmental scanning electron microscopy (ESEM)
Astrocytes plated on hydrogels after 2 days were washed with phosphate-buffered saline (PBS) for 5 min and fixed with 4% (w/v) paraformaldehyde for 15 min. Samples were dehydrated using an alcohol scale. Specifically, samples were incubated with progressively higher ethanol concentrations (i.e. 30%, 50%, 70%, 90% and 100% v/v), each of them twice and for 10 minutes, then were left under the hood overnight. Samples were mounted on aluminum stubs with carbon tape and gold sputtered by a Polaron sputter coater E5100 (Polaron Equipment, Watford, Hertfordshire, UK). Samples were analyzed with an environmental scanning electron microscope (ESEM, Quanta 600 FEG, FEI Company, Hillsboro, OR).2.12 Statistical analysis
Statistical analysis and graph elaboration were performed using GraphPad Prism 9.0.0 (GraphPad Software, San Diego, CA). Unpaired Student's t test was performed to evaluate differences between two groups. Differences were considered significant for p-values less than 0.05.3. Results and discussion
3.1 Genipin-assisted gelation of gelatin and gelatin-PEDOT:PSS
In order to promote gelatin gelation in a suitable time at physiological temperature, genipin was exploited as the crosslinker. Genipin is a naturally occurring crosslinking agent able to bind primary amine groups of a large variety of biomolecules and behaves in a temperature and time dependent manner, as previously reported for other polymers.47–51 Genipin was mixed with a gelatin solution, forming a colorless liquid mixture. Upon incubation at the physiological temperature of 37 °C, for 24 h, genipin promoted the formation of a dark blue hydrogel (Scheme 1A, Fig. 1A and S2†). The addition of PEDOT:PSS to the mixture did not impair the crosslinking process, allowing instead the interaction between the conductive polymer and the guanidinium groups of the gelatin backbone, providing the formation of a homogeneous black liquid mixture.52 The conductive polymer was embedded within the gelatin network and the final hydrogel displayed a black/dark blue color (Scheme 1B, Fig. 1A and S2†). Comparable black hydrogels were previously reported by Annabi and co-workers for hydrogels based on UV-crosslinked methacryloyl gelatin and PEDOT:PSS.18,19 Nevertheless, UV-crosslinking usually displays low biocompatibility. Conversely, the approach reported by the present work is suitable to devise injectable hydrogels and does not require additional steps of gelatin methcrylation and of UV-curing. | ||
| Fig. 1 (A) Upside-down bottles for a visual analysis of gelatin-based matrices at different timeframes, i.e. immediately after genipin addition (time 0) and after 1, 3, 6 and 24 h. (B) Dependence of the loss tangents, on time at two different frequencies, i.e. 5 Hz (red dots) and 3 Hz (blue dots), for the sample in the presence of PEDOT:PSS 0.3% (w/w) in time oscillatory sweep tests by rheology. (C) Dependence of the gelation time on the concentration of PEDOT:PSS (%, w/w) in time oscillatory sweep tests by rheology. (D) Dependence of storage modulus (G′) on applied deformation, γ, for gelatin-based hydrogels with different amounts of PEDOT:PSS [PEDOT:PSS] = 0% (w/w) (red), [PEDOT:PSS] = 0.1% (w/w) (blue), and [PEDOT:PSS] = 0.3% (w/w) (green) by rheology. Solid lines are the best fit of experimental points according to eqn (4). (E) Dependence of the critical strain, γC, on the amount of PEDOT by rheology. γC values were determined according to eqn (5). Data are reported as means ± standard deviations (SD) of at least three measurements. The dotted line is drawn to guide the eye. (F) Dependence of the work at critical strain, WG,yC, on the amount of PEDOT by rheology. Data are reported as means ± standard deviations (SD) of at least three measurements. The dotted line is drawn to guide the eye. (G) Dependence of compression stress by dynamic mechanical analysis (DMA) on applied strain for gelatin-based hydrogels with different amounts of PEDOT:PSS: [PEDOT:PSS] = 0% (w/w) (red), [PEDOT:PSS] = 0.1% (w/w) (blue), and [PEDOT:PSS] = 0.3% (w/w) (green). (H) Dependence of the Young modulus, E, on the amount of PEDOT:PSS, by dynamic mechanical analysis (DMA). Data are reported as means ± standard deviations (SD) of six measurements. The dotted line is drawn to guide the eye. All dotted lines are drawn to guide the eye. Experimental conditions for all tests: [gelatin] = 10% (w/w respect to the total weight of the sample), [genipin] = 1% (w/w respect to gelatin), and [PEDOT:PSS] = 0–0.3% (w/w respect to the total weight of the sample). | ||
The gelation process was investigated by rheological tests. In detail, the loss tangent ( ) was recorded as a function of time at different frequencies (Fig. 1B). The progressive decay of loss tangents suggested the transition from a viscous solution towards a hydrogel upon time. Gelation times were calculated as the intersection of loss tangents at different frequencies.53–55
) was recorded as a function of time at different frequencies (Fig. 1B). The progressive decay of loss tangents suggested the transition from a viscous solution towards a hydrogel upon time. Gelation times were calculated as the intersection of loss tangents at different frequencies.53–55
For the control sample, the gelation time was equal to 3 h and 20 min ± 15 min (Fig. 1C). In the presence of PEDOT:PSS a marked gelation time reduction was detected. Specifically, in the presence of a limited amount of PEDOT:PSS, i.e. with PEDOT:PSS 0.1%, gelation time was close to 1 h. By further increasing PEDOT:PSS concentration from 0.1% w/w to 0.3% w/w, gelation time further decreased to 27 ± 8 min. This behavior can be attributed to the ability of a conductive polymer to behave as a nucleation site for network assembly. Thus, the presence of a conductive polymer greatly enhanced gelatin network gelation. The final results of the process were homogeneous hydrogels based on gelatin embedding PEDOT:PSS. Similar findings, such as a reduction in the gelation time in the presence of colloids, were previously reported for fibrin hydrogels embedding magnetic nanoparticles.56 Systems with similar gelation times, i.e. within 60 min at physiological temperature, resulted to be appropriate for regenerative medicine purposes.57–59
3.2 Mechanical performance of hydrogels
In order to study the role played by PEDOT:PSS in modulating the mechanical properties of networks, rheological measurements and dynamic mechanical analysis (DMA) tests were performed on systems with different amounts of conductive polymer.Mechanical spectra (frequency sweep tests) were acquired at constant shear stress and different frequency values. Mechanical spectra (Fig. S3A†) pointed out that storage (G′) moduli were at least one order of magnitude higher than loss moduli (G′′) for at least two decades of frequency, thus suggesting the “classic” behavior of all hydrogels.60 Both G′ and G′′ experimental data of mechanical spectra were fitted by a combination of Maxwell elements, composed by a sequence of springs and dashpots in parallel, according to eqn (1) and (2):
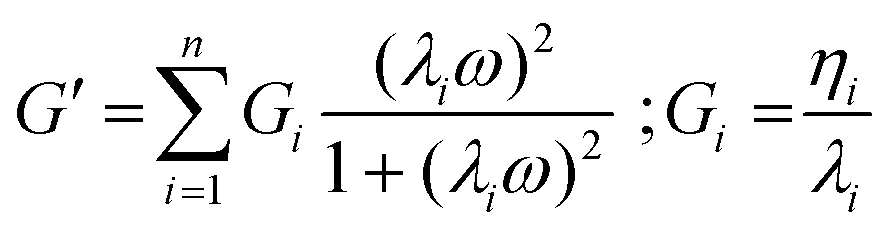 | (1) |
 | (2) |
The use of Maxwell model enabled to determine the shear modulus, G, which reflects the stiffness of hydrogels under a constant stress at small deformations, according to eqn (3):
 | (3) |
By increasing the amount of PEDOT:PSS, a progressive increase in the shear modulus G was detected (Fig. S3B†). The highest mechanical response to the shear stress was detected using the 0.3% w/w PEDOT:PSS amount. The presence of PEDOT:PSS greatly enhances the stiffness of resulting hydrogels. This behavior can be explained considering the gelation process. Indeed, PEDOT:PSS is able to strongly interact with guanidium groups of arginine residues of the gelatin.52 Consequently, during gelation, conductive polymer results physically embedded within the gelatin network (Scheme 1B).
