
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle sample preparation for the simultaneous extraction of drugs, pharmaceuticals, cannabinoids and endogenous steroids in hair†
Clementine
Scholz
a,
Markus R.
Baumgartner
 a,
Thomas
Kraemer
b and
Tina M.
Binz
*a
a,
Thomas
Kraemer
b and
Tina M.
Binz
*a
aCenter for Forensic Hair Analysis, Zurich Institute of Forensic Medicine, University of Zurich, Kurvenstrasse, 17 8006 Zürich, Switzerland. E-mail: TinaMaria.Binz@irm.uzh.ch
bDepartment of Forensic Pharmacology and Toxicology, Zurich Institute of Forensic Medicine, University of Zurich, Zurich, Switzerland
First published on 24th October 2022
Abstract
Recently, we published a multi-analyte method for the simultaneous analysis of 116 drugs and pharmaceuticals including different substance groups like opioids, stimulants, benzodiazepines, z-drugs, antidepressants and neuroleptics based on a single sample workup followed by a single analytical measurement with LC-MS/MS. However, in some cases, additional analysis of further substance groups, such as cannabinoids and endogenous steroids, is required, which are analyzed in our laboratory using separate sample preparation and separate analytical methods. The goal of this study was to use the knowledge from the different sample preparations and combine them into a single sample preparation and extraction workflow for the simultaneous extraction of drugs, pharmaceuticals, cannabinoids, and endogenous steroids to be analyzed with the appropriate analytical methods. A partial validation of selected parameters such as selectivity, linearity, limit of quantification (LOQ), accuracy, precision and robustness for the different analytical methods was carried out and revalidated. In addition, comparative measurements of quality controls and authentic pools were performed and statistically evaluated using the unpaired t-test or the non-parametric Mann–Whitney test. The results using the newly established sample preparation and extraction were in good agreement with the original data. In conclusion, the newly established sample preparation is suitable for the combined extraction of drugs, pharmaceuticals, cannabinoids and endogenous steroids, and gives reliable results for quantification of various substances.
Introduction
A challenge in hair analysis is the often limited amount of hair sample while the analysis of a large number of structurally different substances within different concentration ranges is required. Therefore, single sample extraction protocols and multi-analyte based LC-MS/MS methods containing various groups of substances are the method of choice to approach this challenge. Drugs, pharmaceuticals and their metabolites are incorporated into the hair matrix after consumption. Trapped in hair, these compounds exhibit long-term stability. Therefore, hair allows for a prolonged retrospective detection window of exposition, making hair analysis useful for forensic questions such as e.g. abstinence controls, workplace drug testing, and custody cases. In addition to the long detection window, head hair has the advantage of enabling a temporal resolution of the consumption behavior based on the segments of hair that are analyzed, whereas 1 cm of head hair represents approximately 1 month (e.g.1 and 2). Sample preparation in hair analysis for the analysis of drugs and pharmaceuticals usually involves a tedious, multi-step procedure. First, a wash procedure has to be applied to eliminate possible external contamination. Published washing procedures usually include organic solvents, aqueous buffers, water, soaps or a combination (e.g.3–7). Decontamination processes take typically between 2 and 10 min (each washing) and the number of cleaning steps varies. The next step in the procedure is the sample extraction. Freeing the analytes bound within the hair matrix is commonly achieved either by acidic or alkaline digestion, enzymatic hydrolysis, or incubation of hair snippets or powder with organic solvents and/or various buffer systems.8 The choice of the extraction conditions depends on the chemical properties of the analytes of interest and is known to have a major impact on the performance of a method.9 In order to clean-up the hair extracts, a pre-concentration step may be included such as e.g. liquid–liquid extraction (LLE) or conventional solid-phase extraction (SPE). Alternatively, direct infusion of diluted extracts may be used in order to simplify sample preparation for high through-put analyses. For the analysis of the extracts, liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) using electrospray ionization (ESI) or atmospheric pressure chemical ionization (APCI) is nowadays widely used in hair analysis.8 Hair testing can also be applied to detect cannabinoids.10,11 However, several limitations are known for the detection of cannabis use in hair. Hair analysis often lacks sensitivity which may lead to false-negative results11,12 and external contamination of hair via hands of cannabis users, sebum/sweat or cannabis smoke has to be taken into account.3,13,14 Despite these limitations, hair testing can be a valuable tool to confirm exposure with cannabis products. The analysis of steroid hormones such as cortisol, cortisone and testosterone in the keratinized matrix hair is used to monitor long-term stress and is applied e.g. in psychoneuroendocrinological stress research (e.g.15–19). However, for endogenous compounds such as steroid hormones, it is often difficult or even impossible to obtain analyte-free authentic matrix which is required for calibration and quality control preparation. The classical approach to circumvent this problem is the use of standard addition,20 but this requires a large amount of sample material, which is usually not available in clinical and forensic hair studies. Other approaches include the use of a surrogate matrix (e.g. synthetic melanin21) or the use of hair containing a low amount of endogenous analytes (e.g. tips from long hair22,23). In our laboratory, the method of choice is the use of surrogate analytes, which are usually stable isotope labeled forms of the original analyte.15,24–28For our routine casework, we use a multi-analyte approach for the analysis of 116 drugs and pharmaceuticals.29 Cannabinoids and endogenous steroids are detected using separate methods.24,27,30 Due to the different sample preparation procedures, the amount of required hair sample increases significantly in those cases where the detection of cannabinoids and endogenous steroids is requested besides drugs and pharmaceuticals. Additionally, segmental analysis of hair is typically performed to monitor changes in consumption patterns within smaller time windows (e.g.31–35) further increasing the demand for sample material. To ensure that the available sample amount suffices, we developed a sample preparation for the combined extraction of drugs, pharmaceuticals, cannabinoids and endogenous steroids introducing several modifications compared to the original methods. These modifications include the type of homogenization (snippets instead of powder) in case of the analysis of drugs and pharmaceuticals and endogenous steroids, the number of deuterated standards added to the hair prior to extraction, and the extraction procedure (e.g. ball mill instead of ultra-sonication) in the case of endogenous steroids. In this study, we present the data of additional experiments to ensure the applicability of the herein established combined sample workup. Eventually, the simultaneous sample preparation for all these substances may additionally reduce cost and required time for hair analysis.
