
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantification of drug loading in polymeric nanoparticles using AFM-IR technique: a novel method to map and evaluate drug distribution in drug nanocarriers†
M. Seray
Ural
a,
Emmanuel
Dartois
a,
Jérémie
Mathurin
 b,
Didier
Desmaële
c,
Philippe
Collery
d,
Alexandre
Dazzi
b,
Ariane
Deniset-Besseau
*b and
Ruxandra
Gref
a
b,
Didier
Desmaële
c,
Philippe
Collery
d,
Alexandre
Dazzi
b,
Ariane
Deniset-Besseau
*b and
Ruxandra
Gref
a
aInstitut de Sciences Moléculaires d'Orsay (ISMO), CNRS UMR 8214, Université Paris-Saclay, 91405, Orsay, France. E-mail: ruxandra.gref@universite-paris-saclay.fr
bInstitut de Chimie Physique (ICP), CNRS UMR 8000, Université Paris-Saclay, 91405, Orsay, France. E-mail: ariane.deniset@universite-paris-saclay.fr
cInstitut Galien (IGPS), CNRS UMR 8612, Université Paris-Sud, Université Paris-Saclay, 91405, Orsay, France
dSociety for the Coordination of Therapeutic Research, 20220, Algajola, France
First published on 31st October 2022
Abstract
Researchers are increasingly thinking smaller to solve some of the biggest challenges in nanomedicine: the control of drug encapsulation. Although recent years have witnessed a significant increase in the development and characterization of polymeric drug nanocarriers, several key features are still to be addressed: Where is the drug located within each nanoparticle (NP)? How much drug does each NP contain? Is the drug loading homogeneous on an individual NP basis? To answer these questions, individual NP characterization was achieved here by using atomic force microscopy-infrared spectroscopy (AFM-IR). A label-free quantification methodology was proposed to estimate with a nanoscale resolution the drug loadings of individual poly(lactic acid) (PLA) NPs loaded with an anticancer drug. First, a drug loading calibration curve was established using conventional IR microspectroscopy employing PLA/drug homogeneous films of well-known compositions. Then, single NPs were investigated by AFM-IR acquiring both IR mappings of PLA and drug as well as local IR spectra. Besides, drug location within single NPs was unravelled. The measured drug loadings were drastically different (0 to 21 wt%) from one NP to another, emphasizing the particular interest of this methodology in providing a simple quantification method for the quality control of nanomedicines.
Introduction
Nanotechnology provides the opportunity to organize matter at the nanometer scale, building supramolecular constructs with particular interest for healthcare. Drug nanocarriers can protect and ferry the active molecules from their administration site to the targeted tissues, organs, or cells.1 Poly(lactic acid) (PLA) and poly(lactic-co-glycolic acid) (PLGA) (co)polymers remain among the most employed biomaterials to engineer drug nanocarriers, due to their well-established safety profiles, biocompatibility and biodegradability.2 However, to date, only a few PLA and PLGA nanoparticle (NP) formulations reached the clinical trials stage.3,4 Indeed, on the journey from bench to bedside, the quality control of NP formulations, encompassing accurate estimations of size distributions and drug incorporation is a challenging step.Drug loading (DL) is one of the key parameters that determine the therapeutic efficacy of NPs. It has to be carefully optimized as an important prerequisite for preclinical development.5 In the majority of the studies, DL was determined by separating the NPs from their suspension media, in which the non-encapsulated drug was quantified by chromatographic or spectroscopic methods. These measurements provide average values on large populations of NPs but cannot monitor individually their heterogeneities in composition. Indeed, individual drug-loaded NPs might be drastically different in terms of DL and/or drug location.6 These factors are not only important for quality control, but they also play a major role in the drug release mechanism and the interplay with biological media. Significant DL variations could render only a fraction of the NPs therapeutically active.7,8 To the best of our knowledge, today, there is no available method able to assess DL and the molecular integrity of the drug when inserted in the NPs, on an individual NP basis.
There is a clear need to develop a characterization methodology that would enable investigating polymeric NPs to: (i) map the location of the drug with high resolution and (ii) perform quantitative analysis of the drug content at the nanoscale level.9
Only a few techniques including high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) enable mapping the constitutive elements of the NPs. However, STEM-HAADF is not adapted to the detection of carbon, hydrogen and oxygen (main components of drug-loaded polymeric NPs) because of the presence of carbon in the grids, trace water, and possible impurities in the NPs. Besides, this method only tracks elements, but not whole molecules, therefore cannot ascertain drug location. Single-particle inductively coupled plasma mass spectrometry (spICP-MS) could provide element mass concentration on single NP but it excludes carbon-based nanomaterials due to the intrinsic low sensitivity of carbon in ICP-MS.10
Electron microscopes (EMs) coupled with energy-dispersive X-ray spectroscopy were also used for chemical mappings, but sensitive polymeric NPs often suffer significant damages under the electron beam.9 In addition, EMs often require cumbersome sample preparation and do not allow three-dimensional imaging. Raman microspectroscopy imaging coupled with chemical analysis is an interesting alternative, but the method remains restricted by the diffraction limit, and its spatial resolution is too low to explore the compositon of NPs smaller than 300 nm.11
Our study aims at mapping the location of a drug within its polymeric NP matrix with a high spatial resolution (around 15 nm) and determining the individual drug loading of a NP batch. These goals were achieved here in the case of PLA NPs loaded with an anticancer drug by using atomic force microscopy (AFM) coupled to infrared spectroscopy (IR). AFM-IR is a method of choice, which combines the chemical sensitivity of IR with the spatial resolution of scanning probe microscopy, AFM. It is based on the detection of the photo-thermal process that occurs within the sample after its illumination via a tunable pulsed IR laser.12–14 Indeed, when molecules absorb at the tuned wavenumber of the IR laser, this results in a fast and highly localized heating leading to a rapid thermal expansion of the sample, detected by the AFM tip in contact with the surface.15 Noteworthy, the AFM-IR signal is proportional to the local IR absorption.12,13 Overall, AFM-IR allows chemical analysis overcoming the classical IR diffraction limit, achieving a nanometer-size resolution (∼15 nm).16 Noteworthy, AFM-IR can be operated either in top-down illumination17 where the laser shines directly on the sample, or in bottom-up configuration18 where the specimen is illuminated via an evanescent field wave through an IR transparent prism. Furthermore, the AFM-IR measurements can be performed using the contact,14 the tapping,19 or the peak force mode20 of the AFM. AFM-IR appears a priori as a versatile technique to be used on almost any organic material under ambient conditions with a high resolution that is promising to quantify the composition of individual drug-loaded NPs.
Pioneering studies in our groups demonstrated that AFM-IR is a reliable, non-destructive and direct method able to precisely locate and chemically characterize polymeric NPs19,21 as well as investigation from another group on liposomal nanocarriers.22 In another study, PLA NPs of about 150 nm in diameter were characterized inside the cells without the need for labelling.23 This was a great advantage, as previous studies reported that labelling with fluorescent dyes or other molecules can significantly alter the physicochemical properties of the NPs and their interactions with biological systems.24 The location of the drug incorporated in PLA NPs was precisely assessed despite the very low loadings (<1 wt%).19 Surprisingly, it was found that the majority of the drug was in the NP shell consisting of poly(vinyl alcohol) (PVA) chains intermingled with the PLA cores. This location correlated well with the fast (“burst”) release of the drug. In our investigations, AFM-IR was used in the tapping mode, which is more appropriated than the contact mode for soft and the loosely adhesive PLA NPs.19
Scarce attempts have been made so far to use AFM-IR for quantification purposes. In 2016, Tang et al.25 investigated the distribution of polyethylene components in high impact polypropylene alloys by using AFM-IR in contact mode, using a set up with bottom-up illumination that necessitates the preparation of thin specimens on prisms and requires a perfect sample to prism adhesion. This study pointed out the necessity of an external calibration to lead to quantitative analysis by AFM-IR. However, the proposed calibration by ATR-FTIR has the inconvenience of the dependence of the analysed depth with the wavelength (the penetration depth of the evanescent wave increases at lower wavenumbers, and it must be adequately considered). This semiquantitative method is therefore challenging to be adapted for calibration purposes. More recently, Ramer et al. conducted measurements using AFM-IR in contact mode, once again with a bottom-up illumination, and analysed the sample parameters (thickness, homogeneity, intensity of absorption bands) in an attempt to achieve a rigorous quantitative measurement on poly(methyl methacrylate) films of 43 to 1243 nm thick. They suggested that thin samples (<500 nm) should be used to maintain a linear dependence of signal intensity with the amount of analysed sample.26
To the best of our knowledge, no study has considered yet local component quantification on NP systems by using AFM-IR, neither in top-down nor in bottom-up configuration. The top-down approach in comparison to bottom-up, simplifies the sample preparation and is less constrained by the physical properties of the sample (size, shape, transparency, etc.) therefore it is adapted to routine analysis.
In this work, AFM-IR was in top-down configuration and operated in the tapping mode as the best configuration to limit deformation or displacements of PLA NPs.19 We present the quantification of the DL on single PLA NPs from a heterogenous preparation deposited on flat surface appropriated to IR study. The quantification methodology by employing AFM-IR was established by using a PLA NPs (size range of 200–300 nm) system loaded with a Re-based anticancer drug. Re(I) tricarbonyl-diselenium complex in addition to its remarkable anticancer activity, efficacy at low doses, and low toxicity27 exhibits through its Re-tricarbonyl chemical pattern a highly specific IR signature.
The methodology proposed here establishes a precise calibration of the infrared band intensities combining conventional IR microspectroscopy and AFM-IR measurements using reference thin films with well-known PLA and drug compositions. Drug loading was assessed on several NPs of a given region of interest through the local AFM-IR spectral analysis. Then, a fast local quantification method was deduced using only two IR maps. This paves the way to perform statistical analysis on numerous individual NPs within the same batch overcoming the slowness of spectral analysis. Unexpectedly, large differences were observed in the NPs' drug loading without any correlation with their sizes.
Experimental section
Chemicals
PLA acid terminated (MW: 6–10 kDa, EXPANSORB® 10P006) was kindly donated by Sequens (Expansorb, Aramon, France). Poly(vinyl alcohol) (PVA) (30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–70000 g mol−1, 88% hydrolyzed), Re(CO)5Cl, cyclohexane and acetone were purchased from Sigma-Aldrich and used as received.
000–70000 g mol−1, 88% hydrolyzed), Re(CO)5Cl, cyclohexane and acetone were purchased from Sigma-Aldrich and used as received.
Calcium fluoride (CaF2) optical windows used as AFM-IR substrates were purchased from Crystal GmbH (Germany). AFM-IR cantilevers (Multi75GB-G, f = 75 kHz 3 N m−1) were supplied from Budged Sensors (Germany). Eppendorf™ membrane filters (0.45 μm) were used to remove precipitates from NP suspensions.
Drug synthesis and characterization
The Re(I) tricarbonyl-diselenium drug was synthesized, as previously reported, by reacting the commercially available Re(CO)5Cl with a selenium containing ligand.28 Briefly, Re(CO)5Cl (0.154 g, 0.43 mmol) was added to the solution of 3,7-diselena-nonanedioic acid (0.135 g, 0.43 mmol) in 15 ml of THF. After stirring at reflux overnight THF was removed under reduced pressure, and the drug was recovered as a white powder. Yield: 0.240 g (90%); MS (positive mode): 589 ([M − Cl]+); IR: 3330–2340 (m), 2029 (s), 1935 (s), 1895 (s), 1731 (s), 1699 (s), 1415 (m), 1351 (m), 1282 (m), 1249 (m), 1206 (s), 1186 (m), 1092 (m), 834 (m), 785 (m), 752 (m), 670 (m), 637 (m), 624 (s), 605 (m) cm−1; 1H NMR (CD3OD): δ 2.6–3.9 (m, 10 H); Anal. Calc. for C10H12O7ClSe2Re: C, 19.25; H, 1.94. Found: C, 19.46; H, 1.97.Preparation of drug loaded PLA films
The physical mixtures of PLA and (Re)-based drug were prepared in acetone with drug:PLA mass ratios 0; 0.03; 0.05; 0.10; 0.15 and 0.20. The PLA concentration was 100 mg mL−1 for all the solutions. The PLA–drug mixtures were vigorously stirred overnight at room temperature to achieve complete dissolution. Then, 15 μL of each solution were casted onto glass plates and the solvent was allowed to evaporate overnight at room temperature. Translucent, homogeneous films were thus obtained and named according to their drug:PLA mass ratios as F(0); F(0.03); F(0.10); F(0.15) and F(0.20). They were further analysed in IR microspectroscopy. For AFM-IR studies, films were prepared and analysed directly on CaF2 windows. To do so, the previous PLA–drug mixtures were diluted ten times in acetone and 5 μL of these solutions were casted and spread on the CaF2 windows, followed by complete solvent evaporation.
Preparation and characterization of drug loaded PLA NPs
Drug-loaded PLA NPs were prepared by nanoprecipitation. Briefly, 0.5 mL of drug–PLA mixtures (drug:PLA mass ratios of 0; 0.03; 0.05; 0.10; 0.15 and 0.20) was added drop-wisely into 5 mL of 0.5% PVA aqueous solution under continuous vigorous magnetic stirring. The rapid solvent diffusion in the aqueous phase led to instantaneous precipitation of the polymer to form NPs. The organic solvent was evaporated at room temperature under gentle magnetic stirring. Control NPs were prepared under the same conditions except that no drug was added. Resulting NPs were named as NP(0); NP(0.03); NP(0.10); NP(0.15) and NP(0.20), according to the drug:PLA mass ratios used in their preparation. NP suspensions were filtrated through a cellulose 0.45 μm membrane to remove precipitates. These NPs were stored at 4 °C. All formulations were prepared at least in triplicate.
DL, a commonly used term to describe drug quantity in drug delivery systems, was defined as the mass fraction of NPs that is composed of drug and was determined according to following eqn (1):
 | (1) |
:PLA mass ratio in the NPs, DL was represented as (2): | (2) |
The NP average hydrodynamic diameter was determined by DLS at 25 °C with an equilibration time of 60 s using a Zetasizer® (Nano ZS90, Malvern Instruments, Worcestershire, UK). All formulations were measured at least in triplicate. Mean diameters were reported as Z average (nm) ± SE (standard error) with the polydispersity index (PdI). The NPs mean diameters were monitored during three months of storage at 25 °C.
TEM
TEM images of NPs were acquired using a JEOL electron microscope (JEM 100 CX II, operating at 120 kV) equipped with an US1000 2k × 2k camera (Gatan). To do so, 5 μL of samples were deposited onto a 400 mesh carbon-coated copper grid beforehand treated with a glow discharge (easiGlow, Ted Pella Inc).ATR-FTIR spectroscopy
ATR-FTIR spectra of pure drug, PLA and PVA were obtained by using a Fourier Transform Bruker Vertex 70 spectrometer (MCT detector, cooled down using liquid nitrogen) and an ATR module (PIKE MIRacle crystal plate diamond ZnSe). Spectra were recorded in the 4000–600 cm−1 range with an accumulation of 100 scans and a spectral resolution of 4 cm−1.IR microspectroscopy
IR microspectroscopy measurements were recorded on a Nicolet Continuum infrared microscope (Thermo Fisher Scientific) equipped with a liquid nitrogen cooled MCT detector, with a ×32 objective. Spectra were recorded in transmission mode covering the 4000 to 650 cm−1 energy range. Spectra were acquired by averaging 128 scans and were recorded at 2 cm−1 spectral resolution. The aperture dimension on the sample was set to 50 × 50 or 75 × 75 μm, depending on the local homogeneity of the area.Films and NPs were analysed as bulk. For a reliable quantitative analysis, the thickness of the analyzed samples was carefully optimized and controlled using a diamond compression cell. Smooth and flat samples in the μm thickness range were obtained.
For each sample, at least five independent areas were analysed. Before each analysis, a background spectrum was collected in a clean region of the diamond substrate without sample. The transmittance spectrum was obtained by dividing the sample spectrum by this background. The spectra were extracted in transmittance, and samples thin film interference fringes were baseline-corrected through a spline function generated in the software Origin Pro 8. Then spectra were converted to absorbance.
AFM-IR
NanoIR-2 (Anasys Instruments, Santa Barbara) in top-down configuration coupled with a multichip QCL source (MIRcat, Daylight Solutions; tuneable repetition rate range of 0–2 MHz; maximal spectral resolution of 0.1 cm−1) covering the range from 890 cm−1 to 1945 cm−1 of the mid-IR region with a tuneable repetition rate was used in tapping mode. The setup was described previously in details.19 The QCL beam was focused onto the gold-coated silicon probe (tapping AFM-IR cantilever, Multi75GB-G, f = 75 kHz 3 N m−1, Budget Sensors) that avoids the effects linked to IR absorption of the silicon cantilever. Measurements were carried out with the tapping oscillation at the fundamental mode of cantilever and IR absorption was detected using the second mode.19 AFM tip was fixed at a specific location to acquire local IR spectra (also line or matrix spectra) by tuning the laser wavenumber with a spectral resolution of 1 cm−1. Topographic images were acquired simultaneously with IR absorption maps at the specific wavenumbers. IR map of single NP was performed at 23 different wavenumbers, precisely at 1700, 1720, 1730, 1740, 1750, 1755, 1760, 1765, 1770, 1780, 1790, 1800, 1810, 1820, 1830, 1840, 1850, 1860, 1880, 1885, 1890, 1900 and 1905 cm−1. They were then analysed using the MountainMap 7.3 software. To each recorded image, a background correction was applied, followed by a Gaussian filtering of 3 × 3 pixels. Threshold was performed by setting non-measured points to grey. Gold palette was then applied to the final images.150 local IR spectra were taken for each film in three different locations and were analysed on Orange 3.27.1. Spectra were filtered using the Savitzky–Golay algorithm (2nd order polynomial using 15 points), followed by pre-processing with extended multiplicative signal correction (EMSC) since it deals with variations in the vertical scale and offsets.29 Resulting spectra were baseline subtracted on the y-axis in order to set the baseline at zero outside the absorption regions, prior to calculate the intensity band ratios.
