
Smartphone clip-on instrument and microfluidic processor for rapid sample-to-answer detection of Zika virus in whole blood using spatial RT-LAMP†
Aaron M.
Jankelow‡
 ab,
Hankeun
Lee‡
bc,
Weijing
Wang‡
bc,
Trung-Hieu
Hoang‡
c,
Amanda
Bacon
bc,
Fu
Sun
bc,
Seol
Chae
bc,
Victoria
Kindratenko
ab,
Katherine
Koprowski
ab,
Robert A.
Stavins
d,
Dylann D.
Ceriani
e,
Zachary W.
Engelder
e,
William P.
King
bd,
Minh N.
Do
c,
Rashid
Bashir
abcdf,
Enrique
Valera
*ab and
Brian T.
Cunningham
*abcf
ab,
Hankeun
Lee‡
bc,
Weijing
Wang‡
bc,
Trung-Hieu
Hoang‡
c,
Amanda
Bacon
bc,
Fu
Sun
bc,
Seol
Chae
bc,
Victoria
Kindratenko
ab,
Katherine
Koprowski
ab,
Robert A.
Stavins
d,
Dylann D.
Ceriani
e,
Zachary W.
Engelder
e,
William P.
King
bd,
Minh N.
Do
c,
Rashid
Bashir
abcdf,
Enrique
Valera
*ab and
Brian T.
Cunningham
*abcf
aDepartment of Bioengineering, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA. E-mail: evalerac@illinois.edu; bcunning@illinois.edu
bNick Holonyak Jr Micro and Nanotechnology Lab, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA
cDepartment of Electrical and Computer Engineering, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA
dDepartment of Mechanical Science and Engineering, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA
eGener8, LLC, Carlsbad, CA 92011, USA
fCenter for Genomic Diagnostics, Woese Institute for Genomic Biology, Urbana, IL 61801, USA
First published on 15th June 2022
Abstract
Rapid, simple, inexpensive, accurate, and sensitive point-of-care (POC) detection of viral pathogens in bodily fluids is a vital component of controlling the spread of infectious diseases. The predominant laboratory-based methods for sample processing and nucleic acid detection face limitations that prevent them from gaining wide adoption for POC applications in low-resource settings and self-testing scenarios. Here, we report the design and characterization of an integrated system for rapid sample-to-answer detection of a viral pathogen in a droplet of whole blood comprised of a 2-stage microfluidic cartridge for sample processing and nucleic acid amplification, and a clip-on detection instrument that interfaces with the image sensor of a smartphone. The cartridge is designed to release viral RNA from Zika virus in whole blood using chemical lysis, followed by mixing with the assay buffer for performing reverse-transcriptase loop-mediated isothermal amplification (RT-LAMP) reactions in six parallel microfluidic compartments. The battery-powered handheld detection instrument uniformly heats the compartments from below, and an array of LEDs illuminates from above, while the generation of fluorescent reporters in the compartments is kinetically monitored by collecting a series of smartphone images. We characterize the assay time and detection limits for detecting Zika RNA and gamma ray-deactivated Zika virus spiked into buffer and whole blood and compare the performance of the same assay when conducted in conventional PCR tubes. Our approach for kinetic monitoring of the fluorescence-generating process in the microfluidic compartments enables spatial analysis of early fluorescent “bloom” events for positive samples, in an approach called “Spatial LAMP” (S-LAMP). We show that S-LAMP image analysis reduces the time required to designate an assay as a positive test, compared to conventional analysis of the average fluorescent intensity of the entire compartment. S-LAMP enables the RT-LAMP process to be as short as 22 minutes, resulting in a total sample-to-answer time in the range of 17–32 minutes to distinguish positive from negative samples, while demonstrating a viral RNA detection as low as 2.70 × 102 copies per μl, and a gamma-irradiated virus of 103 virus particles in a single 12.5 μl droplet blood sample.
1. Introduction
Zika virus (ZIKV) is a flavivirus that was first isolated in Uganda, Africa in 1947 and is known to cause fetal microcephaly, a developmental disorder of newborn babies that is a consequence of mothers becoming infected during early pregnancy.1–3 ZIKV is transmitted mostly by the bites of infected Aedes aegypti mosquitos, but sexual, perinatal, and blood-transfusion transmission have also been documented.4–9 Although its presence was of little concern at first as the virus spread sporadically within the countries of continental South America and Asia, the 2015 ZIKV epidemic in Brazil progressed quickly, raising global awareness of the infectious disease, leading to a declaration of global health emergency by the World Health Organization in early 2016.10 Now, ZIKV has become one of the neurotropic viruses that circulate in more than 87 countries, still actively infecting thousands of people annually.11 In 2020, a total of 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 923 cases were reported worldwide among which 18941 cases were from Brazil and 2215 cases were from countries in Central America.12 Given the wealth of evidence indicating the apparent danger posed by the ZIKV, immediate action must be taken to stop the propagation of the virus and prevent future outbreaks.
923 cases were reported worldwide among which 18941 cases were from Brazil and 2215 cases were from countries in Central America.12 Given the wealth of evidence indicating the apparent danger posed by the ZIKV, immediate action must be taken to stop the propagation of the virus and prevent future outbreaks.
Controlling and monitoring ZIKV and other insect-borne viral pathogens have been particularly challenging since most infection cases are asymptomatic, while even the symptomatic infections have been reported to show non-specific acute febrile illness with symptoms that are identical to that of other common viral infections such as Dengue virus and Chikungunya virus.13–15 All these viruses result in diseases that present clinically with similar symptoms, including fever, fatigue, headache, rash, arthralgia, myalgia, and conjunctivitis.16 As a result of these characteristics, viral containment and early detection have been especially difficult in underdeveloped countries where limited access to testing facilities, shortage of trained personnel, and under-funded medical infrastructures have hindered patients from receiving a proper diagnosis and prompt effective treatment. This disproportionate distribution of the burden of infectious diseases is most prominent in the poorest countries and regions, motivating the development of more portable and cost-effective POC diagnosing tools for viral detection that can provide equivalent sensitivity and accuracy as conventional laboratory-based methods.17
Various technologies are currently available for ZIKV detection but have limitations that hinder their application to POC scenarios. The gold standard molecular testing method, quantitative reverse transcription PCR (RT-qPCR) has been extensively exploited to detect the presence of genomic contents of ZIKV through the utilization of the reverse transcriptase enzyme to amplify viral RNA copies from patient samples, including blood,18,19 urine,20–22 saliva,20 semen,23,24 and amniotic fluid.25 Although PCR-based nucleic acid amplification provides detection limits as low as 190 copies per μl when detecting extracted ZIKA RNA spiked in whole blood,26 the assays require sample preparation and instruments in laboratory settings, using protocols that require benchtop instrumentation, highly trained personnel to perform sample purification, viral isolation, complex genome extractions, and interpretation of complex time-resolved calibration curves.27 Moreover, due to the necessity for precise thermal control, the acquisition of bulky and/or expensive equipment is not optional, making the method unsuitable for POC testing.28–32 An alternative to nucleic acid testing is enzyme-linked immunosorbent assay (ELISA), which detects viral proteins, such as antigens or antibodies. Recognized as being sensitive and robust, ELISA has been used in several studies for detecting low concentrations of Zika-specific proteins.33,34 However, ELISA also requires benchtop instrumentation, complex workflow, and suffers from cross-reactivity, long sample-to-answer time (∼4 h.), washing steps, and stringent sample preparation procedures, resulting in an assay that is difficult to deploy in resource-limited environments. Further, the antibody reagents for ELISA require careful handling and strict storage conditions to preserve their function against denaturation and aggregation.35
For these reasons, nucleic acid tests using isothermal amplification such as Loop-Mediated Isothermal Amplification (LAMP) have recently generated significant attention for virus detection due to the simplicity of assay preparation (single step) and the ease of translation to POC devices. LAMP circumvents the need for thermal cycling by executing enzymatic amplification at a constant temperature, which results in simplification of the assay protocol, reduction in the instrument size through removal of extra circuitry for thermal control systems, and high amplification efficiency.36–42 Compared to PCR, RT-LAMP uses a larger number of primers (typically 4–6), while Bst DNA polymerase provides increased amplification speed and yield through its superior thermal stability compared to Taq polymerase. As a result, RT-LAMP is associated with significantly decreased non-specific amplification and enhanced resistance to interference from inhibitors in biological samples.43–46
This paper describes the design and characterization of a single-use, plastic, 3D printed microfluidic POC diagnostic cartridge that is used in conjunction with an inexpensive, battery-powered, handheld detection instrument that clips on to a smartphone to utilize its rear-facing camera as the sensor. The cartridge is comprised of two stages, in which the first stage performs sample pre-processing from a droplet of whole blood, while the second stage contains six separate microfluidic compartments for performing independent RT-LAMP assays. The liquids in the RT-LAMP compartments are allowed to spread laterally to provide a large surface area for two-dimensional image analysis of fluorescent “clusters” that occur during the LAMP enzymatic amplification process when the target nucleic acid sequence is present. Spatial analysis of positive LAMP amplification events (called “Spatial LAMP” (S-LAMP)) allows the system to identify a positive reaction in a compartment in a shorter time, compared to waiting for the entire compartment to reach a saturated fluorescent state. Reduction in the time required to indicate a positive test is especially pronounced for low concentrations of the target nucleic acid sequence. Importantly, the sample processing cartridge and the detection instrument are intended to accurately and repeatably replicate the reagent volumes, mixing times, and temperature used in the laboratory-based version of the assay.
