
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh efficiency capture of biomarker miRNA15a for noninvasive diagnosis of malignant kidney tumors†
Alexander M.
Renner
 a,
Christina
Derichsweiler
a,
Shaista
Ilyas
a,
Isabel
Gessner
a,
Jochen W. U.
Fries
b and
Sanjay
Mathur
*a
a,
Christina
Derichsweiler
a,
Shaista
Ilyas
a,
Isabel
Gessner
a,
Jochen W. U.
Fries
b and
Sanjay
Mathur
*a
aInstitute of Inorganic Chemistry, University of Cologne, Greinstr. 6, 50939 Cologne, Germany. E-mail: sanjay.mathur@uni-koeln.de
bInstitute of Urology/Pathology, University Hospital of Cologne, Kerpenerstr. 62, 50924 Cologne, Germany
First published on 20th January 2022
Abstract
To date, there are no preoperative and quantitative dynamics in clinical practice that can reliably differentiate between a benign and malignant renal cell carcinoma (RCC). For monitoring different analytes in body fluids, more than 40 different molecular biomarkers have been identified, however, they are associated with limited clinical sensitivity and/or non-optimal specificity due to their leaky nature. Previous work on RCC demonstrated the miRNA15a to be reliable and novel biomarker with 98.1% specificity and 100% sensitivity. Despite the high potential of miRNA15a biomarker, its clinical application is considerably hampered by the insensitive nature of the detection methods and low concentration of biomarker in samples that is aggravated by the high level of contamination due to other solutes present in body fluids. In this work, a non-invasive quantitative approach is demonstrated to overcome such diagnostics issues through biotin–streptavidin binding and fluorescence active magnetic nanocarriers that ensured prompt isolation, enrichment and purification of the biomarker miRNA15a from urine. The study demonstrates that detectable low levels of these miRNAs through miRNA capturing nanocarriers can potentially function as advanced diagnostic markers for the non-invasive investigation and early detection of renal cancer.
Introduction
Renal cell carcinoma (RCC), the most common neoplasm of the adult kidney accounts for about 3% of adult cancers1,2 with the highest mortality rate at over 40%.3,4 Four major different histologic types have been described,5 of which the clear renal cell carcinoma (cRCC) is the most frequent subtype representing approximately 80–85% of metastatic RCC. Lack of efficacy in chemotherapy and radiation therapy,6 as well as a high recurrence rate of up to 40% after surgical resection,7 are the most dreaded consequences. Most importantly, the on-time diagnosis has been severely impeded since there are currently no biomarkers available for clinical diagnosis, early detection and follow-up of the disease. In this situation, microRNA (miRNA) diagnostic reflecting the underlying molecular and genetic mechanisms for RCC initiation and development, would enable early diagnosis and potentially the prediction of early metastasis after curative nephrectomy as well.miRNAs represent an abundant and important class of small noncoding single-strand RNAs responsible for regulating gene expression. miRNAs play significant roles in different cellular function by targeting mRNA sequences of protein-coding transcripts.8 miRNAs elicit the help of Argonaute proteins to repress mRNA translation and stability. The classic, canonical interaction occurs by base pairing of the miRNA seed region (equivalent of nucleotides 2–7), to sites in mRNA 3′ untranslated regions.9–11 We have recently described a defined role for miRNAs and Endothelin-1 (ET-1) in the kidney:12 a signaling pathway induced by ET-1 via its ETA-receptor leading to maturation of miRNA15a and its release into urine in renal cell carcinoma13,14 and proteinuric renal disease.15 This is due to a novel mechanism of miRNA and DNA interaction in which miRNA15a acts directly via N-terminal truncation at the DNA level in the intron 10 region of MAPKp38alpha,16 generating Mxi-2 that no longer has a nuclear localization sequence. This prohibits the formation of the transcription complex in resting cells of NFkBp65/MAPKp38alpha, preventing PKC alpha from entering the nucleus. Meanwhile, NF-κB can enter uninhibited and trigger transcription of inflammatory genes.17 Since PKC alpha can no longer bind pri-miRNA, an increased amount of mature miRNA15a is produced and can be detected in urine, creating a negative feedback loop.18,19 Mxi-2 is further involved in downregulation of p53 in a complex with Ago-2 and miRNA1285.20 It also plays a role in increased p16INK4a and p21waf1/cip1 via a complex consisting of ETS1, ERK2 and Mxi2 in renal tumor cells.21 Furthermore, potential therapeutic approaches have been described with either specific ETA-receptor blockers or the monoamine oxidase inhibitor selegeline,22 which upregulates PKCalpha. Its efficacy has been tested in vitro and in animal models of proteinuria such as in Adriamycin nephropathy,15 where miRNA level were downregulated within hours of selegeline application or in the mouse Alport model after applying an ETA-receptor blocker (unpublished data).
In view of the above, testing of urinary miRNA15a levels is a safe, direct and efficient option to identify proximal tubule activation and analyse therapeutic efficacy that has the potential to control patients’ compliance. However, till date, the development on sensitive diagnostic tools to assess the expression of miRNA from body fluids remains elusive and there are only two reports available on the usefulness of this miRNA.19,23 Traditionally, the identification and quantification of miRNA rely on the use of expensive and time-consuming quantitative real-time polymerase chain reaction (qRT-PCR).24 To enhance the capture and reliable detection of miRNA at low concentrations, nanocarrier (NC) based approaches offer unprecedented progress in early prognosis of different diseases.25,26 Thus, the aim of this study was to evaluate the detection limit and capture miRNA15a expression in human urine in single digit picomolar range (Fig. 1). Herein, the novelty of our approach involves the chemically engineered magnetic NCs equipped with surface-attached ss-oligonucleotides, which can be used to separate tumor-related miRNA15a within a few hours. The as-developed approach provides an innovative, inexpensive and quick diagnostic tool which can be applied on RCC patients for quick evaluation of their body fluid analysis as compared to existing methods such as qRT-PCR.
