
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMethod for identification and quantification of intact teduglutide peptide using (RP)UHPLC-UV-(HESI/ORBITRAP)MS
Raquel
Pérez-Robles
 abc,
Antonio
Salmerón-García
ad,
Susana
Clemente-Bautista
e,
Inés
Jiménez-Lozano
e,
María Josep
Cabañas-Poy
e,
Jose
Cabeza
ad and
Natalia
Navas
*ab
abc,
Antonio
Salmerón-García
ad,
Susana
Clemente-Bautista
e,
Inés
Jiménez-Lozano
e,
María Josep
Cabañas-Poy
e,
Jose
Cabeza
ad and
Natalia
Navas
*ab
aInstituto de Investigación Biosanitaria de Granada (ibs.GRANADA), Granada, Spain
bDepartment of Analytical Chemistry, Science Faculty, University of Granada, Granada, Spain
cFundación para la Investigación Biosanitaria de Andalucía Oriental-Alejandro Otero, Granada, Spain
dDepartment of Clinical Pharmacy, San Cecilio University Hospital, Granada, Spain
eMaternal and Child Pharmacy Service, Vall d’Hebron Hospital, Pharmacy, Barcelona, Spain
First published on 10th October 2022
Abstract
Teduglutide (Revestive®, 10 mg mL−1) is a recombinant human glucagon-like peptide 2 analogue, used in the treatment of short bowel syndrome, a serious and highly disabling condition which results from either too small a length of intestine or loss of critical intestinal function. The determination of therapeutic compounds of protein-nature is always challenging due to their complex structure. In this work, we present a fast, straightforward reversed phase (RP)UHPLC-UV-(HESI/ORBITRAP)MS method for the identification and quantification of the intact teduglutide peptide. The method has been developed and validated in accordance with the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) guidelines; therefore, linearity, limits of detection and quantification, accuracy (precision and trueness), robustness, system suitability and specificity using the signal from the UV and MS, have been evaluated. The validation performance parameters obtained from the UV and MS signals were compared throughout the work, to select the most suitable. To study the specificity of the method and the impact of medicine mishandling under hospital conditions, force degradation studies were performed, i.e. thermal (40 °C and 60 °C), shaking (mechanical) and light (accelerated exposition) effects. Identification by the exact mass of teduglutide was achieved and it was confirmed that the peptide does not undergo any post-translational modifications (PTMs). To the best of our knowledge, the present work reports the first method developed for the simultaneous identification, structural characterization, and quantification of the therapeutic teduglutide peptide. Finally, the proposed method is able to indicate stability when quantifying the intact teduglutide since detects and characterises the exact mass of the degradation/modification products.
1. Introduction
Peptide drugs have been in use for almost a century for the treatment of a wide range of diseases, including diabetes, cancer, osteoporosis, HIV infection, chronic pain, etc.1 Over 150 peptides are in clinical development and another 400–600 are undergoing preclinical trials.2 Peptide drugs occupy a distinct pharmaceutical space between small-molecule drugs and biologics, they account for 5% of the global pharmaceutical market in terms of global sales.3 The majority of peptide drugs on the market and in development are analogues that build on the intrinsic activity of native hormones with improved pharmaceutical properties.4 Therefore, a rigorous analytical method for the study of these medicines is of great interest and demand.Teduglutide (TGT) is a recombinant human glucagon-like peptide 2 (GLP-2) analogue – a naturally occurring peptide which is secreted primarily by the lower gastrointestinal tract. TGT (C164H252N44O55S) is expressed by a genetically modified strain of E. coli. Structurally it constitutes 33 aminoacidic residues in a single chain, which correspond to a molecular weight of 3752![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 0919 Da, it has no disulfide bonds, no glycosylation sites, and no post-translational modifications5 (Fig. 1). TGT differs from GLP-2 by an alanine to glycine substitution in the second position of the N-terminus. This substitution renders the peptide resistant to in vivo degradation by dipeptidyl peptidase IV, and increases its half-life from just 7 minutes to approximately 2 to 3 hours.6 TGT is indicated for the treatment of short bowel syndrome (SBS), a serious and highly disabling condition, which results from either too small a length of intestine or loss of critical intestinal function, whereby the amount of remaining functional gut is too short to allow for adequate absorption of nutrients and fluids.7 Patients must be treated with long-term parenteral support (PN) and/or intravenous (IV) fluids, which can cause life threatening complications, such as intestinal failure-associated liver disease, central line-associated blood stream infections, and cerebral venous thrombosis.8 TGT is approved in the United States and Europe for the treatment of patients with SBS that are older than 1 year.9 The indicated dosage is 0.05 mg kg−1, administered subcutaneously once a day. Patients must be stable following a period of intestinal adaptation after surgery before they can be treated.10 The administration of TGT reduces the need for the PN/IV support, including in some cases independence from PN/IV support altogether.11