A similar trend, i.e. an increase of shear modulus as a function of different colloid amounts embedded within hydrogels, was previously reported by other authors.56,61
Stress sweep tests were carried out at constant frequency of 1 Hz while progressively increasing the shear stress (τ), and therefore the corresponding strain (γ). Experimental data in Fig. 1D show that the elastic modulus, G′, for hydrogels with different amounts of PEDOT:PSS is independent of deformation up to at least γ = 0.5. Beyond that value (comprised within the range of γ 0.5–2 depending on PEDOT:PSS amounts), a marked decrease of G′ was detected, suggesting strain softening and hydrogel fracture for larger strain values. Experimental data were nicely fitted by the Soskey–Winter equation,62 according to eqn (4):
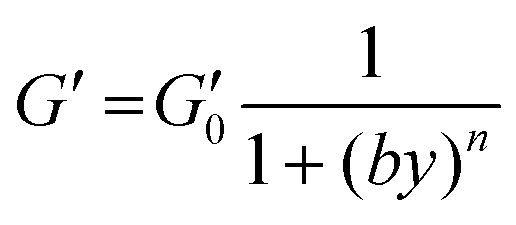 | (4) |
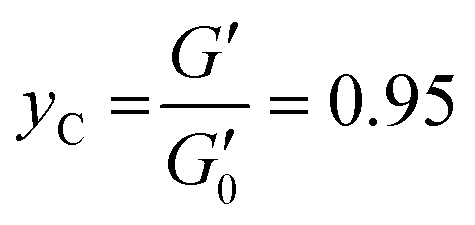 | (5) |
The trend of yCvs. PEDOT:PSS amount is reported in Fig. 1E. A progressive decrease in yC, proportional to PEDOT:PSS amount, was detected. A similar trend in the corresponding critical stress, τC, was detected (data not shown). An inverse trend of the extensibility of hydrogels as function of PEDOT:PSS amount was reported by Annabi and collaborators.19 They attributed this phenomena to electrostatic interactions between PSS and UV-crosslinked methacryloyl gelatin. The different outcome reported for UV-cured hydrogels can be attributed to the lower UV light penetration, and thus lower crosslinking density, in the presence of PEDOT.
To further investigate the strength of hydrogels made of gelatin with different PEDOT:PSS amounts, the work at yC, WG,yC, was calculated according to eqn (6):64
 | (6) |
The energy required to elicit the onset of strain softening was found to decrease upon PEDOT:PSS addition (Fig. 1F). Thus, albeit hydrogels embedding PEDOT:PSS were stiffer, a lower work, compared to hydrogels without conductive polymer, was needed to promote network breakage. A reduction of the critical strain and the energy required to elicit strain softening recently emerged as able to enhance cell adhesion and spreading.65 Thus, the presence of the conductive polymer within gelatin networks could enhance the interaction between the resulting networks and cells.
Findings about yC, τC, and WG,yC can be explained considering the gelation process (Scheme 1). Indeed, the conductive polymer was embedded within gelatin networks. The presence of PEDOT:PSS within gelatin networks partially prevented the bending and stretching of polymer chains. These phenomena entail that the presence of a conductive polymer partially limited the deformation of the resulting hydrogels. Thus, hydrogels embedding conductive polymers display a reduction of the strain necessary for the onset of strain softening, i.e. yC. These findings can be explained considering the gelation mechanism, according to which PEDOT:PSS is physically embedded within gelatin network (Scheme 1). Although the embedded conductive polymer enhanced hydrogel stiffness, it also partially impairs polymer bending and stretching, thus reducing network deformability. A reduction of yC entails a decrease of the corresponding stress, i.e. τC. Thus, a limited amount of energy, i.e. WG,yC, was needed to elicit strain softening in the presence of PEDOT:PSS.
Similar trends in the mechanical performance (taking into account yC, τC, and WG,yC) were previously reported for chitosan-based hydrogels with different chemical composition.64 These trends were explained considering the different contribution of elastic (entrapped chains) and non-elastic chains (loose ends) within hydrogels.
Stress–strain tests were then performed by dynamic mechanical analysis (DMA). In Fig. 1G are reported the compression stress values as a function of the applied strain. Young moduli (E) were determined as the angular coefficient of the stress–strain response from 0 to 10% strain. Control hydrogels displayed a Young modulus (E) equal to 11.3 ± 1.5 kPa. A similar Young modulus was previously reported by Annabi and collaborators for hydrogels based on UV-crosslinked methacryloyl gelatin suitable for myoblast encapsulation.19
The addition of PEDOT:PSS improved the stiffness of the resulting hydrogels. In detail, using a concentration of PEDOT:PSS equal to 0.3% w/w, Young modulus was increased up to 17.7 ± 1.7 kPa. Specifically, Young modulus resulted to be proportional to the amount of PEDOT:PSS (Fig. 1H). A similar trend was detected for the dependence of the shear modulus (Fig. S3B†) as a function of the amount of PEDOT:PSS. An inverse trend, i.e. a decrease in the Young modulus as a function of the PEDOT:PSS amount, was previously reported by Annabi and co-workers and by Zhang and collaborators for hydrogels based on UV-crosslinked methacryloyl gelatin and the same amounts of PEDOT:PSS.18,19,39 They attributed these findings to the lower ability of UV light penetration through the gelatin network in the presence of the conductive polymer, entailing lower crosslinks of the resulting hydrogels.
Similar Young moduli were previously reported for other electroconductive materials devised for neural tissue engineering and as sensors/actuators.32,66,67 Indeed, Young modulus of all fabricated hydrogels is in the range of the native nervous tissue (E ∼ 0.1–20 kPa).68 Furthermore, materials with similar ranges of Young moduli were reported to promote differentiation of mesenchymal stem cells towards neural and muscular lineage.69,70 Taking into account these pivotal reports, hydrogels reported in the present work can be considered as biomimetics of the mechanics of the central nervous system.