Experimental
Chemicals, reagents
Methanolic or acetonitrilic solutions of the standards and deuterated standards were purchased from Lipomed (Arlesheim, Switzerland) or Cerilliant (delivered by ReseaChem GmbH, Burgdorf, Switzerland) or ElSohly Laboratories, Inc. (Oxford, MS, USA) as previously reported.24,27,29,30 Water for decontamination of hair samples was processed by a PURELAB Option-Q system by ELGA LabWater (Labtec Services AG, Villmergen, Switzerland). Acetone (p.a., ≥99.5%), acetonitrile (LC-MS grade), formic acid (p.a., ≥98%), ammonium formate (≥99%), hexane (p.a., ≥99%), methanol (LC-MS grade or p.a., ≥99.8%), and water (LC-MS grade or p.a., ≥99.8%) were purchased from Sigma-Aldrich (St Louis, MO, USA).Preparation of internal standard solution (IScombi), calibrators and quality control
A stock solution containing internal standards (IScombi) was prepared. According to the multi-analyte approach for drugs and pharmaceuticals, it contained 43 deuterated standards with a concentration of 0.04 or 0.2 μg ml−1 depending on the corresponding IS group (1 or 2).29 For the herein described combined sample workup, the IScombi solution additionally contained 0.6 μg per ml THC-D3 and 0.02 μg per ml cortisone-D7. Calibrators were prepared using pooled drug free scalp hair which were tested negative using the presented LC-MS/MS methods.27,29,30 The calibration curve was prepared by adding calibrator solutions and 100 μl of the IScombi solution into approx. 20 mg of drug free hair. Further, a quality control was prepared by adding 100 μl of the IScombi solution into approx. 20 mg of a pooled drug-positive hair sample.Sample preparation
The sample preparation procedure was performed according to the fully validated routine methodologies27,29,30 including several alterations as shown in Fig. 1. Briefly, hair samples were successively washed, dried, and chopped into snippets. Approximately 20 mg of the snippets were exactly weighed into an Eppendorf tube. The first extraction was achieved by shaking the hair sample in the presence of one tungsten carbide ball (Ø 7 mm, 3 g, Retsch) in 1.4 ml of methanol and 0.1 ml of IScombi solution in the ball mill (Type MM 400, Retsch GmbH & Co. KG, Haan, Germany) at 10 Hz for 90 min. After centrifugation, 75 μl of the supernatant were transferred into an LC-vial with a silanized glass inlet (Thermo Scientific, Waltham, MA) and 75 μl of a solution of 2 mM aqueous ammonium formate were added prior to the analysis of cannabinoids by LC-MS/MS. The remaining supernatant of the first extraction was collected and dried at 35 °C under nitrogen.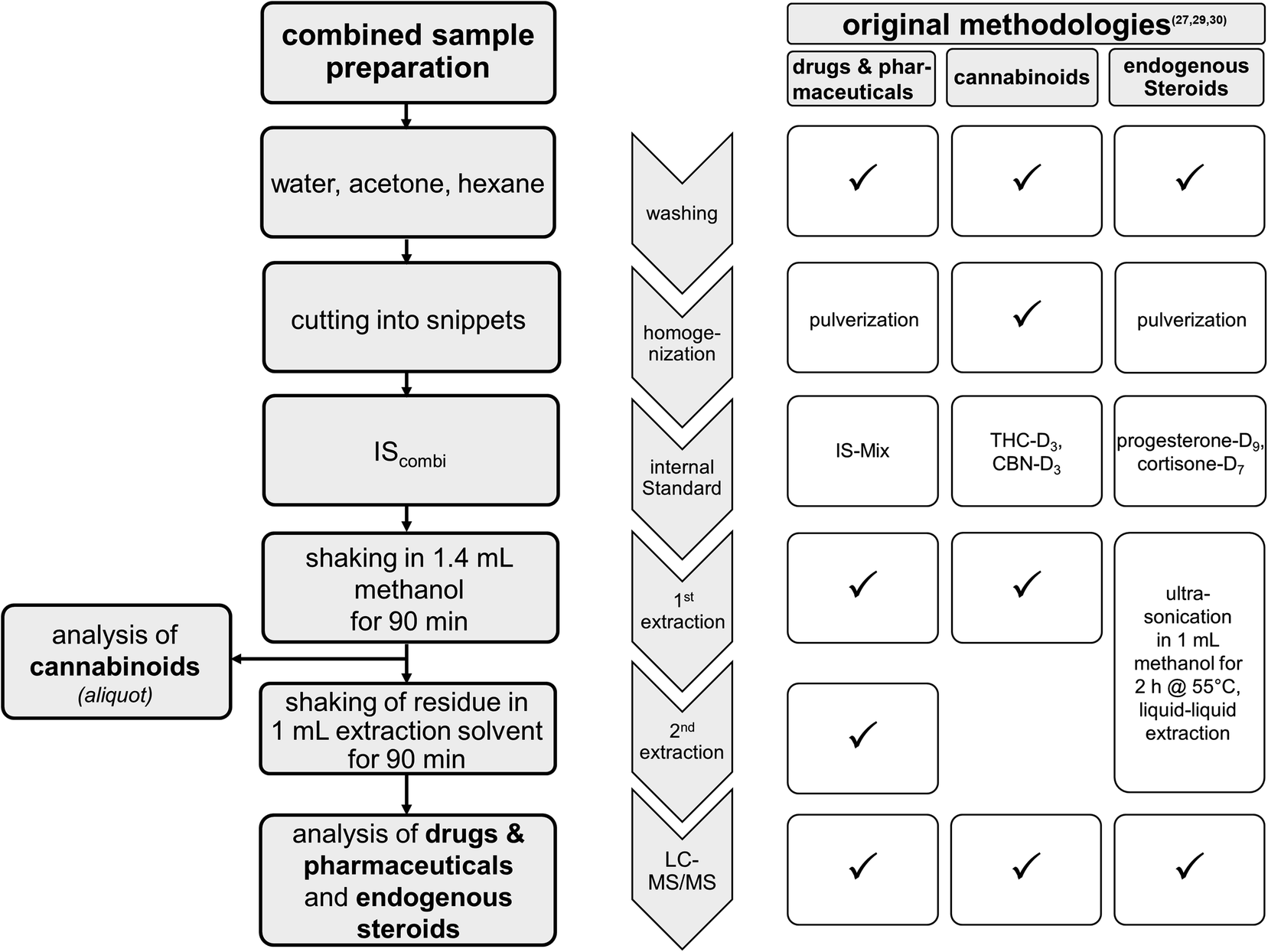 | ||
| Fig. 1 Scheme for the combined sample workup established for the analysis of drugs and pharmaceuticals, cannabinoids and endogenous steroids. Information on modifications compared to the original methods is given in boxes on the right-hand side, with “✓” indicating no modification (IS = Internal Standard; IS-Mix = mixture containing 43 deuterated drugs and pharmaceuticals as previously published,29 IScombi = IS-Mix plus THC-D3, CBN-D3 and cortisone-D7). | ||
For the second micro-extraction step, 1 ml of extraction solvent containing 1 mM aqueous ammonium formate containing 0.1% formic acid/methanol (1/1, v/v) was added to the remaining hair snippets followed by shaking at 10 Hz for 90 min in the ball mill. The resulting supernatant was combined with the dried supernatant of the first step and dried at 35 °C under nitrogen. The dried residues were reconstituted in 150 μl methanol, vortexed and 350 μl of a solution of 2 mM aqueous ammonium formate was added. The solutions were measured by LC-MS/MS for the detection of endogenous steroids and drugs and pharmaceuticals.
Analysis of THC, CBN, CBD
Ten microliters of the diluted first extract were injected into an LC-MS/MS system consisting of a Shimadzu Prominence high performance liquid chromatography system (Shimadzu, Duisburg, Germany) and a QTrap 5500 mass spectrometer (Sciex, Darmstadt, Germany). Separation was achieved as previously described.30 Briefly, a Kinetex® C18 column (100 mm × 2.1 mm, 100 Å, 1.7 μm, Phenomenex) was operated at a flow rate of 0.5 mL min−1 with a total run time of 15 min. A linear gradient consisting of a mobile phase A (water containing ammonium formate [1 mM] and formic acid [0.1%]) and mobile phase B (acetonitrile containing ammonium formate [1 mM] and formic acid [1 mM]) was used. The gradient was programmed as follows: 0.01–1.5 min, 40% eluent B; 1.5–1.6 min increasing to 60% eluent B; 1.6–10 min increasing to 65% eluent B; 10–11 min, 95% eluent B; 11–12 min hold at 95% eluent B; 12–12.01 min decreasing to 40% eluent B, 12.01–15 min starting conditions (40% eluent B). The MS instrument was operated using Atmospheric Pressure Chemical Ionization (APCI) in positive mode. The samples were acquired in the “scheduled Multiple Reaction Monitoring” (sMRM) using MS parameters for THC, CBN, CBD, tetrahydrocannabinol-D3 (THC-D3) and cannabinol-D3 (CBD-D3) as previously published.30Analysis of drugs and pharmaceuticals (multi-analyte approach)