0919 Da, it has no disulfide bonds, no glycosylation sites, and no post-translational modifications5 (Fig. 1). TGT differs from GLP-2 by an alanine to glycine substitution in the second position of the N-terminus. This substitution renders the peptide resistant to in vivo degradation by dipeptidyl peptidase IV, and increases its half-life from just 7 minutes to approximately 2 to 3 hours.6 TGT is indicated for the treatment of short bowel syndrome (SBS), a serious and highly disabling condition, which results from either too small a length of intestine or loss of critical intestinal function, whereby the amount of remaining functional gut is too short to allow for adequate absorption of nutrients and fluids.7 Patients must be treated with long-term parenteral support (PN) and/or intravenous (IV) fluids, which can cause life threatening complications, such as intestinal failure-associated liver disease, central line-associated blood stream infections, and cerebral venous thrombosis.8 TGT is approved in the United States and Europe for the treatment of patients with SBS that are older than 1 year.9 The indicated dosage is 0.05 mg kg−1, administered subcutaneously once a day. Patients must be stable following a period of intestinal adaptation after surgery before they can be treated.10 The administration of TGT reduces the need for the PN/IV support, including in some cases independence from PN/IV support altogether.11
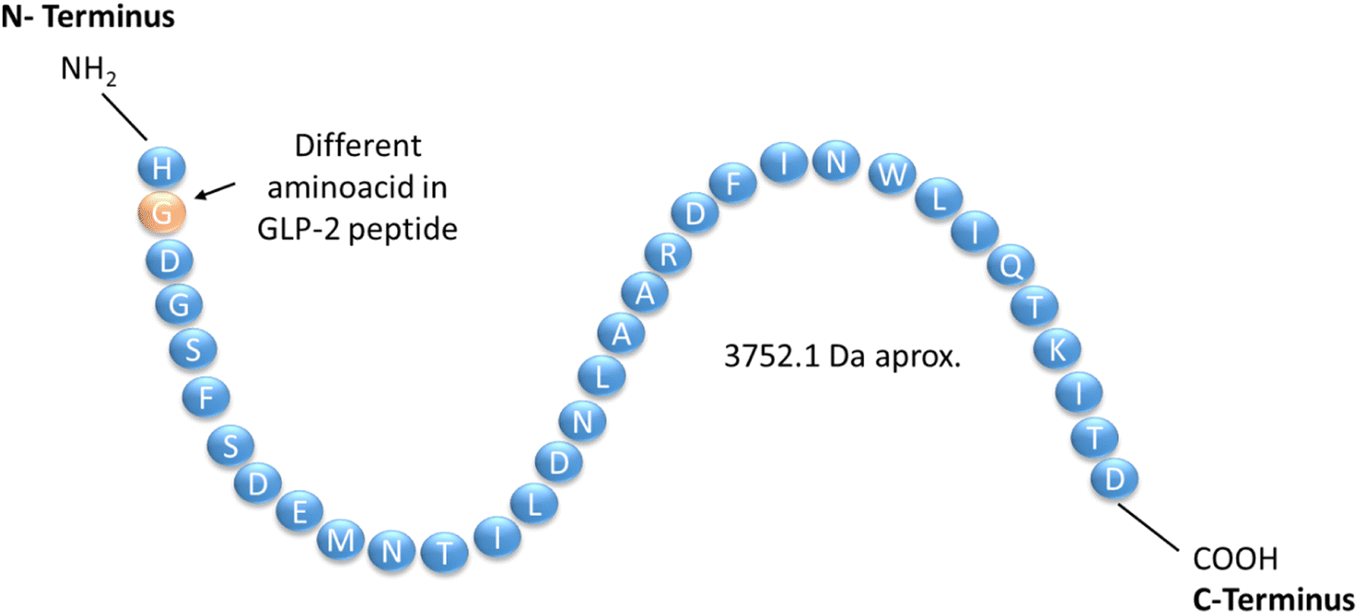 | ||
| Fig. 1 Diagram of the chemical primary sequence of teduglutide. | ||
Analytical techniques play an important role in the life cycle of bio-drugs – the discovery, development, production and post-marketing – as they allow understanding of different parameters and attributes. This is essential for selecting and designing pharmaceuticals – evaluating stability, quantifying, identifying and/or evaluating the toxicity profiles of synthesis impurities or degradation products.12 However, the characterization of therapeutic peptides poses many challenges compared to the characterization of traditional chemical drugs because of their inherent complexity. Different analytical techniques (orthogonal techniques) based on different principles are required to fully characterize the proteinaceous nature of molecules (peptides, monoclonal antibodies, fusion proteins, etc.).13,14 Chromatographic techniques are well-established for protein and peptide analysis in any of their modalities i.e., size exclusion (SEC), ion exchange (IEX) or reverse-phase chromatography (RPLC).
RPLC is generally considered more efficient and more sensitive for the analysis of intact biotherapeutic proteins, compared to other modes of chromatography, due to the high selectivity, low limit of detection and quantification, robustness and high sensitivity.15,16 The developed reverse phase chromatographic stationary phases (porous and monolithic phases) make this mode a very effective technique for the analysis of intact proteins and fragments. On other hand, mass spectrometry (MS) is also an excellent analytical tool for studying the properties and behaviour, and the characterization, of proteinaceous natural molecules i.e., membrane proteins,17 peptide drugs,18,19, mAbs,20 fusion proteins21 and antibody drug conjugates,22 under native or denatured conditions. MS-based methods are particularly useful for studying the structural aspects of proteins such as primary sequence characterization,23 post-translational modification (PTM),24 degradation patterns,25etc. Reverse phase ultra high-pressure liquid chromatography-mass spectrometry detection ((RP)UHPLC-MS), enhances this mode of chromatography to a gold standard technique to identify and quantify proteins.
In the pharmaceutical field, taking into account the complexity of the drugs composed of proteins or peptides, the development of rigorous and reliable analytical methods to determine their quality is one of the most urgent tasks to ensure product security, efficacy and quality. Also, it is essential to develop such analytical methods under the specifications and requirements stated by official organizations. The International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH), the Food and Drug Administration Agency (FDA) and the United States Pharmacopeia (USP), publish specific guidelines for analytical method validation in the pharmaceutical field. From the point of view of the analytical validation of biopharmaceuticals, the ICH Q6B guideline26 indicates that the validation of analytical procedures used in the quantification of biotechnological products (such as therapeutic peptides) should be performed in compliance with the ICH Q2(R1) guideline.27 Also, the Analytical Procedures and Methods Validation for Drugs and Biologics guideline from the FDA,28 and the <1225> USP guideline,29 also describe recommendations regarding pharmaceutical analytical method validation.
In this work, we present a (RP)UHPLC-UV-(HESI/ORBITRAP)MS method for the identification and quantification of the intact TGT peptide in the medicine, Revestive®. In terms of analytical quality assurance, the method has been validated for quantification purposes and qualified for the detection of peptide modification/degradation. The validation has been performed in accordance with ICH guidelines for biotechnological products, also taking into account FDA recommendations and those of Hsu and Chien.30 The performance parameters such as linearity, accuracy (precision and trueness), detection limits, quantification limits, robustness, system suitability and specificity were all evaluated. Two signal sources (UV and MS) were used for quantification purposes. Accordingly, a comparative study concerning the quality of the validation performance characteristics was conducted regarding the signal used for the quantification. The TGT identification was carried out by mass spectrometry, allowing the intact TGT structure mass characterization. In addition, we carried out a stress study to test the feasibility of using the developed method in the presence of modified or degraded products. To the best of our knowledge, this represents the first method developed for the simultaneous identification, characterization and quantification of the teduglutide peptide.
