
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, characterisation and evaluation of hyperbranched N-(2-hydroxypropyl) methacrylamides for transport and delivery in pancreatic cell lines in vitro and in vivo†
Akosua B.
Anane-Adjei
 a,
Nicholas L.
Fletcher
bc,
Robert J.
Cavanagh
a,
Zachary H.
Houston
bc,
Theodore
Crawford
b,
Amanda K.
Pearce
a,
Vincenzo
Taresco
a,
Alison A.
Ritchie
d,
Phillip
Clarke
d,
Anna M.
Grabowska
d,
Paul R.
Gellert
e,
Marianne B.
Ashford
f,
Barrie
Kellam
a,
Kristofer J.
Thurecht
*bc and
Cameron
Alexander
*a
a,
Nicholas L.
Fletcher
bc,
Robert J.
Cavanagh
a,
Zachary H.
Houston
bc,
Theodore
Crawford
b,
Amanda K.
Pearce
a,
Vincenzo
Taresco
a,
Alison A.
Ritchie
d,
Phillip
Clarke
d,
Anna M.
Grabowska
d,
Paul R.
Gellert
e,
Marianne B.
Ashford
f,
Barrie
Kellam
a,
Kristofer J.
Thurecht
*bc and
Cameron
Alexander
*a
aDivision of Molecular Therapeutics and Formulation, School of Pharmacy, University of Nottingham, NG7 2RD, UK. E-mail: cameron.alexander@nottingham.ac.uk
bCentre for Advanced Imaging, University of Queensland, Brisbane, Australia. E-mail: k.thurecht@uq.edu.au
cAustralian Institute for Bioengineering and Nanotechnology, University of Queensland, Brisbane, Australia
dSchool of Medicine, University of Nottingham, NG7 2RD, UK
eProduct Technology & Development, Operations, AstraZeneca, Macclesfield, UK
fAdvanced Drug Delivery, Pharmaceutical Sciences, R&D AstraZeneca, Macclesfield, UK
First published on 5th April 2022
Abstract
Hyperbranched polymers have many promising features for drug delivery, owing to their ease of synthesis, multiple functional group content, and potential for high drug loading with retention of solubility. Here we prepared hyperbranched N-(2-hydroxypropyl)methacrylamide (HPMA) polymers with a range of molar masses and particle sizes, and with attached dyes, radiolabel or the anticancer drug gemcitabine. Reversible addition–fragmentation chain transfer (RAFT) polymerisation enabled the synthesis of pHPMA polymers and a gemcitabine-comonomer functionalised pHPMA polymer pro-drug, with diameters of the polymer particles ranging from 7–40 nm. The non-drug loaded polymers were well-tolerated in cancer cell lines and macrophages, and were rapidly internalised in 2D cell culture and transported efficiently to the centre of dense pancreatic cancer 3D spheroids. The gemcitabine-loaded polymer pro-drug was found to be toxic both to 2D cultures of MIA PaCa-2 cells and also in reducing the volume of MIA PaCa-2 spheroids. The non-drug loaded polymers caused no short-term adverse effects in healthy mice following systemic injection, and derivatives of these polymers labelled with 89Zr-were tracked for their distribution in the organs of healthy and MIA PaCa-2 xenograft bearing Balb/c nude mice. Tumour accumulation, although variable across the samples, was highest in individual animals for the pHPMA polymer of ∼20 nm size, and accordingly a gemcitabine pHPMA polymer pro-drug of ∼18 nm diameter was evaluated for efficacy in the tumour-bearing animals. The efficacy of the pHPMA polymer pro-drug was very similar to that of free gemcitabine in terms of tumour growth retardation, and although there was a survival benefit after 70 days for the polymer pro-drug, there was no difference at day 80. These data suggest that while polymer pro-drugs of this type can be effective, better tumour targeting and enhanced in situ release remain as key obstacles to clinical translation even for relatively simple polymers such as pHPMA.
Introduction
Polymer-based therapeutics are now established clinically in several indications where their improved pharmacokinetic and pharmacodynamic properties compared to conventional drug molecules lead to patient benefits.1 The requirement in many cases for polymer-based therapeutics stems from the need to maximise therapeutic activity and minimise side effects.2–4 Polymeric carriers can significantly improve the transport of many drug molecules in aqueous environments, as complexation or conjugation of the drug to a water-soluble polymer effectively renders the solubility of the carrier as the key factor in retaining the drug in the body, and this has been exploited in multiple therapeutic settings.5–9 This is particularly important for new drugs in the discovery process, as it has been estimated that approx. 40–60% of drugs in development exhibit low aqueous solubility, thus limiting their dosing in vivo.10,11 Amongst the classes of polymer investigated as therapeutics carriers, materials based on poly(2-hydroxypropylmethacrylamide) (pHPMA) are amongst the most widely studied, owing to their relative ease of synthesis, the potential for drug conjugation and good tolerance in human clinical trials.12–14 Since the initial pioneering studies of pHPMA,15–17 which progressed to candidate therapeutics for cancers, there have been attempts to develop the materials chemistry platform of pHPMA and related polymers for greater efficacy.18–22 The backbone of pHPMA is not biodegradable but there have been no safety concerns with its use in humans to date, and the main reason for its failure to be adopted clinically was limited tumour concentration of the delivered payload from the initially developed materials.15 There have accordingly been increasing reports of pHPMA materials conjugated with targeting agents to improve therapeutic window, and pHPMA derivatives with higher drug loadings and combinations of drugs attached to the backbone to improve clinical efficacy.23–28 More recently, there have been studies of hyperbranched pHPMA structures, as these materials have multiple end-groups to attach labels and/or targeting agents, thus conferring a greater sensitivity to detection or ability to engage receptors compared to their linear analogues.29–31 A key additional advantage is that the hyperbranched architectures can confer shape persistence in solution, thus enabling the polymers to behave as unimolecular micelles with high drug content ‘cores’ while retaining solubility. From an industrial perspective, the synthetic challenges and costs of developing hyperbranched polymers are considerably lower than for comparable dendrimers, which also feature high functionality for drug loading, making them highly appealing for use in polymer–drug conjugate delivery systems.32,33In this work, we evaluate the in vitro and in vivo properties of hyperbranched pHPMA materials, as a first step towards polymeric prodrugs for pancreatic cancer therapy. The specific need for polymer therapeutics for pancreatic cancer arises from the dense stromal tissue surrounding pancreatic tumours, which affects the delivery and thus efficacy of current drugs.33,34 There is accordingly a trade-off between carrier size and efficacy, as high doses of cytotoxic drugs are needed to induce tumour cell death, implying the need for larger carriers, but smaller polymers provide greater penetration through the tumour matrix.35 We accordingly chose to study hyperbranched architectures, as these should provide maximum loading of drugs on the polymer backbone without inducing significant changes in overall solubility or the formation of large micellar aggregates, which is often a limiting factor with linear polymers containing high drug loadings. In the first instance, our goal was to study polymers with dyes as drug analogues, to establish the fundamental trafficking properties of hyperbranched materials. For this we prepared three different hyperbranched materials from N-(2-hydroxypropyl)methacrylamide (HPMA) via reversible addition–fragmentation chain-transfer (RAFT) polymerisation. We evaluated transport in 2D cell culture and co-cultures of MIA PaCa-2 3D spheroids with mesenchymal stem cells as mimics of stromal tissues surrounding pancreatic tumours. We then derivatised the materials further with ligands to allow radiolabelling and monitored their biodistribution over time in healthy and pancreatic cancer-bearing mice. We also prepared a fourth polymer which included a gemcitabine-functionalised co-monomer. For in vitro and in vivo efficacy studies we used polymer which was of a similar size, as measured by DLS and TEM, to the non-drug loaded polymer which showed the highest accumulation in the tumours of the MIA PaCa-2 tumour-bearing mice. We subsequently evaluated the effects of this polymer on MIA PaCa-2 cells in 2D and 3D culture, and in xenografted mice in comparison to the free drug.
Results and discussion
Synthesis and physical characterisation of HB-HPMA polymers
The set of hyperbranched N-(2-hydroxypropyl)methacrylamide (HB-HPMA) polymers were designed and synthesised using the same starting materials but varying the conditions to provide different physical properties in terms of molecular weight and particle size (Fig. 1[A–C], Fig. S1 and S2A, ESI,† and Table 1). A hyperbranched architecture was chosen for this work due to the presence of abundant functional groups for drug loading and the demonstrated potential for tumour targeting and in vivo particle uptake.36,37 The syntheses of the HB polymers were achieved via RAFT polymerisation, utilising an alkyne terminated RAFT agent, a bifunctional ethylene dimethacrylamide (EDMA) monomer, and AIBN as the initiator as previously described.38 HPMA was synthesised as previously described by Kopeček et al. and was obtained in a 65% yield after purification by recrystallisation.39 The bifunctional methacrylamide unit (EDMA), incorporated into the polymerisation to induce branching in the macromolecules and generate unimolecular globular particles, was synthesised through the reaction of methacrylic anhydride and ethylenediamine in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio. Monomer conversion was calculated from the 1H NMR spectrum of the crude product by comparing the ratio between the vinyl protons and the polymer resonances at 3.82–3.57 ppm (Fig. S1, ESI†). Varying monomer conversions were achieved for the polymer syntheses (90–97%) and the resultant hyperbranched polymers were characterised (Fig. 1; S1 and S2A. ESI;† and Table 1) for their purity (1H NMR), molecular weight (aqueous SEC-MALLS), hydrodynamic radius (DLS), and physical size and morphology (TEM). To probe the reproducibility of the synthesis, the procedures were repeated (Table S2, ESI†) and no significant difference was observed with the DLS sizes between the different batches. Variations were seen between the molar masses across the repeat sets, suggesting that the sizes reported by light scattering are less sensitive to variations in molar mass distribution than those derived from chromatography. All the polymers were confirmed to contain no detectable residual monomer after purification due to the absence of vinyl peaks at 5.72 and 5.32 ppm (Fig. S1, ESI†) as well as the characteristic broadening of monomer resonances in the 1H NMR spectrum, indicative of polymeric materials. The final hyperbranched pHPMA-GEM polymer conjugate was also synthesised through RAFT polymerisation in 50:50 water/DMAc. For drug loading, we chose in the first instance to prepare a pro-drug monomer with gemcitabine (GEM), which is a standard-of-care for pancreatic cancer therapies, and for which there was reported efficacy of an analogue when incorporated into a linear polymer backbone.27 For this we reacted HPMA monomer with succinic anhydride to provide a carboxylic acid functional group, which was subsequently coupled to gemcitabine through the formation of an amide bond with the N-4 amino group (Fig. S3, ESI†). The target functional GEM monomer compared to HPMA content per polymer chain was set at 20 mol/mol% to provide a sufficient dose while maintaining the overall water solubility and stealth properties of HPMA polymers.40 High monomer conversion was achieved also for the HPMA-GEM polymer synthesis (94%) and the resultant hyperbranched polymer was characterised by 19F NMR and 1H NMR (Fig. 1G), and FT-IR (Fig. S4, ESI†). The sizes of these polymers determined from DLS, (Fig. 1H and I, Table 1) indicated different hydrodynamic diameters of approx. 7 nm, 20 nm, and 40 nm for the non-functional polymers (HB-HPMA-7, HB-HPMA-20, and HB-HPMA-40 respectively) and ∼16 nm for the pHPMA-GEM. The sizes of the polymers, as well as the morphology, were further investigated using transmission electron microscopy (TEM). From the images (Fig. 1C–F), it is evident that the polymers exist in a distinct globular structure with sizes largely similar to those seen in the DLS, albeit slightly smaller for HB-HPMA 20 and HB-HPMA-40 polymers. The slight decrease in apparent radius in TEM may in part be attributed to the shrinkage of the polymer particle on drying in the TEM chamber.