10 μL of the combined extracts were injected into an LC-MS/MS system consisting of a Shimadzu Prominence high performance liquid chromatography system (Shimadzu, Duisburg, Germany) and a QTrap 5500 mass spectrometer (Sciex, Darmstadt, Germany). Separation was achieved as previously described.29 Briefly, a Kinetex® F5 column (100 mm × 2.1 mm, 100 Å, 2.6 μm, Phenomenex) was operated at a flow rate was set at 0.6 mL min−1. A linear gradient consisting of a mobile phase A consisting of mobile phase A (water containing ammonium formate [1 mM] and formic acid [0.1%]) and mobile phase B (acetonitrile containing ammonium formate [1 mM] and formic acid [1 mM]) was used. The gradient was programmed as follows: 0.01–1.5 min, 3% eluent B; 1.5–9 min increasing to 60% eluent B; 9–10 min increasing to 95% eluent B; 10–11 min, 95% eluent B; 11–11.1 min decreasing to 3% eluent B, 11.1–12 min starting conditions (3% eluent B). The MS instrument was operated using ESI in positive mode. The samples were acquired in the “Scheduled MRM™ Algorithm Pro” mode using MS parameters for the 116 analytes as previously published.29Analysis of endogenous steroids: cortisol, cortisone and testosterone
10 μL of the combined extracts were injected into an LC-MS/MS system consisting of a Shimadzu Prominence high performance liquid chromatography system (Shimadzu, Duisburg, Germany) and a QTrap 6500+ mass spectrometer (Sciex, Darmstadt, Germany). Separation was achieved as previously described.27 Briefly, a Kinetex® XB-C18 column (50 mm × 2.1 mm, 100 Å, 2.6 μm, Phenomenex) was operated at a flow rate of 0.45 mL min−1. The mobile phase consisted of 0.2 mM NH4F in water/methanol 97/3 v/v (A) and 0.2 mM NH4F in water/methanol 3/97 v/v (B). The gradient was set follows: 0–40% B for 0–0.1 min, 40–50% B from 0.1 to 5 min, isocratic 50% from 5 to 8 min, 50–90% B from 8 to 11 min, isocratic 90% B from 11 to 14 min, 90–40% B from 14 to 15 min followed by an equilibration step of 1 min. For the quantification of steroid hormones in hair, the surrogate analyte approach in adaption to our previous work was used.15,24,26–28 In this approach, 13C3-cortisol, 13C3-cortisone and 13C3-testosterone were used for calibration of steroids. The MS parameters have been published previously.27Validation experiments
Validation has previously been performed for the methods analyzing drugs and pharmaceuticals,29 cannabinoids30 and endogenous steroids.27 In the herein established work, several modifications have been introduced to establish a combined sample preparation (Fig. 1). Therefore, selective parameters such as selectivity, linearity, LOQ and accuracy were revalidated.Linearity and selectivity
Because of the presence of endogenous steroids in blank hair, the surrogate analyte approach was used for quantification of steroids according to our previous work.15,24–27 For calibration, nine calibration levels were used in different concentration ranges that correspond to typically observed concentration levels in hair. The resulting concentrations in hair correspond for drugs and pharmaceuticals to the published values.29 For THC, CBN and CBD, the calibration levels 5, 10, 20, 30, 40, 100, 400, 2000 and 4000 pg mg−1 were used. Additionally, the calibration levels 1.0, 2.5, 5.0, 10, 25, 50, 100, 250 and 500 pg mg−1 were analyzed for 13C3-cortisol and 13C3-cortisone, respectively, and the calibration levels 0.1, 0.25, 0.5, 1.0, 2.5, 5.0, 10, 25 and 50 pg mg−1 were analyzed for 13C3-testosterone, respectively. Two replicates for each calibration level were analyzed. The calibration curve was estimated by least-squares regression procedure. For selectivity, 2 different blank hair samples from non-users were extracted according to the combined sample preparation with and without the addition of IScombi. All samples were analyzed to exclude any interfering signals. As steroid hormones are endogenous compounds which are always present in hair, selectivity was tested for 13C3-cortisol, 13C3-cortisone and 13C3-testosterone in the 2 blank hair samples.Limit of quantification (endogenous steroids)
For endogenous steroids, limits of quantification (LOQ) were determined by analyzing spiked hair samples in the low concentration range. A signal-to-noise ratio equal or greater than 10/1 was considered as valid. The limits of detection (LODs) were not determined.Accuracy, precision and robustness
Two replicates of a QC sample at medium concentration levels and two replicates of an authentic hair pool were measured on 8 days. For the QC sample, the bias (accuracy) was determined by calculating the percent deviation (RSD) of the mean of all calculated concentration values at a specific level from the respective nominal concentration. For the QC sample and the authentic hair sample, the relative standard deviation for time-different intermediate measurements (precision) and within-days (repeatability) were calculated as previously described.36 Robustness of the combined sample preparation was evaluated by calculating the standard deviations for the 16 replicate measurements of the QC sample and the authentic hair sample, respectively.Comparative measurements and statistical analysis