3.3 Physical–chemical characterization of hydrogels
FTIR-ATR analyses were performed to investigate the physical–chemical properties of hydrogels (Fig. 2A and Fig. S4† for the shifted spectra). Specifically, genipin-crosslinked gelatin hydrogels with different amounts of PEDOT:PSS, and their singular components, i.e. gelatin and PEDOT:PSS, were considered.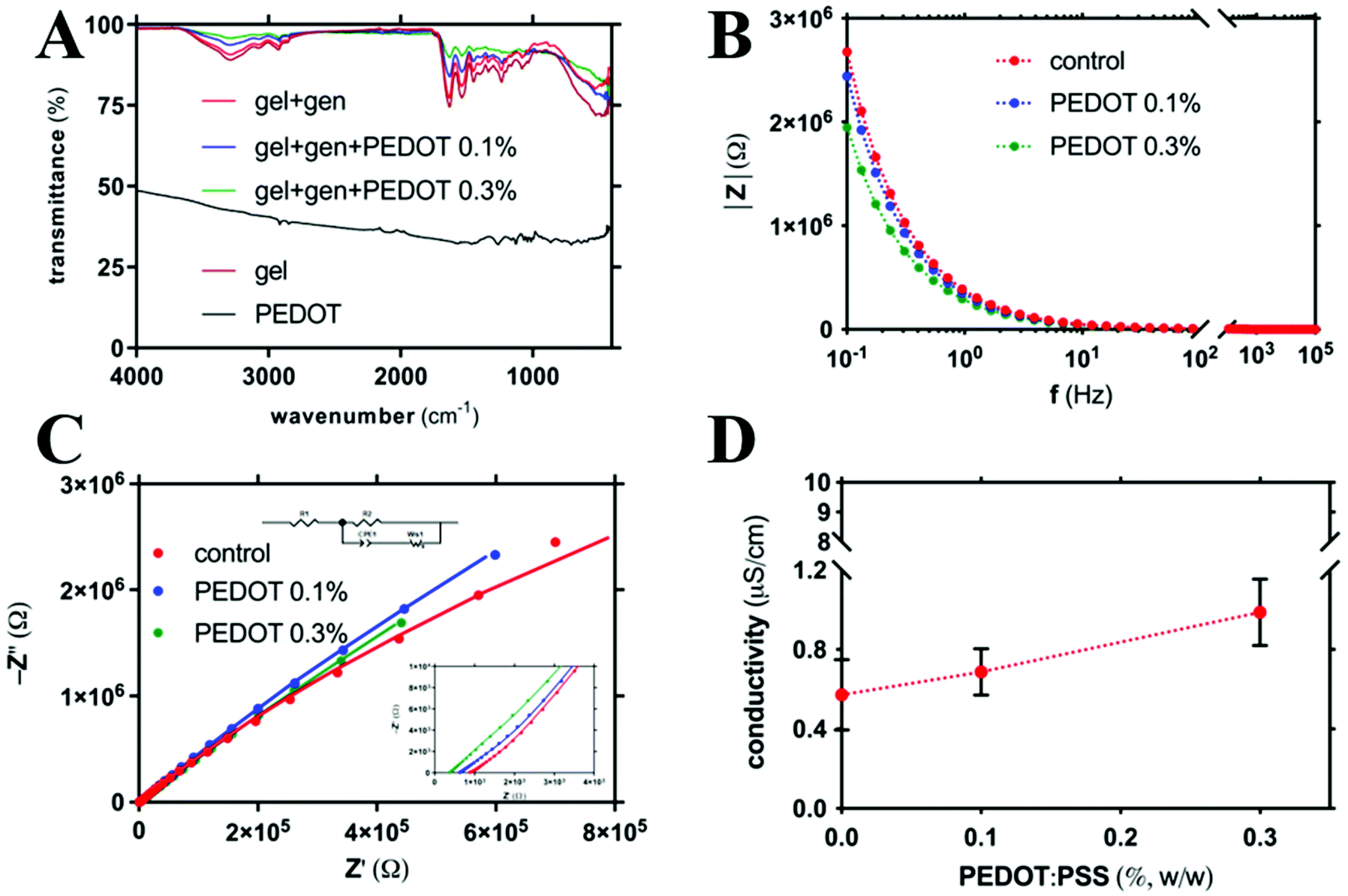 | ||
| Fig. 2 (A) FTIR-ATR of freeze-dried gelatin-based hydrogels with different amounts of PEDOT: [PEDOT:PSS] = 0% (w/w) (red), [PEDOT:PSS] = 0.1% (w/w) (blue), and [PEDOT:PSS] = 0.3% (w/w) (green). Spectra of freeze-dried gelatin (brown) and PEDOT:PSS (black) are also reported for comparison. (B) Electrochemical impedance spectroscopy measurements of gelatin-based hydrogels with different amounts of PEDOT: [PEDOT:PSS] = 0% (w/w) (red), [PEDOT:PSS] = 0.1% (w/w) (blue), and [PEDOT:PSS] = 0.3% (w/w) (green). (C) Nyquist plot for gelatin-based hydrogels with different amounts of PEDOT: [PEDOT:PSS] = 0% (w/w) (red), [PEDOT:PSS] = 0.1% (w/w) (blue), and [PEDOT:PSS] = 0.3% (w/w) (green). Colored solid lines are the best fit of experimental points. (D) Dependence of the electronic conductivity on the amount of PEDOT:PSS. Data are reported as means ± standard deviations (SD) of seven measurements. All dotted lines are drawn to guide the eye. Experimental conditions for all tests: [gelatin] = 10% (w/w respect to the total weight of the sample), [genipin] = 1% (w/w respect to gelatin), and [PEDOT:PSS] = 0–0.3% (w/w respect to the total weight of the sample). | ||
Considering the PEDOT:PSS spectra, the peak at 1064 cm−1 can be attributed to the ethylenedioxy group stretching vibration and to the S–C phenyl bonds in sulfonic acid but the peak at 1259 cm−1 to the C–O–C stretching of PEDOT.71,72 On the other hand, the peaks at 1127, 1039, and 1011 cm−1 can be attributed to the –SO3– groups of PSS.73 Specifically, the peak at 1039 cm−1 can be attributed to the –SO3– symmetric stretching.74 Other peaks, including the vibrations of the C–S bond of the thiophene ring (commonly found at 823–655 cm−1)75 and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–C stretching vibrations of the quinonoid structure of the thiophene ring (usually found at 1637 cm−1) are partially covered by broad absorption of the PEDOT:PSS network.71,72,76
C and C–C stretching vibrations of the quinonoid structure of the thiophene ring (usually found at 1637 cm−1) are partially covered by broad absorption of the PEDOT:PSS network.71,72,76
Taking into account the gelatin-based materials numerous characteristic peaks were detected.77 In all gelatin-based samples a broad peak at 3287–3289 cm−1, in the amide A region, indicates hydrogen bonding and N–H vibration of the amine group overlapped with O–H stretching vibration of hydroxyl groups.77 A peak at 2917–2921 cm−1, in the amide B region, corresponding to CH stretching and –NH3+, was also detected.78–80 Additionally, similar major bands at 1627–1634 cm−1, 1536–1538 cm−1 and 1235–1236 cm−1 were detected. These bands are amide-I (CO stretching and hydrogen bonding coupled with COO), amide-II (bending vibration of the N–H groups and stretching vibration of the C–N groups) and amide-III (vibration of the C–N and N–H groups of bound amide and vibration of the CH2 groups of glycine), respectively.71,78,79,81–83 In the presence of genipin, the shift from 1629 to 1634 in the peak was attributed to the role of CO in the formation of secondary amide formation deriving from the binding of amino groups of gelatin with the carboxymethyl groups of genipin.83–85 Indeed, genipin is able to bind primary amine groups of gelatin, as previously reported for other polymers.47–51 All samples, except the sample with the highest PEDOT:PSS concentration (i.e. in the presence of PEDOT:PSS 0.3%) displayed a prominent peak at 1080 cm−1 which can be attributed to ring C–H in-plane bending and C–O stretching of the primary alcohol groups.86 Similar results for genipin-crosslinked gelatin hydrogels were previously reported by Kasapis and collaborators.77
In the samples based on gelatin, genipin and PEDOT:PSS the amplitude of almost all the peaks decreased. When PEDOT:PSS was added into the hydrogel, the peak of gelatin at 1634 cm−1 corresponding to the C–N stretching shifted to 1629 cm−1 and this can be attributed to the interaction between the NH2 and OH side groups of gelatin and the side groups on PEDOT:PSS. In the presence of PEDOT:PSS a shift in the amide B region, from 2921 to 2917 cm−1, was detected. Additionally, in the presence of PEDOT:PSS a progressive shift of the amide-III band, from 1236 to 1233 cm−1 (with PEDOT:PSS 0.1%), and then disappearance (in the presence of PEDOT:PSS 0.3%), was detected. These shifts suggest the interaction between positively charged –NH3+ of gelatin and negatively charged –SO3– groups of PSS. Indeed, in the samples based on gelatin, genipin and PEDOT:PSS the intensities of peaks from wavenumbers ∼1000 cm−1 to ∼1400 cm−1 became flatter and peaks at 1127, 1039, and 1011 cm−1 were not detected. This scenario can be attributed to the breaking of thiophene groups and the formation of bonds with the amide groups of gelatin. Similar findings were previously reported for hydrogels based on chitosan, gelatin, agar and PEDOT:PSS.87 Thus, the FTIR spectra confirmed the encapsulation of PEDOT:PSS within the gelatin-based hydrogel.