:1 stoichiometric ratio. Monomer conversion was calculated from the 1H NMR spectrum of the crude product by comparing the ratio between the vinyl protons and the polymer resonances at 3.82–3.57 ppm (Fig. S1, ESI†). Varying monomer conversions were achieved for the polymer syntheses (90–97%) and the resultant hyperbranched polymers were characterised (Fig. 1; S1 and S2A. ESI;† and Table 1) for their purity (1H NMR), molecular weight (aqueous SEC-MALLS), hydrodynamic radius (DLS), and physical size and morphology (TEM). To probe the reproducibility of the synthesis, the procedures were repeated (Table S2, ESI†) and no significant difference was observed with the DLS sizes between the different batches. Variations were seen between the molar masses across the repeat sets, suggesting that the sizes reported by light scattering are less sensitive to variations in molar mass distribution than those derived from chromatography. All the polymers were confirmed to contain no detectable residual monomer after purification due to the absence of vinyl peaks at 5.72 and 5.32 ppm (Fig. S1, ESI†) as well as the characteristic broadening of monomer resonances in the 1H NMR spectrum, indicative of polymeric materials. The final hyperbranched pHPMA-GEM polymer conjugate was also synthesised through RAFT polymerisation in 50:50 water/DMAc. For drug loading, we chose in the first instance to prepare a pro-drug monomer with gemcitabine (GEM), which is a standard-of-care for pancreatic cancer therapies, and for which there was reported efficacy of an analogue when incorporated into a linear polymer backbone.27 For this we reacted HPMA monomer with succinic anhydride to provide a carboxylic acid functional group, which was subsequently coupled to gemcitabine through the formation of an amide bond with the N-4 amino group (Fig. S3, ESI†). The target functional GEM monomer compared to HPMA content per polymer chain was set at 20 mol/mol% to provide a sufficient dose while maintaining the overall water solubility and stealth properties of HPMA polymers.40 High monomer conversion was achieved also for the HPMA-GEM polymer synthesis (94%) and the resultant hyperbranched polymer was characterised by 19F NMR and 1H NMR (Fig. 1G), and FT-IR (Fig. S4, ESI†). The sizes of these polymers determined from DLS, (Fig. 1H and I, Table 1) indicated different hydrodynamic diameters of approx. 7 nm, 20 nm, and 40 nm for the non-functional polymers (HB-HPMA-7, HB-HPMA-20, and HB-HPMA-40 respectively) and ∼16 nm for the pHPMA-GEM. The sizes of the polymers, as well as the morphology, were further investigated using transmission electron microscopy (TEM). From the images (Fig. 1C–F), it is evident that the polymers exist in a distinct globular structure with sizes largely similar to those seen in the DLS, albeit slightly smaller for HB-HPMA 20 and HB-HPMA-40 polymers. The slight decrease in apparent radius in TEM may in part be attributed to the shrinkage of the polymer particle on drying in the TEM chamber.
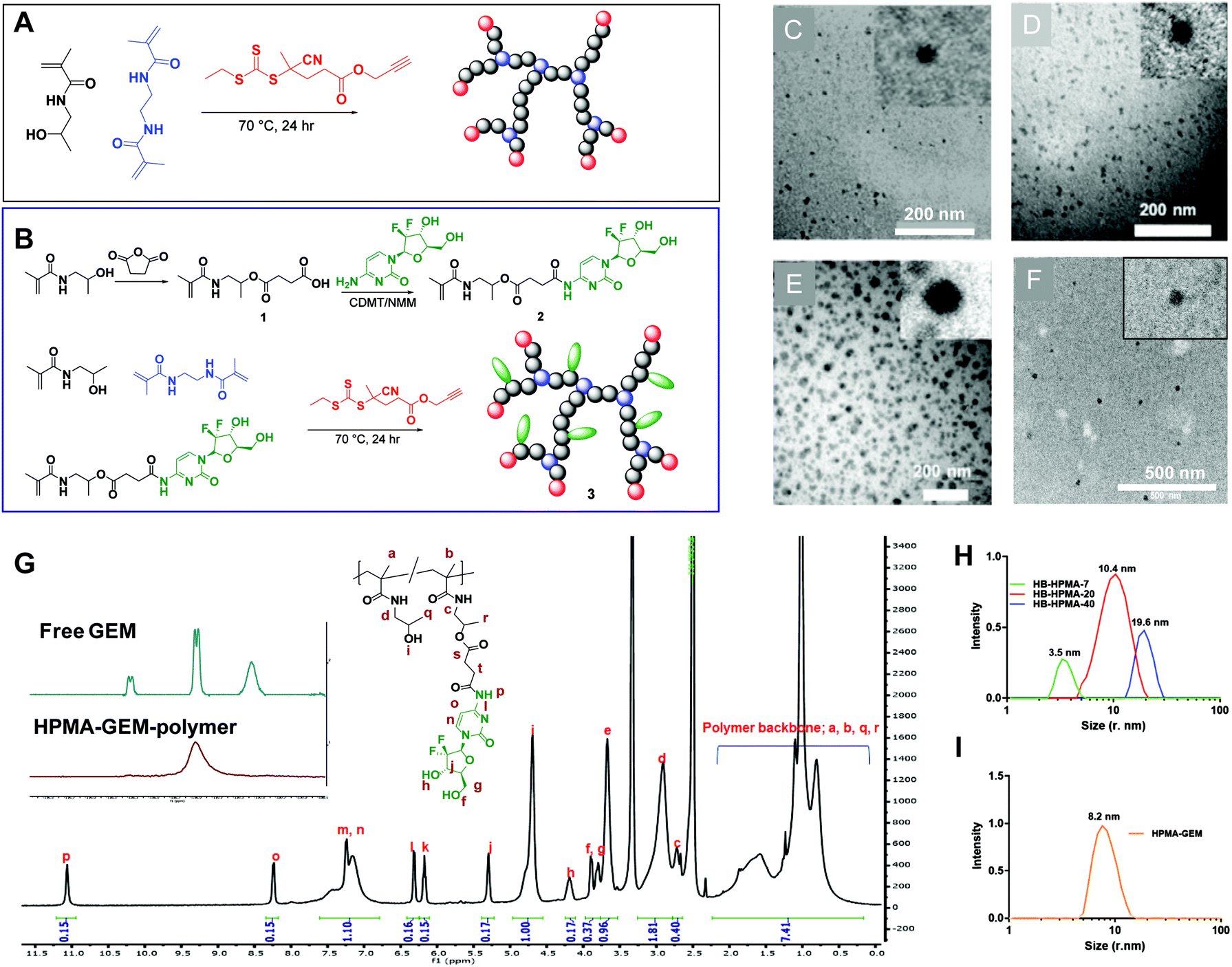 | ||
| Fig. 1 (A and B) HB-HPMA polymers were synthesised using Reversible Addition Fragmentation Chain Transfer (RAFT) polymerisation. characterisation of HB-HPMA polymers (C–F); transmission electron microscopy of HB-HPMA-7 (C), HB-HPMA-20 (D), HB-HPMA-40 (E) (scale bar = 200 nm) and HB-HPMA-GEM polymer (scale bar = 500 nm), (G) 19F NMR (inset) of GEM and of HB-HPMA-GEM polymer, and 1H NMR spectra of HB-HPMA-GEM polymer after purification. (H and I) Representative DLS intensity/size curves for HB-HPMA-7, HB-HPMA-20, HB-HPMA-40 and HB-HPMA-GEM polymer. | ||
| Polymer | HB-HPMA-7 | HB-HPMA-20 | HB-HPMA-40 | HB-HPMA-GEM |
|---|---|---|---|---|
| a dn/dc = 0.1589. | ||||
| M n by SEC-MALLSa (g mol−1) | 1.8 × 104 | 1.1 × 105 | 1.8 × 106 | 4.9 × 104 |
| Đ | 2.1 | 2.5 | 3.1 | 2.1 |
| Size by DLS (nm) | 7 | 20 (±0.6) | 40 (±0.7) | 16.4 (±1.8) |
| Size by TEM (nm) | 7 | 15 (±1.9) | 30 (±9.5) | 17.9 (±3.5) |
| Cy5/HB polymer (UV-Vis) wt% | 5.7 | 5.1 | 2.2 | |
| GEM loading (NMR) mol% | 18 | |||
To provide a fluorescent label for the initial biological screening of these materials, an azide-functional Cy5 dye was conjugated to the polymers via copper catalysed azide–alkyne 1,3-dipolar cycloaddition to the alkyne RAFT end group. The physical properties of the dye-hyperbranched polymers are summarised in Table 1.
In vitro toxicity and cellular uptake of synthesised polymers
Initial experiments utilised the dye-labelled polymers with the MIA PaCa-2 pancreatic cancer cell line in both 2D and 3D cell culture models to evaluate internalisation, transport through cell layers, and inherent cytocompatibility. The trypan blue dye exclusion test was used to determine the effects of the polymers in 2D cell culture over 72 h. The polymers did not induce any marked detrimental effects on the MIA PaCa-2 cell line at the concentrations tested (0.1–1000 μg mL−1, Fig. S5, ESI†). This observation was expected due to previously reported literature on HPMA polymers in both in vitro and in vivo settings,19,41–43 but confirmed that no toxic residues from synthesis or modification procedures remained in the polymer samples.To study the internalisation of these polymers, the Cy5-labelled polymers were incubated with the 2D cultured MIA PaCa-2 cells for timepoints up to 4 hours (Fig. 2A and B). Live cell imaging and spectrometric based experiments revealed time-dependent uptake of all polymers. Quantitative analysis (Fig. 2B) indicated rapid initial uptake of all polymers between 5–60 minutes, but a much reduced rate of internalisation after 2 hours. The data showed the highest fluorescence due to internalisation of HB-HPMA-20 (Fig. 2B), while two way ANOVA analysis revealed significant differences between fluorescence due to HB-HPMA-40 and HB-HPMA-7 (P < 0.001), and HB-HPMA-20 and HB-HPMA-7 (P < 0.0001) at 120 and 240 time points. However, no significant differences were observed between HB-HPMA-20 and HB-HPMA-40 at these points. Next, the internalisation mechanisms were investigated in assays whereby polymers were incubated with MiaPaCa-2 cells at different temperatures, and in the presence or absence of specific endocytosis inhibitors (Fig. 2C and D). These data indicated that all polymers were internalised in an energy-dependent manner, as evidenced by the significantly reduced uptake at 4 °C compared to 37 °C. Furthermore, co-localisation studies demonstrated that there was lysosomal accumulation following internalisation with all polymers: Pearson's Correlation Coefficient (PCC) values were calculated at 0.69 ± 0.05, 0.76 ± 0.05 and 0.72 ± 0.05 for HB-HPMA-7, HB-HPMA.20, and HB-HPMA-40 respectively.