Comparative measurements between the original method and the combined sample preparation were carried out for selective analytes in authentic hair samples and statistically evaluated using Prism 6 (GraphPad Software, CA, USA). Analyzed data sets were either normally or not normally distributed as indicated by the Shapiro–Wilks test. Normal distributions and not normal distributions were analyzed by the unpaired t-test and the non-parametric Mann–Whitney test, respectively. The level of significance was set at p < 0.05.Results and discussion
Among others, three validated methods in hair analysis are routinely used in our laboratory: (1) a multi-analyte approach for the analysis of drugs and pharmaceuticals,29 (2) a method for the analysis of the cannabinoids THC, CBN and CBD30 and (3) a method for the analysis of the endogenous steroids cortisol, cortisone and testosterone.27 For various reasons, cannabinoids and endogenous steroids have not been included in the multi-analyte approach.29 While hair pulverization resulted in better yields for drugs and pharmaceuticals, extraction of cannabinoids from powdered hair was only slightly more efficient. Therefore, cannabinoids are routinely extracted from hair snippets instead. The measurement of cortisol, cortisone and testosterone was validated in our laboratory after extraction from hair powder,27 while the measurement of cortisol and cortisone was also validated for hair snippets.15,24 Further, cannabinoids and steroid hormones are already extracted in sufficient yields after a single methanolic extraction step, whereas the additional extraction step using a mixture of acidic water and methanol is used to enhance extraction yields for several drugs and pharmaceuticals such as e.g. opiates.9In cases for which the analysis of drugs, pharmaceuticals, cannabinoids and endogenous steroids is requested, three independent sample preparations would be required which is associated with a high demand for sample material and further entails a tedious and complex sample workup. Therefore, a strategy for the combined extraction of these substances was established introducing several modifications in the sample preparation process as shown in Fig. 1. Compared to the original methodologies, additional internal standards are used. No further modifications resulted for the sample workup of cannabinoids. Regarding the analysis of drugs and pharmaceuticals, extraction was achieved after pulverization of the hair samples in the original method29 whereas hair snippets are used in the combined sample preparation. In the combined sample preparation and extraction workflow, most of the changes occurred in the analysis of endogenous steroids. Using the combined sample preparation, extraction of endogenous steroids was achieved by shaking hair snippets in 1.4 mL methanol in the presence of a tungsten ball in the ball mill followed by a second extraction step, rather than after ultra-sonication of hair snippets in 1 mL methanol for 2 hours according to the original method.27 Due to these modifications and although each of the methods involved has been fully validated previously, several experiments were performed to test the applicability for the combined sample workup. The results of these experiments are presented in the following.
Linearity and selectivity
Calibration curves were evaluated for both MRM transitions of all analytes as previously reported.27,29,30 The regressions were calculated using a linear model with 1/x weighting. According to the original method for steroid hormones, calibration was carried out for 13C3-cortisol, 13C3-cortisone and 13C3-testosterone.27 For cannabinoids and steroid hormones, regressions were linear which is in agreement with previous data.27,30 For drugs and pharmaceuticals, quadratic or modified quadratic (Wagner) regression models were applied for those analytes which did not fulfill the criteria (regression factor r ≥ 0.98; accuracy ≥ 70% and ≤130%), as reported previously.29Compared to the original methods, additional internal standards were spiked into the hair samples using the combined sample workup (Fig. 1). The use of additional internal standards entails the possibility for new interfering signals. However, the analysis of two blank hair samples from non-users prepared with and without internal standard solution (IScombi) showed that drugs and pharmaceuticals as well as cannabinoids eluted free of interfering peaks. While no interfering peaks for cortisol, cortisone and testosterone were detected in one of the two blank hair samples, the second blank hair still contained endogenous steroids. Therefore, selectivity for 13C3-cortisol, 13C3-cortisone and 13C3-testosterone was tested in this sample and no interfering peaks were detected.