In previous reports, we demonstrated the detection of ZIKV with an RT-LAMP assay performed in narrow, low-volume silicon microfluidic channels that required a syringe pump to precisely control flow rates and tubing connections to transport fluids.47 In this work, the microfluidic cartridge is designed for precise metering of the volumes of the blood, lysis buffer, and assay buffer. All fluid manipulation is performed through a sliding valve manifold with pneumatic pressure supplied by manual rotation of a threaded screw with a spiral-shaped microfluidic cavity. All functions of the cartridge are performed manually, without the need for external power or complementary devices. The detection instrument is intended to facilitate simple clip-on interfacing with mobile computing devices such as smartphones and tablets with a rear-facing camera. The instrument utilizes battery power to operate an array of LEDs to uniformly illuminate the LAMP assay compartments and a thermoelectric heater to rapidly and uniformly bring the liquids in the LAMP compartments to a temperature of 65 °C, required for the amplification reaction. The instrument also contains a fluorescence emission filter and macro lens that enables the mobile device's camera to function as a broad-area fluorescence microscope that can gather a sequence of images during the LAMP process from above, while the compartments are heated from below. We share measurements of the liquid volume metering repeatability provided by the cartridge and the temperature uniformity provided by the heater. Through the utilization of our system, precise and repetitive LAMP assay preparation can be performed.
In this work, we characterize the performance of the LAMP assay by comparing the kinetic fluorescent amplification measurements conducted in conventional PCR tubes and a laboratory-based thermal cycler with the same assay performed in the microfluidic compartments measured with the smartphone instrument. We initially characterize the assay (in microfluidic and PCR-tube format) using the target viral RNA sequence spiked into buffer, followed by further characterization performed by spiking gamma ray-deactivated Zika virus into whole blood. In summary, we demonstrate that our system (cartridge and clip-on smartphone instrument) achieves a detection of 2.70 × 102 copies per μl for viral RNA spiked into buffer and a 103 virions per μl for gamma-irradiated Zika virus spiked into whole blood. The detection limits achieved with the microfluidic-based sample handling and smartphone-based readout are equivalent to performing the same assay in PCR tubes with laboratory instruments. Utilizing S-LAMP, the time required for positive detection is 22 min for the lowest detectable virus concentration, compared to 31 min using measurement of the entire microfluidic assay volume, and 39 min for detection in a PCR tube. Overall, the sample-to-answer time is 17–32 min with the system to distinguish positive from negative samples.
While the system is capable of estimating viral load with the use of calibration standards, we envision the approach as a means to provide rapid POC discrimination of negative samples from positive samples when viral concentrations are above the clinically relevant threshold of pre-symptomatic patients (ranging from 3 × 103–5.2 × 106, mean 1.1 × 106 copies per mL) which would contribute to the early detection of infection.48 Likewise, it has been reported that in the case of observed chronic infection of ZIKV-FLR in C6/36 cells, the concentration of viral RNA peaks at 4 × 109 copies per mL.49 Importantly, analysis of whole blood samples rather than plasma samples may be crucial, as data has shown that Zika virus RNA persisted in whole blood after it disappeared in plasma.50 While the functions of the present microfluidic cartridge are segregated between two physically separate modules, we envision integrating them together into a single unit that would be inexpensively manufacturable by injection molding, compared to the 3-D printing technology used to fabricate the devices in this report.
2. Experimental
2.1. Assay workflow for point of care sample pre-processing and RT-LAMP analysis
Fig. 1a illustrates the workflow of the microfluidic cartridge sample processing and smartphone-based readout with the clip-on instrument. The cartridge functions are comprised of two physically separate modules. Module “A” performs the sample lysis and mixing with an RT-LAMP reaction mix (composed of buffers, RT-LAMP primers, polymerase, and reverse transcriptase enzymes), and Module “B” contains six physically separated 25 μl volume liquid compartments that are filled from separate inlets. Prior to sample analysis, the cartridge is prepared by drawing the lysis buffer (0.12% Formic Acid (v/v) & 0.05% Saponin (w/v) in DI H2O) and LAMP reaction mix through separate inlets to fill internal fluid chambers with precisely metered volumes. To initiate sample analysis, a drop of unprocessed whole blood is deposited onto the sample inlet port of Module A. By manually rotating the threaded screw to push and pull the combined liquids through hourglass-shaped mixing channels, a 12.5 μl portion of the blood sample is mixed with the lysis buffer for 1 minute. By repositioning the sliding valve, the fluid manifold opens a path between the lysed blood and the LAMP reaction mix, enabling rotation of the same threaded screw to push and pull the combined liquids through the mixing channels a second time. After 1 minute of mixing all three assay components (blood, lysis buffer, and RT-LAMP reaction mix) the resulting solution is drawn fully into the spiral fluid channel within the threaded screw. The final action performed in Module A is to move the sliding valve to a new position, so the fluid manifold connects the threaded screw fluid channel to the outlet. Rotation of the threaded screw pushes the approximately 200 μl mixed solution into the outlet reservoir. For the assays reported here, the fluid is withdrawn from the outlet reservoir by pipette, and immediately injected into the inlet holes of Module B, where the reactions take place and are recorded using a smartphone CCD camera in real-time. Second, the prepared assay solution is evenly distributed into each 25 μl volume compartment using a pipette followed by sealing the inlet and outlet holes with a silicone-based adhesive tape to prevent the evaporation of fluid during RT-LAMP heating. For characterization purposes, 75 μl of the final mixture from Module A was inserted into three individual compartments in Module B while the remaining three compartments were filled with negative controls prepared separately using whole blood from healthy donors. The remaining volume from Module A was tested in a benchtop thermocycler to confirm the results obtained with the device. The prepared Module B is placed inside the instrument (Fig. S1†) where it makes physical contact with a Peltier module for heating. Finally, the smartphone camera is aligned and clipped onto the detection instrument to obtain a collection of images of the fluid compartments, with the LED illumination, image collection frequency, and total acquisition time controlled by an app. After image collection, the data is wirelessly transferred to an off-camera database for temporal and spatial analysis by Python. | ||
| Fig. 1 Point-of-care testing workflow of the smartphone-based Spatial RT-LAMP ZIKV detection system. (1) The patient blood droplet is collected and loaded into the inlet port of Module A; (2) the blood is thoroughly mixed with lysing buffer and RT-LAMP reagents using the mixing channel and threaded syringe of Module A; (3) the final assay mixture is extracted from Module A and loaded into Module B, followed by sealing of inlet and outlet holes; (4) the prepared Module B is inserted into the detection instrument where the amplification reaction occurs, and a smartphone is locked in position using a clip to align the rear-facing camera with the instrument; (5) using an audio cable connection between the smartphone and the instrument (not shown in the figure), the app is initiated to take consecutive images of the reaction compartments at 10-seconds intervals, producing a collection of images that are subsequently analyzed by software. | ||
Starting with a prepared Module A with metered volumes of lysis buffer and RT-LAMP reaction mix in their respective fluid channels, the sample is prepared for LAMP analysis in 10 min. Following a 4.5 min for Module B to reach the target temperature of 65 °C, we run the LAMP reaction for a total of 40 min. However, using S-LAMP, we show that only 7–22 min of LAMP reaction time is required to differentiate positive from negative tests for all concentrations tested. Therefore, the total sample-to-answer time is approximately 17–32 min.