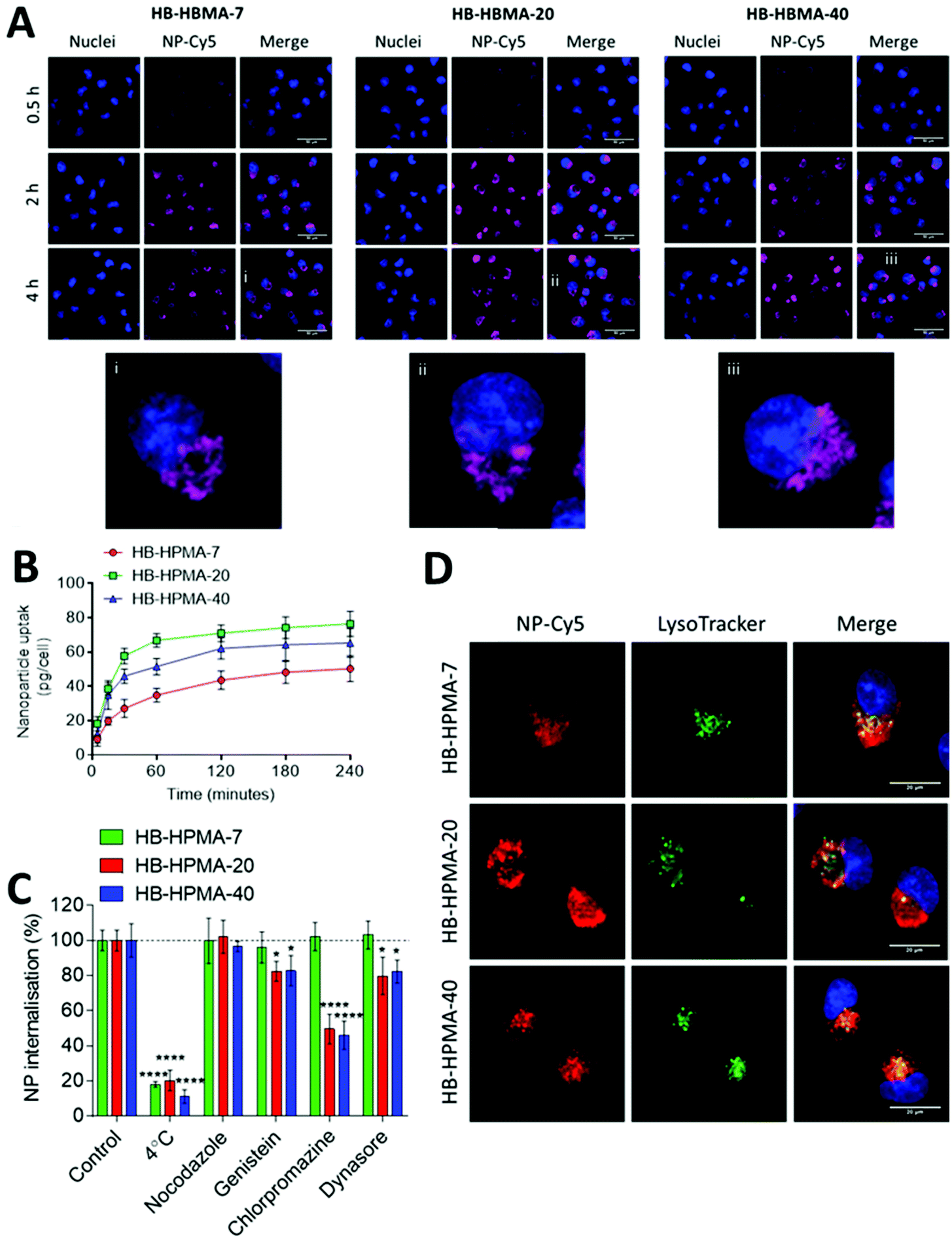 | ||
| Fig. 2 Polymer internalisation (A) fluorescent micrographs of nanoparticle internalization in MIA PaCa-2 cells. (i, ii, iii) Enlarged areas highlighted by white boxes for HB-HPMA-7, HB-HPMA-20, and HB-HPMA-40 respectively. Scale bar = 50 μm. (B) Assessment of NP-Cy5 uptake kinetics evaluated by fluorescence quantification. (C) Effect of endocytosis inhibitors on particle internalisation. (D) Co-localization studies of particles in MiaPaCa-2 cells following 2 h incubation with particles. Cy5 signal of NP-Cy5 was false coloured to red to aid in visualisation of co-localisation with green lysosomal signal. Merged images include NP-Cy5 signal, lysosome stain and Hoechst 33342 staining of nuclei. Scale bar = 20 μm. Data represent mean ± S.D (n = 3). Statistical significance determined by two-way ANOVA (*, p < 0.05; ****, p < 0.0001). | ||
Application of the inhibitor genistein reduced the uptake of HB-HPMA-20 and HB-HPMA-40 in a statistically significant manner compared to HB-HPMA-7, suggesting a greater role of caveolin-mediated endocytosis for the larger polymers. Additionally, the internalisation of HB-HPMA-20 and HB-HPMA-40 was also significantly decreased relative to HB-HPMA-7 when the cells were treated with CPZ, suggesting both the larger polymers were also processed via clathrin-mediated routes. Inhibition of dynamin with dynosore also resulted in significantly reduced uptake levels of HB-HPMA-20 and HB-HPMA-40 compared to HB-HPMA-7, providing evidence for the involvement of dynamin-dependent caveolin- and/or clathrin-mediated pathways during the internalisation of these polymers.
As apparent from the above data, the uptake of the smallest polymeric nanoparticles investigated, HB-HPMA-7, was not affected by endocytosis inhibitors targeted against caveolin-, clathrin-, or dynamin-mediated internalisation. Therefore, although HB-HPMA-7 entered the MiaPaCa-2 cells via an energy-dependent route, and for which lysosome accumulation occurred (Fig. 2C and D), the internalisation of this polymer took place by alternative, dynamin-independent endocytosis pathways. Clathrin- and caveolin-mediated endocytosis have previously been reported to be restricted with smaller nanoparticles.44 This was attributed to an insufficient thermodynamic driving force for membrane wrapping, and the docking of a small single particle to an endocytotic receptor not generating enough free energy to wrap the particles to the membrane surface.45–47 Indeed, previous evidence has indicated that NPs of <20 nm diameter may exploit slower, clathrin- and caveolin-independent routes of cellular internalisation48,49 and the results observed here are in accord with these findings. Uptake of all three polymers into the interior of co-cultured spheroids of MIA PaCa-2 and bone-marrow-derived mesenchymal stem cells (MSCs) was also observed (Fig. 3A). Flow cytometry assessments revealed no significant differences in the number of Cy5 positive cells following treatment with each of the polymers in both 2D and 3D MIA PaCa-2 monoculture (Fig. 3B and D). However, statistically, significant differences were observed between the uptake of HB-HPMA-40 compared to HB-HPMA-7 and HB-HPMA-20 polymers in the co-culture experiments, with HB-HPMA-40 being internalised less in the MSCs compared to MIA-PaCa-2 cells in both the 2D and 3D experiments (Fig. 3C and E).
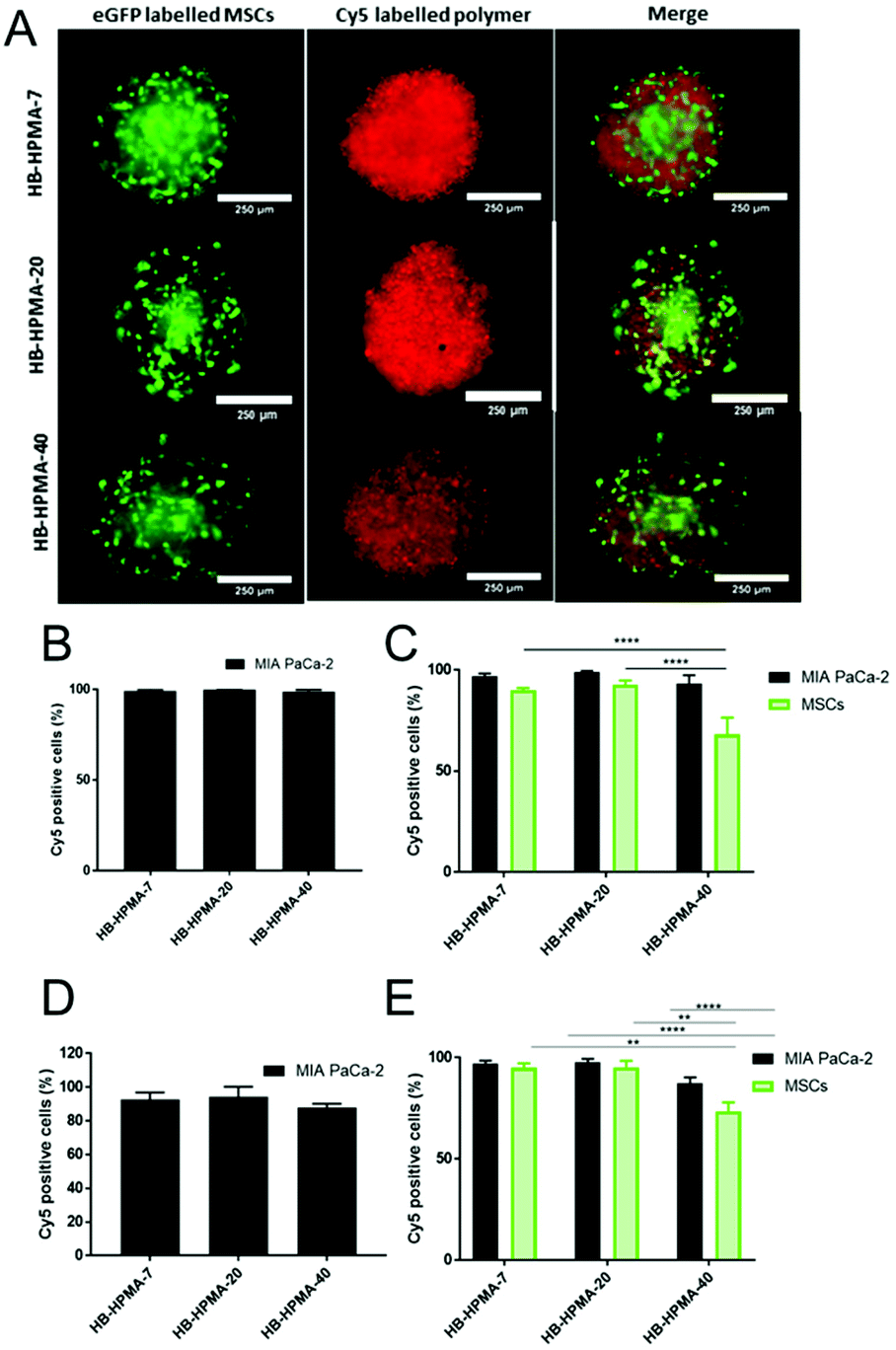 | ||
| Fig. 3 (A) Representative microscope images of cellular uptake of HB-HPMA polymers in co-culture spheroids (MSC: green; Cy5 polymer: red). Percentage of Cy5 positive cells in FACS analyses of HB-HPMA polymers in (B) 2D monoculture cells, (C) 2D co-cultured cells, (D) 3D monoculture spheroids, and (E) 3D co-cultured spheroids. Data are representative of three experiments (N = 3) (****p ≤ 0.0001 two-way ANOVA with Tukey's multiple comparisons test). | ||
These data implied that the co-culture spheroids possessed greater barriers to internalisation of the larger nanoparticles, and thus supported their adoption as better mimics for tumours than the 2D systems, albeit without vasculature or more organised tissue components.
Interactions of the polymers with the murine macrophage cell line, RAW 264.7, were also investigated as a precursor to biodistribution experiments.50,51 The PrestoBlue™ cell metabolism assay and the LDH release assay were used to confirm that none of the three polymers tested caused observable loss of metabolic activity or membrane damage (Fig. S6, ESI†).
Analysis of organ distribution of polymers in healthy mice
The series of HPMA polymers were then assessed in preclinical models as a screening step in advance of any efficacy studies. Accordingly, a dose (500 μg mL−1) was chosen to allow for easy visualisation and minimal toxicity. First, the distributions of the hyperbranched polymers in the organs of immune-deficient tumour naïve CD1 mice were determined up to 24 hours following systemic injection. Six healthy female CD-1 nude mice were injected via the tail vein with each Cy5-labelled polymer. Fluorescence images were acquired at 1, 4- and 24-hours post-injection (Fig. 4). Ex vivo images were utilised to overcome the limited penetration abilities of in vivo fluorescence imaging. Two mice from each group were culled at different time points and ex vivo images were taken of the organs (Fig. 4) for analysis of polymer accumulation with a focus on the organs involved in particle clearance from the bloodstream. | ||
| Fig. 4 (A) Representative in vivo images (4 h post-injection), hollow and solid arrow heads highlight accumulation of polymers in the liver and kidneys, respectively. (B) Ex vivo images at 1 h, 4 h and 24 h post-injection (Br = brain, H = heart, Lu = lungs, Li = liver, P = pancreas, Sp = spleen, K = kidney, U = urine, Bl = bladder). (C) Quantitative data to highlight fluorescence signal in nanoparticle clearance organs as a percentage of total organ signal (n = 2) Data presented as mean ± S.D. Statistical significance was accepted at a level of p < 0.05. (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001). | ||
The fluorescence signal of each organ was analysed by expressing it as a percentage of the total organ signal (Fig. 4C). From the in vivo images of the 4-hour time point, it can be seen that the polymers in all cases accumulated predominantly in the kidneys and livers of the mice, with the smaller HB-HPMA-7 particles present to a greater extent in the kidneys than the larger HB-HPMA-40 polymers (Fig. 4A).
