
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioAdhere: tailor-made bioadhesives for epiretinal visual prostheses
Kai-Wolfgang
Hintzen
ab,
Christian
Simons
b,
Kim
Schaffrath
c,
Gernot
Roessler
c,
Sandra
Johnen
c,
Felix
Jakob
ab,
Peter
Walter
c,
Ulrich
Schwaneberg
 ab and
Tibor
Lohmann
*c
ab and
Tibor
Lohmann
*c
aInstitute of Biotechnology, RWTH Aachen University, Aachen, Germany
bDWI – Leibniz-Institute for Interactive Materials, Aachen, Germany
cDepartment of Ophthalmology, RWTH Aachen University, Aachen, Germany. E-mail: tlohmann@ukaachen.de; Fax: +49-241-8082408; Tel: +49-241-8088191
First published on 18th May 2022
Abstract
Introduction: Visual prostheses, i.e. epiretinal stimulating arrays, are a promising therapy in treating retinal dystrophies and degenerations. In the wake of a new generation of devices, an innovative method for epiretinal fixation of stimulator arrays is required. We present the development of tailor-made bioadhesive peptides (peptesives) for fixating epiretinal stimulating arrays omitting the use of traumatic retinal tacks. Materials and methods: Binding motifs on the stimulating array (poly[chloro-p-xylylene] (Parylene C)) and in the extracellular matrix of the retinal surface (collagens I and IV, laminin, fibronectin) were identified. The anchor peptides cecropin A (CecA), KH1, KH2 (author's initials) and osteopontin (OPN) were genetically fused to reporter proteins to assess their binding behavior to coated microtiter plates via fluorescence-based assays. Domain Z (DZ) of staphylococcal protein A was used as a separator to generate a bioadhesive peptide. Following ISO 10993 “biological evaluation of medical materials”, direct and non-direct cytotoxicity testing (L-929 and R28 retinal progenitor cells) was performed. Lastly, the fixating capabilities of the peptesives were tested in proof-of-principle experiments. Results: The generation of the bioadhesive peptide required evaluation of the N- and C-anchoring of investigated APs. The YmPh–CecA construct showed the highest activity on Parylene C in comparison with the wildtype phytase without the anchor peptide. eGFP–OPN was binding to all four investigated ECM proteins (collagen I, laminin > collagen IV, fibronectin). The strongest binding to collagen I was observed for eGFP–KH1, while the strongest binding to fibronectin was observed for eGFP–KH2. The selectivity of binding was checked by incubating eGFP–CecA and eGFP–OPN on ECM proteins and on Parylene C, respectively. Direct and non-direct cytotoxicity testing of the peptide cecropin-A–DZ–OPN using L-929 and R28 cells showed good biocompatibility properties. Proof-of-concept experiments in post-mortem rabbit eyes suggested an increased adhesion of CecA–DZ–OPN–coated stimulating arrays. Conclusion: This is the first study to prove the applicability and biocompatibility of peptesives for the fixation of macroscopic objects.
Introduction
To this day, retinal degenerations and dystrophies remain leading causes of severe visual impairment and blindness, causing up to 50% of new-onset blindness in developed countries.1–3 Treatment remains difficult, especially in retinal dystrophies (i.e., retinitis pigmentosa). While efforts in gene therapy,4 optogenetic therapy5 and stem cell therapy6 have been made, the therapeutic approach with the longest history and most treated patients remains visual prostheses.7–9 In principle, the efficacy has been shown by epiretinal, subretinal and suprachoroidal stimulating arrays,10 while an epiretinal stimulating array is most frequently used in patients.11 To grant epiretinal fixation, titanium retinal tacks were introduced.12–14 While holding the epiretinal array in place, the tack causes unavoidable retinal damage and reactive gliosis.15,16 Also, a single retinal tack does not grant sufficient retinal alignment, especially in larger epiretinal arrays.16 Lastly, retinal tacks render the controlled and safe removal of an epiretinal device difficult.17 In the wake of a new generation of retinal prostheses, an innovative and effective fixating method is needed.The immobilization of functional moieties or biochemical groups or the fixation of microgels on surfaces has been achieved using bioadhesives, namely anchor peptides (APs). The functionalized surfaces include polymers such as polystyrene (PS), polypropylene (PP), polyethylene terephthalate (PET), polyester-polyurethane and polycaprolactone (PCL), metals such as stainless steel and gold, amorphous materials like silicon wafers and glass, and natural surfaces like cucumber and apple leaves.18–22 Some, but not all, APs have been derived from naturally occurring anti-microbial peptides (AMPs), like the AP Liquid Chromatography Peak I of Bacillus subtilis (LCI).23 Several modes of action are known, explaining the antimicrobial activity of AMPs by e.g., the self-assembled formation of pores in bacterial cell walls.24,25
APs bind to surfaces from aqueous solutions at ambient temperatures and under mild conditions.23 The binding properties are determined by the chemical composition of the APs and the material properties of the surface.26 It has been shown that AP binding to surfaces is governed by π–π, hydrophobic and electrostatic interactions as well as hydrogen bonding.27–29 Further influencing factors are the size and primary and secondary structure of the APs as well as the internal rigidity.26,27,30
Hence, binding can be fine-tuned using protein engineering methods to tailor the binding properties to a specific application. The substitution of negatively charged amino acids in the AP Liquid Chromatography Peak I of Bacillus subtilis (LCI) was shown to improve the binding on PP surfaces. Further, the introduction of positively charged amino acids and an increase in the grand average of hydropathy (GRAVY) led to improved binding, underlining the importance of electrostatic as well as hydrophobic interactions for AP binding. It was also established that more than two substitutions would weaken the binding ability to PP, suggesting that larger changes to the secondary structure have a negative influence on binding.31
APs can be exploited to functionalize synthetic and natural polymeric membranes with ease. Poly[chloro-p-xylylene] (Parylene C) is a commonly used polymeric coating in biomedical applications and, more recently, in organic electronics due to its biocompatibility.32,33 Its optical transparency and flexibility make it a suitable coating for retinal stimulating arrays.34 With currently limited available and feasible methods for epiretinal fixation, we investigated a recently reported application of APs for suitability: a bioadhesive peptide (peptesive) has been reported to join two chemically different materials – stainless steel and PCL.18 Therein, APs with different material binding properties – dermaseptin S1 (DS1) (stainless steel binder) and LCI (PCL binder) – are genetically fused to opposing ends of a spacer peptide, Domain Z (DZ). The DS1–DZ–LCI peptesive successfully demonstrated the ability to immobilize PCL micro-containers on stainless steel.18
The aim of this study was to demonstrate the proof-of-concept for the “tack-free” fixation of an epiretinal very large array for retinal stimulation (VLARS)16,34 using tailor-made peptesives. As targets for epiretinal adhesion, the extracellular matrix (ECM) proteins collagen I (col I), collagen IV (col IV), laminin, and fibronectin of the retinal inner limiting membrane (ILM), a basal membrane separating the vitreous body and underlying retinal cells, were identified.35–39 Parylene C coating was chosen as the binding target on the epiretinal stimulating arrays. Due to their hydrophobic properties, APs were chosen that reportedly also bind to other hydrophobic polymer surfaces such as PP (LCI and CecA)40 and PS (TA2, LCI and CecA).41 Like PS, Parylene C also harbors an aromatic ring and is hydrophobic. Hence, TA2, LCI and CecA were evaluated for their binding capabilities to Parylene C, exploiting potentially similar binding mechanisms like π–π and hydrophobic interactions.
The APs were identified based upon the reported biochemical and physical properties of APs and Parylene C. They were evaluated for their binding strength to the respective surface by either detection of the fluorescent reporter enhanced green fluorescent protein (eGFP) or activity detection of the reporter protein Yersinia mollaretii ATCC 43969 phytase (YmPhytase). eGFP constructs were used for detection on Parylene C-coated microtiter plates (MTP). YmPhytase constructs were used for detection on the Parylene C-coated VLARS. Here, autofluorescence of the array rendered eGFP detection infeasible. APs showing binding were then genetically assembled to peptesives binding both the retinal surface and the coated surface of the stimulating arrays. Afterwards, the biocompatibility of the peptesives was evaluated using standardized direct and non-direct cytotoxicity testing following ISO 10993, “Biological evaluation of medical materials”, part 5 (tests for in vitro cytotoxicity) and part 12 (sample preparation and reference material). Lastly, the fixating capabilities of the peptesives were tested in proof-of-principle experiments in post-mortem rabbit eyes.
Materials and methods
Plasmids and strains
Plasmid pET28a(+) (Novagen, Darmstadt, Germany) was used as the expression vector for AP-enhanced green fluorescent protein (eGFP) fusion constructs. Plasmid pALXtreme-5b was used for the expression of AP–phytase fusion constructs as previously reported.19,42E. coli strain DH5α (Agilent Technologies Inc., Santa Clara, USA) was used as a host for the cloning of DNA constructs. E. coli strain BL21 Gold (DE3) (Agilent Technologies Inc.) was used for the expression of eGFP fusion constructs. E. coli strain BL21 Gold (DE3) LacIQ1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 43 was used for the expression of phytase fusion constructs as previously reported.19
43 was used for the expression of phytase fusion constructs as previously reported.19
Generation of anchor peptide fusion constructs
APs were genetically fused to reporter proteins to assess their binding behavior via fluorescence-based assays. eGFP and the Yersinia mollaretii ATCC 43969 phytase (GenBank: JF911533.1; YmPhytase) were used as reporter proteins in this study.42 The consensus sequence H–V–F/W–Q/M–P/A–P/K has been reported as highly affine and specific for col I binding.44 In this study, the CBM of OPN (GLRSKSKKFRRPDIQYPDATDEDITSHM) and the peptides HVFQPP (KH1, author's initials) and HVWMAK (KH2) were investigated. The KH1 and KH2 sequences were chosen to include each amino acid of the consensus sequence, respectively. AP fusion constructs KH1–eGFP and eGFP–KH2 were generated via phosphorothioate-based ligase-independent gene cloning (PLICing).43 KH1 and KH2 were fused to the N- and C-terminus, respectively, to investigate the influence on binding behavior. The AP fusion constructs eGFP–osteopontin–collagen binding motif (OPN–CBM) and eGFP–cecropin A (CecA) were purchased in pET28a(+) from GenScript Biotech B.V. (Leiden, The Netherlands). N-terminal AP constructs contain an N-terminal AP, followed by a stiff 17 amino acid helix as a spacer (17H, AEAAAKEAAAKEAAKA)45 between the AP and the C-terminal phytase or eGFP reporter. C-terminal AP constructs contain an N-terminal eGFP reporter, followed by the 17H spacer domain and a tobacco etch virus (TEV) cleavage site (ENLYFQG)46 separating the C-terminal AP. The constructs 17H–TEV–eGFP and 17H–TEV–phytase were used as negative controls, respectively.The AP–phytase constructs were generated as previously reported.19 The AP–eGFP constructs were expressed as previously reported.23 After incubation, the cells were harvested by centrifugation (3200g, 20 min, 4 °C, Eppendorf centrifuge 5810 R, Eppendorf AG, Hamburg, Germany). Cell pellets were stored at −20 °C. The AP–eGFP constructs were purified as previously reported.23
Generation, expression, and purification of Domain-Z constructs
The DZ of staphylococcal protein A47 was used as a separator between two APs to generate a bioadhesive peptide for retinal and Parylene C binding. CecA–DZ–OPN–CBM and CecA–4 × DZ–OPN–CBM were ordered in the pET28(a) vector and expressed in E. coli BL21 Gold (DE3) according to the previously published procedure.48 An empty pET28(a) vector with no insert was expressed accordingly and used as negative control. The DZ constructs were purified using IgG Sepharose 6 Fast Flow, 10 mL columns (GE Healthcare Life Science, Chalfont St Giles, UK).Binding of AP–eGFP fusion constructs on ECM protein-coated microtiter plates
MTPs coated with col I, col IV, fibronectin and laminin respectively were purchased (Corning, New York, USA). MTPs coated with Parylene C were prepared as described below. The AP–eGFP fusion construct and the eGFP control containing cell pellets were resuspended in 5 mL gcell weight−1 phosphate buffered saline (PBS) buffer, pH 7.4. Cells were disrupted with ultrasound (5 min total time, 15 s on/off pulses, 60% amplitude, VCX 130 ultrasonic processor, Sonics & Materials Inc., Newton, USA) and lysate was clarified using centrifugation (21130g, 20 min, 4 °C, Eppendorf Centrifuge 5424R). The 50 μL volume of cell-free extract (CFE) was supplemented with 50 μL of PBS buffer in a black polypropylene MTP (Greiner Bio-One International GmbH, Frickenhausen, Germany). The fluorescence was measured using a 96-well MTP reader, Tecan Infinite® M1000Pro (Tecan Group, Männedorf, Switzerland; λexcitation = 488 nm, λemission = 509 nm, gain 50, 50 flashes, 400 Hz flash frequency). The CFEs were fluorescence normalized to 500 RFU by dilution with PBS.