2.2. ZIKV genomic RNA and viruses
Genomic RNA for Zika virus strain PRVABC59 (NR-50244), genomic RNA for Zika virus strain R103451 (NR-50358), and gamma-irradiated inactivated Zika virus strain PRVABC59 (NR-50547) were obtained from BEI Resources and stored at −80 °C after aliquoting. The stocks were then diluted to the desired concentrations in either TE buffer or whole blood.2.3. Whole blood samples
Human whole blood from healthy individuals was obtained from BioIVT (USA) in 10 mL K2EDTA tubes. The blood was stored at 4 °C and under stirring until used. Storage of blood at 4 °C and under stirring is standard practice to prevent blood coagulation.2.4. RT-LAMP composition
All RT-LAMP assays were comprised of the following components: 10X isothermal amplification buffer from New England Biolabs (NEB), MgSO4 (100 mM, NEB), betaine (5 M, Sigma-Aldrich), dNTP (10 mM each), Bst 2.0 WarmStart DNA Polymerase (8 U μl−1, New England Biolabs), WarmStart RTX reverse transcriptase (15 U μl−1, NEB), Evagreen dye (25 μM, Biotium), 20 μg μl−1 of Bovine Serum Albumin (NEB). In addition to these buffer components, a primer mix was added to achieve the following primer concentrations in the final reaction: F3 and B3 (0.3 μM), FIP and BIP (1.8 μM), and LoopF and LoopB (0.7 μM). Primer sequences were sourced from the literature43 to target unique genes that code for the capsid protein of ZIKV, as illustrated in Fig. S2,† and synthetized by Integrated DNA Technologies (IDT). The two most outer capsid gene sequences were selected as the targets of the forward primer F3 and the backward primer B3. The two most inner capsid gene sequences were selected as the targets of the second primer pair, the forward inner primer FIP and backward inner primer BIP. The remaining primer pair, the forward loop primer LF and backward loop primer LB, were designed to target the unique sequences found in between the F3 & FIP and B3 & BIP. All LAMP reagents were stored at −20 °C except for betaine (stored at 4 °C). Accugene molecular biology grade water was used to make up the remaining volume in the reaction mix to 14 μl, then 2 μl of the sample was mixed into a final reaction volume of 16 μl (7:1, reaction mix:sample ratio). For on-cartridge reactions, the volume was scaled-up as needed while maintaining the 7:1 ratio.
2.5. Dual module microfluidic cartridge design and fabrication by 3D printing
The cartridge is comprised of two physically separate components: Module A for sample pre-processing and Module B for LAMP reactions and assay readout. While our longer-term goal is to integrate both modules into a single unit, in which the output of Module A fills the compartments in Module B, keeping them separated enabled us to optimize the design of each independently, while facilitating prototype fabrication by 3D printing and assay characterization. Module A consists of three major elements: the cartridge body, a sliding valve manifold, and a threaded syringe. As discussed in the following section, Module A is fabricated using a transparent material to enable the user to visually observe the location of the assay fluid during the lysing and mixing procedure. The cartridge enables the user to manually perform the assay with the same volumes and homogenous mixing as the laboratory-based method, while avoiding the use of external pumps or motors. A detailed schematic drawing of Module A is shown in Fig. 2a and S3.† The cartridge incorporates eight holes (0.7 mm radius): three inlet ports (for lysis buffer, LAMP reagents, and whole blood sample), two outlet ports for reagent metering (lysis buffer and LAMP reagents), two vent holes, and the outlet well for the final mixed solution used for LAMP amplification. All the holes have connecting channels that are extended toward the center of the cartridge body at specific positions to allow a continuous flow of the fluid from inlet ports, through the channels of the sliding valve, and to the outlet ports. Thus, the sliding valve serves as a fluid manifold, whose lateral position connects specific inlets to specific outlets for each step of cartridge preparation and sample preparation. The mixing channel utilizes an hourglass shape to alternatively compress and expand the fluid, to induce chaotic advection of the solution for rapid homogenization.51 The numbered arrows represent the exact positions of the sliding valve in each step of the assay preparation where positions 1, 2, and 3 are determined by the right sliding valve arrow while positions 4, 5, and 6 are determined by the left sliding valve arrow. The sliding valve has a total of four channels, two of which meter the lysing buffer and LAMP reagent volumes, while the others serve to connect channels across the sliding valve. The metered channels reside on the far-left and far-right sides of the sliding valve (referred to as lysis buffer and RT-LAMP reagent channels) and are designed to hold volumes of 12.5 μl and 175 μl respectively. The metered lysis buffer is mixed with the blood sample in a 1:1 ratio. Thus, the lysed sample and RT-LAMP reagent follow the 1:7 ratio as optimized for off-cartridge performance. By pushing the solution through the channels until the fluid reaches the outlet ports, the proper ratio of the reagents is ensured. The bottom of the sliding valve has circular grooves (1 mm wide, 3 mm inner diameter) for O-rings (McMaster-Carr, IL, US) to prevent leakage at the junctions of the channels. The hand-operated 800 μl hollow threaded screw is used to push and pull the solution through the mixing channel, and to push the mixed solution into the Modula A outlet. To ensure the correct positioning of the sliding valve for each step of the cartridge preparation and sample processing, two “keys” with numbered lugs corresponding to each position (1 through 6) were 3D printed. To position the sliding valve into the correct position for its corresponding step, the key is simply pressed into the indicated side of the cartridge, and the length of the lug pushes the sliding valve into the correct location. The sliding valve, threaded screw pump, and transparent materials were utilized to enable manual operation of the cartridge functions, without the use of on-board actuators, complex micromachined pumps/valves, or sensors that contribute to increased cost and complexity. We also envision the potential for a simple desktop mechanical instrument that could interface with an inserted cartridge to automate the processes of rotating the threaded pump and sliding the valve in the correct sequence of positions. A video showing how to assemble the Module A microfluidic cartridge can be viewed in Video S1 (ESI†).
 | ||
| Fig. 2 PathTracker detection concept of operation. (a) Module A consists of three major components: cartridge body, sliding valve, and threaded syringe. The sliding valve has two metered channels to ensure the proper ratio between lysis buffer, RT-LAMP reagents, and the blood sample. The threaded syringe provides pneumatic force for mixing and fluid movement to the outlet; (b) Module B has six separate compartments that each hold 25 μl. Transparent tape is applied to both top and bottom surfaces to seal the compartments; (c) a photograph of the complete detection system; (d) spatial analysis of fluorescence generation in the fluid compartments is used to identify the initial formation and growth of fluorescent clusters during the RT-LAMP, using an “activated intensity” metric to more rapidly identify a positive test. For brevity, only 3 consecutive image frames in the middle of a recording are visualized. | ||
Maintaining the proper ratio between reagents is critical for optimal RT-LAMP assay performance, and with our cartridge, we seek to accurately and repeatedly replicate the volumes and ratios of the laboratory-based version of the assay. Five volume tests were performed with deionized (DI) water for each metering channel and for the volume added to the sample inlet port of a single Module A. Results are shown in Fig. 3b–d. The 3D-printed sliding valves yielded average volumes of 12.79 μl and 174.79 μl for lysis buffer and RT-LAMP reagent metering channels, respectively. The percent errors for each channel were calculated to be 2.3% and 0.1%. Moreover, the 3D-printed sliding valves showed a sample recovery rate of 12.17 μl from 12.5 μl of the input sample (2.61% error). Thus, all the measured volumes were within our design tolerance of <5% volume error.
 | ||
| Fig. 3 Device characterization. (a) Temperature characterization curve for Module B compartments in contact with the instrument heater. The gray dotted line denotes the target temperature is 65 °C; (b and c) Repeatability characterization of (b) blood volume metering (Module A) and (c) lysis buffer metering (Module A), with a target volume of 12.5 μl in both cases; (d) Repeatability characterization of RT-LAMP reaction mix metering (Module A), with a target volume of 175 μl; (e) bar plot showing amplification time versus Zika virus concentration spiked in whole blood. Module A mixing and off-cartridge amplification. The numbers over the bars indicate the number of amplifying replicates; (f) the average amplification time of the results from (e); (g) normalized amplification curves of the results from (f); (h) Comparison of on- and off-cartridge mixing (off-cartridge amplification). | ||
The body of Module B, shown in Fig. 2b, is 3D printed from an opaque material to reduce the potential for optical crosstalk between adjacent channels, and to reduce the potential for external illumination to reach the image sensor. The component integrates six independent vertically oriented fluid compartments (0.5 depth × 20 length × 2.5 mm width) designed to contain 25 μl of fluid. The thickness of the Module B body was selected to minimize the mechanical bowing of the device during the 65 °C LAMP assay heating process. Module B assembly includes attaching transparent biocompatible tape (ARSeal, Adhesive Research) to both the top and bottom surface of the device, followed by drilling inlet/outlet holes. The transparent tape allows for optical imaging of the LAMP reaction and visual inspection of bubble-free filling. Assembled modules are stored at room temperature in a sealed container until use. A video showing how to seal Module B can be viewed in Video S2 (ESI†).