 | ||
| Fig. 1 Schematic structure of IONCs and their application as a non-invasive diagnostic tool for RCC. (A) NC with a ssDNA strand, complementary to miRNA15a located on the particle surface, are incubated with miRNA15a which is excreted in the human urine leading of a hybridization of both strands. The particles were extracted from urine and purified by magnetic separation to remove all cell debris and potential disruptive factors. After fluorescence labeling by the second ss-oligonucleotides, the amount of miRNA15a can be quantified based on its emission signal in an ELISA-reader. (B) SAV modified IONCs as transporter enables binding of biotin labeled ss-oligonucleotides complementary to 16 nucleotides starting from the 5′-end of target miRNA15a (22 nt). The six remaining nucleotides are hybridized by a second ss-oligonucleotides which is marked with a fluorescence dye. | ||
Experimental section
Materials
Iron(III) chloride (FeCl3; 98%), sodium acetate (NaOAc; ≥98%) and triethanolamine (TEOA; 97%) were obtained from Fisher Scientific. Biotin-4-Fluorescein (B4F) was purchased from biomol. N-Ethyl-N′-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC; 98%) was achieved from abcr. Streptavidin, streptomyces avidinii (SAV) was obtained from Alfa Aesar. 3,4-Dihydroxyhydrocinnamic acid (DHCA; 98%), N-hydroxysulfosuccinimide sodium salt (sulfo-NHS; ≥98%), 2-morpholinoethanesulfonic acid monohydrate (MES; ≥ 99%) and Tris-EDTA buffer solution pH 8.0 (TE-buffer; BioUltra) were obtained from Sigma Aldrich. All oligonucleotide were purchased from Biomers.net. All chemicals were used as received.Synthesis of Fe3O4@DHCA NCs
Fe3O4@DHCA NCs were synthesized according to the literature with minor modifications.28 0.245 g FeCl3 (1.51 mmol) was dissolved in 30 mL ethylene glycol and stirred for 1 h. Afterwards, 1.969 g NaOAc (24 mmol) were added and the reaction mixture was stirred for 15 min. At least, 0.110 g DHCA (0.63 mmol) was added and the solution was stirred for another 10 min before being transferred to a Teflon-lined stainless-steel autoclave at 200 °C for 24 h. The reaction mixture was then allowed to settle. The NCs were washed with a H2O/EtOH mixture at 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min and also magnetically separated several times.
000 rpm for 10 min and also magnetically separated several times.
Surface functionalization of Fe3O4@DHCA NCs with SAV
Fe3O4@SAV were synthesized according to a published procedures with minor modifications.35 In a typical reaction, a dispersion of 5 mg mL−1 Fe3O4@DHCA NCs in water was prepared. 400 μL of this dispersion was taken and washed three times with 400 μL MES-buffer (50 mmol L−1). The NCs were collected with the use of an external magnet. Afterwards, the NCs were dispersed in 400 μL of MES-buffer with 200 μL sulfo-NHS (57.57 μmol mL−1; in MES-buffer) and 200 μL EDC (32.21 μmol mL−1; in MES-buffer). The reaction mixture was stirred for 1 h at ambient temperature protected from light. The particles were collected with a magnet and the supernatant discard. The NCs were dispersed in 1 mL PBS and 50 μL SAV (2 mg mL−1) was added. The reaction was stirred for additional 3 h. At least, Fe3O4@SAV NCs were washed several times with PBS using an external magnet and stored in 100 μL PBS at 4 °C.Quantification of binding sites of Fe3O4@SAV NCs
The binding sites of SAV were quantified by using B4F following a procedure reported by G. Kada et al., with minor modifications.36 1 mL Fe3O4@SAV NCs (2 mg mL−1) were washed three times with 1 mL buffer solution (TEOA; 0.129 mmol mL−1) and divided into 10 tubes containing of 0.02 mg dispersed in 150 μL buffer solution. Different volumes of B4F (1.6 μM) were then added and the reaction mixture was incubated for 40 min under gentle shaking. The NCs were separated using an external magnet and 100 μL of the supernatant was transferred into a 96 well-plate for fluorescence measurements.ssDNA15a extraction and purification (analogous for FITC-15a, FITC-mut15a-soft and FITC-mut15a-hard)
A dispersion of 100 μL Fe3O4@SAV NCs (20 mg mL−1) was taken for all experiments The NCs were washed three times with 200 μL TE-buffer by magnet. Homogenous dispersion was ensured by an ultrasonic-bathing for 10 s before using the NCs. In a typical procedure, 60 μL of the prepared dispersion was transferred to a 0.2 mL PCR-tube and treated with 7.5 μL Compl.-TEG-BIO (9.12 nmol mL−1). The reaction mixture was incubated at ambient temperature for 40 min with occasional shaking. The NCs were then collected with a magnet and washed with 200 μL buffer (TE-buffer including 1.8 M NaCl). In the next step, ss-oligonucleotides (respectively FITC-15a, FITC-mut15a-soft or FITC-mut15a-hard) were diluted in 110 μL hybridization buffer (TE-buffer including 0.9 M NaCl) and added to Fe3O4@SAV-Compl.-TEG-BIO NCs. In case of urine samples, 55 μL washing buffer was mixed with 55 μL ssDNA-15a diluted in urine. The reaction mixture was incubated at RT for 1 h with occasional shaking. The NCs were washed with 100 μL hybridization buffer via magnet. At least, the NCs were dispersed in 110 μL hybridization buffer including 10 pmol FITC-C-tail. The PCR-Tubes were located in an aluminium block on ice. After 1 h, the NCs were collected via ice-cooled magnet and 100 μL supernatant were taken for measurement. All experiments were performed in accordance with the guidelines of ethical standards formulated in the Declaration of Helsinki 1975, with preapproval of the Ethics Committee at the University Hospital, Cologne (reference no. 09-232). Informed consents were obtained from human participants of this study.Characterization of Fe3O4@SAV
