
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Detection of small-sized DNA fragments in a glassy nanopore by utilization of CRISPR-Cas12a as a converter system†
Shumin
Zhang
a,
Minyi
Liu
a,
Haofa
Cui
a,
Muhammad Asad
Ziaee
a,
Rongwei
Sun
a,
Liting
Chen
a,
Daqi
Chen
*a,
Denis
Garoli
*bc and
Jiahai
Wang
 *a
*a
aSchool of Mechanical and Electrical Engineering, School of Chemistry and Chemical Engineering, Guangzhou University, Guangzhou, 510006, China. E-mail: daqichen@gzhu.edu.cn; jiahaiwang@gzhu.edu.cn; Tel: +86-15068805047 Tel: +86-18816801579
bIstituto Italiano di Tecnologia, Via Morego 30, 16136 Genova, Italy. E-mail: denis.garoli@iit.it
cLiberà Università di Bolzano, Piazza Università 1, 39100 Bolzano, Italy
First published on 20th January 2022
Abstract
The fabrication of nanopores with a matched pore size, and the existence of multiple interferents make the reproducible detection of small-sized molecules by means of solid-state nanopores still challenging. A useful method to solve these problems is based on the detection of large DNA nanostructures related to the existence of small-sized targets. In particular, a DNA tetrahedron with a well-defined 3D nanostructure is the ideal candidate for use as a signal transducer. Here, we demonstrate the detection of an L1-encoding gene of HPV18 as a test DNA target sequence in a reaction buffer solution, where long single-stranded DNA linking DNA tetrahedra onto the surface of the magnetic beads is cleaved by a target DNA-activated CRISPR-cas12 system. The DNA tetrahedra are subsequently released and can be detected by the current pulse in a glassy nanopore. This approach has several advantages: (1) one signal transducer can be used to detect different targets; (2) a glassy nanopore with a pore size much larger than the target DNA fragment can boost the tolerance of the contaminants and interferents which often degrade the performance of a nanopore sensor.
Introduction
Nanopores have become a promising sensing platform for biomolecular analysis due to their easy operation, single-molecule resolution and label-free sensing.1–6 After the fruitful development in the past twenty years, two main kinds of nanopores, including engineered biological nanopores and artificial solid-state nanopores, have been demonstrated as powerful tools for single molecule experiments. Biological, or protein, nanopores have extremely high resolution in the analysis of small-sized targets, which can be practically used in gene sequencing and in investigating molecular interactions.7–13 However, the available pore size of this family of nanopores is limited. Moreover, the mechanical robustness of the biological nanopores is poor, which limits their application range. Recently, solid-state nanopores have received more and more research interest due to their multiple fabrication methods, mechanical stability, low cost and adjustable pore size.14–23 Among the family of solid-state nanopores, glass nanopores have been reported as a versatile platform that enables multiple single-molecule investigations.24–30 Glass nanopores can be easily fabricated without the use of costly nanolithographic methods, with the main strategy for fabrication being based on pulling a glass nanopipette with controlled force. A distinct advantage of the glass nanopore is that a wide range of pore sizes, spanning from ten nanometers up to a hundred micrometers, is easy to achieve.5,25,28,31–34 However, fabrication of glass nanopores with a pore size smaller than 10 nm is challenging and variation in the pore size under the same pulling parameter is still not an ignorable issue.Consequently, the size target range for nucleic acid detection by means of a solid-state glassy nanopore is limited to 500–1000 nt bp−1. In fact, the detection of small species is one of the main challenges that still limits the applications of solid-state nanopores. In order to achieve the capability of detecting small-sized target molecules, strategies comprising the functionalization of the nanopore with DNA nanostructures or other molecules have been proposed.25,28,33,35 Other methods employ indirect transduction strategies via modifying the nanopores to enhance the interactions between the small analytes and the nanopores. However, those methods all require the use of small pore sizes matched with small-sized target molecules.
