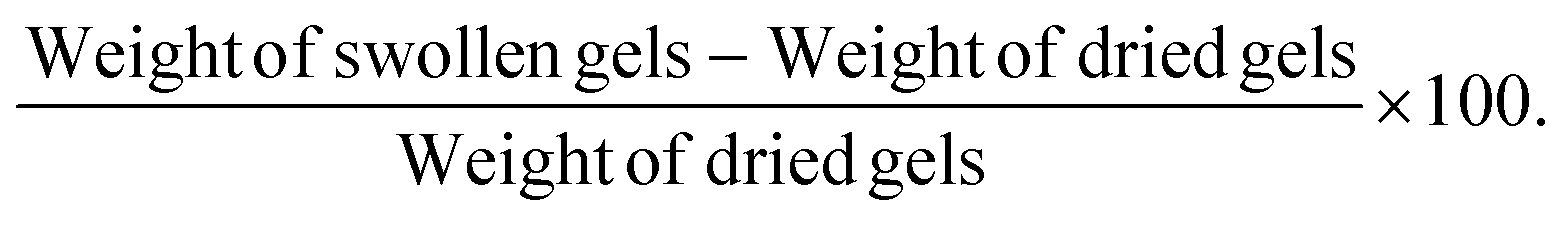DOI:
10.1039/D2BM00022A
(Paper)
Biomater. Sci., 2022,
10, 4492-4507
Development of gelatin methacrylate (GelMa) hydrogels for versatile intracavitary applications†
Received
6th January 2022
, Accepted 9th June 2022
First published on 23rd June 2022
Abstract
Applicability of hydrogels as drug delivery systems is on the rise due to their highly tunable degree of polymeric crosslinking to attain varying rates of payload release. Sustaining the release of therapeutic payloads at certain physiological sites has been the need of the hour to treat disorders such as peritoneal or pleural malignancies. These disorders can be targeted via intracavitary administration of hydrogels, providing localized therapy. In this study, a gelatin methacrylate (GelMa) hydrogel with tunable physicochemical traits is developed and characterized. A hydrogel-based depot system was curated using GelMa as backbone, a photo-initiator (lithium phenyl-2,4,6-trimethylbenzoylphosphinate) and a chemical crosslinker (N,N-methylenebisacrylamide). Hydrogels were optimized using a 23 factorial design, by testing for their gelling time, injectability, viscosity change, elasticity, bio-adhesion, swelling-index, in vitro degradation, in vitro release, and biocompatibility. Gelling time for hydrogel formulations was found to be <60 seconds with gelling being achieved in as fast as 24 seconds. Bio-adhesion studies revealed that formulations with higher concentrations of both crosslinkers had more adhesion to guinea pig lung tissues. Hydrogels with higher swelling showcased a more sustained release. Biocompatibility studies for hydrogel formulations was done by evaluating formulation performance in MTT, live/dead, and apoptosis assays performed using non-malignant Human embryonic kidney cells (HEK-293). The optimized hydrogel formulations were biocompatible, yielding >90% cellular viability over 72 hours. This delivery system prototype may be used to deliver potent chemotherapeutics locally, reducing off target effects and improving therapeutic benefits.
Introduction
The human body comprises multiple cavities, which are hollow physiological spaces that hold and protect delicate organs such as the brain, heart, lungs, etc. Based on their location, major cavities can be classified as either ventral or dorsal.1 Ventral cavities include the thoracic (heart and lungs) and abdominopelvic (spleen, kidneys, bladder, and reproductive organs) cavities, whereas dorsal cavities include the cranial (brain) and vertebral (spinal cord) cavities.1 Apart from the essential function of protecting these physiological organs, cavities may also act as gateways, allowing a direct access to essential organs, often to treat certain disorders.2 Intracavitary therapy is often exercised when all other conventional therapies have failed to achieve the desired therapeutic outcome.3 Intracavitary therapy provides a local effect, which not only enhances therapeutic benefits4 but also may potentially reduce dose and associated off-target effects. These intracavitary therapies include intracavitary brachytherapy, intracavitary radioimmunotherapy, and intracavitary therapy for multiple malignancies, etc.5,6 However, physiological cavities are difficult to access and require invasive procedures for therapeutic delivery, or for withdrawing intracavitary fluid (which lubricates the cavity walls) which is exacerbated in disease conditions.7 This often leads to patient incompliance, making frequent dosing troublesome. To overcome the frequent accessibility issue, designing a sustained release depot system that resides in the physiological cavity can be a viable option. This depot system will reside in close proximity to the diseased tissues, and will release therapeutic payloads over a period, circumventing frequent dosing and invasive procedures.
A sustained delivery of therapeutic payloads can be achieved by formulating a delivery system with certain characteristics that can retard the diffusion of drug through the delivery carrier. Mainly, polymeric matrix-based delivery systems have been exceedingly used in sustained drug release applications as the matrices provide excellent drug dispersibility, along with providing first-order release kinetics.8,9 One such well-studied polymeric matrix-based system is hydrogel. Hydrogels are 3D-networks of crosslinked hydrophilic polymers made up of >90% water,10 and have been traditionally used as substitutes for fibrin glues for applications such as wound healing or wound closure.11 Hydrogels have also been extensively studied as tissue substitutes and have been reported to enhance tissue regeneration, physiologically.12 In recent times, applicability of hydrogels has grown multiple fold with their potential as a post-surgical local drug delivery system being tapped. This is possible as hydrogels exhibit a macroporous matrix structure, which ensures even distribution of therapeutic agents along with providing a controlled release of cargo.13 Hydrogels have certain innate properties which vary based on the polymeric backbone, chosen to form gels. These polymeric backbones include materials like alginates, chitosan, hyaluronic acid, collagen derivatives, etc.14 One of the most commonly used collagen derivatives is gelatin, which is a hydrolyzed form of collagen type I.15 Gelatin has been reported to have excellent hydration properties along with being biocompatible.16 Moreover, gelatin is inherently bio-adhesive, which can be attributed to its numerous arginine–glycine–aspartic acid (RGD) residues that naturally promote cell and tissue binding.16 Even though gelatin gels at lower temperatures, it fails to form gels at physiological temperatures. Moreover, gelatin poses poor mechanical strength along with being easily degraded by matrix metalloproteinases.17 The lack of sufficient mechanical strength and early degradation makes gelatin a poor candidate for sustained drug delivery applications. However, derivatization of gelatin into semi-synthetic compounds such as gelatin methacrylate (GelMa) may help overcome these drawbacks and can be a viable option for extended drug delivery applications.16
Modification of gelatin with methacryloyl substituents at specific amino acid residues, first reported by Van Den Bulcke et al. in 2000, imparts gelatin with the property of photo-crosslinking in the presence of a photoinitiator and a suitable source of light.18 This can be attributed to the inherent photopolymerization characteristic of methacryloyl substituents that can form covalent crosslinks with the photoinitiator.18 This covalent crosslinking relatively increases the mechanical strength of GelMa in comparison to gelatin.18 Moreover, the degree of substitution of methacryloyl substituents with amino acid residues that are essential for cellular binding and biodegradation is low enough (<5%) to not affect the aforementioned bio-adhesion and biodegradation ability of gelatin.16,18,19 A photoinitiator such as 2-hydroxy-1-[4-(2-hydroxyethoxy)phenyl]-2-methyl-1-propanone (Irgacure-2959) has been conventionally used along with GelMa to form photo-crosslinkable hydrogels in presence of UV light at 365 nm wavelength.20 However, Irgacure 2959 has limited water solubility (5 mg ml−1)21 and is recently being replaced with lithium acylphosphinate salts which exhibit much higher aqueous solubility along with a higher molar extinction coefficient at 365 nm, making it more efficient in terms of photo-crosslinking with GelMa.22 Further, GelMa hydrogels are highly tunable in terms of their pore size and degree of crosslinking with the photoinitiator, making tailored release profiles for therapeutic agents possible.23 While GelMa was originally fabricated to be a minimally invasive injectable system to deliver cells physiologically,24 its injectability has promoted its exploration for intracavitary applications as an external UV source can gel GelMa hydrogels once injected inside physiological cavities.25 One such clinical exploration (phase-III) for intracavitary application of hydrogels was performed by Kokorovic and Matin in 2020 where they tested the applicability of mitomycin gel for the treatment of low-grade upper tract urothelial carcinoma (ClinicalTrials.gov identifier: NCT02793128).26 The intention of formulating a hydrogel-based formulation of mitomycin was to increase the urinary dwell time of mitomycin. The group achieved promising results to treat lower-grade upper tract urothelial carcinoma indicating the potential of hydrogel systems to be beneficial, especially as a local intracavitary therapy.
With these advantages in mind, this study dwells in developing an injectable GelMa hydrogel system using lithium acylphosphinate (LAP) salt as a photoinitiator. Moreover, to improve crosslinking efficiency and promote bio-adhesion, N,N-methylenebisacrylamide (BIS) has been used as a chemical crosslinker. A design of experiment (DoE) approach has been deployed to ascertain ideal concentrations of hydrogel excipients. Extensive biophysical characterization and biocompatibility assessment of the fabricated hydrogel system demonstrates its potential to be used as an intracavitary depot system to release cargo inside physiological cavities.
Materials
Photogel methacrylated gelatin (GelMa; sterile) was procured from Advanced Biomatrix Inc. (Carlsbad, CA, USA). The photoinitiator used to crosslink with GelMa was lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP), procured from Sigma-Aldrich (St Louis, MO, USA), N,N-methylenebisacrylamide (BIS) was procured from Acros Organics B.V.B.A. (Geel, Belgium). UV source with an intensity of 760 μW cm−2 was procured from Analytik Jena (Upland, CA, USA). Guinea pig lungs, used for bio-adhesion studies, were procured frozen from Lampire Biological Laboratories Inc. (Pipersville, PA, USA). Collagenase type IV was used in in vitro degradation studies and was procured from MP Biomedicals, LLC (Irvine, CA, USA). Sulforhodamine 101 (Texas red) was used as a model small molecule to assess drug release profile from fabricated hydrogels and was procured from Tocris Bioscience (Bristol, United Kingdom). HEK-293 cells were used to assess biocompatibility of fabricated hydrogels and were procured from ATCC (Manassas, VA, USA). 3-(4,5-Dimethylthiazol-2-yl) (MTT) was procured from Acros Organics B.V.B.A. (Geel, Belgium). Live/dead cytotoxicity assay kit was used to study the viability of HEK-293 cells encapsulated inside hydrogels and was procured from Biotium Inc. (Fremont, CA, USA). Annexin-V FITC, propidium iodide and Annexin-V binding buffer were used to study apoptosis of HEK-293 cells when incubated along with hydrogels and were procured from Nexcelom Bioscience (Lawrence, MA, USA).
Methods
Optimization of hydrogel formulation variables by design of experiment approach (DoE)
To optimize the formulation variables in methacrylate gelatin (GelMa) hydrogels, a design of experiment (DoE) approach was utilized. A 23 factorial design was prepared using Minitab statistical software (Minitab release 17, State College, PA, USA) comprising of 2 levels of concentrations for each formulation variable, i.e., GelMa (5% and 10% w/v), LAP (0.2% and 0.5% w/v), and BIS (1.25% and 2.5% w/v). Eight hydrogel formulations were established by Minitab to ascertain the effect of each formulation variable on biophysical characteristics of hydrogels (Fig. 1B). Target parameters for optimized hydrogel formulation was determined to be gelling time (faster gelling desired), viscosity (optimum viscosity increase with time), injectability (precursor solutions with lesser force for injection), bio-adhesion (higher bio adhesion desired), and tensile strength (higher tensile strength desired).