PET-CT biodistribution study in tumour naïve and tumour bearing mice
For real-time analysis of polymer biodistribution, the polymers were labelled with 89Zr for quantitative longitudinal PET imaging. For these materials, we succinylated a proportion of the hydroxyl groups on the HPMA repeat units and then reacted with deferoxamine to install the Zr-chelating groups (Table 2).| Polymer | DFO units (1H NMR) (% mol per mol HPMA) |
|---|---|
| HB-HPMA-7 | 18 |
| HB-HPMA-20 | 12 |
| HB-HPMA-40 | 9 |
Radiolabelled polymers were injected intravenously into Balb/c nude MIA PaCa-2 xenograft mice and tumour naïve mice (n = 4 per group). The non-tumour bearing mice study was conducted to gain an understanding of the basic distribution of the polymers and to ascertain the effect of the tumour on the distribution of the particles. PET-CT images were acquired at different time points for both assays for each polymer (Fig. S7 and S8, ESI†). These data showed that at 3 hours post-injection, in all cases, the majority of the particles were in circulation with little accumulation in tissues such as the liver, kidneys, and spleen. However, the number of circulating particles decreased by 27 and 72 hours mostly due to liver and spleen retention. The tumour 89Zr signal appeared to increase over the 72-hour time point with HB-HPMA-20 showing higher 89Zr signals, indicative of enhanced particle accumulation, particularly at 72 hours post-injection. The longitudinal in vivo biodistribution behaviour of these particles was also quantified from the region of interest (ROI) analysis of the 89Zr-PET-CT images for both assays. The graphical presentations of the distribution patterns of the different polymers in the tissues of interest for tumour bearing and tumour naïve assays are shown in Fig. 5. The heart ROI, which is assumed to represent the concentration of circulating particles on the assumption there is no cardiac tissue perfusion, exhibited similar biodistribution profiles for all three polymers with the majority of the materials being cleared from the bloodstream over the course of the assay. The renal profiles of the three polymers resembled those of the heart profiles, further confirming the gradual clearance of the particles from circulation. As observed from the images, high hepatic and splenic accumulation of the materials were also seen from the ROI analysis of the PET-CT images, particularly at the earlier time points. The overall biodistribution profiles of the particles in the tumour-bearing and non-tumour bearing mice were very similar, indicating that the presence of the tumours had no observable effect on the distribution pattern of these materials in the various tissues.
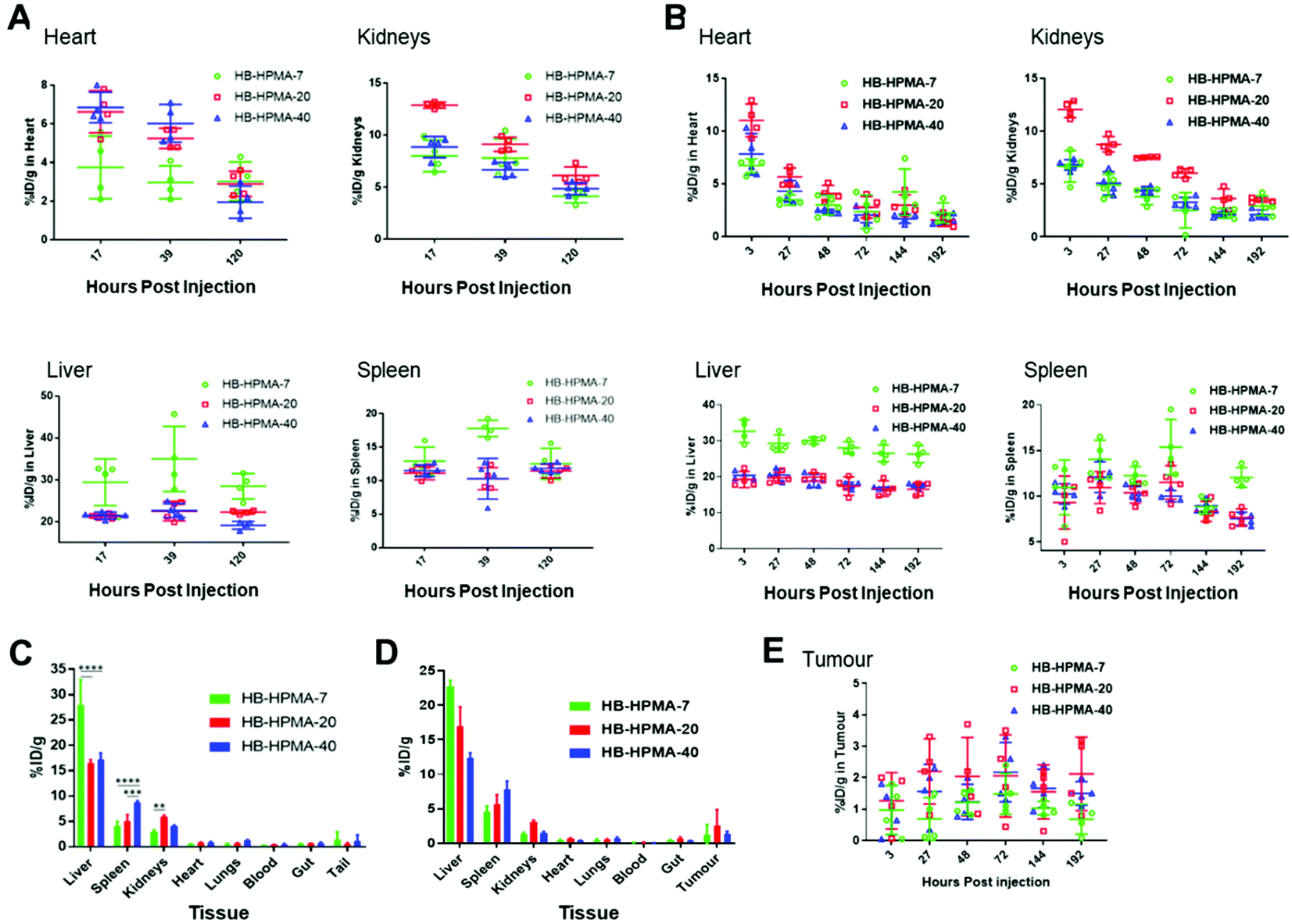 | ||
| Fig. 5 PET ROI analysis of in vivo tissues accumulation of HB-HPMA polymers in MIA PaCa-2 xenograft model. (A) Graphs of hyperbranched HPMA polymer distribution over time in heart, kidneys, liver and spleen, respectively in tumour-naïve mice, and (B) in tumour-bearing mice. In (C) and (D) the ex vivo λ-scintillation biodistribution data provide the total percentages of the injected dose in the organs of tumour-naïve and tumour-bearing mice, respectively, while in (E) the percentage of dose over time in the tumours is given. Data are presented as mean ± S.D. | ||
In accordance with the end-fate polymer accumulation data, HB-HPMA-7 displayed the highest percentage of material retained in the liver compared with HB-HPMA-20 and HB-HPMA-40. However, for renal uptake, the highest values were observed for HB-HPMA-20, which may have been due to conformational changes of the polymers in solution after conjugation of DFO. It is known that the conjugation of hydrophobic drugs and labels to the backbone of water-soluble polymers can perturb their solution structures, causing conformational changes that can affect the polymer's associative properties, solution stabilities,52 and likely also their transport and uptake in tissues. To determine if the enhanced accumulation of the particles in the splenic and hepatic tissues might be due to increased internalisation by macrophages, uptake studies in the RAW 264.7 cells were done with non-functionalised HB-HPMA particles and DFO functionalised HB-HPMA particles. The results (Fig. S9, ESI†) showed that the conjugation of DFO to the backbone of the polymer had no significant effect on the uptake of the polymers by the macrophage cells compared to Cy5 labelled polymers. Consequently, in the absence of detailed information on the structures and conformations of the polymers in tissue, it is not possible to assign a clear mechanism to explain the difference in tissue distribution of the Cy5-labelled HB-HPMA compared to the Zr-DFO-labelled polymers.
After 8 days post-injection, there was no significant difference between the tumour accumulation profiles of the different sized particles, and the percentage of accumulated particles in the tumour for all three polymers was comparable with literature findings for nontargeted nanoparticles.53–55 However, as observed from the PET-CT images (Fig. S7 and S8, ESI†), and the ROI analysis, for some of the data points, the HB-HPMA-20 polymer appeared to have slightly higher tumour accumulation than HB-HPMA-7 and HB-HPMA-40.
Evaluation of HB-HPMA-GEM efficacy against pancreatic cells in vitro and in vivo
To probe therapeutic applications of these polymers, we evaluated a hyperbranched polymer prepared with the HPMA-GEM pro-drug monomer in comparison with the free drug in subsequent assays with pancreatic cancer cells in vitro and in vivo. The design of the polymer pro-drug included an amide-linker to the N4 position of gemcitabine, for which there was a prior report of gemcitabine release in vitro.27 Furthermore, we hypothesised that upregulation of proteases in the stroma associated with pancreatic cancer might trigger enhanced local release of the drug from the polymer backbone after entry into diseased regions in vivo. The polymer pro-drug synthesised (HB-HPMA-GEM) was most similar in molar mass to HB-HPMA-7 but its size by DLS (16 nm) and TEM (18 nm) was closest to that of HB-HPMA-20, hence we conducted initial spheroid comparative tolerability assays with these polymers too. The assay was carried out in the MIA PaCa-2 3D cell culture model whereby changes in spheroid volume and cellular ATP levels, assessed via CellTiter-Glo® 3D assays were used to determine cytocompatibility. Similar to the 2D assays, no adverse effects were observed in the 3D models (Fig. 6A and B). The volumes of the spheroids in both the control and treated wells increased at a constant rate throughout the assay and the cellular ATP levels (Fig. S10, and S11, ESI†) remained above 90% following the polymer dosing, further indicating that the polymers alone were not toxic to the cancer cells.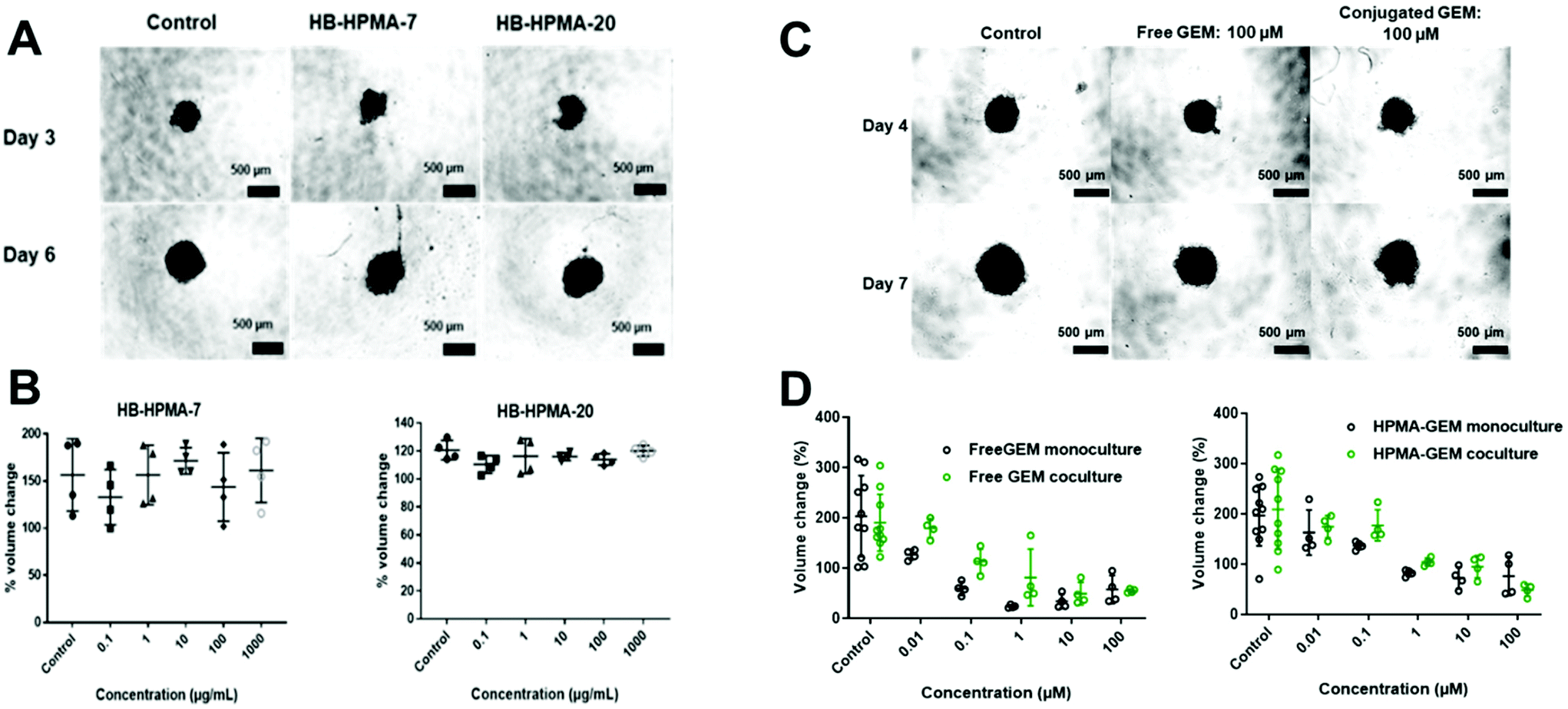 | ||
| Fig. 6 (A) Representative brightfield images of control and treated spheroids (1000 μg mL−1), demonstrating volume increase over the assay period, (N = 4). (B) Percentage volume change between day 3 (pre-treatment) and day 6 (72 hours post-treatment). Data presented as mean ± S.D. Statistical significance was accepted at a level of p < 0.05. (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001). (C). Representative microscope images of different drug concentrations to show differences in volume on day 4 and day 7 of the co-cultured population, (N = 4). (D) Graph showing changes in the spheroid volume between days 4 and 7 expressed as a percentage; (1) free GEM and (2) HPMA-GEM. The scale bar represents 500 μM. | ||
In contrast, for both free GEM and the polymer-conjugated HB-HPMA-GEM there was a decrease in cell viability as indicated by changes in the volume of the spheroid throughout the treatment (Fig. 6C and S12, ESI†), as well as the CellTiter-Glo® 3D cell viability assay at the end of the study (Fig. S13, ESI†) In addition to measuring the effects of the drug and polymer pro-drug on 3D MIA PaCa-2 monoculture spheroids, the assays were repeated with spheroids from MIA PaCa-2 cells co-cultured with eGFP labelled MSCs to investigate whether stromal cells would have an impact on the cytotoxicity profiles of the free and conjugated drug. Baseline microscopy images of the spheroids were taken on day 4 of growth immediately before treatment. Images were then taken every day for 72 h of treatment, and the volume was calculated for each spheroid (Fig. S14, ESI†). For the lowest concentration of the free drug and the 2 lowest doses of HB-HPMA-GEM, there was an increase in the volume of the mono-cultured spheroids during the assay but there was an observed reduction in cell viability at higher concentrations. When comparing the effect of the conjugated drug on the spheroid volume in both mono and co-culture, there were no significant differences in treatment efficacy (Fig. 6D).