IR microspectroscopy and AFM-IR data treatment
In order to establish comparability between classical IR-microspectroscopy and AFM-IR spectra, we took into consideration two factors. The first is that the measured AFM-IR bands appeared slightly broader than the FTIR microspectroscopy. A simple broadening correction could enable to superimpose the two carbonyl bands of PLA from both measurements. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O band of PLA (at 1760 cm−1) of each measured film was then normalized to unity.
O band of PLA (at 1760 cm−1) of each measured film was then normalized to unity.
The second consideration was related to the spectral analysis range in AFM-IR. The quantum cascade laser (QCL) used in AFM-IR limits the measurements to about 1908 cm−1, where the laser power starts dropping significantly (Fig. SI1†). The useful spectral acquisition range was thus ended at 1908 cm−1.
To obtain a calibration curve, the intensity ratio I1900/I1760 (the intensity of the peaks corresponding to the drug (1900 cm−1) and to the PLA (1670 cm−1)) was plotted as a function of drug:PLA mass ratios (mdrug/mPLA).
IR spectra of NP (as bulk) were measured and ratios were evaluated using the intensities of the carbonyl absorption in selected peaks corresponding to PLA and the drug. In this way, the drug:PLA mass ratios in each NP formulation was calculated from the calibration curve, and then converted to the DL (wt%).
Results and discussion
Preparation of NPs and films
The NPs were prepared with a precise drug:PLA mass ratio, from 0 (pure PLA) to 0.20. Empty NP(0), and drug-loaded PLA NPs were successfully obtained by nanoprecipitation using PVA as an emulsifier. They were named according to the drug:PLA mass ratios used in their preparation, as NP(0.03); NP(0.10); NP(0.15) and NP(0.20). After removing aggregates by filtration, the NPs were characterized (Table 1). Mean hydrodynamic diameters of the NPs ranged from 249 to 264 nm, and polydispersities from 0.1 to 0.3. Noteworthy, the drug content in the formulations had no significant impact on the NP size. The NPs suspensions were stable for 3 months after their preparation, with less than 5% size variations.
:PLA mass ratio)
| Name | Drug:PLA mass ratio |
Mean hydrodynamic diameter (nm) | PDI |
|---|---|---|---|
| NP(0) | 0 | 255 ± 10 | 0.32 ± 0.01 |
| NP(0.03) | 0.03 | 249 ± 10 | 0.21 ± 0.09 |
| NP(0.10) | 0.10 | 264 ± 12 | 0.09 ± 0.04 |
| NP(0.15) | 0.15 | 250 ± 18 | 0.30 ± 0.05 |
| NP(0.20) | 0.20 | 255 ± 10 | 0.29 ± 0.05 |
To complement the study, films were cast using exactly the same drug:polymer solutions which were used for NP preparation. The use of physical mixtures of drug and PLA with a well-known drug:polymer mass ratios enabled to obtain the signal of bulk materials, avoiding the issue of aggregate formation as was the case with NPs. The films were named according to their drug:PLA mass ratios as F(0); F(0.03); F(0.10); F(0.15) and F(0.20). All the films were translucent with a homogenous aspect.
TEM images showed that the NPs had spherical shapes, with sizes in the range of around 200–400 nm (Fig. 1), in agreement with DLS data (Table 1). The NP morphology was unaffected by the loaded drug.
 | ||
| Fig. 1 Typical TEM images of PLA NPs: (A) NP(0) and (B) NP(0.20). Scale bars are 500 nm. | ||
AFM-IR methodology
The films and NPs were characterized by AFM-IR in tapping mode which is schematically represented in Fig. 2. In the top-down configuration used here, a tuneable IR radiation is shed onto the sample (Fig. 2A). When a compound absorbs the IR source an immediate expansion occurs in a small volume and is detected by the tip in intermittent contact with the sample. The main idea was to irradiate the sample with specific wavelengths at which only one of the NP components (either drug or polymer) will respond (Fig. 2C and D). In this way, IR maps of both drug and polymer can be recorded. To do so, it was necessary to first investigate the IR spectrum of each component of the NP to identify their signature.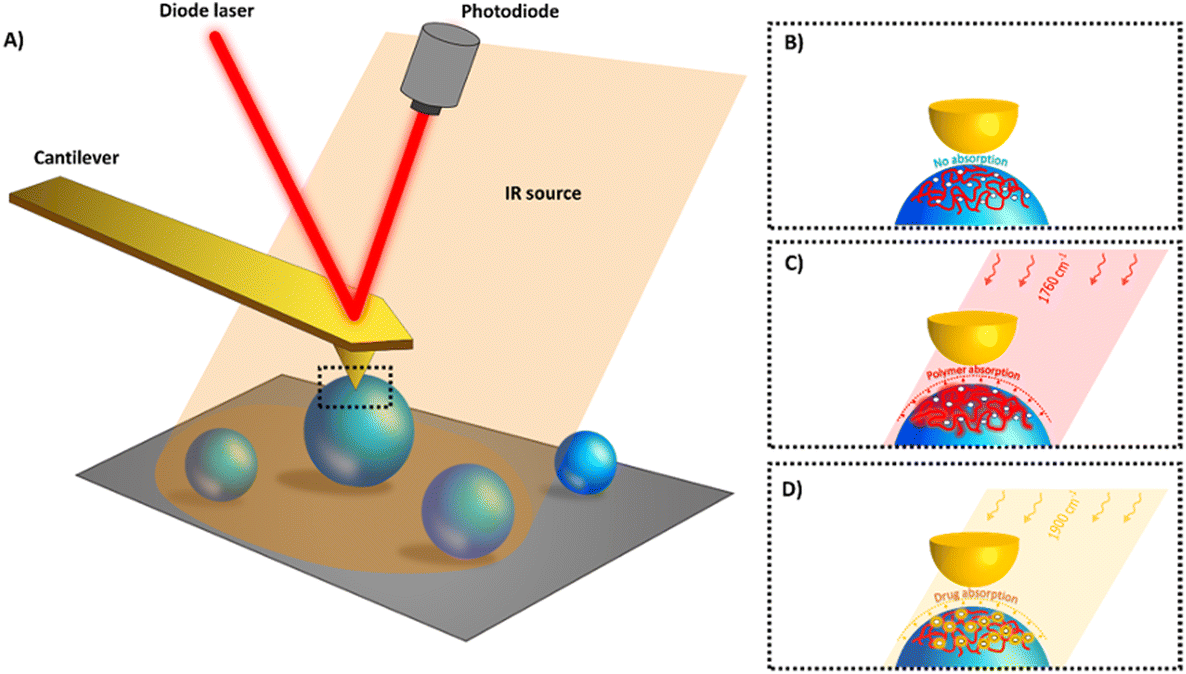 | ||
| Fig. 2 In AFM-IR with top-down configuration pulsed, tuneable laser light is focused onto the sample at the AFM tip location (A) enabling to analyse the surface of individual NPs (B). When the IR matches the drug (C) or the polymer (D) absorption band (“fingerprint”) a rapid thermal expansion occurs due to heating (few kelvins19,30). The picometric expansion affects the cantilever oscillations. IR maps at specific wavelengths corresponding to the polymer and/or the drug absorptions can be acquired. | ||
IR characterization
To specifically map each element constitutive of the films and NPs, isolated IR bands are needed, to avoid overlapping/mixing of the response with contributions from other components. The films and NPs mixtures were characterized by ATR-FTIR to determine their absorption frequencies, focusing in particular on the carbonyl bands. To begin with, the IR absorptions of each component (PLA, drug and PVA) were recorded separately and their corresponding IR bands were attributed (Fig. SI2†).