3.4 Electroconductive properties of hydrogels
In order to study the role played by PEDOT:PSS in modulating the electroconductive properties of networks, electrochemical impedance spectroscopy (EIS) tests were performed on hydrogels with three different PEDOT:PSS concentrations, namely 0, 0.1 and 0.3% w/w.In Fig. 2B are reported the Bode plots of hydrogels with different amounts of PEDOT:PSS. Conductive properties of the resulting hydrogels were found to be proportional to the amount of PEDOT:PSS. A similar trend of impedance values as a function of PEDOT:PSS amount was previously reported by Annabi and co-workers for hydrogels based on methacrylate gelatin.18,19
EIS spectra were plotted also according to the Nyquist plot (Fig. 2C, with magnification in the high frequencies region). In order to calculate the hydrogel conductivity, Nyquist plots were fitted using the equivalent circuit reported in Fig. 2C where R1 describe the overall electrical conductivity, R2 and CPE1 describe respectively the resistance and the charge accumulation capacitance of the interface between the hydrogel pellet and the metal electrodes, and finally WS-1 describe the Warburg impedance related to ions diffusion. According to this approach, it was possible to distinguish between the electronic and ionic properties of electroconductive materials.88 Hydrogel electronic conductivity (σ) was determined according to eqn (7):
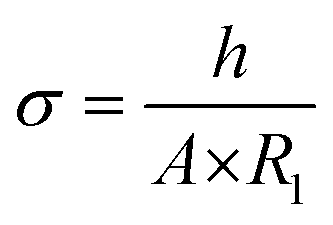 | (7) |
Electronic conductivity of hydrogels resulted to be proportional to the amount of PEDOT:PSS (Fig. 2D). Specifically, the presence of PEDOT:PSS within gelatin networks greatly enhanced the electronic conductivity of resulting hydrogels. For the highest PEDOT:PSS amount, similar conductivity values were reported by Magistris and collaborators for poly(ethylene oxide)-based polymer electrolytes.89
The ionic conductivity was similar in all samples (observed at medium-low frequencies of Nyquist plots in Fig. 2C). These findings can be explained considering that hydrogels are mainly made of deionized water (∼90%), which behaves as a good insulant medium (0.055 μS cm−1, 25 °C). Indeed, no additional ions were introduced during hydrogel fabrication and this entails that ion transport within hydrogels is partially impaired. Nevertheless, cells live in an ion-rich environment in which the ionic conductivity could not be prevented.90 In order to test this hypothesis, hydrogels were fabricated by using phosphate buffered saline as solvent. This medium was selected since it is able to mimic physiological pH and osmolarity. In this case a similar trend in the conductivity was detected (Fig. S5†). Specifically, also in the presence of physiologically relevant ions the electronic conductivity of hydrogels resulted to be proportional to the amount of PEDOT:PSS.
Hydrogels were fabricated also using PEDOT:PSS sterilized by means of different techniques. This step is necessary in order to find the best sterilization technique for the preparation of hydrogels for biological applications. Gamma radiation treatment on liquid and freeze-dried PEDOT:PSS was proved to not significantly affect the electroconductivity of resulting hydrogels contrary to autoclaving (Fig. S6†). A similar decrease in conductivity of electrophysiological devices based on PEDOT:PSS upon autoclaving was previously reported by Malliaras and co-workers.91 On the other hand, other authors reported a change in PEDOT:PSS-based material conductivity upon gamma radiation exposure.30,92 Specifically, Kim and co-workers reported that electrical-conductivity of pristine PEDOT:PSS films decreased, whereas that of ethylene diamine-treated films increased upon gamma irradiation.30 Again, Schrote and Fray reported a decrease in electroconductivity of PEDOT:PSS nanofibers after gamma radiation exposure.92 The different experimental results of the present work can be due to the different gamma radiation intensity and to the different state and chemical environment of the conductive polymer.
3.5 Hydrogel stability
In order to study the role played by PEDOT:PSS in modulating the stability of networks, swelling and degradation tests were performed on hydrogels with different amounts of the conductive polymer.After 24 h in PBS, control hydrogels were able to uptake a large amount of solvent (their weight increase of about 70%) (Fig. S7†). At later timeframes – up to 21 days – their solvent uptake slightly increased up to almost doubling their weight (increase of about 100%). A faster and higher swelling ability – up to 300% in PBS in a couple of hours – was previously reported by Rubini and collaborators for genipin crosslinked gelatin films.93 A similar swelling ability was reported by Chen and collaborators for hydrogels based on gelatin and the same amount of genipin soaked in deionized water.94 They also reported higher swelling ability by lowering the genipin amount due to the lower crosslinking density of the resulting networks.
Hydrogels embedding PEDOT:PSS displayed a reduced – and proportional to the conductive polymer concentration – swelling ability (Fig. S7†). Specifically, for hydrogels with a concentration of PEDOT:PSS equal to 0.3% w/w the swelling of the resulting hydrogels after 21 days was equal to about 50%. This behavior can be attributed to the partial hindrance of network deformation due to the conductive polymer presence, as previously discussed in the mechanics section (section 3.2). Consequently, networks embedding conductive polymers displayed a lower solvent uptake ability. A similar swelling trend was reported by Annabi and collaborators for lyophilized hydrogels (scaffolds) based on UV-crosslinked methacryloyl gelatin embedding PEDOT:PSS soaked in water.19
Hydrogels were then also used for degradation tests. Specifically, hydrogels were lyophilized after the incubation in DPBS for different timeframes to assess their stability. All hydrogels – with and without PEDOT:PSS – displayed a mass increase up to 10% after the incubation in DPBS for 1 week (Fig. S8†). The sample with the highest PEDOT:PSS concentration (in the presence of PEDOT:PSS 0.3%) showed the lowest mass increase, indeed it was also able to uptake the lowest amount of solvent (Fig. S7†). All samples displayed an increase up to 20% after 21 days in DPBS (Fig. S8†). The mass increase can be attributed to the swelling ability of hydrogels in PBS. Indeed, all hydrogels were able to uptake a large amount of DPBS – and consequently of salts like sodium chloride and phosphate ions – within the polymer network. The similar trend in mass increase of samples can be attributed to the higher ability of PBS salts to interact with PEDOT:PSS compared to non-conductive polymer networks (i.e. genipin-crosslinked gelatin networks). Specifically, albeit hydrogels fabricated in the presence of PEDOT:PSS showed a reduced ability to uptake solvent, after freeze-drying an higher amount of salts was entrapped within the networks.
FTIR-ATR analyses on the same samples used for degradation tests were performed to confirm the sample stability (Fig. S9, S10 and S11†). In all samples the amplitude of almost all the peaks slightly decreased suggesting that DPBS salts entrapped within the polymer network partially reduced the IR spectra intensity. The good stability of these hydrogels was then confirmed by indirect analyses. Specifically, UV-vis analyses on DPBS incubated with hydrogels at different timeframes suggest that it was not possible to detect the presence of free PEDOT:PSS in the conditioning media. Thus, all hydrogels displayed an excellent stability after 21 days in physiological conditions of pH and osmolarity.
A degradation trend was previously reported by Chen and co-workers for hydrogels based on gelatin and the same amount of genipin in deionized water.94 They also reported higher degradation rates for hydrogels with a lower crosslinking degree. An higher degradation trend in PBS – proportional to PEDOT:PSS – was reported by Annabi and collaborators for hydrogels based on UV-crosslinked methacryloyl gelatin and the same amounts of PEDOT:PSS.19
3.6 Effects of PEDOT:PSS hydrogels on the viability of primary astrocytes
It has been recently highlighted that the interaction of implantable materials and devices with astrocytes is critical for the long-term stability and to determine the successful outcome of the implantation remedy in the brain.95 Given the potential of conductive materials in neuroregenerative medicine targeting brain repair, we next sought to investigate the effect of electroconductive hydrogels on primary astrocytes.In this respect, to determine the impact of hydrogels on astrocytes adhesion, morphology and viability, confluent primary rat cortical astrocytes were re-plated on electroconductive hydrogels (i.e. fabricated with PEDOT:PSS). The latter is a validated primary cell culture model used to screen and study the interaction of neural interface with glial cells. Given the well-known biocompatibility of gelatin and genipin,42,96 hydrogels without electroconductive polymer were used as control.