Coated MTP wells were washed with 200 μL of PBS buffer (5 min, room temperature (RT), 600 rpm) and rinsed with 200 μL of ddH2O (5 min, RT, 600 rpm; Microplate Shaker TiMiX 5, Edmund Bühler GmbH, Bodelshausen, Germany). The volume of 50 μL of fluorescence normalized CFE (eGFP–AP and eGFP control) was supplemented with 50 μL of PBS buffer and incubated in coated MTP wells (10 min, RT, 600 rpm). Binding tests with purified proteins were conducted with a normalized protein concentration of 1 mg mL−1. The protein concentration was determined using a Pierce™ BCA Protein Assay Kit (ThermoFisher Scientific Inc., Waltham, USA). The incubated solution was removed and the wells were washed twice with PBS buffer (200 μL, 5 min, RT, 600 rpm). The residual eGFP fluorescence was measured as described above. Triplicate blank measurements of “empty” coated MTP wells were included to take the auto-fluorescence of coating proteins into consideration. The average blank was subtracted as noise before further analysis of eGFP fluorescence measurements. Statistical significance was analyzed using an unpaired t-test with Welch's correction with data consisting of the mean and standard deviation of all triplicate measurements.
Parylene C coating of stimulating array and microtiter plates
Parylene C (Parylene Dimer DPX-C; Specialty Coating Systems, Indianapolis, USA) was deposited onto the VLARS using a chemical vapor deposition (CVD) process (PDS 2010 LABCOTER 2; Specialty Coating Systems) with a final thickness of 3 μm and patterned using a dry-etching process with oxygen plasma. The Parylene C encapsulation serves as an additional moisture barrier due to its hydrophobic behavior. Additionally, Parylene C is moldable under the influence of elevated temperatures, allowing for adaption to a specific surface.34 MTP wells were similarly coated using CVD with a final Parylene C thickness of 4.5 μm.Binding of phytase constructs on Parylene C
AP–phytase fusion constructs and phytase control containing cell pellets were resuspended in 6 mL gcell pellet−1 50 mM Tris/HCl buffer, pH 7.4. Cells were disrupted with ultrasound (5 min total time, 15 s on/off pulses, 60% amplitude, VCX 130 ultrasonic processor, Sonics & Materials Inc.) and lysate was clarified using centrifugation (21130g, 20 min, 4 °C, Eppendorf Centrifuge 5424R; Eppendorf AG). The activity of AP–phytase fusion constructs and phytase control in solution and on Parylene C as a binding substrate was evaluated using the 4-methylumbelliferyl-β-D-phosphate (4-MUP) assay.42 Therefore, a 10 mM 4-MUP stock was prepared in 250 mM sodium acetate buffer (pH 5.5, 250 mM sodium acetate, 0.01% Tweem-20, 1 mM CaCl2) and diluted to 0.5 mM 4-MUP in 250 mM sodium acetate buffer during the activity assay. CFEs were diluted 1:100 in 50 mM Tris/HCl buffer, pH 7.4. Next, 60 μL of diluted CFEs was supplemented with 140 μL of 250 mM sodium acetate buffer. Of this solution, 25 μL was added to a black PP MTP and 25 μL of 4-MUP was added to a final concentration of 0.5 mM. The activities of phytase constructs were evaluated in a Tecan Infinite® M1000Pro (Tecan Group, λexcitation = 360 nm, λemission = 465 nm, gain 100, 50 flashes, 400 Hz flash frequency). AP–phytase fusion constructs and phytase control were normalized according to their activity by dilution in 50 mM Tris/HCl buffer, pH 7.4.
VLARS made from polyimide 2611 coated with Parylene C were provided by the Department of Ophthalmology (University Hospital RWTH Aachen) and the Institute of Materials in Electrical Engineering 1 (RWTH Aachen University).16,34 The VLARS structures were cut into 5 by 5 mm square pieces and added to sample vials with snap-caps (VWR International, Radnor, USA). Samples were coated by the addition of 60 μL of normalized AP–phytase constructs and phytase controls and incubated for 10 min at 600 rpm and RT. The solution was removed, and the samples were rinsed once with 100 μL PBS (10 min, 600 rpm, RT) and washed twice with 100 μL 0.125 mM sodium dodecylbenzenesulfonate (LAS) to remove any non-specifically bound protein and to reduce phytase control binding. Next, the activity assay was started by the addition of 600 μL of 0.5 mM 4-MUP in 250 mM sodium acetate buffer. The activity was monitored by transferring 50 μL of the reaction solution at 30 min intervals for 3 h to a black PP MTP, diluting with 50 μL of 250 mM sodium acetate buffer, measuring the fluorescence using a Tecan Infinite® M1000Pro (Tecan Group, λexcitation = 360 nm, λemission = 465 nm, gain 100, 50 flashes, 400 Hz flash frequency). The statistical significance was analyzed using an unpaired t-test with Welch's correction with data consisting of the mean and standard deviation of all triplicate measurements.
Direct and non-direct cytotoxicity testing
Corresponding to the standard ISO10993 “Biological evaluation of medicinal devices” parts 5 and 12 and to previous work published by Johnen et al., biocompatibility was tested by analyzing the cell growth after direct contact as well as non-direct contact, for which cells were incubated with the respective extractive media.49 Following the standard ISO protocol, sensitive L-929 cells were used. Additionally, the retinal precursor cell line R28 was used to mimic the targeted neurosensory retinal tissue.50The L-929 cells (ATCC No. CCL-1) were maintained in minimum essential medium (MEM) with Earle's salts (PAN-Biotech GmbH, Aidenbach, Germany) supplemented with 10% fetal bovine serum (FBS; PAN-Biotech GmbH), 2 mM L-glutamine (Sigma-Aldrich, St Louis, USA), 80 μ mL−1 penicillin, and 80 μg mL−1 streptomycin (Sigma-Aldrich) at 37 °C in a humidified atmosphere of 95% air and 5% CO2. The retinal precursor cell line R28, originating from an immortalized postnatal day six rat retinal culture, was maintained in Dulbecco's modified Eagle's medium (DMEM; PAN-Biotech) supplemented with 10% FBS (PAN-Biotech), 5.5 mL of 100× MEM vitamins (ThermoFisher Scientific Inc.), non-essential amino acids (Biochrom, Berlin, Germany), 80 μ mL−1 penicillin, and 80 μg mL−1 streptomycin (Sigma-Aldrich) at 37 °C in a humidified atmosphere of 95% air and 5% CO2. The medium was changed twice a week. The cells were passaged once a week at a ratio of 1:10.
In direct cytotoxicity testing, L-929 and R28 cells were cultivated on glass or peptesive-coated polyethylene at a density of 31250 cells per cm2 or 10000 cells per cm2, respectively. Cell viability was measured after 72 h of cultivation using fluorescein diacetate (FDA; 5 μg mL−1 in PBS 0.1% acetone, Sigma-Aldrich), which stains living cells green, and propidium iodide (PI; 10 μg mL−1 in PBS 0.1% acetone, Sigma-Aldrich), staining dead cells red. Immediately after staining, cell viability was evaluated using fluorescence microscopy (Leica DM RX Microscope, Leica Microsystems, Wetzlar, Germany) in several randomly selected microscopic fields. In total five runs were conducted, each containing two cultures grown on peptesive-coated polyethylene and one culture grown on glass as the corresponding control. The total cell number was normalized to the respective glass control. The number of dead cells was calculated as the percentage of the corresponding total cell number. Data (mean ± standard deviation) were compared with the glass control and analyzed using an unpaired two-tailed t-test.
In non-direct cytotoxicity testing, materials were incubated in MEM with Earle's salts (PAN-Biotech GmbH) for L-929 cells and DMEM (PAN-Biotech) for R28 cells at 37 °C for 72 h in a humidified atmosphere of 95% air and 5% CO2. The extractive media were diluted (1:1, 1:2, and 1:4) and applied to L-929 and R28 cells that were seeded at a density of 31250 cells per cm2 and pre-incubated at 37 °C for 24 h. Viability was analyzed using the CellTiter-Glo luminescent Cell Viability Assay (Promega, Madison, WI, USA), which is based on the quantification of adenosine triphosphate, indicating the presence of metabolically active cells, according to the manufacturer's protocol. Positive and negative reference materials (RM; Hatano Research Institute, Hadano, Japan) were used as controls: the positive RM A shows a moderate level of cytotoxicity and consists of a polyurethane film containing 0.1% zinc ethyldithiocarbamate; RM B contains 0.25% zinc dibutyldithiocarbamate and has a weaker cytotoxic effect; the negative RM C consists of a high-density polyethylene film, which is not cytotoxic. Moreover, cell culture medium containing 10% dimethylsulfoxide (DMSO 10%) was used as an additional positive cytotoxic reference. Cells cultivated on glass were used as another negative control. Each material was examined twice for every dilution (1:1, 1:2, and 1:4) in a total of five runs. The mean values for every dilution were taken for each run and used for the statistical analysis (ordinary one-way ANOVA (Dunnett's multiple comparison test)).