Fig. 3 presents the IR absorption bands in the region 1675–1908 cm−1 in which the AFM-IR experiments are carried on. The optimal bands selected to discriminate the PLA from the drug are located at 1760 cm−1 for PLA and 1900 cm−1 for the drug. The former band corresponds to the CO stretching of the carbonyl of the ester function of PLA while the latter correspond to the A′ Cs stretching mode of the octahedral fac-Re(CO)3 moiety.31 In addition, this choice ascertains that the presence of eventual residual PVA in the NPs formulations does not interfere with the detection of the PLA matrix and that of the incorporated drug, as PVA presents in the selected region a CO stretching absorption around 1724 cm−1 (Fig. 3).
 | ||
| Fig. 3 Chemical structures and ATR-FTIR spectra of PLA (black), PVA (blue) and drug (red). | ||
Calibration by IR microspectroscopy
After determining the drug and PLA IR signature, the next goal was to establish the relationship between the intensities of the respective IR signals of drug and polymer and their concentration inside the analysed samples, films and NPs, in order to establish a calibration curve to be used throughout the study, for the proof of concept and for drug quantification by AFM-IR. To do so, IR microspectroscopy was employed as an accurate analytical tool to gain spatially resolved chemical information. It allowed investigating films and NP samples that were gently compressed and flattened to a fine and uniform thickness (∼1 μm) ensuring non-saturated absorptions. First, PLA–drug films were prepared with known respective concentrations of each component. Analysed by IR microspectroscopy, the casted films exhibited typical Fabry–Perot interference fringes. The fringes appeared as sinusoidal waves that are characteristic of thin, non-scattering samples with uniform thicknesses.32 Chemical analysis of at least 5 independent domains of 50 × 50 μm or 75 × 75 μm (rectangular aperture) on each film preparation was performed to gain insights into the homogeneity of bulk samples at the spatial scale typically probed by classical IR microspectroscopy, i.e. of a few tens of microns. The domains analysed showed reproducible spectra demonstrating that the samples were homogeneous at such analysis scale.
Fig. 4A shows typical spectral features of drug–PLA films, F(0)–F(0.20), which were normalized to the PLA band intensity at 1760 cm−1 (I1760). Information in this region was comparable to ATR-FTIR data (Fig. 3). Band intensity at 1900 cm−1 (I1900) steadily increased with the amount of drug incorporated in the film (Fig. 4A). A linear relationship (R2 = 0.98) was found by plotting the ratios of the intensities of the specific carbonyl bands of drug and polymer (I1900/I1760) against the drug:polymer mass ratios (mdrug/mPLAFig. 4B). As the amount of PLA (mPLA) was kept constant throughout the experiments, this means that I1900/I1760 is proportional to the drug content in this explored drug concentration range. This linear relationship was further used as a calibration curve.
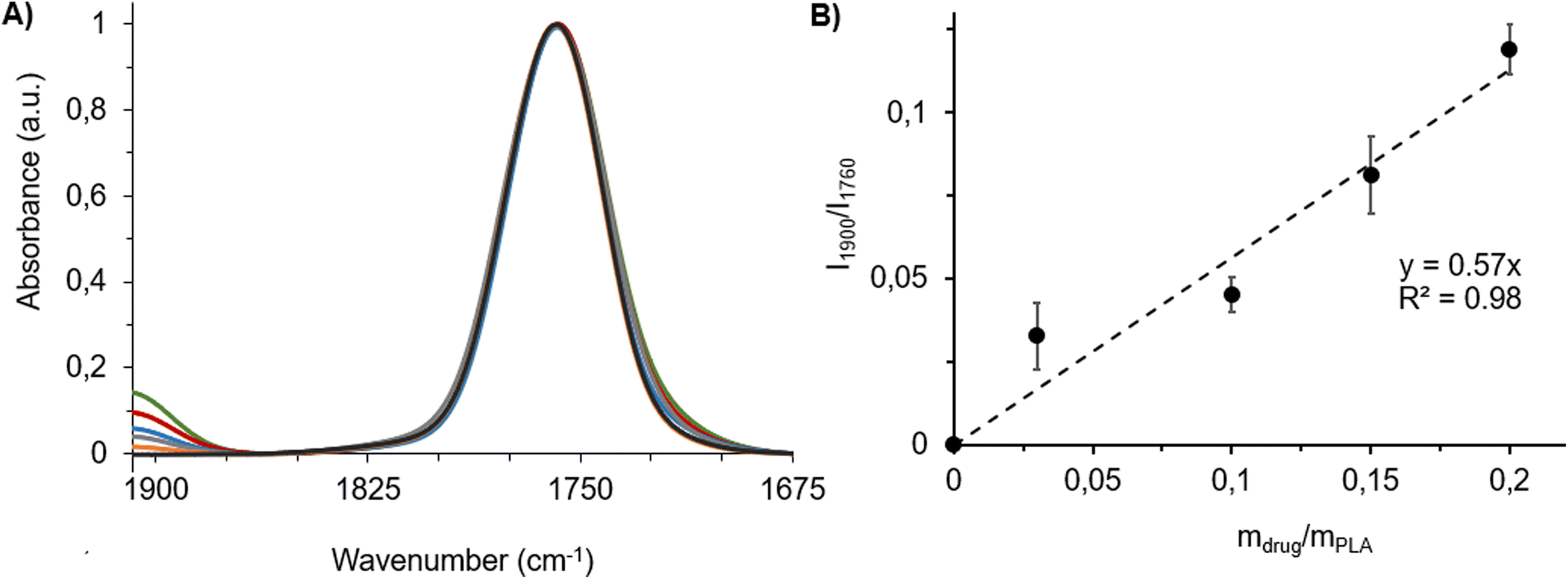 | ||
| Fig. 4 (A) IR absorption spectra of F(0) (black), F(0.03) (orange), F(0.10) (blue), F(0.15) (red) and F(0.20) (green) films recorded by IR microspectroscopy, normalised to the PLA carbonyl intensity. (B) Calibration curve and its respective calibration line. Drug:polymer mass ratios in the films were represented as mdrug/mPLA. Intensity ratios of carbonyl band of the drug to polymer were represented as I1900/I1760. The AFM-IR working range was between 1675 and 1908 cm−1 as detailed in the Experimental section. | ||
Fig. 5A represents the absorbance spectra of six NPs formulations prepared with different drug:polymer mass ratios. Carbonyl band intensity ratios of drug and polymer (I1900/I1760) were plotted as a function of the drug:polymer mass ratios (mdrug/mPLA) (Fig. 5B). The increase of the drug absorption (1900 cm−1) was evident upon the increase in its mass ratio. This indicates the loading of the drug into the NPs. The presence of residual PVA as well as the other absorption bands of the drug in the formulation were detected at around 1712 cm−1 as a shoulder contribution to the main PLA carbonyl band. The I1900/I1760 values were used to calculate the drug:polymer mass ratios for each NP, i.e. NP(0)–NP(0.20), by using the calibration curve established above (Fig. 4B). Table 2 presents the drug:polymer mass ratios, measured as 0.05 ± 0.02; 0.14 ± 0.03; 0.17 ± 0.04; 0.34 ± 0.04 and 0.35 ± 0.04 for NP(0); NP(0.03); NP(0.10); NP(0.15) and NP(0.20), respectively. The AFM-IR working range was in between 1675 and 1908 cm−1 as detailed in the Experimental section.
 | ||
| Fig. 5 (A) IR absorption spectra of NPs prepared with initial drug concentration of NP(0) (black), NP(0.03) (orange), NP(0.10) (blue), NP(0.15) (red) and NP(0.20) (green). (B) Intensity ratios of carbonyl bands (I1900/I1760) as a function of the drug:polymer mass ratios used in the NP preparation and calibration curve obtained with films of known mdrug/mPLA ratios (Fig. 4B). In addition, analysis of drug-loaded NPs was performed by IR microspectroscopy. | ||
:polymer mass ratios used in the preparation procedure (mdrug/mPLA), measured drug:polymer mass ratios and measured DLs of NPs: NP(0)–NP(0.20)
| NP(0) | NP(0.03) | NP(0.10) | NP(0.15) | NP(0.20) | |
|---|---|---|---|---|---|
| (mdrug/mPLA) | 0 | 0.03 | 0.10 | 0.15 | 0.20 |
| (mdrug/mPLA)meas. | 0 | 0.04 ± 0.01 | 0.24 ± 0.02 | 0.31 ± 0.04 | 0.36 ± 0.07 |
| DL (wt%) | 0 | 4.3 ± 1.2 | 19.5 ± 2.3 | 23.5 ± 3.8 | 26.5 ± 6.6 |
Table 2 shows that the measured drug:polymer mass ratios in the NPs are higher than the ones used for their preparation. This could be explained by the fact that, during NP formation, part of the polymer precipitated without forming NPs and did not entrap the drug. The “bulk” DLs of the NP formulations, calculated according to eqn (2), increased from 4.3 to 26.5 wt% with the drug:PLA mass ratio used for their preparation (Table 2). Noteworthy, as precipitates were formed during NP formation, the data altogether question the homogeneity of the NPs and point out the interest to investigate and determine the DL of individual NPs within a formulation. Before achieving this goal, the proof of concept of quantification from AFM-IR measurements was established on drug–PLA films with known compositions and proven to be homogeneous by IR microspectroscopy.
AFM-IR calibration curve using drug–PLA films
The aim was to establish the link between quantitative conventional IR microspectroscopy and AFM-IR, using the drug–PLA films; F(0); F(0.03); F(0.10); F(0.15) and F(0.20). Two main goals were pursued. First, the homogeneity of films was probed with the nanometric AFM resolution, to check if the analysed regions of interest are representative of the whole sample. To do so, 150 local absorption spectra were recorded in three different locations on each of the five investigated films. The IR intensities of both PLA and drug fingerprints (the I1900/I1760 values were used) were analysed and compared. Second, it was important to determine the sample thickness range in which quantification could be carried on by AFM-IR. Too thin samples could exhibit a low signal-to-noise ratio, thus not allowing an accurate analysis. Whereas, too thick sample may present a non-homogenous response: the AFM-IR signal might arise only from the part of the sample probed and not the whole sample due to the penetration depth of the detection, and this can lead to misinterpretations of the sample overall composition. Previous quantifications of film compositions by AFM-IR were all carried on in contact mode with a bottom-up configuration,25,26 and the AFM-IR signal was proven to be linear up to about 1 μm sample thickness.26,33,34 However, contact mode and bottom-up illumination were shown to be inadequate for NP investigation.19 It was thus essential to quantify sample composition by AFM-IR used in tapping mode with a top-down illumination. Films were cast with thicknesses lower than 500 nm, to cover the range of NP's thickness. Special attention was given to the analysis of regions of the film which offered an extended range of thicknesses, increasing progressively but not sharply. In addition, films with a wide range of compositions were analyzed to study the relationship between the AFM-IR signal and the relative concentration of each of their components. Indeed, a linear relationship is a requirement for quantitative analysis by AFM-IR.
Fig. 6 summarizes the strategy pursued for the films analysis, focusing on the example of F(0.15). The AFM topography image was recorded at the edge of the film to study the IR signal versus the thickness (Fig. 6A). Local spectra acquisition was performed on a region of 5 μm length represented by the green line on the topography. The corresponding height profile shown in Fig. 6B shows that the film thickness ranges from 35 nm to 425 nm. The IR intensity values of PLA (1760 cm−1) and drug (1900 cm−1) were plotted in function of the film thickness in Fig. 6C and D, revealing that the intensity values increase linearly with the sample thickness in the range from 60 to 420 nm sample thickness. The deviation from the linearity below 60 nm was related to the electromagnetic enhancement induced by gold-coated cantilever tip which may prevail for the lowest thickness. Gold tips are mandatory in the top-down illumination to limit self-absorption of the AFM probe. Noteworthy, the enhancement induced by the gold-coating may impair the measurement for sample with a thickness below ∼50 nm: previous studies show that results obtained on thin samples presented non-linearity in the signal intensity as well as possible artefacts during the analysis.33,34 Those effects were negligible for samples with a thickness above 50 nm like the NPs. The ratio of the two carbonyl intensities (PLA and drug) plotted in a secondary axis (orange circles) in function of the thickness (Fig. 6C and D), showed constant values at around 0.22 ± 0.02. This clearly indicates the homogenous distribution of both drug and PLA within the film. In a nutshell, these investigations showed the linearity of the IR intensities of both drug and polymer as a function of the sample thickness up to 420 nm, thus setting the ground of our methodology. Typical local spectra collected along the studied film were plotted in Fig. 6D, showing as expected, characteristic peaks of both PLA and the drug, at 1760 cm−1 and 1900 cm−1, respectively. Normalization of the spectral profiles as shown in Fig. 6E reveals the self-similar nature of these curves that is also a signature of the homogeneity of each analysed region. The mean spectrum gives a value of the band ratios (drug:polymer) of 0.22. The same methodology was applied for all the films. Fig. 7A shows mean spectra of the five films F(0)–F(0.20), normalized to the PLA carbonyl band. The intensity of the drug carbonyl band at 1900 cm−1 increases with the drug concentration. For each studied film, the ratio of the intensities of the carbonyl bands of the drug peaking at 1900 cm−1 and the PLA one at 1760 cm−1 was plotted against the mdrug/mPLA mass ratio (Fig. 7B). A linear fit was obtained with a correlation coefficient R2 = 0.98. Nevertheless, the most significant inference from Fig. 7B is the observed linearity between intensity ratios and the concentration, thereby enabling us to perform a quantitative analysis based on intensity ratio measurements, in the sample thickness range from 60 to 420 nm.
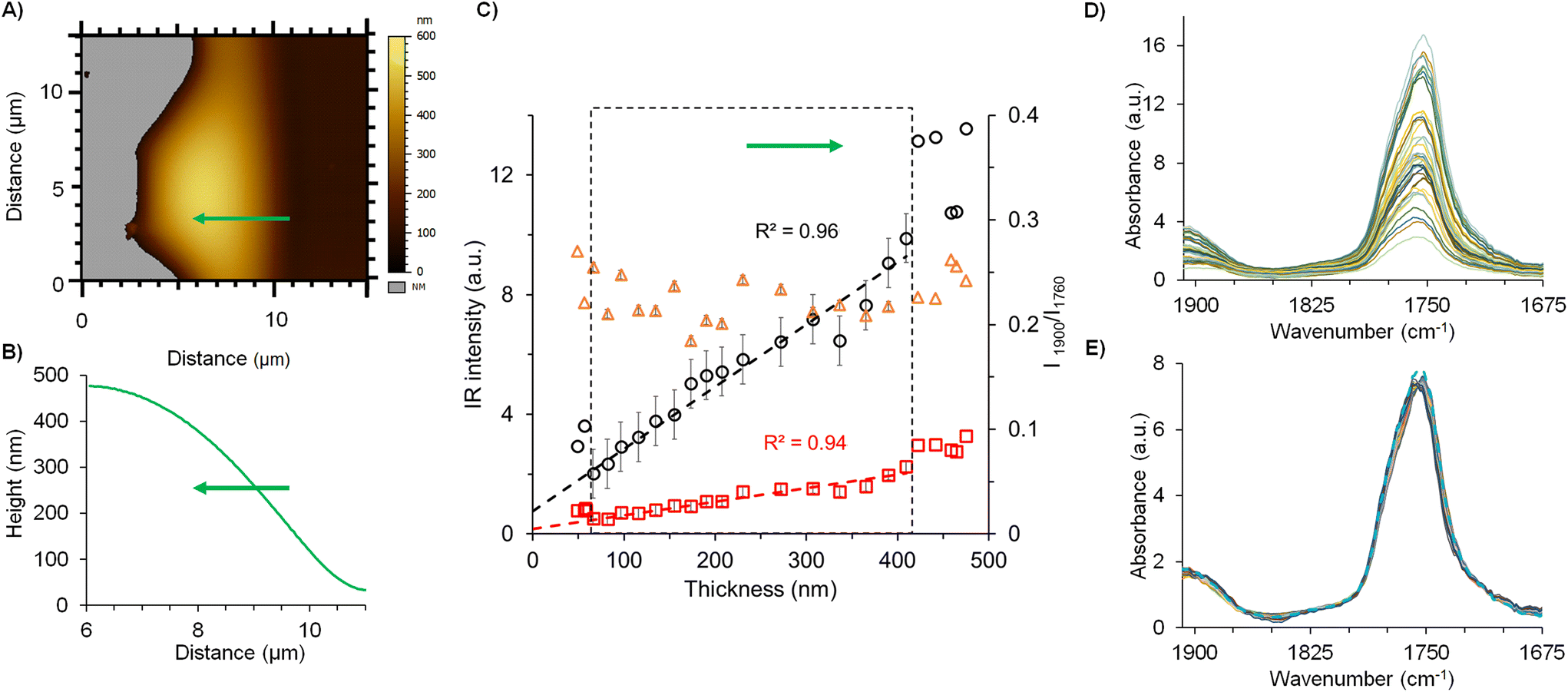 | ||
| Fig. 6 (A) AFM topography image of F(0.15). The green line shows the measured spectral line map of 5 μm length. Grey regions represent the substrate (signal below the threshold). (B) Height profile of the studied region. (C) IR intensities at 1760 (black circle) and 1900 cm−1 (red square) were collected from 23 local spectra (average of 4 single point spectra) on the green line with spatial intervals of 200 nm and plotted as a function of film thickness. Intensity ratios (1900 to 1760 cm−1i.e. I1900/I1760) are plotted (orange triangle) and value refers to the secondary Y-axis. (D) 23 local IR spectra recorded on the film in the topography image shown in A across the 5 μm region with 200 nm steps. (E) Normalized local IR spectra and mean spectrum of spectra (blue dashed line). The AFM-IR working range was between 1675 and 1908 cm−1 as detailed in the Experimental section. | ||
 | ||
| Fig. 7 (A) Normalized mean spectra of films analysed by AFM-IR; F(0) (black), F(0.03) (orange), F(0.10) (blue), F(0.15) (red) and F(0.20) (green). (B) A linear curve was derived by the ratiometric analysis of the respective PLA and drug band intensities obtained by AFM-IR as a function of the mdrug/mPLA mass ratio. The AFM-IR working range was in between 1675 and 1908 cm−1 as detailed in the Experimental section. | ||
The proof of concept can now be continued with a comparative analysis of the two techniques (quantitative IR-microspectoscopy and AFM-IR) with respect to their calibration curves (Fig. 4B and 7B). As listed in Table SI1,† the I1900/I1760 ratio in IR-microspectroscopy were 0, 0.03, 0.05, 0.08 and 0.12 for F(0); F(0.03); F(0.10); F(0.15) and F(0.20), respectively with a slope of 0.57. In the case of AFM-IR, the slope of the fitting curve equals to 1.40, and I1900/I1760 ratio for the same films were 0.08, 0.15, 0.23, 0.25 respectively. This reveals a scale factor of 2.7 ± 0.5 between the two techniques. This scale factor was subsequently implemented to rescale AFM-IR spectra of each film. The rescaled AFM-IR spectra were overlaid with those of IR-microspectroscopy. This comparison was represented in Fig. 8A for the case of F(0.15) and in Fig. SI3† for each film. Considering this scaling factor, the IR microspectroscopy calibration curve (Fig. 4B) can be used for quantification by AFM-IR. Given the good agreement between the AFM-IR and IR microspectroscopy investigations, the next step was the quantitative analysis of individual NPs.
 | ||
| Fig. 8 Comparison of (A) AFM-IR spectra (solid line) to IR-microspectroscopy spectra (dashed lines) of F(0.15) (B) data set and calibration curve obtained from IR microspectroscopy (black) in comparison to AFM-IR (blue) data set of the same films. A scaling factor of 2.7 was applied to AFM-IR data. The AFM-IR working range was in between 1675 and 1908 cm−1 as detailed in Experimental section. | ||
Methodology for quantitative analysis of individual NPs by AFM-IR in tapping mode
Following the demonstrated proof of concept and the correlation between IR microspectroscopy and tapping AFM-IR, we present here the methodology to perform quantitative analysis of the local DL on individual NPs by using any of the two acquisition modes in AFM-IR: local IR absorption spectra or IR maps.The NP(0.20) formulation (Fig. 9) was used to illustrate the methodology. A topography image (Fig. 9A) was recorded on a 5 μm × 5 μm area. The NPs size ranged from 200 to 500 nm, in agreement with DLS and TEM investigations. Fig. 9B presents the local IR spectra of individual NPs. The absence of signal on the support (orange arrow, Fig. 9B) clearly shows that the drug was only associated to the NPs and that no residual polymer was on the support.
 | ||
| Fig. 9 (A) Topography image of NPs(0.20). (B) Local IR absorption spectra of NPs and background recorded on the regions of interest indicated with arrows in A. Orange arrow presents the analysis on the region without NP. The AFM-IR working range was in between 1675 and 1908 cm−1 as detailed in the Experimental section. | ||
The ratio of the intensities of the drug and PLA IR bands (I1900/I1760) were calculated from the NPs’ local IR spectra (Fig. 9B). The drug:PLA mass ratios were determined from the calibration curve equation established by IR microspectroscopy (Fig. 4B). Mass ratios were then converted to DLs by using eqn (2). For the four randomly chosen NPs, the DL were respectively 4% (violet arrow, NP of around 390 nm), 10% (grey arrow, NP of 300 nm), 21% (red or green arrows; NP of 425 nm) and 10% (black arrow; NP of 570 nm). The DLs differed strikingly from one NP to another (4; 10; 21%). However, the DLs measured on the same NP at different locations were consistent (21%, red and green arrows).
Interestingly, the NP size and the DL (Fig. 9) do not correlate. It can be hypothesized that the large heterogeneities in DLs (factor of 5) are due to a random segregation between the drug and the polymer occurring during the process of NP formation. Indeed, the NP preparation involves a dispersion step, consisting in pouring a drug–polymer organic solution in water. Immediately after, the common solvent of the drug and the polymer diffuse in the aqueous dispersion medium. As a consequence, the polymer precipitates under the form of NPs, while aggregates are also formed. The nanoprecipitation process was investigated35 and it was found that aggregate amounts depend on the experimental conditions and the physicochemical properties of both the drug and the polymer. Possibly, the large DL heterogeneities of individual NPs observed here are related to affinity differences between the drug and the polymer, leading to phase separation at the nanoscale during the fast nanoprecipitation process. To the best of our knowledge, this is the first report of large discrepancies in terms of individual NP composition. More interestingly, these differences are not related to NP diameter.
Drug quantification using the IR absorbance spectra provides a very precise local quantitative analysis on a region of around 10–15 nm depending on the AFM tip geometry. However, the investigation of a large number of NPs to provide statistically relevant data is time consuming. Therefore, an alternative method was developed here, based on the capability of AFM-IR to perform IR maps of both drug and PLA, at one specific wavenumber corresponding to the absorption band of these compounds. AFM-IR provides access to the IR intensity at any point on the maps. Then, by performing ratiometric analysis (dividing two maps corresponding to drug absorption and PLA absorption), DL can be assessed at any point. The method has the advantage to probe large area (micrometers square), thus analysing simultaneously several NPs.
To validate this approach, a direct transposition from a local spectrum to an IR map was needed. Therefore, the strategy was to compare the initial local AFM-IR spectra at a specific point with the IR spectra reconstructed from a series of IR maps. At first, local IR spectra were recorded at 11 different points on an individual NP (Fig. 10A). When normalized, all the 11 local spectra overlapped and their average spectrum (represented by the red curve in Fig. 10C) was further used. Then, a series of 23 IR maps (in the spectral range between 1670 and 1920 cm−1, for details see the Experimental section) was recorded on the region containing the same NP.
 | ||
| Fig. 10 (A) Topography image of drug-loaded NPs. One NP (indicated with an arrow) was selected. (B) Local IR spectra were recorded by AFM-IR at 11 randomly chosen positions on this NP. (C) Overlay of the average spectrum of the 11 local IR spectra recorded by AFM-IR (red line) and the reconstructed IR profile (black line) from the 23 IR maps series (precisely at 1700, 1720, 1730, 1740, 1750, 1755, 1760, 1765, 1770, 1780, 1790, 1800, 1810, 1820, 1830, 1840, 1850, 1860, 1880, 1885, 1890, 1900 and 1905 cm−1). 3 reconstructed spectra were obtained from random locations on the chosen NP and averaged. The AFM-IR working range was in between 1675 and 1908 cm−1 as detailed in the Experimental section. | ||
From each IR map recorded, the IR intensities of a selected point were retrieved. Then, the local IR spectra were obtained by plotting the intensity values against their corresponding wavenumbers. The resulting average local spectrum extracted from the maps (black line in Fig. 10C) was superimposed to the high-resolution measured spectra at specific positions (red line in Fig. 10C). Both are similar which validates that a quantitative study can be made simply by recording a series of IR map. Eventually, from these average local spectra, intensities of the carbonyl bands were determined to calculate the drug:PLA mass ratio from the calibration curve equation by using the scale factor previously determined. The DL calculated was 28 ± 6 wt%.
The same NP was further analysed focusing on IR maps. Its height (Fig. 11) was around 90 nm (Fig. 11B) and it's apparent diameter around 320 nm. IR maps recorded at 1760 cm−1 and 1900 cm−1 (Fig. 11C and D) revealed the location of both the polymer and the drug, although a lack of intensity with a crescent shape was observed in the upper part of the NP. This phenomenon, previously reported for PLA NPs, was attributed to a phase shift during the contact between the tip and the NPs in tapping AFM.19,21
 | ||
| Fig. 11 (A) AFM topography image of a single NP. (B) Height profile of the NP recorded on the blue line. (C) IR map recorded at 1760 cm−1 and (D) 1900 cm−1 to detect PLA and the drug, respectively. (E) IR intensity profile at 1760 cm−1 (PLA, black) and 1900 cm−1 (drug, red) and the ratios of IR intensities (1900 to 1760 cm−1) (orange) recorded on the line as a function of thickness of NP and plotted on the secondary Y axis. Grey regions represent the AFM support (signal below the threshold). The AFM-IR working range was in between 1675 and 1908 cm−1 as detailed in the Experimental section. | ||
The areas, where a phase shift is observed, are excluded for the DL calculation. Furthermore, it is possible to estimate the percentage of the NP volume, where this phase shift artifact appears: it is around 10 to 20% of the NP volume. However, as this artifact generally occurs at the edge of the NP, the excluded area can contain either only PLA or PLA and drug. This can lead to either overestimating or underestimating the DL.