Both modules were designed using 3D Studio software and 3D printed by a Form2 desktop stereolithography 3D printer (Fig. 2c). BioMed Clear resin, designated for biocompatible applications, was used to print Module A at 100-micron resolution, and black resin was used to produce Module B at a 25-micron printing resolution. To ensure a smooth surface finish, the parts were oriented vertically on the build platform. After printing, the parts were immediately washed with isopropyl alcohol to remove excess resin inside the channels, ports, and through holes to prevent blockage. After an isopropyl alcohol bath (10 min), the parts were dried (room temperature, 30 min) and put into the post-curing chamber (Form Cure, FormLabs; 1 h., 60 °C).
Fabrication of two complete sets of Module A requires approximately 12 hours of printing time (∼8 hours for the cartridge body and threaded syringes, and ∼4 hours for the sliding valves). The parts were printed separately to obtain the most accurate and repeatable finished dimensions to ensure correct volumes. Fabrication of three Module Bs requires approximately ∼8 hours of printing time. To avoid the potential for nucleic acid contamination between tests, each Module A and Module B was only used for one assay.
To assemble a complete Module A from its component parts, silicon lubricant was lightly applied to the O-rings on the bottom of the sliding valve, after which the valve was inserted into the cartridge body with the valve arrow pointing toward the position indicators. The threaded syringe was prepared by placing the O-ring at the bottom of the plunger located at the inner center of the syringe cap and sliding it into the chamber after applying silicone lubricant on the inner surface. The assembled threaded syringe was inserted into the cartridge body with an additional O-ring placed in the surface junction to prevent leakage. The assembled module was stored at room temperature in a sealed container until use.
2.6. Design, fabrication, and testing of the clip-on detection instrument
The detection instrument was designed to accomplish the following objectives: 1. To provide rapid, uniform, and accurate heating of Module B; 2. To provide consistent intensity and uniform illumination of the ∼490 nm wavelength excitation to match the 485 nm peak absorption of the Evagreen fluorescent dye; and 3. To enable the collection of fluorescence emission images of all six compartments using a smartphone camera in a single field of view. To achieve the desired thermal conditions (65 °C ± 1 °C) in each reaction chamber, a Peltier thermoelectric module was used (30 × 30 × 5.3 mm3, 5 V, 1 A, Adafruit) and attached to a copper sheet (1 mm, McMaster) by applying thermal paste (IC Diamond, Innovation Cooling) in between the contact surfaces. The Peltier module was mounted in the center of the lower base of the instrument on a disc-shaped platform with a threaded rotating lid. The rotating lid is used to press the Peltier module against Module B to achieve excellent thermal contact for repeatable heat transfer. While the Peltier module presses against Module B from below, a housing with a rectangular window holds Module B firmly in place from above, while exposing the reaction chambers for illumination and optical imaging. Providing mechanical force around the perimeter of Module B during the LAMP heating process also serves to minimize the potential for bowing of the part, which would result in loss of uniform contact to the heater, thus inducing temperature variation between the fluid compartments. To verify uniform heat distribution, an infrared thermal imager (FLK-TIS55, Fluke Corporation, WA, USA) was used to measure the fluid temperature of each compartment of Module B (Fig. S4a†). In addition, we constructed a “test” Module B that enabled us to measure the temperature of the fluid within each compartment for the maximum duration of an assay (40 min) and to measure the repeatability of the temperature stabilization. The test Module B was produced by drilling holes through the transparent tape, for insertion of K-Type thermocouple probes (TL0260 K-Type Thermocouple, 0.13 mm Diameter, Perfect Prime, NY, USA) into each compartment followed by sealing of the holes with a gel-type super glue (Super Glue Gel control, Loctite, Ohio, USA) to prevent fluid evaporation. The submerged thermocouple probes measure the fluid temperature at 1 second intervals using a thermocouple thermometer (TC0520, Thermocouple Thermometer, Perfect Prime, NY, USA) (Fig. S4b†). The Peltier module offered thermal homogeneity throughout the six compartments in Module B. As shown in Fig. 3a, the temperature of each reaction compartment reached 60 °C within 4.5 min on average and stayed under 66 °C for the rest of the incubation time. Because an average of 4.5 min was spent on bringing the solution to the optimal temperature range, the reaction amplification time was started after this time.To provide uniform illumination, we designed and fabricated a custom printed circuit board (PCB) with eight 458 nm blue LEDs (XPEBBL, Cree, NC, USA) and a custom constant current LED driver module. We control the circuit's applied voltage with a signal supplied by the audio (headphone) jack of the smartphone, and a software app. A standard wired audio cable was used to connect the audio terminal on the LED controller circuit board to the smartphone. The custom constant current LED driver module ensured a consistent excitation light intensity across Module B, which was captured at 10 seconds intervals and with 2 seconds illumination duration in each interval. The PCB was fitted to a mechanical fixture to hold four 490 nm short-pass fluorescence emission filters (Asahi Spectra, Tokyo, Japan) arranged with square symmetry. The LEDs were arranged on the PCB in a circular pattern surrounding Module B and oriented downwards, so that each emission filter covers two LEDs. To verify uniform excitation light illumination, images of Module B were taken without the emission filter and the light intensity of each pixel was analyzed using MatLab. Fig. S5† shows that the system achieved uniform excitation illumination across the surface of Module B. Two AAA batteries provide power to the LEDs and a 9 V battery powers the heater. With two AAA batteries, the system is capable of approximately 10 h. of image recording, sufficient for 20–30 tests. The 9 V battery is capable of performing approximately 1.5 hours of heating.
Finally, the top part of the instrument houses multiple components, including a 510 nm pass filter to permit only the emission wavelength to reach the smartphone image sensor, a macro lens (B00XXK4AN2, 12X Super Macro Lens, Techo) for close-up photography, and a clip to securely hold the smartphone. The instrument weighs approximately 15 ounces and has the dimensions of ∼87 × 60 × 50 mm3. Two switches located in the middle layer of the instrument turn the circuitry on and off. The detailed schematic of the instrument is illustrated in Fig. S1a† while Fig. S1b† illustrates the working principles. The components used to produce the instrument are all commercially available with a total cost of ∼$385.