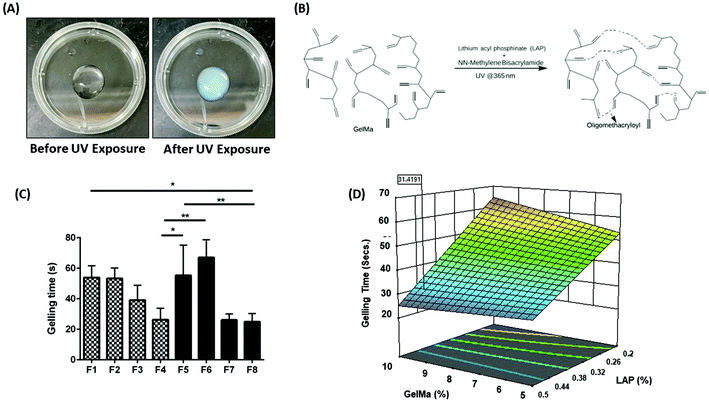 |
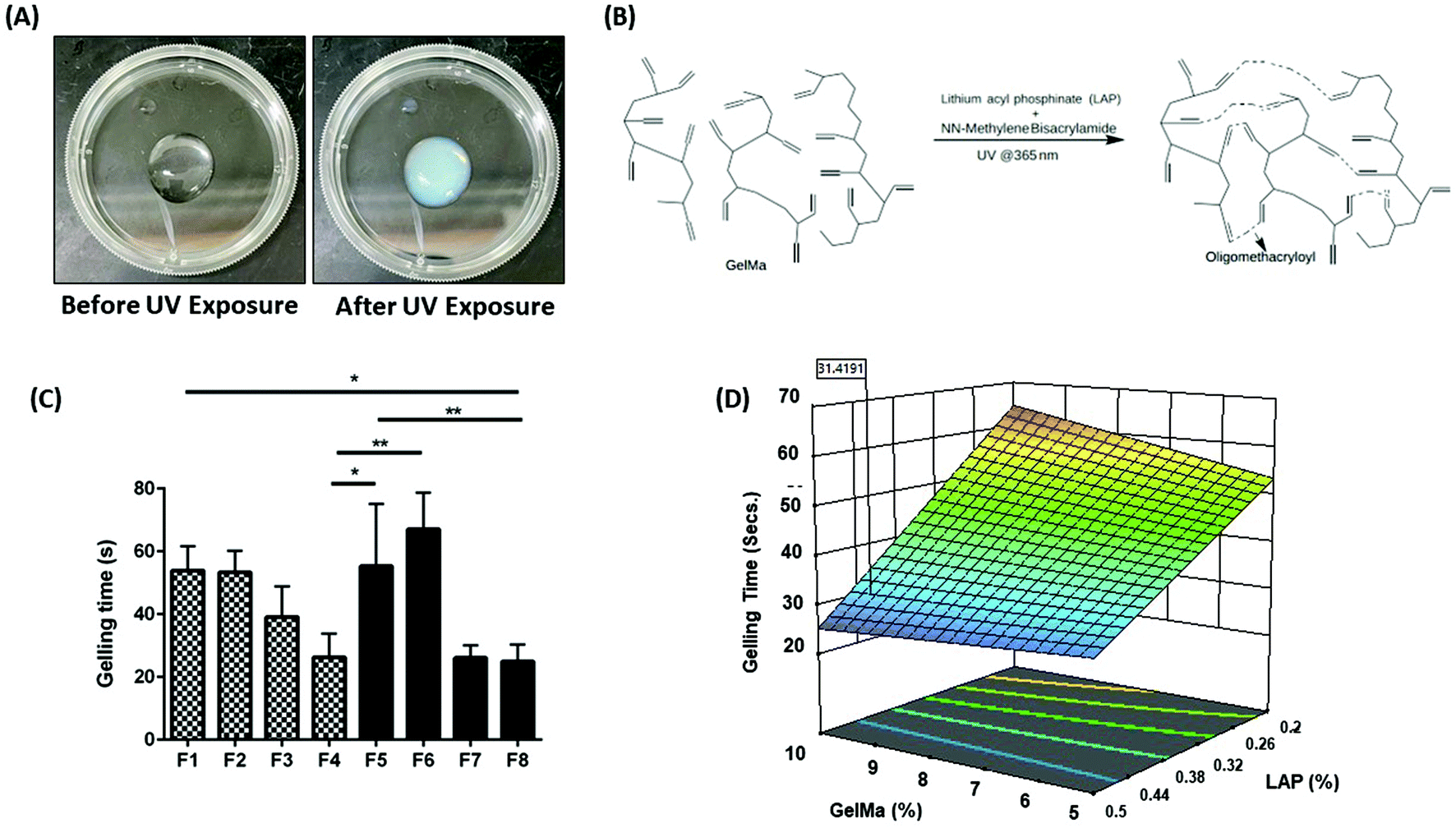
| | Fig. 1 (A) Representation of hydrogel precursor solution (PS) before and after UV exposure of 365 nm wavelength. A clear sol–gel transition can be observed after UV exposure, indicated by the opacity of the PS. (B) Schematic representation of the chemical crosslinking reaction between GelMa, LAP and BIS on exposure to UV light at 365 nm wavelength. (C) The gelling time required for a complete transition of sol–gel for hydrogel PS after exposure to UV light at 365 nm wavelength. It can be seen that formulations F3, F4, F7, and F8 had relatively faster gelling time as compared to other formulations. This faster gelling time can be attributed to the higher concentration of LAP present in these four formulations. (D) 3D surface plot represents the effect of LAP and GelMa concentrations on gelling time. It can be inferred that concentration of LAP affects the rate of gelling, i.e., an increase in LAP concentration drastically reduces the gelling time. However, concentration of GelMa and BIS did not drastically affect the gelling time. Data represent mean ± SD with n = 3. *p < 0.05, **p < 0.01. | |
Fabrication of hydrogel precursor solution (PS)
Hydrogel precursor solution (PS) was formed by mixing GelMa, LAP and BIS at their respective concentrations, as obtained from DoE, and dissolving each component in phosphate buffered saline (pH 7.4). Further, formulations were subjected to bath sonication at 37 °C for 30 minutes followed by overnight shaking at 180 rpm and 37 °C to ensure a completely homogenous system. The precursor solution was stored at 4 °C, until further usage.
Gelling time
Hydrogel PS were incubated at 37 °C for 30 min to ascertain physiological temperature. Further, 0.5 ml of PS was placed in a 60 mm Petri dish and subjected to UV light at 365 nm wavelength with an intensity of 760 μW cm−2 (Analytik Jena, Upland, CA, USA) with the UV lamp being placed at a distance of three inches from the samples (common for all further experiments). The time required for all PS to form stable gels was noted. Preliminary endpoint to ascertain sol–gel conversion was a drip test, i.e., the time required for PS to not flow/drip when inverted.27 Based on the gelling time, effective concentrations for both crosslinkers were narrowed down.
Viscosity
Viscosity analysis for GelMa hydrogels was performed using Brookfield viscometer DV II+ (Brookfield Engineering Laboratories Inc., Middleboro, MA, USA). Briefly, PSs were maintained at 37 °C and were subjected to simultaneous UV and viscosity measurement. As the crosslinking with traditional 760 μW cm−2 UV source (used for gelling time experiments) was found to be expeditious, the intensity of UV light was drastically to 68 μW cm−2, so as to retard the crosslinking rate. Retarding the rate of crosslinking slowed the gelling time, which in turn allowed for a better understanding of the changes in viscosity. Shaft S06 rotating at 30 rpm was used for measuring the change in viscosity over time. Centipoise versus time plot was used to analyze the change in viscosity with UV exposure.
Texture analysis
Injectability.
To evaluate the ability of PS to be injected into physiological cavities via catheter, injectability for hydrogel PS was done using TA.XT Plus texture analyzer (Stable MicroSystems, MA, USA). Briefly, 0.5 ml of hydrogel PS was loaded in a 1 ml syringe, fitted with a 26-gauge needle, that mimics the inner diameter of traditionally used catheters. The PS loaded syringes were fitted on to the texture analyzer, which was pre-set with the following parameters: pre-test and test speed for the movement of the probe was set to 1 mm s−1, post-test speed was set to 10 mm s−1 and the probe's travel distance was set to 3 mm.
Bio-adhesion
Evaluation of bio-adhesion for four hydrogel formulations was done using frozen guinea pig lung tissues (Lampire Biological Laboratories, Pipersville, PA, USA). Briefly, guinea pig lungs were activated by immersing them in 1× PBS (pH 7.4) at 40 °C. Following day, a clean tissue section, approximately 15 mm wide and 3 mm in height was placed and secured in a A-MUC mucoadhesion rig (Stable MicroSystems, Surrey, UK). The rig was immersed in a water bath, maintained at 37 °C. Hydrogel PSs were applied on the tissue and were immediately subjected to UV light exposure at 365 nm. A bio-adhesion cylindrical probe, TA-10ss, was attached to the TA.XT Plus to measure the force of detachment of the gel from the tissue (Fig. 3A). The bio-adhesion test protocol loaded on to the TA.XT Plus had the following optimized parameters: test speed and detachment speed were maintained at 0.5 mm s−1, post-test speed was maintained at 5 mm s−1, a force of 50 g was applied on the tissue to evaluate the force required to detach the probe. A plot of force versus time was plotted to obtain the absolute positive force required to detach the gel from the tissue surface.
Tensile strength
Tensile strength of hydrogel formulations was evaluated using TA.XT Plus. Briefly, 3.5 mm × 14 mm hydrogel pieces were prepared by gelling PS in an appropriate mold. The hydrogels were then subjected to breaking using a TA-52 puncture probe and the force required to completely break the hydrogel formulations was recorded. The following parameters were loaded onto the texture analyzer; test speed was maintained at 0.5 mm s−1, distance that the probe would travel was maintained at 5 mm, post-test speed was maintained at 10 mm s−1. Young's modulus was calculated using exponent component software by evaluating the ratio of shear stress and strain (MPa).
Scanning electron microscopy
Mesh-network for hydrogel formulations was visualized using a Helios NanoLab 660 Dualbeam FIB (FEI, Hillsboro, Oregon, USA). Briefly, hydrogel PSs were gelled and frozen at −80 °C for 4 hours followed by lyophilization using a FreeZone 6 freeze dry system (Labconco Corporation, Kansas City, MO, USA). Lyophilized hydrogels were carefully sectioned using a standard scalpel (1–2 mm thickness) to expose the macropores and sections were carefully attached on the SEM pin stub (Ted Pella, Inc., Redding, CA, USA) using an electron microscopy compatible double adhesive tape. Further, lyophilized hydrogel sections were gold sputter coated (coat thickness 30 nm); and were imaged at 2 kV. SEM micrographs were subjected to pore diameter analysis. Briefly, micrographs were exported to ImageJ software (National Institutes of Health, Bethesda, MD, USA), and a total of 150 pores were analyzed for their diameters per formulation (30 pores with n = 5 micrographs per formulation).
Swelling index
Swelling index for two hydrogel formulations (F4 and F8) was evaluated to predict their potential drug release patterns. Briefly, hydrogel PSs (0.5 ml) were gelled and freeze dried to remove all water content. Dry weight of the gels was recorded followed by immersing dried gels in 70% ethanol for 2 hours. Weight of swollen gels was recorded and the % swelling was calculated using the following equation:| | 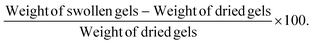 | (1) |
In vitro degradation
In vitro degradation of hydrogel formulations was determined based on the amount of intact gel remaining over a period of time on incubation with collagenase type IV enzyme. Briefly, 0.5 ml of hydrogel PSs were gelled and placed in 1 ml of collagenase solution (50 μg ml−1), placed at 37 °C for 28 days. At each time interval (every 72 hours), hydrogels were thoroughly dried with extra-low linting Kimwipes, to remove excess liquid and weighed to determine the % of hydrogel degraded as compared to day 0. A plot of % hydrogel remaining versus time was plotted to understand the probable physiological degradation behavior.
In vitro release
To determine the in vitro release profile of small molecules from hydrogel formulations (F4 and F8), sulforhodamine 101 (Texas red; SR101) was used as a model fluorescent small molecule. Briefly, 100 μg of SR101 was mixed with 0.3 ml hydrogel PSs. Solutions were gelled and placed in a 60 mm Petri dish along with 1 ml of 1× PBS solution. The release media was completely replaced at every time point, and the amount of SR101 released from hydrogels was analyzed by evaluating the fluorescence intensity (595 ex/635 em).
Additionally, in vitro release in collagenase solution (50 μg ml−1) was also analyzed to determine the effect of degradation on the release of SR101. For these experiments, all release parameters were kept constant apart from the release medium, which was modified to 1 ml of 1× PBS spiked with collagenase to a final concentration of 50 μg ml−1. Release kinetic models (zero order, first order, Higuchi model, Korsmeyer–Peppas model and Hixson–Crowell model) were evaluated to ascertain the type of modified release of SR101 from formulations F4 and F8 in 1× PBS.
Effect of gelling time on in vitro release
For GelMa hydrogels that have been gelled by an external stimulus such as UV exposure, the exposure time determines the degree of crosslinking, which in turn may determine the rate of drug release from the gel. To test this hypothesis, an experiment was designed where hydrogels were subjected to gelling at multiple UV exposure times. Release of SR101 from these hydrogels was analyzed when hydrogels were gelled at their original gelling time (25 seconds), two times the gelling time (2×) (50 seconds), and four times the gelling time (4×) (100 seconds). 1× PBS was used as the release medium and released SR101 was analyzed by evaluating the fluorescence intensity (595 ex/635 em), as described earlier.
Biocompatibility
Live/dead assay.
In this assay, the viability of HEK-293 cells was evaluated by encapsulating cells within the hydrogel matrix and incubating for 24- and 72-hours. Briefly, 1 × 104 cells were mixed with 25 μL of hydrogel precursor solution. This solution was then placed in a black TC-treated 96-well plate followed by curing using UV at 365 nm. Once gelled, 100 μL of fresh cell nutrition media was added to the wells and cells were further incubated until desired time points were reached. At each time point, cells were labeled with calcein AM (live cells) and Ethd-III (dead cells). Cells were then imaged using an EVOS-FL fluorescence microscope (Thermo Fisher Scientific, Waltham, MA, USA) at 4× magnification for qualitative analysis of live/dead cells.