The cytotoxicity was further evaluated by the CellTiter-Glo® 3D cell viability assay with free GEM and conjugated GEM in both mono and co-cultured (with MSC) MIA PaCa-2 spheroid models. From the data (Fig. 7) it was clear that the free GEM was less toxic against the cocultured cells compared to the monoculture system as significant differences were observed between the mono and cocultured systems of free GEM (Fig. 7A) whereas no significant difference was seen between both systems with the HB-HPMA-GEM polymer (Fig. 7B). As the MSCs were employed in the cocultured system to be a mimic for the stromal outer layer present in pancreatic cancer, it can be concluded that the presence of the stroma negatively affected the efficacy of free GEM compared with the HB-HPMA-GEM polymer conjugate.
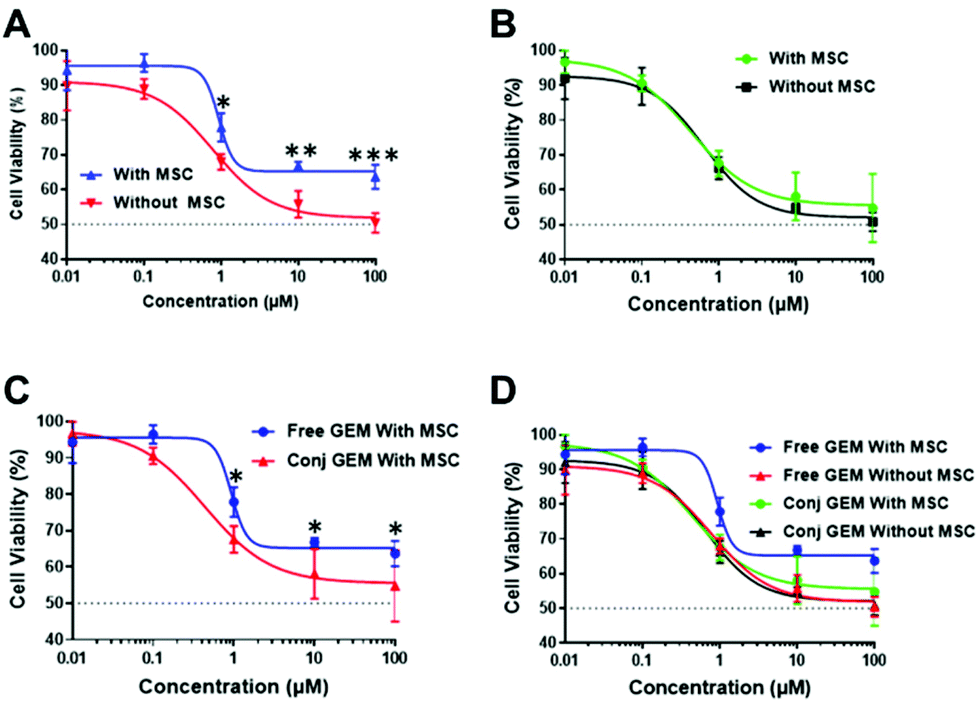 | ||
| Fig. 7 CellTiter-Glo® 3D cell viability assays of MIA PaCa-2 cells in mono and cocultured systems with (A) free GEM and (B) conjugated HB-HPMA-GEM polymer. (C) MSC effect on free and conjugated HPMA-GEM polymer; and (D) 3D spheroid cytotoxicity. Error bars show standard deviation (N = 4). (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, two-way ANOVA with Sidak's multiple comparison test). | ||
In addition, significant differences were seen between the data for conjugated and free GEM in the cocultured system (Fig. 7C), whereby the conjugated GEM was more potent at the same concentration and incubation time.
We subsequently dosed the HB-HPMA-GEM polymer and GEM systemically via tail vein injection against subcutaneous human pancreatic cancer xenografts established in BALB/c nude mice. For these studies, we administered 40 mg GEM per kg to the mice once per week for 5 weeks to give a total of 5 injections. As apparent from Fig. 8 while there were clear differences between the mouse body weights, tumour volumes and survival probabilities for drug and polymer pro-drug compared to saline control, the differences between HB-HPMA-GEM and free GEM were not statistically significant in terms of tumour volume or overall survival. Therefore, although the polymer pro-drug was effective in cell culture, at the dosing levels and schedules we tested there was no enhancement of efficacy over that of the free drug. We were unable, due to Covid-19 restrictions to follow up the initial in vivo efficacy studies with modifications to the dosing protocols, and similar restrictions prevented the growth of more clinically relevant orthotopic pancreatic tumours in mice.
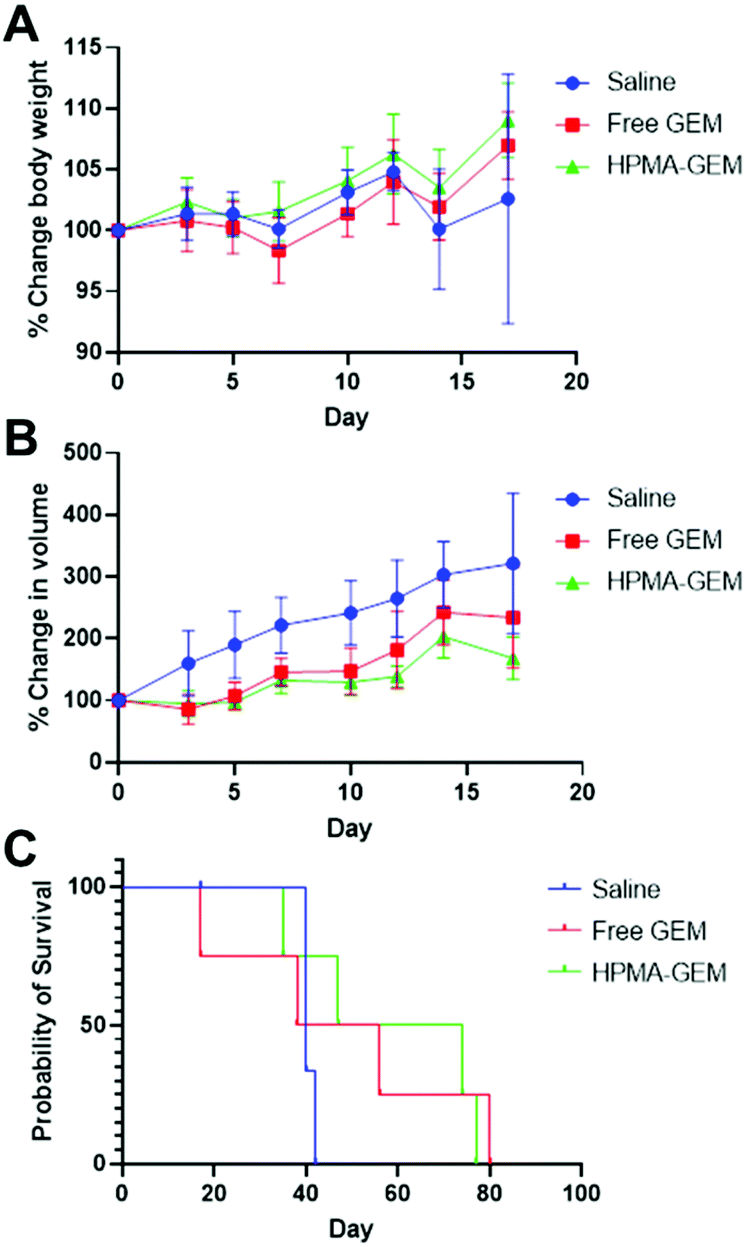 | ||
| Fig. 8 (A) Changes in mouse body weight and (B) tumour volume following dosing of BALB/c nude pancreatic cancer xenograft mice with saline control, or free GEM or HB-HPMA-GEM at 40 mg kg−1 once per week for 5 weeks. (C) survival plots for dosing with saline, GEM and HB-HPMA-GEM over the same schedule as (A and B). | ||
When considered together, these data show that the hyperbranched pHPMA materials were internalised efficiently in pancreatic cancer cells in both 2D and 3D cultures and that in the absence of attached drugs, were well-tolerated in vitro and in vivo. Since the linear pHPMA platform has already been shown to be safe in multiple prior studies, it is reassuring that the introduction of the branching co-monomer and architecture did not result in observable detrimental effects in our experiments. However, extrapolation of short-term cell culture and pre-clinical assays in mice towards trials in humans is not valid and many further assays would be required to establish a full safety profile of these materials prior to human studies.