Ex vivo proof-of-principle experiments evaluating the adhesive capabilities in post-mortem rabbit eyes
To test the adhesive capabilities, the peptesive-coated VLARS were implanted in post-mortem rabbit eyes, which were obtained from the Institute of Laboratory Animal Science (RWTH Aachen University). To reach the posterior pole, open-sky access was performed by removing the anterior segment of the eye at the pars plana using a surgical scalpel (No. 11; Feather Safety Razor Co. Ltd, Osaka, Japan) and curved corneal scissors (Geuder AG, Heidelberg, Germany). Using a 5 ml syringe (B. Braun Melsungen AG, Melsungen, Germany), the vitreous body was carefully removed in its entirety, revealing the posterior retinal pole. The coated arrays were carefully placed on the bare retinal surface and incubated for 60 min at room temperature without a liquid intraocular tamponade. The adhesion was tested by carefully lifting the edges of the array from the retinal surface using surgical forceps (Geuder AG, Heidelberg, Germany). The results were documented with a binocular microscope camera system (M80; Leica Microsystems, Wetzlar, Germany).Additionally, spectral-domain optical coherence (SD-OCT) and infrared imaging (Spectralis-OCT, Heidelberg Engineering GmbH, Heidelberg, Germany; Spectralis software version 6.16.2; high-resolution mode; scan focus 41.12 D; camera objective, wide field 2; scan angle, 55°; pattern size, 55 × 15°; distance between B-scans, 145 μm; automatic real time (ART) mode enabled) was performed on enucleated rabbit eyes after open-sky lentectomy and vitrectomy.
Statistical analysis
All statistical analyses were performed with GraphPad Prism (GraphPad Prism V7, San Diego, USA). Specific information on the statistical tests used can be found in the corresponding paragraphs.Results
Anchor peptide binding to extracellular matrix proteins
Binding of AP–eGFP fusion constructs eGFP–OPN–CBM, eGFP–KH2 and KH1–eGFP to ECM protein (col I, col IV, laminin and fibronectin) coated MTPs was analyzed using fluorescence measurements. The eGFP control was used to take any unspecific adsorption of the reporter protein into consideration. After elimination of unspecific eGFP–reporter protein adsorption and autofluorescence of the ECM-coated MTP wells, eGFP–OPN–CBM showed strong adsorption to col I (76.0 ± 18.0 residual fluorescence units (RFU)), col IV (26.0 ± 8.0 RFU), laminin (57.0 ± 18.0 RFU), and fibronectin (42.0 ± 7.0 RFU). Binding to col IV and fibronectin appeared to be slightly reduced in comparison with binding to col I and laminin. The strongest binding to col I was observed for KH1–eGFP (160.0 ± 12.0 RFU), which otherwise showed only weak binding to laminin (6.0 ± 5.0 RFU) and no observable binding to col IV or fibronectin. The strongest fibronectin binding was observed for eGFP–KH2 (171.0 ± 36.0 RFU), which also showed binding to col I (67.0 ± 21.0 RFU) and a reduced binding to col IV (12.0 ± 3.0 RFU), but no binding to laminin (Fig. 1). To sum up, eGFP–OPN–CBM was the only AP that showed binding capabilities to all four investigated ECM proteins.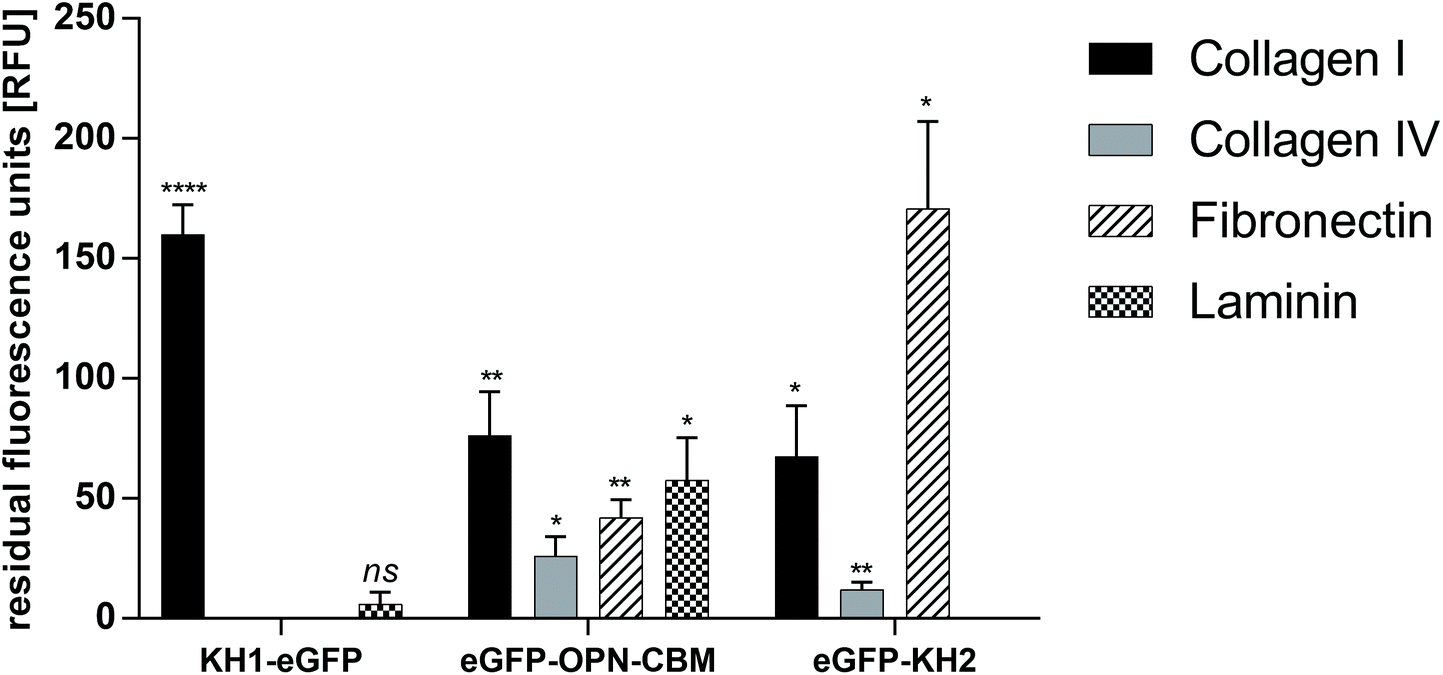 | ||
| Fig. 1 Results of the eGFP–AP screening on ECM-protein-coated MTP plates. Blank and eGFP control values were subtracted from each data point prior to analysis to compensate for autofluorescence of the ECM coating and unspecific binding of the eGFP reporter protein. KH1–eGFP shows strong binding to collagen I, no significant binding to collagen IV and fibronectin and weak binding to laminin. eGFP–osteopontin binds to all four tested ECM proteins, strongest on collagen 4 and laminin. eGFP–KH2 shows strong binding to fibronectin, binding to collagen IV and weak binding to collagen IV. eGFP = enhanced green fluorescent protein, KH1/KH2 = HVFQQPP (KH1)/HVWMQAK (KH2), author's initials, OPN–CBM = osteopontin collagen binding motif. n = 3, data are represented as mean ± standard deviation. Statistical analysis with unpaired t-test with Welch's correction. * p < 0.05, ** p < 0.01, **** p < 0.0001, ns = not significant. Statistical significance is given for the comparison of the eGFP control with KH1–eGFP, eGFP–OPN–CBM and eGFP–KH2 on ECM protein-coated MTPs respectively. | ||
Anchor peptide binding to Parylene C
Binding of AP–phytase fusion constructs LCI–YmPh, TA2–YmPh and CecA–YmPh to Parylene C was determined using the 4-MUP assay. Phytase without the anchor peptide was used as a control to account for any unspecific adsorption of the reporter protein. The reduction of unspecific protein adsorption was achieved by washing the Parylene C coated arrays twice with 0.125 mM LAS detergent. The activities of each immobilized construct were measured over time and the increase in the fluorescence signal was determined. The respective slopes of the fluorescence signal over time were compared with the wildtype phytase without AP. An increase in the slope ratio Variant/WT indicated a higher increment in fluorescence over time, suggesting a better and more wash-resistant immobilization of phytase on Parylene C.Constructs employing LCI, TA2 and CecA showed an increased adsorption to Parylene C of 1.2-, 2.9-, and 2.2-fold, respectively. All constructs showed binding to Parylene C (Fig. 2).