The identification of crystalline phase of as-prepared NCs was performed by powder XRD on a STADI-MP (STOE & Cie. GmbH) with Mo Kα (λ = 0.7093 Å) as incident radiation with an operating voltage of 50 kV and an electric current of 30 mA. For detection of the reflexes, a linear position sensitive detector was used with a scan range between 5 and 40° and a step size of 0.05° with a holding time of 4 s. The morphology of NCs was investigated by SEM, which were recorded on Nova NanoSEM (FEI) using a highly diluted dispersion of NCs in EtOH. The surface groups of modified NCs were characterized by FTIR measurements, performed on an FTIR spectrometer IRAffinity-1 (Shimadzu) under air conditions and ambient temperature. UV/Vis spectra of prepared NCs were recorded on a UV-1600PC (VWR) at ambient temperature. All NCs were dispersed in dest. H2O before measurement. Fluorescence measurements were performed on a FLUOstar OPTIMA (BMG Labtech). A bottom optic was used with a position delay of 0.2 s and 20 flashes per well. An Excitation filter at 485 nm and an emission filter at 520 nm were used. The gain of the laser beam was set on 1000 for B4F and 1100 for FITC labeled samples. For sample preparation, 100 μL solution were transferred into a 96 well plate before measurement.Results and discussion
Iron oxide nanocarriers (IONCs) consisting of a magnetite core (Fe3O4) and 3,4-dihydroxyhydrocinnamic acid (DHCA) as surface-modifier were synthesized by solvothermal decomposition of an Fe(III) salt in the presence of DHCA and sodium acetate.19 The terminal carboxyl groups present on the IONCs were activated for a covalent attachment of the protein streptavidin (SAV) via carbodiimide coupling reaction. This enabled the selective and facile capture of ssDNA strands that consisted of biotin and 16 nucleotides (complementary to miRNA15a), which were both linked together via triethylene glycol (TEG), due to the strong binding affinity between biotin and SAV, located on the particle surface of IONC (Fe3O4@SAV-Compl.-TEG-BIO).27The accessibility of the binding sites were evaluated by conjugating biotin-4-fluorescein (B4F) as schematically shown in Fig. 2A and B. Subsequently, these Fe3O4@SAV-Compl.-TEG-BIO carriers were hybridized with ssDNA-15a (22 nucleotides; analogous to miRNA) enabling a magnetic separation of the target strand. For this purpose, ssDNA was used since it has a higher stability compared to miRNA, however exhibiting similar chemical and binding behaviour. In the presence of the connecting central ssDNA-15a strand, the remaining nucleotides were hybridized with a third, fluorescence labeled ssDNA strand (6 nucleotides; complementary to miRNA15a) that served the purpose of quantification (Fig. 2C).
 | ||
| Fig. 2 Schematic overview of the synthesis of IONCs and ssDNA hybridizations steps. (A) Interaction of surface grafted carboxyl-groups with SAV enabled biotin–streptavidin binding between IONCs and biotin-labeled molecules. Quantification of free binding sites was performed using B4F, which loses its fluorescence after biotin–streptavidin binding. (B) Biotin labeled ss-oligonucleotides can be attached on the surface of particle with complementary base sequence to target miRNA strands. (C) Nomenclature and hybridization sequence of ss-oligonucleotides strands to prepare Fe3O4@SAV-FITC-15a nanocarriers. | ||
Design and surface functionalization of Fe3O4@SAV nanocarriers
DHCA modified iron oxide nanocarrier (IONCs) were synthesized according to the solvothermal procedures described by H. Zhu et al. (Fig. 3A).28 SEM images of as-prepared IONCs showed a homogeneous distribution of nanoparticles with a spherical morphology (Fig. 3B and C). Based on these results, the average particle size was determined to be 142.59 ± 0.45 nm (Fig. 3D). The powder X-ray diffraction (PXRD) peaks confirmed the as-synthesized IONCs to be crystalline magnetite particles present in the cubic space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (a = b = c = 8.396 Å; α = β = γ = 90°; PDF no. C0190629) (Fig. 3E). In the FTIR spectra, surface grafted DHCA molecules were identified by the C
m (a = b = c = 8.396 Å; α = β = γ = 90°; PDF no. C0190629) (Fig. 3E). In the FTIR spectra, surface grafted DHCA molecules were identified by the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C vibration of the aromatic ring at 1547 cm−1 and 1417 cm−1 (Fig. 3F). In addition, two prominent bands at 1262 cm−1 and 1047 cm−1 were attributed to catechol moiety, whereas the absorption bands at 876 cm−1 and 808 cm−1 possibly belong to the out-of-plane bending of C–H vibrations.29 The Fe–O vibration frequency predictable for IONCs was observed at 545 cm−1.30 The surface-conjugation step involving conversion of carboxyl function of Fe3O4@DHCA NCs via carbodiimide coupling reaction was verified by the specific absorption band due to SAV that was found to appear at 1637 cm−1 corresponding to the amide groups located at the eight antiparallel β-strands of SAV.31,32 Additionally, we evaluated the magnetic separation of IONCs in water to achieve complete purification within 3 min (Fig. 3G).
C vibration of the aromatic ring at 1547 cm−1 and 1417 cm−1 (Fig. 3F). In addition, two prominent bands at 1262 cm−1 and 1047 cm−1 were attributed to catechol moiety, whereas the absorption bands at 876 cm−1 and 808 cm−1 possibly belong to the out-of-plane bending of C–H vibrations.29 The Fe–O vibration frequency predictable for IONCs was observed at 545 cm−1.30 The surface-conjugation step involving conversion of carboxyl function of Fe3O4@DHCA NCs via carbodiimide coupling reaction was verified by the specific absorption band due to SAV that was found to appear at 1637 cm−1 corresponding to the amide groups located at the eight antiparallel β-strands of SAV.31,32 Additionally, we evaluated the magnetic separation of IONCs in water to achieve complete purification within 3 min (Fig. 3G).