Limit of quantification (LOQ) for endogenous steroids
Compared to the original method for endogenous steroids, a modified extraction procedure has been used in the present approach (Fig. 1). With this combined sample preparation, the limits of quantifications were determined as 1 pg mg−1 for 13C3-cortisol and 13C3-cortisone and 0.3 pg mg−1 for 13C3-testosterone which is in the same range as reported from the original method.27Evaluation of accuracy, precision and robustness
In the present study, accuracy and precision experiments were performed for a QC sample at medium concentration levels for eight days. Additionally, precision was also determined for an authentic hair sample. Recommended acceptance intervals of the bias are 15% and RSD ≤ 15% for the within-days precision (repeatability) and intermediate precision.36 Due to the high complexity of the hair matrix, and the extremely wide dynamic range, acceptance criteria were modified as follows: ±30% RSD for the bias, RSDR ≤ 30%, and RSDT ≤ 30% for the repeatability and the intermediate precision, such as described earlier.29 These criteria were met for the majority of the analytes; deviations are marked in bold (Table 1). The accuracy (bias) for the QC sample was within the allowed range, with exception of tramadol for which the bias was 35%. Notable deviations in the precision were found for methylphenidate in the authentic hair sample, which can be attributed to the high inhomogeneity of the non-powdered hair sample. Taking into account the high complexity of the hair matrix, these results were considered acceptable. The robustness of the combined sample preparation was evaluated by calculating the percent deviation (RSD) of the mean of all 16 replicative measurements of the QC sample and the authentic hair sample. The RSD was for the majority of the analytes within the accepted range of ±30%, and varied between 4.6 to 26% and between 12 to 35% for the QC sample and the authentic hair sample, respectively (Table 1). Overall, these data indicate good robustness of the combined sample preparation.| Analyte | QC sample | Authentic hair sample | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Target value (pg) | Accuracy | Precision | Characteristics | Precision | Characteristics | |||||||
| Intra-day | Inter-day | Intra-day | Inter-day | |||||||||
| Bias [%] | RSDR [%] | RSDT [%] | Mean (pg) | SD (pg) | RSD [%] | RSDR [%] | RSDT [%] | Mean (pg) | SD (pg) | RSD [%] | ||
| a n.d.: not detected. | ||||||||||||
| Morphine | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
3.6 | 8.0 | 8.6 | 10356 |
858 | 8.3 | 19 | 20 | 663 | 130 | 20 |
| Acetylmorphine | 10000 |
5.3 | 21 | 24 | 10525 |
2383 | 23 | 18 | 16 | 586 | 91 | 15 |
| Acetylcodeine | 10000 |
4.8 | 5.8 | 24 | 10481 |
2363 | 23 | 16 | 29 | 20 | 5.3 | 27 |
| Codeine | 10000 |
14 | 16 | 15 | 11419 |
1685 | 15 | 31 | 26 | 85 | 22 | 26 |
| Oxycodone | 10000 |
6.9 | 15 | 13 | 10694 |
1314 | 12 | 11 | 13 | 459 | 56 | 12 |
| Fentanyl | 500 | 18 | 9.4 | 8.0 | 592 | 46 | 7.8 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Norfentanyl | 500 | −21 | 16 | 25 | 398 | 93 | 23 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Pethidine | 500 | −1.4 | 18 | 16 | 493 | 77 | 16 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Tapentadol | 10000 |
7.3 | 23 | 24 | 10725 |
2431 | 23 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Tilidine | 10000 |
2.8 | 9.3 | 10 | 10275 |
1001 | 9.7 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Tramadol | 10000 |
35 | 13 | 19 | 13450 |
2374 | 18 | 17 | 23 | 436 | 94 | 22 |
| Nortramadol | 10000 |
−11 | 11 | 23 | 8913 | 1900 | 21 | 14 | 31 | 104 | 31 | 29 |
| Dextromethorphan | 10000 |
1.3 | 14 | 20 | 10125 |
1953 | 19 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Methadone | 50000 |
11 | 8.5 | 15 | 55688 |
7760 | 14 | 15 | 19 | 356 | 66 | 19 |
| EDDP | 10000 |
12 | 9.3 | 11 | 11181 |
1191 | 11 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Buprenorphine | 500 | 4.6 | 23 | 25 | 523 | 124 | 24 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Norbuprenorphine | 10000 |
6.4 | 18 | 25 | 10644 |
2552 | 24 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Cocaine | 50000 |
14 | 4.5 | 4.8 | 57125 |
2643 | 4.6 | 11 | 12 | 767 | 89 | 12 |
| Benzoylecgonine | 10000 |
13 | 16 | 14 | 11250 |
1552 | 14 | 11 | 13 | 700 | 89 | 13 |
| Norcocaine | 500 | 14 | 4.8 | 9.2 | 571 | 50 | 8.7 | 17 | 20 | 15 | 3 | 19 |
| Cocaethylene | 10000 |
10 | 9.8 | 14 | 11031 |
1489 | 14 | 15 | 28 | 5.1 | 1.4 | 27 |
| p-Hydroxycocaine | 500 | 7.0 | 7.2 | 8.2 | 535 | 42 | 7.8 | 24 | 24 | 3.8 | 0.9 | 23 |
| m-Hydroxycocaine | 500 | 2.4 | 7.8 | 8.4 | 512 | 41 | 8.1 | 21 | 24 | 6.8 | 1.5 | 23 |
| Amphetamine | 10000 |
0.8 | 20 | 20 | 10075 |
1925 | 19 | 13 | 16 | 596 | 89 | 15 |
| Methamphetamine | 10000 |
−0.2 | 20 | 21 | 9981 | 1990 | 20 | 16 | 21 | 796 | 160 | 20 |
| MDMA | 10000 |
−1.3 | 15 | 15 | 9869 | 1423 | 14 | 18 | 18 | 1024 | 178 | 17 |
| MDA | 10000 |
0.3 | 18 | 20 | 10025 |
1885 | 19 | 24 | 26 | 58 | 15 | 25 |
| Methylphenidate | 10000 |
−5.3 | 15 | 18 | 9469 | 1642 | 17 | 30 | 36 | 163 | 56 | 35 |
| Ketamine | 10000 |
18 | 18 | 18 | 11844 |
1536 | 13 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Norketamine | 10000 |
16 | 27 | 14 | 11550 |
3035 | 26 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Alprazolam | 10000 |
22 | 7.9 | 7.4 | 12188 |
882 | 7.2 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Clobazam | 500 | 16 | 8.1 | 17 | 582 | 94 | 16 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Norclobazam | 500 | 24 | 19 | 21 | 622 | 124 | 20 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Clonazepam | 10000 |
29 | 7.3 | 11 | 12875 |
1317 | 10 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Diazepam | 10000 |
21 | 7.5 | 7.1 | 12063 |
827 | 6.9 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Nordazepam | 10000 |
13 | 6.5 | 7.9 | 11338 |
857 | 7.6 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Oxazepam | 10000 |
29 | 13 | 14 | 12863 |
1676 | 13 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Temazepam | 500 | 9.5 | 18 | 22 | 548 | 116 | 21 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Flunitrazepam | 500 | 12 | 4.9 | 7.3 | 562 | 39 | 7.0 | n.d. | n.d. | n.d. | n.d. | n.d. |
| 7-Amino flunitrazepam | 500 | 20 | 13 | 16 | 599 | 91 | 15 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Flurazepam | 500 | 12 | 8.2 | 11 | 559 | 59 | 11 | n.d. | n.d. | n.d. | n.d. | n.d. |
| N-Desalkylflurazepam | 500 | 0.8 | 16 | 16 | 504 | 76 | 15 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Lorazepam | 500 | 19 | 7.2 | 9 | 593 | 52 | 8.8 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Lormetazepam | 500 | 30 | 18 | 28 | 648 | 171 | 26 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Midazolam | 10000 |
22 | 7.4 | 8.7 | 12188 |
1014 | 8.3 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Hydroxymidazolam | 500 | 19 | 7.9 | 9.1 | 593 | 52 | 8.8 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Nitrazepam | 500 | 3.4 | 12 | 14 | 517 | 69 | 13 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Phenazepam | 500 | −0.3 | 16 | 14 | 499 | 70 | 14 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Prazepam | 500 | 3.6 | 7.0 | 12 | 518 | 59 | 11 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Tetrazepam | 500 | 2.0 | 8.4 | 12 | 510 | 58 | 12 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Triazepam | 500 | 11 | 7.6 | 8.4 | 553 | 45 | 8.1 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Zalepam | 500 | 11 | 12 | 11 | 553 | 61 | 11 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Zolpidem | 10000 |
10 | 7.2 | 9.2 | 11044 |
974 | 8.8 | 22 | 26 | 21 | 5 | 25 |
| Zopiclone | 10000 |
−2.8 | 9.6 | 20 | 9725 | 1791 | 18 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Citalopram | 50000 |
4.9 | 8.8 | 11 | 52469 |
5658 | 11 | 10 | 16 | 763 | 114 | 15 |
| Duloxetine | 500 | −12 | 8.4 | 12 | 439 | 49 | 11 | 18 | 25 | 14 | 3 | 24 |
| Fluoxetine | 50000 |
0.9 | 22 | 22 | 50438 |
10873 |
22 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Mirtazepam | 10000 |
−2.2 | 13 | 17 | 9784 | 1548 | 16 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Paroxetine | 500 | −14 | 12 | 8.8 | 432 | 37 | 8.6 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Sertraline | 10000 |
−13 | 4.5 | 7.2 | 8697 | 593 | 6.8 | 19 | 18 | 833 | 144 | 17 |
| Trazodone | 50000 |
−2.0 | 5.4 | 6.5 | 49000 |
3036 | 6.2 | 16 | 22 | 802 | 167 | 21 |
| Venlafaxine | 50000 |
−3.7 | 28 | 28 | 48156 |
12912 |
17 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Doxylamine | 10000 |
−12 | 21 | 21 | 8769 | 1753 | 20 | 22 | 26 | 559 | 140 | 25 |
| Diphenhydramine | 10000 |
−11 | 9.2 | 9.9 | 8888 | 848 | 9.5 | 9.2 | 23 | 14 | 3.1 | 22 |
| Lamotrigine | 50000 |
−24 | 12 | 20 | 38250 |
7250 | 19 | n.d. | n.d. | n.d. | n.d. | n.d. |
| Tizanidine | 10000 |
18 | 8.2 | 15 | 11831 |
1698 | 14 | n.d. | n.d. | n.d. | n.d. | n.d. |
| CBD | 5000 | 0.5 | 7.0 | 11 | 5025 | 541 | 11 | 27 | 30 | 42 | 12 | 29 |
| CBN | 5000 | −1.0 | 8.6 | 10 | 4950 | 491 | 9.9 | 26 | 28 | 242 | 65 | 27 |
| THC | 5000 | −1.4 | 9.4 | 8.8 | 4931 | 424 | 8.6 | 28 | 28 | 434 | 116 | 27 |
| Cortisol | 5.0 | −12 | 20 | 27 | 4.4 | 1.1 | 26 | 14 | 16 | 14 | 2.2 | 15 |
| Cortisone | 5.0 | 25 | 12 | 16 | 6.3 | 0.9 | 15 | 12 | 12 | 19 | 2.2 | 11 |
| Testosterone | 0.5 | 8.8 | 19 | 29 | 0.5 | 0.1 | 28 | n.d. | n.d. | n.d. | n.d. | n.d. |
Comparative measurements
Using the combined sample preparation, drugs, pharmaceuticals and endogenous steroids were extracted from hair snippets instead powder in contrast to the original methods (Fig. 1). This may affect the extraction yields and therefore also the measured concentration levels. In order to evaluate this effect, comparative measurements between both methods were carried out for selective analytes in authentic hair samples. Noteworthy, the evaluation for rare or low-concentrated metabolites was omitted as only the most abundant drugs were relevant for our sample case-work.For 58% of the analytes, the medians of the calculated concentrations for the combined sample preparation are lower compared to the original methods which may be attributed to higher yields for the extraction from powdered hair compared to snippets (e.g.37 and 38).