 | ||
| Fig. 2 Comparison of RFU min−1 slopes of each phytase construct in relation to WT control after 2 × 0.125 mM LAS washing. The C-anchored construct of YmPh–CecA shows a 6.4-fold higher slope. This indicates a 6.4-fold increased immobilization of phytase and hence shows that anchor peptide cecropin A is able to bind to Parylene C. N-anchored constructs of LCI–YmPh, TA2–YmPh and CecA–YmPh show 1.25-, 2.95- and 2.2-fold increased immobilization of phytase. The C-anchored CecA AP is the strongest binder. WT = wildtype Yersinia molaretti phytase, no anchor peptide; LCI–YmPh = liquid-chromatography peak 1 – Yersinia molaretti phytase construct; TA2–YmPh = tachystatin A 2 – Yersinia molaretti phytase construct; CecA–YmPh = cecropin A – Yersinia molaretti phytase construct; YmPh – CecA = Yersinia molaretti phytase – cecropin A construct. n = 3, data are represented as slope ratio ± standard deviation. Statistical analysis with unpaired t-test with Welch's correction. ** p < 0.01, ns = not significant. Statistical significance is given for the comparison of LCI–YmPH, TA2-YmPH, CecA–YmPH and YmPh–CecA to WT YmPh on Parylene C respectively. | ||
Selectivity of AP binding to ECM proteins and Parylene C
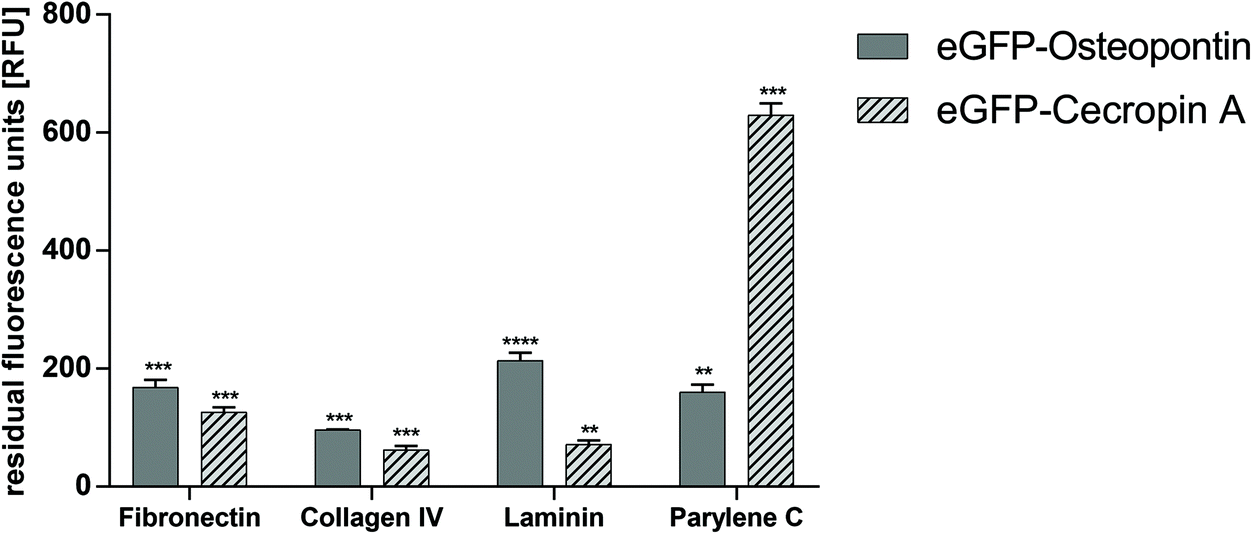
The selectivity of binding was checked by incubating eGFP–CecA and eGFP–OPN–CBM on ECM proteins and on Parylene C, respectively. The strongest binding to Parylene C was observed for the eGFP–CecA construct (630.0 ± 20.0 RFU). A significantly decreased binding to Parylene C was observed for OPN–CBM (160.0 ± 13.0 RFU). The binding of OPN–CBM was observed on fibronectin (168.0 ± 13.0 RFU), col IV (96.0 ± 1.0 RFU), and laminin (214.0 ± 13.0 RFU) (Fig. 3).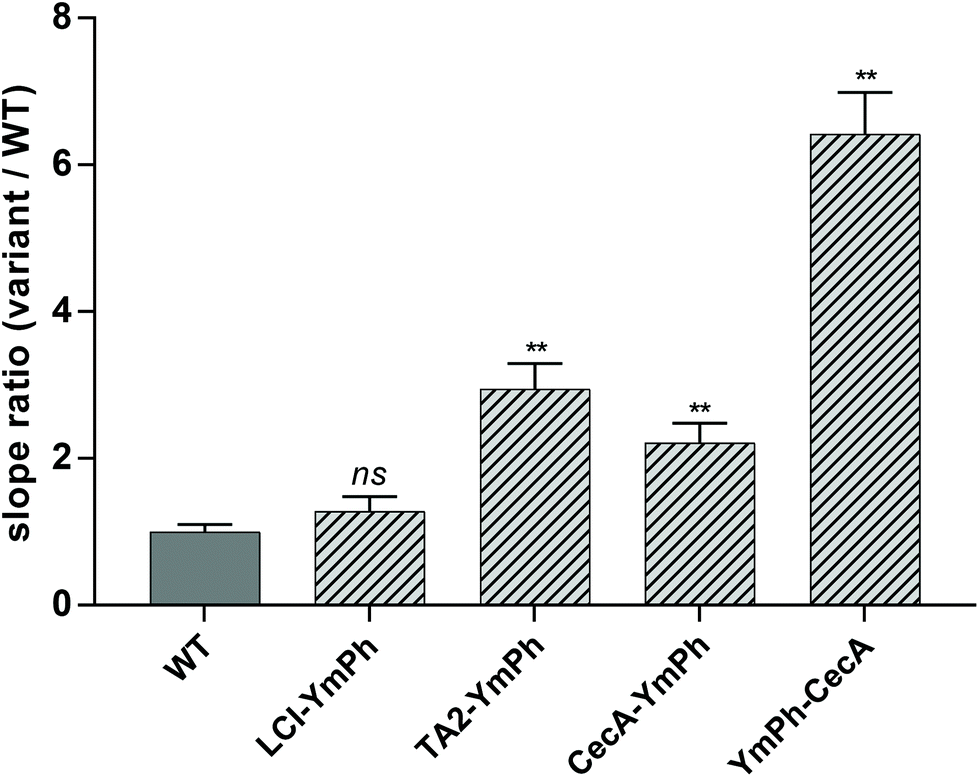 | ||
| Fig. 3 Results of selectivity screening of CecA and OPN–CBM on ECM and Parylene C. Blank and eGFP–control values were subtracted from each data point prior to analysis to compensate for autofluorescence of the ECM and Parylene C coating and unspecific binding of the eGFP reporter protein. Cecropin A shows the strongest binding to Parylene C and significantly weaker binding to ECM components. OPN–CBM shows binding to all ECM proteins and significantly weaker binding to Parylene C compared with cecropin A. eGFP = enhanced green fluorescent protein, OPN–CBM = osteopontin collagen binding motif. n = 3, data are represented as mean ± standard deviation. Statistical analysis with unpaired t-test with Welch's correction. ** p < 0.01, *** p < 0.001, **** p < 0.0001. Statistical significance is given for the comparison of eGFP control with eGFP–OPN–CBM and eGFP–CecA on ECM protein coated MTPs respectively. | ||
Effects of direct contact on cell viability
Both L-929 and R28 cells were able to grow on polyethylene coated with the peptesive CecA–DZ–OPN–CBM. While L-929 cells grew uniformly on the glass and coated polyethylene (Fig. 4A/B), R28 cells showed cluster growth (Fig. 4C/D). | ||
| Fig. 4 Effects of direct and indirect contact on cell viability. A/B: Viability of L-929 cells in direct contact with peptide CecA–DZ–OPN–CBM. In fluorescence microscopy vital cells are green, dead cells are red. A: L-929-cells on a CecA–DZ–OPN–CBM-coated wafer. B: L-929-cells on glass as a control. The L-929-cells grow evenly distributed. Note the low quantity of dead cells (red). C/D: Viability of R28 cells in direct contact with peptide CecA–DZ–OPN–CBM. In fluorescence microscopy vital cells are green, dead cells are red. C: R28-cells on a CecA–DZ–OPN–CBM-coated wafer. D: R28-cells on glass as a control. The R28 cells grow in patches. Again, only a few dead cells can be observed (red). E: Effects on the total cell number of L-929 cells in direct contact. Cell viability measured after 72 h of exposure. Total cell number of L-929-cells normalized to cells plated on glass control. No significant difference in cell number detected (glass, n = 5; CecA–DZ–OPN–CBM, n = 10). F: Effects on total cell number of R28 cells in direct contact. Cell viability measured after 72 h of exposure. Total cell number of R28 cells normalized to cells plated on glass control. No significant difference in cell number detected (glass, n = 5; CecA–DZ–OPN–CBM, n = 8). Data are represented as mean ± standard deviation. ns = not significant. G: Percentage of dead L-929 cells. No significant difference in cell death detected. H: Percentage of dead R28-cells. No significant difference in cell death detected. I: Effects of indirect contact on cell viability of L-929 cells (n = 5). Cell viability measured after 24 h of exposure. Tests of 1:1 dilution of extracted media from CecA–DZ–OPN–CBM, 10% dimethylsulfoxide (DMSO 10%) as a negative control and certified positive (RM A, RM B) and negative (RM C) reference materials. Extractive medium from glass was used as an internal control; its mean values are marked by the dotted lines. Significant decrease in luminance for DMSO 10% (p < 0.0001), RM A (p < 0.0001) and RM B (p < 0.0001). No difference in luminance for CecA–DZ–OPN–CBM (p = 0.9302) and RM C (p = 0.9980) compared with the glass control. J: Effects of indirect contact on cell viability of R28 cells (n = 5). Cell viability measured after 24 h of exposure. Tests of 1:1 dilution of extracted media from CecA–DZ–OPN–CBM, 10% dimethylsulfoxide (DMSO 10%) as a negative control and certified positive (RM A, RM B) and negative (RM C) reference materials. Extractive medium from glass was used as internal control; its mean values are marked by the dotted lines. Significant decrease in luminance for DMSO 10% (p < 0.0001), RM A (p < 0.0001) and RM B (p < 0.0001). No difference in luminance for CecA–DZ–OPN–CBM (p > 0.9999) and RM C (p = 0.9915) compared with the glass control. Data are represented as mean ± standard deviation. Statistical analysis with ordinary one-way ANOVA (Dunnett's multiple comparison test). **** p < 0.0001, ns = not significant. | ||
In the case of the L-929 cells, the total number of cells cultivated on glass was similar to the number cultivated on the peptide-coated polyethylene (glass, 1543.9 ± 726.9; CecA–DZ–OPN–CBM, 1561.8 ± 597.9; Fig. 4E). The percentage of dead cells was 1.1 ± 1.7% on glass and 1.1 ± 1.3% on coated polyethylene (Fig. 4G). There was no statistically significant difference in cell viability between the two groups (glass vs. CecA–DZ–OPN–CBM, p = 0.893) or in the percentage of dead cells (glass vs. CecA–DZ–OPN–CBM, p = 0.809).
In the case of the R28 cells, the total number of cells cultivated on glass was similar to the number cultivated on coated polyethylene (glass, 1023.2 ± 542.4; CecA–DZ–OPN–CBM, 1105.0 ± 561.7; Fig. 4F). The percentage of dead cells was 0.9 ± 1.2% for the glass control and 1.9 ± 3.7% for coated polyethylene (Fig. 4H). There was no statistically significant difference in cell viability between the two groups (glass vs. CecA–DZ–OPN–CBM, p = 0.839) or in the percentage of dead cells (glass vs. CecA–DZ–OPN–CBM, p = 0.088).
Effects of non-direct contact on cell survival
The effects of the extractive media, obtained by incubation of MEM with Earle's salts (PAN-Biotech GmbH) for L-929 cells and DMEM (PAN-Biotech) for R28 cells in contact with CecA–DZ–OPN, glass, DMSO 10% and RM A, RM B and RM C, on cell survival of L-929 cells showed no difference in cell survival compared with the glass control (CecA–DZ–OPN, 2.76 × 106 ± 2.97 × 105 RLU vs. glass, 2.88 × 106 ± 3.22 × 105; p = 0.931; Fig. 4I). There was no difference in cell survival comparing the extractive media from the peptesive-coated polyethylene with the extractive media from the glass control or extractive media from the negative control RM-C (RM-C, 2.82 × 106 ± 2.69 × 105 RLU vs. glass, 2.88 × 106 ± 3.22 × 105; p = 0.998; Fig. 4I). The positive (cytotoxic) controls DMSO 10%, RM A and RM B showed a reduction of luminescence compared with the glass control (DMSO 10%, 1.19 × 106 ± 4.04 × 105 RLU; p = <0.001; RM A, 5.43 × 103 ± 6.12 × 102 RLU; p = <0.001; RM B, 6.38 × 104 ± 1.11 × 105 RLU; p = <0.001; Fig. 4I). The reduction of luminescence in the positive (cytotoxic) controls decreased with increased dilution corresponding to a reduced cytotoxicity (data for 1:2 and 1:4 dilution not shown).