We first performed environmental scanning electron microscopy (ESEM) 2 and 7 days after seeding cells on the gel-gen and gel-gen + PEDOT:PSS samples. We observed an abundant number of astrocytes on the gel-gen and gel-gen + PEDOT:PSS samples. Notably, after 2 days cells display a rounded-up morphology, typical of adhering cells after 2 days. On the other hand, astrocytes on both type of hydrogels appear with numerous extensions, a possible indication of permissive and efficient interaction with both the substrates (Fig. 3). Notably, fluorescence microscopy for the staining of the samples with DAPI, a fluorescent nuclear marker, (Fig. S12†) confirmed the numerous amounts of primary cells at both time points either in gel-gen and in gel-gen + PEDOT:PSS.
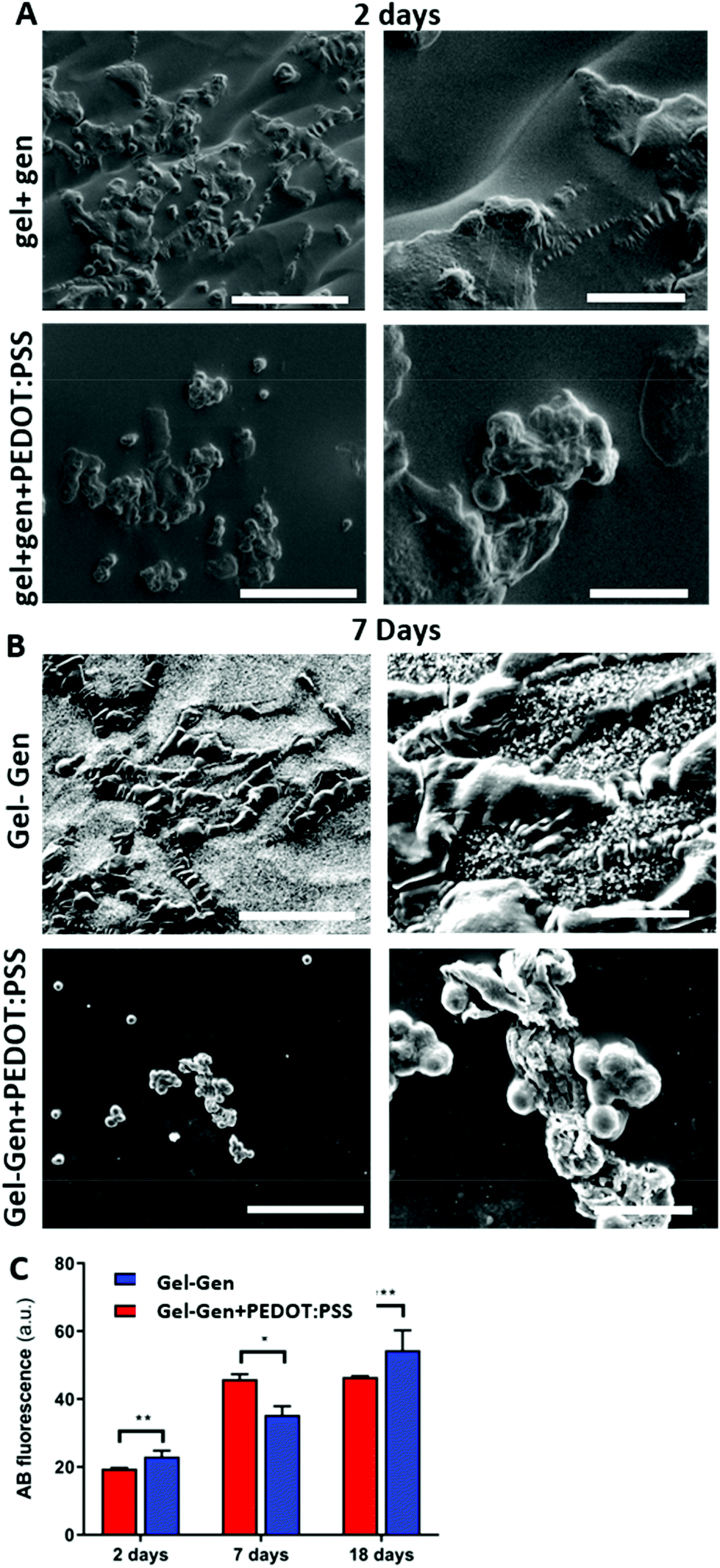 | ||
| Fig. 3 (A) Environmental scanning electron microscopy (ESEM) images of astrocytes, collected at different magnification (1000× and 4000×) 2 and 7 days after cells seeding on gelatin-genipin (gel-gen, control hydrogels, upper panels) (A) and electroconductive hydrogels (gel-gen + PEDOT:PSS, lower panels) (B). The scale bar stands for 100 μm in left panels and 20 μm in right panels. Experimental conditions: [gelatin] = 10% (w/w respect to the total weight of the sample), [genipin] = 1% (w/w respect to gelatin), and [PEDOT:PSS] = 0 or 0.3% (w/w respect to the total weight of the sample). (C) Time course of astrocytes viability on electroconductive and control hydrogels obtained by Alamar Blue (AB) assay performed after 2, 7 and 18 days in vitro from cells seeding on the substrates. Data are plotted as the averaged percentages of reduced AB ± Standard Error (SE). Unpaired Student's t test was performed to compare samples (*: p value <0.05; **: p value <0.01; ***: p value <0.001). | ||
To quantify and compare the cell viability on hydrogels samples over time, we performed Alamar Blue (AB) assay after 2, 7 and 18 days in vitro from cells seeding on the substrates.
The bar plot shown in Fig. 3C reports the averaged percentage of reduced AB, with respect to the oxidized one observed in different samples tested at different time points equal to 2, 7 and 18 days after replating. The values reported are proportional to the metabolic activity of the cells and, on turn, correlate with the presence of viable cells in the substrate.97
Notably, the results demonstrate that, at 2 days, the viability was significantly higher on electroconductive samples (Gel-Gen + PEDOT:PSS) than on genipin crosslinked gelatin samples (Fig. 3C), that we used as the internal control. In particular, electroconductive samples promote the highest adhesion of astrocytes after 2 days. These data are in line with other in vitro and in vivo studies showing good biocompatibility of PEDOT:PSS films and bidimensional substrates with other cell types, including brain cells.98 We found that the adhesion as well as the long-term viability of the cells are ameliorated in the electroconductive hydrogels, gel-gen + PEDOT:PSS. Indeed, the cell viability increases over time in hydrogels up to 7 days, while the increase is prolonged to 18 days in gel-gen + PEDOT:PSS hydrogels (Fig. S13†). It is remarkable that, while the growth of astrocytes on control samples reaches a plateau after 7 days, astrocytes continue to grow in electroconductive hydrogels up to 18 days.
Collectively, these in vitro analyses revealed that electroconductive hydrogels display a very good biocompatibility with primary astrocytes and thus might be a suitable candidate as neural interface or for neural engineering. These results suggest that the presence of electroconductive polymer enhance a sustained cell growth and colonization.
As described previously, conductivity of the samples might promote cell viability or impact on cell differentiation. However, mechanical properties can also influence the adhesion and growth of astrocytes (for a review see ref. 95) and of other cell types. In the reported samples, the difference in stiffness is doubled in the presence of conductive polymer, thus potentially it may contribute to the increased adhesion and growth observed in conductive hydrogels. However, previous studies showed that difference in the substrate's stiffness can impact on cell behavior only if higher than one order of magnitude.99,100 On the other hand, small differences in the electrical conductivity of samples can promote a different cell behavior.18,19 Consequently, the higher adhesion and growth of astrocytes can be mainly attributed to the higher electroconductivity of PEDOT:PSS samples.
4. Conclusions
In the present contribution we have proposed an original method to fabricate electroconductive hydrogels based on gelatin, a natural biopolymer selected to recreate the microenvironment of the extracellular matrix, and PEDOT:PSS, a biocompatible conductive polymer. Genipin, in synergy with PEDOT:PSS, demonstrate the ability to promote a homogenous reticulation of the resulting composite networks and consolidation of the hydrogel. Physical–chemical properties – including mechanical performance, electroconductive properties, swelling and degradation behavior – of resulting hydrogels were proved to be finely tunable through the amount of conductive polymer added. In detail, hydrogels embedding conductive polymer displayed an increase of the shear modulus but were less resistant to deformation. This behavior was attributed to the partial hindrance of hydrogel bending and stretching in the presence of the conductive polymer. Furthermore, the presence of PEDOT:PSS reduced gelation time of the proposed system. All hydrogels resulted to be biocompatible with primary rat astrocytes, an essential condition for materials intended as neural probes or for neuroregenerative medicine. Notably, we reported the unprecedented result that the presence of conductive polymer PEDOT:PSS in a 3D scaffold prolong the growth of astrocytes over long-term, indicating the suitability of such preparation for therapeutic approaches in neurology.The resulting hydrogels can be proposed as biomaterials in the field of tissue engineering, being potentially in vivo injectable and in situ cross-likable thanks to its unique properties, especially for regeneration of electrically conductive tissues, e.g. muscular and neural tissues, and as conductive interfaces with human tissues, e.g. as neural probes. Furthermore, this system could be promising for the development of 3D (bio)printed electroconductive biomaterials for regenerative medicine, which will be the subject for future evaluation and optimization.
Author contributions
Conceived and designed the experiments: FF, MM, NS, ES, VB, MS. Performed the experiments: FF, MM, NS, ES. Analyzed the data: FF, NS, MM, ES. Discussed the data: FF, NS, MM, ES, EC, AS, VB, AT, SP, MS. Wrote the paper: FF.All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
The authors disclose no actual or potential conflict of interest.Acknowledgements
The skillful assistance of Dr. Davide Gardini in rheological experiments is gratefully acknowledged. M. Caprini, A. Minardi, and F. Formaggio from FABIT Department, University of Bologna are kindly acknowledged for their assistance in cell preparation and maintenance.This work was supported by the Mat2Rep project financed by POR-FESR 2014-20 and FSC of Emilia-Romagna. The work is also supported by Air Force Office of Scientific Research - AFOSR Research Projects FA9550-19-1-0370 (V. B.), FA9550-18-1-0255 (V. B.) and FA9550-20-1-0386 (V. B.), and MSCA-ITN-2020-ASTROTECH (GA956325) (V. B.). E.S is supported by POR-FESR 2014-2020 of Emilia Romagna Region, MAT-2-REP- FSC and by AFOSR ASTROLIGHT.
References
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS PubMed.
- Y. S. Zhang and A. Khademhosseini, Science, 2017, 356, 6337 Search PubMed.
- Y. Li, J. Rodrigues and H. Tomás, Chem. Soc. Rev., 2012, 41, 2193–2221 RSC.
- Y. Jiang, J. Chen, C. Deng, E. J. Suuronen and Z. Zhong, Biomaterials, 2014, 35, 4969–4985 CrossRef CAS PubMed.
- T. G. Kim, H. Shin and D. W. Lim, Adv. Funct. Mater., 2012, 22, 2446–2468 CrossRef CAS.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2015, 15, 13–26 CrossRef PubMed.
- B. M. Holzapfel, J. C. Reichert, J. T. Schantz, U. Gbureck, L. Rackwitz, U. Nöth, F. Jakob, M. Rudert, J. Groll and D. W. Hutmacher, Adv. Drug Delivery Rev., 2013, 65, 581–603 CrossRef CAS PubMed.
- Q. He, Y. Huang and S. Wang, Adv. Funct. Mater., 2018, 28, 1–10 Search PubMed.
- M. D. Baumann, J. C. Stanwick, I. E. Donaghue and M. S. Shoichet, Biomaterials for spinal cord repair, Elsevier Ltd., 2011, vol. 6 Search PubMed.
- X. Li, Z. Xiao, J. Han, L. Chen, H. Xiao, F. Ma, X. Hou, X. Li, J. Sun, W. Ding, Y. Zhao, B. Chen and J. Dai, Biomaterials, 2013, 34, 5107–5116 CrossRef CAS PubMed.
- F. S. Wong and A. C. Lo, J. Stem Cell Res. Ther., 2015, 5(2), 1000267 Search PubMed.
- S. Sirivisoot, R. Pareta and B. S. Harrison, Interface Focus, 2013, 4, 20130050 CrossRef PubMed.
- D. Eyre, Arthritis Res., 2002, 4, 30–35 CrossRef CAS PubMed.
- T. Distler and A. R. Boccaccini, Acta Biomater., 2020, 101, 1–13 CrossRef CAS PubMed.
- S. Zhang, Y. Chen, H. Liu, Z. Wang, H. Ling, C. Wang, J. Ni, B. Çelebi-Saltik, X. Wang, X. Meng, H. J. Kim, A. Baidya, S. Ahadian, N. Ashammakhi, M. R. Dokmeci, J. Travas-Sejdic and A. Khademhosseini, Adv. Mater., 2020, 32, 1–7 Search PubMed.
- J. R. Aggas, S. Abasi, J. F. Phipps, D. A. Podstawczyk and A. Guiseppi-Elie, Biosens. Bioelectron., 2020, 168, 112568 CrossRef CAS PubMed.
- S. Usmani, A. F. Biagioni, M. Medelin, D. Scaini, R. Casani, E. R. Aurand, D. Padro, A. Egimendia, P. R. Cabrer, M. Scarselli, M. De Crescenzi, M. Prato and L. Ballerini, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 25212–25218 CrossRef CAS PubMed.
- A. R. Spencer, E. Shirzaei Sani, J. R. Soucy, C. C. Corbet, A. Primbetova, R. A. Koppes and N. Annabi, ACS Appl. Mater. Interfaces, 2019, 11, 30518–30533 CrossRef CAS PubMed.
- A. R. Spencer, A. Primbetova, A. N. Koppes, R. A. Koppes, H. Fenniri and N. Annabi, ACS Biomater. Sci. Eng., 2018, 4, 1558–1567 CAS.
- V. Kuzmenko, T. Kalogeropoulos, J. Thunberg, S. Johannesson, D. Hägg, P. Enoksson and P. Gatenholm, Mater. Sci. Eng., C, 2016, 58, 14–23 CrossRef CAS PubMed.
- Z. Bagher, Z. Atoufi, R. Alizadeh, M. Farhadi, P. Zarrintaj, L. Moroni, M. Setayeshmehr, A. Komeili and S. K. Kamrava, Mater. Sci. Eng., C, 2019, 101, 243–253 CrossRef CAS PubMed.
- S. Wang, S. Guan, J. Xu, W. Li, D. Ge, C. Sun, T. Liu and X. Ma, Biomater. Sci., 2017, 5, 2024–2034 RSC.
- S. Wang, S. Guan, W. Li, D. Ge, J. Xu, C. Sun, T. Liu and X. Ma, Mater. Sci. Eng., C, 2018, 93, 890–901 CrossRef CAS PubMed.
- V. M. Tysseling and J. A. Kessler, Biomaterials for central nervous system regeneration, Elsevier Ltd., 2011, vol. 5 Search PubMed.
- J. Guo, Y. Yu, H. Wang, H. Zhang, X. Zhang and Y. Zhao, Small, 2019, 15, 1–7 Search PubMed.
- T. Nezakati, A. Seifalian, A. Tan and A. M. Seifalian, Chem. Rev., 2018, 118, 6766–6843 CrossRef CAS PubMed.
- H. Yano, K. Kudo, K. Marumo and H. Okuzaki, Sci. Adv., 2019, 5, 1–10 Search PubMed.
- H. Yuk, B. Lu, S. Lin, K. Qu, J. Xu, J. Luo and X. Zhao, Nat. Commun., 2020, 11, 4–11 CrossRef PubMed.
- W. Teng, Q. Zhou, X. Wang, H. Che, P. Hu, H. Li and J. Wang, Chem. Eng. J., 2020, 390, 124569 CrossRef CAS.
- H. Jang, J. Park and F. S. Kim, Phys. Status Solidi A, 2019, 216, 1–7 CrossRef.
- N. Alegret, A. Dominguez-Alfaro, M. Salsamendi, I. J. Gomez, J. Calvo, D. Mecerreyes and M. Prato, Inorg. Chim. Acta, 2017, 468, 239–244 CrossRef CAS.
- Y. Wu, Y. X. Chen, J. Yan, D. Quinn, P. Dong, S. W. Sawyer and P. Soman, Acta Biomater., 2016, 33, 122–130 CrossRef CAS PubMed.
- B. Lu, H. Yuk, S. Lin, N. Jian, K. Qu, J. Xu and X. Zhao, Nat. Commun., 2019, 10, 1–10 CrossRef CAS PubMed.
- B. Yang, F. Yao, T. Hao, W. Fang, L. Ye, Y. Zhang, Y. Wang, J. Li and C. Wang, Adv. Healthcare Mater., 2016, 5, 474–488 CrossRef CAS PubMed.
- N. Alegret, A. Dominguez-Alfaro, J. M. González-Domínguez, B. Arnaiz, U. Cossío, S. Bosi, E. Vázquez, P. Ramos-Cabrer, D. Mecerreyes and M. Prato, ACS Appl. Mater. Interfaces, 2018, 10, 43904–43914 CrossRef CAS PubMed.
- L. Ferlauto, A. N. D'Angelo, P. Vagni, M. J. I. A. Leccardi, F. M. Mor, E. A. Cuttaz, M. O. Heuschkel, L. Stoppini and D. Ghezzi, Front. Neurosci., 2018, 12, 1–10 Search PubMed.
- N. Alegret, A. Dominguez-Alfaro and D. Mecerreyes, Biomacromolecules, 2019, 20, 73–89 CrossRef CAS PubMed.
- L. Ghasemi-Mobarakeh, M. P. Prabhakaran, M. Morshed, M. H. Nasr-Esfahani, H. Baharvand, S. Kiani, S. S. Al-Deyab and S. Ramakrishnav, J. Tissue Eng. Regener. Med., 2011, 17–35 CrossRef PubMed.
- D. N. Heo, S. J. Lee, R. Timsina, X. Qiu, N. J. Castro and L. G. Zhang, Mater. Sci. Eng., C, 2019, 99, 582–590 CrossRef CAS PubMed.
- L. V. Kayser and D. J. Lipomi, Adv. Mater., 2019, 31, 1–13 CrossRef PubMed.
- T. Vishnoi, A. Singh, A. K. Teotia and A. Kumar, ACS Biomater. Sci. Eng., 2019, 5, 3007–3021 CrossRef CAS PubMed.
- D. J. Macaya, K. Hayakawa, K. Arai and M. Spector, Biomaterials, 2013, 34, 3591 CrossRef CAS PubMed.
- J. W. Salatino, K. A. Ludwig, T. D. Y. Kozai and E. K. Purcell, Nat. Biomed. Eng., 2017, 1, 862–877 CrossRef CAS PubMed.
- T. Posati, A. Pistone, E. Saracino, F. Formaggio, M. G. Mola, E. Troni, A. Sagnella, M. Nocchetti, M. Barbalinardo, F. Valle, S. Bonetti, M. Caprini, G. P. Nicchia, R. Zamboni, M. Muccini and V. Benfenati, Sci. Rep., 2016, 6, 1–16 CrossRef PubMed.
- M. Durso, A. I. Borrachero-Conejo, C. Bettini, E. Treossi, A. Scidà, E. Saracino, M. Gazzano, M. Christian, V. Morandi, G. Tuci, G. Giambastiani, L. Ottaviano, F. Perrozzi, V. Benfenati, M. Melucci and V. Palermo, J. Mater. Chem. B, 2018, 6, 5335–5342 RSC.
- E. Saracino, L. Maiolo, D. Polese, M. Semprini, A. I. Borrachero-Conejo, J. Gasparetto, S. Murtagh, M. Sola, L. Tomasi, F. Valle, L. Pazzini, F. Formaggio, M. Chiappalone, S. Hussain, M. Caprini, M. Muccini, L. Ambrosio, G. Fortunato, R. Zamboni, A. Convertino and V. Benfenati, Adv. Biosyst., 2020, 4(4) DOI:10.1002/adbi.201900264.
- L. Bi, Z. Cao, Y. Hu, Y. Song, L. Yu, B. Yang, J. Mu, Z. Huang and Y. Han, J. Mater. Sci. Mater. Med., 2011, 22, 51–62 CrossRef CAS PubMed.
- J. S. Barbosa, A. Ribeiro, A. M. Testera, M. Alonso, F. J. Arias, J. C. Rodríguez-Cabello and J. F. Mano, Adv. Eng. Mater., 2010, 12, 37–44 CrossRef.
- G. Fessel, J. Cadby, S. Wunderli, R. Van Weeren and J. G. Snedeker, Acta Biomater., 2014, 10, 1897–1906 CrossRef CAS PubMed.
- S. Dimida, C. Demitri, V. M. De Benedictis, F. Scalera, F. Gervaso and A. Sannino, J. Appl. Polym. Sci., 2015, 132, 1–8 CrossRef.
- P. Sacco, F. Furlani, A. Marfoglia, M. Cok, C. Pizzolitto, E. Marsich and I. Donati, Macromol. Biosci., 2020, 2000236, 1–7 Search PubMed.
- N. J. Burke, A. D. Burrows, M. F. Mahon and S. J. Teat, CrystEngComm, 2004, 6, 429–436 RSC.
- L. A. Ditta, D. Bulone, P. L. S. Biagio, R. Marino, D. Giacomazza and R. Lapasin, Int. J. Biol. Macromol., 2020, 158, 985–993 CrossRef CAS PubMed.
- A. Maleki, A. L. Kjøniksen and B. Nyström, Carbohydr. Res., 2007, 342, 2776–2792 CrossRef CAS PubMed.
- E. E. Holly, S. K. Venkataraman, F. Chambon and H. Henning Winter, J. Non-Newtonian Fluid Mech., 1988, 27, 17–26 CrossRef CAS.
- A. B. Bonhome-Espinosa, F. Campos, I. A. Rodriguez, V. Carriel, J. A. Marins, A. Zubarev, J. D. G. Duran and M. T. Lopez-Lopez, Soft Matter, 2017, 13, 2928–2941 RSC.
- S. Supper, N. Anton, N. Seidel, M. Riemenschnitter, C. Curdy and T. Vandamme, Expert Opin. Drug Delivery, 2014, 11, 249–267 CrossRef CAS PubMed.
- K. M. Park, S. Y. Lee, Y. K. Joung, J. S. Na, M. C. Lee and K. D. Park, Acta Biomater., 2009, 5(6) DOI:10.1016/j.actbio.2009.01.040.
- T. C. Tseng, L. Tao, F. Y. Hsieh, Y. Wei, I. M. Chiu and S. H. Hsu, Adv. Mater., 2018, 30(25) DOI:10.1002/adma.201500762.
- R. Lapasin, Polysaccharide Hydrogels: Characterization and Biomedical Applications, Pan Stanford, Singapore, 2016, pp. 83–137 Search PubMed.
- G. Scionti, L. Rodriguez-Arco, M. T. Lopez-Lopez, A. L. Medina-Castillo, I. Garzón, M. Alaminos, M. Toledano and R. Osorio, J. Biomed. Mater. Res., Part A, 2018, 106, 738–745 CrossRef CAS PubMed.
- P. R. Soskey and H. H. Winter, J. Rheol., 1984, 28, 625–645 CrossRef CAS.
- E. Marsich, A. Travan, M. Feresini, R. Lapasin, S. Paoletti and I. Donati, Macromol. Chem. Phys., 2013, 214, 1309–1320 CrossRef CAS.
- P. Sacco, M. Cok, F. Asaro, S. Paoletti and I. Donati, Carbohydr. Polym., 2018, 196, 405–413 CrossRef CAS PubMed.
- P. Sacco, G. Baj, F. Asaro, E. Marsich and I. Donati, Adv. Funct. Mater., 2020, 30(31) DOI:10.1002/adfm.202001977.
- J. Odent, T. J. Wallin, W. Pan, K. Kruemplestaedter, R. F. Shepherd and E. P. Giannelis, Adv. Funct. Mater., 2017, 27, 1–10 CrossRef.
- B. Zhang, S. Li, H. Hingorani, A. Serjouei, L. Larush, A. A. Pawar, W. H. Goh, A. H. Sakhaei, M. Hashimoto, K. Kowsari, S. Magdassi and Q. Ge, J. Mater. Chem. B, 2018, 6, 3246–3253 RSC.
- W. J. Tyler, Nat. Rev. Neurosci., 2012, 13, 867–878 CrossRef CAS PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- W. L. Murphy, T. C. McDevitt and A. J. Engler, Nat. Mater., 2014, 13, 547–557 CrossRef CAS PubMed.
- A. Shahini, M. Yazdimamaghani, K. J. Walker, M. A. Eastman, H. Hatami-Marbini, B. J. Smith, J. L. Ricci, S. V. Madihally, D. Vashaee and L. Tayebi, Int. J. Nanomed., 2013, 9, 167–181 Search PubMed.
- K. Yin and Z. Zhu, Synth. Met., 2010, 160, 1115–1118 CrossRef CAS.
- Y. Wang, Y. Shen, Y. Zhang, B. Yue and C. Wu, J. Macromol. Sci., Part B: Phys., 2006, 45, 563–571 CrossRef CAS.
- Y. Han, M. Shen, J. Zhu, Y. Wu and X. Zhang, Polym. Compos., 2013, 34, 989–996 CrossRef CAS.
- S. Oktay and N. Alemdar, J. Appl. Polym. Sci., 2019, 136, 1–8 CrossRef.
- M. Lay, M. À. Pèlach, N. Pellicer, J. A. Tarrés, K. N. Bun and F. Vilaseca, Carbohydr. Polym., 2017, 165, 86–95 CrossRef CAS PubMed.
- F. A. Whitehead, S. A. Young and S. Kasapis, Int. J. Biol. Macromol., 2019, 141, 867–875 CrossRef CAS PubMed.
- P. Tongnuanchan, S. Benjakul and T. Prodpran, J. Food Eng., 2013, 117, 350–360 CrossRef CAS.
- P. Bergo and P. J. A. Sobral, Food Hydrocolloids, 2007, 21, 1285–1289 CrossRef CAS.
- N. Samsalee and R. Sothornvit, Int. J. Food Sci. Technol., 2017, 52, 964–971 CrossRef CAS.
- C. Xiao, H. Liu, Y. Lu and L. Zhang, J. Macromol. Sci., Part A: Pure Appl.Chem., 2001, 38, 317–328 CrossRef.
- Y. Li, H. Jia, Q. Cheng, F. Pan and Z. Jiang, J. Membr. Sci., 2011, 375, 304–312 CrossRef CAS.
- S. D. García Schejtman, R. Toselli, M. C. Strumia and M. Martinelli, Polym. Bull., 2015, 72, 3043–3062 CrossRef.
- W. Zhang, G. Ren, H. Xu, J. Zhang, H. Liu, S. Mu, X. Cai and T. Wu, J. Polym. Res., 2016, 23(8) DOI:10.1007/s10965-016-1059-5.
- S. Dimida, A. Barca, N. Cancelli, V. De Benedictis, M. G. Raucci and C. Demitri, Int. J. Polym. Sci., 2017, 8410750 Search PubMed.
- H. Khan, R. N. Shukla and A. K. Bajpai, Mater. Sci. Eng., C, 2016, 61, 457–465 CrossRef CAS PubMed.
- D. A. Ahmad Ruzaidi, M. M. Mahat, Z. Mohamed Sofian, N. A. Nor Hashim, H. Osman, M. A. Nawawi, R. Ramli, K. A. Jantan, M. F. Aizamddin, H. H. Azman, Y. H. R. Chang and H. H. Hamzah, Polymers, 2021, 13, 1–20 Search PubMed.
- S. Lee and C. A. Randall, Solid State Ionics, 2013, 249–250, 86–92 CAS.
- C. Capiglia, P. Mustarelli, E. Quartarone, C. Tomasi and A. Magistris, Solid State Ionics, 1999, 118, 73–79 CrossRef CAS.
- S. Zhao, P. Tseng, J. Grasman, Y. Wang, W. Li, B. Napier, B. Yavuz, Y. Chen, L. Howell, J. Rincon, F. G. Omenetto and D. L. Kaplan, Adv. Mater., 30(25) DOI:10.1002/adma.201800598.
- I. Uguz, M. Ganji, A. Hama, A. Tanaka, S. Inal, A. Youssef, R. M. Owens, P. P. Quilichini, A. Ghestem, C. Bernard, S. A. Dayeh and G. G. Malliaras, Adv. Healthcare Mater., 2016, 5, 3094–3098 CrossRef CAS PubMed.
- K. Schrote and M. W. Frey, Polymer, 2013, 54, 737–742 CrossRef CAS.
- A. Bigi, G. Cojazzi, S. Panzavolta, N. Roveri and K. Rubini, Biomaterials, 2002, 23, 4827–4832 CrossRef CAS PubMed.
- C. H. Yao, B. S. Liu, C. J. Chang, S. H. Hsu and Y. S. Chen, Mater. Chem. Phys., 2003, 83, 204–208 CrossRef.
- L. Maiolo, V. Guarino, E. Saracino, A. Convertino, M. Melucci, M. Muccini, L. Ambrosio, R. Zamboni and V. Benfenati, Adv. Healthcare Mater., 2021, 10(1) DOI:10.1002/adhm.202001268.
- E. Saracino, V. Cirillo, M. Marrese, V. Guarino, V. Benfenati, R. Zamboni and L. Ambrosio, Mater. Sci. Eng., C, 2021, 118, 111363 CrossRef CAS PubMed.
- A. Sagnella, A. Pistone, S. Bonetti, A. Donnadio, E. Saracino, M. Nocchetti, C. Dionigi, G. Ruani, M. Muccini, T. Posati, V. Benfenati and R. Zamboni, RSC Adv., 2016, 6, 9304–9314 RSC.
- Y. Liang, A. Offenhäusser, S. Ingebrandt and D. Mayer, Adv. Healthcare Mater., 2021, 10(11) DOI:10.1002/adhm.202100061.
- J. Oliver-De La Cruz, G. Nardone, J. Vrbsky, A. Pompeiano, A. R. Perestrelo, F. Capradossi, K. Melajová, P. Filipensky and G. Forte, Biomaterials, 2019, 205, 64–80 CrossRef CAS PubMed.
- D. E. Discher, P. Janmey and Y. L. Wang, Science, 2005, 310, 1139–1143 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: (i) picture and schematic representation of the electrochemical cell; (ii) visual analyses of hydrogels fabricated with different amounts of PEDOT:PSS at different timeframes; (iv) additional rheological data; (v) conductivity tests on hydrogels fabricated with sterilized components; (vi) conductivity tests on hydrogels fabricated by using phosphate buffered saline as solvent; (vii) degradation tests; (viii) fluorophore-labelled cells on the hydrogels; (ix) proliferation rate of cells on the hydrogels. See DOI: 10.1039/d2bm00116k |
| This journal is © The Royal Society of Chemistry 2022 |