
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A minimal standardized human bone marrow microphysiological system to assess resident cell behavior during normal and pathological processes†
Thibault
Voeltzel
 *abcde,
Gaëlle
Fossard
abcdf,
Michaël
Degaud
abcdf,
Kevin
Geistlich
abcdg,
Nicolas
Gadot
abch,
Sandrine
Jeanpierre
abcdg,
Ivan
Mikaelian
abcd,
Marie
Brevet
i,
Adrienne
Anginot
je,
Marie-Caroline
Le Bousse-Kerdilès
je,
Valérie
Trichet
ke,
Sylvain
Lefort
*abcde and
Véronique
Maguer-Satta
*abcdeg
*abcde,
Gaëlle
Fossard
abcdf,
Michaël
Degaud
abcdf,
Kevin
Geistlich
abcdg,
Nicolas
Gadot
abch,
Sandrine
Jeanpierre
abcdg,
Ivan
Mikaelian
abcd,
Marie
Brevet
i,
Adrienne
Anginot
je,
Marie-Caroline
Le Bousse-Kerdilès
je,
Valérie
Trichet
ke,
Sylvain
Lefort
*abcde and
Véronique
Maguer-Satta
*abcdeg
aCNRS UMR5286, Centre de Recherche en Cancérologie de Lyon, 69000 Lyon, France
bInserm U1052, Centre de Recherche en Cancérologie de Lyon, 69000 Lyon, France
cUniversité de Lyon, 69000, Lyon, France
dDepartment of Cancer Initiation and Tumor cell Identity and Lyon, France
eCNRS GDR 3697 MicroNiT, Tours, France. E-mail: thibault.voeltzel@inserm.fr; Sylvain.LEFORT@lyon.unicancer.fr; veronique.maguer-satta@lyon.unicancer.fr
fHospices Civils de Lyon, Hematology Department, Centre Hospitalier Lyon Sud, F-69495 Pierre Bénite, France
gCentre Léon Bérard, Lyon, France
hResearch Pathology Platform, Department of Translational Research and Innovation, Centre Léon Bérard, Lyon, France
iPathology Department, Hospices Civils de Lyon, Bron F-69500, France
jUMR1197, Université Paris-Saclay, 94800 Villejuif, France
kINSERM, UMR 1238, PHYOS, Faculty of Medicine, University of Nantes, Nantes, France
First published on 14th December 2021
Abstract
Bone marrow is a complex and dynamic microenvironment that provides essential cues to resident cells. We developed a standardized three-dimensional (3D) model to decipher mechanisms that control human cells during hematological and non-hematological processes. Our simple 3D-model is constituted of a biphasic calcium phosphate-based scaffold and human cell lines to ensure a high reproducibility. We obtained a minimal well-organized bone marrow-like structure in which various cell types and secreted extracellular matrix can be observed and characterized by in situ imaging or following viable cell retrieval. The complexity of the system can be increased and customized, with each cellular component being independently modulated according to the issue investigated. Introduction of pathological elements in this 3D-system accurately reproduced changes observed in patient bone marrow. Hence, we have developed a handy and flexible standardized microphysiological system that mimics human bone marrow, allowing histological analysis and functional assays on collected cells.
Introduction
The importance of substituting in vivo experiments for deciphering human pathophysiological mechanisms is becoming increasingly evident.1,2 This became particularly important in the field of human healthcare, such as drug testing. Indeed, regulatory and governmental agencies, as well as academic and pharmaceutical industries, frequently report that animal models poorly predict human responses. This likely reflects the fact that, even if animal models can mimic disease features following human-like engineered genetic alterations for example, underlying mechanisms are often different. This includes divergent molecular, cellular and physiological parameters, such as the immune response and major differences that distinguish mouse and human pluripotent cells.3 New models mimicking human tissue could particularly be relevant to design and assay innovative therapies. Conversely to in vivo assays, in vitro studies fail to reproduce the complex and dynamic 3-dimensional (3D) microenvironment structure of human bone marrow, such as integrating vascular flow and physico-chemical aspects. In this context, 3D modeling of human tissues is necessary in order to reliably decipher cell–cell and cell–matrix interactions, for example. For this purpose, human microphysiological systems, like Organ-on-Chips, have been developed by different laboratories and were demonstrated to accurately mimic pathophysiological responses observed in humans, which have so far never been detected in preclinical animal studies.4The bone marrow (BM) is found within the central cavities of axial and long bone accounting for 4% of the human body weight and contains distinct non-cellular and cellular components, including stem cells of hematopoietic (HSC) and stromal lineages and a variety of more differentiated hematopoietic cells (like immune cells), as well as non-hematopoietic cells (adventitial reticular cells, endothelial cells, nerve cells, fibroblasts, adipocytes, bone lining cells-osteoblasts) and elements of the extracellular matrix (such as collagens and fibronectin).5,6 Cells are not randomly spatially distributed and display a particular organization of hematopoietic tissue islands (called hematons) and of adipose cells surrounded by sinusoids interspersed within trabecular bone.7,8 Owing to its complex, dynamic and heterogeneous cellular and non-cellular composition, modeling of the human BM tissue has remained challenging for many years.9,10 Indeed, it is important to recapitulate biological functions of that tissue including the regulation of HSC (survival, adhesion, quiescence, homing, and differentiation) in response to multiple physiological or non-physiological stresses (like soluble factors, hypoxia), but also to mimic other aspects such as structural parameters, cell–cell or cell–matrix interactions, other non-hematopoietic cells or pathogen homing and regulation.11,12 The modeling of human BM is also of utmost importance as this tissue hosts a variety of hematological and non-hematological pathologies, the underlying mechanisms of which remain obscure.11,13–17 For instance, several studies have focused on the BM as a host tissue for migrating cancer cells detected in situ and identified to persist in this new microenvironment without growing.16,18–20 Of main clinical relevance, BM-driven dormancy of cancer or infected cells has become a broad concern as their detection is associated with a poor prognosis related to relapse or resistance to treatment, thus highlighting BM as an important therapeutic target in many contexts.21–25
Several technological developments have led to the generation of reliable 3D models that mimic the in vivo human BM.26–33 Most common approaches use scaffold-based 3D biomaterials (hydrogels, colloids or collagens)26,34 or decellularized bone27 associated with primary human BM stromal cells (BM-SCs) based on their ability to differentiate in situ into osteoblasts. However, no consensual model has yet been designed to simultaneously meet different requirements of a user-friendly and standardized system. Indeed, current systems mainly rely on primary cells and consequently suffer from limited access, availability and considerable heterogeneity of human samples. Moreover, such models remain highly technical and complex to set up for a large number of laboratories.
Here, our objective was to develop an easy-to-access, reliable and reproducible microphysiological human BM minimal system from which cells could be studied and recovered, to investigate diverse phenomena occurring in the BM. To do so, we have chosen to use biphasic calcium phosphate (BCP) particles associated with human cell lines derived from endothelial and bone marrow stromal cells which represent major cell types of the bone marrow and produce structural non-cellular components of the extracellular matrix and cytokines involved in the regulation of homeostasis and differentiation. Furthermore, osteoblastic differentiation was triggered by the addition of specific medium and reinforced by the presence of BCP particles which also promote 3D growth, as previously shown.35,36 In the current study, we present evidence that our new BM model support normal hematopoiesis and present dynamic evolution of matrix features upon challenges with various cell types.
Experimental procedures
Cell culture
The HS27A cell line was immortalized from healthy BM cells37 and displayed properties of immature bone marrow stromal cells (BM-SC)34,37–39 such as long-term hematopoiesis support.40,41 KG1A (ATCC; Ref# CRL2496; Acute Myelogenous Leukemia), MEG-01 (ATCC; Ref# CRL2021; megakaryoblastic) and MDA-MB-231 (ATCC; Ref# HTB-26; epithelial Breast Cancer) cells were cultured in RPMI1640 with 10% Fetal Bovine Serum (FBS) and 1% penicillin–streptomycin. T47D (ATCC; Ref# HTB-133; Breast cancer) cells were grown in DMEM supplemented with 10% FBS and 1% penicillin–streptomycin. MS-5 (DSMZ; Ref ACC 441; murine stromal cells) were maintained in 90% alpha-MEM with 10% Fetal Bovine Serum (FBS) and 1% penicillin–streptomycin. HMEC-1 (Cell Culture Development; Ref#CDC·HMEC-1) human microvascular endothelial cell line42 was grown in MCDB 131 medium (Gibco), supplemented with 10% FBS, 1% penicillin–streptomycin, 1% glutamax 100× (Gibco), 1 mg L−1 hydrocortisone (Sigma) and 10 μg L−1 epidermal growth factor (Sigma). Osteoblastic differentiation medium was composed of DMEM with 10% FBS, dexamethasone (0.1 μM; Gibco), β-glycerophosphate (10 mM; Sigma) and ascorbic acid (250 μM; Sigma).Fluorescent cell lines were derived by introducing lentiviral vectors to produce a turquoise fluorescent protein (visible at 458 nm) for HS27A, a Kusabira orange fluorescent protein (visible at 554 nm) for HMEC-1, and a EGFP fluorescent protein (visible at 488 nm) for KG1A. Lentivectors PGK-GFP, CSII-mKO2 (kindly provided by Dr Atsushi Miyawaki43 and CSII-mTurquoise2-Tubulin (Addgene plasmid #36202)44 were produced in 293T cells by co-transfection with helper plasmids pMD2.G and psPAX2 (Addgene plasmids #12259 and #12260). Recipient cells were infected at a low multiplicity of infection (moi < 1) and sorted on a BD FACSAria II SORP cell sorter.
Primary cells
BM samples were obtained either from healthy BM donors for allogeneic transplants or from Acute Myeloid Leukemia (AML) patients prior to treatment. All donors and patients provided written informed consent in accordance with the Declaration of Helsinki. Studies were approved by local ethics committee bylaws. CD34 immunomagnetic separation (StemCell Technologies) was performed as previously described45 and reached an average purity of 90%.Functional assays
For CFU-F assay, cells from fresh bone marrow were seeded at a concentration of 0.1 million in a well of 6 well plate with StemMACS MSC expansion media (130-104-182, Miltenyi). At day 10, cells were fixed with pure Methanol during 10 minutes and stained with Giemsa solution (15940, Electron Microscopy Science) for 2 minutes. Giemsa was discarded and stained cells were washed 3 times with water.CFC (colony forming cells) and LTC-IC (long term culture-initiating cells) assays were performed as previously described46 on hematological cells collected after suspension culture, 2D co-culture with HS27A cells or in the 3D system and cell sorting. For LTC-IC CD34+/CD38− cells were co-cultured with murine MS5 cells feeder in human long-term culture medium (StemCell Technologies) supplemented with freshly dissolved 10−6 M hydrocortisone sodium hemisuccinate (Sigma-Aldrich), with weekly half-medium change. After 5 weeks, both non-adherent and adherent cells were harvested, pooled, and washed; and the number of CFCs produced was expressed as W5-CFC/1000 initial CD34+CD38− cells and correlated with the number of primitive progenitors in the original input suspension.