2.7. PathTracker app
We designed our own mobile software (the PathTracker app), to obtain images and to analyze the on-cartridge experiments. The PathTracker app gathers and records fluorescence emission images of Module B at user-determined intervals. The user-friendly interface allows the user to make manual adjustments to the camera parameters, including zoom factor, focus mode, exposure time, and ISO. All images are compressed into High Efficiency Image File Format (HEIF) before transfer to cloud storage for offline image processing.2.8. On-cartridge measurement procedures
2.9. Spatial RT-LAMP image analysis
Although the fluorescence formation process is continuous, our image processing module performs its analysis on a discrete sequence of images with a sampling interval equal to 10 seconds. The pipeline is categorized into two main functional modules: image processing and amplification analysis. The overview of our image analysis is described in Fig. 2d. The image processing module systematically performs preprocessing of images, thresholding pixels, detecting amplification clusters, and tracking temporal and spatial changes of identified clusters.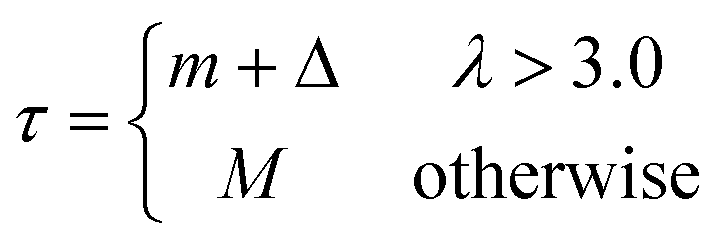 | (1) |
Where m and M denote lower and upper global threshold values while Δ and λ denote margin of error and growth factor, respectively. Both m and M are calculated separately from two individual sets of six consecutive frames that represent the darkest and brightest moments of a recording. The value of M is taken from the median intensity value of all the pixels confined by an ROI in the last six images of a recording where all the target nucleic acids have amplified, if any. Likewise, the value of m is taken from the median intensity value of all the pixels confined by an ROI in the darkest moment of a recording, which represents the timing where the intercalating dye has become inactivated due to the denaturation of primers as the temperature gradually increases. To account for any intensity fluctuation, 10% of the difference between M and m was added to the final lower threshold value as a margin of error Δ.
| Δ = 0.1(M − m) | (2) |
Growth factor λ decides whether the identified cluster is an active amplification of target nucleic acid or background signal, such as reflections of LEDs.
| λ = M/m | (3) |
Given the information obtained from the image processing module, the amplification analysis module enables temporal and spatial characterization of clusters. Two distinct types of fluorescent intensities are mainly investigated: global and activated. The global intensity (GI) measures the normalized average fluorescent intensity of all pixels within an ROI at a given amplification time, while the activated intensity (AI) measures the normalized average fluorescent intensity of all the pixels that delineate amplified nucleic acid clusters within the ROI at a given amplification time.
3. Results and discussion
3.1. Assay characterization with gamma-irradiated ZIKV in buffer and whole blood using RT-LAMP performed off-cartridge in PCR tubes
To evaluate the performance of selected primers, off-cartridge RT-LAMP experiments were performed on a standard thermocycler (QuantStudio 3, Applied Biosystems) with various concentrations (ranging from 3 × 103 to 3 × 106 copies per μl, and blank samples) of gamma-irradiated ZIKV strain PRVABC59 in buffer solutions. In all the experiments, the standard RT-LAMP protocol was followed using pipettes to mix and transfer solutions. The amplification curves and threshold times for the experiments are shown in Fig. S6† (ESI†). Based on the collected data, the assay can detect ZIKV at concentrations ≥3 × 103 copies per μl with non-nonspecific amplification (reported for 6 blank samples tested) occurring after 40 minutes.Next, off-cartridge RT-LAMP experiments were performed, under the same conditions but spiking the gamma-irradiated virus into whole blood samples instead of buffer. The assay showed consistent performance in both blood and buffer media. To further verify the statistical similarity, t-tests were performed between the results from both buffer and whole blood samples. We found that the p-value across all the concentrations was greater than 0.05, indicating no significant differences in the threshold times between the two distinct types of samples. The raw data for each amplification can be found in Fig. S7 (ESI†). The comparison data for amplification in blood and buffer are provided in Fig. S8 (ESI†).
3.2. Assessment of Module A lysis and mixing, using off-cartridge RT-LAMP reactions
Module A's viral lysis and LAMP reagent mixing performance was assessed by performing the sample preparation steps in Module A, followed by RT-LAMP reactions in conventional PCR tubes in a thermocycler. Gamma-irradiated Zika virus was spiked to whole blood samples in concentrations ranging from 3 × 103 to 3 × 106 copies per μl (n = 3). These results were compared side-by-side with the previously performed RT-LAMP experiments conducted completely off-cartridge. Each replicate was tested in a newly fabricated Module A. Since a single Module A produced multiple 16 μl reactions, we considered the whole batch to be a true positive if at least one replicate showed amplification. The final amplification time was determined by averaging the time of amplified replicates.Results demonstrated that the amplification times for a given concentration were consistent between different Module As (Fig. 3e–g). There were no significant differences between the assays prepared with and without Module A as the p-values were >0.05 (0.751, 0.095, and 0.733 for 3 × 104, 3 × 105, and 3 × 106 copies per μl, respectively) (Fig. 3h). Moreover, the mixing performed with Module A showed improved consistency at higher concentration in terms of amplification time than the assay prepared off-cartridge using pipettes and PCR tubes (Fig. 3h). This also demonstrates that the presence of the lubricant has no significant effects on the amplification time.
3.3. Spatial RT-LAMP assay characterization in whole blood samples using Module A, Module B, and the PathTracker app
Finally, the full on-cartridge assay (Module A for mixing, Module B for amplification, and PathTracker app for imaging and analysis) was characterized by determining amplification time and amplification location for each reaction compartment. Two separate Module As were used to prepare the experimental control groups for every concentration being tested (0, 103, 3 × 103, 6 × 103, 3 × 104, 3 × 104, 3 × 105, and 3 × 106 copies per μl). For positive samples with concentrations ≥6 × 103 copies per μl, 75 μl out of 200 μl was extracted from each Module A, for on-cartridge reactions in Module B (25 μl for each three compartments). Hence, the remaining volumes were applied to the standard thermocycler as controls to evaluate the accuracy of on-cartridge testing (characterization purposes). For positive samples with concentrations ≤3 × 103 copies per μl, the total volume (200 μl) was extracted from each Module A, for on-cartridge reactions in two Module Bs (25 μl for each three compartments). In all Module Bs, three compartments were filled with the negative control solution. The prepared Module Bs were then loaded onto the instrument, immediately followed by a recording of the amplification event using the PathTracker app. For all our experiments, the camera settings were set to an imaging interval = 10 s., 400 ISO, integration time = 1 s, 4X zoom factor, and auto focus mode. Upon completion of recording, the image analysis was performed on the collected images to calculate the amplification threshold time of each reaction compartment.Fig. 4a and b show the smartphone images taken at 0, 12, 18, 24, and 30 min of an on-cartridge Spatial RT-LAMP experiment with 0, 3 × 104, and 3 × 106 copies per μl of gamma-irradiated ZIKV spiked in whole blood sample. Videos showing the amplification events for other concentrations tested can be found in the ESI (Video S5, ESI†). Visual inspection of the images reveals that, for all the concentration ranges, the nucleic acid clusters were clearly observable in more than one compartment. We also observe that the time required for initial cluster formation increased as the virus concentration decreased. The number of clusters also shows a decreasing trend as the virus concentration is reduced. All the negative control groups (0 copies per μl) had no signs of cluster formation within 35 minutes of the incubation, indicating the high specificity of the assay when performed in the cartridge. Minimal to no fluid evaporation was observed in all compartments throughout the experiment, and we conclude that evaporation has no effect on the quantitative positive/negative determination of the reaction. Lastly, we observe no signs of cross-contamination between the compartments, indicating secure isolation of compartments for multiplexing analysis.
 | ||
| Fig. 4 On-cartridge analysis and detection results with gamma-irradiated virus spiked in whole blood. Images taken by the smartphone are analyzed by the PathTracker app, which tracks fluorescence intensity and clusters of amplification; (a and b) Smartphone images of on-cartridge amplification of (a) 0 and 3 × 106 copies per μl, and (b) 0 and 3 × 104 copies per μl of gamma-irradiated Zika virus in whole blood at t = 0, 12, 18, 24, and 30 min. Images marked to indicate area of focus for each compartment in Module B; (c) amplification times versus concentration bar plot for global intensity and activated intensity analysis from the PathTracker app. T-tests performed between the two analysis types show significant differences in the two methods in all concentrations. Numbers over bars indicate how many compartments out of all tested amplified for a given concentration; (d) Zika detection calibration curves obtained from both analysis methods. | ||
Next, the amplification times of full on-cartridge reactions (Module A & B) were compared with full off-cartridge results previously obtained using the standard commercial thermocycler. As Fig. S9 (ESI†) shows, we obtain consistent amplification for all concentrations down to 3 × 104 copies per μl where all replicates amplified for both on and off cartridge assays. Additionally, the on-cartridge amplification results display shorter amplification threshold times with statistically significant (p-values < 0.05) across all concentrations. However, for on-cartridge with 6 × 103 copies per μl, 5 out of 6 replicates amplified while the off-cartridge result showed amplification on all replicates. The on-cartridge assays with 103 and 3 × 103 copies per μl had 1 out of 6 replicates amplified while the off-cartridge assay with 3 × 103 copies per μl had amplification on 1 out of 3 replicates.