It should be noted that, while there are advantages to hyperbranched polymers in terms of the relative ease of synthesis, potential for high loading of hydrophobic drugs while retaining solubility, there are also disadvantages. The high polydispersity in some hyperbranched polymers presents a confounding factor when interpreting biological data, as within a population of these polymers there will be a very wide range of individual molar masses. Accordingly, attempting to derive structure–function relations for the transport of these polymers into cells and tissues, and especially for their biodistribution and penetration into tumours, is challenging. For example, the smaller particles in any population will be excreted by renal filtration to a greater extent than the larger particles in each batch, and would similarly be excluded from the cores of dense tumours. In such cases, hyperbranched polymers of different average molar masses may not show any differences in efficacy if their ‘tumour penetrating’ sub-populations are similar. In addition, the conformational flexibility of the hyperbranched polymers may profoundly influence their transport in the body. Work by Duro-Castano et al.56 has shown that star polymers based on polyglutamate can self-associate and this results in supramolecular conformations with different bioaccumulation patterns than their initial hydrodynamic radii would suggest. In our case, we measured the sizes of the hyperbranched polymers by both DLS and TEM, and the observed diameters were much larger than the Mn values would suggest if the polymers were indeed present as unimolecular nanoparticles. This was most pronounced for the HB-HPMA-GEM polymer, which was much larger by TEM than the unsubstituted HB-HPMA-7 polymer, but of lower Mn. It is likely therefore that all the polymers in the study were present in solution as self-assembled superstructures, and the substitution of end-groups with Cy5 and monomer side-chain hydroxyls with DFO and the GEM-succinimide pro-drug likely contributed to this self-association process. Detailed analysis by Lammers et al.57 has previously demonstrated that for linear HPMA systems of molar masses between 20–60 kDa, modification of side-chains with hydrophobic labels and drugs decreased circulation time in tumour-bearing mice with higher renal accumulation and subsequent clearance, but did not alter distribution to other organs significantly. However, these studies did not evaluate whether any self-assembled structures were present in the solution which may have altered biodistribution. More recent work by Allmeroth and co-workers58 evaluated the behaviour of HPMA polymers which assembled into ‘compound micelles’ of 30–40 nm, and thus of similar size initially to the hyperbranched polymers in this work. In this case, PET-imaging of 18F-labelled polymers allowed live imaging of biodistribution in mice, and kidney accumulation again dominated, but the micellar-like polymers were retained for much longer in circulation than smaller, non-micelle-forming analogues. Sadekar et al. prepared PAMAM dendrimers to compare their biodistribution with pHPMA polymers of 26 kDa and 52 kDa and found greater renal accumulation again in tumour-bearing mice of the pHPMA systems,59 but even the largest polymers in these studies(RH ∼3–4 nm) were significantly smaller than the assemblies of HB-HPMA polymers we describe here. At the other end of the size range, Weiss and co-workers prepared semi-telechelic pHPMA and attached this to pre-formed poly(glycerol adipate) nanoparticles, generating pHPMA-coated particles of ∼136 nm diameter.60 These were found to accumulate very markedly in the livers of healthy and tumour-bearing mice, with a much lesser, albeit non-negligible spleen and kidney signal.
The other question related to the clinical translation of hyperbranched polymers made by free radical polymerisation is the control and repeatability of the synthesis. We made several batches of the non-drug-loaded polymers and the data showed very good reproducibility (>95%) in regards to the solution diameters of the polymers (Table S1, ESI†). However, the Mn and Đ values were less consistent, and with much higher dispersities for the larger polymers. Some batch-to-batch variation was to be expected as we did not attempt to optimise the synthetic routes, and even with these variations the sizes of the self-assembled particles in solution were similar, but for a clinically acceptable medicine, a much tighter product profile would be needed.
For the efficacy studies the analysis is even more complex, as although the polymer pro-drug was slightly more effective than the free drug in the 2D and 3D cell culture assays, there was no biologically meaningful difference in tumour reduction or probability of survival. These studies were carried out in a xenograft rather than an orthotopic model, and the dosing schedules were not changeable in the study period we had available for experiments, however, the data were clear that the polymer pro-drug strategy was not advantageous in this protocol compared to the free drug.
These initial experiments have shown that hyperbranched pHPMA polymers can be produced easily and show good in vitro performance. However, we suggest that future synthesis should focus on truly well-defined polymers with properties that can be completely characterised such that full structure–function relations can be determined. In turn, this will affect the subsequent synthesis of drug-carrying analogues and the evaluation of their behaviour in vitro and in vivo.
Conclusions
In this work, we have demonstrated the successful synthesis of four hyperbranched HPMA polymer scaffolds via RAFT polymerisation. These polymers were characterised by NMR, SEC-MALLS, DLS, TEM etc. and were found to form nanoparticles in solution with dimensions ranging from ∼7–40 nm. The resultant materials displayed desirable in vitro cytocompatibility and cellular uptake in both 2D and 3D MIA PaCa-2 cell culture models and were well-tolerated in healthy mice. In studies up to 24 h, Cy5-labelled polymers were present mostly in the kidneys, while PET analysis of 89Zr-labelled derivatives of the HB-HPMAs indicated that the polymers were found to accumulate more in the liver than in other organs, in both MIA PaCa-2 xenograft and tumour-naïve mice. The HB-HPMA-GEM analogue of the non-drug loaded HPMAs showed greater efficacy against pancreatic cancer cells in vitro compared to the free drug, but there were no biologically meaningful differences in vivo. The lack of in vitro/in vivo correlation may have been due to a number of factors. These include insufficient retention of the polymer in the circulation and thus low accumulation in the tumour, a sub-optimal local concentration of Gemcitabine released into the xenograft due to slow amide hydrolysis kinetics in the cancerous cells, or simply less effective penetration of the polymer pro-drug nanoparticle into dense-packed cells in the tumour compared to the free drug. Further in vivo studies with receptor-targeted polymers, more labile linkers and high resolution tissue analysis following delivery will be required to address these questions in future investigations.Experimental
Materials
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 140.03 (C–CH), 120.47 (HCCH), 67.54 (CO), 47.56 (CH2), 21.31 (CH3), 19.04 (CH3). FT-IR: ν (cm−1) = 3263, 3089, 2974, 1654, 1614, 1545. These data correspond with reported literature on the synthesis of HPMA monomer.39,61–63
O), 139.81(CCH), 120.72 (HCCH), 40.90 (CH2), 19.02 (CH3). FT-IR:ν (cm−1) = 3328, 1652, 1611, 1215. The data presented correspond to reported literature on ethylene dimethacrylamide synthesis.64
:50 hexane:ethyl acetate mobile phase) to yield 4-cyano-4-(ethylsulfanylthiocarbonyl) sulfanyl pentanoic acid (CEPA) as an orange-red oil (81%). 1H NMR (400 MHz, CDCl3) δ 3.38 (q, J = 7.4 Hz, 2H), 2.76–2.68 (m, 2H), 2.63–2.52 (m, 1H), 2.43 (ddd, J = 14.2, 9.3, 6.9 Hz, 1H), 1.91 (s, 3H), 1.39 (t, J = 7.4 Hz, 3H). ESI-TOF-MS: m/z: [M + H]+ C9H13NO2S3 calculated, 263.01; found, 264.02. The data is in agreement with a previously reported synthesis of CEPA.65
O), 171.67 (CO), 167.85 (CO), 139.87 (CH), 119.08 (HCCH), 69.16 (CO), 43.05 (CH2), 28.86 (CH2), 18.63(CH3), 17.47 (CH3). ESI-TOF-MS: m/z: [M − H]− C11H17NO5 calculated, 242.11; found, 242.10.
O), 172.12 (CO), 168.28 (CO), 162.99 (CN), 154.64 (CO), 145.20 (CCH), 140.31 (CF), 119.52 (HCCH), 96.35 (CH), 81.48 (CO), 69.66 (CO), 43.53 (CO), 31.81 (CH2), 28.83 (CH2), 19.09 (CH3), 17.93 (CH3). ESI-TOF-MS:m/z: C20H26F2N4O8 [M + H]+ calculated, 489.17; found, 489.18.
:1 ratio of N,N-dimethylacetamide (DMAc) and water in a 2 mL tube equipped with a magnetic stirrer bar. The reaction mixture was purged with argon for 30 minutes, the vial was sealed tightly and stirred in an oil bath at 70 °C for 24 h. After 24 h, the polymer was precipitated dropwise into excess acetone three times, followed by dissolving in Milli Q water and dialysing for 48 hours in a 3.5 kDa molecular weight cut-off pleated snakeskin tubing and subsequently lyophilised.
:50 N,N-dimethylacetamide (DMAc) and deionised water (1.3 mL) in a 2 mL tube equipped with a magnetic stirrer bar. The tube was part-sealed, and the reaction mixture purged with argon for 30 minutes. The vial was placed in an oil bath at 70 °C for 24 h. After 24 h, the polymer was precipitated dropwise into excess acetone (x3), followed by dissolving in Milli Q water and dialysing for 48 hours in a 3.5 kDa molecular weight cut-off pleated snakeskin tubing and subsequently lyophilised. The polymer was then characterised by NMR, GPC, DLS and FT-IR.
O), 140.03 (C–CH), 120.47 (HCCH), 67.54 (CO), 47.56 (CH2), 21.31 (CH3), 19.04 (CH3). FT-IR: ν (cm−1) = 3263, 3089, 2974, 1654, 1614, 1545. These data correspond with reported literature on the synthesis of HPMA monomer.39,61–63
O), 139.81(CCH), 120.72 (HCCH), 40.90 (CH2), 19.02 (CH3). FT-IR:ν (cm−1) = 3328, 1652, 1611, 1215. The data presented correspond to reported literature on ethylene dimethacrylamide synthesis.64
:50 hexane:ethyl acetate mobile phase) to yield 4-cyano-4-(ethylsulfanylthiocarbonyl) sulfanyl pentanoic acid (CEPA) as an orange-red oil (81%). 1H NMR (400 MHz, CDCl3) δ 3.38 (q, J = 7.4 Hz, 2H), 2.76–2.68 (m, 2H), 2.63–2.52 (m, 1H), 2.43 (ddd, J = 14.2, 9.3, 6.9 Hz, 1H), 1.91 (s, 3H), 1.39 (t, J = 7.4 Hz, 3H). ESI-TOF-MS: m/z: [M + H]+ C9H13NO2S3 calculated, 263.01; found, 264.02. The data is in agreement with a previously reported synthesis of CEPA.65
O), 171.67 (CO), 167.85 (CO), 139.87 (CH), 119.08 (HCCH), 69.16 (CO), 43.05 (CH2), 28.86 (CH2), 18.63(CH3), 17.47 (CH3). ESI-TOF-MS: m/z: [M − H]− C11H17NO5 calculated, 242.11; found, 242.10.
O), 172.12 (CO), 168.28 (CO), 162.99 (CN), 154.64 (CO), 145.20 (CCH), 140.31 (CF), 119.52 (HCCH), 96.35 (CH), 81.48 (CO), 69.66 (CO), 43.53 (CO), 31.81 (CH2), 28.83 (CH2), 19.09 (CH3), 17.93 (CH3). ESI-TOF-MS:m/z: C20H26F2N4O8 [M + H]+ calculated, 489.17; found, 489.18.
:1 ratio of N,N-dimethylacetamide (DMAc) and water in a 2 mL tube equipped with a magnetic stirrer bar. The reaction mixture was purged with argon for 30 minutes, the vial was sealed tightly and stirred in an oil bath at 70 °C for 24 h. After 24 h, the polymer was precipitated dropwise into excess acetone three times, followed by dissolving in Milli Q water and dialysing for 48 hours in a 3.5 kDa molecular weight cut-off pleated snakeskin tubing and subsequently lyophilised.
:50 N,N-dimethylacetamide (DMAc) and deionised water (1.3 mL) in a 2 mL tube equipped with a magnetic stirrer bar. The tube was part-sealed, and the reaction mixture purged with argon for 30 minutes. The vial was placed in an oil bath at 70 °C for 24 h. After 24 h, the polymer was precipitated dropwise into excess acetone (x3), followed by dissolving in Milli Q water and dialysing for 48 hours in a 3.5 kDa molecular weight cut-off pleated snakeskin tubing and subsequently lyophilised. The polymer was then characterised by NMR, GPC, DLS and FT-IR.