Intensity profiles were extracted through a line along the NP (black and red line Fig. 11C and D) and plotted versus thickness (Fig. 11E). The evolutions of the intensity profiles at 1760 cm−1 (black) and 1900 cm−1 (red) suggested a direct proportionality of IR intensity with thickness. Both intensities were linear (R2 ≥ 0.96) when the thickness was above 60 nm, in good agreement with the lower limits found in the proof of concept. The ratios of the intensities at 1908 to 1760 cm−1 were calculated, showing a constant value of 0.74 ± 0.06 except at the borders of the NP (orange dots Fig. 11E). This is an indication of good NP composition homogeneity. The deviations observed at the NP edges are probably due to the sensitivity limit of the AFM-IR technique. Noteworthy, the constant value of 0.74 ± 0.06 was the average of 17 measurements, above the thickness threshold of 60 nm. The corresponding DL was then calculated and obtained as 32 ± 6%. This value is in line with the DL calculated by local IR spectra (28 ± 6 wt%, Fig. 10) emphasizing the accuracy of the proposed quantification method.
Next, 15 profiles were extracted from the NP's ratio map (1900/1760) (Fig. 12). The ratio map appeared to be almost constant. A linear trend was obtained for each analysed profile (Fig. 12B), which suggests the NP's overall homogeneity. Noticeably, the calculated ratios of intensities were around 0.7, as the violet line represents the average profile that is in close agreement with the above findings (average of 0.74 ± 0.06, Fig. 11E).
 | ||
| Fig. 12 (A) Ratio of the IR maps recorded at 1900 cm−1 (drug) to the one at 1760 cm−1 (polymer) as shown in Fig. 11D and C, respectively. Detailed profile analyses were performed along a distance of 200 nm on 15 different regions represented in black lines on the NP. (B) The ratio of intensities (1900/1760) was recorded from the 15 profiles analysis (A), as a function of distance. Each grey curve represents one profile analysis and the average curve is shown in purple. | ||
Finally, we can propose a simple protocol for local quantitative analysis of a large set of individual NPs: a topography image was recorded (Fig. 13A) while simultaneously acquiring chemical maps at 1760 cm−1 (Fig. 13B) and 1900 cm−1 (Fig. 13C), to detect the PLA polymer and the drug, respectively.
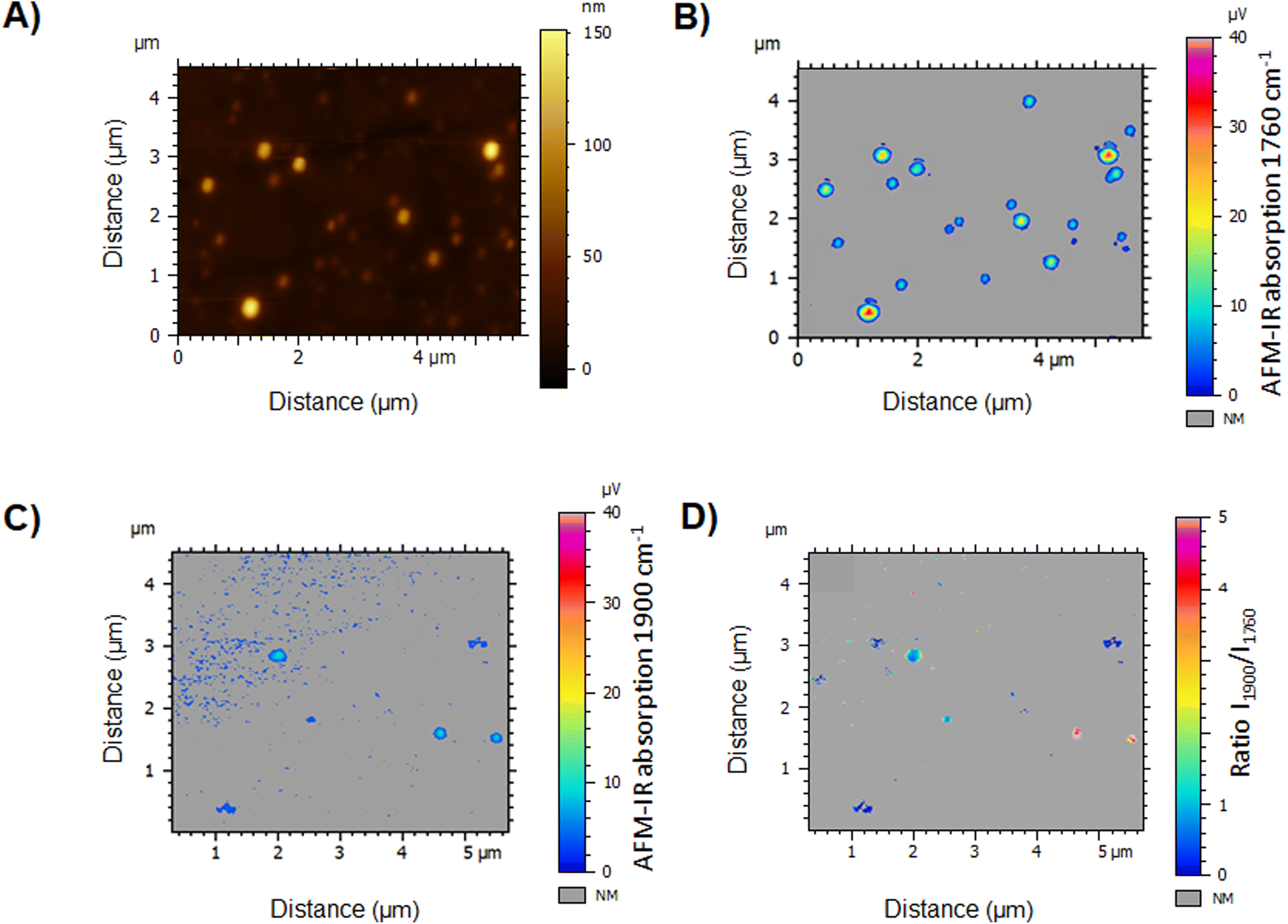 | ||
| Fig. 13 (A) AFM topography image of NP(0.20). IR maps recorded by AFM-IR at (B) 1760 cm−1 and (C) 1900 cm−1 to detect the polymer and the drug, respectively. (D) Ratio of the IR maps recorded at 1900 cm−1 (drug) to the one at 1760 cm−1 (polymer). | ||
The IR map at 1760 cm−1 displays the location of the polymer, thus the NPs. While the NPs appear to vary in size (80 to 370 nm), even the tiniest ones could be detected in the IR PLA map (Fig. 13B) with regard to the topography image (Fig. 13A). However, the drug map at 1900 cm−1 revealed, that only few NPs are loaded with the drug (Fig. 13C). This clearly points out the NPs’ chemical heterogeneity and emphasizes the interest of performing NP characterization individually. Advantageously, the ratio of two IR maps (Fig. 13D) allows the analysis of more than 20 NPs overcoming the slowness of the spectral analysis. Results showed that only 9 of the NPs were loaded, in which the DLs varied from 10 to 90 wt% with no NP size dependence. We hypothesized that the discrepancies in terms of NP DLs were due to their fast precipitation in the preparation process, inducing drug/polymer segregation. Since NP function is tightly regulated by the physicochemical properties bestowed by their composition, such significant DL variations could render only a low fraction of the NPs therapeutically active. While heterogeneities in terms of sizes of NPs obtained by nanoprecipitation were widely reported, up to our knowledge, this study presents for the first-time large heterogeneities in terms of drug content in individual polymeric NPs.
Conclusions
A stringent correlation was proven between IR microspectroscopy and AFM-IR in tapping mode with a top-down illumination. A correlation between these two techniques was shown, enabling to perform quantitative chemical analysis by AFM-IR in tapping mode with a top-down illumination with AFM resolution. The method was shown to be robust. The acquisition of local AFM-IR spectra allows to obtain drug mass fraction with standard error of 5 to 6%. Calibration curves determined by two different methods, showed no statistical differences even if the IR map standard deviation was not directly evaluated (each IR map is only acquired one time). By performing local AFM-IR on an individual NP, it was possible to investigate its chemical homogeneity at nanoscale (10–20 nm) and to calculate its drug mass fraction within 60 to 420 nm sample thickness.However, but this procedure does not take in account two points: the possible degradation of the tip during acquisition and the phase shift artifact. This can lead to either an overestimation or an underestimation of the total NP DL. In the future, this problem may be solved by using a double-phase locking loop system that is actually lacking in the actual commercial AFM-IR systems. We hope those technical points will be addressed in the future to further improve the quantification method.