Results were statistically evaluated using paired t-test evaluating the difference of the concentrations for the original and modified method. The number of pairs, medians and p-values are presented in Table 2. Depending on whether normal or non-normal distribution was present, data was analyzed using either the unpaired t-test or the non-parametric Mann–Whitney test (Table 2). The resulting p-values were >0.05 which was considered as not significant (Table 2). According to this finding, it was shown in boxplots that the medians of the calculated concentrations measured by the original sample method did not differ significantly for the modified approach of the combined sample preparation (ESI†). Nevertheless, the results between the original methodologies and the combined sample preparation can differ to some extent as discussed above and shown in the ESI.† The observed quantitative differences were generally lower than the measurement uncertainty, which is often set at a harmonized value of 30% for hair analyses. Results of the combined sample preparation may need to be interpreted even more carefully and critically. The combined sample preparation is intended to be used in cases were only a small sample amount is available and multiple analytes have to be tested. This can be especially interesting in research studies or for special forensic cases.
| Analyte | Number of paired measurements | Original method | Combined sample preparation | p-Value |
|---|---|---|---|---|
| Concentration (pg mg−1) | ||||
| Median | Median | |||
| a Mann–Whitney test. b Unpaired t-test. | ||||
| Morphine | 19 | 3500 | 2800 | 0.4743a |
| Acetylmorphine | 16 | 428 | 350 | 0.8300a |
| Acetylcodeine | 6 | 130 | 92 | 0.8442a |
| Codeine | 10 | 334 | 440 | 0.9648b |
| Methadone | 8 | 1800 | 1685 | 0.6847b |
| EDDP | 7 | 185 | 210 | 0.4981b |
| Cocaine | 22 | 6498 | 4600 | 0.6671a |
| Benzoylecgonine | 23 | 4050 | 4250 | 0.9783a |
| Norcocaine | 18 | 620 | 328 | 0.3508a |
| Amphetamine | 8 | 540 | 473 | 0.9005a |
| Methamphetamine | 6 | 393 | 310 | 0.6741b |
| MDMA | 12 | 338 | 190 | 0.2717a |
| Methylphenidate | 8 | 528 | 553 | 0.9826a |
| Ketamine | 5 | 110 | 104 | 0.6905a |
| Diazepam | 10 | 117 | 135 | 0.8388a |
| Nordazepam | 10 | 93 | 180 | 0.9557a |
| Midazolam | 8 | 340 | 288 | 0.9408a |
| α-Hydroxymidazolam | 5 | 34 | 37 | 0.5238a |
| Citalopram | 8 | 51 | 44 | 0.5737a |
| Mirtazapine | 7 | 172 | 150 | 0.6200a |
| Trazodone | 7 | 305 | 545 | 0.6200a |
| Paracetamol | 14 | 4176 | 5350 | 0.9367a |
| Cortisol | 8 | 6.3 | 5.7 | 0.9591a |
| Cortisone | 9 | 22 | 22 | 0.467b |
Conclusion
A combined sample preparation for the simultaneous extraction of drugs, pharmaceuticals, cannabinoids and endogenous steroids was developed based on three independent fully validated analytical methods. Due to modifications in sample preparation and extraction compared to the original methods, re-evaluation was successfully performed including validation experiments and the statistical analysis of comparative measurements. It was shown that results for the original methods and the combined sample preparation method did not differ significantly in terms of limits of quantification, accuracy, precision and robustness. Overall, the combined sample preparation enables the analysis of a wide range of substances thus reducing the demand for sample material and preparation time. Therefore, this method is suitable for analysis in cases with limited sample amounts and/or for segmental analysis.Author contributions
Clementine Scholz: methodology, validation, formal analysis, investigation, writing – original draft, visualization. Markus R. Baumgartner: conceptualization, resources, writing – review & editing, supervision. Thomas Kraemer: writing – review & editing, supervision. Tina M. Binz: methodology validation, data curation, writing – original draft, supervision, project administration.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We express our gratitude to Emma Louise Kessler, MD for her generous legacy she donated to the Institute of Forensic Medicine at the University of Zurich, Switzerland for research purposes.References
- S. Hayashi, I. Miyamoto and K. Takeda, Br. J. Dermatol., 1991, 125, 123–129 CrossRef PubMed.
- F. Pragst, M. Rothe, K. Spiegel and F. Sporkert, Forensic Sci. Rev., 1998, 10, 81–111 Search PubMed.
- F. Musshoff and B. Madea, Anal. Bioanal. Chem., 2007, 388, 1475–1494 CrossRef PubMed.
- C. L. Morris-Kukoski, M. A. Montgomery and R. L. Hammer, J. Anal. Toxicol., 2014, 38, 628–636 CrossRef PubMed.
- L. C. Bossers, R. Paul, A. J. Berry, R. Kingston, C. Middendorp and A. J. Guwy, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 953–954, 115–119 CrossRef PubMed.
- M. A. Balikova and V. Habrdova, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2003, 789, 93–100 CrossRef.
- A. F. Hamel, J. S. Meyer, E. Henchey, A. M. Dettmer, S. J. Suomi and M. A. Novak, Clin. Chim. Acta, 2011, 412, 382–385 CrossRef PubMed.
- T. Baciu, F. Borrull, C. Aguilar and M. Calull, Anal. Chim. Acta, 2015, 856, 1–26 Search PubMed.