Cytotoxicity towards HEK-293 cells
Cytotoxicity of blank hydrogel formulations and individual hydrogel formulation components was evaluated to determine the biocompatibility of the formulations. Briefly, HEK-293 non-malignant mammalian cells were seeded in a 24-well TC-treated plate at a density of 2 × 104 cells per well and were allowed to adhere overnight. Following day, 25 μL of hydrogel PSs were gelled and carefully incubated along with cells for 24- and 72-hours. At each time interval, gel pieces and cell nutrition media were aspirated and 1 mg ml−1 MTT solution was added to each well and incubated at 37 °C/5% CO2 for 2 hours. Subsequently, MTT solution was aspirated and DMSO was added to each well to dissolve the formazan crystals. Absorbance values for each well were calculated at 570 nm and % cell viability for hydrogel incubated cell groups was determined relative to a no-treatment (media only) control group of cells.
Annexin V FITC based apoptosis assay
Analysis of apoptosis for cells incubated with hydrogel pieces was done using an Annexin-V FITC apoptosis assay (Nexcelom Bioscience LLC, Lawrence, MA, USA). Briefly, 1 × 105 HEK-293 cells were seeded into a TC-treated 6-well plate and were allowed to adhere overnight. Following day, cells were incubated with 25 μL of hydrogel pieces for 24 hours. After 24-hours, gel pieces were removed, and cells were trypsinized and washed with 1× PBS twice. Further, cells were re-dispersed in Annexin-V binding buffer along with Annexin-V FITC (1 mg ml−1) and propidium iodide (1 mg ml−1) for 15 minutes in dark. Excess dye was removed, and cells were re-dispersed in Annexin-V binding buffer. Cells were analyzed using Nexcelom Cellometer Vision fluorescent cell counter (Nexcelom Bioscience LLC, Lawrence, MA, USA) to visualize Annexin-V and propidium iodide positive cells. Propidium iodide stains cells with a compromised cell membrane to visualize necrotic cells. Annexin-V and propidium iodide positive cells were classified as apoptotic cells.
Statistical analysis
All data presented here are mean ± SD or SEM (n = 3–6), unless otherwise stated. Cytotoxicity studies represent average of 3 independent trials (n = 3 for each trial). Unpaired Student's t-test was used to compare two groups whereas to compare more than two groups one-way ANOVA followed by Tukey's post hoc multiple comparison test was used. P value <0.05 was considered statistically significant.
Results & discussion
Development of eight GelMa hydrogel formulations using a design of experiment (DoE) approach
The concept of Design of Experiment (DoE) dates back to 1920 and is a vital statistical concept developed by statistician Sir Ronald Fisher.28 The DoE approach was first utilized by Marlowe and Shangraw in 1967 to assess the dissolution of salicylic acid from tablets.29 Generally speaking, a DoE approach is an organized way to evaluate the effect, certain formulation excipients may have on the process or formulation characteristics.28 By understanding the effect of each formulation excipient, prediction of formulation performance becomes possible. This strategy has been used to formulate hydrogels with excellent physicochemical characteristics. In 2020, Li et al. demonstrated the effectiveness of using DoE approach to develop PEG nanoparticle loaded GelMa hydrogels.30 The team developed a 34 factorial design to test the effect of both the formulation excipients and process on overall outcome of the delivery system. The team focused on systematically developing drug-loaded nano carriers which helped them narrow down on excipient concentration and process parameters to achieve the best possible outcome in terms of particle size and drug entrapment within the nanoformulation. However, the team failed to take into consideration the development of GelMa hydrogels using DoE, which could have helped in achieving better therapeutic outcomes in terms of nanoparticle loading and payload release from the hydrogels. With that in mind, in this study, an in-depth analysis of hydrogel excipients has been attempted to extract the maximum potential of GelMa hydrogels in terms of its physical traits. Hydrogel PS were allowed to gel using UV exposure from about 0.5-inch distance. Fig. 1A demonstrates the pre- and post-UV exposure hydrogels. It can be seen that pre-exposure, hydrogel precursor solution is a clear free flowing liquid which is converted into a gel, following a chemical reaction indicated in Fig. 1B, indicated by a turbid appearance and non-flowability, post-exposure (Fig. 1A). Eight (8) potential hydrogel formulations were theorized by the Minitab software with varying concentrations of two crosslinkers (LAP and BIS) and two varying concentrations of polymeric backbone of GelMa using a 23 factorial design. Table 1 represents the eight formulations (F1–F8) that were tested extensively for their physical traits. Formulations were narrowed down systematically based on their target profiles such as gelling time, injectability, bio-adhesion, etc. to ascertain a hydrogel formulation with optimum properties desired for intracavitary applications. In further studies, the effect of each excipient on physical traits of hydrogels was analyzed.
Table 1 Representation of eight formulations that were developed after deploying a 23 factorial design using Minitab. Two levels (excipient concentrations) for three factors (concentrations of GelMa, LAP and BIS) were evaluated
| Formulation |
X
1
|
X
2
|
X
3
|
| F1 |
−1 |
−1 |
−1 |
| F2 |
−1 |
−1 |
1 |
| F3 |
−1 |
1 |
−1 |
| F4 |
−1 |
1 |
1 |
| F5 |
1 |
−1 |
−1 |
| F6 |
1 |
−1 |
1 |
| F7 |
1 |
1 |
−1 |
| F8 |
1 |
1 |
1 |
| Coded values |
GelMa (%) |
LAP (%) |
BIS (%) |
|
X
1
|
X
2
|
X
3
|
| −1 |
5 |
0.2 |
1.25 |
| 1 |
10 |
0.5 |
2.5 |
LAP is essential for efficient gelling rate and crosslinking of GelMa hydrogels
With the derivatization of gelatin into GelMa, forming covalent crosslinks to obtain stable hardened gelatin hydrogels became possible.16 These covalent crosslinks were possible in presence of certain compounds that would photopolymerize (photoinitiators).31 These photoinitiators have been used in conjunction with GelMa to form hydrogels suitable for tissue regeneration, wound healing, drug delivery, etc.32,33 Irgacure 2959 is the most commonly used photoinitiator with GelMa as it has moderate water solubility along with being biocompatible with a low degree of immunogenicity.34 Recently, LAP has been a preferred choice of photoinitiator to crosslink with GelMa and this can be attributed to its higher aqueous solubility in comparison to Irgacure along with a higher molar extinction coefficient which results in more efficient crosslinking with GelMa at UV exposure of 365 nm wavelength.22 Xu et al. compared both crosslinkers along with GelMa for developing 3D-printed vascular constructs and reported that LAP performed better in terms of biocompatibility as compared to Irgacure.35 Moreover, the team reported that Irgacure-cured GelMa hydrogels had faster degradation rate which is unfavorable for designing a long-term depot therapy. Also the team reported to achieve a larger pore size with Irgacure linked hydrogels which will contribute towards a relatively faster release of drug payloads as compared to a smaller pore size.13 With this in mind, LAP was screened to understand its applicability for an intracavitary application, that would require slower degradation and sustained drug release.
Gelling time is essential to determine hydrogel gelling efficacy, which in turn governs various biophysical characteristics of hydrogels such as degree of crosslinking, in vitro drug release, bio-adhesion, etc. Using the DoE approach, eight hydrogel formulations were fabricated and tested for their gelling time and the time required for a complete gel–sol transition was recorded. It was observed that gelling time for all hydrogel formulations was between 24 and 67 seconds. Formulation F1 (53.9 ± 7.7 s), F2 (53.4 ± 6.9 s), F5 (55.3 ± 19.8 s), and F6 (67.1 ± 11.6 s) (Fig. 1C) were observed to have higher gelling times as compared to the other formulations. A slower gelling rate can be attributed to lower concentration of the photoinitiator LAP (0.2%) in these formulations, which is the primary crosslinking agent. Formulations F3 (39.1 ± 9.8 s), F4 (26.2 ± 7.6 s), F7 (26.1 ± 4.0 s) and F8 (24.9 ± 5.4 s) had significantly higher gelling rate due to a relatively higher concentration of LAP present in the formulations (0.5%) (Fig. 1C). The effect of each component of hydrogels on gelling time was evaluated by plotting a surface response plot for gelling time versus concentrations of GelMa and LAP (Fig. 1D). A surface plot of GelMa and BIS with respect to gelling time was plotted to ascertain the impact of BIS on gelling time. However, lowering or increasing BIS concentration did not alter the gelling time (ESI Fig. S1†). Gelling time seems to be independent of GelMa concentrations but tends to increase as LAP concentration is decreased. Thus, it can be inferred that LAP is essential for efficient crosslinking and faster gelling rate. Faster gelling is essential for injectable hydrogels for intracavitary applications that gel via external stimuli such as UV exposure. Based on these observations, formulations F1, F2, F5 and F6 were eliminated and were not ideal candidates for intracavitary applications.
Viscosity of hydrogels increases exponentially during the gelling process
For intracavitary application of injectable hydrogels needing an external stimulus for gelling, it is essential to understand their rate of viscosity change during gelling. This knowledge may help in determining the extent of hydrogel displacement inside the cavity during the gelling process. Faster increase in viscosity is desirable for all injected hydrogel solutions as that would ensure intracavitary presence and minimal clearance via the lymphatic drainage systems clearing out intracavitary fluids. Change in viscosity for formulations optimized based on gelling time, F3, F4, F7 and F8 was performed using DVII+ Brookfield viscometer (Brookfield Engineering Laboratories Inc., Middleboro, MA, USA). An UV lamp with lower intensity was used for this experiment as traditional higher intensity lamps yielded higher gelling rates, resulting in an instantaneous change in viscosity, making the evaluation of viscosity change extremely difficult. Fig. 2A represents the exponential rise in viscosity (centipoise, cP) with respect to time and as can be seen, all formulations had a similar rise in viscosity over time with no significant difference in the rate of viscosity change. However, it was observed that formulation F3 had lower rate of viscosity change at the 5-minutes mark (166.7 ± 40.7 cP) as compared to other formulations, which can be attributed to the slower gelling time and a less efficient degree of crosslinking relative to the other three formulations (Fig. 2A). No formulation was eliminated during this study owing to the similarity in the rate of change of viscosity for all four formulations.