Lastly, the performance of the global and activated intensity analyses are compared in Fig. 4c and d. The calculated amplification times at the end of the amplification event for both GI and AI analyses are compared in Fig. 4c. For all the concentrations tested, the amplification times obtained with the AI technique were statistically lower (p-values < 0.05) than the amplification times obtained with the GI technique. Importantly, all six negative controls did not amplify. Using the amplification times obtained for each virus concentration and analysis technique, the calibration curves are shown in Fig. 4d. These curves also highlight the faster amplification times obtained when using AI analysis, as well as the lower variability of this technique at lower concentrations.
3.4. Discussion
In this study, gamma-irradiated ZIKV was used as a simulant for infectious ZIKV to characterize the developed POC system performance, as clinical samples or patient samples with real ZIKV are challenging to obtain and will be the subject of or follow-on study with international collaborators. As can be seen in Fig. 4c and d, the lowest ZIKV concentration detected was 103 copies per μl, which roughly corresponds to 1560 target gamma-irradiated viral particles molecules in each 25 μl compartment in Module B. It is important to point out that several studies report that gamma-irradiation damages the genetic material of a virus,52 which can affect the sensitivity of nucleic acid tests when the target RNA sequence within the virus is no longer intact. This phenomenon is clearly observed in the off-cartridge assays, where the lowest concentration we have detected above zero of 270 RNA copies per μl is obtained, corresponding to a gamma-irradiated virus detection limit of 3 × 103 copies per μl. This factor of approximately one order of magnitude represents the expected loss in sensitivity when testing gamma-irradiated viruses instead of intact live viruses. For instance, a recent study on SARS-CoV-2 (also an RNA virus) showed lost sensitivity by almost an order of magnitude in PCR tests when gamma-irradiated virus was used as the target.53 Therefore, the sensitivity of our assay was determined not using gamma-irradiated virus as the target, but two different strains of ZIKV genomic RNA (Fig. S10, ESI†). As shown in this figure, the sensitivity improved one order of magnitude as the obtained Limits of Detection (LOD) of the assays were 400 and 270 copies per μl for PVRABC58 and R103451 strains, respectively. This sensitivity is similar to the other previously reported ZIKA RT-LAMP assays.54 Likewise, when comparing the amplification times obtained with genomic RNA and gamma-irradiated virus, the RNA amplification times were of the same order as gamma-irradiated virus samples with a concentration approximately two orders of magnitude higher (270 copies per μl of RNA (R103451 strain) amplified in ∼27 min, while 3 × 104 per μl of gamma-irradiated virus amplified in ∼24 min; targets spiked in buffer and experiments performed off-cartridge).As mentioned in Section 3.3, the assay solution produced by Module A was fragmented. While part of the final assay solution from Module A was employed for on-chip experimentation, the rest of the solution was used for side-by-side off-cartridge control. Likewise, three compartments were used as negative controls in all the Module Bs analyzed. This approach is utilized because, in future implementations of this device, we plan to achieve multiplexing by incorporating RT-LAMP primers for different viruses into each compartment, so that a single cartridge could simultaneously determine the presence of up to four viruses, while utilizing the remaining two compartments for experimental controls to ensure assay validity. Multiplexing via sample splitting has the unfortunate effect of increasing detection limits, although the detection limits demonstrated here are expected to be suitable for clinically relevant testing scenarios.
Regarding the analysis technique, this paper compares two methods, the analysis of the global intensity and the analysis of the activated intensity using the Spatial LAMP approach. The GI resembles the standard quantification method used in a conventional thermocycler where overall fluorescent intensity from a micro-tube represents the amplification signal. Whereas the AI quantification method detects individual amplification within ROI and computes the normalized average fluorescent intensities of only the identified clusters. Using GI quantification, a compartment is considered amplified if its normalized average fluorescent exceeds 20% of the intensity gap between the lower and upper global threshold values (m and M). In the case of AI quantification, a compartment is considered amplified when a first cluster is detected. To further increase the stability and robustness of the pipeline in detecting clusters from outliers, a Savitzky–Golay filter with size 11 is applied to the results before being approximated by a sigmoid function in a non-linear least-squares fashion.
The results from Fig. 4c and d demonstrate the advantages of the Spatial LAMP approach (activated intensity) over the traditional global intensity method. The benefits of AI analysis increase at lower virus concentrations, as greater reductions in amplification time were observed. All samples analyzed with the activated intensity Spatial LAMP approach showed amplification before 20 minutes. Therefore, the AI method offers faster virus detection and is especially useful for screening purposes (yes/no detection). Thus, with this method, we can differentiate positive from negative samples as little as 20 min for the amplification reaction. However, the steeper slope in the calibration curve obtained using the GI method highlights the greater quantification capacity of this method. The raw data from Fig. 4c and d can be found in Fig. S11 and S12 (ESI†). Likewise, the criteria used by the PathTracker app to differentiate positive samples from negative ones can be found in the ESI† (Results 1).
Despite advances in the development of POC devices for ZIKV detection in recent decades, many critical challenges remain to be addressed in the application of these devices. Recently, Faria et al. demonstrated ZIKV viral RNA detection in the nM range using gold electrodes anchored with capture DNA probes.55 Although achieving low LOD is a powerful aspect of a system for early diagnosis and controlling and monitoring infectious viruses, the measurement relies on RNA extraction and purification, which mandates laboratory equipment, which subsequently increases the complexity of the workflow. Likewise, the fabrication of electrodes requires cleanroom fabrication techniques, which increase the overall device cost. In another example, Afsahi et al. showed label-free detection of ZIKV (LOD = 0.45 nM) by immobilizing anti-ZIKV NS1 antibodies on the surface of a graphene electrode.56 However, the measurements were performed in PBS or diluted serum. Other reported assays demonstrated ZIKV detection by replacing the chemical RNA extraction with thermal lysing when using serum as the sample matrix.57 Similarly, to the previous example, this device was not capable of working with unprocessed blood as they require a centrifugation step to obtain serum. Likewise, the total assay time was 3 hours. While our device can achieve the detection of the virus in <32 min from sample to answer. Rong et al. developed a lateral flow immunoassay that could detect ZIKV NS1 (LOD = 0.045 ng ml−1) in 20 minutes.58 However, sensitivity was drastically reduced in serum (LOD = 0.15 ng ml−1) and was not shown in whole blood, meaning once again centrifugation was a necessary step beforehand thus limiting POC applications. RT-RPA has been used to detect Sars-CoV-2, another RNA virus, at 1 copy per μl in 15 minutes in a point-of-care device made by Liu et al.59 However, this required a previous RNA extraction step beforehand and thus similar to previous examples is less suitable for POC applications.
In Table 1 we compare our device to other RT-LAMP POC devices used for the detection of Zika virus. It demonstrates how we were able to achieve a fast sample-to-answer time of 32 minutes and a high level of sensitivity without the need for any purification steps. Additionally, we demonstrated our assay's performance with whole blood samples. This further simplifies the workflow since it removes the need to process blood through centrifugation or filtration to obtain plasma before running our assay.