2. Materials and method
2.1. Chemicals and reagents
Reverse-osmosis-quality water was purified with a Milli-Q station from Merck Millipore (Darmstadt, Germany). The reagents used were of LC-MS purity grade. Acetonitrile (ACN) was purchased from VWR International Eurolab, S.L. (Barcelona, Spain) and formic acid (FA) was supplied by Thermo Fisher Scientific (Geel, Belgium). Water for solvent parenteral injections use Meinsol® (Barcelona, Spain).2.2. Teduglutide samples
Revestive® (Shire Pharmaceuticals Ireland Limited, Dublin, Ireland) is presented as a powder for solution, each vial contains 5 mg of teduglutide (TGT). After reconstitution in 0.5 mL of water for injections for parenteral use, a solution of 10 mg mL−1 (Bach T1805F/10) was obtained. The medicine vials used throughout this study were kindly supplied by the Pharmacy Unit of the University Hospital Vall d’Hebron (Barcelona, Spain).2.3. Force degradation
Force degradation conditions were performed on the medicine Revestive® when reconstituted at the final concentration of 10 mg mL−1, by subjecting the samples (0.2 mL) to particular stress conditions. Three forced degradation conditions were tested: (i) exposure to 40 °C and 60 °C for 3 hours (using amber glass vials) in a thermomixer chamber (Eppendorf, Madrid, Spain), (ii) exposure to light irradiation (250 W m−2) for 24 hours (using a glass vial) in an aging chamber (Solarbox 3000e RH, Cofomegra, Milan, Italy), (iii) mechanical smooth shaking for 3 hours (using amber glass vials) at room temperature (Eppendorf Thermo mixer, Madrid, Spain).2.4. Teduglutide intact analysis by reverse phase UHPLC-UV-(HESI/ORBITRAP)MS
A suitable analytical platform (Thermo Scientific, Waltham, MA, USA) was used to perform the method. A chromatograph equipped with two ternary pumps, a degasser, an autosampler, a thermostatted column compartment, and a multiple-wavelength detector (MWD-3000(RS)UV-Vis detector) was coupled in line with a Q-Exactive hybrid quadrupole-ORBITRAP mass spectrometer. The ionization was performed using a heated electro spray ionization (HESI) source. The chromatographic instrument was managed by Xcalibur® 4.0 software and the mass spectrometer by Tune® Software.For the chromatographic separation an Acclaim Vanquish C18, 2.2 mm, 2.1 mm × 250 mm column (Thermo Fisher Scientific, Waltham, MA, USA) was used. The flow rate was 0.3 mL min−1 and 5 μl of samples were injected into the column. The column temperature was set at 25 °C. The eluent system was composed of 0.1% FA in deionized water (mobile phase A) and 0.1% FA in ACN (mobile phase B). The column was equilibrated with 30% of eluent B for 5 min. Then, a linear gradient was applied from 30% to 90% of eluent B for 5 min, and kept constant for 2 min. To recondition the column, the gradient was reduced to 30% of eluent B for 1 min. Total analysis run time was therefore 13 min.
The UV chromatograms were registered at 214 nm, using 360 ± 10 nm as the reference wavelength. The MS instrument was operated in positive mode (M-H+) in a mass range of 300 to 4000 m/z using 17500 resolution. The subsequent MS settings were as follows: spray voltage 3.8 kV, sheath gas flow rate 40 AU, auxiliary gas flow rate 10 AU, capillary temperature 320 °C, AGC target value 3 × 106, S-Lens RF Level 50, max injection time 100 ms and number of micro-scans 1.
The peptide data processing, quantitation and identification were performed using an Xcalibur® QualBrowser 4.0 for signal integration (Thermo Scientific®). The deconvoluted mass spectrum signals were performed manually.
2.5. Method validation for quantification using UV and MS signals
Validation was performed according to ICH recommendations,27 particularly following guideline ICH Q2(R1), and taking into account criteria for acceptance from the FDA28 and Hsu and Chien’s recommendations.30 Performance parameters such as linearity, accuracy (precision and trueness), detection limits (LOD), quantification (LOQ) limits, robustness, system suitability and specificity, were evaluated. The validation of the method was carried out calculating the performance parameters using the UV and MS signals. The objective of this double validation was to compare the results obtained by the two detectors to determine the most adequate for the quantification of intact TGT, or to propose a strategy using both signals to fulfil the validation requirement as published by the ICH.
Validation data treatment was performed using Startgraphics Centurion (v XVI.II) software.
3. Results and discussion
3.1. Method optimization
The chromatographic conditions were optimized in a previous work for peptide mapping analysis for several marketed monoclonal antibodies20 and were used here as the start conditions to optimize our method for the analysis of TGT, since it is a small peptide similar to the peptides resulting from protein enzymatic digestion. Then, an Acclaim Vanquish C18, 2.2 mm, 2.1 mm × 250 mm column was used, as it is suitable for peptide analysis. The mobile phase composition was based in a combination of water, acetonitrile (ACN) and formic acid (FA). This mobile phase is usually required for the analysis of proteinaceous molecules to reduce column interactions,31 and also FA is an ion-pairing agent required to enhance the peptide ionization using HESI ionization.The chromatograms were recorded at two different wavelengths, i.e., λ = 214 nm and 280 nm, using λ = 350 ± 10 nm as reference in both cases. The analytical parameters of the method were calculated using λ at 214 nm since this was the maximum absorption of the proteinaceous molecules such as TGT. The 280 nm signal was used to corroborate the proteinaceous nature of the chromatographic peaks. Regarding the MS detector, the HESI ionization source parameters in ref. 20 for peptide mapping analysis were checked for the analysis of TGT. Suitable signals were obtained by increasing the resolution to 17500 in full scan mode.
The experimental chromatographic conditions were optimized to shorten the time of analysis from the starting method.35 Several experiments were carried out testing different times for the gradient (5 and 10 min) and different compositions of mobile phase B (from 2% to 85%, from 40% to 100% and from 30% to 90%). The best results were obtained using a gradient from 30% to 90% during 5 min, considering the retention time (6.22 min) of TGT and the shape of the peak.
The column temperature (25 °C, 40 °C, 60 °C) was checked and did not affect the TGT chromatographic figures of merit, therefore, it was set low at 25 °C to protect the column and avoid undesirable modifications to the TGT structure. The flow was fixed at 0.3 mL min−1 to obtain the shape of TGT chromatographic peaks. Fig. 2 shows a representative chromatogram of a standard sample of TGT recorded with these optimised conditions. TGT elutes at 6.22 ± 0.03 min and 6.27 ± 0.03 min when detected using UV and MS signals, respectively, with a total analysis time of 8 min. As expected, the UV-chromatograms are more affected by the mobile phase compositions, with a front signal between 1.5 and 2.5 min and a drift of the gradient baseline. In contrast, the MS-chromatograms recorded are clearer chromatograms with no noise or drift of the baseline. Nevertheless, both signals could be used for the analysis of TGT, once validated as discussed next. In addition, these conditions allow for rapid results, which is important for routine quality control analysis.
 | ||
| Fig. 2 TGT standard solution. (1) UV absorption chromatograms: (e) 10 mg L; (f) 15 mg L; and (g) 25 mg L−1. (2) MS signal chromatograms: (a) 10 mg L; (b) 15 mg L; (c) 25 mg L−1, and (d) TGT mass spectrum characterized by the presence of 3 ions i.e. 938.95 m/z, 1251.60 m/z and 1876.90 m/z. | ||
3.2. TGT mass profile using the (RP)UHPLC-UV-(HESI/ORBITRAP)MS
Using the above optimized conditions of the (RP)UHPLC-UV-(HESI/ORBITRAP)MS method, the TGT mass profile was obtained for control samples (no-degraded samples or “fresh” samples) and for samples submitted to controlled degradation (samples stressed by heat at 40 °C and 60 °C, mechanical agitation and accelerated exposition to light). In the case of the control samples, the intact mass analysis of TGT was performed to obtain the experimental exact mass of the peptide and to check the presence of isoforms (PTMs). Stressed samples were used with two objectives: to evaluate the ability of this method to detect and identify TGT mass modifications; and to assess the impact of the stress factors on Revestide®, factors which the peptide may be subjected between manufacture and patient administration. The comparison of the mass spectra from the stressed samples with the control TGT samples allowed detection of the unequivocal identification of chemical structural degradation/modifications. Fig. 3 shows the raw mass spectra obtained for the control/fresh and stressed TGT samples. | ||
| Fig. 3 Representative raw mass spectra of teduglutide during stress studies. 15 mg L−1 solution of TGT was injected into the chromatograph system. (A) Control/fresh teduglutide, (B) teduglutide under shake stress, (C) teduglutide under temperature stress (40 °C), (D) teduglutide under temperature stress (60 °C), (E) teduglutide under light stress. | ||
To correlate the experimental mass and interpret the mass data generated, the theoretical average mass of the expected TGT were calculated based on the theoretical sequence of the peptide, provided from different bibliographic sources,32,33 and this was calculated as 3752.0919 Da. Table 1 lists the ion mass detected in control/fresh and stressed TGT samples.
| Sample | Ion mass (Da) | Charge | Deconvoluted mass (Da) | Modification | Average theoretical mass (Da) | Mass difference (Da) |
|---|---|---|---|---|---|---|
| Control/Fresh | 1876.8967 | +2 | 3751.7934 | None | 3752.0919 | 0.30 |
| 1251.6025 | +3 | 3751.8075 | None | 3752.0919 | 0.28 | |
| 938.9531 | +4 | 3751.8124 | None | 3752.0919 | 0.28 | |
| Shake stress | 1876.8961 | +2 | 3751.7922 | None | 3752.0919 | 0.30 |
| 1251.6008 | +3 | 3751.8024 | None | 3752.0919 | 0.29 | |
| 938.9521 | +4 | 3751.8084 | None | 3752.0919 | 0.28 | |
| Temperature (40 °C) stress | 1876.8945 | +2 | 3751.7890 | None | 3752.0919 | 0.30 |
| 1251.5996 | +3 | 3751.7988 | None | 3752.0919 | 0.29 | |
| 938.9514 | +4 | 3751.8056 | None | 3752.0919 | 0.29 | |
| Temperature (60 °C) stress | 1876.8944 | +2 | 3751.7888 | None | 3752.0919 | 0.30 |
| 1251.5992 | +3 | 3751.7976 | None | 3752.0919 | 0.29 | |
| 938.9515 | +4 | 3751.8060 | None | 3752.0919 | 0.29 | |
| Light stress | 1251.6052 | +3 | 3751.8156 | None | 3752.0919 | 0.28 |
| 1256.9372 | +3 | 3767.8116 | +1 oxidation | 3768.0913 | 0.28 | |
| 1262.6005 | +3 | 3784.8015 | +2 oxidations | 3784.0907 | −0.71 | |
| 1267.6005 | +3 | 3799.8015 | +3 oxidations | 3800.0901 | 0.29 | |
| 1272.5971 | +3 | 3814.7913 | Unknown | — | — | |
| 1277.9306 | +3 | 3830.7918 | Unknown | — | — | |
| 1313.2860 | +3 | 3936.8580 | Unknown | — | — | |
| 1323.9496 | +3 | 3968.8488 | Unknown | — | — | |
| 1328.9471 | +3 | 3983.8410 | Unknown | — | — | |
| 938.9557 | +4 | 3751.8228 | None | 3752.0919 | 0.27 | |
| 942.9541 | +4 | 3767.8164 | +1 oxidation | 3768.09129 | 0.27 | |
| 946.9530 | +4 | 3783.8120 | +2 oxidations | 3784.0907 | 0.28 | |
| 950.9500 | +4 | 3799.8000 | +3 oxidations | 3800.0901 | 0.29 | |
| 954.9506 | +4 | 3815.8024 | Unknown | — | — | |
| 958.7070 | +4 | 3830.8280 | Unknown | — | — | |
| 985.2157 | +4 | 3936.8628 | Unknown | — | — | |
| 993.2137 | +4 | 3968.8548 | Unknown | — | — | |
| 997.2136 | +4 | 3984.8544 | Unknown | — | — |
The control/fresh TGT mass spectrum is characterized by the presence of 3 major ions i.e. 938.9531, 1251.6025 and 1876.8967 m/z which correspond with ion charge +4, +3 and +2, respectively. These ions match with a deconvoluted mass of 3752.0919 Da which corresponds with the theoretical average mass of TGT without PMTs, as indicated in the Revestive® Assessment Report.10
The thermal stress was evaluated for a temperature exposition of 40 °C and 60 °C. No modifications with respect to the control/fresh sample were observed in the mass spectra. As in the control/fresh TGT sample, 3 three major ions were observed (1876.8945 m/z, 1251.5996 m/z, and 938.9514 m/z) which correspond to an average deconvoluted mass of 3752.0919 Da. Therefore, no structure modifications or PTMs were found when TGT samples were subjected to thermal stresses.
Regarding the effect of mechanical stress on TGT, similar results were achieved when TGT samples were subjected to shaking for 3 h (see Fig. 3 and Table 1). No new mass signals were detected with respect to the control/fresh TGT, therefore, no changes in the chemical structure or PTMs were identified.
For the TGT samples exposed to light, two contiguous chromatographic peaks at 6.06 and 6.37 min were detected, indicating that TGT degradation took place. The MS spectra show 9 different masses after deconvolution, all of them from two TGT charge states, i.e., +3 and +4 (see Table 1). One of these matched the theoretical mass of intact TGT (3752.0919 Da); three matched oxidized TGT (+1 oxidation: 3768.09129 Da, +2 oxidation: 3784.0907 Da and +3 oxidation: 3800.0901 Da); and the other 5 were not identified since no PTMs could be related to them, and adduct formation is suggestion as the likely source of these unknown ions. Regarding the oxidation, we highlight that light induces amino acid oxidation particularly in the Met and Trp residues. Trp is susceptible to one oxidation (+16 Da) meanwhile Met could be oxidized once (methionine sulfoxide, +16 Da) or twice (methionine sulfone, +32 Da). The TGT primary sequence has only one Met and one Trp, therefore the experimental mass matched with the three different possibilities of TGT oxidation. The most intense ion corresponds to the theoretical mass of TGT +1 oxidation that could have occurred in the Met or Trp residue.
3.3. Method validation
Once the method performance was established, and the experimental parameters set, the method was validated in accordance with the ICH Q2(R1). Since the ICH guidelines do not establish any criteria for acceptance, those from FAD28 and Hsu and Chien30 were followed.
Throughout the validation study, Revestive® was used as the TGT standard to prepare the standard working solutions. This is a common practice in the pharmaceutical field when no proper standards are available.
The signals from the two detectors, UV and MS, were considered to study the linearity. Both calibration curves were fitted by an ordinary residual least-squares regression (OLS). The linearity of the curves was evaluated by the coefficient of determination (R2) and the calculation of the linearity index (LIN) is described by eqn (1).
 | (1) |
The calibration performance characteristics of the UHPLC-UV and the UHPLC-MS signals are summarized in Table 2. The parameters which evaluate the quality of the regressions (R2 and LIN) showed satisfactory results for the method linearity for the two signals used for the quantification (UV or MS). The established accepted criteria fulfilled requirements, with R2 and LIN ≥ 95%.21 The sensibility, evaluated by the slope of the linear function, was greater when using the MS signal.
| Parameter | UHPLC-UV | UHPLC-MS |
|---|---|---|
| a Arbitrary units. | ||
| Slope, b (AUa/(mg L−1)) | 7.90 × 103 | 9.95 × 108 |
| Standard deviation of the slope, sb (AUa/(mg L−1)) | 1.02 × 102 | 1.88 × 107 |
| Intercept, a (AUa) | −1.88 × 104 | −1.48 × 109 |
| Standard deviation of the intercept, sa (AUa) | 1.69 × 103 | 3.12 × 108 |
| R 2 (%) | 99.80 | 99.54 |
| LIN (%) | 98.70 | 98.10 |
| Linear interval (mg L−1) | 5 to 25 | 5 to 25 |
| LOD (mg L−1) | 0.64 | 0.94 |
| LOQ (mg L−1) | 2.14 | 3.14 |
 | (2) |
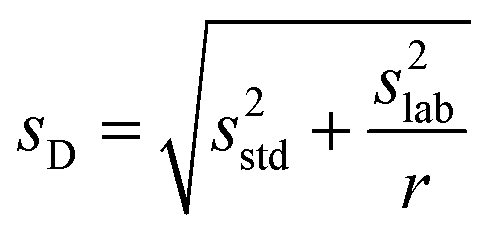 | (3) |
The repeatability was determined from the results of the analysis of TGT standard solutions prepared at the same concentration on the same day. Nine samples – low concentration levels (3 samples of 5 mg L−1), medium concentration levels (3 samples of 15 mg L−1) and high concentration levels (3 samples of 25 mg L−1) – were used for this purpose. The intermediate precision was determined from the analysis of standard solutions at the same concentration levels as the repeatability (5, 15 and 25 mg L−1), over three consecutive days (as is recommended in the ICH guidelines). Three independent samples of each concentration were prepared and analysed daily. The values of precision (repeatability and intermediate precision) obtained from the UV and MS signals were accepted since all RSD (5) obtained were less than 5% of the fixed criteria value (Table 3).
| Signal | Reference (mg L−1) | Relative error (%) | Recovery (%) | Relative standard deviation (%), interday precision | Relative standard deviation (%), repeatability |
|---|---|---|---|---|---|
| UHPLC-UV | 5 | 2.0 | 102.0 | 1.8 | 2.7 |
| 15 | 4.7 | 104.4 | 1.4 | 4.6 | |
| 25 | 4.4 | 104.4 | 0.5 | 3.4 | |
| UHPLC-MS | 5 | −4.0 | 102.0 | 2.4 | 4.2 |
| 15 | −4.7 | 95.3 | 4.6 | 0.8 | |
| 25 | −6.0 | 94.0 | 3.7 | 1.9 |
The accuracy was determined by analysing three standard solutions at the low, medium and high concentration levels from the calibration curve (5, 15 and 25 mg mL−1) in triplicate (as is described in the ICH guidelines), Table 3 shows the results obtained. The overall t-student test shows non-significant differences between the reference and found concentrations (P-value 0.82 for MS signals and 0.88 for the UV signals). In addition, the ISO-test showed that all the single differences were less than the critical value.
According to these results, the precision and trueness values regardless of the signal used for the quantification, fulfilled the acceptance criteria and therefore were acceptable.
| Run | Flow (mL min−1) | Initial proportion of eluent B (%) | Column temperature (°C) | |||
|---|---|---|---|---|---|---|
| 1 | 0.30 | (0) | 30 | (0) | 25 | (0) |
| 2 | 0.27 | (−1) | 27 | (−1) | 27 | (1) |
| 3 | 0.33 | (1) | 27 | (−1) | 23 | (−1) |
| 4 | 0.30 | (0) | 30 | (0) | 25 | (0) |
| 5 | 0.27 | (−1) | 33 | (1) | 23 | (−1) |
| 6 | 0.33 | (1) | 33 | (1) | 27 | (1) |
| 7 | 0.30 | (0) | 30 | (0) | 25 | (0) |
The total effect analysis and the analysis of the variance (ANOVA) of the DoE variables were carried out. The total effect analysis showed that no significant values were obtained for any of the variables in the range studied (Fig. 4) for the two signals used, UV and MS. The DoE ANOVA results of each factor were as follow: considering the UV signal, flow P-value = 0.0727, initial proportion of eluent B P-value = 0.0761 and column temperature P-value = 0.0764; and regarding the MS signal, flow P-value = 0.3904, initial proportion of eluent B P-value = 0.6588 and column temperature P-value = 0.5249. P-values were not significant for the two signals, indicating also that the total effect analysis and the ANOVA results were similar. In conclusion, none of the values of the variables studies (within the experimental domain) affected the robustness of the method, therefore, indicating satisfactory method robustness.
 | ||
| Fig. 4 Bar-chart showing the total effect from the UV signal (A) and MS signal (B) of the variables analysed in the robustness study. 15 mg L−1 TGT sample were injected in the chromatograph system. The horizontal line denotes the effect significance threshold which is calculated from the standard deviation related to the estimate of the effects. | ||
Since the method is based on the chromatographic analysis of TGT, the system suitability test was carried out by calculating the chromatographic parameters theoretical plate number (N) and the symmetry factor (k′). Also, the instrumental injection repeatability evaluated as RSD (%) was checked. For this purpose, TGT standard samples of 15 mg L−1 were analyzed.
As previously indicated, the ICH Q2 (R1) guidelines do not indicate any criteria to evaluate the method system suitability, therefore we followed the FDA criteria for HPLC methods, which indicate the N (criterium N > 2000) and k′ (criterium k′ > 2.0) and the criterium in ref. 36 to evaluate the instrumental injection repeatability (RSD < 5% for biologics). The results (Table 5) demonstrate the fulfilment of the criteria, and therefore the system suitability.
| Signal | Retention time (min) | N | k′ | RSD (%) |
|---|---|---|---|---|
| UHPLC-UV | 6.21 | 26512.19 |
3.05 | 4.6 |
| UHPLC-MS | 6.28 | 2347.59 | 2.93 | 0.8 |
Regarding the UV signal source, identical chromatograms were obtained for the control/fresh TGT samples and for the TGT samples subjected to temperatures of 40 °C and 60 °C and to mechanical stress by shaking (Fig. 5A). TGT samples subjected to light were heavily degraded and no chromatographic peak was detected; the chromatographic peak corresponding to fresh TGT when the sample was submitted to light stress indicated that light stress induces high degradation in the peptide structure, as indicated in Section 3.2.
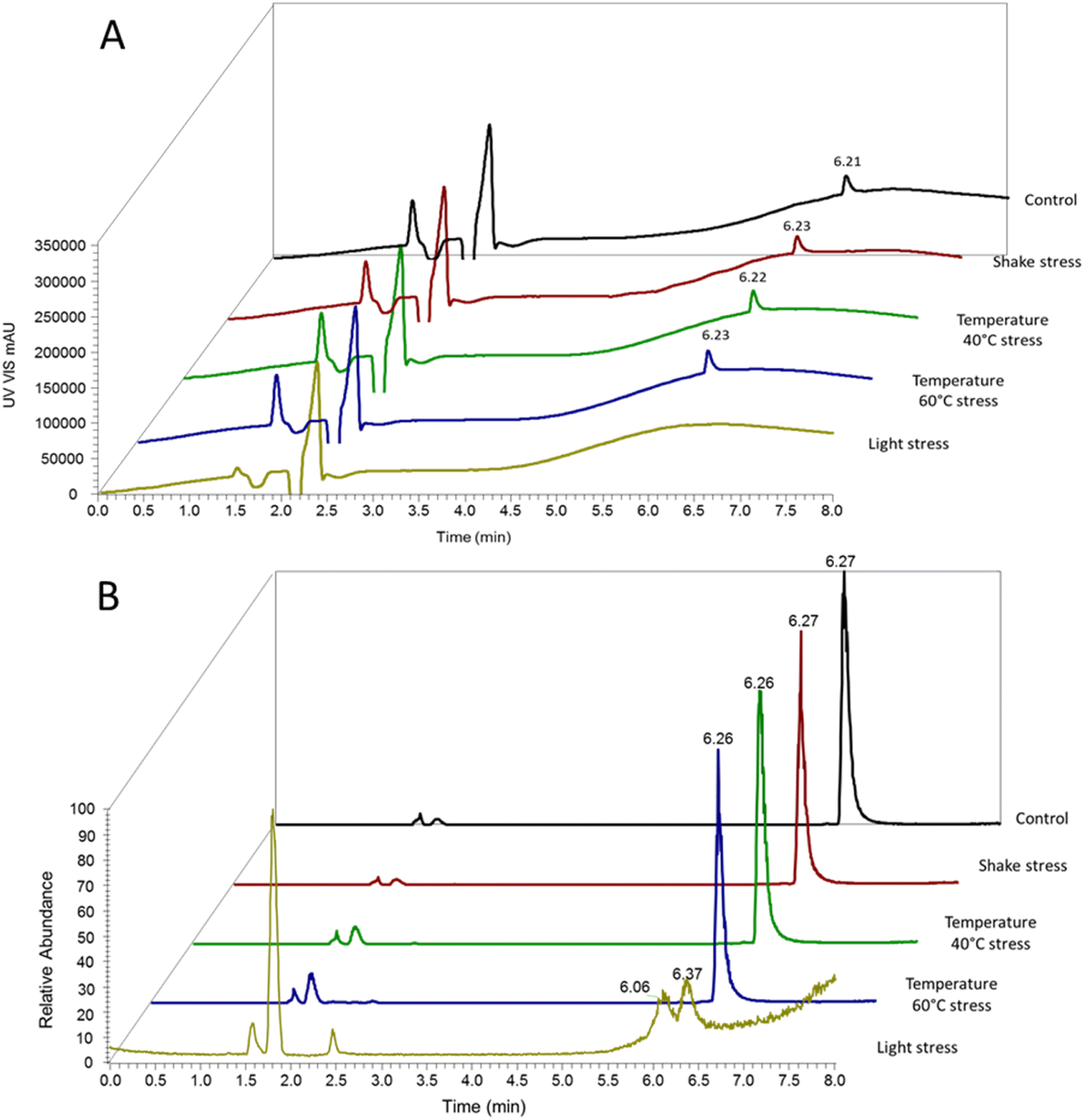 | ||
| Fig. 5 Representative chromatograms of the teduglutide stress study to evaluate the proposed (RP)UHPLC-UV-(HESI/ORBITRAP)-MS method. 15 mg L−1 solutions of TGT were injected in the chromatograph system. (A) UV signal chromatograms and (B) MS signal chromatograms. The chromatograms are displayed as normalized from the main peak. | ||
The MS results confirmed the UV-results, i.e. no chromatographic differences among control/fresh and samples subjected to temperatures of 40 °C and 60 °C, or mechanical shake stressed samples, therefore indicating no degradation of the peptide; which was also confirmed by the mass obtained, in all cases it was the mass of intact TGT (3752.0919 Da) (see Section 3.2.). Regarding light stress, the chromatogram showed two new chromatographic peaks slightly shifted around the TGT retention time (from 626 min to 6.06 min and to 6.37 min) but with an important decrease of the signal intensity. This evidenced degradation of the peptide by producing degraded compounds retained in the column (Fig. 5B). Those degraded compounds eluted at 6.06 min and 6.37 min have been discussed in Section 3.2.
From the point of view of the specificity of the (RP)UHPLC-UV-(HESI/ORBITRAP)MS method, the results from this controlled degradation study indicate that the degraded/modified TGT could not be chromatographically separated from non-degraded intact TGT, as the different forms of TGT eluted at similar retention times in the MS-chromatogram and were not detected in the UV-chromatogram. However, the use of the MS detector allowed characterization of TGT from the exact mass, and could be used to detect modifications (PTMs) when they occur from the degradation of pharmaceutical samples (e.g., reconstituted medicine Revestive® or similar). Our method is therefore suitable for detecting degradation/modification of TGT but can only be used to quantify TGT up to modification/degradation; once degraded, the remaining TGT cannot be evaluated to the required level of accuracy using MS.
4. Conclusions
In this research, for the first time a (RP)UHPLC-UV-(HESI/ORBITRAP)MS method for the identification and quantification of the marketed peptide TGT is presented. The method was developed and validated according to ICH guidelines Q2(R1); therefore, it was validated in terms of linearity, limits of detection and quantification, accuracy (precision and trueness), robustness, system suitability and specificity; and FDA criteria and those reported by Hsu and Chien were used. All parameters were studied using both UV and MS detectors. Except for the specificity, the results indicate the applicability and reliability of our method for quantification purposes, regardless of the signal used (UV or MS), as it is accurate and robust, and shows satisfactory detection and quantification limits. For specificity, the MS signal is essential. The unequivocal identification by the exact mass of TGT and the degraded products achieved using the signal from an MS detector is required for the method to be specific, since degradation components are not chromatographically separated. Therefore, our method incorporating MS signals is able to detect the degradation/modification of TGT but can only be used to quantify TGT up to modification/degradation. The method using UV signals does not fulfil any requirement for specificity, and it could be used only for quantification purposes in cases in which TGT cannot have been degraded.Regarding TGT structure, we confirmed the absence of PTMs in the formulated medicine Revestive® when reconstituted. Also, it was demonstrated that light has a critical environmental effect as it induces degradation in the TGT structure, mainly through oxidation. Accordingly, TGT solution should be protected from light exposure as much as possible. In contrast, TGT is resistant to modification when subjected to temperatures of 40 °C and 60 °C for one hour and when smoothly shaken for three hours.
This work is part of a wider project that aims to propose rigorous analytical methods for the full characterization of teduglutide, focusing on the stability of the peptide. This method represents one of the analyses (peptide) required for full teduglutide characterisation in pharmaceutical solutions.
Author contributions
Raquel Pérez-Robles: conceptualization, formal analysis, investigation, writing-original draft, visualization, supervision. Antonio Salmerón-García: conceptualization, formal analysis, resources, funding acquisition, visualization, investigation. Susana Clemente-Bautista: conceptualization, resources, visualization. Inés Jiménez-Lozano: conceptualization, resources, visualization. Maria Josep Cabañas-Poy: conceptualization, resources, visualization. Jose Cabeza: conceptualization, resources, funding acquisition, visualization. Natalia Navas: conceptualization, methodology, formal analysis, writing – review and editing, supervision, funding acquisition, project administration, resources.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
This study was funded by the Fundación Andaluza de Farmacia Hospitalaria (Spain), TEC01 from Ibs. Granada and FQM 118 from University of Granada research groups. Raquel Pérez-Robles currently holds a postdoctoral position granted by the Junta de Andalucía (PAIDI 2020), Spain (DOC_01694). The authors would like to thank the Hospital Paediatrics Pharmacy Unit of the Hospital Vall d’ Hebron (Barcelona, Spain) for kindly supplying the medicine samples. Funding for open access charge: Universidad de Granada.References
- J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700–2707 CrossRef CAS.
- M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Nat. Rev. Drug Discovery, 2021, 20, 309–325 CrossRef CAS PubMed.
- Global Information Inc, Global Peptide Therapeutics Sales Market Report 2020, QY Research, 2020 Search PubMed.