 | ||
| Fig. 3 Characterization of as-prepared IONC. (A) Schematic illustration of Fe3O4@DHCA NCs. (B–D) SEM shows a homogeneous distribution of particles with size of 142.59 ± 0.45 nm. (E) XRD pattern confirms magnetite phase. (F) Comparison between Fe3O4@DHCA and Fe3O4@SAV leads to additional bands in FTIR spectra. (G) Magnetically responsive efficient nanocarriers: a magnetic separation process is used to isolate the functionalized NCs from distilled water. | ||
Quantification of available binding slots of Fe3O4@SAV NCs
Fluorescence assays using B4F (λex = 493 nm, λem = 520 nm) are fast and easy method to quantify the available binding sites of SAV.33 The fluorescence properties of B4F are quenched after its strong binding in a binding pocket of streptavidin until all B4F molecules are occupied, leading to a subsequent change in the slope of the titration curve (Fig. S1†). To avoid scattering effects of the particles, an external magnetic field was applied to collect and separate NCs from solvent before the measurement of remaining B4F in the supernatant. As a control, we showed that the pure core-material Fe3O4 had no effect on the fluorescence of B4F (Fig. 4A). In this context, we investigated the synthesis reaction time and optimized the reaction condition in terms of the yield of Fe3O4@SAV NCs. Here, 12.36 pmol of B4F was bound to 0.2 mg of SAV-modified IONCs synthesized during a 3 h reaction, which was found to be the optimal reaction condition compared with a 1 h or 16 h synthetic route (Fig. 4B). The reaction was easily increased six fold, keeping all parameters constant except the concentration of IONCs used (Fig. 4C and D). Encouragingly, Fe3O4@SAV NCs synthesized on a larger batch showed more available binding sites for biotin-labeled molecules. This suggests that SAV molecules are more widely distributed on the particle surface so that the binding sites are not blocked. In this way, 0.2 mg Fe3O4@SAV NCs (synthesized in a 5 times larger scale) with 16 pmol available binding sites were obtained and used in the following experiments. | ||
| Fig. 4 Quantification of free binding slots of Fe3O4@SAV using B4F. (A) Fe3O4@DHCA NCs have no effect on the fluorescence of B4F, while (B) Fe3O4@SAV NCs with a reaction time of 3 h during the carbodiimide coupling reaction with SAV show the highest yield compared to 1 h and 16 h. (C and D) Quantification of the binding sites of synthesized Fe3O4@SAV NCs when the amount of Fe3O4@DHCA NCs (2 mg to 20 mg) was varied during synthesis. For all measurements, 0.2 mg Fe3O4@SAV NCs were dispersed in TEOA/H2O solution and incubated with organic dye B4F at different concentrations for 40 min. The particles were separated and the supernatant was measured. | ||
Binding of biotin labeled ss-oligonucleotides
The binding affinities of biotin labeled ss-oligonucleotides towards streptavidin conjugated nanocarriers (Fe3O4@SAV) were explored for various sequences of ss-oligonucleotides listed with their corresponding nomenclature in Table 1. The Fe3O4@SAV NCs were incubated with different amounts of complementary molecules (Compl.-TEG-BIO) to extract the conjugates by a subsequent washing step. The remaining binding sites of SAV-modified IONCs were quantitatively estimated by titrating the samples with fluorescent B4F, as described before.| Sample | ssDNA sequence |
|---|---|
| Compl.-TEG-BIO | 5′ C CAT TAT GTG CTG CTA 3′-TEG-biotin |
| Capping agent | 5′ TAG CAG CAC ATA ATG G 3′ |
| ssDNA-15a | 5′ TAG CAG CAC ATA ATG GTT TGT G 3′ |
| FITC-15a | 5′ TAG CAG CAC ATA ATG GTT TGT G 3′-FITC |
| FITC-mut15a-soft | 5′ TAG CTG CAC ATT ATG GTT TGT G 3′-FITC |
| FITC-mut15a-hard | 5′ TAC CAG GAC TTA TTG GTT TGT G 3′-FITC |
| FITC-C-tail | FITC-5′ CCC CCC CAC AAA 3′ |
Interestingly, despite the larger molecular size of conjugates, when compared to B4F, more than 80% of all binding slots were found to be occupied with Compl.-TEG-BIO (Fig. 5A and B). This indicated that chemical specificity of molecular recognition predominantly supersedes the steric hindrance of the residue bound on biotin molecule. The attachment of the biotin labeled ss-oligonucleotides strands on Fe3O4@SAV NCs evidently followed a second order reaction. However, since the reaction kinetics are logarithmic in nature, the occupancy rises only marginally above a certain value, thus high quantities of Compl.-TEG-BIO would be needed to achieve a occupancy above 80%. As a potential alternative, 22.79 pmol Compl.-TEG-BIO (for 0.2 mg SAV modified IONCs) was selected for further experiments, while maintaining 70% occupancy of surface sites available on nanocarriers. This corresponded to 11.2 pmol ss-oligonucleotides attached on the particle surface for 0.2 mg Fe3O4@SAV NCs.
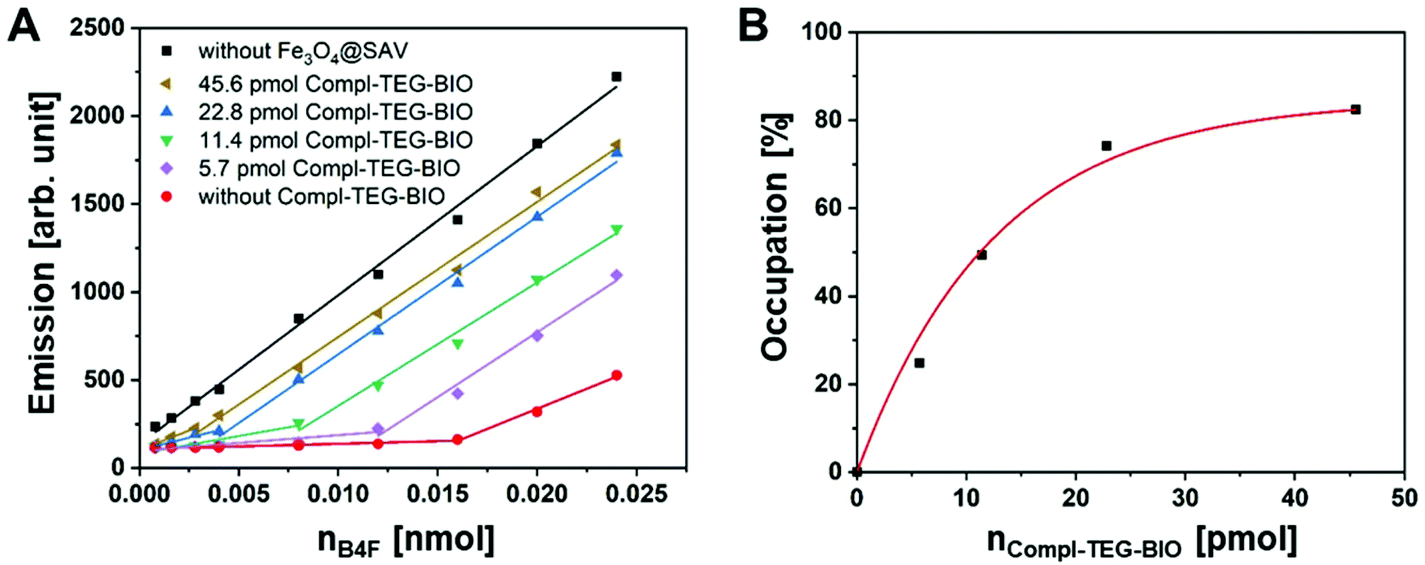 | ||
| Fig. 5 Quantification of remaining binding slots of Fe3O4@SAV after incubation with Compl.-TEG-BIO strand using B4F. (A) Emission spectra of B4F at different concentrations incubated with 0.2 mg Fe3O4@SAV-Compl.-TEG-BIO NCs dispersed in TEOA/H2O solution for 40 min. (B) Occupancy of free binding sites of Fe3O4@SAV with Compl.-TEG-BIO. For all measurements, the particles were magnetically separated and the supernatant was measured. | ||
Binding and purification of ssDNA-15a