| Pre-treatment and mixing of samples | Sample | Reaction time (min) | RNA extraction method | Limit of detection | Reading method | Ref |
|---|---|---|---|---|---|---|
| Magnetic actuation through microfluidic Device | Plasma | 50 | Extraction kit processed on-chip | 100 copies per ml | Colorimetric | 44 |
| Automated syringe pump based mixing | Whole blood | 50 | Chemical lysis on-chip | 1.56 × 105 PFU per ml | Smartphone-based reader (fluorescence) | 47 |
| LAMP buffer pipetted onto paper-chip after extraction | Serum | ∼60 | Chemical lysis, hybridization, and elution on-chip | 10 copies per μl | ChemiDoc MP imaging system | 60 |
| Microfluidic device with metering capabilities, no pumps or motors needed | Whole blood | 32 | Chemical lysis on-chip | 103 copies per μl | Smartphone-based reader (fluorescence) | This paper |
Field-ready assay systems demand minimal to no intervention of laboratory equipment to process the sample, perform the assay, and receive results with a short sample-to-answer time. In this regard, our smartphone-based microfluidic system satisfies the conditions for field employment as the total time necessary to perform a complete Spatial-LAMP assay is currently <32 min: (1) sample injection and mixing with reagents using Module A (5 min); (2) loading and sealing Module B (4 min); (3) amplification and reaction site recording (22 min); and (4) image analysis and diagnosis (0.5 min). The current design for Module A allows for simplified mixing and metering of the lysing buffer and reagents, and only requires minimal use of pipettes to inject the prepared solution for sample treatment and transfer the final mixture to Module B. This study demonstrates the precise and repeatable RT-LAMP assay preparation at low cost with minimal involvement of laboratory equipment, which makes the device suitable for POC application. The preparation time can further be reduced by physically connecting Module A and B to eliminate the manual loading and sealing of the chip. We envision that a consolidated Module would also be equipped with buffer pod reservoirs that contain reagents to establish a single-use, one-step assay system only requiring the acquisition of a whole blood sample without the utilization of centrifuges, pumps, motors, and incubators. We are actively developing injection molded versions of the cartridge and instrument, which can greatly reduce the production cost while allowing for inexpensive mass-production of the system. Lastly, this compact, portable pathogen detection system takes advantage of readily available mobile devices as the image sensor for optical measurement, data analysis, and results sharing/transmission, which renders on-site diagnosis possible in various POC settings, and integration with cloud-based telehealth systems for result interpretation, reporting to health authorities, and facilitated access to physicians for development of a treatment plan.
4. Conclusion
We have demonstrated the fast detection (<32 min) of Zika virus using the developed Spatial RT-LAMP approach. The activated intensity Spatial LAMP approach can differentiate positive from negative samples as quickly as 22 min. The presented point-of-care device allows for the analysis of a drop whole blood sample with minimal external intervention, as the device meters the lysing buffer and LAMP reagents and performs mixing without using external pump or pipettes. Likewise, the assay does not require RNA purification and can work with unprocessed whole blood.Moreover, the microfluidic cartridge fabricated using 3D printing enables low-cost, rapid, and simple assay preparation, making the system attractive for various POC settings. Together with broad availability of smartphones and an inexpensive clip-on instrument that utilizes the rear-facing image sensor, we hope to provide a cost-effective device capable of bringing pathogen diagnostics to resource-poor regions.
Likewise, the spatially separated detection compartments open the door to multiplexing capabilities. Thus, soon we plan to incorporate assays for other blood-based viruses that present similar symptoms as Zika virus, such as Dengue and Chikungunya. Also, we will integrate the two existing modules into a single device where the mixed solution from Module A flows directly into Module B without needing to pipette between two distinct devices.
Author contributions
A. M. J., H. L., W. W. and T.-H. H. contributed equally to this work. B. T. C., E. V., R. B., M. N. D. and W. P. K. designed the research. A. M. J., H. L., W. W., T.-H. H., A. B., F. S., S. C., V. K., K. K., R. A. S., D. D. C. and Z. W. E. performed the research. A. M. J, H. L., W. W., T.-H. H., E. V., and B. C. analyzed data. H. L., A. M. J., W. W., T.-H. H., R. B., E. V., and B. T. C. wrote the paper.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The following reagents were obtained through BEI Resources, NIAID, NIH: (1) Genomic RNA from Zika Virus, PRVABC59, NR-50244; (2) Genomic RNA from Zika Virus, R103451, NR-50358; (3) Zika Virus, PRVABC59, Infected Cell Lysate, Gamma-Irradiated, NR-50547. The authors are grateful for financial support from the National Institutes of Health (R01AI139401) and from the National Science Foundation Partnership for Innovation program (PFI-TT 1919015).References
- A. R. Plourde and E. M. Bloch, A Literature Review of Zika Virus, Emerg. Infect. Dis., 2016, 22(7), 1185–1192 CrossRef CAS PubMed.
- J. J. Waggoner and B. A. Pinsky, Zika Virus: Diagnostics for an Emerging Pandemic Threat, J. Clin. Microbiol., 2016, 54(4), 860–867 CrossRef CAS PubMed.
- E. Antoniou, et al., Zika Virus and the Risk of Developing Microcephaly in Infants: A Systematic Review, Int. J. Environ. Res. Public Health, 2020, 17(11), 3806 CrossRef PubMed.
- J. F. Chan, et al., Zika fever and congenital Zika syndrome: An unexpected emerging arboviral disease, J. Infect., 2016, 72(5), 507–524 CrossRef PubMed.
- D. T. Deckard, et al., Male-to-Male Sexual Transmission of Zika Virus–Texas, January 2016, Morb. Mortal. Wkly. Rep., 2016, 65(14), 372–374 CrossRef PubMed.
- G. Venturi, et al., An autochthonous case of Zika due to possible sexual transmission, Florence, Italy, 2014, Eurosurveillance, 2016, 21(8), 30148 CrossRef PubMed.
- B. D. Foy, et al., Probable non-vector-borne transmission of Zika virus, Colorado, USA, Emerging Infect. Dis., 2011, 17(5), 880–882 CrossRef PubMed.
- M. Besnard, et al., Evidence of perinatal transmission of Zika virus, French Polynesia, December 2013 and February 2014, Eurosurveillance, 2014, 19(13), 20751 CrossRef PubMed.
- M. M. Magnus, et al., Risk of Zika virus transmission by blood donations in Brazil, Hematol. Transfus. Cell Ther., 2018, 40(3), 250–254 CrossRef PubMed.
- C. Brown, Zika virus outbreaks in Asia and South America, Can. Med. Assoc. J, 2016, 188(2), E34–E34 CrossRef PubMed.
- P. Pielnaa, et al., Zika virus-spread, epidemiology, genome, transmission cycle, clinical manifestation, associated challenges, vaccine and antiviral drug development, Virology, 2020, 543, 34–42 CrossRef CAS PubMed.
- Organization, P.A.H., Cases of Zika Virus Disease by Country or Territory, 2020.
- M. R. Duffy, et al., Zika virus outbreak on Yap Island, Federated States of Micronesia, N. Engl. J. Med., 2009, 360(24), 2536–2543 CrossRef CAS PubMed.
- S. N. Ladhani, et al., Outbreak of Zika virus disease in the Americas and the association with microcephaly, congenital malformations and Guillain-Barré syndrome, Arch. Dis. Child., 2016, 101(7), 600–602 CrossRef PubMed.
- Centers-for-Disease-Control-and-Prevention, Zika Virus.
- M. Miller, et al., What Are People Tweeting About Zika? An Exploratory Study Concerning Its Symptoms, Treatment, Transmission, and Prevention, J. Med. Internet. Res., 2017, 3(2), e38–e38 Search PubMed.
- E. Valera, et al., COVID-19 Point-of-Care Diagnostics: Present and Future, ACS Nano, 2021, 15(5), 7899–7906 CrossRef CAS PubMed.
- J. J. C. Voermans, et al., Whole-Blood Testing for Diagnosis of Acute Zika Virus Infections in Routine Diagnostic Setting, Emerg. Infect. Dis., 2019, 25(7), 1394–1396 CrossRef CAS PubMed.
- T. T. Bui, et al., Development of Universal and Lineage-Specific Primer Sets for Rapid Detection of the Zika Virus (ZIKV) in Blood and Urine Samples Using One-Step Reverse Transcription Loop-Mediated Isothermal Amplification (RT-LAMP), Jpn. J. Infect. Dis., 2020, 73(2), 153–156 CrossRef CAS PubMed.
- B.-T. Teoh, et al., A reverse transcription loop-mediated isothermal amplification for broad coverage detection of Asian and African Zika virus lineages, BMC Infect. Dis., 2020, 20(1), 947 CrossRef CAS PubMed.
- A. E. Calvert, et al., Rapid colorimetric detection of Zika virus from serum and urine specimens by reverse transcription loop-mediated isothermal amplification (RT-LAMP), PLoS One, 2017, 12(9), e0185340 CrossRef PubMed.
- L. E. Lamb, et al., Rapid Detection of Zika Virus in Urine Samples and Infected Mosquitos by Reverse Transcription-Loop-Mediated Isothermal Amplification, Sci. Rep., 2018, 8(1), 3803 CrossRef PubMed.