For the survival of R28 cells similar results were obtained, with no difference comparing CecA–DZ–OPN and the glass control (CecA–DZ–OPN, 2.84 × 106 ± 4.37 × 105 RLU vs. glass, 2.84 × 106 ± 5.05 × 105; p = >0.999; Fig. 4J). No difference in cell survival comparing the extractive media from the peptesive coated polyethylene with the extractive media from glass control or extractive media from the negative control RM C was detected (RM C, 2.95 × 106 ± 4.03 × 105 RLU vs. glass, 2.84 × 106 ± 5.05 × 105; p = 0.992; Fig. 4J). The positive (cytotoxic) controls DMSO 10%, RM A and RM B showed a reduction of luminescence compared with the glass control (DMSO 10%, 8.11 × 104 ± 4.40 × 104 RLU; p = <0.001; RM A, 2.55 × 103 ± 6.69 × 102 RLU; p = <0.001; RM B, 1.65 × 104 ± 1.60 × 104 RLU; p = <0.001; Fig. 4J). Again, reduction of luminescence in the positive (cytotoxic) controls decreased with increased dilution corresponding to reduced cytotoxicity (data for 1:2 and 1:4 dilutions not shown).
Retinal adhesion in post-mortem rabbit eyes
Via an open-sky approach, the vitreous cavity was accessed safely. Using a syringe, the vitreous body was completely removed. After removal of the vitreous, the coated arrays were safely placed on the posterior retinal pole. Incubating for 60 min at room temperature simulated a post-surgical air tamponade in an in vivo procedure.Overall, the retina was attached to the coated array when the array's wings were carefully lifted from the surface with a surgical forceps. Coating with the peptide CecA–1 × DZ–OPN–CBM showed epiretinal adhesion, as seen in the light reflections and elevated intraretinal blood vessels (Fig. 5A, black arrows). Greater traction would separate the array eventually, without permanently damaging the retina underneath. The CecA–4 × DZ–OPN–CBM coating showed epiretinal attachment (Fig. 5B). There was a visible ridge in the retina caused by its attachment to the lifted wing of the coated array (Fig. 5B, white arrows; note the sharp turn in retinal vessels). Yet again, greater forces applied with the surgical forceps lifted the array of the retina without causing retinal damage. Having a larger spacer domain (4 × DZ being four times as large) did not have noticeable benefits for the adhesion in this experiment. Fig. 5C shows the EV-coated control array. Here, epiretinal adhesion was not achieved, as the array peeled off without adhering to the retinal surface. SD-OCT imaging showed signs of closer epiretinal adhesion in CecA–DZ–OPN–CBM-coated arrays (Fig. 5D/E) compared with an EV-coated array (Fig. 5F). While multiple points of adhesion were seen in the CecA–DZ–OPN–CBM-coated array (Fig. 5D/E, white arrows), larger epiretinal spaces were apparent in the EV-coated array (Fig. 5F, white asterisk). Central elevation of the sclera, choroid and retina occurred due to the lack of vitreous after open-sky vitrectomy, resulting in a decreased ocular posture.
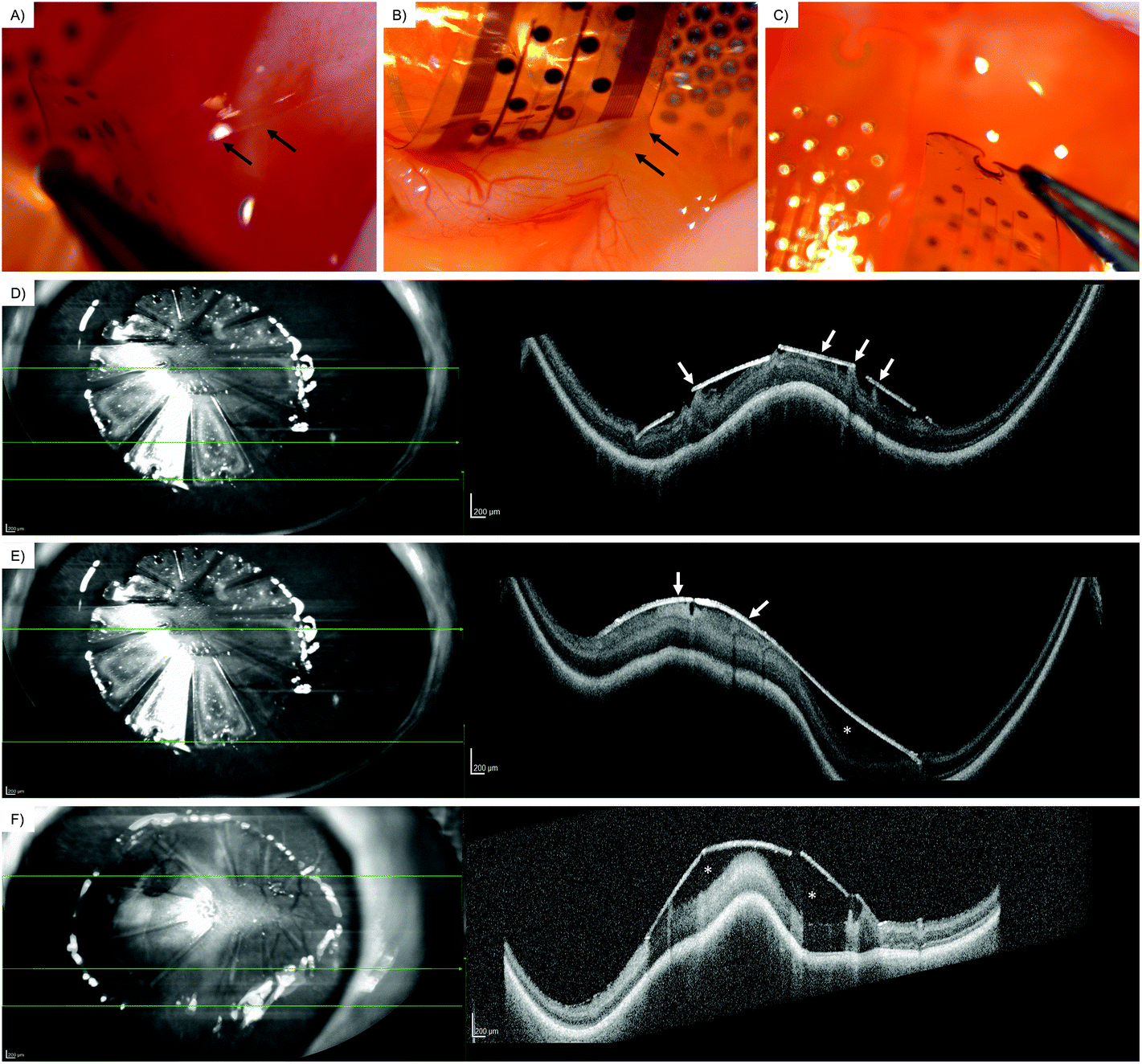 | ||
| Fig. 5 Retinal adhesion of the coated array in post-mortem rabbit eyes. A: Epiretinal array coated with CecA-1 × DZ-OPN–CBM on retinal surface after open-sky vitrectomy. The arrows show the retinal adhesion. B: Epiretinal array coated with CecA-4 × DZ-OPN–CBM on retinal surface after open-sky vitrectomy. The arrows show the retinal adhesion. Note the lifted retina and crease in retinal blood vessels. C: Array coated with empty vector (EV). No epiretinal adhesion was seen. D/E: En-face infrared imaging (left) and spectral-domain optical coherence tomography (SD-OCT) (right) of the CecA-4 × DZ-OPN–CBM-coated array on the posterior retinal pole. The green arrow shows the localization of the OCT B-scan. The white arrows display retinal adhesion to the coated array's surface. The white asterisk shows the space between the array and the retinal surface. F: En-face infrared imaging (left) and spectral-domain optical coherence tomography (SD-OCT) (right) of the empty vector (EV)-coated array on the posterior retinal pole. The green arrow shows the localization of the OCT B-scan. The empty vector-coated array is positioned on the posterior retinal pole. The white arrows display retinal adhesion to the coated array's surface. The white asterisk shows space between the array and the retinal surface. | ||
Discussion
While using retinal tacks to fixate epiretinal stimulating arrays is the established method of choice, the disadvantages are evident and have been repeatedly shown in literature.15–17 Inevitable damage to the retina, insufficient alignment to the retinal surface and a traumatic removal procedure emphasize the need for a novel approach in epiretinal fixation and led us to investigate the AP-based peptesive concept.15–17 In the BioAdhere study we developed a selectively binding, biocompatible peptesive showing promising results in ex vivo proof-of-concept experiments.Adhesives for ocular use ideally are non-toxic and non-inflammatory, have sufficient adhesive properties, are easy to apply and function shortly after application.51 A certain reversibility is also of use, especially in the context of retinal prostheses, where safe removal in the case of malfunction is crucial. With methyl-2-cyanoacrylate Bloomfield et al. first introduced an adhesive in ophthalmology, successfully closing limbal conjunctival wounds in rabbits.52 Further studies introduced the intraocular use of cyanoacrylate derivatives in the treatment of retinal detachment and macular holes in humans.53–58 However, cytotoxicity and difficult application due to rapid polymerization disqualified cyanoacrylate for fixating epiretinal arrays.51,59,60 Synthetic polymeric adhesives additionally tend to release heat during their rapid exothermic formation process, possibly causing damage to the retina.59 More recent polymeric approaches such as plasma polymerized-N-iso-propyl acrylamide (ppNIPAM) showed promising results for fixating Parylene C coated poly(dimethyl siloxane) (PDMS) implants on the retinal surface, yet with a detachment rate of approx. 30% sufficient adhesion remains questionable.51,61 While pegylated hydrogels showed promising results concerning the strength of adhesion and consistency, moderate toxicity to retinal photoreceptors was found in some subtypes.59,62 With fibrin glue, cross-linked gelatin, sea mussel adhesive (Cell-Tak; secreted by sea mussel Mytilus edulis to attach to rocky ground) and transforming growth factor beta (TGF-beta) bioadhesives were introduced.59,63–68 In the case of Cell-Tak cases of ocular inflammation and unsatisfactory adhesive behavior were reported; fibrin glue, cross-linked gelatin and TGF-beta lacked adhesive capabilities while showing mixed biocompatibility.51,59 Roessler et al. showed intriguing results using immobilized laminin peptide composites to fixate polyimide microstructures.69 While good biocompatibility and adhesive properties were suggested, a conclusive report on the matter has not been published yet. While a few studies have established quantitative methods to measure adhesion, none of these studies has applied the approach in a physiological setting inside the ocular bulb.59,61,65 Most studies on the matter chose to report qualitative findings on the adhesive properties of the applied substances.51,53,56–58,60,62,63,66,68Table 1 sums up the few reported quantitative findings of adhesive forces in adhesives used on the retinal surface (Table 1). Note, that due to differences in the experimental set-ups, comparing results is not possible. Overall, the established adhesives did not fully meet the requirements for fixating epiretinal arrays. Thus, we further explored the possibility of using APs.
| Adhesive tested | Adhesive force reported | Adhesion test method | |
|---|---|---|---|
| SS-PEG, succinimidyl ester of polyethylene glycol-bis-succinic acid; SPA-PEG, succinimidyl ester of polyethylene glylcol-bis-propionic acid. ST-PEG, succinimidyl-glutaryl + thiol-polyethylene glycol.a Product nomenclature by producer. | |||
| Tunc et al., 200761 | Plasma polymerized N-isopropyl acrylamide (pNIPAM) | 148.0 mN | Pull test |
| Margalit et al., 200059 | Hydrogels | ||
| SS-PEG | 90.7 mN | Strain gauge test | |
| SPA-PEG | 154.5 mN | ||
| ST-PEG | 546.6 mN | ||
| Fibrin sealants | |||
| Commercial fibrin | 37.0 mN | ||
| Autologous fibrin | 14.2 mN | ||
| Photocurable glues | |||
| 3321a | 42.0 mN | ||
| Lm219a | 20.5 mN | ||
| M181a | 42.0 mN | ||
| Cell-Tak | 0.0 mN | ||
| Chen et al., 200665 | Gelatin microbial Transglutaminase (Gelatin-mTG) | 1.2–2.3 N cm−2 | Lap-shear strength test |
APs were shown to bind to a variety of surfaces, natural and synthetic, and lent themselves as a suitable approach to immobilize synthetic epiretinal arrays on the natural retinal structure. Immobilization of functional biological moieties at the nano-scale (enzymes, proteins)19,23 as well as at the micro-scale (microgels)21 employing APs as adhesion promotors was reported. The APs LCI, TA2 and CecA were reported to bind to polymeric surfaces – LCI and CecA to PP23 and TA2 to PS.41 Hence, these APs were investigated in this study as potential binders to Parylene C. The concept of peptesives has been proven at the micro-scale by immobilization of 5–25 μm sized PCL containers on stainless steel.18 Herein, we report the first application of AP-based peptesives at the macro-scale by fixation of epiretinal arrays on the retinal surface.
Therefore, ECM binding peptide sequences were selected from the literature reports. Currently known collagen binding sequences are naturally occurring collagen binding proteins such as collagen-binding adhesion protein 35 (CNA35),70 peptides of the van Willebrand Factor71 or the CBM of OPN, a protein which is involved in the mineralization of bones.72 Additionally, phage display libraries were screened for collagen binding peptides and the consensus sequence H–V–F/W–Q/M–P/A–P/K was reported as highly affine and specific for col I binding.44 In this study, the CBM of OPN and the peptides KH1 and KH2 were tested for ECM protein binding. As determined herein, KH1 and KH2 peptides showed significantly stronger binding to col I compared with col IV. This finding confirms the reports of Helms et al.44 and shows the applicability of the eGFP-based MTP screening protocol used for the identification of ECM binding peptides. Different binding behaviors of KH1 and KH2 were observed for fibronectin binding. Where KH2 showed the strongest binding to fibronectin, KH1 did not show any fibronectin binding activity. This finding suggests that the composition of the reported consensus sequence influences the binding specificity of the peptide towards further ECM components. Both peptides differ in three out of six amino acids. These differences in binding behavior show the potential of the limited six amino acid sequence space which yielded the two investigated peptides. Hence, methods like protein engineering can be applied to further fine tune the binding behavior of the identified peptides by, for example, engineering them for increased binding strength.31,73 Both KH1 and KH2 showed little to no binding towards laminin, making KH1 a very specific col I-binding peptide and KH2 a more promiscuous variant with slight col IV- and strong fibronectin-binding abilities. The CBM of OPN showed binding to all four tested ECM components. This finding is consistent with reports of OPN binding to Col I72,74,75 and fibronectin.76 Herein, we report further binding capabilities of OPN–CBM to col IV and laminin. Due to its promiscuous binding to all tested ECM components, OPN–CBM was chosen as the AP for ECM binding in the final peptesive variant. The binding mechanism of OPN–CBM to the tested ECM components, especially collagens, is based upon protein–protein interactions.77 These interactions derive from a diverse interplay of physical and chemical forces. OPN–CBM harbors several hydrophobic amino acids such as Met, Phe and Tyr, which govern hydrophobic protein–protein interactions.78 Further, the positively charged amino acids within OPN–CBM, Arg, Lys and His can govern electrostatic attraction with negatively charged residues, e.g. Asp in the Arg–Gly–Asp cell binding site of fibronectin and laminin and glycans thereof.79
Retinal stimulators such as the VLARS structure require the use of a flexible multielectrode array that allows insertion of the large structure into the eye. The VLARS structure is encapsulated in Parylene C, which serves as a flexible moisture barrier to prolong the device's lifespan in the moist conditions of the inner eye.34 As this outmost layer of the VLARS encounters the retina directly, it was identified as primary binding partner for the attachment of the peptesive to the retinal stimulating array. Identification of a Parylene C binding AP was performed using a phytase reporter protein as previously reported.19 The investigated AP LCI, TA2 and CecA have been reported to bind to synthetic polymers such as PP and PS.19,41,73 Hence, these three APs were also investigated for their Parylene C binding ability. All three tested APs showed binding, with CecA displaying the strongest binding. In the bound state the APs, which are also reported as AMPs, are no longer available to permeabilize bacterial or fungal membranes, hence obstructing their antimicrobial activities. With AP CecA the orientation of the AP on the reporter protein was investigated. CecA showed binding to Parylene C as both CecA–YmPh and YmPh–CecA constructs. As the orientation of the AP on the reporter protein did not influence the binding ability of the AP, it is deduced that the binding domains are not obstructed by attachment to larger proteins. This finding suggests that attachment to the DZ separator protein will also not alter the binding ability. As both orientations of the APs are potential candidates for Parylene C binding in the final peptesive, the choice of final orientation was made based upon the orientation of the ECM binding AP. The results indicate that the hypothesis of CecA Parylene C binding capabilities remains true. The binding mechanism can be compared with the described AP–surface interactions such as π–π interactions. Parylene C consists of an aromatic xylylene with delocalized π electrons which can interact with CecA's aromatic amino acid residues such as Trp and Phe via π–π interactions.31,80,81 Further similarities between CecA binding to Parylene C and previously reported surfaces are noted: CecA reportedly binds to hydrophobic surfaces such as PP through hydrophobic interactions and to the hydrophobic and phenyl-containing PS.40 Hence, the binding mechanism of CecA to Parylene C is expected to be a combination of π–π and hydrophobic interactions between peptide and polymer.
Taking the results of ECM and Parylene C screening into account, it was decided to construct a bioadhesive peptide containing the C-anchored OPN–CBM to promote ECM protein adsorption as well as the N-anchored CecA to promote Parylene C adsorption. Both APs were separated by DZ as a spacer group.
Both chosen APs were tested for selectivity between Parylene C and ECM components. Within these experiments, both APs were normalized to a concentration of 1 mg mL−1. Notably, binding of the OPN–CBM construct to ECM components is slightly stronger than binding of the CecA construct (Fig. 3). This difference in binding behavior is most prominent for the example of laminin. Here, OPN–CBM shows significantly stronger binding compared with CecA. Likewise, binding of CecA is stronger on Parylene C compared with binding of OPN–CBM. These results indicate a preferred orientation of CecA towards Parylene C. Hence, it is likely that in the final peptesive construct directed immobilization will occur, orienting the CecA towards the epiretinal array while the OPN–CBM is oriented towards the retinal ECM. As the array will be coated prior to implantation, the higher binding strength of CecA to Parylene C suggests that the ECM binding OPN–CBM will not be obstructed after coating of the array.
Diverse other studies have reported peptide-based adhesives. However, no peptide-based adhesive has been reported for intra-ocular use. The reported adhesive cannot be compared directly, due to the use of different test methods as well as different binding targets. Single molecule force spectroscopy (SMFS) measurements of peptide-based adhesives report binding forces per molecule. Surface force apparatus (SFA) measurements report binding energies per area. For intra-ocular adhesives, binding forces for the evaluated adhesive are reported as the binding force for the adhesive in total. Peptide-based adhesives are applied to hard surfaces (Li et al.82), wet organic surfaces (Levine et al.83) and organic porcine tissue (Ma et al.84). Adhesives have been derived from mussel foot proteins (Lys-DOPA peptide and MFP-3s peptide).82,83 Others are supercharged polypeptides (SUPs), genetically engineered elastin variants with high net charge through the introduction of lysine residues in the elastin pentapeptide repeat unit. Thereon, the aromatic surfactant sodium dodecylbenzene sulfonate (SDBS) has been added via electrochemical interactions, generating an adhesive with a variety of interaction potentials.84
The peptesive we have described in our study also harbors lysine residues (15 in total, compared with 18 in SUP-SDBS) as well as tyrosine, the proteinogenic precursor amino acid to L-DOPA, which is also present in MFPs. We expect similar binding strength to MFP-3 to hydrophobic SAMs for the binding towards hydrophobic Parylene C due to the presence of lysine and tyrosine residues in our peptesive. The presence of a similar count of charged residues as in SUP-SDBS appears to inflict stronger binding to the retinal tissue, as shown for SUP-SDBS to porcine tissues. The reported peptesive can also interact electrostatically through lysine residues, hydrophobically through hydrophobic residues Met, Phe and Tyr and via π–π, and π–cation interactions between aromatic and charged groups.
The resulting peptesive CecA–DZ–OPN–CBM was subjected to in vitro cytotoxicity experiments. The use of L-929 and R28-cells as sensitive and retina-specific cell lines in cytotoxicity experiments has been documented.49,50 L-929 cells are fibroblast cells from subcutaneous connective tissue of an adult C3H mouse.85 The L-929 cell line is the oldest continuous cell line, having been established in 1948 by Earle.85 In a comparative study, L-929 cells were shown to be particularly sensitive to a cytotoxic reference material (polyetherurethane film containing 0.1% zinc diethyldithiocarbamate).86 Immortalized R28 retinal precursor cells have successfully been used in studies on, inter alia, retinal transplantation, growth factor effects and electrophysiology.50 As they retain neuronal and retinal properties, they are especially useful for simulating a physiological ocular environment.49,50 In correspondence to the ISO 10993 “Biological evaluation of medical materials” part 5 (tests for in vitro cytotoxicity) qualitative grading system for cellular reactivity, the tested CecA–DZ–OPN–CBM received grade 0, showing no signs of intracellular morphologic changes, cytolysis or decrease in cellular growth. The quantitative analysis showed approx. 1–2% dead cells for L-929 and R28 cell lines without an increase compared with the control. A decrease in cellular viability of greater than 30% is recognized as a cytotoxic effect of a tested substance. In non-direct cytotoxicity testing, there were no significant differences in luminance, representing cell deaths, compared with the control. Overall, cytotoxicity was ruled out. Further tissue interaction must be evaluated in in vivo experiments.
In the cell culture experiments R28 cells showed a more clustered growth pattern both on glass and on coated wells. Johnen et al. also reported a clustered growth of R28 cells, yet they saw a pronounced cluster growth of R28 cells on wafers coated with vertically aligned multiwalled carbon nanotubes.49 In our experiment, CecA–DZ–OPN–CBM coating did not further affect the R28 cell growth pattern.
The ex vivo proof-of-concept experiments using post-mortem rabbit eyes showed epiretinal adhesion, even though the arrays could be lifted off the retinal surface when applying traction. While in vivo the eye is exposed to vibration and the retinal array experiences shear forces, lifting the array of the retina in an ex vivo setting using a forceps does not represent physiological stress.87 Also, the retinal adhesion to the retinal pigment epithelium must be considered. Retinal adhesion is higher in vivo compared with ex vivo.88,89 The open-sky approach allows easy access to the posterior segment. In flat, large epiretinal arrays the scleral curvature can lead to gaps between the surface and the array, especially when using a single central tack.16 The coated arrays align to the said curvature, granting an advantage for that method of fixation. In further experiments, i.e., in an in vivo setting, pars-plana vitrectomy with a vitreous cutter would be desirable, especially to grant controlled vitreous detachment and removal of cortical vitreous from the retinal surface. The influence of repeated DZ spacer groups was investigated in a post-mortem rabbit eye experiment, but did not result in improved retinal adhesion, suggesting the APs are responsible for the binding strength rather than the DZ spacer. Due to the lack of clinically available adhesives for retinal applications we could not add a positive comparator in our study.
Within this study we identified peptides with ECM and Parylene C binding abilities and constructed a bioadhesive peptide (peptesive). We evaluated it as safe for intraocular use as well as establishing the peptesive as an advantageous method of fixation for epiretinal arrays compared with the currently used tack fixations. Due to the oriented immobilization of the peptesive on the epiretinal array, ECM binding is not hindered. Therefore, this is the first study to prove the applicability of peptesives for the fixation of macroscopic objects.
Author contributions
KH and TL contributed equally to this manuscript. KH: conceptualization, data curation, formal analysis, investigation, methodology, writing – original draft. CS: methodology. KS: methodology. GR: methodology. SJ: funding acquisition methodology, writing – review/editing. FJ: funding acquisition methodology, writing – review/editing. PW: writing – review/editing. US: writing – review/editing. TL: funding acquisition, conceptualization, investigation, data curation, fomal analysis, methodology, writing – original draft.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank AK and SI for Parylene C-coated MTP wells and VLARS arrays. We thank AF for her support in conducting cell culture experiments. RWTH Aachen University ERS Seed Fund (OPSF473) is acknowledged.References
- J. B. Jonas, R. R. Bourne, R. A. White, S. R. Flaxman, J. Keeffe, J. Leasher, K. Naidoo, K. Pesudovs, H. Price, T. Y. Wong, S. Resnikoff and H. R. Taylor, Am. J. Ophthalmol., 2014, 158, 808–815 CrossRef PubMed.
- I. Kocur and S. Resnikoff, Br. J. Ophthalmol., 2002, 86, 716–722 CrossRef CAS PubMed.
- R. P. Finger, B. Bertram, C. Wolfram and F. G. Holz, Dtsch. Ärztebl. Int., 2012, 109, 484–489 Search PubMed.
- S. Russell, J. Bennett, J. A. Wellman, D. C. Chung, Z. F. Yu, A. Tillman, J. Wittes, J. Pappas, O. Elci, S. McCague, D. Cross, K. A. Marshall, J. Walshire, T. L. Kehoe, H. Reichert, M. Davis, L. Raffini, L. A. George, F. P. Hudson, L. Dingfield, X. Zhu, J. A. Haller, E. H. Sohn, V. B. Mahajan, W. Pfeifer, M. Weckmann, C. Johnson, D. Gewaily, A. Drack, E. Stone, K. Wachtel, F. Simonelli, B. P. Leroy, J. F. Wright, K. A. High and A. M. Maguire, Lancet, 2017, 390, 849–860 CrossRef CAS.
- M. Garita-Hernandez, M. Lampic, A. Chaffiol, L. Guibbal, F. Routet, T. Santos-Ferreira, S. Gasparini, O. Borsch, G. Gagliardi, S. Reichman, S. Picaud, J. A. Sahel, O. Goureau, M. Ader, D. Dalkara and J. Duebel, Nat. Commun., 2019, 10, 4524 CrossRef PubMed.
- A. Gonzalez-Cordero, E. L. West, R. A. Pearson, Y. Duran, L. S. Carvalho, C. J. Chu, A. Naeem, S. J. I. Blackford, A. Georgiadis, J. Lakowski, M. Hubank, A. J. Smith, J. W. B. Bainbridge, J. C. Sowden and R. R. Ali, Nat. Biotechnol., 2013, 31, 741–747 CrossRef CAS PubMed.
- M. S. Humayun, M. Prince, E. de Juan Jr., Y. Barron, M. Moskowitz, I. B. Klock and A. H. Milam, Invest. Ophthalmol. Visual Sci., 1999, 40, 143–148 CAS.
- M. S. Humayun, E. de Juan Jr., J. D. Weiland, G. Dagnelie, S. Katona, R. Greenberg and S. Suzuki, Vision research, 1999, 39, 2569–2576 CrossRef CAS PubMed.
- B. W. Jones, R. L. Pfeiffer, W. D. Ferrell, C. B. Watt, M. Marmor and R. E. Marc, Exp. Eye Res., 2016, 150, 149–165 CrossRef CAS PubMed.
- T. C. Lin, H. M. Chang, C. C. Hsu, K. H. Hung, Y. T. Chen, S. Y. Chen and S. J. Chen, J. Chin. Med. Assoc., 2015, 78, 501–505 CrossRef PubMed.
- A. V. Rachitskaya and A. Yuan, Ophthalmic Genet., 2016, 37, 260–266 CrossRef CAS PubMed.
- P. Walter, Z. F. Kisvarday, M. Gortz, N. Alteheld, G. Rossler, T. Stieglitz and U. T. Eysel, Invest. Ophthalmol. Visual Sci., 2005, 46, 1780–1785 CrossRef PubMed.
- P. Walter, P. Szurman, M. Vobig, H. Berk, H. C. Ludtke-Handjery, H. Richter, C. Mittermayer, K. Heimann and B. Sellhaus, Retina, 1999, 19, 546–552 CrossRef CAS PubMed.
- Y. H. Luo and L. da Cruz, Prog. Retinal Eye Res., 2016, 50, 89–107 CrossRef PubMed.
- J. Menzel-Severing, B. Sellhaus, T. Laube, C. Brockmann, N. Bornfeld, P. Walter and G. Roessler, Ophthalmic Res., 2011, 46, 192–198 CrossRef PubMed.
- T. K. Lohmann, F. Haiss, K. Schaffrath, A. C. Schnitzler, F. Waschkowski, C. Barz, A. M. van der Meer, C. Werner, S. Johnen, T. Laube, N. Bornfeld, B. E. Mazinani, G. Rossler, W. Mokwa and P. Walter, J. Neural Eng., 2019, 16, 066031 CrossRef PubMed.
- G. Roessler, T. Laube, C. Brockmann, T. Kirschkamp, B. Mazinani, M. Goertz, C. Koch, I. Krisch, B. Sellhaus, H. K. Trieu, J. Weis, N. Bornfeld, H. Rothgen, A. Messner, W. Mokwa and P. Walter, Invest. Ophthalmol. Visual Sci., 2009, 50, 3003–3008 CrossRef PubMed.
- L. Apitius, S. Buschmann, C. Bergs, D. Schönauer, F. Jakob, A. Pich and U. Schwaneberg, Macromol. Biosci., 2019, 19(9), e1900125 CrossRef PubMed.
- S. Dedisch, A. Wiens, M. D. Davari, D. Söder, C. Rodriguez-Emmenegger, F. Jakob and U. Schwaneberg, Biotechnol. Bioeng., 2020, 117, 49–61 CrossRef CAS PubMed.
- S. Islam, L. Apitius, F. Jakob and U. Schwaneberg, Environ. Int., 2019, 123, 428–435 CrossRef CAS PubMed.
- R. A. Meurer, S. Kemper, S. Knopp, T. Eichert, F. Jakob, H. E. Goldbach, U. Schwaneberg and A. Pich, Angew. Chem., Int. Ed., 2017, 56, 7380–7386 CrossRef CAS PubMed.
- S. Pariyar, F. Jakob, L. Apitius, U. Schwaneberg, M. Hunsche and G. Noga, Acta Hortic, 2020, 1292, 175–180 CrossRef.
- K. Rübsam, B. Stomps, A. Böker, F. Jakob and U. Schwaneberg, Polymer, 2017, 116, 124–132 CrossRef.
- N. Rangarajan, S. Bakshi and J. C. Weisshaar, Biochemistry, 2013, 52, 6584–6594 CrossRef CAS PubMed.
- M. Zasloff, Nature, 2002, 415, 389–395 CrossRef CAS PubMed.
- M. Hnilova, E. E. Oren, U. O. Seker, B. R. Wilson, S. Collino, J. S. Evans, C. Tamerler and M. Sarikaya, Langmuir, 2008, 24, 12440–12445 CrossRef CAS PubMed.
- T. Serizawa, T. Sawada, H. Matsuno, T. Matsubara and T. Sato, J. Am. Chem. Soc., 2005, 127, 13780–13781 CrossRef CAS PubMed.
- H. Ejima, H. Matsuno and T. Serizawa, Langmuir, 2010, 26, 17278–17285 CrossRef CAS PubMed.
- D. J. Lee, H. S. Park, K. Koo, J. Y. Lee, Y. S. Nam, W. Lee and M. Y. Yang, Langmuir, 2019, 35, 522–528 CrossRef CAS PubMed.
- J. Chen, T. Serizawa and M. Komiyama, J. Pept. Sci., 2011, 17, 163–168 CrossRef CAS PubMed.
- K. Rübsam, M. D. Davari, F. Jakob and U. Schwaneberg, Polymer, 2018, 423 DOI:10.3390/polym10040423.
- B. J. Kim and E. Meng, Polym. Adv. Technol., 2016, 27, 564–576 CrossRef CAS.
- T. Marszalek, M. Gazicki-Lipman and J. Ulanski, Beilstein J. Nanotechnol., 2017, 8, 1532–1545 CrossRef CAS PubMed.
- F. Waschkowski, S. Hesse, A. C. Rieck, T. Lohmann, C. Brockmann, T. Laube, N. Bornfeld, G. Thumann, P. Walter, W. Mokwa, S. Johnen and G. Roessler, Biomed. Eng. Online, 2014, 13(1), 11 CrossRef PubMed.
- W. Halfter, J. Candiello, H. Hu, P. Zhang, E. Schreiber and M. Balasubramani, Cell Adhes. Migr., 2013, 7, 64–71 CrossRef PubMed.
- S. C. Bu, R. Kuijer, R. J. van der Worp, X. R. Li, J. M. Hooymans and L. I. Los, PLoS One, 2015, 10, e0134325 CrossRef PubMed.
- W. Halfter, C. Monnier, D. Müller, P. Oertle, G. Uechi, M. Balasubramani, F. Safi, R. Lim, M. Loparic and P. B. Henrich, PLoS One, 2013, 8, e67660 CrossRef CAS PubMed.
- J. Candiello, M. Balasubramani, E. M. Schreiber, G. J. Cole, U. Mayer, W. Halfter and H. Lin, FEBS J., 2007, 274, 2897–2908 CrossRef CAS PubMed.
- T. Ihanamäki, L. J. Pelliniemi and E. Vuorio, Prog. Retinal Eye Res., 2004, 23, 403–434 CrossRef PubMed.
- S. Dedisch, A. Wiens, M. D. Davari, D. Soder, C. Rodriguez-Emmenegger, F. Jakob and U. Schwaneberg, Biotechnol. Bioeng., 2020, 117, 49–61 CrossRef CAS PubMed.
- K. Rübsam, L. Weber, F. Jakob and U. Schwaneberg, Biotechnol. Bioeng., 2018, 115, 321–330 CrossRef PubMed.
- A. V. Shivange, A. Serwe, A. Dennig, D. Roccatano, S. Haefner and U. Schwaneberg, Appl. Microbiol. Biotechnol., 2012, 95, 405–418 CrossRef CAS PubMed.
- M. Blanusa, A. Schenk, H. Sadeghi, J. Marienhagen and U. Schwaneberg, Anal. Biochem., 2010, 406, 141–146 CrossRef CAS PubMed.
- B. A. Helms, S. W. Reulen, S. Nijhuis, P. T. de Graaf-Heuvelmans, M. Merkx and E. W. Meijer, J. Am. Chem. Soc., 2009, 131, 11683–11685 CrossRef CAS PubMed.
- R. Arai, H. Ueda, A. Kitayama, N. Kamiya and T. Nagamune, Protein Eng., 2001, 14, 529–532 CrossRef CAS PubMed.
- R. B. Kapust, J. Tozser, J. D. Fox, D. E. Anderson, S. Cherry, T. D. Copeland and D. S. Waugh, Protein Eng., 2001, 14, 993–1000 CrossRef CAS PubMed.
- M. Tashiro, R. Tejero, D. E. Zimmerman, B. Celda, B. Nilsson and G. T. Montelione, J. Mol. Biol., 1997, 272, 573–590 CrossRef CAS PubMed.
- L. Apitius, S. Buschmann, C. Bergs, D. Schonauer, F. Jakob, A. Pich and U. Schwaneberg, Macromol. Biosci., 2019, 19, e1900125 CrossRef PubMed.
- S. Johnen, F. Meissner, M. Krug, T. Baltz, I. Endler, W. Mokwa and P. Walter, J. Ophthalmol., 2016, 2016, 2371021 Search PubMed.
- G. M. Seigel, W. Sun, J. Wang, D. H. Hershberger, L. M. Campbell and R. J. Salvi, Curr. Eye Res., 2004, 28, 257–269 CrossRef CAS PubMed.
- M. Tunc, M. Humayun, X. Cheng and B. D. Ratner, Retina, 2008, 28, 1338–1343 CrossRef PubMed.
- S. Bloomfield, A. H. Barnert and P. Kanter, Am. J. Ophthalmol., 1963, 55, 946–953 CrossRef CAS PubMed.
- B. Ricci and F. Ricci, Acta Ophthalmol. Scand., 2001, 79, 506–508 CrossRef CAS PubMed.
- R. Reyniers, S. Boekhoorn, M. Veckeneer and J. van Meurs, Graefe's Arch. Clin. Exp. Ophthalmol., 2012, 250, 311–312 CrossRef PubMed.
- B. W. McCuen 2nd, T. Hida, S. M. Sheta and R. Machemer, Trans. Am. Ophthalmol. Soc., 1987, 85, 195–207 CAS.
- M. A. Tilanus and A. F. Deutman, Int. Ophthalmol., 1994, 18, 355–358 CrossRef PubMed.
- S. M. Sheta, T. Hida and B. W. McCuen 2nd, Am. J. Ophthalmol., 1990, 109, 28–32 CrossRef CAS PubMed.
- J. Faulborn and H. Witschel, Graefe's Arch. Clin. Exp. Ophthalmol., 1978, 207, 15–20 CrossRef CAS PubMed.
- E. Margalit, G. Y. Fujii, J. C. Lai, P. Gupta, S. J. Chen, J. S. Shyu, D. V. Piyathaisere, J. D. Weiland, E. De Juan Jr. and M. S. Humayun, Retina, 2000, 20, 469–477 CrossRef CAS PubMed.
- M. Spitznas, H. Lassagk, M. Vogel and F. Joussen, Graefe's Arch. Clin. Exp. Ophthalmol., 1973, 187, 102–110 CrossRef CAS PubMed.
- M. Tunc, X. Cheng, B. D. Ratner, E. Meng and M. Humayun, Retina, 2007, 27, 938–942 CrossRef PubMed.
- J. P. Hubschman, A. Govetto, M. Farajzadeh, T. Sato, S. Askari and B. Glasgow, Clin. Exp. Ophthalmol., 2017, 45, 708–716 CrossRef PubMed.
- Q. Wang, J. Zhao, Q. Xu, C. Han and B. Hou, Retina, 2020, 40, 718–724 CrossRef PubMed.
- I. Bahar, D. Weinberger, M. Lusky, R. Avisar, A. Robinson and D. Gaton, Curr. Eye Res., 2006, 31, 31–36 CrossRef CAS PubMed.
- T. Chen, R. Janjua, M. K. McDermott, S. L. Bernstein, S. M. Steidl and G. F. Payne, J. Biomed. Mater. Res., Part B, 2006, 77, 416–422 CrossRef PubMed.
- B. M. Glaser, R. G. Michels, B. D. Kuppermann, R. N. Sjaarda and R. A. Pena, Ophthalmology, 1992, 99, 1162–1172 CrossRef CAS PubMed ; discussion 1173.
- A. R. Margherio, Curr. Opin. Ophthalmol., 2000, 11, 186–190 CrossRef CAS PubMed.
- D. J. Coleman, B. C. Lucas, J. A. Fleischman, P. H. Dennis Jr., S. Chang, T. Iwamoto and R. M. Nalbandian, Retina, 1988, 8, 250–256 CrossRef CAS PubMed.
- G. Roessler, W. Mokwa, B. Sellhaus, B. Mazinani and P. Walter, Invest. Ophthalmol. Visual Sci., 2014, 55, 3003–3008 CrossRef PubMed.
- K. N. Krahn, C. V. Bouten, S. van Tuijl, M. A. van Zandvoort and M. Merkx, Anal. Biochem., 2006, 350, 177–185 CrossRef CAS PubMed.
- J. Takagi, H. Asai and Y. Saito, Biochemistry, 1992, 31, 8530–8534 CrossRef CAS PubMed.
- J. Y. Lee, J. E. Choo, Y. S. Choi, J. B. Park, D. S. Min, S. J. Lee, H. K. Rhyu, I. H. Jo, C. P. Chung and Y. J. Park, Biomaterials, 2007, 28, 4257–4267 CrossRef CAS PubMed.
- L. Apitius, K. Rubsam, C. Jakesch, F. Jakob and U. Schwaneberg, Biotechnol. Bioeng., 2019, 116, 1856–1867 CrossRef CAS PubMed.
- Y. Chen, B. S. Bal and J. P. Gorski, J. Biol. Chem., 1992, 267, 24871–24878 CrossRef CAS PubMed.
- S. M. Martin, J. L. Schwartz, C. M. Giachelli and B. D. Ratner, J. Biomed. Mater. Res., Part A, 2004, 70, 10–19 CrossRef PubMed.
- B. B. Mukherjee, M. Nemir, S. Beninati, E. Cordella-Miele, K. Singh, I. Chackalaparampil, V. Shanmugam, M. W. DeVouge and A. B. Mukherjee, Ann. N. Y. Acad. Sci., 1995, 760, 201–212 CrossRef CAS PubMed.
- J. Y. Lee, J. E. Choo, H. J. Park, J. B. Park, S. C. Lee, I. Jo, S. J. Lee, C. P. Chung and Y. J. Park, Biochem. Biophys. Res. Commun., 2007, 357, 68–74 CrossRef CAS PubMed.
- B. Y. Ma, T. Elkayam, H. Wolfson and R. Nussinov, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5772–5777 CrossRef CAS PubMed.
- J. E. Schwarzbauer and D. W. DeSimone, Cold Spring Harbor Perspect. Biol., 2011, 3(7), a005041 Search PubMed.
- C. G. Salzmann, M. A. H. Ward, R. M. J. Jacobs, G. Tobias and M. L. H. Green, J. Phys. Chem. C, 2007, 111, 18520–18524 CrossRef CAS.
- C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS.
- Y. R. Li, J. Cheng, P. Delparastan, H. Q. Wang, S. J. Sigg, K. G. DeFrates, Y. Cao and P. B. Messersmith, Nat. Commun., 2020, 11, 3895 CrossRef PubMed.
- Z. A. Levine, M. V. Rapp, W. Wei, R. G. Mullen, C. Wu, G. H. Zerze, J. Mittal, J. H. Waite, J. N. Israelachvili and J. E. Shea, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4332–4337 CrossRef CAS PubMed.
- C. Ma, J. Sun, B. Li, Y. Feng, Y. Sun, L. Xiang, B. H. Wu, L. L. Xiao, B. M. Liu, V. S. Petrovskii, B. Liu, J. R. Zhang, Z. L. Wang, H. Y. Li, L. Zhang, J. J. Li, F. Wang, R. Goestl, I. I. Potemkin, D. Chen, H. B. Zeng, H. J. Zhang, K. Liu and A. Herrmann, Nat. Commun., 2021, 12, 3613 CrossRef CAS PubMed.
- K. K. Sanford, W. R. Earle and G. D. Likely, J. Natl. Cancer Inst., 1948, 9, 229–246 CAS.
- J. C. Park, B. J. Park, D. H. Lee, H. Suh, D. G. Kim and O. H. Kwon, Yonsei Med. J., 2002, 43, 518–526 CrossRef CAS PubMed.
- R. I. Angunawela, A. Azarbadegan, G. W. Aylward and I. Eames, Invest. Ophthalmol. Visual Sci., 2011, 52, 7046–7051 CrossRef PubMed.
- M. Kita and M. F. Marmor, Invest. Ophthalmol. Visual Sci., 1992, 33, 1879–1882 CAS.
- M. Kita, A. Negi, S. Kawano, Y. Honda and S. Maegawa, Invest. Ophthalmol. Visual Sci., 1990, 31, 624–628 CAS.
| This journal is © The Royal Society of Chemistry 2022 |