An important finding here was to evidence huge heterogeneities in terms of individual NP composition, by contrast to films prepared using the same components which were found to be homogeneous. These studies proved the usefulness of AFM-IR in nanomedicine, by enabling a precise chemical analysis and quantification at the nanoscale. We believe that these findings go steps beyond the commonly used characterization methods, by affording new tools to better understand the supramolecular organization of NPs and their composition with a nanometric resolution. The methodology proposed here can set up the ground towards quality assessment of drug nanocarriers. Moreover, it could also help understanding the NP formation process and help optimizing the formulations protocols. Further studies will comprise AFM-IR investigations of other type of NPs such as hybrid organic–inorganic ones and more complex hybrid structures resulting from the association of polymeric and metal NPs. It is worth investigating the limits of the methods in terms of minimal NP size that can be analysed as a function of the nature of the materials that are used. Furthermore, the analysis spectral domain of AFM-IR could be enlarged given that novel lasers can be employed, opening the way to study a larger set of available drugs and polymers.
Author contributions
M. Seray Ural prepared PLA NPs and films, performed IR spectroscopy and TEM measurements, implemented data treatment, prepared figures and wrote the manuscript. Emmanuel Dartois performed IR micro-spectroscopy, designed and implemented data treatment, and corrected the manuscript. Jérémie Mathurin performed AFM-IR measurements, designed and implemented data treatment, and corrected the manuscript. Didier Desmaële participated to the design of some experiments and corrected the manuscript. Philippe Collery provided the rhenium derived compounds. Alexandre Dazzi participated to the design of some experiments. Ariane Deniset-Besseau lead the project, performed AFM-IR measurements, designed the experiments and corrected the manuscript. Ruxandra Gref lead the project, designed the experiments, wrote and corrected the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research received support from the French National Research Agency (ANR-14-CE08-0017 and ANR-20-CE19-0020) and by Labex NanoSaclay (ANR-10-LABX-0035). The present work has benefited from Imagerie-Gif core facility supported by ANR (ANR-11-EQPX-0029/Morphoscope, ANR-10-INBS-04/FranceBioImaging; ANR-11-IDEX-0003-02/Saclay Plant Sciences).References
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- D. Essa, P. P. D. Kondiah, Y. E. Choonara and V. Pillay, Front. Bioeng. Biotechnol., 2020, 8, 1–20 CrossRef PubMed.
- Z. Li, S. Tan, S. Li, Q. Shen and K. Wang, Oncol. Rep., 2017, 38, 611–624 CrossRef CAS PubMed.
- Y. Wang, W. Qu and S. H. Choi, FDA's regulatory science program for generic PLA/PLGA-based drug products, 2017, vol. 20 Search PubMed.
- S. Rezvantalab, N. I. Drude, M. K. Moraveji, N. Güvener, E. K. Koons, Y. Shi, T. Lammers and F. Kiessling, Front. Pharmacol., 2018, 9, 1260 CrossRef CAS PubMed.
- T. L. Andresen and J. B. Larsen, Acta Biomater., 2020, 118, 207–214 CrossRef CAS PubMed.
- S. M. Stavis, J. A. Fagan, M. Stopa and J. A. Liddle, ACS Appl. Nano Mater., 2018, 1, 4358–4385 CrossRef CAS.
- J. M. Rabanel, V. Adibnia, S. F. Tehrani, S. Sanche, P. Hildgen, X. Banquy and C. Ramassamy, Nanoscale, 2019, 11, 383–406 RSC.
- M. Chaupard, M. de Frutos and R. Gref, Part. Part. Syst. Charact., 2021, 38 Search PubMed.
- F. Laborda, E. Bolea and J. Jiménez-Lamana, Anal. Chem., 2014, 86, 2270–2278 CrossRef CAS PubMed.
- D. Kurouski, A. Dazzi, R. Zenobi and A. Centrone, Chem. Soc. Rev., 2020, 49, 3315–3347 RSC.
- A. Dazzi, F. Glotin and R. Carminati, J. Appl. Phys., 2010, 107, 124519 CrossRef.
- A. Dazzi, C. B. Prater, Q. Hu, D. B. Chase, J. F. Rabolt and C. Marcott, Appl. Spectrosc., 2012, 66, 1365–1384 CrossRef CAS PubMed.
- A. Dazzi, R. Prazeres, F. Glotin and J. M. Ortega, Opt. Lett., 2005, 30, 2388–2390 CrossRef CAS PubMed.
- A. Dazzi and C. B. Prater, Chem. Rev., 2017, 117, 5146–5173 CrossRef CAS PubMed.
- F. Lu and M. A. Belkin, Opt. Express, 2011, 19, 19942 CrossRef CAS PubMed.
- G. A. Hill, J. H. Rice, S. R. Meech, D. Q. M. Craig, P. Kuo, K. Vodopyanov and M. Reading, Opt. Lett., 2009, 34, 431 CrossRef CAS PubMed.
- A. Dazzi, R. Prazeres, F. Glotin and J. M. Ortega, Ultramicroscopy, 2007, 107, 1194–1200 CrossRef CAS PubMed.
- J. Mathurin, E. Pancani, A. Deniset-Besseau, K. Kjoller, C. B. Prater, R. Gref and A. Dazzi, Analyst, 2018, 143, 5940–5949 RSC.
- L. Wang, H. Wang, M. Wagner, Y. Yan, D. S. Jakob and X. G. Xu, Sci. Adv., 2017, 3, e1700255 CrossRef PubMed.
- M. S. Ural, M. Menéndez-Miranda, G. Salzano, J. Mathurin, E. N. Aybeke, A. Deniset-Besseau, A. Dazzi, M. Porcino, C. Martineau-Corcos and R. Gref, Pharmaceutics, 2021, 13, 1992 CrossRef CAS PubMed.
- K. Wieland, G. Ramer, V. U. Weiss, G. Allmaier, B. Lendl and A. Centrone, Nano Res., 2019, 12, 197–203 CrossRef CAS PubMed.
- E. Pancani, J. Mathurin, S. Bilent, M.-F. Bernet-Camard, A. Dazzi, A. Deniset-Besseau and R. Gref, Part. Part. Syst. Charact., 2018, 35, 1700457 CrossRef.
- P. Álamo, V. Pallarès, M. V. Céspedes, A. Falgàs, J. M. Sanchez, N. Serna, L. Sánchez-garcía, E. Voltà-duràn, G. A. Morris, A. Sánchez-chardi, I. Casanova, R. Mangues, E. Vazquez, A. Villaverde and U. Unzueta, Pharmaceutics, 2020, 12, 1–18 CrossRef PubMed.
- F. Tang, P. Bao and Z. Su, Anal. Chem., 2016, 88, 4926–4930 CrossRef CAS PubMed.
- G. Ramer, V. A. Aksyuk and A. Centrone, Anal. Chem., 2017, 89, 13524–13531 CrossRef CAS PubMed.
- P. Collery, D. Desmaele and V. Vijaykumar, Curr. Pharm. Des., 2019, 25, 3306–3322 CrossRef CAS PubMed.
- A. Kermagoret, G. Morgant, J. D'Angelo, A. Tomas, P. Roussel, G. Bastian, P. Collery and D. Desmaële, Polyhedron, 2011, 30, 347–353 CrossRef CAS.
- J. Skogholt, K. H. Liland and U. G. Indahl, J. Raman Spectrosc., 2019, 50, 407–417 CrossRef CAS.
- A. Mancini, V. Giliberti, A. Alabastri, E. Calandrini, F. De Angelis, D. Garoli and M. Ortolani, J. Phys. Chem. C, 2018, 122, 13072–13081 CrossRef CAS.
- B. S. Ault, T. M. Becker, G. Q. Li and M. Orchin, Spectrochim. Acta, Part A, 2004, 60, 2567–2572 CrossRef PubMed.
- J. M. Chalmers, in Handbook of Vibrational Spectroscopy, ed. J. M. Chalmers, John Wiley & Sons, Ltd, Chichester, 2006 Search PubMed.
- P. Nguyen-Tri, P. Ghassemi, P. Carriere, S. Nanda, A. A. Assadi and D. D. Nguyen, Polymers, 2020, 12, 1142 CrossRef CAS PubMed.
- B. Lahiri, G. Holland and A. Centrone, Small, 2013, 9, 439–445 CrossRef CAS PubMed.
- P. Legrand, S. Lesieur, A. Bochot, R. Gref, W. Raatjes, G. Barratt and C. Vauthier, Int. J. Pharm., 2007, 344, 33–43 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2an01079h |
| This journal is © The Royal Society of Chemistry 2022 |