Development of the 3D culture system
The 3D culture model is based on the culture of cells in the presence of ceramic biphasic calcium phosphate (BCP) particles and adapted from ref. 35. BCP particles composed of hydroxyapatite [Ca10(PO4)6(OH)2](60%) and β-tricalcium phosphate [Ca3(PO4)2](40%), calibrated between 40 and 80 μm (obtained from Graftys SARL-France or CaP Biomaterials LLC-USA) were sterilized by heating at 180 °C for 2 h. The 3D culture was performed in 8 mm diameter cell culture inserts with a polycarbonate membrane of 3 μm pores (Nunc, Thermofisher, Ref# 140627). BCP particles (35 mg) were loaded into each insert which was placed in a culture well of a 6-well plate. BCP particles were washed several times with PBS and then incubated 2 days with RPMI medium with 10% FBS. After washing, cells (2 × 106 HS27A with or without 1 × 106 HMEC1 as indicated) were resuspended in 500 μL of medium (control or osteoblastic differentiation medium as mentioned) were added into the insert and 4.5 mL of medium was added into the well around the insert. Cells and BCP particles were mixed by gentle pipetting and incubated at 37 °C and 5%CO2. After 24 h, the supernatant in the insert was replaced, before BCP particles and cells were again gently mixed. The whole medium was changed the day after and then twice a week. In order to allow HS27A differentiation, cells were maintained in osteoblastic differentiation medium. For co-culture of HS27A and HMEC-1, cells were maintained in a (50/50) combination of osteoblastic differentiation medium and HMEC-1 specific medium. According to the experiments, a third cell type was introduced after 3 weeks once a native bone marrow microenvironment was formed due to advanced osteoblastic differentiation, thus favoring cells/cells interaction with third cell type. In this case, the initial medium is replaced by a mix of HMEC-1 medium and the medium necessary for this third cell type (DMEM, RPMI1640 or IMDM with 10% FBS). At indicated time, rigid disks were retrieved by cutting the membrane at the bottom of the insert (Fig. S2B†).Histological analysis
The disk containing particles and cells, or half of it, was fixed 3 h in 4% paraformaldehyde (Sigma). Disks were then decalcified through 4 incubations of 48 h in EDTA (0.5 M, pH8) and embedded in paraffin ((Sakura, Ref# 4511). Sections (4 μm-thick) of formalin-fixed, paraffin-embedded tissue were prepared according to conventional procedures. Sections were stained with Hematoxylin–Phloxine–Saffron (HPS) (Hematoxylin (Diapath, Ref#. EP813)-Phloxine (RAL Diagnostic Ref# 361470-0025)-Saffron (VWR Ref# 27481.105). Sections from samples containing cells expressing fluorescent proteins were maintained at 4 °C and obscurity all along the process until imaging.Immunohistochemistry (IHC) was performed on an automated immunostainer (Ventana Benchmark ultra, Roche) using Ultra View DAB Kit. Sections were incubated with specific antibodies targeting: CD31 (clone JC70A, M0823-01 Agilent), CD45 (clone 2B11+PD7/23, M0701-01 Agilent), cytokeratins (Pan CK; clone AE1/AE3, M3515 Agilent) to identify endothelial, hematopoietic or epithelial cells. Antibodies targeting Collagen 1 (ab90395 Abcam), Collagen III (ab7778 Abcam) and Fibronectin (Cell Signaling #26836S) were used to identify extracellular matrix production. Antibodies targeting CD45 (Agilent M0701), CD34 (Agilent M7165), and CD38 (Cell Signaling #51000S) characterized hematopoietic cells while E-Cadherin (ZYTOMED BRB047), Vimentin (Agilent M0725) and CD44 (Thermo Fisher MA5-13890) characterize mammary cells. KI67 (clone Mib1, M7240 Agilent) and H3K27Me3 (Cell Signaling #9733) identified proliferating and dormant cells respectively.47 Staining was revealed by an anti-rabbit or -mouse -HRP and visualized with 3,3′-diaminobenzidine as a chromogenic substrate and counterstained with Gill's-hematoxylin. Multiplexed immunofluorescence was performed on Bond RX automated immunostainer (Leica biosystems) using OPAL detection kits (AKOYA bioscience). Sequential immunofluorescences were performed using OPAL 520 (CD34, green), OPAL 570 (CD45, yellow), OPAL 690 (ki67, red) and cells were counterstained with DAPI. Following IHC and specific coloration slides were mounted using Pertex (Histolab, Ref# 00801-EX). For fluorescent slides were mounted using Prolong™ Gold Antifad Reagent (Invitrogen, Ref# P36930).
Differentiation of stromal cells
Osteoblastic differention of stromal cells was assayed by plating 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well of 12 well plates and cultured in StemMacs OsteoDiff medium (Miltinyi, Ref# 130-091-678). After 12 days, Alizarin Red staining was performed to locate calcium deposition. Therefore, wells containing cells were washed with PBS 1× and cells fixed using paraformaldehyde 4% for 24 hours. Wells were then rinsed twice with water and incubated 2 minutes in Alizarin red solution (2%; Merck) and rinsed again 3 times with water. Adipocytic differentiation was performed using StemMacs AdipoDiff media (Miltinyi Ref# 130-091-677). Stromal cells were plated (40000 cells per well of 12 well plates) and incubated in differentiation medium for 8 days. After fixation using PFA4% for 30 min cells were incubated with Oil Red O (3 mg mL−1; Polyscience; Ref# 06317) for 15 min. Wells were then rinse twice with water.
000 cells per well of 12 well plates and cultured in StemMacs OsteoDiff medium (Miltinyi, Ref# 130-091-678). After 12 days, Alizarin Red staining was performed to locate calcium deposition. Therefore, wells containing cells were washed with PBS 1× and cells fixed using paraformaldehyde 4% for 24 hours. Wells were then rinsed twice with water and incubated 2 minutes in Alizarin red solution (2%; Merck) and rinsed again 3 times with water. Adipocytic differentiation was performed using StemMacs AdipoDiff media (Miltinyi Ref# 130-091-677). Stromal cells were plated (40000 cells per well of 12 well plates) and incubated in differentiation medium for 8 days. After fixation using PFA4% for 30 min cells were incubated with Oil Red O (3 mg mL−1; Polyscience; Ref# 06317) for 15 min. Wells were then rinse twice with water.
Microscopy
Fluorescent slides were analyzed by microscopy (upright Nikon microscope). To analyze standard coloration (HPS), specific coloration and immunohistochemistry, the Primovert microscope Zeiss was used, and images were collected using Axiocam 105 color as Camera and ZEN Lite 2012 as software. For global analyses, sections were scanned with panoramic scan II (3D Histech, Hungary) at 40× for IHC and using the Vectra POLARIS (Akoya) device. Captured images were processed using HALO software (Indica Lab, V3.2.1851) and adapted algorithms for quantification: area quantification v2.1.3, cytonuclear v2.0.8 and cytonuclear FL v2.0.6. Images from scanned slices, were generated using softwares: Caseviewer (3Dhistotech 2.3.0.99276) for coloration and IHC, and Phenochart (Akoya 1.0.12) for fluorescent images.Viable cell retrieval and analysis
After recovery, disks formed at the bottom of the insert were washed twice with PBS. Disks were first incubated for 11 min at 37 °C in mL of collagenase (0.4 mg mL−1, Sigma) then centrifuged prior before 1 min incubation in 300 μL of trypsin (in EDTA 0.05%, Gibco) at 37 °C. Remaining clots were finally dislocated by flushing and the reaction was stopped with 400 μL of FBS. After centrifugation recovered cells were resuspended in PBS containing 10% FBS and viability counted using trypan blue. Cell populations were then either plated or analyzed.Cytometry
Phenotypic comparison of HS27A cells and BM-SC were performed using LSRFortessa (Becton Dickinson (BD)) cytometer and the following markers: CD73-APC (BD Ref#: 560847)/CD90-FITC (BD Ref#: 555595)/CD105-PE (BD Ref#: 560839)/CD146-R718 (BD Ref#: 752158)/SSEA4-PE CF-594 (BD Ref#: 562487). Phenotypic analysis of hematological cells was performed on LSRFortessa cytometer using the following markers: CD45-V500 (BD Ref#: 560777/CD34-APC (BD Ref#: 345804)/CD38-pe·Cy7 (BD Ref#: 335825) and CD14-PE (BD Ref#: 562691)/DAPI (Chemotec Ref#: 910-312). The software Diva was used for acquisition and Flow Jo for analysis.Following suspension, 2D co-culture or 3D culture steps, hematological cells were sorted according to their maturity using the FACS ARIA II (BD) with CD45+/CD34+/CD38−/DAPI− cells as very immature cells and CD45+/CD34+/CD38+/DAPI− cells as progenitors.
For selection, harvested cell suspensions were filtered through a 40 μm mesh and hematopoietic cells sorted by FACS according to their CD45+CD34+ phenotype or based on mean fluorescence intensity of Turquoise, Kusabira or Orange when fluorescent cell lines were used.