 |
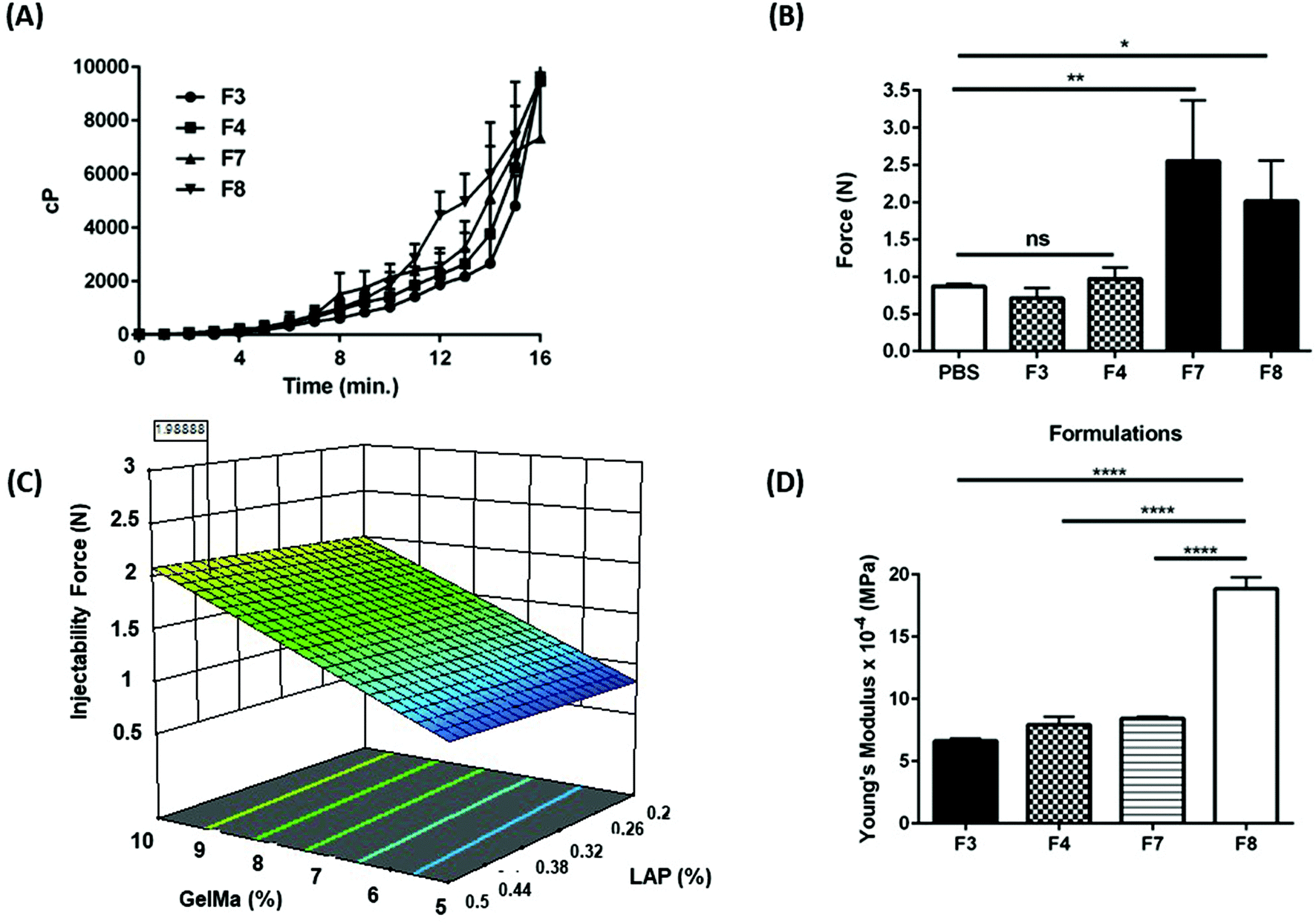
| | Fig. 2 (A) Change in viscosity over time for formulations F3, F4, F7, and F8 using Brookfield viscometer with an external low intensity 365 nm UV source. It can be seen that over time, viscosity of all formulations increases in tandem and there was no significant difference between the rate of change of viscosity for any formulations. (B) Force required for injecting hydrogel PS for formulations F3, F4, F7, and F8, assessed using texture analyzer equipped with a 5 kg load cell. There was no significant difference between the force of injection for formulations F3 and F4 relative to a standard 1× PBS solution. However, formulations F7 and F8 required a significantly higher force for being injected through a 26-gauge needle. (C) 3D surface plot representing the effect of GelMa and LAP concentrations on injectability of hydrogel PS. It can be validated that injectability force requirement increases with an increase in GelMa concentration and this can be attributed to the fact that higher concentration of GelMa (10%) is more viscous as compared to lower concentration (5%). (D) Plot represents elasticity of formulations F3, F4, F7, and F8 measured using texture analyzer. As indicated by Young's modulus, formulation F8 had highest brittleness as compared to all other formulations which were revealed to be relatively more elastic. Data represent mean ± SD with n = 3. *p < 0.05, **p < 0.01, ****p < 0.001. | |
Precursor solutions for GelMa hydrogels are sufficiently injectable for intracavitary applications
For local delivery of therapeutically active ingredients, injecting payloads in physiological cavities can provide local therapy for certain tumors such as malignant mesothelioma.36 Intracavitary injections are possible either via direct insertion of payloads into the cavity or by means of a catheter, that is usually placed on patients to drain excess build-up of intracavitary fluid (pleural effusions, ascites, hydrocephalus, etc.). For providing hydrogels as a local intracavitary therapy, it is essential to evaluate the injectability of hydrogel precursor solutions, which can be done by analyzing the force required to inject the PSs. Although there have been multiple reports that demonstrate the excellent injectability of GelMa hydrogels,37–39 none of them till date has established the injectability of hydrogels through a catheter tube potentially for an intracavitary implant. In this study, a 26-gauze needle with an internal diameter of 0.26 mm was used to study the injectability using TA.XT Plus texture analyzer. It was observed that all four tested PSs require an initial force for injection of less than or around 2 N. All initial forces to initiate injection were noted and compared with a 1× PBS solution as reference. It was observed that 10% GelMa formulations (F7 and F8) have a relatively higher initial force requirement as compared to 5% GelMa hydrogels (F3 and F4) (Fig. 2B). F3 (0.7 ± 0.1 N) and F4 (1.0 ± 0.1 N) PSs required a force similar to 1× PBS injection (0.9 ± 0.0.03 N) whereas F7 and F8 had an initial force requirement of 2.6 ± 0.8 N and 2.0 ± 0.6 N for injection, respectively (Fig. 2B). A surface response plot for injectability versus concentrations of GelMa and LAP revealed the role of GelMa in ease or difficulty in injection (Fig. 2C). As GelMa concentration is increased, force required for injecting PSs is increased and it is independent of concentration of LAP. A surface plot of GelMa and BIS with respect to injectability force was plotted to ascertain the impact of BIS on precursor solution injectability. It was seen that increasing BIS concentration increased the force required to inject precursor solutions, as seen in ESI Fig. S2.† However, no formulations were eliminated after this study as a force of about 2 N is fairly injectable for intracavitary applications. Thus, it was established that all PSs were easily injectable and would not pose an issue for the healthcare professional injecting this therapy.
Brittleness of GelMa hydrogels increases with an increase in GelMa concentration
Hydrogels intended to reside in physiological cavities may endure dynamic conditions (inflation and deflation of the lungs in the pleural cavity, fluid exchange in the peritoneal cavity, etc.40) and thus elasticity of hydrogels is essential to evaluate. Elasticity for F3, F4, F7 and F8 was evaluated using TA.XT Plus texture analyzer, by measuring the force required to puncture hydrogels with respect to the distance traveled by the probe (Fig. 3A). Young's modulus (MPa) values were computed to determine the extent of elasticity. Young's modulus for F3 (6.6 ± 0.2 × 10−4 MPa) and F4 (7.9 ± 0.6 × 10−4 MPa) was observed to be significantly lesser than for formulations F7 (8.4 ± 0.1 × 10−4 MPa) and F8 (18.9 ± 0.9 × 10−4 MPa, p < 0.0001 relative to all other formulations) (Fig. 2D). However, all values were found to be extremely low (<0.1) indicating that all formulations are very elastic, and thus will not cause any obstruction in normal functioning of the organ that sits in the cavity of interest.
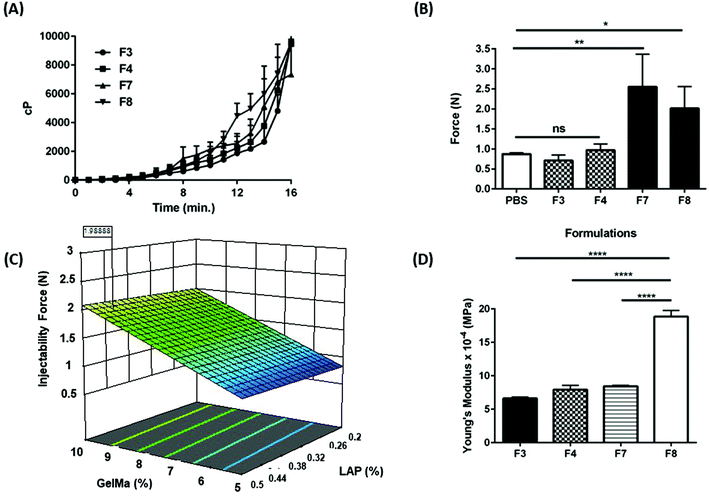
 |
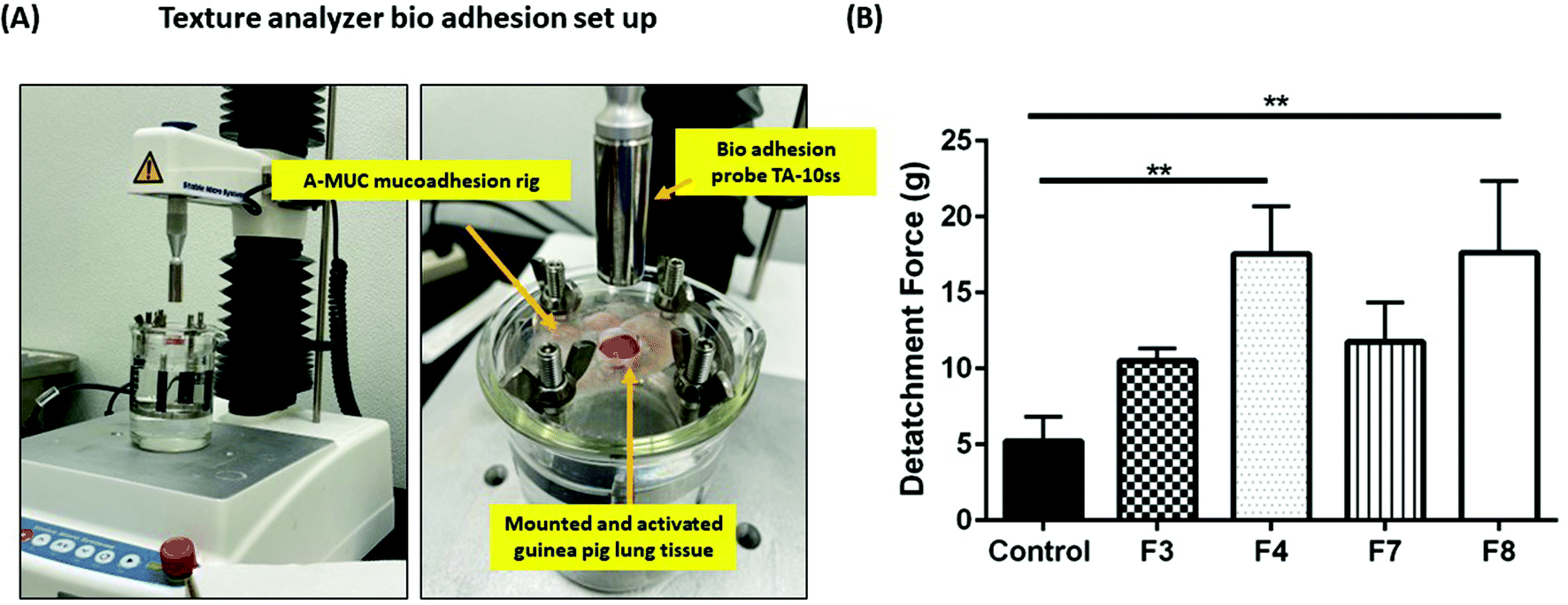
| | Fig. 3 (A) Image representing bio-adhesion assembly that was deployed for measurement of force of detachment of formulations F3, F4, F7, and F8 from guinea pig lung tissues. The tissue was mounted in a bio adhesion rig which was maintained at 37 °C in a water bath. Hydrogel PS were applied on the tissue and gelled followed by measurement of force required to detach the gels from tissue surface. (B) The force required to detach hydrogels from guinea pig lung tissues. It can be seen that the force required to detach formulations F4 and F8 was significantly higher relative to control (1× PBS solution). Data represent mean ± SD with n = 3. **p < 0.01. | |
GelMa hydrogels are sufficiently bio-adhesive for intracavitary applications
Intracavitary local delivery of payloads can be therapeutically and practically enhanced if the delivery system can release payloads over a sustained period. To be able to release the payload locally over a sustained period, long term residence in physiological cavities is necessitated. This can be achieved by delivery systems which have a strong adhesion to the mucus layer of physiological cavity. GelMa is inherently a tissue adhesive,41 and it can further be modified to attain higher degree of adhesiveness. The techniques to improve adhesion may include addition of a bio-adhesive adjunct such as polyacrylic acid and its derivates, or chitosan.42,43 Bio-adhesion can be tested in vitro using bio-adhesion tests that determine the force required to detach systems from a tissue or a biosimilar material.44 To study the extent of bio-adhesion of GelMa hydrogels, texture analyzer (TA.XT Plus) was used with guinea pig lungs as reference tissue. Bio-adhesion was performed for four hydrogel formulations, shortlisted from previous studies, and it was revealed that formulations F3 (10.5 ± 0.8 g) and F7 (11.8 ± 2.6 g) had lower force requirement for detachment from guinea pig lungs as compared to formulations F4 (17.55 ± 3.2 g) and F8 (17.62 ± 4.8 g) (Fig. 3B). Moreover, F4 and F8 had significantly more adhesion (p < 0.01) to the tissue as compared to a reference 1× PBS adhesion while formulations F3 and F7 failed to provide any relative significant adhesion to the tissue. This higher bio-adhesion may be attributed to the concentration of BIS, the secondary crosslinker in the four formulations. Formulations F4 and F8 have a higher concentration of BIS (2.5% w/v), and this relatively higher concentration may be vital in imparting higher bio-adhesion. There have been reports of compounds derived from acrylic acid to have a degree of bio-adhesion45 and that property of BIS may be contributing toward higher adhesivity of hydrogels. Keeping the relatively lower bio-adhesion in mind, formulations F3 and F7 were eliminated, and all further studies were performed using formulations F4 and F8.