Effects of polymers measured by dye exclusion in MIA PaCa-2 cells
The effects of the polymers on MIA PaCa-2 cells were measured over 72 h using a trypan blue dye exclusion test as a marker for cell viability. The cells were cultured in 24-well plates at a seeding density of 2.5 × 105 cells per well for 24 h. The cells were treated with polymers dissolved in phenol red-free DMEM culture medium at different concentrations. The culture medium in the well plates was replaced with 500 μL of sample solutions. After 72 h incubation, the medium was removed, cells treated with 400 μL phenol red-free TrypLe Express Enzyme (1X) for approximately 5 min, and diluted with 600 μL fresh medium. The suspension was then centrifuged and redissolved in 100 μL of fresh medium. An equal volume of 0.4% (v/v) trypan blue was added to each cell suspension and the extent of dye exclusion was determined on a TC20 automated cell counter. Percentage cell viability was calculated as follows:68| % viable cell = (NT/Nc) × 100 | (1) |
Effects of polymers on 3D MIA PaCa-2 spheroids and co-cultures
Corning 7007 Ultra-low attachment (ULA) 96-well round-bottom plates were used to culture the 3D spheroids. 80% confluent MIA PaCa-2 and eGFP MSCS monolayer cells were detached, collected and the cell number determined using the Biorad TC20 automated cell counter. A single and co-cultured cell suspensions were diluted in the appropriate culture medium with the addition of Cultrex basement membrane extract (Cultrex-BME, 100 μg mL−1) and cells seeded at 1000 cells per well (mono-culture) and 2000 cells per well (co-culture, 1:1 MIA PaCa-2 to MSC) to generate the spheroids (final volume of cell suspension in each well was 100 μL). The plates were then centrifuged at 300 RCF for 5 min and incubated for approx. 3 days until visible spheroids formation.
The polymers were prepared as solutions in cell culture medium at x2 the concentration of 2D cell culture. An aliquot (100 μL) of the sample was added to each well containing a spheroid in 100 μL of the medium, taking the total volume to 200 μL and the plates incubated for 72 h. Images were taken immediately before treatment and every day thereafter for 3 days for spheroid volume analysis. The volume of the spheroids was analysed by measuring their cross-sectional area using the in-house open-source macro for the Fiji distribution of ImageJ written by Ivanov.69 The measured area (S) from the macro data of the 2D projection of the spheroid was then used to calculate the radius, 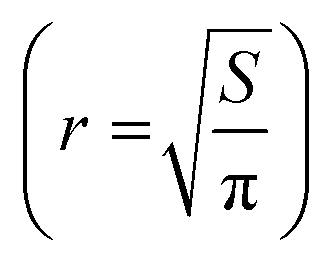 and subsequently, the volume,
and subsequently, the volume, 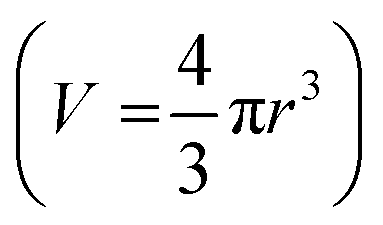 of an equivalent sphere.
of an equivalent sphere.
At 72 h post-treatment of the spheroids, the plates were equilibrated to room temperature for approximately 30 minutes. 100 μL of cell culture medium was taken from each well and replaced with an equal volume of assay-ready CellTiter-Glo® 3D solution. The contents of the plates were mixed vigorously for 5 minutes to induce cell lysis, the plate was incubated at room temperature for an additional 25 minutes and analysed for luminescence.
For co-culture experiments, MIA PaCa-2 and MSC cells were seeded at 1 × 103 cells per well. Seeding density and times were predetermined via optimisation of the MIA PaCa-2 and MIA PaCa-2/MSC co-culture spheroid formation.
The internalisation of polymers in 2D MIA PaCa-2 cell culture
The uptake of Cy5-labelled HB-HPMA was studied using the 2D cell culture model. Briefly, cells were cultured in 6-well plates at a seeding density of 2.5 × 105 cells per well for 24 h. The medium was replaced with a 200 μg mL−1 solution of Cy5-labelled polymer and the plate was incubated for 1 h. The cells were then washed thrice with fresh medium and Hoechst 33342 stain was applied and incubated for 30 minutes. Uptake of the polymer by cells was visualised using a Nikon Ti Eclipse inverted microscopy and quantified using flow cytometry.The internalisation of polymers in 3D MIA PaCa-2 spheroids
The cells were incubated for 4 days during which time they aggregated and formed spheroids with diameters of approximately 500 μm (mono-culture) and 620 μm (co-culture). 100 μL of a 400 μg mL−1 Cy5 labelled HB-HPMA polymers were added to the spheroids (n = 4), taking the concentration to 200 μg mL−1 and the volume of each well to 200 μL. The plate was then incubated for 2 h, after which the spheroids were washed (x3) with fresh medium and Hoechst 33342 stain was applied and incubated for 2 hours. Uptake of the polymer by cells was visualised using a Nikon Ti Eclipse inverted microscope and quantified using flow cytometry (n = 10). 3D MIA PaCa-2 mono and co-culture (with MSCs) spheroids were used for this experiment.Evaluation of polymer cytocompatibility in macrophages
The lactate dehydrogenase (LDH) release assay (Sigma Aldrich, TOX7 kit) and the PrestoBlue™ cell viability assay (Thermo Fisher Scientific) were performed to assess particle cytotoxicity. RAW 264.7 cells were seeded at 1 × 104 cells per well in 96 well plates and cultured for 24 hours before assaying. HB-HPMA polymers were exposed to cells for 24 hours and applied in 100 μL phenol red-free DMEM containing 10% (v/v) FBS and 2 mM L-glutamine. Triton X-100 applied at 1% (v/v) in phenol red-free medium was used as a cell death (positive) control and a vehicle control containing no nanoparticles was used as a negative control. Following exposure, 50 μL of supernatant was collected per well for analysis of LDH content. Cells were then washed twice with warm PBS and 100 μL 10% (v/v) PrestoBlue™ reagent diluted in phenol red-free medium applied per well for 60 minutes. The resulting fluorescence was measured at 560/600 nm (λex/λem). Relative metabolic activity was calculated by setting values from the negative control as 100% and positive control values as 0% metabolic activity. Assessment of LDH release was performed according to the manufacturer's instructions and involved adding 100 μl LDH reagent to collected supernatant samples and incubating at room temperature shielded from light for 25 minutes. Absorbance was then measured at 492 nm. Relative LDH release was calculated with the negative control absorbance at 492 nm taken as 0%, and the positive control assumed to cause total cell lysis, as 100%.Evaluation of polymer internalisation in macrophages
RAW 264.7 macrophages were plated in 12 well plates at a seeding density of 1.2 × 105 cells per well and cultured for 48 hours. The culture medium was then removed, and 1 mL of nanoparticles were applied in a phenol red-free medium. The time-dependent uptake was assessed with a polymer concentration of 50 μg mL−1. Following exposure, polymer solutions were removed, and cells were washed three times with ice-cold PBS. Five hundred microlitres of 1% (v/v) Triton X-100 solution applied in PBS was then added per well for 10 minutes at 37 °C. To assess nanoparticle uptake, 100 μL of the resulting solution was collected per well and transferred to 96 well plates for the measurement of fluorescence at 640/680 nm (λex/λem). Quantification of nanoparticles uptake was achieved via calibration curves of known nanoparticle concentrations diluted in 1% (v/v) Triton X-100 in PBS solution. Values were normalised to viable cell numbers per well determined by the trypan blue exclusion test and cell counting on a haemocytometer.CellTiter-Glo® 3D cell viability assay
CellTiter-Glo® 3D cell viability assay determines cell viability through quantification of adenosine triphosphate (ATP) levels as a marker of metabolically active cells. At a period of 72 h post-treatment of the spheroids, the plate was equilibrated to room temperature for approximately 30 minutes. 100 μL of cell culture medium was taken from each well and replaced with an equal volume of assay-ready CellTiter-Glo® 3D solution. The contents of the plate were mixed vigorously for 5 minutes to induce cell lysis, the plate was incubated at room temperature for an additional 25 minutes and analysed for luminescence.Organ distribution study in tumour naïve mice
The experiments were conducted under the UK Home Office Licence number PPL P435A9CF8. In addition, LASA good practice guidelines, FELASA working group on pain and distress guidelines and ARRIVE reporting guidelines were also followed. In these studies, 8–9-week-old female immunodeficient CD-1 NuNu mice purchased from Charles River UK were maintained in Individually Ventilated Cages (Tecniplast UK) within a barriered unit, illuminated by fluorescent light set to give a 12-hour light–dark cycle (on 07.00, off 19:00). The air temperature range of the room was maintained at 21 ± 2 °C with a humidity of 55% ± 10%. During the study, the mice were housed in social groups, 3 per cage, with irradiated bedding and autoclaved nesting materials and environmental enrichment (Datesand UK). Sterile irradiated 5V5R rodent diet (IPS Ltd, UK) and autoclaved water were offered ad libitum. The condition of the animals was monitored by an experienced technician throughout the study. The mice were randomly assigned to the study groups (6 mice per polymer) by weight after a week's acclimatisation. No power calculation was required for a simple biodistribution assay. After acclimatisation, the mice were warmed in a thermostatically controlled heating box (Datesand UK) and injected intravenously via the tail vein with 100 μL of a 500 μg mL−1 solution of the relevant polymer in PBS. There were no adverse effects observed post-injection and throughout the study. The concentrations of the experimental formulations were determined from a balance of non-toxicity from in vitro analysis as well as sufficient fluorescent intensity for imaging in vivo. Images were taken at 1 h, 4 h and 24 h post-injection and prior to each study time point, the mice were anaesthetised with an injectable anaesthetic combination (Anaestemine [ketamine]/Sedastart [medetomadine], Animalcare Ltd UK). Two mice in each group were culled by cervical dislocation, organs dissected out, and imaged ex vivo at these time points. The other mice were then allowed to recover from the anaesthetic with appropriate post-procedural monitoring and therapy; including placing mice on a heating pad and provided fluid replacement via wet mash once awake. An aliquot (25 μL) of urine along with the excised organs (kidney, liver, spleen, pancreas, lung, heart, bladder, brain and subiliac lymph nodes) were imaged. All images were collected using the IVIS® Spectrum imaging system, PerkinElmer (MA, USA) and fluorescent signals were quantified using Regions of Interest (ROIs) and quantified as photons emitted using Living Image/Igor Pro Software (Caliper Life Sciences).PET-CT biodistribution study in tumour naïve and tumour bearing mice
In vivo efficacy studies
Mice were anaesthetized with 2% isoflurane in oxygen at a flow rate of 2 mL min−1. Subcutaneous human pancreatic cancer xenografts were established in BALB/c nude mice (Australian Resources Centre, Australia) by subcutaneous injection (27G, 50 μL saline) of 6 × 106 MIA PaCa-2 cells and tumour growth was monitored by calliper measurements three times per week.After 4 weeks of cell growth when the tumours had reached ∼200 mm3, in vivo tumour regression studies were performed. Three groups were used in the study: vehicle control (saline), free GEM and HPMA-GEM. In each case, the formulations were injected through the tail vein using a 29G needle in ∼150 μL saline. To enable the comparison of the different formulations, with the exception of the saline control, each injection contained a dose of 40 mg GEM per kg for each mouse. Each treatment group consisted of 4 mice which were administered once per week for 5 weeks; 5 injections in total. The tumour sizes were measured using an electronic digital calliper, and the mouse body weight was monitored throughout the experiment. Mice were humanely sacrificed when tumours exceeded 1000 mm3. The tumour volume was calculated using the formula: tumour volume = 0.5 × length × width2.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council [grant numbers EP/N006615/1; EP/N03371X/1; EP/H005625/1; EP/L013835/1]. This work was also funded by the Royal Society [Wolfson Research Merit Award WM150086] to CA. The authors thank the Nanoscale and Microscale Research Centre (nmRC) for providing access to instrumentation, Paul Cooling and Douglas Crackett for expert technical help, and Sian Rankin-Turner and Carol Turrill for excellent administrative support.References
- R. Duncan, J. Drug Targeting, 2017, 25, 759–780 CrossRef CAS PubMed.
- E. Ben-Akiva, S. E. Witte, R. A. Meyer, K. R. Rhodes and J. J. Green, Biomater. Sci., 2019, 7, 14–30 RSC.
- H. Cabral and K. Kataoka, J. Controlled Release, 2014, 190, 465–476 CrossRef CAS PubMed.