Molecular biology
For RT-PCR analysis, the half-disk was resuspended in 1 mL of Trizol Reagent (Life technologies™) and performed using standard protocols. Total RNA was purified using mini-RNA extraction kit (Qiagen). For RT-qPCR, cDNA was produced using Superscript II (Invitrogen) and quantified using Sybr-green (Quantifast, Qiagen) and Real-Time PCR system (Roche, LC480). TATA-binding protein (TBP) and hypoxanthine-guanine phosphoribosyl transferase (HPRT) genes were used for normalization. Arbitrary Unit (AU) corresponds to the ratio of expression between samples and a single normal sample used as a reference in each PCR. The primer sequences were the following ones: HPRT (F: TGACCTTGATTTATTTTGCATACC; R: CGAGCAAGACGTTCAGTCCT), TBP (F: CAGCGAACCACGGCACTGATT; R: TTTTCTTGCTGCCAGTCTGGAC), COL1A (COLLAGEN, TYPE 1, ALPHA-1), (F: CTGGACCTAAAGGTGCTGCT; R: GCTCCAGCCTCTCCATCTTT), BSP (BONE SIALOPROTEIN) (F: CGAATACACGGGCGTCAATG; R: GTAGCTGTACTCATCTTCATAGGC), GATA2 (F: GGCTCGTTCCTGTTCAGAAG; R: CTTGGGCTTGATGAGTGGTC, OSC (OSTEOCALCIN) F: GGCGCTACCTGTATCAATGG; R: TCAGCCAACTCGTCACAGTC.Statistical analysis
Unless otherwise specified, statistical analysis was performed using the parametric Student-T test. Mann–Whitney non-parametric test was used for comparison between two groups when data was not normally distributed. The statistical analyses and graphs were performed with Graphpad prism (version 6). Significant P values are given in the text or symbolized by asterisks (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001).Results
The human stromal cell line HS27A is a standardized substitute to generate a MSC-derived minimal bone-like structure
Developing a bone marrow 3D model first required the generation of a scaffold mimicking the rigid bone architecture consisting of a meshwork of trabecular bone. We thus associated BM-SCs and BCP particles (Fig. 1A), reported to facilitate rigid bone formation using primary osteoblast cells and allow the constitution of calcified 3D structures.35 In order to avoid issues associated with the use of primary stromal cells and obtain a reproducible 3D model, we used a human cell line of BM-SCs, HS27A cells.37 These cells display classical BM-SC markers, such as CD90, CD73 and CD105 and others more recently reported (like SSEA4 and CD14648) (Fig. S1A†), differentiate into osteoblasts and adipocytes (ref. 37 and Fig. S1B†) and generate CFU-F activity similarly to normal BM-SCs (Fig. S1C†). Sterilized BCP particles were placed on the microporous polycarbonate membrane of an insert then placed in a culture plate (Fig. 1A and S2A†).35 Two million HS27A cells were added to the BCP particles in the presence of osteoblastic differentiation medium. After 3 weeks, a solid disk lying on the polycarbonate membrane could be retrieved from the system and processed for analyses similar to those typically conducted on bone samples (Fig. S2A and S2B†).
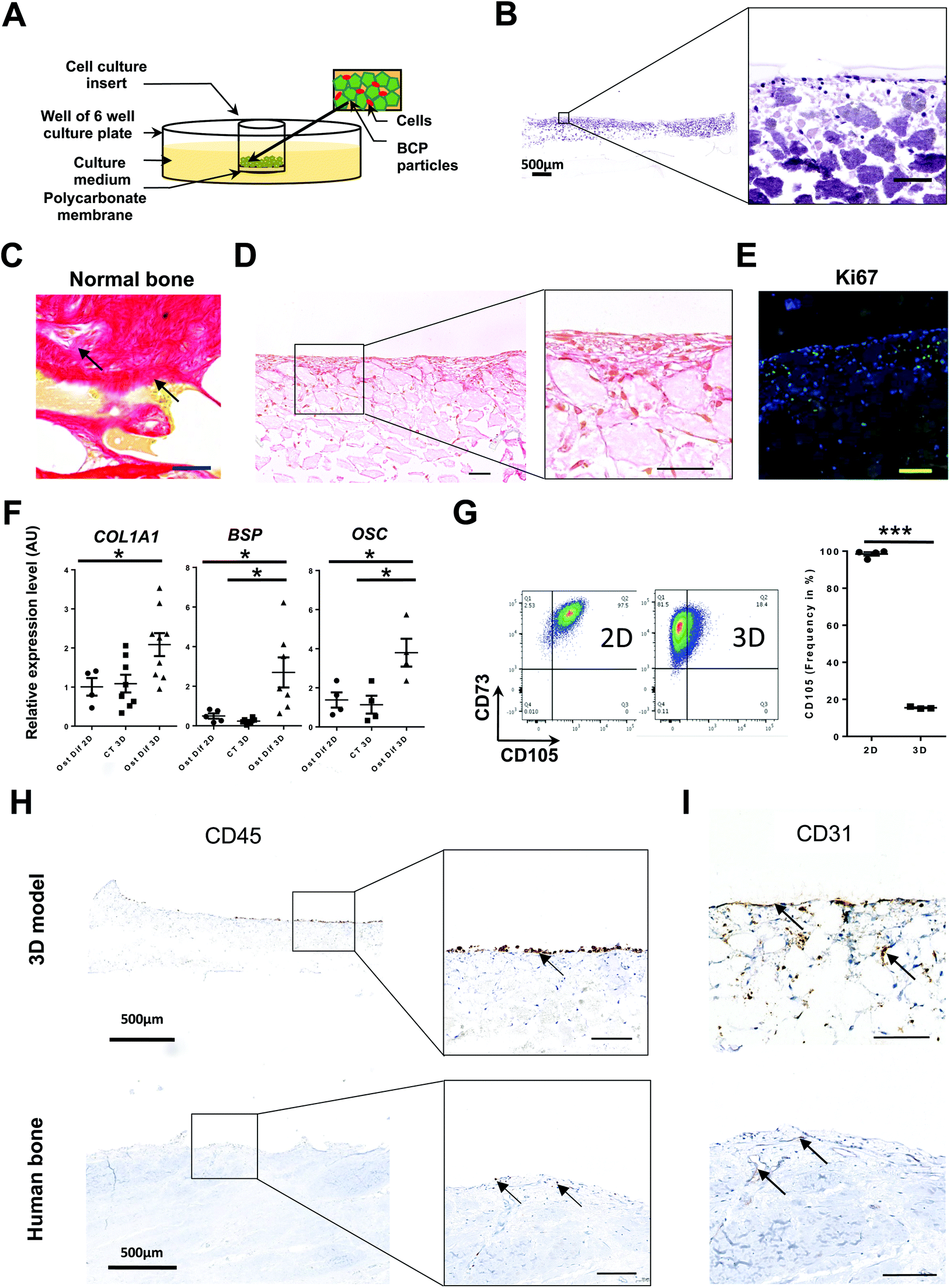 | ||
| Fig. 1 Establishment of a rigid bone structure by in situ osteoblastic differentiation of HS27A cells. (A) Schematic diagram of the initial setup of the system (see also Fig. S2A and B†). Unless specified, all structures were recovered at day 21 and then proceed for the indicated analysis. (B) Staining by hematoxylin–phloxine–saffron (HPS) of systems generated with HS27A cells in culture with BCP particles and osteoblastic differentiation medium (see also Fig. S2B†) (n = 12). Scale bar, (left panel, 500 μm; right panel, 100 μm). (C) Sirius Red staining from human bone sections (scale bar, 100 μm). (D) Sirius Red staining from 3D-structures derived in osteoblastic differentiation medium (n = 12) (scale bar, 100 μm) (see also Fig. S2C†) (E) Ki67 immunostaining from structures derived in osteoblastic differentiation medium (n = 6). Scale bar, 100 μm. (F) Expression of differentiation markers measured by RT-qPCR for COLLAGEN Type 1 Alpha-1 (COL1A1), BONE SIALOPROTEIN (BSP) and OSTEOCALCIN (OSC) after 28 days in control medium (CT) or osteoblastic differentiation medium (Ost Dif) in 2D or 3D culture conditions (n = 4 to 9). Mann–Whitney non-parametric test, *P < 0.05. (G) Representative plots of CD105/CD73 staining of HS27A following 2D or 3D culture. Data displayed CD105 frequency (% of positive cells) (n = 3) (left panel). Individual data from independent experiments are presented with the mean value as a dark line (right panel). P-Values were determined by unpaired Student's t-test, ***P < 0.001. (H and I) Upper panels: systems were collected after 3 weeks of 3D co-culture with HS27A and HMEC-1 and the addition of KG1A cells after 1week (n = 28) (see also Fig. S3†). Lower panels: normal human bone section. Staining by IHC for CD45 (H) or CD31 (I). Arrows indicate either CD45 or CD31 positive cells according to the staining. Unspecified scale bars: 100 μm. | ||
Macroscopic observations and manipulations of the disk indicated that only cells cultured for a minimum of 3 weeks in osteoblastic differentiation medium gave rise to easy-to-handle discoid structures (Fig. S2B†). Histological sagittal sections of the solid disk followed by HPS (hematoxylin–phloxine–saffron) staining allowed visualization of areas presenting different aspects within the formed structure. A layer of cells was observed in the upper part of the disk where we can see BCP particles and cells distributed heterogeneously according to the area observed (Fig. 1B). In addition, Sirius red staining of collagen fibers, representative of the matrix network found in human normal bone sections (Fig. 1C), highlighted in situ production within the 3D-derived discoid structures (Fig. 1D and S2C†). Finally, Alizarin staining of non-decalcified sections of HS27A cells cultured for 21 days in our 3D system show the deposit of Calcium beside stromal cells and BCP particles (Fig. S2C,† right panel). This indicates that HS27A-derived cells secrete extracellular matrix within the 3D culture system in the established culture conditions.
We observed calcification with clusters of cells either within or at the contact of the matrix (Fig. 1D). Within the calcified part, most HS27A stained cells (collagen producing) were very elongated, forming lines on BCP-beads, while other less stained cells remained rounder and more clustered (Fig. S2C†). A similar morphology of collagen-producing cells and cellular clusters were observed in sections of human bone (Fig. 1C). Performing KI67 staining on 3D structures revealed the heterogeneity of HS27A in terms of cell cycle as a portion of cells remained proliferating in situ, indicating that this system is dynamic (Fig. 1E). As expected, molecular analysis of harvested HS27A cells confirmed that the relative expression levels of osteoblastic differentiation genes (COL1A1, BSP and OSC) were higher when cultures were performed in osteoblastic differentiation medium than in control medium and in 3D as compared to 2D culture (Fig. 1F). Accordingly, flow cytometric analysis revealed a marked reduction of CD105 frequency and a slight decrease in CD73 intensity after 3D culture (Fig. 1G), illustrating in situ differentiation of HS27A like reported for primary BM-SCs.49
To obtain a more comprehensive model of the bone medullar microenvironment, we then successively introduced human endothelial and hematopoietic cells. The human microvascular endothelial immortalized cell line (HMEC-1) was used since this cell line retains primary endothelial cell features and was more stable than other cell lines tested such as BMEC-1 cells (data not shown).42 We introduced immature hematopoietic cells using the KG1A cell line. This leukemic cell line model represents immature hematopoietic cells identified by their CD34+CD38− status, ability to generate colony-forming cells (CFC) and high levels of embryonic markers.50 Optimization of media (Fig. S3A†) and timing of cell introduction led us to the conclusion that both HS27A and HMEC-1 cells could be introduced at day 0 and KG1A cells at day 21 (Fig. S3B†). Indeed, at day 21 the calcified BM-like structure was obtained suggesting advanced osteoblastic differentiation that could promote hematopoietic cells interactions with their niche. At day 28, we observed the presence of hematopoietic and endothelial cells at the surface and inside the rigid structure, as assessed by CD45 and CD31 IHC staining, respectively (Fig. 1H and I). Interestingly, similar analyses performed on native adult BM samples showed that CD45+ cells are also located in contact of the BM surface and that blood vessels are formed within BM structure but also near the BM surface (Fig. 1H and I). Fluorescent cell lines (HS27A (blue), HMEC-1 (red) and KG1A (green)) were used to identify in situ interactions of the three cell types without further processing and highlight the proximity of KG1A cells with either endothelial or stromal cells (Fig. S3C†). Similar results could be obtained using non-fluorescent cell lines via immunofluorescent staining, which revealed that most KG1A cells in contact with the surface of the rigid structure are CD45+CD34+ (Fig. S3D†). Remarkably, aside from its use to analyze cells in situ, our 3D system also enabled us to recover single cell suspensions through enzymatic dissociation, using Collagenase and Trypsin. Cells could then be analyzed by flow cytometry (Fig. S3E†) or further cultured (Fig. S3F†), demonstrating that each cell type had been viably recovered. These results illustrate those different techniques can be used to successfully analyze both hematopoietic and non-hematopoietic compartments after 3D culture in this system.
Altogether, our results indicate that, using BCP particles and a specific osteoblastic differentiation medium, the BM-SC model based on HS27A cells provided an organized and rigid entity with neo-synthetized collagen-enriched matrix, BM-SC and differentiated cells that suitably represents a minimal human BM structure.
A minimal functional BM-like unit to host and sustain normal hematopoiesis
We next investigated the capacity of our microphysiological system to perform functional, molecular and/or analytical assays on live sorted cells following their exposure to physiological-like conditions. We evaluated whether this system could maintain the properties of HSC arising from BM samples. To do so, we substituted the KG1A hematopoietic cell line with primary BM mononuclear hematopoietic unsorted cells (MNC) or sorted immature CD34+ cells isolated from adult human BM from healthy donors. In situ staining after 7 days confirmed that most of the hematopoietic cells in contact with the bone-like structure were CD34+, indicating the maintenance in the system of immature primary hematopoietic cells (Fig. 2A and S4A†). Moreover, even using primary cells we observed a high reproducibility in terms of CD34+ and CD34− maintenance using 3 different bone marrow samples (Fig. S4A†), attesting of the adequate standardization of this system. In addition, since some HSC have been described to become quiescent in their BM niche,51 we validated that most CD45+ cells were Ki67− or H3K27me3+ (ref. 47) (Fig. S4B and S4C†). The survival and enrichment of immature cells was further confirmed with initial seeding in the 3D system of either MNC or selected CD34+. Following cell retrieval from the system and sorting, we identified HSC and their derived progenitors by CD34 and CD38 expression and functional assays (Fig. 2B). The detection of the CD34+CD38− fraction indicated the higher maintenance of HSC in the 3D system as compared to other culture conditions (Fig. 2C and D) and was further validated owing to an enrichment in cells expressing GATA2 in this sorted subpopulation (Fig. S4D†). The production following culture in the 3D-system of more mature cells was also observed by flow cytometry analysis of indicators such as CD14 (monocytes) (Fig. S4E and F†). At the functional level, we evaluated the capacity of 3D-seeded cells to support maintenance and production of progenitors and HSC in situ using respectively the colony-forming cell assay (CFC) and long-term initiating cell (LTC-IC) assay that quantify their derived progenitors by colony-forming cell assay (W5-CFC). Data obtained highlighted a trend towards an increase in the total number of CFC colonies formed when cultured in the presence of stroma (Fig. 2E). More importantly, identification of the different hematopoietic subtypes45 revealed the ability of the 3D model to maintain and favor the generation of all early and late hematopoietic progenitor subsets (Fig. 2F). Results from the LTC-IC assay showed a better maintenance of HSC derived colonies (W5-CFC) when cultured in the 3D system, whereas they were lost when co-cultured with HS27A cells or in suspension (Fig. 2G).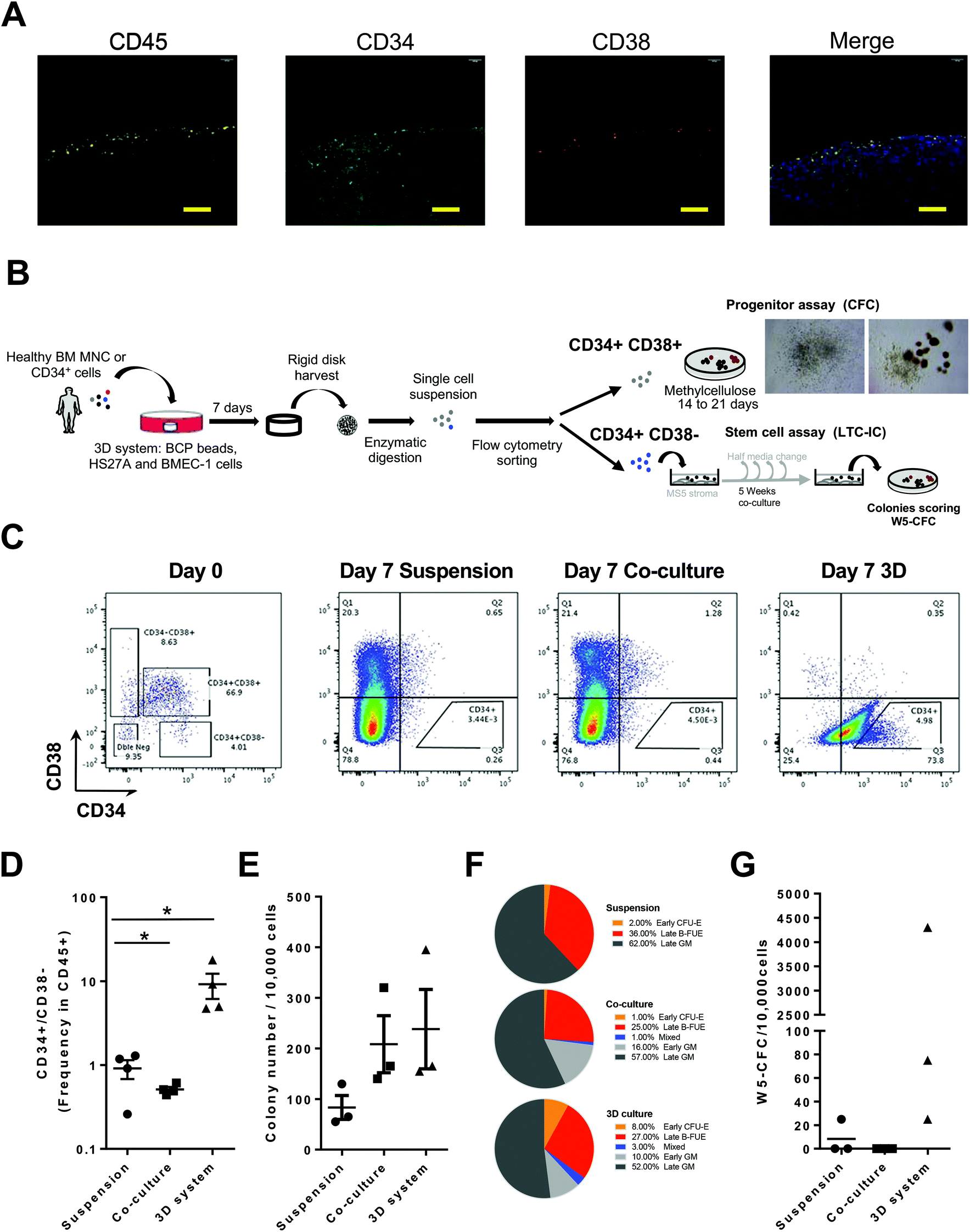 | ||
| Fig. 2 Recovery of viable and functional cells after culture in the BM-like 3D model. (A) Immunostaining of systems after one week of incubation with primary bone marrow mononuclear cells (BM MNC) for CD45, CD34 and CD38 as hematopoietic markers (n = 12). Scale bars, 100 μm (see also Fig. S4A and B†). (B) Experimental procedure to assay progenitor ability for progenitors (CD34+CD38+) and HSC (CD34+CD38−) after culture in the 3D-system using cell sorting and CFC assay or LTC-IC assay, respectively. On the right upper part: bright field pictures representative of recovered CFCs. (C) Flow cytometry plots illustrating CD34/CD38 expression at seeding (day 0, left panel) or recovery (day 7) from suspension, co-culture with HS27A in 2D or in the 3D system. (D) CD34+CD38− frequency among CD45+ subsets as determined by flow cytometry from cells taken after 7 days of suspension culture, co-culture or 3D culture. (E, F and G) Cells collected after 7 days of suspension culture, co-culture with HS27A in 2D or within the 3D system were sorted according to CD34/CD38 expression and characterized by functional assays (n = 3). Frequency (E) of CFC was determined among CD34+CD38+ sorted cells from each culture method. (F) Pie charts showing proportions of means of identified colonies subtypes of progenitors counted in the CFC assay (Orange for Early CFU-E; red for Late BFU-E, blue for mixed colonies; light grey for Early CFU-GM and dark grey for Late CFU-GM). (G) Proportion of LTC-IC obtained from CD34+CD38− cells recovered after 7 days of suspension culture, co-culture or 3D culture and quantified by their W5-CFC output (n = 3). P-Values were determined by Student's t-test, *P < 0.05. | ||
Altogether these data demonstrate that the 3D-system developed is suitable to preserved HSC and progenitor properties, and to observe their differentiation towards more mature hematopoietic cells, both in situ or after cell harvesting. Importantly, this system showed a reliable capacity to reflect the diversity of hematopoietic progenitors, as observed in the human bone marrow.
A BM-like system to host metastatic cancer cells to study dormancy and microenvironment remodeling
Bones constitute frequent sites for metastasis of several solid cancers. Hence, we investigated whether our standardized human BM-like 3D model was relevant to study processes taking place in this microenvironment like dormancy, expansion or invasion. We compared the behavior of two breast cancer (BC) cell lines with different properties (unlike T47D cells, MDA-MB-231 cells generates bone metastasis in mouse models52). The BC cells were seeded onto 3-week-old 3D-structures, and disks were collected 1 week later. We confirmed by IHC analysis that T47D cells displayed luminal properties (E-cadherin+), whereas MDA-MB-231 displayed basal-like features (CD44+Vimentin+) (Fig. S5A†). We characterized the impact of BC cell lines on the surrounding microenvironment. At a structural level, no significant difference was observed by IHC staining of different extracellular matrix molecules (Fig. 3A, B and S5B, C†). We quantified the number of cytokeratin-positive cells that were localized within the bone-like structure and more frequently observed for metastatic MBA-MB-231 cells as compared to non-metastatic T47D cells (Fig. 3C and D), which mostly remained at the surface of the solid structure like hematopoietic cells (Fig. 1H). This is in accordance with the known production and secretion of matrix metalloproteinases by MDA-MB231 and related to their higher invasive potential.53 Indeed, pan-cytokeratin staining identified localized regions where MDA-MB231 cells have infiltrated the lower part of the structure (Fig. 3E) and which show a lower level of collagen 1 (Fig. 3F) reflecting the ability of these cells to penetrate the bone structure and degrade it. In addition, Ki67 staining indicated that MDA-MB-231 cells proliferate more than T47D cells in situ (Fig. 3G). This is further supported by an increased H3K27me3 staining in T47D cells (Fig. 3H) that will promote gene-silencing.54 Hence, these non-metastatic breast cancer cells could become dormant through their interaction with the bone marrow microenvironment as previously reported.21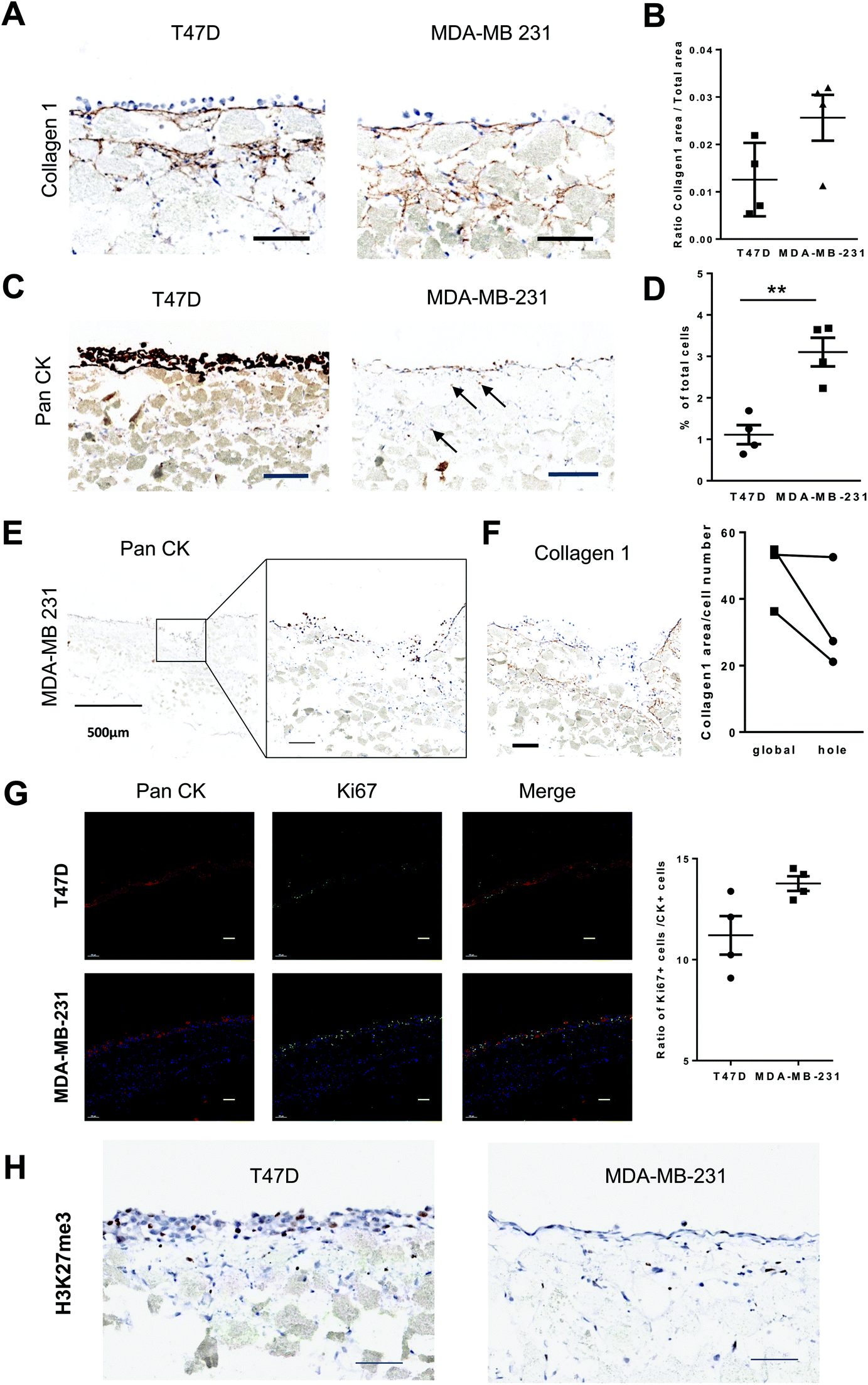 | ||
| Fig. 3 Use of the microphysiological BM system to evaluate microenvironmental remodeling by disseminated cancer cells. (A) Representative images of Collagen 1 stained sections of 3D structures after one week in the presence of either T47D (left panel) or MDA-MB-231 (right panel) cells (see also Fig. S5†). (B) Proportion of Collagen 1-positive area in the structure after one week in the presence of T47D (left or MDA-MB-231 cells (n = 4). (C) Representative images of Pan-cytokeratin stained sections of 3D structures after one week in the presence of either T47D (left panel) or MDA-MB-231 (right panel) cells. Arrows indicate BC cells that migrate within the calcified area of the system. (D) Percentage of Pan-cytokeratin cells observed at least 50 μm below the upper surface of the structure (n = 4) (right panel). P-Values were determined by Student's t-test, **P < 0.01. (E) Representative image of Pan-cytokeratin stained sections of 3D structures after one week in the presence of MDA-MB-231, corresponding to areas where epithelial cells have entered the upper part of the structure. (F) Representative image of Collagen 1 stained sections of 3D structures after one week in the presence of MDA-MB-231, corresponding to areas where epithelial cells have entered the upper part of the structure. (left panel). Quantification of Collagen 1-positive area inside portions of the structure invaded by MDA-MB-231 cells (n = 3) (right panel). (G) Slides of 3D systems supplemented with T47D or MDA-MB-231 and stained by immunofluorescence for Pan-cytokeratin and Ki67 markers. Proportion of Ki67-positive cells among Pan-cytokeratin-positive cells (right panel). P-Values were determined by Student's t-test, *P < 0.5. (H) IHC of T47D or MDA-MB-231 for H3K27me3. All Scale bars represent 100 μm except when specified. | ||
These observations show that this microphysiological system constitutes a suitable model to address issues related to the impact of pathogenic elements on BM microenvironment remodeling and intra-heterogeneity of cancer models.
A BM-like system that mimics a pathological microenvironment with matrix remodeling
BM fibrosis that is associated with a number of hematological and non-hematological disorders55 characterized by the increased deposition of collagen III (reticulin) and collagen 1A fibers. Since Acute Myeloid Leukemia (AML) is often associated with BM fibrosis and collagen alterations,55 we evaluated whether leukemic AML cells implantation in this 3D system could impact in situ collagen production. The introduction of non-AML (MEG-01) or AML (KG1A), hematopoietic cell lines identified by CD45 staining, led to modifications in extracellular matrix production and distribution. The overall 3D-architecture revealed by collagen 1, and to a lesser extent by fibronectin staining, appeared denser and more organized after the addition of KG1A than in systems generated with MEG-01 alone (Fig. 4A, B and S6A, B†). This likely reflects the fact that the addition of leukemic cells (KG1A) to the co-culture of BM-SC (HS27A cells) may increase collagen 1 production during the last 7 days of culture. We introduced primary hematopoietic cells (MNC or CD34+ cells) isolated from either healthy or AML BM samples and observed that CD45-positive cells from both normal BM (NBM) or AML samples adhered to the surface of the BM-like structure (Fig. 4C). Analysis of matrix components on sections of structures obtained after 7 days revealed a significant increase in collagen III components (but not fibronectin), specifically in the presence of AML CD34+ cells, but not with NBM CD34+ cells (Fig. 4D and S6C, D†).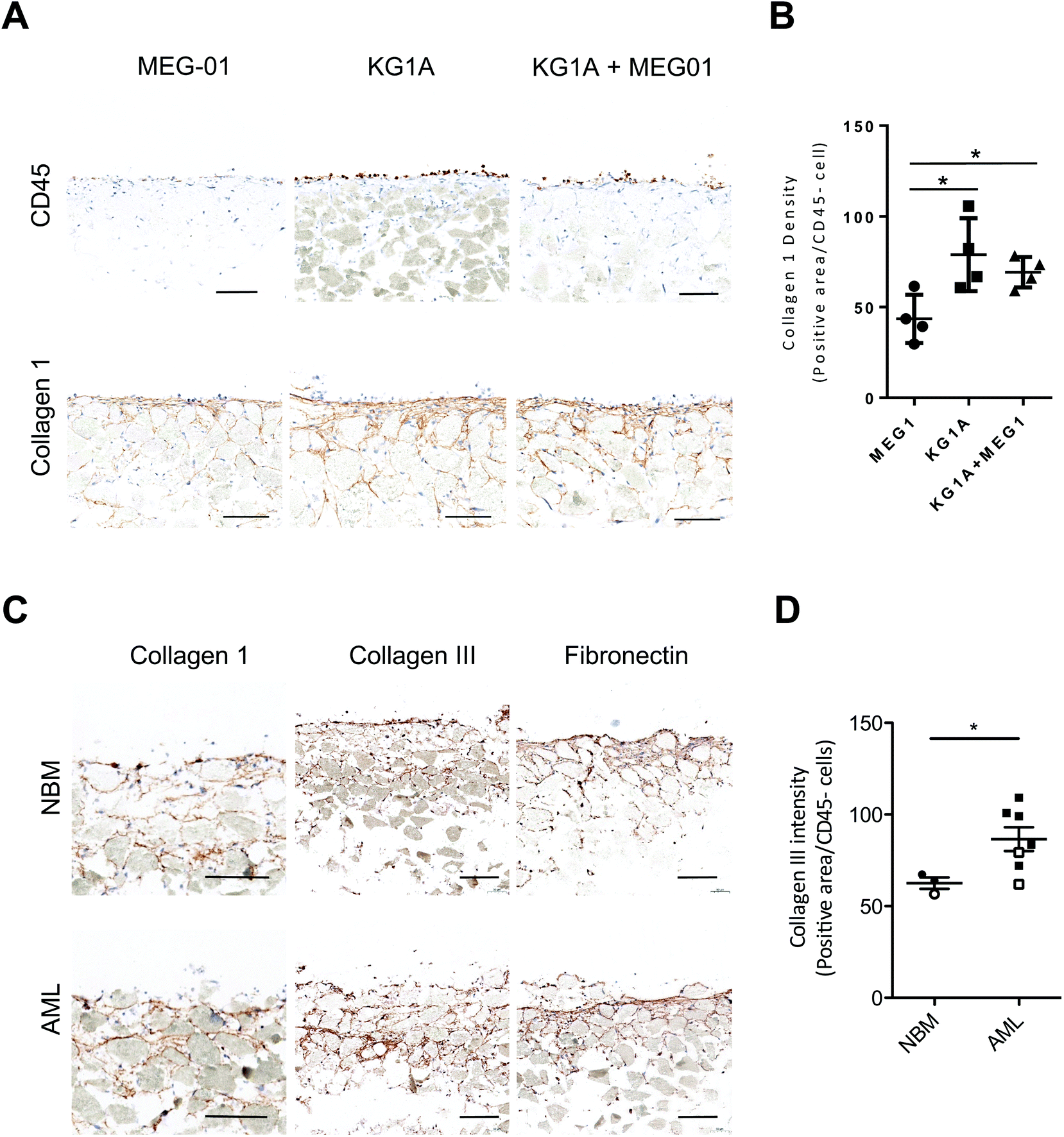 | ||
| Fig. 4 Leukemic cells alter the matrix of the bone marrow niche model. (A) Representative images of CD45 and Collagen 1-stained sections of 3D systems produced with MEG-01 (left panel), KG1A (middle panel) or both (right panel) cells (n = 3). Scale bar, 100 μm (see also Fig. S6†). (B) Dot plot indicating the proportion of Collagen 1-positive area per CD45− cells after one week in the presence of hematopoietic cells. (C) Representative images of Collagen 1, Collagen III and fibronectin-stained sections of 3D systems produced with CD34+ from NBM (top panel, n = 3) or AML (bottom panel, n = 7) cells. Scale bar, 100 μm. (D) Dot plot showing measured levels of Collagen III (staining intensity) with MNC (open circles/squares) or CD34+ (closed circles/squares). P-Values were determined by Student's t-test, *P < 0.05. | ||
Collectively, these data demonstrate the ability to alter non-cellular components of the hematopoietic microenvironment using either cell lines or primary hematopoietic cells and to reliably reproduce normal or pathophysiological changes of the BM microenvironment.
Discussion
The BM microenvironment hosts several key physiological processes such as hematopoiesis but also bone regeneration, in particular through in situ differentiation of stem cells. In addition, the human BM has emerged as a major site for the development of a variety of hematological and non-hematological pathologies that directly involve dysregulations of resident cells that display specific features.11,17 Therefore, the BM constitutes a remarkable and complex system to extensively study the regulation of various cells. In this context, a large community of researchers in many different fields would largely benefit from a simple, reproducible and cost-effective minimal microphysiological system of the human BM. We have developed a 3D-model that ensures a remarkable level of reproducibility, along with a maximum flexibility for a broad range of applications. Bone Marrow stromal cells (BM-SCs) constitute a key element in the constitution of the BM and its environment because they can differentiate into different cellular components such as osteoblasts and adipocytes and are the source of numerous molecules of the extracellular matrix.56,57 In order to gain reproducibility, we substituted primary BM-SC with a human BM-SC cell line (HS27A) that displays similar properties to primary BM-SC,34,37–39 including long term support of hematopoiesis in 2D culture,40,41 which we confirmed here in 3D conditions. Indeed, we show that the HS27A-based 3D-like BM system allows us to maintain, recover and expand CD34+CD38− HSC, and to generate more differentiated progenitors and mature cells. In our model, we induced osteoblastic rather than adipocytic differentiation since preosteoblasts and osteoblasts have been described as part of the endosteal niche.58 The use of BCP particles also participates in osteoblastic differentiation, which leads to the formation of a rigid architecture allowing to localize each of the cellular and matrix components. In addition, rigidity plays a role in bone marrow functions such as supporting hematopoietic differentiation, as shown by several studies.59,60 Histological analysis of these rigid 3D constructs confirmed the generation of a neo-synthesized dense network of matrix with physical features of bone formation.6 Therefore, we established that HS27A cells could efficiently be used as substitutes to primary BM-SC. This provides unlimited access to a highly reproducible basic bone-like structure.As sinusoid vessels and associated niches play a major role in BM homeostasis and resident (stem) cell regulation, we introduced into the system a microvascular cell line HMEC-1 that reliably represents the endothelial compartment.42 Even though we could not formally identify vessels, cellular alignment of HMEC-1 cells was present and, when added, leukemic cells (KG1A) were frequently observed in close interaction with endothelial cells (HMEC-1), corroborating reports in AML patient bone marrow or in AML mouse models.61,62 Furthermore, beyond their role as structuring vessels, endothelial cells also have a major role in regulating homeostasis and differentiation (for example through the supply of soluble factors or cell–cell interactions). The fact that HMEC-1 cells are maintained viable in our system all along the culture indicates that they can fulfill some of their key functions within our model. Different solutions to obtain functional vessels have been developed by different groups.26,27,63 Nevertheless, it is to consider that building such vascularized model will likely generate a much more complex system and therefore not be accessible to all laboratories. This will fulfill different objectives and request more technical aspects than the currently described 3D system.
To further evaluate the potential of this 3D-BM like system by increasing the level of complexity, we introduced a mature hematopoietic cell line representing the megakaryocytic lineage (MEG-01). We were able to identify functional changes by in situ analysis of the different types of fibers. This indicates the suitability of this system to address pathophysiological questions related to the impact not only on HSC but also on the other major components of the microenvironment including the extracellular matrix. Indeed, it should be possible to use this standardized 3D-BM system to evaluate, for example, the causal importance of the number of platelet-producing cells to initiate fibrosis, as reported in essential thrombocythemia, myelofibrosis or during leukemogenesis. In order to test the flexibility of the system, we substituted the hematopoietic cell line with primary hematopoietic mononuclear cells or sorted stem/progenitor cells isolated from healthy donors or AML patients. This step is of particular importance for stem cell-based therapies as it is well documented that major differences distinguish mouse and human pluripotent cells precluding in some cases the use of animal models to obtained reliable preclinical data.3,4 One remarkable illustration of the latter is the ongoing controversy and complexity in deciphering the function of the hematopoietic-related stem cell marker, the sialomucin CD34, in both healthy and leukemic contexts. While the CD34 expression pattern in HSC displays similarities between human and mice, its spatio-temporal regulation in the BM strongly differs and is somehow related to distinct functional stem cell subsets.64 For example, interactions between HSC and their regulating stromal BM niche, partly mediated by CD34 binding to selectin, could impact different mechanisms between the two species. Therefore, deciphering the importance of this adhesion molecule in controlling HSC or their leukemic counterparts could be facilitated by the new 3D-model that we propose herein. Indeed, using either CD34-expressing cell lines or primary sorted CD34+ cells, this system offers multiparametric analyses such as in situ cell identification, localization and functional properties (cell cycle, epigenetic modification), and also viable cell harvest. This latter outcome provides the opportunity to analyze the consequences of several days of exposure to a human BM-like microenvironment using multiple readouts (such as flow cytometry, molecular analysis or functional assays), and to broaden our perspectives when studying hematological and non-hematological cells in the BM context. We then achieved the development of 3D culture systems with human cells that secrete most of non-cellular components of the matrix provides the advantage of being able to be implemented with different cell types, which models involving hydrogels do not allow.
Indeed, this system has the advantage of remaining partially open and potentially scalable. For instance, sequential addition of adipocytic or chondrocytic cells derived from the same MSC cell line or a diversity of different cell types can be added depending on the issues that will be addressed. However, this aspect is constrained by the ability of each cell type to be cultivated in a comon cell culture medium.
Clinical applications of this model are wide ranging and it could address issues in normal hematopoiesis or pathological contexts such as infection by pathogens, immune response, pre-neoplastic stages, leukemia, multiple myelomas, osteosarcoma or bone metastasis.65–68 Importantly, we were also able to reproduce the specific impact of leukemic cells on in situ collagen production as reported in the bone marrow of AML patients, conversely to normal hematopoietic cells.55,69 Replacing each cell type by primary cells, normal or pathological, or adding other cell types (like immune cells, adipocytes or fibroblasts) will increase biomimetic properties and complexity of the system in order to achieve a better representativeness of the human BM. This will in turn facilitate investigations into a broad range of issues on either non-stromal or stromal cellular components and matrix-related aspects, such as physical properties, and could be performed in the context of a local BM inflammation or to explore resistance mechanisms or events to test innovative therapeutic strategies including immunotherapy. Lastly, this standardized BM 3D system also appears to be suitable to address current issues on the specific relationships between cancer cells disseminated from solid tumors toward the BM microenvironment. In fact, the “seed and soil” concept, known for decades and now formally demonstrated, strongly relies on the microenvironment at all steps of cancer progression.70,71 This includes initiation of cell transformation72 such as in donor-induced leukemia73 or breast cancer,74 and likely involves different mechanisms21,75 even cell reprogramming.76 For instance, we illustrate herein that this 3D-BM model reliably reproduces metastatic breast cancer cell bone remodeling, since the highly metastatic MDA-MB231 cells were able to degrade the matrix in situ as shown by collagen type 1 and III quantification and unlike T47D model, a less aggressive breast cancer cell line.