Fabricated GelMa hydrogels have uniform mesh-size
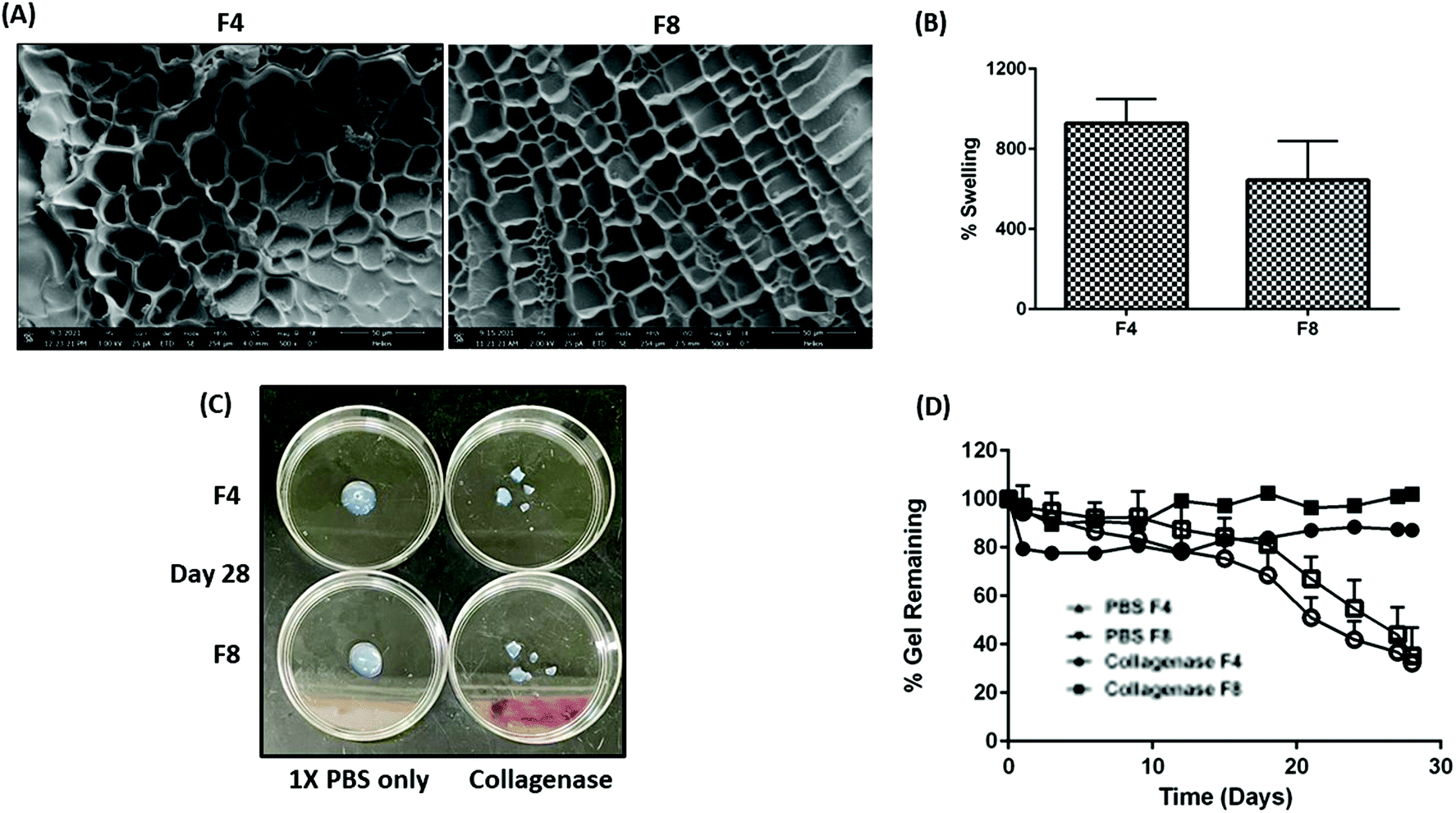
Hydrogels exhibit a microporous structure with varying pore sizes based on the efficiency of crosslinking between the polymeric backbone and crosslinkers.13 The presence or absence of micropores governs multiple physical traits for hydrogels such as mechanical strength, rate of payload release from within the hydrophilic matrices, elasticity of hydrogels, etc.13 Pores with larger diameter offer less resistance to drug diffusion and hence yield a relatively faster payload release as compared to smaller pore sizes within hydrogels.13 A uniform mesh-size can be seen in formulations F4 and F8 with an average mesh size of 62.2 ± 15.3 μm and 62.4 ± 11.8 μm, respectively (Fig. 4A). There was no significant difference in the average mesh size, observed between both formulations.
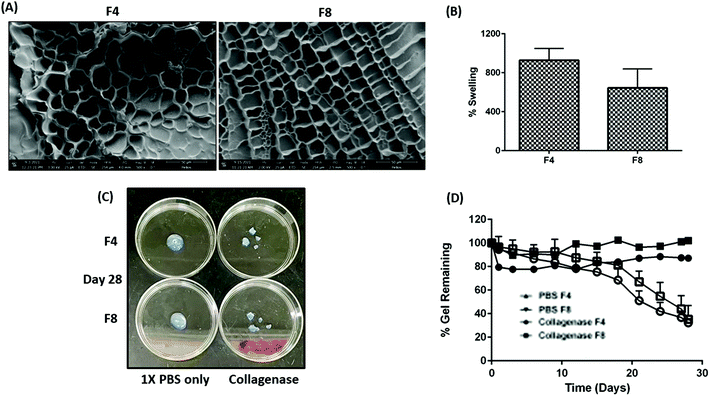 |
| | Fig. 4 (A) Micrographs represent the mesh-like hydrophilic polymeric network within GelMa hydrogels. Formulations F4 and F8 represent similar mesh structure with a similar average pore diameter. Briefly, 150 pores were measured across ten micrographs for formulations F4 and F8. (B) % swelling for formulations F4 and F8. It can be seen that formulation F4 swells about 900% of its original weight while formulations F8 swells about 700% of its original weight. This difference, although non-significant, can be attributed to efficient crosslinking in formulations F4 which made a higher percentage of water retention possible. (C) Images represent formulations F4 and F8 on day 28 of degradation assessment. Degradation of both formulations can be clearly seen after incubation with collagenase as compared to a solution of 1× PBS. (D) Plot represents the % gel remaining in terms of hydrogel weight after incubating formulations F4 and F8 along with a solution of collagenase and a control group of formulations incubated with 1× PBS. A drastic decrease in hydrogel weight was observed after 15 days of incubation indicating the MMP-9 cleavage to be slow. This in turn proves the ability of both formulations to reside in physiological cavities for more than a month. Data represents mean ± SD with n = 3. | |
Swelling index for fabricated hydrogels decreases with an increase in GelMa concentration
Inherently, hydrogels can swell multiple-fold and retain relatively large amounts of water without compromising their physical crosslinks and matrices.46 This property of hydrogels naturally retards the release of payloads trapped within the hydrogel matrices, which is one of the reasons for extensive hydrogel use for designing sustained release therapies.13 Moreover, the extent of hydrogel swelling can be modified to tailor the sustainability of payload release.13 Lai et al. demonstrated the ability of GelMa hydrogels to swell up to 1000% of their original weight and the team also reported that swelling behavior can be improved by addition of external moieties.47 Moreover, conditions such as temperature, pH, and light can affect the degree of swelling.48–50 With that in mind, formulating GelMa hydrogels with multiple crosslinkers necessitated an evaluation of swelling index to ascertain retention of inherent GelMa swelling properties. Swelling index for formulations F4 and F8 revealed that formulations F4 (928.8 ± 119.7%) had slightly higher swelling that formulation F8 (644.9 ± 195.1%) (Fig. 4B). A surface plot of GelMa, LAP, and BIS with respect to swelling index was plotted to ascertain the impact of excipients on swelling behavior of hydrogels. It was revealed that GelMa concentration is the contributing factor for swelling while altering the concentrations of LAP and BIS does not affect the swelling behavior of hydrogels, as seen in ESI Fig. S3.† However, there was no significant difference in swelling for the fabricated GelMa hydrogels. This phenomenon of similar swelling for two different concentrations of GelMa can be attributed to the degree of crosslinking of LAP and BIS with GelMa. Hypothetically, lower concentration of GelMa has a more efficient crosslinking as compared to the higher concentrations, resulting in higher water retention, and swelling.
GelMa hydrogels demonstrate excellent stability in presence of collagenase type IV over 28 days
For a complete evaluation of GelMa hydrogels as a sustained local therapy, it is vital to predict the stability profile of these systems in presence of enzymes such as collagenase (present in certain physiological cavities such as pleural cavity51), which are known to break hydrogel matrices and collapse gel structure. Gelatin inherently has MMP-9 cleavable sites which are unaltered when derivatized into its methacrylate substitute (GelMa).16 These MMP-9 cleavable sites promote physiological hydrogel degradation and multiple studies have reported the extent of degradation over a period.47,52,53 Wang et al. reported that in presence of an external crosslinker such as polyethylene glycol diacrylate (PEGDA), GelMa hydrogels degrade at a slower rate as compared to GelMa hydrogels without PEGDA.53 This study indicated that adjuvants such as chemical crosslinkers were able to alter physical traits of hydrogels. In this study, BIS was used as a chemical crosslinker and to study its effect on degradation and predict the probable residence time of GelMa hydrogels in physiological cavities, formulations F4 and F8 were incubated with 50 μg ml−1 of collagenase type IV, as previously reported,47 and % intact gel remaining over a period was computed based on hydrogel weight. The stability of formulations F4 and F8 incubated with collagenase type IV was compared with formulations incubated with 1× PBS as reference. A long-term incubation with collagenase type IV revealed tremendous stability for both F4 and F8, with more than 75% of gel intact after 15 days of incubation (Fig. 4C). On day 21, 51.1 ± 8.2% of formulation F4 was remaining while 67.1 ± 9.1% of formulation F8 was remaining relative to their initial swollen weights. The study was carried out for 28 days and on the final day, 32.3 ± 3.4% of formulation F4 and 35.2 ± 11.7% of formulations F8 relative to their initial swollen weights was remaining, indicating excellent stability in presence of collagenase enzyme (Fig. 4D). On the other hand, formulations incubated with 1× PBS showed negligible deterioration after 28 days (>85% hydrogels intact). This study indicates the ability of both formulations, F4 and F8, to be used for long term local therapeutic intracavitary applications.
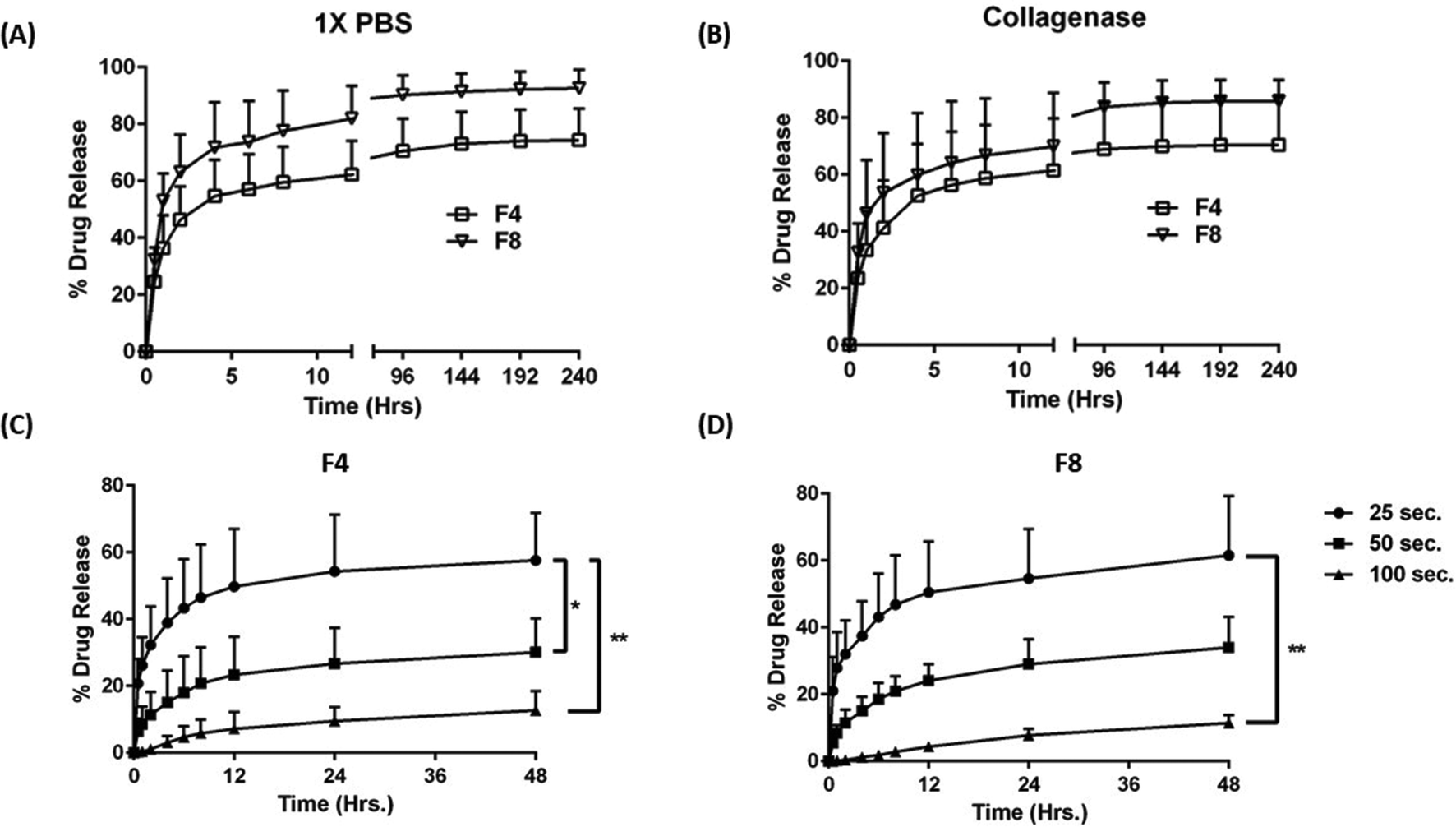
GelMa hydrogels sustain the release of Texas red (SR101) over 10 days
A sustained release of therapeutic payloads is beneficial from patients’ perspective as it would reduce the dosing frequency. Lower dosing frequency is desired, especially while designing an intracavitary delivery as an invasive procedure is deployed to deliver payloads (with an exception for catheter-assisted delivery where invasive procedure is required only at the beginning of therapy). Hydrogels are well-known release retardants, and this ability can be attributed to the degree of swelling which retards the rate of therapeutic payload diffusion through hydrogel matrices.13 Along with swelling, other parameters that can control the rate of payload release from hydrogels include deformation of hydrogel matrix, interactions between payload and polymeric matrix, etc.54,55 A common phenomenon of burst drug release is observed when hydrophilic payloads are loaded onto hydrogels.56 This burst release can be controlled by improving the degree of crosslinking, or by coating hydrogels with polymers that can further provide resistance to drug diffusion.57 One such study was reported by Han et al. where the burst release of drug-loaded microspheres could be suppressed by dip coating GelMa hydrogels with DMA–MPC polymer.57 Another way to curb burst release has been attempted in this study by improving the degree of crosslinking by enhancing the gelling time.58 In an earlier report, Teng et al. suggested that physical crosslinking between polymers may retard the release of payloads from films.58 This report suggests that a similar phenomenon can be demonstrated with hydrogels to curb the extensive burst release of hydrophilic payloads.
To evaluate release from formulations F4 and F8, Texas red (sulforhodamine 101; SR101), a hydrophilic fluorescent dye, was used as a model payload representing small molecules. 100 μg of SR101 dye was loaded on to both formulations and release profile was evaluated over 10 days. Two release media were used in this experiment, 1× PBS (Fig. 5A) with pH 7.4; and 1× PBS spiked with 50 μg ml−1 collagenase type IV (Fig. 5B) to evaluate the effect of the enzyme on drug release. As can be seen, there is an initial burst release of SR101, when evaluated with both release media with formulation F4 and F8 releasing 46.4 ± 11.7% and 63.1 ± 13.1% Texas red within the first two hours in 1× PBS, respectively. In collagenase spiked medium, formulation F4 released 41.2 ± 16.2% Texas red while formulation F8 released 53.6 ± 21.2% Texas red within the first two hours, indicating high burst release profiles. Thereafter, the drug release was sustained with formulation F4 releasing 74.4 ± 11.0% and F8 releasing 92.6 ± 6.5% Texas red after 10 days of incubation with 1× PBS (Fig. 5A). With collagenase medium, similar results were obtained for formulations F4 (70.4 ± 16.5%) and F8 (85.9 ± 7.6%) (Fig. 5B), which can be attributed to the delayed effect of collagenase on hydrogel degradation as reported in in vitro degradation study, where collagenase degraded hydrogels heavily after 15 days of incubation, at a concentration of 50 μg ml−1 (Fig. 4D).
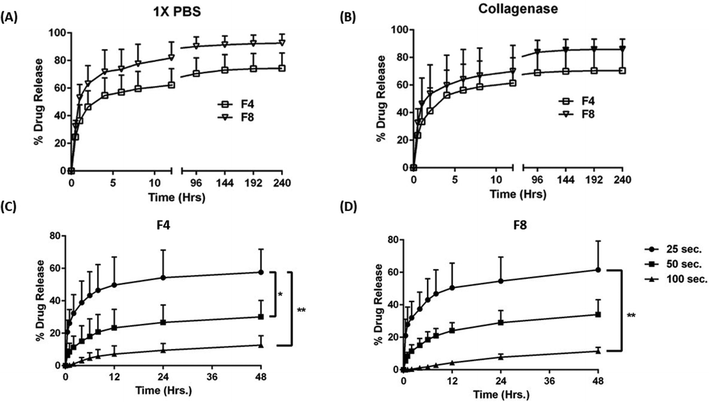 |
| | Fig. 5 (A) SR101 (Texas red) release profile of formulations F4 and F8 with 1× PBS as the release media over a period of 10 days. An initial burst release is observed for both formulations which is followed by a gradual sustained release of Texas red over 10 days. (B) Release profile of formulations F4 and F8 with 1× PBS spiked with collagenase (50 μg ml−1) as the release media over a period of 10 days. A similar initial burst release to plot (A) is observed with a sustained release of Texas red over 10 days. However, there is no degradation related escalation in release profile for Texas red in both formulations and this can be attributed to the fact that collagenase degrades formulations F4 and F8 slowly and drastic degradation in formulations begin after 15 days of incubation with collagenase. Plots (C) & (D) represent the extent of Texas red release on modulating the gelling time for formulations F4 and F8, respectively. Increasing the gelling time significantly retarded the rate of Texas red release from both formulations. This can be attributed to the higher degree of crosslinking associated with longer gelling time. Data represent mean ± SD with n = 3. *p < 0.05, **p < 0.01. | |
Further, to ascertain the order of release of SR101 from formulations F4 and F8, release kinetic evaluation was convened. Table 2 represents the linear equation and correlation coefficient (R2) values for the release of SR101 in 1× PBS from formulations F4 and F8. As can be seen, both formulations F4 and F8 followed a Korsmeyer–Peppas model for release of SR101 over 48- and 240-hours with R2 values for formulations F4 being 0.8535 over 48 hours and 0.8029 over 240 hours while formulation F8 has R2 values of 0.9213 over 48 hours and 0.7422 over 240 hours (Table 2). This indicates that the release of SR101 from developed hydrogels is not only diffusion dependent but also dependent on erosion of hydrogels over time.
Table 2 Release kinetic models with their linear equation and correlation coefficient (R2) values for SR101 (Texas red) release in the first 48 hours and over 240 hours with 1× PBS as the release media
| Release kinetic model |
48 hours |
240 hours |
| F4 |
F8 |
F4 |
F8 |
| Equation |
R
2
|
Equation |
R
2
|
Equation |
(R2) |
Equation |
(R2) |
| Zero order |
y = 0.8234x + 38.487 |
0.3402 |
y = 1.0873x + 51.345 |
0.3448 |
y = 0.1612x + 45.607 |
0.3674 |
y = 0.1819x + 60.939 |
0.2989 |
| First order |
y = 0.0053x + 1.6393 |
0.3328 |
y = 0.0052x + 1.7661 |
0.3365 |
y = 0.0021x + 1.5082 |
0.1235 |
0.1235y = 0.002x + 1.6212 |
0.1029 |
| Higuchi |
y = 7.9869x + 26.655 |
0.6222 |
y = 10.472x + 35.915 |
0.6216 |
y = 3.042x + 37.864 |
0.5517 |
y = 3.545x + 51.582 |
0.4789 |
| Korsmeyer–Peppas |
y = 0.1465x + 1.6171 |
0.8535 |
y = 0.1299x + 1.7576 |
0.9213 |
y = 0.1408x + 1.5863 |
0.8029 |
y = 0.1261x + 1.7208 |
0.7422 |
| Hixson and Crowell |
y = −0.1023x + 2.711 |
0.7994 |
y = −0.1248x + 2.9183 |
0.7766 |
y = −0.013x + 2.9938 |
0.814 |
y = −0.0133x + 2.9997 |
0.7264 |
Effect of gelling time on Texas red (SR101) release
Interestingly, it was observed that the extent of burst release of Texas red from formulations F4 and F8 can be controlled by controlling the gelling time, i.e., by modulating the length of exposure of UV light prior to the introduction of release medium. This was tested by exposing Texas red-loaded F4 and F8 to three gelling times (computed gelling time (∼25 s), 2 times the computed time (∼50 s) and 4 times the computed time (∼100 s)). Fig. 5C represents the effect of gelling time on Texas red release from F4; and Fig. 5D represents the effect of gelling time on Texas red release from F8. As can be seen, 2-times and 4-times gelling time significantly reduced the burst release from both the formulations, F4 and F8, in 1× PBS. Formulations F4 released 32.3 ± 11.5% Texas red within first two hours when gelled for 25 seconds but the release went down to 11.3 ± 6.9% when gelled for 50 seconds and it was further diminished to 1.10 ± 0.99% when gelled for 100 seconds (Fig. 5C). Similar results were obtained for formulation F8 after two hours, 32.0 ± 10.1% (25 seconds), 11.5 ± 3.9% (50 seconds), and 0.4 ± 0.2% (100 seconds) (Fig. 5D). This indicated the ability of formulations F4 and F8 to attain tailored drug release based on the gelling time, which in turn governs the degree of crosslinking, and thus may instigate a resistance in release for Texas red through the hydrogel matrix. A recent study by Vigata et al. demonstrated the release profile of Cefazolin encapsulated in GelMa hydrogels. The group utilized 10% GelMa to fabricate hydrogels and a complete drug release was observed within 12 hours. In our study, we discovered a technique to sustain drug release by increasing the UV exposure time which resulted in controlling the burst release and in turn prolonging the release of a model small molecule from hydrogels over 10 days. We hypothesize that this sustained drug release is due to the improved crosslinking between GelMa and the crosslinkers.
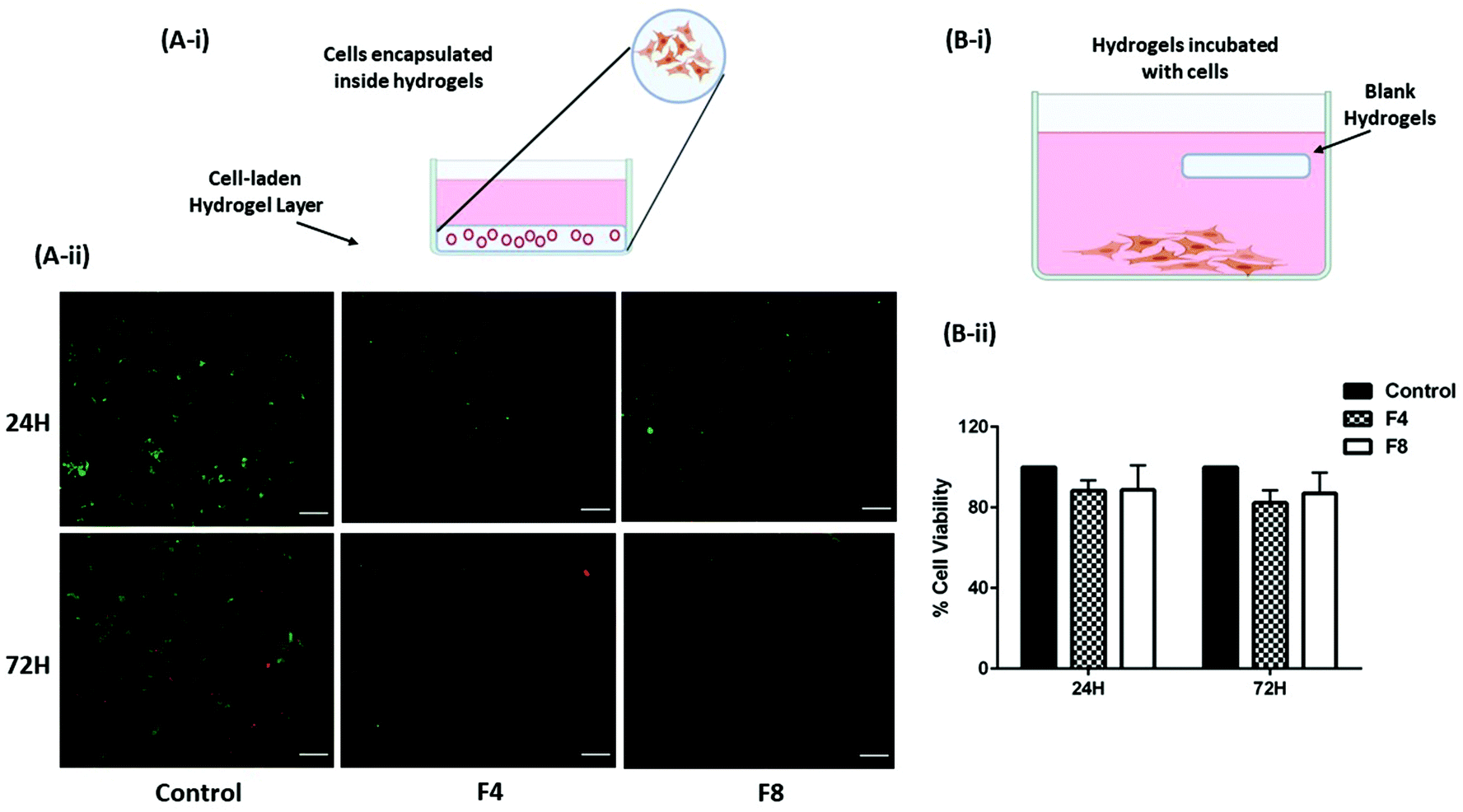
Normal human cells survive when encapsulated within blank hydrogels
GelMa hydrogels have the tendency to promote cell proliferation and differentiation59 and there is a probability that cells lining the mucus membrane of the physiological cavities may get encapsulated inside hydrogels in the process of cell growth (Fig. 6A-i). However, in such a scenario, the cell viability becomes very crucial as toxicity to cells while being encapsulated inside hydrogels would be deleterious to the homeostasis of the physiological cavities. Moreover, macroporous structure of hydrogels would promote an easy exchange of nutrients provided by the pleural fluid to promote regular cell growth. This was tested in vitro by encapsulating non-malignant HEK-293 cells inside hydrogels and staining the cells with calcein AM and Ethd-III staining the live cells green and dead cells red, respectively. This fluorescent staining was visualized using Evos FL fluorescence microscope (Life Technologies, Carlsbad, CA, USA). As can be seen in Fig. 6A-ii, the intensity of red fluorescence is least for control group for both 24- and 72-hours treatment; and is comparable to cells encapsulated within formulations F4 and F8. These data are further supported by cytotoxicity data computed by incubating formulations F4 and F8 with HEK-293 cells.