- M. M. Madry, T. Kraemer and M. R. Baumgartner, Forensic Sci. Int., 2018, 282, 137–143 CrossRef CAS.
- E. Beasley, S. Francese and T. Bassindale, Anal. Chem., 2016, 88, 10328–10334 CrossRef CAS.
- M. A. Huestis, R. A. Gustafson, E. T. Moolchan, A. Barnes, J. A. Bourland, S. A. Sweeney, E. F. Hayes, P. M. Carpenter and M. L. Smith, Forensic Sci. Int., 2007, 169, 129–136 CrossRef CAS PubMed.
- M. Taylor, R. Lees, G. Henderson, A. Lingford-Hughes, J. Macleod, J. Sullivan and M. Hickman, Drug Alcohol Rev., 2017, 36, 220–226 CrossRef PubMed.
- B. Moosmann, N. Roth and V. Auwarter, Sci. Rep., 2015, 5, 14906 CrossRef CAS PubMed.
- B. Moosmann, N. Roth, M. Hastedt, A. Jacobsen-Bauer, F. Pragst and V. Auwarter, Drug Test. Anal., 2015, 7, 349–357 CrossRef CAS.
- T. M. Binz, F. Gaehler, C. D. Voegel, M. Hofmann, M. R. Baumgartner and T. Kraemer, Anal. Bioanal. Chem., 2018, 410, 4895–4903 CrossRef CAS PubMed.
- C. Kirschbaum, A. Tietze, N. Skoluda and L. Dettenborn, Psychoneuroendocrinology, 2009, 34, 32–37 CrossRef CAS.
- T. Stalder, S. Steudte-Schmiedgen, N. Alexander, T. Klucken, A. Vater, S. Wichmann, C. Kirschbaum and R. Miller, Psychoneuroendocrinology, 2017, 77, 261–274 CrossRef CAS PubMed.
- J. H. Lanfear, C. D. Voegel, T. M. Binz and R. A. Paul, Steroids, 2020, 163, 108712 CrossRef CAS.
- S. L. Kroll, D. P. Williams, M. Thoma, M. Staib, T. M. Binz, M. R. Baumgartner, C. Kirschbaum, J. F. Thayer and B. B. Quednow, Psychoneuroendocrinology, 2019, 100, 264–275 CrossRef CAS.
- F. Vaiano, G. Serpelloni, S. Furlanetto, D. Palumbo, F. Mari, A. Fioravanti and E. Bertol, J. Pharm. Biomed. Anal., 2016, 118, 161–166 CrossRef CAS PubMed.
- P. Kintz, V. Cirimele, C. Jamey and B. Ludes, J. Forensic Sci., 2003, 48, 195–200 CAS.
- N. Quinete, J. Bertram, M. Reska, J. Lang and T. Kraus, Talanta, 2015, 134, 310–316 CrossRef CAS PubMed.
- W. Gao, T. Stalder and C. Kirschbaum, Talanta, 2015, 143, 353–358 CrossRef CAS.
- T. M. Binz, U. Braun, M. R. Baumgartner and T. Kraemer, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1033–1034, 65–72 CrossRef CAS.
- T. M. Binz, L. Rietschel, F. Streit, M. Hofmann, J. Gehrke, M. Herdener, B. B. Quednow, N. G. Martin, M. Rietschel, T. Kraemer and M. R. Baumgartner, Forensic Sci. Int., 2018, 284, 33–38 CrossRef CAS PubMed.
- C. D. Voegel, M. R. Baumgartner, T. Kraemer, S. Wust and T. M. Binz, Talanta, 2021, 222, 121499 CrossRef CAS.
- C. D. Voegel, M. Hofmann, T. Kraemer, M. R. Baumgartner and T. M. Binz, Steroids, 2020, 154, 108547 CrossRef CAS.
- C. D. Voegel, P. La Marca-Ghaemmaghami, U. Ehlert, M. R. Baumgartner, T. Kraemer and T. M. Binz, Steroids, 2018, 140, 144–150 CrossRef CAS.
- C. Scholz, J. Cabalzar, T. Kraemer and M. R. Baumgartner, J. Anal. Toxicol., 2021, 45, 701–712 CrossRef CAS PubMed.
- C. Scholz, M. M. Madry, T. Kraemer and M. R. Baumgartner, J. Anal. Toxicol., 2022, 46, 504–511 CrossRef CAS.
- M. K. Nielsen, S. S. Johansen and K. Linnet, Forensic Sci. Int., 2015, 248, 134–139 CrossRef CAS PubMed.
- X. Wang, Y. Zhuo, X. Tang, H. Qiang, W. Liu, H. Wu, P. Xiang, G. Duan and M. Shen, Drug Test. Anal., 2020, 12, 472–484 CrossRef CAS.
- X. Wang, S. S. Johansen, M. K. K. Nielsen and K. Linnet, J. Forensic Sci., 2019, 64, 950–955 CrossRef CAS PubMed.
- T. Wang, B. Shen, H. Wu, J. Gu, M. Shen and P. Xiang, J. Anal. Toxicol., 2020, 44, 596–600 CrossRef CAS.
- K. N. Gunther, J. Banner, K. Linnet and S. S. Johansen, J. Pharm. Biomed. Anal., 2020, 190, 113510 CrossRef CAS PubMed.
- F. T. Peters, M. Hartung, M. Herbold, G. Schmitt, T. Daldrup and F. Musshoff, Toxichem. Krimtech., 2009, 76, 185 Search PubMed.
- A. Salomone, M. R. Baumgartner, T. Lombardo, E. Alladio, D. Di Corcia and M. Vincenti, Forensic Sci. Int., 2016, 267, 60–65 CrossRef CAS PubMed.
- M. E. Albermann, F. Musshoff, L. Aengenheister and B. Madea, Anal. Bioanal. Chem., 2012, 403, 769–776 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ay01325h |
| This journal is © The Royal Society of Chemistry 2022 |