Conclusions
Our study delivers to the community, an easy-to-implement standardized microphysiological system of the human BM, approaching the complexity of the in vivo structure. This model is unique, flexible and innovative. As it is based on histological analyses and/or cell recovery, this human 3D bone marrow model is pertinent to finely study a broad range of normal and pathological events taking place in the BM. This also includes the assessment of the importance of the BM microenvironment like matrix parameters as it integrates a minimal level of complexity and heterogeneity of BM cell types and reproduces a matrix very similar to that found in human long bones.Author contributions
T. V. and S. L. designed and performed the project and experiments, provided financial support, analyzed the data and wrote the manuscript. G. F., M. D. performed experiments, analyzed the data and participated in writing the manuscript. K. G., I. M., N. G., S. J. and M. B. collaborated on some experiments and data analysis. A. A., M. C. L. B. K. and V. T. collaborated on scientific discussions and in writing the manuscript. V. M. S. designed the project and experiments, analyzed the data, provided administrative and financial support and wrote the manuscript.Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
The authors are grateful to Nathalie Rochet who provided BCP particles, numerus and useful advices. We thank P. Battiston-Montagne and C. Vanbelle, CRCL-PIC cytometry and imaging platform; J. Valantin and C. Le Nevé, Research Pathology Platform, Centre Léon Bérard and B. Manship for the English edition. This work was funded by the association Ligue contre le Cancer (Comités du Puy de Dôme, Drôme, Loire, Saone et Loire and Rhone) to T. V. and V. M.-S. ALTE-SMP; FI-LMC, “Fondation de France” 2014-0047501 and 2017-00076282/Fondation Ramona Ehrman Amador and Agence Nationale de la Recherche: ANR : Institut Convergence PLAsCAN, ANR-17-CONV-0002 grants to V. M. S. and S. L. K. G. was funded on a INCA-ARC-Ligue-11905 grant to V. M.-S. on BM-SC characterization. G. F. and M. D. were funded by Agence Régionale de Santé Rhône-Alpes Auvergne.References
- L. A. Low, C. Mummery, B. R. Berridge, C. P. Austin and D. A. Tagle, Nat. Rev. Drug Discovery, 2021, 20(5), 345–361 CrossRef CAS PubMed.
- D. E. Ingber, Adv. Sci., 2020, 7, 2002030 CrossRef CAS PubMed.
- A. Schnerch, C. Cerdan and M. Bhatia, Stem Cells, 2010, 28, 419–430 CrossRef PubMed.
- D. Huh, H. J. Kim, J. P. Fraser, D. E. Shea, M. Khan, A. Bahinski, G. A. Hamilton and D. E. Ingber, Nat. Protoc., 2013, 8, 2135–2157 CrossRef CAS PubMed.
- G. S. Travlos, Toxicol. Pathol., 2006, 34, 566–598 CrossRef PubMed.
- G. S. Travlos, Toxicol. Pathol., 2006, 34, 548–565 CrossRef PubMed.
- I. Blazsek, X. H. Liu, A. Anjo, P. Quittet, M. Comisso, B. Kim-Triana and J. L. Misset, Exp. Hematol., 1995, 23, 309–319 CAS.
- A. Janel, J. Berger, C. Bourgne, R. Lemal, N. Boiret-Dupre, F. Dubois-Galopin, P. Dechelotte, C. Bothorel, E. Hermet, S. Chabi, J. O. Bay, C. Lambert, B. Pereira, F. Pflumio, R. Haddad and M. G. Berger, Am. J. Hematol., 2017, 92, 1020–1031 CrossRef CAS PubMed.
- A. N. Tikhonova, I. Dolgalev, H. Hu, K. K. Sivaraj, E. Hoxha, A. Cuesta-Dominguez, S. Pinho, I. Akhmetzyanova, J. Gao, M. Witkowski, M. Guillamot, M. C. Gutkin, Y. Zhang, C. Marier, C. Diefenbach, S. Kousteni, A. Heguy, H. Zhong, D. R. Fooksman, J. M. Butler, A. Economides, P. S. Frenette, R. H. Adams, R. Satija, A. Tsirigos and I. Aifantis, Nature, 2019, 569, 222–228 CrossRef CAS PubMed.
- G. Tjin, E. Flores-Figueroa, D. Duarte, L. Straszkowski, M. Scott, R. A. Khorshed, L. E. Purton and C. Lo Celso, Bone, 2019, 119, 19–35 CrossRef CAS PubMed.
- A. Batsivari, M. L. R. Haltalli, D. Passaro, C. Pospori, C. Lo Celso and D. Bonnet, Nat. Cell Biol., 2020, 22, 7–17 CrossRef CAS PubMed.
- S. J. Morrison and D. T. Scadden, Nature, 2014, 505, 327–334 CrossRef CAS PubMed.
- T. Badar, D. R. Handisides, J. M. Benito, M. A. Richie, G. Borthakur, E. Jabbour, K. Harutyunyan, S. Konoplev, S. Faderl, S. Kroll, M. Andreeff, T. Pearce, H. M. Kantarjian, J. E. Cortes, D. A. Thomas and M. Konopleva, Am. J. Hematol., 2016, 91, 800–805 CrossRef CAS PubMed.
- T. M. Cooper, E. A. R. Sison, S. D. Baker, L. Li, A. Ahmed, T. Trippett, L. Gore, M. E. Macy, A. Narendran, K. August, M. J. Absalon, J. Boklan, J. Pollard, D. Magoon and P. A. Brown, Pediatr. Blood Cancer, 2017, 64, e26414 CrossRef PubMed.
- T. R. Cox, J. T. Erler and R. M. H. Rumney, Calcif. Tissue Int., 2018, 102, 163–173 CrossRef CAS PubMed.
- S. Jeanpierre, K. Arizkane, S. Thongjuea, E. Grockowiak, K. Geistlich, L. Barral, T. Voeltzel, A. Guillemin, S. Gonin-Giraud, O. Gandrillon, F. E. Nicolini, A. J. Mead, V. Maguer-Satta and S. Lefort, Haematologica, 2021, 106(1), 111–122 CrossRef CAS PubMed.
- A. L. MacLean, M. A. Smith, J. Liepe, A. Sim, R. Khorshed, N. M. Rashidi, N. Scherf, A. Krinner, I. Roeder, C. Lo Celso and M. P. H. Stumpf, Stem Cells, 2017, 35, 2292–2304 CrossRef CAS PubMed.
- A. Giustacchini, S. Thongjuea, N. Barkas, P. S. Woll, B. J. Povinelli, C. A. G. Booth, P. Sopp, R. Norfo, A. Rodriguez-Meira, N. Ashley, L. Jamieson, P. Vyas, K. Anderson, A. Segerstolpe, H. Qian, U. Olsson-Stromberg, S. Mustjoki, R. Sandberg, S. E. W. Jacobsen and A. J. Mead, Nat. Med., 2017, 23, 692–702 CrossRef CAS PubMed.
- N. Linde, G. Fluegen and J. A. Aguirre-Ghiso, Adv. Cancer Res., 2016, 132, 45–71 CrossRef CAS PubMed.
- M. Ono, N. Kosaka, N. Tominaga, Y. Yoshioka, F. Takeshita, R. U. Takahashi, M. Yoshida, H. Tsuda, K. Tamura and T. Ochiya, Sci. Signaling, 2014, 7, ra63 CrossRef PubMed.
- E. Risson, A. R. Nobre, V. Maguer-Satta and J. A. Aguirre-Ghiso, Nat. Cancer, 2020 DOI:10.1038/s43018-020-0088-5.
- P. Avril, L. R. Le Nail, M. A. Brennan, P. Rosset, G. De Pinieux, P. Layrolle, D. Heymann, P. Perrot and V. Trichet, J. Bone Oncol., 2016, 5, 5–14 CrossRef PubMed.
- C. Liu, Y. Sun and Z. Shao, Curr. Pharm. Des., 2019, 25, 236–241 CrossRef CAS PubMed.
- P. Perrot, J. Rousseau, A. L. Bouffaut, F. Redini, E. Cassagnau, F. Deschaseaux, M. F. Heymann, D. Heymann, F. Duteille, V. Trichet and F. Gouin, PLoS One, 2010, 5, e10999 CrossRef PubMed.
- D. B. Widner, S. H. Park, M. R. Eber and Y. Shiozawa, Curr. Osteoporos. Rep., 2018, 16, 596–602 CrossRef PubMed.
- D. B. Chou, V. Frismantas, Y. Milton, R. David, P. Pop-Damkov, D. Ferguson, A. MacDonald, O. Vargel Bolukbasi, C. E. Joyce, L. S. Moreira Teixeira, A. Rech, A. Jiang, E. Calamari, S. Jalili-Firoozinezhad, B. A. Furlong, L. R. O'Sullivan, C. F. Ng, Y. Choe, S. Marquez, K. C. Myers, O. K. Weinberg, R. P. Hasserjian, R. Novak, O. Levy, R. Prantil-Baun, C. D. Novina, A. Shimamura, L. Ewart and D. E. Ingber, Nat. Biomed. Eng., 2020, 4, 394–406 CrossRef PubMed.
- A. Marturano-Kruik, M. M. Nava, K. Yeager, A. Chramiec, L. Hao, S. Robinson, E. Guo, M. T. Raimondi and G. Vunjak-Novakovic, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 1256–1261 CrossRef CAS PubMed.
- A. B. Bello, H. Park and S. H. Lee, Acta Biomater., 2018, 72, 1–15 CrossRef CAS PubMed.
- D. Discher, C. Dong, J. J. Fredberg, F. Guilak, D. Ingber, P. Janmey, R. D. Kamm, G. W. Schmid-Schonbein and S. Weinbaum, Ann. Biomed. Eng., 2009, 37, 847–859 CrossRef PubMed.
- M. A. Brennan, A. Renaud, A. L. Gamblin, C. D'Arros, S. Nedellec, V. Trichet and P. Layrolle, Biomed. Mater., 2015, 10, 045019 CrossRef PubMed.
- A. L. Gamblin, A. Renaud, C. Charrier, P. Hulin, G. Louarn, D. Heymann, V. Trichet and P. Layrolle, Acta Biomater., 2014, 10, 5139–5147 CrossRef CAS PubMed.
- A. Raic, L. Rodling, H. Kalbacher and C. Lee-Thedieck, Biomaterials, 2014, 35, 929–940 CrossRef CAS PubMed.
- M. A. Walasek, R. van Os and G. de Haan, Ann. N. Y. Acad. Sci., 2012, 1266, 138–150 CrossRef CAS PubMed.
- S. S. Kotha, B. J. Hayes, K. T. Phong, M. A. Redd, K. Bomsztyk, A. Ramakrishnan, B. Torok-Storb and Y. Zheng, Stem Cell Res. Ther., 2018, 9, 77 CrossRef CAS PubMed.
- F. Boukhechba, T. Balaguer, J. F. Michiels, K. Ackermann, D. Quincey, J. M. Bouler, W. Pyerin, G. F. Carle and N. Rochet, J. Bone Miner. Res., 2009, 24, 1927–1935 CrossRef CAS PubMed.
- Y. Li, T. Jiang, L. Zheng and J. Zhao, Mater. Sci. Eng., C, 2017, 80, 296–300 CrossRef CAS PubMed.
- B. A. Roecklein and B. Torok-Storb, Blood, 1995, 85, 997–1005 CrossRef CAS PubMed.
- M. Iwata, R. S. Sandstrom, J. J. Delrow, J. A. Stamatoyannopoulos and B. Torok-Storb, Stem Cells Dev., 2014, 23, 729–740 CrossRef CAS PubMed.
- S. Vallet, S. Pozzi, K. Patel, N. Vaghela, M. T. Fulciniti, P. Veiby, T. Hideshima, L. Santo, D. Cirstea, D. T. Scadden, K. C. Anderson and N. Raje, Leukemia, 2011, 25, 1174–1181 CrossRef CAS PubMed.
- L. Li, L. A. Milner, Y. Deng, M. Iwata, A. Banta, L. Graf, S. Marcovina, C. Friedman, B. J. Trask, L. Hood and B. Torok-Storb, Immunity, 1998, 8, 43–55 CrossRef CAS PubMed.
- B. Torok-Storb, M. Iwata, L. Graf, J. Gianotti, H. Horton and M. C. Byrne, Ann. N. Y. Acad. Sci., 1999, 872, 164–170 CrossRef CAS PubMed.
- E. W. Ades, F. J. Candal, R. A. Swerlick, V. G. George, S. Summers, D. C. Bosse and T. J. Lawley, J. Invest. Dermatol., 1992, 99, 683–690 CrossRef CAS PubMed.
- A. Sakaue-Sawano, K. Ohtawa, H. Hama, M. Kawano, M. Ogawa and A. Miyawaki, Chem. Biol., 2008, 15, 1243–1248 CrossRef CAS PubMed.
- J. Goedhart, D. von Stetten, M. Noirclerc-Savoye, M. Lelimousin, L. Joosen, M. A. Hink, L. van Weeren, T. W. Gadella Jr. and A. Royant, Nat. Commun., 2012, 3, 751 CrossRef PubMed.
- B. Laperrousaz, S. Jeanpierre, K. Sagorny, T. Voeltzel, S. Ramas, B. Kaniewski, M. Ffrench, S. Salesse, F. E. Nicolini and V. Maguer-Satta, Blood, 2013, 122, 3767–3777 CrossRef CAS PubMed.
- S. Jeanpierre, F. E. Nicolini, B. Kaniewski, C. Dumontet, R. Rimokh, A. Puisieux and V. Maguer-Satta, Blood, 2008, 112, 3154–3163 CrossRef CAS PubMed.
- J. Lee, S. Kang, K. C. Lilja, K. J. Colletier, C. J. Scheitz, Y. V. Zhang and T. Tumbar, Nat. Commun., 2016, 7, 11278 CrossRef CAS PubMed.
- T. H. Ambrosi, M. T. Longaker and C. K. F. Chan, Front. Cell Dev. Biol., 2019, 7, 189 CrossRef PubMed.
- H. J. Jin, S. K. Park, W. Oh, Y. S. Yang, S. W. Kim and S. J. Choi, Biochem. Biophys. Res. Commun., 2009, 381, 676–681 CrossRef CAS PubMed.
- T. Picot, C. M. Aanei, A. Fayard, P. Flandrin-Gresta, S. Tondeur, M. Gouttenoire, E. Tavernier-Tardy, E. Wattel, D. Guyotat and L. Campos, Tumor Biol., 2017, 39, 1010428317716629 Search PubMed.
- E. O'Reilly, H. A. Zeinabad and E. Szegezdi, Blood Rev., 2021, 100850, DOI:10.1016/j.blre.2021.100850.
- R. M. Neve, K. Chin, J. Fridlyand, J. Yeh, F. L. Baehner, T. Fevr, L. Clark, N. Bayani, J. P. Coppe, F. Tong, T. Speed, P. T. Spellman, S. DeVries, A. Lapuk, N. J. Wang, W. L. Kuo, J. L. Stilwell, D. Pinkel, D. G. Albertson, F. M. Waldman, F. McCormick, R. B. Dickson, M. D. Johnson, M. Lippman, S. Ethier, A. Gazdar and J. W. Gray, Cancer Cell, 2006, 10, 515–527 CrossRef CAS PubMed.
- M. Balduyck, F. Zerimech, V. Gouyer, R. Lemaire, B. Hemon, G. Grard, C. Thiebaut, V. Lemaire, E. Dacquembronne, T. Duhem, A. Lebrun, M. J. Dejonghe and G. Huet, Clin. Exp. Metastasis, 2000, 18, 171–178 CrossRef CAS PubMed.
- D. Zhao, L. Zhang, M. Zhang, B. Xia, J. Lv, X. Gao, G. Wang, Q. Meng, Y. Yi, S. Zhu, A. S. Tomoiaga, M. G. Lee, J. P. Cooke, Q. Cao and K. Chen, Nat. Commun., 2020, 11, 5560 CrossRef CAS PubMed.
- A. A. Zahr, M. E. Salama, N. Carreau, D. Tremblay, S. Verstovsek, R. Mesa, R. Hoffman and J. Mascarenhas, Haematologica, 2016, 101, 660–671 CrossRef PubMed.
- Y. Han, X. Li, Y. Zhang, Y. Han, F. Chang and J. Ding, Cells, 2019, 8(8), 886 CrossRef CAS PubMed.
- H. Le, W. Xu, X. Zhuang, F. Chang, Y. Wang and J. Ding, J. Tissue Eng., 2020, 11, 2041731420943839 Search PubMed.
- M. Galan-Diez and S. Kousteni, Curr. Mol. Biol. Rep., 2017, 3, 53–62 CrossRef PubMed.
- J. Holst, S. Watson, M. S. Lord, S. S. Eamegdool, D. V. Bax, L. B. Nivison-Smith, A. Kondyurin, L. Ma, A. F. Oberhauser, A. S. Weiss and J. E. Rasko, Nat. Biotechnol., 2010, 28, 1123–1128 CrossRef CAS PubMed.
- P. Zhang, C. Zhang, J. Li, J. Han, X. Liu and H. Yang, Stem Cell Res. Ther., 2019, 10, 327 CrossRef PubMed.
- L. Behrmann, J. Wellbrock and W. Fiedler, Front. Oncol., 2018, 8, 444 CrossRef PubMed.
- C. R. Cogle, D. C. Goldman, G. J. Madlambayan, R. P. Leon, A. A. Masri, H. A. Clark, S. A. Asbaghi, J. W. Tyner, J. Dunlap, G. Fan, T. Kovacsovics, Q. Liu, A. Meacham, K. L. Hamlin, R. A. Hromas, E. W. Scott and W. H. Fleming, Leukemia, 2014, 28, 1978–1987 CrossRef PubMed.
- A. Sobrino, D. T. Phan, R. Datta, X. Wang, S. J. Hachey, M. Romero-Lopez, E. Gratton, A. P. Lee, S. C. George and C. C. Hughes, Sci. Rep., 2016, 6, 31589 CrossRef CAS PubMed.
- M. R. Hughes, D. Canals Hernaez, J. Cait, I. Refaeli, B. C. Lo, C. D. Roskelley and K. M. McNagny, Exp. Hematol., 2020, 86, 1–14 CrossRef CAS PubMed.
- J. Aleman, S. K. George, S. Herberg, M. Devarasetty, C. D. Porada, A. Skardal and G. Almeida-Porada, Small, 2019, 15, e1902971 CrossRef PubMed.
- M. Pavlou, M. Shah, P. Gikas, T. Briggs, S. J. Roberts and U. Cheema, Acta Biomater., 2019, 96, 247–257 CrossRef CAS PubMed.
- P. de la Puente, B. Muz, R. C. Gilson, F. Azab, M. Luderer, J. King, S. Achilefu, R. Vij and A. K. Azab, Biomaterials, 2015, 73, 70–84 CrossRef CAS PubMed.
- D. N. Tavakol, J. Tratwal, F. Bonini, M. Genta, V. Campos, P. Burch, S. Hoehnel, A. Beduer, M. Alessandrini, O. Naveiras and T. Braschler, Biomaterials, 2020, 232, 119665 CrossRef CAS PubMed.
- A. J. Favreau, C. P. Vary, P. C. Brooks and P. Sathyanarayana, Cancer Med., 2014, 3, 265–272 CrossRef CAS PubMed.
- M. Akhtar, A. Haider, S. Rashid and A. Al-Nabet, Adv. Anat. Pathol., 2019, 26, 69–74 CrossRef CAS PubMed.
- s. Paget, Lancet, 1889, 133, 571–573 CrossRef.
- A. Kode, J. S. Manavalan, I. Mosialou, G. Bhagat, C. V. Rathinam, N. Luo, H. Khiabanian, A. Lee, V. V. Murty, R. Friedman, A. Brum, D. Park, N. Galili, S. Mukherjee, J. Teruya-Feldstein, A. Raza, R. Rabadan, E. Berman and S. Kousteni, Nature, 2014, 506, 240–244 CrossRef CAS PubMed.
- C. M. Flynn and D. S. Kaufman, Blood, 2007, 109, 2688–2692 CrossRef CAS PubMed.
- M. Chapellier, E. Bachelard-Cascales, X. Schmidt, F. Clement, I. Treilleux, E. Delay, A. Jammot, C. Menetrier-Caux, G. Pochon, R. Besancon, T. Voeltzel, C. Caron de Fromentel, C. Caux, J. Y. Blay, R. Iggo and V. Maguer-Satta, Stem Cell Rep., 2015, 4, 239–254 CrossRef CAS PubMed.
- V. Maguer-satta, in Cancer stem cells, ed. S. Stanley, Intech, 2011, vol. Intech, pp. 215–240 Search PubMed, http://www.intechopen.com/articles/show/title/the-stem-cell-niche-the-black-master.
- T. Voeltzel, M. Flores-Violante, F. Zylbersztejn, S. Lefort, M. Billandon, S. Jeanpierre, S. Joly, G. Fossard, M. Milenkov, F. Mazurier, A. Nehme, A. Belhabri, E. Paubelle, X. Thomas, M. Michallet, F. Louache, F. E. Nicolini, C. Caron de Fromentel and V. Maguer-Satta, Cell Death Dis., 2018, 9, 1011 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 6 figures. See DOI: 10.1039/d1bm01098k |
| This journal is © The Royal Society of Chemistry 2022 |