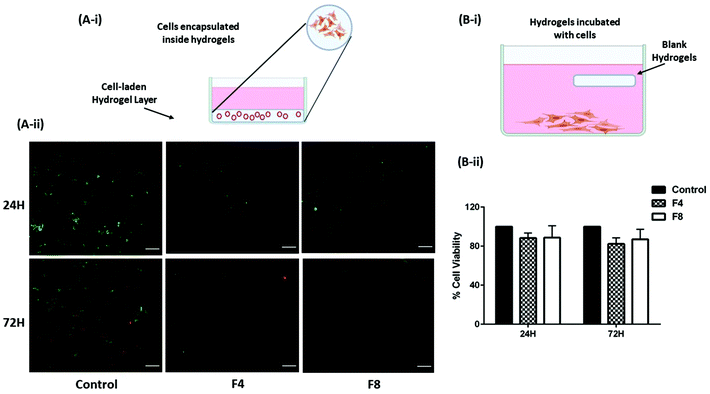 |
| | Fig. 6 (A-i) Representation of live/dead cell assay performed on non-malignant human HEK-293 cells. Briefly, cells were suspended with hydrogel PS for formulations F4 and F8 and were seeded in 96-well plates followed by curing hydrogel PS. Cells were then supplied with fresh cell media to supplement cell growth. (A-ii) Images representing live cells stained in green and dead cells stained in red after 24- and 72-hours of incubation. Formulations F4 and F8 did not exhibit significant dead cells relative to control group of cells. This indicates that cells are not necrotic even after encapsulation within hydrogels. (B-i) Representation of MTT assay protocol. Briefly, HEK-293 cells were seeded and allowed to adhere in a 24-well plate format. Following day, blank hydrogel pieces were incubated with cells and incubated further for 24- and 72-hours. (B-ii) Graph represents % cell viability assessed using MTT assay. No significant toxicity was observed with formulations F4 and F8 that were incubated with cells. Data represents mean ± SD with n = 3. | |
Blank hydrogels and individual hydrogel formulation components are not cytotoxic toward non-malignant human cells in vitro
To assess the biocompatibility of formulations F4 and F8, their toxicity against representative non-malignant human embryonic kidney cells (HEK-293) was tested. Hydrogels were incubated along with adherent HEK-293 cells which represents intracavitary physiological residence of hydrogels as (Fig. 6B-i). Moreover, potential toxicity of hydrogel excipients that leach out over time would be assessed using this method. Two timepoints were fixed, 24- and 72-hours, and it was observed that no significant toxicity was caused in HEK-293 cells when incubated with formulations F4 and F8. After 24 hours, formulation F4 had 88.3 ± 5.2% cells viable whereas formulation F8 had 88.7 ± 12.2% cells viable relative to untreated control HEK-293 cells (Fig. 6B-ii). After a longer incubation period of 72-hours, cells incubated with formulation F4 were 82.3 ± 6.2% viable whereas cells incubated with formulation F8 were 86.9 ± 10.3% viable relative to untreated control HEK-293 cells (Fig. 6B-ii). A viability of >80% even after an incubation of 72-hours indicates negligible toxicity from blank formulations F4 and F8.
Fig. S4 and S5† represent the toxicity profile of blank hydrogel components, tested from a concentration range of the maximum component amount in formulation and diluted composition concentrations at 24 hours and 72 hours exposure, respectively. No apparent toxicity was observed from individual components after testing on non-malignant HEK-293 cells for 24- and 72-hours.
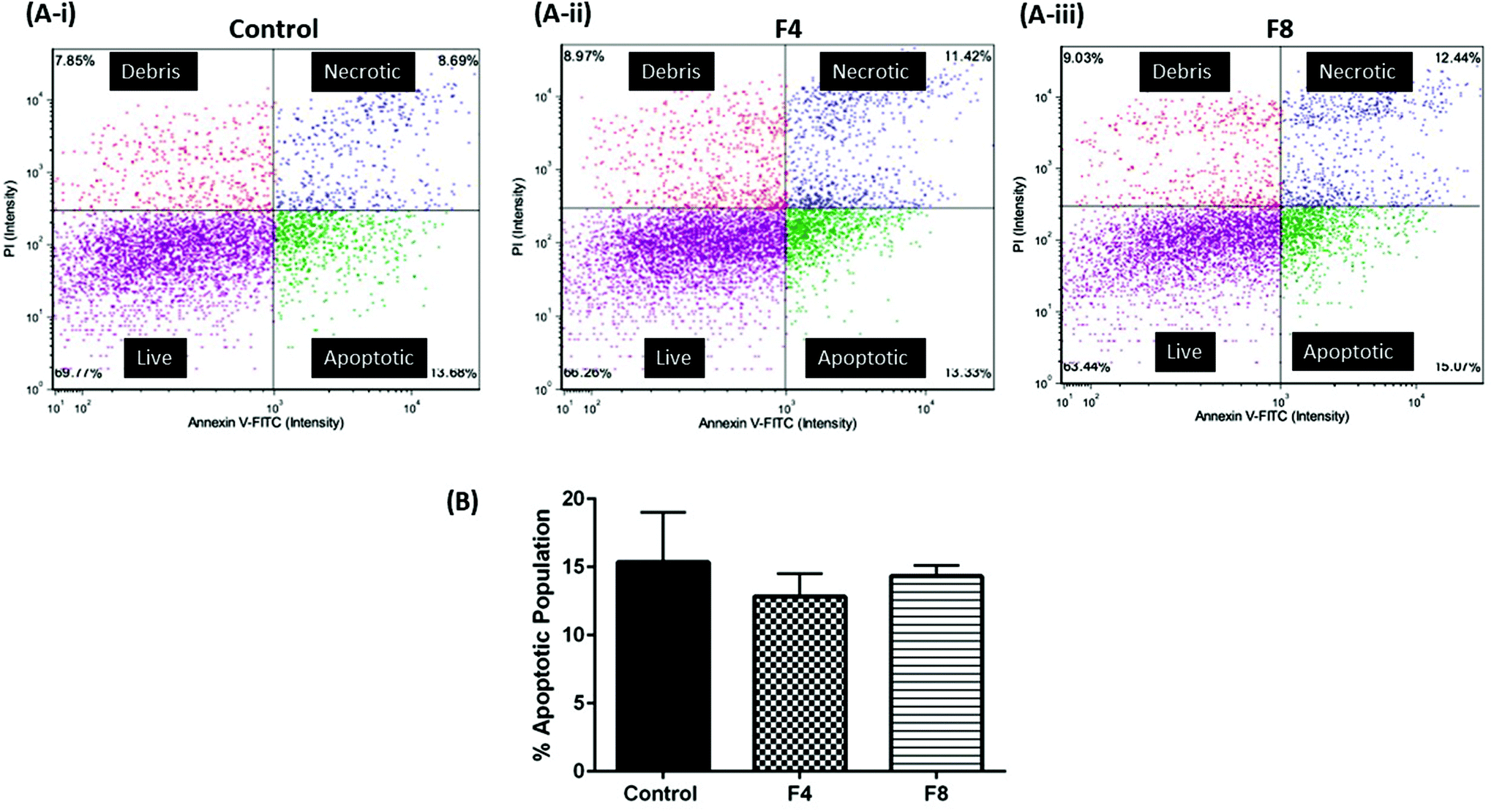
No significant apoptotic events occur when blank hydrogels are incubated with non-malignant human cells
Assessment of cellular viability may not represent the extent of apoptotic cells within a cell population. Thus, we evaluated the extent of apoptosis among cell population that was incubated with F4 and F8. Annexin-V is an identified marker for apoptosis and induction of Annexin-V levels is an identification of apoptotic induction. Annexin-V levels were evaluated in vitro in HEK-293 cells incubated with formulations F4 and F8 cells using Annexin-V FITC/propidium iodide assay (Nexcelom Bioscience, LLC, Lawrence, MA, USA). Briefly, compromised (apoptotic) cells would have increased levels of Annexin-V which would be stained green due to presence of FITC dye while necrotic cells with a compromised cell membrane would internalize propidium iodide, thus staining them red. As can be seen in Fig. 7, incubation with hydrogels for 24 hours resulted in no significant apoptosis relative to untreated control in HEK-293 cells. Fig. 7A shows representative images of cell population distribution, following treatments, based on their fluorescence staining. Fig. 7B quantifies the % apoptosis that occurred in cell populations incubated with formulations F4 and F8, compared with % apoptosis in non-malignant untreated HEK-293 cells. As can be seen, untreated cells exhibited a 15.4 ± 3.7% apoptotic population while formulation F4 resulted in 12.8 ± 1.7% apoptotic population and formulation F8 exhibited 14.3 ± 0.8% apoptotic population, with no significant difference observed among groups, which indicated toward no exacerbated apoptotic events post-hydrogel incubation in HEK-293 cells (Fig. 7B).
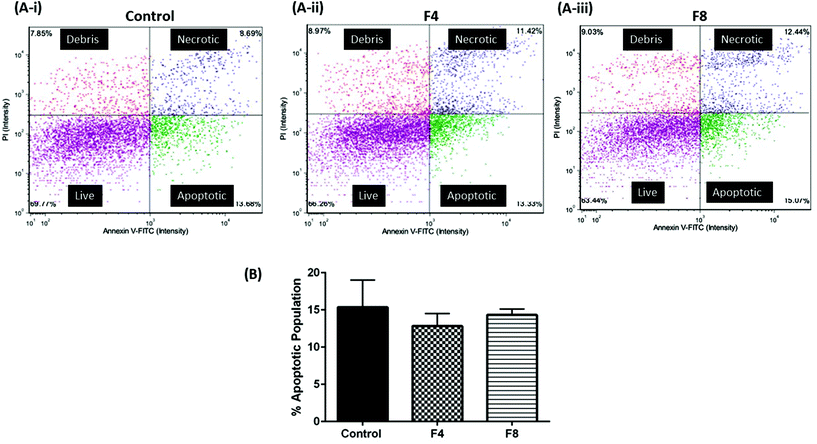 |
| | Fig. 7 (A-i), (A-ii) & (A-iii) Plots represent cell population categorized as live cells, apoptotic, necrotic and cell debris. Annexin-V FITC was used to stain apoptotic populations along with propidium iodide. (B) % apoptotic population of HEK-293 cells after 24-hour incubation with formulations F4 and F8. Apoptosis evaluation compliments viability assay and indicates no severe apoptotic events after hydrogel incubation relative to the untreated control group of cells. Data represents mean ± SD with n = 3. | |
Conclusion
While promising, this study presents a preliminary step in a long journey prior to clinical translation toward intracavitary applications of hydrogels. The current study develops and optimizes a GelMa based hydrogel capable of therapeutic delivery to physiological cavities, however, further pre-clinical studies are necessitated to establish the potency of this therapy. However, this comprehensive in vitro evaluation of GelMa based hydrogels along with LAP and BIS as crosslinkers is the first report to reveal key biophysical traits of this system. It was observed that GelMa, LAP and BIS contribute toward the physical characteristics and performance of hydrogels. GelMa influences the injectability of hydrogel precursor solution, which in turn governs the feasibility of intracavitary use. The amount of LAP in hydrogels governs the crosslinking time with GelMa, which in turn correlates to the time required for a complete sol–gel conversion. BIS aides this crosslinking process and improves the degree of crosslinking of hydrogels, leading to improved bio adhesion and sustained release of payloads. Gelling time not only determines the rate of gelling but also determines the efficacy of crosslinking which in turn may help tailor payload release profiles from hydrogels. An important feature of this formulation is the ability to have tunable release profiles and the ability to control burst release of payloads. Development of this sustained release therapy may not only provide patients with relief from frequent dosing but also improves the off target toxic effects, often associated with systemic administration. Developed hydrogel-based delivery systems, formulations F4 (5% w/v GelMa, 0.5% w/v LAP and 2.5% w/v BIS) and F8 (10% w/v GelMa, 0.5% w/v LAP and 2.5% w/v BIS), showcased promising characteristics and this prototype system can be used to deliver approved chemotherapies to extract the maximum therapeutic benefits.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This study was supported by funds provided to VG by College of Pharmacy and Health Sciences (CPHS), St. John's University. NSK was supported by research assistantship provided by an NIH Research Enhancement Award (R15), 1R15HL138606-01A1 to Dr Vivek Gupta. GC was supported by graduate teaching assistantship provided by College of Pharmacy and Health Sciences (CPHS), St. John's University. The authors would like to acknowledge the Imaging Facility of CUNY Advanced Science Research Center for instrument use, as well as scientific and technical assistance.
References
-
G. Tiarks, Dorsal and Ventral, https://www.osmosis.org/answers/dorsal-and-ventral.
- D. J. Sugarbaker, R. R. Gill, B. Y. Yeap, A. S. Wolf, M. C. DaSilva, E. H. Baldini, R. Bueno and W. G. Richards, J. Thorac. Cardiovasc. Surg., 2013, 145, 955–963 CrossRef CAS PubMed.
-
E. A. Monaco, Z. Tempel, A. Niranjan and L. D. Lunsford, Craniopharyngiomas, Elsevier, 2015, pp. 391–403 Search PubMed.
- P. Bertoglio, V. Aprile, M. C. Ambrogi, A. Mussi and M. Lucchi, J. Thorac. Dis., 2018, 10, S293–S297 CrossRef PubMed.
- Y. Mukai, Y. Minagawa, H. Inoue, A. Sato, K. Matsui, T. Fukuda, K. Onuma, H. Hongo, R. Shirata, H. Nagata, H. Hashimoto, T. Inoue, M. Hata and M. Omura, In Vivo, 2020, 34, 3387–3398 CrossRef PubMed.
- V. Murthy, K. Mangalick and D. H. Sterman, Clin. Chest Med., 2018, 39, 195–209 CrossRef PubMed.
- P. J. Pickhardt and S. Bhalla, RadioGraphics, 2005, 25, 719–730 CrossRef PubMed.
- M. T. Teixeira, L. L. Sa-Barreto, S. F. Taveira, T. Gratieri, G. M. Gelfuso, R. N. Marreto, I. C. Silva and M. Cunha-Filho, AAPS PharmSciTech, 2019, 21, 8 CrossRef PubMed.
- S. Turturro, S. Sunoqrot, H. Ying, S. Hong and B. Y. J. T. Yue, Mol. Pharm., 2013, 10, 3023–3032 CrossRef CAS PubMed.
- C. Norioka, Y. Inamoto, C. Hajime, A. Kawamura and T. Miyata, NPG Asia Mater., 2021, 13, 34 CrossRef CAS.
- Y. Hong, F. Zhou, Y. Hua, X. Zhang, C. Ni, D. Pan, Y. Zhang, D. Jiang, L. Yang, Q. Lin, Y. Zou, D. Yu, D. E. Arnot, X. Zou, L. Zhu, S. Zhang and H. Ouyang, Nat. Commun., 2019, 10, 2060 CrossRef PubMed.
- M. M. Benmassaoud, K. A. Gultian, M. DiCerbo and S. L. Vega, Ann. N. Y. Acad. Sci., 2020, 1460, 25–42 CrossRef CAS PubMed.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 16071 CrossRef CAS PubMed.
- N. Oliva, J. Conde, K. Wang and N. Artzi, Acc. Chem. Res., 2017, 50, 669–679 CrossRef CAS PubMed.
- M. Vigata, C. Meinert, S. Pahoff, N. Bock and D. W. Hutmacher, Polymers, 2020, 12, 501 CrossRef CAS PubMed.
- K. Yue, G. Trujillo-de Santiago, M. M. Alvarez, A. Tamayol, N. Annabi and A. Khademhosseini, Biomaterials, 2015, 73, 254–271 CrossRef CAS PubMed.
-
T. U. Rashid, S. Sharmeen, S. Biswas, T. Ahmed, A. K. Mallik, Md. Shahruzzaman, Md. N. Sakib, P. Haque and M. M. Rahman, in Cellulose-Based Superabsorbent Hydrogels, ed. Md. I. H. Mondal, Springer International Publishing, Cham, 2019, pp. 1601–1641 Search PubMed.
- A. I. Van Den Bulcke, B. Bogdanov, N. De Rooze, E. H. Schacht, M. Cornelissen and H. Berghmans, Biomacromolecules, 2000, 1, 31–38 CrossRef CAS PubMed.
- J. W. Nichol, S. T. Koshy, H. Bae, C. M. Hwang, S. Yamanlar and A. Khademhosseini, Biomaterials, 2010, 31, 5536–5544 CrossRef CAS PubMed.
- J. A. Benton, C. A. DeForest, V. Vivekanandan and K. S. Anseth, Tissue Eng., Part A, 2009, 15, 3221–3230 CrossRef CAS PubMed.
- J. A. Benton, B. D. Fairbanks and K. S. Anseth, Biomaterials, 2009, 30, 6593–6603 CrossRef CAS PubMed.
- B. D. Fairbanks, M. P. Schwartz, C. N. Bowman and K. S. Anseth, Biomaterials, 2009, 30, 6702–6707 CrossRef CAS PubMed.
- S. Van Vlierberghe, V. Cnudde, P. Dubruel, B. Masschaele, A. Cosijns, I. De Paepe, P. J. S. Jacobs, L. Van Hoorebeke, J. P. Remon and E. Schacht, Biomacromolecules, 2007, 8, 331–337 CrossRef CAS PubMed.
- Y. Piao, H. You, T. Xu, H.-P. Bei, I. Z. Piwko, Y. Y. Kwan and X. Zhao, Eng. Regen., 2021, 2, 47–56 Search PubMed.
- I. Noshadi, S. Hong, K. E. Sullivan, E. Shirzaei Sani, R. Portillo-Lara, A. Tamayol, S. R. Shin, A. E. Gao, W. L. Stoppel, L. D. Black III, A. Khademhosseini and N. Annabi, Biomater. Sci., 2017, 5, 2093–2105 RSC.
- A. Kokorovic and S. F. Matin, Ther. Adv. Med. Oncol., 2020, 12, 175883592093795 CrossRef.
- B. Sarker, D. G. Papageorgiou, R. Silva, T. Zehnder, F. Gul-E-Noor, M. Bertmer, J. Kaschta, K. Chrissafis, R. Detsch and A. R. Boccaccini, J. Mater. Chem. B, 2014, 2, 1470 RSC.
- S. N. Politis, P. Colombo, G. Colombo and D. M. Rekkas, Drug Dev. Ind. Pharm., 2017, 43, 889–901 CrossRef CAS PubMed.
- E. Marlowe and R. F. Shangraw, J. Pharm. Sci., 1967, 56, 498–504 CrossRef CAS PubMed.
- X. Li, Y. Wang, F. Xu, F. Zhang, Y. Xu, L. Tang and T. J. Webster, Int. J. Nanomed., 2020, 15, 4591–4606 CrossRef CAS PubMed.
- Y. Liu and M. B. Chan-Park, Biomaterials, 2010, 31, 1158–1170 CrossRef CAS PubMed.
- M. A. Sakr, K. Sakthivel, T. Hossain, S. R. Shin, S. Siddiqua, J. Kim and K. Kim, J. Biomed. Mater. Res., Part A, 2021, 110(3), 708–724 CrossRef.
- J. Cheng, Z. Chen, C. Liu, M. Zhong, S. Wang, Y. Sun, H. Wen and T. Shu, Nanomedicine, 2021, 16, 1567–1579 CrossRef CAS PubMed.
- A. D. Rouillard, C. M. Berglund, J. Y. Lee, W. J. Polacheck, Y. Tsui, L. J. Bonassar and B. J. Kirby, Tissue Eng., Part C, 2011, 17, 173–179 CrossRef CAS.
- H. Xu, J. Casillas, S. Krishnamoorthy and C. Xu, Biomed. Mater., 2020, 15, 055021 CrossRef CAS PubMed.
- M. Szkorupa, D. Klos, J. Chudacek, J. Hanuliak, M. Stasek, O. Fischer and R. Lemstrová, Rozhl. Chir., 2020, 99, 456–461 Search PubMed.
- A. Paul, A. Hasan, H. A. Kindi, A. K. Gaharwar, V. T. S. Rao, M. Nikkhah, S. R. Shin, D. Krafft, M. R. Dokmeci, D. Shum-Tim and A. Khademhosseini, ACS Nano, 2014, 8, 8050–8062 CrossRef CAS PubMed.
- W.-C. Chang, A.-Z. Tai, N.-Y. Tsai and Y.-C. E. Li, Polymers, 2021, 13, 2386 CrossRef CAS PubMed.
- H. Zhang, J. Xu and Saijilafu, Ann. Transl. Med., 2021, 9, 1147–1147 CrossRef CAS PubMed.
-
S. Varadhan, L. Hampton, K. Jackson and U. Chukwuemeka, Lung Compliance, https://www.physio-pedia.com/Lung_Compliance.
- J. R. Choi, K. W. Yong, J. Y. Choi and A. C. Cowie, BioTechniques, 2019, 66, 40–53 CrossRef CAS PubMed.
- J. Oh and B. Kim, Korea Aust. Rheol. J., 2020, 32, 41–46 CrossRef.
- H. Suo, D. Zhang, J. Yin, J. Qian, Z. L. Wu and J. Fu, Mater. Sci. Eng., C, 2018, 92, 612–620 CrossRef CAS PubMed.
- V. Gupta, B. H. Hwang, J. Lee, A. C. Anselmo, N. Doshi and S. Mitragotri, J. Controlled Release, 2013, 172, 753–762 CrossRef CAS PubMed.
- H. T. Lam, O. Zupančič, F. Laffleur and A. Bernkop-Schnürch, Int. J. Adhes. Adhes., 2021, 107, 102857 CrossRef CAS.
-
H. Holback, Y. Yeo and K. Park, Biomedical Hydrogels, Elsevier, 2011, pp. 3–24 Search PubMed.
- T. C. Lai, J. Yu and W. B. Tsai, J. Mater. Chem. B, 2016, 4, 2304–2313 RSC.
- B. Yan, J.-C. Boyer, D. Habault, N. R. Branda and Y. Zhao, J. Am. Chem. Soc., 2012, 134, 16558–16561 CrossRef CAS PubMed.
- S. Zhang, A. M. Bellinger, D. L. Glettig, R. Barman, Y.-A. L. Lee, J. Zhu, C. Cleveland, V. A. Montgomery, L. Gu, L. D. Nash, D. J. Maitland, R. Langer and G. Traverso, Nat. Mater., 2015, 14, 1065–1071 CrossRef CAS PubMed.
- Y. Hirokawa and T. Tanaka, J. Chem. Phys., 1984, 81, 6379–6380 CrossRef.
- A. N. Hurewitz, S. Zucker, P. Mancuso, C. L. Wu, B. Dimassimo, R. M. Lysik and D. Moutsiakis, Chest, 1992, 102, 1808–1814 CrossRef CAS PubMed.
- Z. Luo, W. Sun, J. Fang, K. Lee, S. Li, Z. Gu, M. R. Dokmeci and A. Khademhosseini, Adv. Healthcare Mater., 2019, 8, 1801054 CrossRef PubMed.
- Y. Wang, M. Ma, J. Wang, W. Zhang, W. Lu, Y. Gao, B. Zhang and Y. Guo, Materials, 2018, 11, 1345 CrossRef PubMed.
- M. Ehrbar, V. G. Djonov, C. Schnell, S. A. Tschanz, G. Martiny-Baron, U. Schenk, J. Wood, P. H. Burri, J. A. Hubbell and A. H. Zisch, Circ. Res., 2004, 94, 1124–1132 CrossRef CAS PubMed.
- N. Huebsch, C. J. Kearney, X. Zhao, J. Kim, C. A. Cezar, Z. Suo and D. J. Mooney, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9762–9767 CrossRef CAS.
- X. Huang and C. S. Brazel, Chem. Eng. Commun., 2003, 190, 519–532 CrossRef CAS.
- Y. Han, J. Yang, W. Zhao, H. Wang, Y. Sun, Y. Chen, J. Luo, L. Deng, X. Xu, W. Cui and H. Zhang, Bioact. Mater., 2021, 6, 3596–3607 CrossRef CAS PubMed.
- W. Teng, J. Cappello and X. Wu, J. Controlled Release, 2011, 156, 186–194 CrossRef CAS PubMed.
- J. Li, W. Wang, M. Li, P. Song, H. Lei, X. Gui, C. Zhou and L. Liu, Front. Bioeng. Biotechnol., 2021, 9, 770049 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |

 *
*