- B. Atkinson, et al., Detection of Zika Virus in Semen, Emerg. Infect. Dis., 2016, 22(5), 940 CrossRef CAS PubMed.
- J. M. Mansuy, et al., Zika virus in semen and spermatozoa, Lancet Infect. Dis., 2016, 16(10), 1106–1107 CrossRef PubMed.
- M. Mercado, et al., Zika virus detection in amniotic fluid and Zika-associated birth defects, Am. J. Obstet. Gynecol., 2020, 222(6), 610.e1–610.e13 CrossRef CAS PubMed.
- L. Li, et al., Development of a direct reverse-transcription quantitative PCR (dirRT-qPCR) assay for clinical Zika diagnosis, Int. J. Infect. Dis., 2019, 85, 167–174 CrossRef CAS PubMed.
- R. N. Charrel, et al., Background review for diagnostic test development for Zika virus infection, Bull. W. H. O., 2016, 94(8), 574–584D CrossRef PubMed.
- C. Zhang and D. Xing, Miniaturized PCR chips for nucleic acid amplification and analysis: latest advances and future trends, Nucleic Acids Res., 2007, 35(13), 4223–4237 CrossRef CAS PubMed.
- S. H. Lee, et al., A polymer lab-on-a-chip for reverse transcription (RT)-PCR based point-of-care clinical diagnostics, Lab Chip, 2008, 8(12), 2121–2127 RSC.
- Z. Chen, et al., A microfluidic system for saliva-based detection of infectious diseases, Ann. N. Y. Acad. Sci., 2007, 1098, 429–436 CrossRef CAS PubMed.
- D. Chen, et al., An integrated, self-contained microfluidic cassette for isolation, amplification, and detection of nucleic acids, Biomed. Microdevices, 2010, 12(4), 705–719 CrossRef CAS PubMed.
- F. Ahmad and S. A. Hashsham, Miniaturized nucleic acid amplification systems for rapid and point-of-care diagnostics: a review, Anal. Chim. Acta, 2012, 733, 1–15 CrossRef CAS PubMed.
- N. Mishra, et al., Zika Virus Peptide ELISA (ZIKV-NS2B-Concat ELISA) for Detection of IgG Antibodies to Zika Virus Infection, Methods Mol. Biol., 2020, 2142, 113–122 CrossRef CAS PubMed.
- K. H. Lee and H. Zeng, Aptamer-Based ELISA Assay for Highly Specific and Sensitive Detection of Zika NS1 Protein, Anal. Chem., 2017, 89(23), 12743–12748 CrossRef CAS PubMed.
- M. A. Alam, et al., Diagnostic approaches for the rapid detection of Zika virus–A review, Process Biochem., 2021, 101, 156–168 CrossRef CAS.
- A. Priye, et al., A smartphone-based diagnostic platform for rapid detection of Zika, chikungunya, and dengue viruses, Sci. Rep., 2017, 7(1), 44778 CrossRef CAS PubMed.
- Y. Kurosaki, et al., Development and evaluation of a rapid molecular diagnostic test for Zika virus infection by reverse transcription loop-mediated isothermal amplification, Sci. Rep., 2017, 7(1), 13503 CrossRef PubMed.
- J. E. Kong, et al., Highly Stable and Sensitive Nucleic Acid Amplification and Cell-Phone-Based Readout, ACS Nano, 2017, 11(3), 2934–2943 CrossRef CAS PubMed.
- A. Ganguli, et al., Rapid isothermal amplification and portable detection system for SARS-CoV-2, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(37), 22727–22735 CrossRef CAS PubMed.
- F. Sun, et al., Smartphone-based multiplex 30-minute nucleic acid test of live virus from nasal swab extract, Lab Chip, 2020, 20(9), 1621–1627 RSC.
- W. Chen, et al., Mobile Platform for Multiplexed Detection and Differentiation of Disease-Specific Nucleic Acid Sequences, Using Microfluidic Loop-Mediated Isothermal Amplification and Smartphone Detection, Anal. Chem., 2017, 89(21), 11219–11226 CrossRef CAS PubMed.
- G. L. Damhorst, et al., Smartphone-Imaged HIV-1 Reverse-Transcription Loop-Mediated Isothermal Amplification (RT-LAMP) on a Chip from Whole Blood, Engineering (Beijing, China), 2015, 1(3), 324–335 Search PubMed.
- M. Sabalza, et al., Detection of Zika virus using reverse-transcription LAMP coupled with reverse dot blot analysis in saliva, PLoS One, 2018, 13(2), e0192398 CrossRef PubMed.
- S. Sharma, M. A. Kabir and W. Asghar, Lab-on-a-Chip Zika Detection With Reverse Transcription Loop-Mediated Isothermal Amplification–Based Assay for Point-of-Care Settings, Arch. Pathol. Lab. Med., 2020, 144(11), 1335–1343 CrossRef CAS PubMed.
- K. Dhama, et al., Loop-mediated isothermal amplification of DNA (LAMP): a new diagnostic tool lights the world of diagnosis of animal and human pathogens: a review, Pak. J. Biol. Sci., 2014, 17(2), 151–166 CrossRef CAS PubMed.
- Z. Chen, et al., A Rapid, Self-confirming Assay for HIV: Simultaneous Detection of Anti-HIV Antibodies and Viral RNA, J. AIDS Clin. Res., 2016, 7(1), 540 CrossRef PubMed.
- A. Ganguli, et al., Hands-free smartphone-based diagnostics for simultaneous detection of Zika, Chikungunya, and Dengue at point-of-care, Biomed. Microdevices, 2017, 19(4), 1–13 CrossRef CAS PubMed.
- D. Musso, et al., Molecular detection of Zika virus in blood and RNA load determination during the French Polynesian outbreak, J. Med. Virol., 2017, 89(9), 1505–1510 CrossRef CAS PubMed.
- A. Lahon, et al., Characterization of a Zika Virus Isolate from Colombia, PLOS Negl. Trop. Dis., 2016, 10(9), e0005019–e0005019 CrossRef PubMed.
- J. M. Mansuy, et al., Zika Virus Infection and Prolonged Viremia in Whole-Blood Specimens, Emerg. Infect. Dis., 2017, 23(5), 863–865 CrossRef PubMed.
- B. Mondal, et al., Numerical study of mixing in wavy micromixers: comparison between raccoon and serpentine mixer, Chem. Eng. Process.: Process Intensif., 2019, 136, 44–61 CrossRef CAS.
- A. J. Hume, et al., Inactivation of RNA Viruses by Gamma Irradiation: A Study on Mitigating Factors, Viruses, 2016, 8(7), 204 CrossRef PubMed.
- M. Durante, et al., Virus Irradiation and COVID-19 Disease, Front. Phys., 2020, 8(415), 565861 CrossRef.
- A. Ganguli, et al., Reverse Transcription Loop-Mediated Isothermal Amplification Assay for Ultrasensitive Detection of SARS-CoV-2 in Saliva and Viral Transport Medium Clinical Samples, Anal. Chem., 2021, 93(22), 7797–7807 CrossRef CAS PubMed.
- H. A. M. Faria and V. Zucolotto, Label-free electrochemical DNA biosensor for zika virus identification, Biosens. Bioelectron., 2019, 131, 149–155 CrossRef CAS PubMed.
- S. Afsahi, et al., Novel graphene-based biosensor for early detection of Zika virus infection, Biosens. Bioelectron., 2018, 100, 85–88 CrossRef CAS PubMed.
- K. Pardee, et al., Rapid, Low-Cost Detection of Zika Virus Using Programmable Biomolecular Components, Cell, 2016, 165(5), 1255–1266 CrossRef CAS PubMed.
- Z. Rong, et al., Smartphone-based fluorescent lateral flow immunoassay platform for highly sensitive point-of-care detection of Zika virus nonstructural protein 1, Anal. Chim. Acta, 2019, 1055, 140–147 CrossRef CAS PubMed.
- D. Liu, et al., A microfluidic-integrated lateral flow recombinase polymerase amplification (MI-IF-RPA) assay for rapid COVID-19 detection, Lab Chip, 2021, 21(10), 2019–2026 RSC.
- B. S. Batule, Y. Seok and M. G. Kim, Paper-based nucleic acid testing system for simple and early diagnosis of mosquito-borne RNA viruses from human serum, Biosens. Bioelectron., 2020, 151, 111998 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2an00438k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |