DOI:
10.1039/D2AY00352J
(Paper)
Anal. Methods, 2022,
14, 1603-1610
A validated method for the rapid quantification of melatonin in over-the-counter hypnotics by the atmospheric pressure solid analysis probe (ASAP)†
Received
1st March 2022
, Accepted 22nd March 2022
First published on 6th April 2022
Abstract
Melatonin is a hormone that regulates the biological day and night cycle. It is mainly produced by the pineal gland during the night. People suffering from insomnia use it as a soporific drug. The aim of this study was to develop a method for the rapid quantification of melatonin in hypnotics. For that purpose, atmospheric pressure solid analysis probe-assisted mass spectrometry was applied, where no chromatographic separation is needed. Thereby, one single analysis takes less than 1 min. Reference measurements were performed with ultra-high-performance liquid chromatography coupled with a quadrupole-time-of-flight mass spectrometer. Both methods were validated and real sample extracts were tested. The coefficients of determination were above 0.97 for both methods. The limits of detection and quantification were below 1 mg kg−1. Both methods gave comparable results. Moreover, the content of melatonin differed from the specified value in many samples. The highest and lowest observed deviations were 78% and 1%, respectively.
1. Introduction
Melatonin (N-acetyl-5-methoxytryptamine) is a hormone that is mainly produced in mammals by the pineal gland. Additionally, it can be synthesized by the skin, lymphocytes and the gastrointestinal tract. Tryptophan acts as the starting product, which is then hydroxylated by tryptophan hydroxylase to form 5-hydroxytryptophan and subsequently decarboxylated by aromatic L-amino acid decarboxylase to form serotonin. Finally, serotonin undergoes acetylation by aralkylamine N-acetyl transferase and methylation by hydroxy indole-O-methyl transferase to form melatonin. This production process is dependent on the day and night cycle. The formation rate decreases with light, whereas it increases in darkness. The absence of melatonin plays a major role in insomnia. Moreover, melatonin is associated with many other diseases such as Alzheimer's disease, diabetes, obesity and cancer.1,2 Melatonin also occurs naturally in fish, wheat, vegetables, fruits, seeds, nuts, wine, tea and edible oils. The highest amount of melatonin is typically found in nuts, germinated legumes, eggs, fish and mushrooms. Consumption of melatonin-containing food increases the amount of melatonin in human serum. Many foods rich in melatonin are characteristic of the Mediterranean diet and thus, it is claimed that they have a direct impact on the lower appearance of chronic degenerative disorders.3,4 Nevertheless, the primary field of application is the treatment of insomnia. Nowadays, plenty of chemical compounds are available for the treatment of insomnia (barbiturates, benzodiazepines and chloral hydrate), but many of them have substantial side effects. In comparison, melatonin did not show any encroachments for probands even at dosages up to 100 mg per day.5 Besides the chemical compounds and melatonin, certain plant extracts are also used for the treatment of insomnia, for instance Valeriana officinalis (valerian), Humulus lupulus (hops), Melissa officinalis (lemon balm), Passiflora incarnata (maypop) and Withania somnifera (winter cherry).6–10 The European Food Safety Authority (EFSA) recommends the intake of at least 1 mg melatonin before bedtime to achieve the claimed effect.11 Melatonin not only improves the quality of sleep, but also has a positive effect on anxiety and depression.12,13 Some effects of melatonin are sex-dependent. In infertile females, administration of melatonin led to an increase in pregnancy rates and fertilization.14 By contrast, the sperm quality of males was improved and the secretion of gonadotropins and testosterone was increased.15 With regard to Alzheimer's disease, melatonin, through its antioxidative properties, is suspected to inhibit the formation of amyloid fibrils by interdependence with β-amyloid.16,17 Furthermore, melatonin plays an increasing role in the treatment of cancer and as a chemotherapeutic agent. Both in vitro and in vivo tests have shown that melatonin is able to inhibit the growth of various tumours and patients were found to tolerate chemotherapy better when they received melatonin.18,19 The anti-obesogenic effect of melatonin can be traced back to its influence on the formation and secretion of insulin. It also acts as a mediator for energy balance, as it regulates the flow from and to the energy stores.20,21 Lastly, melatonin is hypothesized to help patients suffering from COVID-19 due to its anti-viral (against other viruses), anti-inflammatory, antioxidant and mitochondria-protective properties.22,23
Due to the high number of positive effects of melatonin on human health and primarily its intensive use in preventing insomnia, analytical methods are needed for the rapid and accurate determination and quantification of melatonin in soporific drugs. For this purpose, many different methods have been implemented in the past, including electrochemical,24–27 bioanalytical28–30 and analytical methods. Analytical methods that were developed encompass gas chromatography – mass spectrometry,31 liquid chromatography – UV/Vis32,33 and liquid chromatography – mass spectrometry.34–37 None of the methods employs ambient mass spectrometry, although it would provide a cheap and fast alternative to conventional methods.38–40 One of these ambient mass spectrometric techniques is the atmospheric pressure solid analysis probe (ASAP), which is related to atmospheric pressure chemical ionization (APCI). In the ASAP, a borosilicate rod is immersed into a sample, no matter if solid or liquid and then inserted into the ionisation chamber. Desorption of the analytes is accomplished by a hot nitrogen stream and ionisation is performed by corona discharge.38 The ASAP for instance can be used for the trace detection of explosives,41 detection of food fraud39 and food analysis.42 Furthermore, the semiquantitative analysis of pharmaceutical agents is described, for instance the quantitation of sildenafil in herbal over-the-counter formulations to prevent medical fraud.43
This paper describes the development of a validated method for the rapid quantification of melatonin in hypnotics with the ASAP. Through design of experiments (DoE) the optimal parameters for the ionization of melatonin have been determined. Numerous hypnotics were purchased online, from which melatonin was extracted. Reference measurements were performed with ultra-high-performance liquid chromatography coupled with a high-resolution quadrupole-time-of-flight mass spectrometer (UHPLC-qTOFMS). To the best of our knowledge, this is the first report of a validated method for the rapid screening and quantification of melatonin in commercially available drugs employing ambient mass spectrometry.
2. Materials and methods
2.1 Materials
Melatonin (certified reference material), caffeine (≥99.0%) and methanol (≥99.0%) were purchased from Sigma-Aldrich (Vienna, Austria). Formic acid (≥99.0%) was obtained from VWR (Vienna, Austria) and acetonitrile (≥99.9%) from Honeywell (Seelze, Germany). Extraction was performed with an USC-TH ultrasonic bath from VWR (Vienna, Austria) and a 5430 R centrifuge from Eppendorf (Vienna, Austria). Ambient mass spectrometry was performed with a RADIAN atmospheric pressure solid analysis probe (ASAP) system from Waters (Milford, MA, USA). The separation of analytes was performed with an Agilent column (EclipsePlus C18, RRHD 1.8 μm, 2.1 × 100 mm) on an UltiMate 3000 UHPLC system from Thermo Scientific (Linz, Austria). The UHPLC was connected to an impact mass spectrometer from Bruker (Bremen, Germany), which utilizes an electrospray ionization (ESI) source and a quadrupole-time-of-flight mass detector (qTOFMS).
2.2 Samples and extraction
A total of 26 different food supplements and herbal formulations (Table 1) that claimed to prevent insomnia were obtained online. Melatonin was listed as an active component in 14 samples. Tablets were ground in a mortar and approximately 100 mg of each sample was transferred into 15 mL falcon tubes. Extraction was performed by addition of 10 mL of 75% methanol in water (v/v) and keeping the samples in an ultrasonic bath for 20 min. Afterwards, the extracts were centrifuged for 5 min at 5000 rpm. The dilution of the samples was performed individually, depending on the approximate melatonin concentration determined in preliminary tests. 1.0 mL of each final sample preparation was pipetted into 1.5 mL vials. The preliminary tests were performed with UHPLC-qTOFMS by injection of standards with different concentrations of melatonin and pure sample extracts without dilution. The amount of melatonin was roughly estimated by comparison of the obtained areas.
Table 1 Overview of all obtained samples including the specified amount of melatonin
| Internal Sample number |
Dosage form |
Units per package |
Recommended dose |
Melatonin (mg) per recommended dose |
| 1 |
Capsules |
60 |
2 |
— |
| 2 |
Capsules |
60 |
3 |
0.27 |
| 3 |
Tablets |
30 |
1 |
1.00 |
| 4 |
Tablets |
30 |
1 |
1.00 |
| 5 |
Capsules |
30 |
2 |
1.00 |
| 6 |
Capsules |
120 |
4 |
— |
| 7 |
Tablets |
30 |
1 |
— |
| 8 |
Capsules |
50 |
1 |
— |
| 9 |
Capsules |
240 |
1 |
1.50 |
| 10 |
Tablets |
30 |
1 |
1.50 |
| 11 |
Capsules |
60 |
2 |
— |
| 12 |
Capsules |
30 |
1 |
3.00 |
| 13 |
Capsules |
60 |
2 |
— |
| 14 |
Capsules |
60 |
2 |
10.00 |
| 15 |
Capsules |
60 |
1 |
— |
| 16 |
Tablets |
90 |
1 |
1.00 |
| 17 |
Capsules |
60 |
2 |
— |
| 18 |
Tablets |
60 |
1 |
0.40 |
| 19 |
Capsules |
60 |
2 |
1.00 |
| 20 |
Capsules |
60 |
2 |
1.00 |
| 21 |
Capsules |
40 |
1 |
1.00 |
| 22 |
Capsules |
30 |
1 |
1.00 |
| 23 |
Tablets |
20 |
1 |
— |
| 24 |
Tablets |
20 |
1 |
— |
| 25 |
Tablets |
20 |
1 |
— |
| 26 |
Tablets |
25 |
1 |
— |
2.3 Standards and the internal standard
Approximately 50 mg of melatonin was weighed into a 50 mL tube. Afterwards, 50 mL of 75% methanol in water (v/v) was added (1000 mg kg−1). The tube was weighed when empty and filled to calculate the amount of melatonin in mg kg−1.1.0 mL of the melatonin solution was transferred into a 100 mL volumetric flask, which was then filled up with 75% methanol in water (v/v) to prepare a 10 mg kg−1 stock solution.From this solution, several different standards were prepared by dilution: 1 mg kg−1, 2 mg kg−1, 4 mg kg−1, 5 mg kg−1, 6 mg kg−1, 8 mg kg−1 and 10 mg kg−1 (without dilution). 1.0 mL of each standard was transferred into 1.5 mL vials.
Caffeine was chosen as the internal standard (ISTD), as it shares structural similarities with melatonin (amine and amide groups which are important for positive ionization), is easily obtainable, cheap and has a similar mass as melatonin (Δm/z = 38). Preliminary tests also showed that no signal with a m/z of 195 could be observed in any of the samples. A caffeine solution with a concentration of 1000 mg kg−1 was prepared in the same way as melatonin. 2.5 mL was transferred into a 100 mL volumetric flask to obtain a solution with 25 mg kg−1.200 μL of the 25 mg kg−1 ISTD was pipetted into each standard and sample (1000 μL standard/sample + 200 μL ISTD).
2.4 Design of experiments (DoE) for the ASAP method
DoE was utilized for the development of the method with Design-Expert (version 11, Stat-Ease Inc., Minneapolis, MN, USA). Three parameters, that were noticed to have the highest influence on the ionization and the data output itself, were varied in the DoE. The gas heater temperature and the cone voltage were changed for the ionization study and the sampling frequency for the data output. A central composite design (response surface methodology) was chosen with one replicate for each factorial and axial point. Additionally, twelve centre points were used, but with slight variations in their cone voltages, to achieve more precise surface shapes in the DoE. The temperature was evaluated from 300 °C to 500 °C, the cone voltage from 5 V to 20 V and the sampling frequency from 5 Hz to 10 Hz. The axial points were at 235 °C, 570 °C, 1 V, 25 V, 2 Hz and 15 Hz. All measurements were performed in positive ionization mode with a source temperature of 150 °C. A scan from 100 m/z to 300 m/z was applied with a total run time of 6 min. Immersion of the borosilicate rod into the sample and desorption inside the ionization chamber were performed five times per sample run (quintuplicate). Melatonin/caffeine mix standards with a concentration of 50 mg kg−1 each were analysed according to the DoE design. For this purpose, 200 μL of each sample was pipetted into 1.5 mL tubes to achieve the same immersion depth as the glass rod and to further improve reproducibility. Two response factors were chosen: the ratio (analyte/ISTD) for the evaluation of the ionisation behaviour of both substances and the sum of signal heights (analyte + ISTD) for the improvement of the limit of detection and the limit of quantification.
2.5 UHPLC-qTOFMS
The injection volume was 1 μL. Separation was performed with a flow rate of 0.4 mL min−1. Formic acid 0.01% in water (v/v) was used as solvent A and acetonitrile as solvent B. The following separation gradient was employed: 5% B for 1 min, followed by an increase to 15% B within 7 min, 30% B within 3 min, 100% B within 5 min (kept constant for 2 min) and finally returning to starting conditions within 0.1 min with an equilibration time of 1.9 min for a total run time of 20 min. The temperature of the column oven was set to 50 °C. Mass detection was performed in positive scan mode from 50 m/z to 800 m/z with a scan rate of 12 Hz. The end plate offset was set to 500 V and the capillary voltage to 4500 V. Calibration of the mass spectrometer was accomplished by using 10 mM sodium formate and each analysis was performed in triplicate.
2.6 Validation
Peak purity, linearity, limit of detection (LOD), limit of quantification (LOQ), accuracy and precision were determined according to ICH guidelines (Validation of Analytical Procedures: Text and Methodology Q2(R1)). Peak purity was accomplished by the screening of sample spectra for overlapping peaks. Linearity was tested by the preparation of two independent stock solutions of melatonin. From each stock solution, seven calibration levels were prepared by dilution (Section 3.3.). The standards were analysed in triplicate for the UHPLC-qTOFMS analysis and in quintuplicate for the ASAP analysis. The evaluation of the linearity was performed by plotting the AMelatonin/ACaffeine (areas) ratio for the chromatographic analysis and the HMelatonin/HCaffeine (signal heights) ratio for the ASAP analysis versus the cMelatonin/cCaffeine ratio. Linear regression analysis was performed for the calculation of the slope, intercept, residual standard deviation and coefficient of determination. The fraction quotient ratio of the residual standard deviation and the slope was multiplied by three for the assessment of the LOD and by ten for the LOQ. Accuracy was determined by spiking sample 3 with different amounts of melatonin stock solution to obtain low, medium and high concentration spikes. This sample was utilised, as the amount of melatonin was quite low in the diluted version and thus, it was easy to spike the sample with standard solutions. Analysis was accomplished in triplicate for UHPLC-qTOFMS and in quadruplicate for ASAP. For the evaluation of precision, three independent extracts from the randomly chosen sample 5 were prepared and measured in triplicate (UHPLC-qTOFMS) or quadruplicate (ASAP) on three subsequent days.
3. Results and discussion
3.1 Results of the DoE for the ASAP method
The results show that a sampling frequency of 8 Hz provides a sufficient amount of data without loss of important information. However, the sampling frequency had no significant influence on the response factors in the DoE (graphs are shown in the ESI†). For the surface model of the ratio (analyte/ISTD) response factor, a quadratic model delivered the best results. The model was significant with a p-value of <0.0001 and a coefficient of determination (R2) of 0.8988. In comparison, the signal height (analyte + ISTD) response factor fitted best with a linear model. The model was significant with a p-value of 0.0029 and a R2 of 0.4129. The 3D graphs of both models are shown in Fig. 1.
 |
| | Fig. 1 3D surface models of the DoE for the (A) ratio (analyte/ISTD) and (B) signal heights (analyte + ISTD) as the response factors. The sampling frequency was kept constant at 8 Hz. | |
According to the DoE, a cone voltage of 5 V and a temperature of 500 °C should be chosen with a desirability of 0.813. The use of low cone voltage can be explained by the fragmentation of melatonin at higher voltages. A fragment of melatonin with 174.1 m/z was observed in all measurements at higher cone voltages. Fig. 2(a) shows a possible mechanism for the generation of the fragment from melatonin and Fig. 2(b), the ratio of the fragment ion and the molecule ion dependent on the different cone voltages.
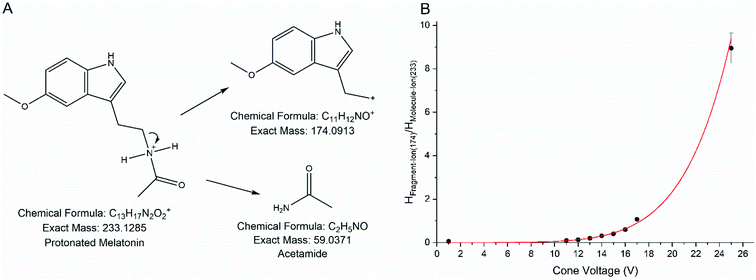 |
| | Fig. 2 (A) Mechanism for the fragmentation of melatonin at higher cone voltages in positive ionization mode. The neutral-exiting group is acetamide. (B) Dependency of the fragment ion/molecule ion ratio from the cone voltage. | |
For the confirmation of the recommended method, five replicates were analysed. For each run a standard mix with 50 mg kg−1 melatonin and 50 mg kg−1 caffeine was used. A two-sided confidence interval with 95% (α = 0.05) was set as an acceptable region and contained all measurements.
3.2 Validation
The validation of both methods (UHPLC-qTOFMS and ASAP) delivered sufficient results (Table 2). The coefficients of determination were above 0.97. Intercepts were small and slightly negative (Fig. 3). The limit of detection (LOD) was below 0.30 mg kg−1 for both methods and the limit of quantification (LOQ) was 0.90 mg kg−1 and 0.88 mg kg−1 for UHPLC-qTOFMS and ASAP methods, respectively which were less than the smallest standard concentration (1.00 mg kg−1 melatonin). Accuracy exhibited a positive deviation, whereas the ASAP values were generally higher than the UHPLC-qTOFMS values. The highest deviation was observed in the ASAP measurements for the spiked sample with the highest concentration (119.7%), but it was still within the desired range of ±20% (close to the linearity range). Due to the results of the accuracy measurements, the occurrence of matrix effects can be ruled out, as otherwise the deviations would be higher. The precision results (intraday and interday) were below 10% for both analytical methods.
Table 2 Validation parameters for the UHPLC-qTOFMS and ASAP methods
| Validation parameter |
UHPLC-qTOFMS |
ASAP |
| Slope |
0.35017 |
0.81507 |
| Intercept |
−0.02743 |
−0.03397 |
| Coefficient of determination (R2) |
0.9811 |
0.9747 |
| Limit of detection (LOD) |
0.27 mg kg−1 |
0.26 mg kg−1 |
| Limit of quantification (LOQ) |
0.90 mg kg−1 |
0.88 mg kg−1 |
| Accuracy – low |
100.0 ± 5.1% |
113.1 ± 3.6% |
| Accuracy – medium |
104.2 ± 7.7% |
109.1 ± 4.3% |
| Accuracy – high |
103.3 ± 0.7% |
119.7 ± 3.7% |
| Intraday precision |
6.3% |
8.1% |
| Interday precision |
5.4% |
6.0% |
 |
| | Fig. 3 Calibration curves generated with (A) the UHPLC-qTOFMS measurements and (B) the ASAP measurements. | |
3.3 Quantification of melatonin in real-world samples
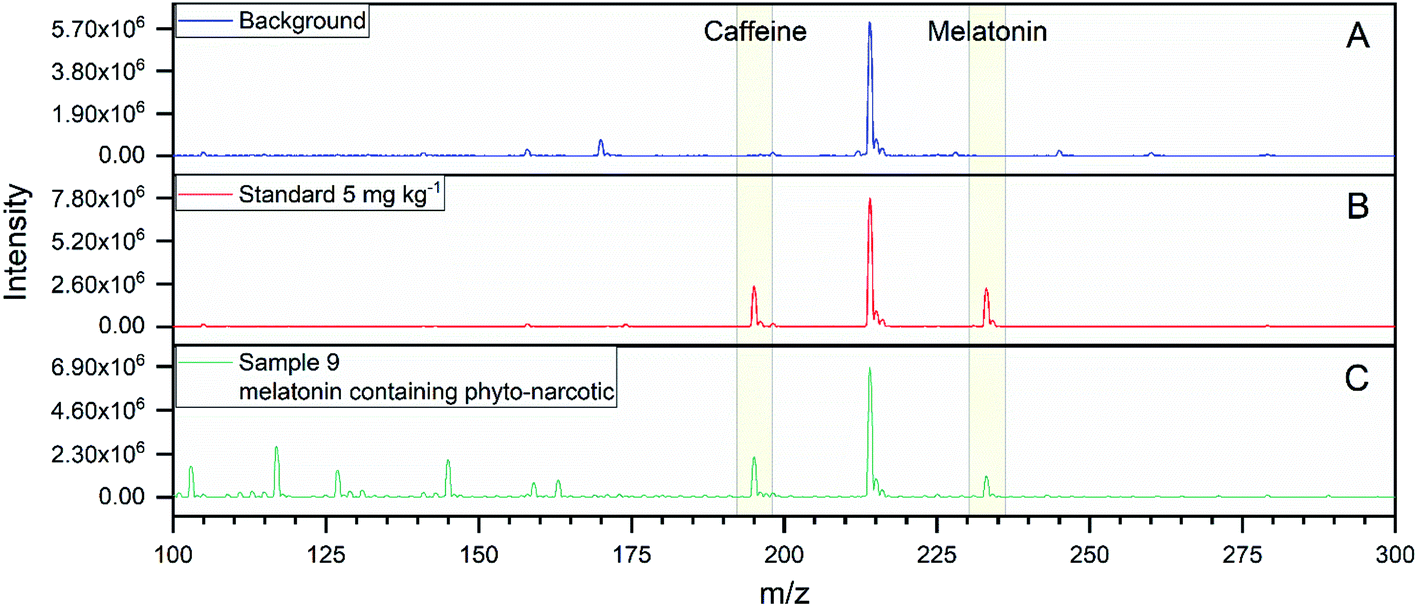
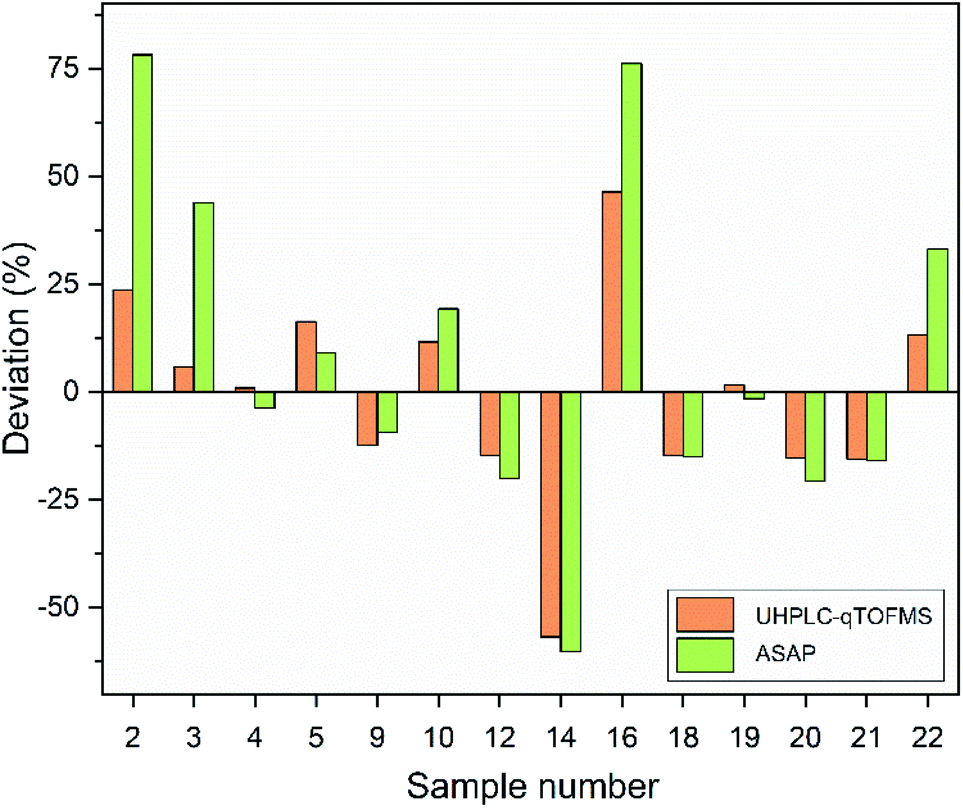
Fig. 4 depicts the mass spectra for the ASAP analysis of the background, a standard with 5 mg kg−1 and sample 9. The comparison of the background and the standard with the melatonin-containing phyto-narcotic shows, that the last-mentioned contains many other compounds due to its more complex constitution. The mass error for the UHPLC-qTOFMS analysis was −5.1 ppm for melatonin (Mtheor. = 233.1290 Da; Mmeas. = 233.1278 Da) and −4.6 ppm for caffeine (Mtheor. = 195.0882 Da; Mmeas. = 195.0873 Da). The amount of melatonin in the samples ranged from 170 mg to 6407 mg per kg drug for the UHPLC-qTOFMS determinations and from 244 mg to 6848 mg per kg drug for the ASAP analysis. The deviation from the nominal value was calculated for each sample containing melatonin and is depicted in Fig. 5. Generally, the mathematical sign for the deviation was the same for every sample, except for sample 4 and sample 19. However, those samples had a slight deviation of less than 4%. In most cases, ASAP measurements delivered higher deviations than the chromatographic measurements, except for samples 5 and 9. The highest deviations from nominal values were observed in samples 2, 14 and 16. Exemplarily in sample 14, less than 50% of the stated amount of 5 mg per capsule was quantified with both analytical methods. In contrast, samples 2 and 16 contained more melatonin than advertised.
 |
| | Fig. 4 ASAP mass spectra; (A) background; (B) standard 5 mg kg−1; (C) sample 9 – a melatonin containing phyto-narcotic. | |
 |
| | Fig. 5 Deviation of the melatonin containing extracts. Specified values compared with the measured values (orange = UHPLC-qTOFMS; green = ASAP). | |
For the direct comparison of the two applied methods, two graphs were generated. Fig. 6(A) shows the measured values of both instruments with a linear regression analysis. The slope was close to unity. Additionally, an intercept of less than 10% of the lowest standard concentration is an indicator of the comparability of the two methods. The coefficient of determination was above 0.94. Fig. 6(B) shows a Bland–Altman plot, where the mean of each method is plotted against the relative difference between the methods. Three outliers have been noticed. Two of them are samples (sample 2, the sample with the lowest concentration and sample 3) and one is a standard (1 mg kg−1, i.e., the lowest concentration standard). All other measurements were within the ± 1.96 SD confidence interval. The relative difference between the mean of all measurements was 3.6%.
 |
| | Fig. 6 Comparison of both applied analytical methods; black squares = samples, red circles = standards: (A) linear regression of UHPLC-qTOFMS and ASAP measurements (concentration of melatonin in mg kg−1); (B) Bland–Altman plot with mean values and the relative difference between both methods. | |
4. Conclusions
Quantification of melatonin with ASAP-MS is a cost-efficient, reliable and fast alternative to conventional methods like UHPLC-qTOFMS. One single analysis with ASAP-MS takes less than 1 min, whereas chromatographic methods typically take 5 min to 30 min. Moreover, ASAP-MS does not require the use of solvents that are potentially hazardous to the environment. The major disadvantage of ASAP-MS is isobars that can be present in the sample. Without a further dimension of separation, it is not possible to distinguish between them. Thus, validation of ASAP-based methods is highly important with a focus on accuracy. This problem could be solved, if high-resolution mass analysers or triple quadrupoles are used instead of low-resolution single quadrupoles. With the former mentioned systems, it would be possible to uncover interferences by evaluating the fragmentation patterns. Moreover, the results showed that the amount of melatonin in over-the-counter products is diverging from the specified value in many cases.
Author contributions
Daniel Moser: conceptualization, data curation, formal analysis, investigation, methodology, software, validation, visualization, writing – original draft. Shah Hussain: conceptualization, methodology, resources, supervision, writing – review & editing. Matthias Rainer: methodology, project administration, supervision, writing – review & editing. Thomas Jakschitz: conceptualization, resources, supervision, writing – review & editing. Günther K. Bonn: project administration, supervision, writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would like to acknowledge and express their gratitude to Nora Engels for grammar and spell checking.
References
- M. Singh and H. R. Jadhav, Melatonin: functions and ligands, Drug Discovery Today, 2014, 19, 1410–1418 CrossRef CAS PubMed.
- J. J. Gooley, K. Chamberlain, K. A. Smith, S. B. S. Khalsa, S. M. W. Rajaratnam, E. van Reen, J. M. Zeitzer, C. A. Czeisler and S. W. Lockley, Exposure to room light before bedtime suppresses melatonin onset and shortens melatonin duration in humans, J. Clin. Endocrinol. Metab., 2011, 96, E463–E472 CrossRef CAS PubMed.
- X. Meng, Y. Li, S. Li, Y. Zhou, R.-Y. Gan, D.-P. Xu and H.-B. Li, Dietary Sources and Bioactivities of Melatonin, Nutrients, 2017, 9(4), 367 CrossRef PubMed.
- M. Iriti, E. M. Varoni and S. Vitalini, Melatonin in traditional Mediterranean diets, J. Pineal Res., 2010, 49, 101–105 CAS.
- Z. Xie, F. Chen, W. A. Li, X. Geng, C. Li, X. Meng, Y. Feng, W. Liu and F. Yu, A review of sleep disorders and melatonin, Neurol. Res., 2017, 39, 559–565 CrossRef CAS PubMed.
- S. Bent, A. Padula, D. Moore, M. Patterson and W. Mehling, Valerian for sleep: a systematic review and meta-analysis, Am. J. Med., 2006, 119, 1005–1012 CrossRef PubMed.
- H. Schiller, A. Forster, C. Vonhoff, M. Hegger, A. Biller and H. Winterhoff, Sedating effects of Humulus lupulus L. extracts, Phytomedicine, 2006, 13, 535–541 CrossRef CAS PubMed.
- A. Soltanpour, F. Alijaniha, M. Naseri, A. Kazemnejad and M. R. Heidari, Effects of Melissa officinalis on anxiety and sleep quality in patients undergoing coronary artery bypass surgery: A double-blind randomized placebo controlled trial, Eur. J. Integr. Med., 2019, 28, 27–32 CrossRef.
- F. A. Guerrero and G. M. Medina, Effect of a medicinal plant (Passiflora incarnata L) on sleep, Sleep Sci., 2017, 10, 96–100 CrossRef PubMed.
- A. Deshpande, N. Irani, R. Balkrishnan and I. R. Benny, A randomized, double blind, placebo controlled study to evaluate the effects of ashwagandha (Withania somnifera) extract on sleep quality in healthy adults, Sleep Med., 2020, 72, 28–36 CrossRef PubMed.
- C. Agostoni, J.-L. Bresson, S. Fairweather-Tait, A. Flynn, I. Golly, H. Korhonen, P. Lagiou, M. Løvik, R. Marchelli, A. Martin, B. Moseley, M. Neuhäuser-Berthold, H. Przyrembel, S. Salminen, Y. Sanz, S. J. J. Strain, S. Strobel, I. Tetens, D. Tomé, H. van Loveren and H. Verhagen, Scientific Opinion on the substantiation of a health claim related to melatonin and reduction of sleep onset latency (ID 1698, 1780, 4080) pursuant to Article 13(1) of Regulation (EC) No 1924/2006, EFSA J., 2011, 9, 2241 CrossRef.
- C. Garzón, J. M. Guerrero, O. Aramburu and T. Guzmán, Effect of melatonin administration on sleep, behavioral disorders and hypnotic drug discontinuation in the elderly: a randomized, double-blind, placebo-controlled study, Aging: Clin. Exp. Res., 2009, 38–42 Search PubMed.
- A. Carrillo-Vico, P. J. Lardone, N. Alvarez-Sánchez, A. Rodríguez-Rodríguez and J. M. Guerrero, Melatonin: buffering the immune system, Int. J. Mol. Sci., 2013, 14, 8638–8683 CrossRef PubMed.
- H. Tamura, A. Takasaki, T. Taketani, M. Tanabe, L. Lee, I. Tamura, R. Maekawa, H. Aasada, Y. Yamagata and N. Sugino, Melatonin and female reproduction, J. Obstet. Gynaecol. Res., 2014, 40, 1–11 CrossRef CAS PubMed.
- C. Li and X. Zhou, Melatonin and male reproduction, Clin. Chim. Acta, 2015, 446, 175–180 CrossRef CAS PubMed.
- L. Lin, Q.-X. Huang, S.-S. Yang, J. Chu, J.-Z. Wang and Q. Tian, Melatonin in Alzheimer’s disease, Int. J. Mol. Sci., 2013, 14, 14575–14593 CrossRef PubMed.
- R. Hardeland, D. P. Cardinali, G. M. Brown and S. R. Pandi-Perumal, Melatonin and brain inflammaging, Prog. Neurobiol., 2015, 127–128, 46–63 CrossRef CAS PubMed.
- A. Cutando, A. López-Valverde, S. Arias-Santiago, J. de Vicente and R. Gómez de Diego, Role of Melatonin in Cancer Treatment, Anticancer Res., 2012, 32, 2747–2754 CAS.
- Y. Li, S. Li, Y. Zhou, X. Meng, J.-J. Zhang, D.-P. Xu and H.-B. Li, Melatonin for the prevention and treatment of cancer, Oncotarget, 2017, 24, 39896–39921 CrossRef PubMed.
- J. Cipolla-Neto, F. G. Amaral, S. C. Afeche, D. X. Tan and R. J. Reiter, Melatonin, energy metabolism, and obesity: a review, J. Pineal Res., 2014, 56, 371–381 CrossRef CAS PubMed.
- A. Karamitri and R. Jockers, Melatonin in type 2 diabetes mellitus and obesity, Nat. Rev. Endocrinol., 2019, 15, 105–125 CrossRef CAS PubMed.
- D. Acuña-Castroviejo, G. Escames, J. C. Figueira, P. de La Oliva, A. M. Borobia and C. Acuña-Fernández, Clinical trial to test the efficacy of melatonin in COVID-19, J. Pineal Res., 2020, 69, e12683 CrossRef PubMed.
- R. J. Reiter, P. Abreu-Gonzalez, P. E. Marik and A. Dominguez-Rodriguez, Therapeutic Algorithm for Use of Melatonin in Patients With COVID-19, Front. Med., 2020, 7, 226 CrossRef PubMed.
- P. W. Stege, L. L. Sombra, G. Messina, L. D. Martinez and M. F. Silva, Determination of melatonin in wine and plant extracts by capillary electrochromatography with immobilized carboxylic multi-walled carbon nanotubes as stationary phase, Electrophoresis, 2010, 31, 2242–2248 CrossRef CAS PubMed.
- E. Molaakbari, A. Mostafavi and H. Beitollahi, Simultaneous electrochemical determination of dopamine, melatonin, methionine and caffeine, Sens. Actuators, B, 2015, 208, 195–203 CrossRef CAS.
- A. Radi and G. E. Bekhiet, Voltammetry of melatonin at carbon electrodes and determination in capsules, Bioelectrochem. Bioenerg., 1998, 45, 275–279 CrossRef CAS.
- S. Burkhardt, D. X. Tan, L. C. Manchester, R. Hardeland and R. J. Reiter, Detection and quantification of the antioxidant melatonin in Montmorency and Balaton tart cherries (Prunus cerasus), J. Agric. Food Chem., 2001, 49, 4898–4902 CrossRef CAS PubMed.
- L. C. Brazaca, C. B. Bramorski, J. Cancino-Bernardi, S. Da Silveira Cruz-Machado, R. P. Markus, B. C. Janegitz and V. Zucolotto, An antibody-based platform for melatonin quantification, Colloids Surf., B, 2018, 171, 94–100 CrossRef CAS PubMed.
- D. J. Kennaway, Measuring melatonin by immunoassay, J. Pineal Res., 2020, 69, e12657 CrossRef CAS PubMed.
- J. Lu, C. Lau, M. Koo Lee and M. Kai, Simple and convenient chemiluminescence method for the determination of melatonin, Anal. Chim. Acta, 2002, 193–198 CrossRef.
- G. Simonin, L. Bru, E. Lelièvre, J.-P. Jeanniot, N. Bromet, B. Walther and C. Boursier-Neyret, Determination of melatonin in biological fluids in the presence of the melatonin agonist S 20098: comparison of immunological techniques and GC-MS methods, J. Pharm. Biomed. Anal., 1999, 21, 591–601 CrossRef CAS PubMed.
- A. B. Cerezo, Á. Leal, M. A. Álvarez-Fernández, R. Hornedo-Ortega, A. M. Troncoso and M. C. García-Parrilla, Quality control and determination of melatonin in food supplements, J. Food Compos. Anal., 2016, 45, 80–86 CrossRef CAS.
- C. Pape and K. Lüning, Quantification of melatonin in phototrophic organisms, J. Pineal Res., 2006, 41, 157–165 CrossRef CAS PubMed.
- K. Eriksson, A. Östin and J.-O. Levin, Quantification of melatonin in human saliva by liquid chromatography–tandem mass spectrometry using stable isotope dilution, J. Chromatogr. B, 2003, 794, 115–123 CrossRef CAS.
- T. Kocadağlı, C. Yılmaz and V. Gökmen, Determination of melatonin and its isomer in foods by liquid chromatography tandem mass spectrometry, Food Chem., 2014, 153, 151–156 CrossRef PubMed.
- D. González-Gómez, M. Lozano, M. F. Fernández-León, M. C. Ayuso, M. J. Bernalte and A. B. Rodríguez, Detection and quantification of melatonin and serotonin in eight Sweet Cherry cultivars (Prunus avium L.), Eur. Food Res. Technol., 2009, 229, 223–229 CrossRef.
- X. Huang and G. Mazza, Application of LC and LC-MS to the analysis of melatonin and serotonin in edible plants, Crit. Rev. Food Sci. Nutr., 2011, 51, 269–284 CrossRef CAS PubMed.
- R. M. Alberici, R. C. Simas, G. B. Sanvido, W. Romão, P. M. Lalli, M. Benassi, I. B. S. Cunha and M. N. Eberlin, Ambient mass spectrometry: bringing MS into the real world, Anal. Bioanal. Chem., 2010, 398, 265–294 CrossRef CAS PubMed.
- C. Black, O. P. Chevallier and C. T. Elliott, The current and potential applications of Ambient Mass Spectrometry in detecting food fraud, TrAC, Trends Anal. Chem., 2016, 82, 268–278 CrossRef CAS.
- M.-Z. Huang, S.-C. Cheng, Y.-T. Cho and J. Shiea, Ambient ionization mass spectrometry: a tutorial, Anal. Chim. Acta, 2011, 702, 1–15 CrossRef CAS PubMed.
- D. Burns, S. Mathias, B. J. McCullough, C. J. Hopley, D. Douce, N. Lumley, S. Bajic and P. Sears, Ambient ionisation mass spectrometry for the trace detection of explosives using a portable mass spectrometer, Int. J. Mass Spectrom., 2022, 471, 116735 CrossRef CAS.
- H. R. Tan, L. Y. Chan, H. H. Lee, Y.-Q. Xu and W. Zhou, Rapid authentication of Chinese oolong teas using atmospheric solids analysis probe-mass spectrometry (ASAP-MS) combined with supervised pattern recognition models, Food Control, 2022, 134, 108736 CrossRef.
- D. Moser, S. Hussain, M. Yaqoob, M. Rainer, T. Jakschitz and G. K. Bonn, Fast and semiquantitative screening for sildenafil in herbal over-the-counter formulations with atmospheric pressure solid analysis probe (ASAP) to prevent medicinal adulteration, J. Pharm. Biomed. Anal., 2022, 214, 114720 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ay00352j |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Shah
Hussain
ab,
Shah
Hussain