- A. C. L. Lee, J. L. Harris, K. K. Khanna and J. H. Hong, Int. J. Mol. Sci., 2019, 20, 1–21 Search PubMed.
- PudChem, https://www.pubchem.ncbi.nlm.nih.gov/compound/Teduglutide#section=Structures, (Accessed May 2021).
- P. B. Jeppesen, Curr. Opin. Gastroenterol., 2014, 30, 182–188 CrossRef CAS PubMed.
- C. B. Burness and P. L. Mccormack, Drugs, 2013, 935–947 CrossRef CAS PubMed.
- K. Chen, F. Mu, J. Xie, S. S. Kelkar, C. Olivier, J. Signorovitch and P. B. Jeppesen, J. Parenter. Enteral Nutr., 2020, 44, 119–128 CrossRef CAS PubMed.
- S. A. Kocoshis, R. J. Merritt, S. Hill, S. Protheroe, B. A. Carter, S. Horslen, S. Hu, S. S. Kaufman, D. F. Mercer, M. P. Pakarinen, R. S. Venick, P. W. Wales and A. A. Grimm, J. Parenter. Enteral Nutr., 2020, 44, 621–631 CrossRef CAS PubMed.
- EMEA/H/C/002345/, European Public Assessment Report (EPAR) Summary for Revestive, Annex I: Summary of Product Characteristics, European Medicines Agency (EMA), London (UK), 2012 Search PubMed.
- P. B. Jeppesen, JPEN, J. Parenter. Enteral Nutr., 2014, 38, 45S–52S CrossRef CAS PubMed.
- D. L. Porto, A. R. R. da Silva, A. de S. Oliveira, F. H. A. Nogueira, M. de F. F. Pedrosa and C. F. S. Aragão, Microchem. J., 2020, 157, 104921 CrossRef CAS.
- J. Hermosilla, R. Sánchez-Martín, R. Pérez-Robles, A. Salmerón-García, S. Casares, J. Cabeza, L. Cuadros-Rodríguez and N. Navas, BioDrugs, 2019, 33, 193–205 CrossRef CAS PubMed.
- J. Hermosilla, R. Pérez-Robles, A. Salmerón-García, S. Casares, J. Cabeza, J. Bones and N. Navas, Sci. Rep., 2020, 10, 1–13 CrossRef PubMed.
- K. Sandra, I. Vandenheede and P. Sandra, J. Chromatogr. A, 2014, 1335, 81–103 CrossRef CAS PubMed.
- S. Grotefend, L. Kaminski, S. Wroblewitz, S. El Deeb, N. Kühn, S. Reichl, M. Limberger, S. Watt and H. Wätzig, J. Pharm. Biomed. Anal., 2012, 71, 127–138 CrossRef CAS.
- J. R. Bolla, F. Fiorentino and C. V. Robinson, Curr. Opin. Struct. Biol., 2021, 70, 53–60 CrossRef CAS.
- B. Fabre, J. P. Combier and S. Plaza, Curr. Opin. Chem. Biol., 2021, 60, 122–130 CrossRef CAS PubMed.
- V. Baghalabadi and A. A. Doucette, Anal. Chim. Acta, 2020, 1138, 38–48 CrossRef CAS PubMed.
- S. Carillo, R. Pérez-Robles, C. Jakes, M. Ribeiro da Silva, S. Millán Martín, A. Farrell, N. Navas and J. Bones, J. Pharm. Anal., 2020, 10, 23–34 CrossRef.
- R. Pérez-Robles, L. Cuadros-Rodríguez, A. Salmerón-García, J. Cabeza-Barrera and N. Navas, J. Pharm. Biomed. Anal., 2020, 185, 113233 CrossRef PubMed.
- A. Martelet, V. Garrigue, Z. Zhang, B. Genet and A. Guttman, J. Pharm. Biomed. Anal., 2021, 201, 114094 CrossRef CAS PubMed.
- R. Black, A. Barkhanskiy, L. A. I. Ramakers, A. Theisen, J. M. Brown, B. Bellina, D. K. Trivedi and P. E. Barran, Int. J. Mass Spectrom., 2021, 464, 116588 CrossRef CAS.
- V. Baghalabadi and A. A. Doucette, Anal. Chim. Acta, 2020, 1138, 38–48 CrossRef CAS PubMed.
- Y. Jin, Y. Yi and B. Yeung, Methods, 2020, 200, 58–66 CrossRef PubMed.
- ICH Q6B, Specifications: test procedures and acceptance criteria for biotechnological/biological products, in Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, Geneva (Switzerland), 1999 Search PubMed.
- ICH Q2(R1), Validation of analytical procedures: text and methodology, in: International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, Geneva (Switzerland), 2005 Search PubMed.
- FDA, Analytical Procedures and Method Validation for Drugs and Biologics Guidance for Industry, US Food and Drug Administration, Maryland (USA), 2015 Search PubMed.
- USP 39, <1225> Validation of Compendial Procedures, United States Pharmacopeia, Maryland (USA), 2016 Search PubMed.
- H. Hsu and C. S. Chien, J. Food Drug Anal., 1994, 2, 161–176 CAS.
- A. Martínez-Ortega, A. Herrera, A. Salmerón-García, J. Cabeza, L. Cuadros-Rodríguez and N. Navas, Int. J. Biol. Macromol., 2018, 116, 993–1003 CrossRef PubMed.
- Drug bank,https://go.drugbank.com/drugs/DB08900, (Accessed May 2022).
- Genome web, https://www.genome.jp/dbget-bin/www_bget?drug:D06053 (Accessed June 2022).
- ISO Guide 33:2015, Reference Materials-Good Practice in Using Reference Materials, International Organization for Standardization, Genève (Switzerland), 2015 Search PubMed.
- R. Pérez-Robles, L. Cuadros-Rodríguez, A. Salmerón-García and N. Navas, J. Pharm. Biomed. Anal., 2018, 159, 437–448 CrossRef PubMed.
- CDER, Reviewer Guidance: Validation of Chromatographic Methods. Center for Drug Evaluation and Research, Rockville (USA), 1994 Search PubMed.
| This journal is © The Royal Society of Chemistry 2022 |