- I. Ekladious, Y. L. Colson and M. W. Grinstaff, Nat. Rev. Drug Discovery, 2019, 18, 273–294 CrossRef CAS PubMed.
- R. Duncan, J. Controlled Release, 2014, 190, 371–380 CrossRef CAS PubMed.
- J. Kopeček, Adv. Drug Delivery Rev., 2013, 65, 49–59 CrossRef PubMed.
- S. A. Elkassih, P. Kos, H. Xiong and D. J. Siegwart, Biomater. Sci., 2019, 7, 607–617 RSC.
- P. Ray, L. Alhalhooly, A. Ghosh, Y. Choi, S. Banerjee, S. Mallik, S. Banerjee and M. Quadir, ACS Biomater. Sci. Eng., 2019, 5, 1354–1365 CrossRef CAS PubMed.
- Z. Wu, Z. Gan, B. Chen, F. Chen, J. Cao and X. Luo, Biomater. Sci., 2019, 7, 3190–3203 RSC.
- C. Lipinski, Am. Pharm. Rev., 2002, 5, 82–85 Search PubMed.
- N. Larson and H. Ghandehari, Chem. Mater., 2012, 24, 840–853 CrossRef CAS PubMed.
- K. Ulbrich and V. Šubr, Adv. Drug Delivery Rev., 2010, 62, 150–166 CrossRef CAS PubMed.
- D. R. Ferry, L. W. Seymour, D. Anderson, S. Hesselwood, P. Julyan, C. Boivin, R. Poyner, P. Guest, J. Doran and D. J. Kerr, Br. J. Cancer, 1999, 80, 413 Search PubMed.
- P. J. Julyan, L. W. Seymour, D. R. Ferry, S. Daryani, C. M. Boivin, J. Doran, M. David, D. Anderson, C. Christodoulou, A. M. Young, S. Hesslewood and D. J. Kerr, J. Controlled Release, 1999, 57, 281–290 CrossRef CAS PubMed.
- R. Duncan and M. J. Vicent, Adv. Drug Delivery Rev., 2010, 62, 272–282 CrossRef CAS PubMed.
- K. Ulbrich and V. Subr, Adv. Drug Delivery Rev., 2010, 62, 150–166 CrossRef CAS PubMed.
- J. Kopecek and P. Kopeckova, Adv. Drug Delivery Rev., 2010, 62, 122–149 CrossRef CAS PubMed.
- Y. Dai, X. L. Ma, Y. H. Zhang, K. Chen, J. Z. Tang, Q. Y. Gong and K. Luo, Biomater. Sci., 2018, 6, 2976–2986 RSC.
- J. Yang and J. Kopeček, Curr. Opin. Colloid Interface Sci., 2017, 31, 30–42 CrossRef CAS PubMed.
- Z.-H. Peng and J. Kopecek, J. Am. Chem. Soc., 2015, 137, 6726–6729 CrossRef CAS PubMed.
- B. S. Tucker, S. G. Getchell, M. R. Hill and B. S. Sumerlin, Polym. Chem., 2015, 6, 4258–4263 RSC.
- H. Wutzel, F. H. Richter, Y. Li, S. S. Sheiko and H.-A. Klok, Polym. Chem., 2014, 5, 1711–1719 RSC.
- E. L. Ferguson, A. Scomparin, H. Hailu and R. Satchi-Fainaro, J. Drug Targeting, 2017, 25, 818–828 CrossRef CAS PubMed.
- M. Golan, V. Feinshtein and A. David, Eur. J. Pharm. Biopharm., 2016, 109, 103–112 CrossRef CAS PubMed.
- Z.-H. Peng and J. Kopeček, J. Am. Chem. Soc., 2015, 137, 6726–6729 CrossRef CAS PubMed.
- J. Yang, R. Zhang, D. C. Radford and J. Kopecek, J. Controlled Release, 2015, 218, 36–44 CrossRef CAS PubMed.
- F. Joubert, L. Martin, S. Perrier and G. Pasparakis, ACS Macro Lett., 2017, 6, 535–540 CrossRef CAS.
- X. Li, L. Sun, X. L. Wei, Q. Luo, H. Cai, X. Y. Xiao, H. Y. Zhu and K. Luo, J. Mater. Chem. B, 2017, 5, 2763–2774 RSC.
- J. A. Alfurhood, H. Sun, P. R. Bachler and B. S. Sumerlin, Polym. Chem., 2016, 7, 2099–2104 RSC.
- X. Zeng, Y. Zhang and A. M. Nyström, Biomacromolecules, 2012, 13, 3814–3822 CrossRef CAS PubMed.
- I. N. Kurniasih, J. Keilitz and R. Haag, Chem. Soc. Rev., 2015, 44, 4145–4164 RSC.
- F. Y. Hern, A. Hill, A. Owen and S. P. Rannard, Polym. Chem., 2018, 9, 1767–1771 RSC.
- A. K. Pearce, J. D. Simpson, N. L. Fletcher, Z. H. Houston, A. V. Fuchs, P. J. Russell, A. K. Whittaker and K. J. Thurecht, Biomaterials, 2017, 141, 330–339 CrossRef CAS PubMed.
- F. Joubert and G. Pasparakis, J. Mater. Chem. B, 2018, 6, 1095–1104 RSC.
- H. Cabral, Y. Matsumoto, K. Mizuno, Q. Chen, M. Murakami, M. Kimura, Y. Terada, M. R. Kano, K. Miyazono, M. Uesaka, N. Nishiyama and K. Kataoka, Nat. Nanotechnol., 2011, 6, 815–823 CrossRef CAS PubMed.
- K. J. Thurecht, I. Blakey, H. Peng, O. Squires, S. Hsu, C. Alexander and A. K. Whittaker, J. Am. Chem. Soc., 2010, 132, 5336–5337 CrossRef CAS PubMed.
- Y. Zheng, S. Li, Z. Weng and C. Gao, Chem. Soc. Rev., 2015, 44, 4091–4130 RSC.
- A. K. Pearce, B. E. Rolfe, P. J. Russell, B. W. C. Tse, A. K. Whittaker, A. V. Fuchs and K. J. Thurecht, Polym. Chem., 2014, 5, 6932–6942 RSC.
- J. Kopeček and H. Baẑilová, Eur. Polym. J., 1973, 9, 7–14 CrossRef.
- B. L. Banik and J. L. Brown, in Natural and Synthetic Biomedical Polymers, ed. S. G. Kumbar, C. T. Laurencin and M. Deng, Elsevier, Oxford, 2014, pp. 387–395, DOI:10.1016/B978-0-12-396983-5.00024-7.
- B. Říhová, Composites, Part B, 2007, 38, 386–397 CrossRef.
- P. A. Vasey, S. B. Kaye, R. Morrison, C. Twelves, P. Wilson, R. Duncan, A. H. Thomson, L. S. Murray, T. E. Hilditch, T. Murray, S. Burtles, D. Fraier, E. Frigerio and J. Cassidy, Clin. Cancer Res., 1999, 5, 83–94 CAS.
- A. K. Pearce, A. B. Anane-Adjei, R. J. Cavanagh, P. F. Monteiro, T. M. Bennett, V. Taresco, P. A. Clarke, A. A. Ritchie, M. R. Alexander, A. M. Grabowska and C. Alexander, Adv. Healthcare Mater., 2020, 9, e2000892 CrossRef PubMed.
- B. D. Chithrani and W. C. Chan, Nano Lett., 2007, 7, 1542–1550 CrossRef CAS PubMed.
- H. Gao, W. Shi and L. B. Freund, Proc. Natl. Acad. Sci., 2005, 102, 9469–9474 CrossRef CAS PubMed.
- G. Bao and X. R. Bao, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 9997–9998 CrossRef CAS PubMed.
- X. Li, J. Appl. Phys., 2012, 111, 024702 CrossRef.
- S. K. Lai, K. Hida, C. Chen and J. Hanes, J. Controlled Release, 2008, 125, 107–111 CrossRef CAS PubMed.
- S. K. Lai, K. Hida, S. T. Man, C. Chen, C. Machamer, T. A. Schroer and J. Hanes, Biomaterials, 2007, 28, 2876–2884 CrossRef CAS PubMed.
- S. Salmaso and P. Caliceti, J. Drug Delivery, 2013, 2013, 374252–374252 Search PubMed.
- Q. Feng, Y. Liu, J. Huang, K. Chen, J. Huang and K. Xiao, Sci. Rep., 2018, 8, 2082 CrossRef PubMed.
- P. K. Choudhury, D. Bagchi, C. S. S. Sangeeth and R. Menon, J. Mater. Chem., 2011, 21, 1607–1614 RSC.
- N. L. Fletcher, Z. H. Houston, J. D. Simpson, R. N. Veedu and K. J. Thurecht, Chem. Commun., 2018, 54, 11538–11541 RSC.
- Q. Yang, L. Li, X. Zhu, W. Sun, Z. Zhou and Y. Huang, RSC Adv., 2015, 5, 14858–14870 RSC.
- E. Koziolová, S. Goel, P. Chytil, O. Janoušková, T. E. Barnhart, W. Cai and T. Etrych, Nanoscale, 2017, 9, 10906–10918 RSC.
- A. Duro-Castano, R. M. England, D. Razola, E. Romero, M. Oteo-Vives, M. A. Morcillo and M. J. Vicent, Mol. Pharm., 2015, 12, 3639–3649 CrossRef CAS PubMed.
- T. Lammers, R. Kühnlein, M. Kissel, V. Subr, T. Etrych, R. Pola, M. Pechar, K. Ulbrich, G. Storm, P. Huber and P. Peschke, J. Controlled Release, 2005, 110, 103–118 CrossRef CAS PubMed.
- M. Allmeroth, D. Moderegger, B. Biesalski, K. Koynov, F. Rösch, O. Thews and R. Zentel, Biomacromolecules, 2011, 12, 2841–2849 CrossRef CAS PubMed.
- S. Sadekar, A. Ray, M. Janat-Amsbury, C. M. Peterson and H. Ghandehari, Biomacromolecules, 2011, 12, 88–96 CrossRef CAS PubMed.
- V. M. Weiss, H. Lucas, T. Mueller, P. Chytil, T. Etrych, T. Naolou, J. Kressler and K. Mäder, Macromol. Biosci., 2018, 18, 1700240 CrossRef PubMed.
- N. Francini, L. Purdie, C. Alexander, G. Mantovani and S. G. Spain, Macromolecules, 2015, 48, 2857–2863 CrossRef CAS.
- C. W. Scales, Y. A. Vasilieva, A. J. Convertine, A. B. Lowe and C. L. McCormick, Biomacromolecules, 2005, 6, 1846–1850 CrossRef CAS PubMed.
- M. F. Ebbesen, D. H. Schaffert, M. L. Crowley, D. Oupický and K. A. Howard, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 5091–5099 CrossRef CAS.
- J. Song, V. Malathong and C. R. Bertozzi, J. Am. Chem. Soc., 2005, 127, 3366–3372 CrossRef CAS PubMed.
- M. C. Arno, M. Inam, Z. Coe, G. Cambridge, L. J. Macdougall, R. Keogh, A. P. Dove and R. K. O'Reilly, J. Am. Chem. Soc., 2017, 139, 16980–16985 CrossRef CAS PubMed.
- B. V. K. J. Schmidt, M. Hetzer, H. Ritter and C. Barner-Kowollik, Macromolecules, 2013, 46, 1054–1065 CrossRef CAS.
- J. C. Knight, S. J. Paisey, A. M. Dabkowski, C. Marculescu, A. S. Williams, C. Marshall and B. Cornelissen, Dalton Trans., 2016, 45, 6343–6347 RSC.
- R. Pardo, A. Lo Ré, C. Archange, A. Ropolo, D. L. Papademetrio, C. D. Gonzalez, E. M. Alvarez, J. L. Iovanna and M. I. Vaccaro, Pancreatology, 2010, 10, 19–26 CrossRef CAS PubMed.
- D. P. Ivanov, T. L. Parker, D. A. Walker, C. Alexander, M. B. Ashford, P. R. Gellert and M. C. Garnett, PLoS One, 2014, 9, e103817 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1bm01548f |
| This journal is © The Royal Society of Chemistry 2022 |