A threefold higher amount of Fe3O4@SAV-Compl.-TEG-BIO was used, when compared to the B4F studies, to achieve a higher ss-oligonucleotide capacity. Upon hybridization of the two strands, the IONCs were magnetically separated, washed several times and the supernatant was analyzed for the residual amount of FITC-15a. For the control reactions, Fe3O4@SAV NCs without a complementary ss-oligonucleotide attached were used. It was observed, that 87.69% of the target ss-oligonucleotides was taken up in the case of Fe3O4@SAV-Compl.-TEG-BIO NCs, whereas the original 17.26 pmol of FITC-15a was still found unaltered in the supernatant of the control particles (Fig. 6A). As proof of concept, the two hybridized strands located on the particle surface of washed NCs where partially separated at 95 °C for 10 min and released FITC-15a could be detected in the solution. The maximum amount of FITC-15a isolated and purified from 0.6 mg NCs (Fig. 6B) was found to be 18.5 pmol. The selectivity of the uptake of FITC-15a was validated by comparing it with two other ss-oligonucleotides showing mutations in the arrangement of base pairs that lead to a 12.5% (FITC-mut15a-soft) or 25% (FITC-mut15a-hard) mismatch. Interestingly, the uptake of these two mutated ss-oligonucleotides was significantly less even at ambient temperature: the FITC-mut15a-soft was taken up in 59.12%, while the FITC-mut15a-hard was only taken up in 4.04% (Fig. 6C). To analyse the influence of these mutated ss-oligonucleotides on the uptake of the genuine FITC-15a oligonucleotide, we mixed the FITC-15a with either mutated oligonucleotide at a ratio of 1:1 or 1:2 to induce a competitive response. As the results show (Fig. 6D), the uptake of FITC-15a at ambient temperature was affected by the presence of FITC-mut15a-soft, as could be expected. Fortunately, it uptake was nearly independent of the concentration of FITC-mut15a-hard. However, we could increase the uptake of the FITC-15a strand again to about 80%, once we changed the hybridization temperature to 41 °C, which is 5 °C lower than the calculated Td of the hybridized double strand (Fig. 6D).
 | ||
| Fig. 6 FITC-15a uptake of Fe3O4@SAV-Compl.-TEG-BIO and two mutants. (A) Uptake of 17.26 pmol FITC-15a as ss-oligonucleotide on Fe3O4@SAV-Compl.-TEG-BIO NCs at ambient temperature for 1 h and release at 95 °C for 10 min. Between uptake and release of ss-oligonucleotid, particles were washed three times. (B) Relation between used and attached amount of FITC-15a on the particle surface. (C) Specific uptake of FITC-15a and two FITC-tagged mutations with 2 (soft) or 4 (hard) mismatched base pairs. (D) Uptake of FITC-15a in presence of mutations with ratio of 1:1 or 1:2, incubated at ambient temperature (orange) and at 41 °C (blue). For all measurements, a solution of 110 μL ss-oligonucleotides was incubated with 0.6 mg Fe3O4@SAV-Compl.-TEG-BIO, and 100 μL of the supernatant was taken for measurement. | ||
FITC labeling of isolated ssDNA-15a in urine samples
Firstly, the isolation and purification of ssDNA-15a was tested under optimal reaction conditions (TE-buffer). After hybridization of ssDNA-15a with Fe3O4@SAV-Compl.-TEG-BIO NCs, particles were magnetically separated, washed with buffer solution, and incubated with FITC-C-tail. The second hybridization of FITC-C-tail and ssDNA-15a was performed at the remaining 6 bases at the 3′end of ssDNA-15a. Due to the fact, that cytosine has the lowest quenching effect on FITC,34 we attached 6 additional cytosine bases between the fluorescent dye FITC and the complementary DNA sequence, which resulted in strong fluorescence of FITC-C-tail, even at smaller volumes and concentrations. During hybridization of FITC-C-tail, the low thermal stability of the second hybridized DNA strand, which has a calculated Td of 14 °C, posed a barrier for the reproducibility of the assay. For this reason, we designed an aluminium block to allow hybridization of the two ss-oligonucleotides and the magnetic separation on ice (Fig. S3†). The handling the IONCs at slightly above 5 °C resulted in high stability of all hybridized strands and ensured reproducible values. As a control, Fe3O4@SAV-Compl.-TEG-BIO NCs was tested that did not contain previously hybridized ssDNA-15a. The results showed that 79% of FITC-C-tail selectively binds to the ssDNA-15a strand compared with 13% unspecific binding on Compl.-TEG-BIO strands. We also evaluated an intermediate step (after the hybridization with ssDNA-15a) in which the remaining Compl.-TEG-BIO groups were blocked with a capping agent, without any detrimental effects (Fig. 7A). For the final quantification of ssDNA-15a, 10 pmol FITC-C-tail was used and the amount of target DNA was gradually varied resulting in a calibration curve. Fig. 7B shows the equation used to determine the sample amount of ssDNA-15a in TE-buffer solutions under optimal conditions. Afterwards, we transferred our system to non-optimal conditions, in which the target strand of ssDNA-15a was dissolved in human urine samples. As expected, only the overall fluorescence of FITC-C-tail was found to increase, whereby the system itself was not affected that validated our initial hypothesis. The equation used to determine the sample amount of ssDNA-15a in the human urine samples examined in this study is shown in Fig. 7C. Comparison of two normalized calibration curves (TE-buffer and urine) further confirmed that both reactions followed the same reaction kinetics (Fig. 7D). Since the composition of the urine may differ depending on the patient-to-patient heterogeneity, 4 pmol ssDNA-15a was added to the urine of three different individuals to evaluate our system and then quantified for the target ss-oligonucleotid. We tested ssDNA-15a in the pmol range, since the exact level of miRNA15a in cRCC patients is unknown. From 77 studies published so far, only 10 used urine as detection material,38 but all of them used qRT-PCR as primary amplification method. To the best of our knowledge, none of these reports analyzed the amount of miRNA in healthy subject, since all these studies were targeting the relative upregulation compared to normal control levels, according to the ddCt method. However, we were able to detect the amount of probe molecules approvable for clinical diagnosis for all the simulated patients, with a deviation of 0.49 pmol (Table 2). In case of a higher level of miRNA15a in RCC patients (μmol range), this has no effect on the performance of the presented diagnostic tool as the urine sample can simply be diluted. However, if the quantity should be lower and in the fmol range, then there are two possibilities: (1) the magnetic properties of IONCs could be used to concentrate miRNA15a from urine samples with higher volume or (2) a new calibration curve could be recorded by using fmol of ssDNA15a to shift the detection range into the fmol range. Future work will be intended to analyze real samples from patients and to define a clear “cut-off” between healthy individuals and RCC patients. | ||
| Fig. 7 FITC-15a uptake of Fe3O4@SAV-ssDNA15a and quantification of ssDNA15a in TE-buffer and human urine. (A) Uptake of FITC-C-tail depending on the presence of ssDNA-15a, whereby a capping agent shows no influence. (B) Emission spectra after uptake of 10 pmol FITC-C-tail as a function of the amount of ssDNA-15a dissolved in TE-buffer or (C) in human urine. (D) Normalized emission spectra of 10 pmol FITC-C-tail as a function of the amount of ssDNA-15a dissolved in TE-buffer or human urine show similar tendency. For all measurements, 0.6 mg Fe3O4@SAV-ssDNA15a were used. The particles were separated and the supernatant was measured. | ||
| Individual | Fluorescence (control; without ssDNA-15a) [arb. unit] | Fluorescence (sample; with 4 pmol ssDNA-15a) [arb. unit] | Calculated amount of ssDNA-15a (sample) via calibration curve [pmol] |
|---|---|---|---|
| A | 2606.5 ± 5.0 | 1951.5 ± 293.4 | 4.1591 |
| B | 2628.0 ± 230.5 | 1886.0 ± 67.9 | 4.5585 |
| C | 2705.0 ± 121.6 | 2052.5 ± 102.5 | 3.5757 |
Conclusions
Application of the biomarker miRNA15a as reliable biomarker for RCC in routine clinical diagnosis relies on the development of a rapid, non-invasive, cost effective, time efficient and highly sensitive detection method that offers reproducible results. Here, we demonstrated an innovative diagnostic tool based on chemically functionalized magnetic nanocarriers for the selective capture of miRNA15a from urine. This uptake strategy based on molecular recognition of surface-attached affinity ligands fused to a magnetic nanocarrier and miRNA molecules allowed magnetic separation of target biomarker (miRNA15a) from body fluids within minutes.In our analysis we used miRNA15a as target molecule as intended diagnostic test. This miRNA seemed particularly suited, since it had been demonstrated as potential biomarker in urine samples from malignant renal carcinomas,13 and is the only miRNA so far which has been evaluated and confirmed in this function by an independent group.23 Furthermore, we have shown that it allows the recognition of small renal masses (<4 cm; pT1a). In contrast to other publications reporting potential miRNAs in the urine as diagnostic markers,37,38 the regulation of this miRNA in renal tumors is known (involving PKC alpha).13 Through binding to the DNA sequence of MAPKp38alpha, miRNA15a generates a truncated, biological functioning protein, Mxi-2.16 This notion is fundamental in the understanding of the function of this miRNA in renal tumors as indicator of malignancy, which is diametrically opposite to its function as tumor suppressor in other tumors: in the progression of CLL, where its deletion or downregulation is observed in more than two thirds of cases39 or in prostate carcinoma, where this miRNA acts to control cell survival, proliferation and invasion.40 While there are only two reports analyzing urine for miRNA,18,23 there are several, in which miRNAs were extracted from the serum of cRCC patients for the diagnosis of renal cancer using RT-PCR. However, currently none of those acclaimed potential biomarkers have been developed into a clinical assay. Analysing their circulating serum levels they seemed either unsuited for cancer diagnostics (miRNA-26a-2, miRNA-191, miRNA-337-3p, miRNA-378);41 their sensitivity was relative low (miRNA-210: 57.5% to 65%);42,43 there was unexplained contradictory evidence of different miRNA levels in spite of the analysis of the same tumor type (i.e. miRNA-378 vs. miRNA-451);41,44 or lack of specificity for RCC (miRNA-221).45
Besides RT-PCR, next generation sequencing (NGS) combined with TaqMan mutation – analysis was tested in a small series of patients with RCCs with von-Hippel–Lindau mutation (VHL).46 No detection of VHL mutation in VHL-RCC blood from 9 patients was found by NGS, and only 1/9 TaqMan assays were positive, while KRAS mutated colon carcinoma by both methods from liquid biopsy was positive in all patients used as positive control for these methods. Results were reportedly being independent of tumor size, metastases, tumor necroses. This negative outcome was explained by reduced tumor shedding or increased clearance of circulating DNA, while these highly sensitive detection systems for known mutation were deemed not suitable for liquid biopsy of RCCs.
To overcome some of the intrinsic limitations of current analytical procedures, we propose the use of a nanotechnology-based detection system for miRNA15a, which has several advantages over the “gold standard” of qRT-PCR in terms of: handling expertise, equipment needed for diagnostic tests, specificity, sensitivity, duration for diagnostic procedure, ease of test manufacturing, and costs. Handling expertise: the implementation of our assay is well within the regular competence of a laboratory assistant. Equipment to perform the diagnostic test: a fluorescent ELISA plate reader and a permanent magnet are the only special equipment needed. Sensitivity: the system was validated by incubating 4 pmol of ssDNA-15a with urine from three different individuals which was successfully re-isolated, purified and quantified. Time for diagnostic procedure: the analysis takes approximately 2.5 h for completion. Specificity: the selectivity of ssDNA-15a uptake was tested by competitive hybridization reactions with two mutants. It was shown that ss-oligonucleotides with 2 mutations can still be taken up at a lower percentage, whereas no interaction could be detected for strands with 4 mutations at ambient temperature. This underscores the specificity of our protocol. However in practice, strands with such a high degree of matching are generally not be present in urine. We also proved that none of the mutants had any effect on the specific uptake of ssDNA-15a by increasing the hybridization temperature up to 41 °C. Costs: the synthetic route of the scavenger material Fe3O4@SAV NCs described in this work, provides an inexpensive alternative to commercially available expensive isolation-kits: while the miRNA analysis using extraction and RT-PCR reagents cost approximately 48 euros per sample, we estimate our costs to be around 22 cent per sample (calculated on a lab-scale setting).
In conclusion, we have presented here a non-invasive diagnostic tool for ssDNA-15a that is analogous to miRNA15a as biomarker for RCC. This offers a time-efficient, cost-effective highly selective, and sensitive quantification of the target ss-oligonucleotide which thus stands out significantly from previous methods (including qPCR). The next steps would be now large-scale studies to be performed in patients with RCC and to establish the presented diagnostic tool in clinical practice.
Author contributions
Conceptualization: A. M. R., S. M., J. W. U. F. and I. G.; methodology: A. M. R., S. M. and J. W. U. F.; formal analysis: A. M. R. and C. D.; investigation and visualization: A. M. R. and C. D.; writing original draft: A. M. R.; writing review and editing: S. I., S. M. and J. W. U. F.; project administration S. M. and J. W. U. F.; all authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the financial support and infrastructure provided through the University of Cologne in the frame of the Excellence Strategy and for supporting the UoC-Forum “Transformative Nanocarriers for RNA Transport and Tracking”. Dr Thomas Fischer is thankfully acknowledged for SEM measurements. We also acknowledge the fruitful discussions with Khan Lê and Arun Ichangi.References
- R. Siegel, J. Ma, Z. Zou and A. Jemal, CA Cancer J. Clin., 2014, 64, 9–29 CrossRef PubMed.
- H. T. Cohen and F. J. McGovern, N. Engl. J. Med., 2005, 353, 2477–2490 CrossRef CAS PubMed.
- B. I. Rini, W. K. Rathmell and P. Godley, Curr. Opin. Oncol., 2008, 20, 300–306 CrossRef PubMed.
- N. K. Janzen, H. L. Kim, R. A. Figlin and A. S. Belldegrun, Urol. Clin. North Am., 2012, 30, 843–852 CrossRef.
- W. Thoenes, S. Störkel and H. J. Rumpelt, Pathol., Res. Pract., 1986, 181, 125–143 CrossRef CAS.
- W. Lilleby and S. D. Fossa, World J. Urol., 2005, 23, 175–179 CrossRef CAS PubMed.
- M. Redova, M. Svoboda and O. Slaby, Biochem. Biophys. Res. Commun., 2011, 405, 153–156 CrossRef CAS PubMed.
- L. B. Chipman and A. E. Pasquinelli, Trends Genet., 2019, 35, 215–222 CrossRef CAS PubMed.
- V. Ambros, Cell, 2003, 113, 673–676 CrossRef CAS PubMed.
- D. P. Bartel, Cell, 2004, 116, 281–297 CrossRef CAS PubMed.
- J. F. Palatnik, E. Allen, X. Wu, C. Schommer, R. Schwab, J. C. Carrington and D. Weigel, Nature, 2003, 425, 257–263 CrossRef CAS PubMed.
- M. von Brandenstein, C. Richter and J. W. U. Fries, Life Sci., 2012, 91, 475–489 CrossRef CAS PubMed.
- M. von Brandenstein, J. J. Pandarakalam, L. Kroon, H. Loeser, J. Herden, G. Braun, K. Wendland, H.-P. Dienes, U. Engelmann and J. W. U. Fries, Am. J. Pathol., 2012, 180, 1787–1797 CrossRef CAS PubMed.
- M. von Brandenstein, J. Herden, B. Köditz, M. Huerta, T. Nestler, A. Heidenreich and J. W. U. Fries, J. Clin. Lab. Anal., 2021, 35, e23762 CAS.
- H. Löser, M. von Brandenstein, V. Burst, C. Richter, R. Buettner, C. Kurschat, B. Hoppe and J. W. U. Fries, J. Mol. Med. Clin. Appl., 2018, 2, 1 Search PubMed.
- M. von Brandenstein, S. H. Bernhart, A. Pansky, C. Richter, T. Kohl, M. Deckert, A. Heidenreich, P. F. Stadler, M. Montesinos-Rongen and J. W. U. Fries, Oncotarget, 2018, 9, 32855–32867 CrossRef PubMed.
- M. G. von Brandenstein, A. N. Abety, R. Depping, T. Roth, M. Koehler, H.-P. Dienes and J. W. U. Fries, Biochim. Biophys. Acta, 2008, 1783, 1613–1622 CrossRef CAS PubMed.
- J. W. U. Fries, Biol. Cell, 2019, 111, 169–186 CrossRef CAS PubMed.
- M. von Brandenstein, R. Depping, E. Schäfer, H.-P. Dienes and J. W. U. Fries, Biochim. Biophys. Acta, 2011, 1813, 1793–1802 CrossRef CAS PubMed.
- B. Köditz, J. W. U. Fries, H. Göbel, P. Paffenholz, K. Richter, A. Heidenreich and M. von Brandenstein, Anticancer Res., 2020, 40, 5539–5544 CrossRef PubMed.
- M. von Brandenstein, M. Schlosser, C. Richter, R. Depping and J. W. U. Fries, Life Sci., 2012, 91, 562–571 CrossRef CAS PubMed.
- K. Dashtipour, J. J. Chen, C. Kani, K. Bahjri and M. Ghamsary, Pharmacotherapy, 2015, 35, 681–686 CrossRef CAS PubMed.
- Y. Mytsyk, V. Dosenko, Y. Borys, A. Kucher, K. Gazdikova, D. Busselberg, M. Caprnda, P. Kruzliak, A. A. Farooqi and M. Lubov, Int. Urol. Nephrol., 2018, 50, 851–859 CrossRef CAS PubMed.
- L. Z. Hong, L. Zhou, R. Zou, C. M. Khoo, A. L. S. Chew, C.-L. Chin and S.-J. Shih, Sci. Rep., 2021, 11, 4435 CrossRef CAS PubMed.
- S. Ilyas, M. Ilyas, R. A. L. van der Hoorn and S. Mathur, ACS Nano, 2013, 7(11), 9655–9663 CrossRef CAS PubMed.
- I. Gessner, J. W. U. Fries, V. Brune and S. Mathur, J. Mater. Chem. B, 2021, 9, 9 RSC.
- M. Sarter, D. Niether, B. W. Koenig, W. Lohstroh, M. Zamponi, N. H. Jalarvo, S. Wiegand, J. Fitter and A. M. Stadler, J. Phys. Chem. B, 2020, 124, 324–335 CrossRef CAS PubMed.
- H. Zhu, C. Hou, Y. Li, G. Zhao, X. Liu, K. Hou and Y. Li, Chem. – Asian J., 2013, 8, 1447–1454 CrossRef CAS PubMed.
- N. Aktaş, N. Sahiner, Ö. Kantoǧlu, B. Salih and A. Tanyolaç, J. Polym. Environ., 2003, 11, 123–128 CrossRef.
- L. Wortmann, S. Ilyas, D. Niznansky, M. Valldor, K. Arroub, N. Berger, K. Rahme, J. Holmes and S. Mathur, ACS Appl. Mater. Interfaces, 2014, 6, 16631–16642 CrossRef CAS PubMed.
- C. Hou, L. Zhang, Y. Wang and Z. Wang, J. Appl. Polym. Sci., 2015, 132, 41560 Search PubMed.
- A. Krüger, A. Bürkle, A. Mangerich and K. Hauser, Biomed. Spectrosc. Imaging, 2018, 7, 1–9 Search PubMed.
- M. J. Waner and D. P. Mascotti, J. Biochem. Biophys. Methods, 2008, 70, 873–877 CrossRef CAS PubMed.
- S. A. E. Marras, F. R. Kramer and S. Tyagi, Nucleic Acids Res., 2002, 30, e122 CrossRef PubMed.
- G. T. Hermanson, Bioconjugate Techniques, Academic Press, London (UK), 3rd edn, 2013, pp. 1–1146 Search PubMed.
- G. Kada, H. Falk and H. J. Gruber, Biochim. Biophys. Acta, 1999, 1427, 33–43 CrossRef CAS.
- A. Di Meo, M. D. Brown, A. Finelli, M. A. S. Jewett, E. P. Diamandis and G. M. Yousef, Clin. Biochem., 2020, 75, 15–22 CrossRef CAS PubMed.
- J. Oto, E. Plana, J. V. Sánchez-González, J. García-Olaverri, A. Fernández-Pardo, F. España, M. Martínez-Sarmiento, C. D. Vera-Donoso, S. Navarro and P. Medina, Curr. Urol. Rep., 2020, 21, 11 CrossRef PubMed.
- S. Lehmann, S. Ogawa, S. D. Raynaud, M. Sanada, Y. Nannya, M. Ticchioni, C. Bastard, N. Kawamata and H. P. Koeffler, Cancer, 2008, 112, 1296–1305 CrossRef CAS PubMed.
- D. Bonci, V. Coppola, M. Musumeci, A. Addario, R. Giuffrida, L. Memeo, L. D'Urso, A. Pagliuca, M. Biffoni, C. Labbaye, M. Bartucci, G. Muto, C. Peschle and R. De Maria, Nat. Med., 2008, 14, 1271–1277 CrossRef CAS PubMed.
- S. Hauser, L. M. Wulfken, S. Holdenrieder, R. Moritz, C. H. Ohlmann, V. Jung, F. Becker, E. Herrmann, G. Walgenbach-Brünagel, A. von Ruecker, S. C. Müller and J. Ellinger, Cancer Epidemiol., 2012, 36, 91–394 Search PubMed.
- G. Li, A. Zhao, M. Péoch, M. Cottier and N. Mottet, Urol. Oncol., 2017, 35, 294–299 CrossRef CAS PubMed.
- H. Iwamoto, Y. Kanda, T. Sejima, M. Osaki, F. Okada and A. Takenakal, Int. J. Oncol., 2014, 44, 53–58 CrossRef CAS PubMed.
- M. Redova, A. Poprach, J. Nekvindova, R. Iliev, L. Radova, R. Lakomy, M. Svoboda, R. Vyzula and O. Slaby, J. Transl. Med., 2012, 10, 55 CrossRef CAS PubMed.
- A. L. Teixeira, M. Ferreira, J. Silva, M. Gomes, F. Dias, J. I. Santos, J. Maurício, F. Lobo and R. Medeiros, Tumor Biol., 2014, 35, 4057–4066 CrossRef CAS PubMed.
- C. Corrò, T. Hejhal, C. Poyet, T. Sulser, T. Hermanns, T. Winder, G. Prager, P. J. Wild, I. Frew, H. Moch and M. Rechsteiner, Exp. Mol. Pathol., 2017, 102, 255–261 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Quenching effect of SAV on B4F and particle scattering effect; calibration line of FITC-15a, FITC-mut15a-soft, FITC-mut15a-hard and FITC-C-tail; photo of designed cooling block. See DOI: 10.1039/d1bm01737c |
| This journal is © The Royal Society of Chemistry 2022 |