The use of small (with diameter <10 nm) nanopores for the detection of small-sized objects has several drawbacks such as time-consuming and costly preparation of the nanopore, interference from contaminants and interferents, and easy blockage of the nanopore. Therefore, poor signal-to-noise ratios, blockage of nanopores and many other resistive-pulses caused by non-target objects hamper the continuous development of solid-state nanopores. A potential strategy to realize solid-state nanopore sensors with good anti-interference ability, high signal-to-noise ratios and repeated usage is to use a large pore size (>10 nm). A pore size much larger than the size of the target and the interferents can meet the requirement that the interferents get no resistive-pulse through the nanopore and can solve the problem of pore plugging. However, the detection of nucleic acids and proteins with relatively large pore size nanopores cannot be achieved with direct measurement.
The combination of a signal converter system and a signal transducer can provide an indirect measurement approach, which can solve the above issues.25,36–39 Among the different signal transducers that have been tested in nanopore experiments, DNA nanostructures with precise shape and adjustable size have the potential to function as the most versatile ones.40–42 DNA nanostructures with a uniform size can be assembled according to the specific pore size. Moreover, the surface of the nucleic acid nanostructures with certain rigidity can also be modified with different nucleic acid probes, small molecules, polypeptides or protein antibodies for selective binding to the target.43 Selectivity, in particular, is a key parameter in the detection of specific small species by means of nanopores. While the selectivity has been demonstrated with functionalized nanopores,25,44–46 an alternative approach could be the use of an external signal converting system. In particular, CRISPR-Cas12a has been demonstrated as an excellent signal converting system,47–53 which entails an RNA-guided enzyme that can specifically bind and cleave a specific target DNA.54 Due to the unique property of trans-cleavage of the single-stranded DNA (ssDNA), Cas12a has been widely applied in the design of biosensors by converting sequence-specific targets into other detectable signals, such as fluorescence, bioluminescence, or colorimetric signals.52,55–58 However, the application of CRISPR-Cas12a in nanopore sensing has been reported only recently.56,59,60
In this work, we extend the application of the CRISPR-Cas12a conversion method by using it in combination with another recently demonstrated signal transducer, i.e. DNA tetrahedron.38 We demonstrated a glass nanopore sensor for the detection of the L1-encoding gene of HPV18 as the target DNA molecule. As is well-known, HPV18 is an important virus that represents the main etiological factor for more than 90% of cervical cancer cases in women.61,62 The DNA tetrahedron used as the signal transducer was linked to magnetic beads via a single stranded DNA. The CRISPR-Cas12a system was applied to relate the DNA tetrahedron to the target DNA. When the target DNA is in the solution, Cas12a will be activated, which digests the ssDNA linker, leaving the corresponding DNA tetrahedron in the solution where it can interact with the electric field that drives the translocation through the pore and produce high signal-to-noise current blockades. Owing to the natural characteristics of the CRISPR-Cas12a system, the sensor ensures high sensitivity and selectivity. Moreover, because of the three-dimensional and adjustable conformation of the DNA nanostructure transporting through the solid-state nanopore, highly reproducible current-pulses can be achieved with different batch-to-batch experiments. This approach can be further developed to incorporate other commonly used DNA amplification approaches (PCR, LAMP, RPA, etc.).22
Results and discussion
Working principle
Fig. 1 shows the working principle of our glass nanopore sensor for target DNA (HPV18) detection using a DNA tetrahedron as a signal transducer based on the CRISPR-Cas12a conversion mechanism. The DNA tetrahedron transducer with a sticky end was linked to streptavidin-coated magnetic beads via an ssDNA (biotin-DNA) with a complementary part to the DNA tetrahedron on one end and a biotin group on the other end, forming a sandwich structure. A magnet placed on the edge of the experiment chamber was used to pull the magnetic beads down to the chamber wall. Cas12a and guide RNA were also added to the solution. If HPV18 existed in the analyte solution, it would specifically bind to the guide RNA, activating the trans-cleavage activity of Cas12a. The activated Cas12a would cleave the ssDNA linkers, leaving the DNA tetrahedra free in the solution, which would then translocate through the nanopore under the electric field, causing current blockages. In contrast, if target DNAs did not exist, the Cas12a would remain inactive and would not degrade the ssDNA linker. DNA tetrahedra would be pulled-down by the magnetic beads, and thus no events can be seen. Therefore, the event rate indicates the quantity of the released DNA tetrahedron transducers as well as the concentration of the target DNA in the analyte.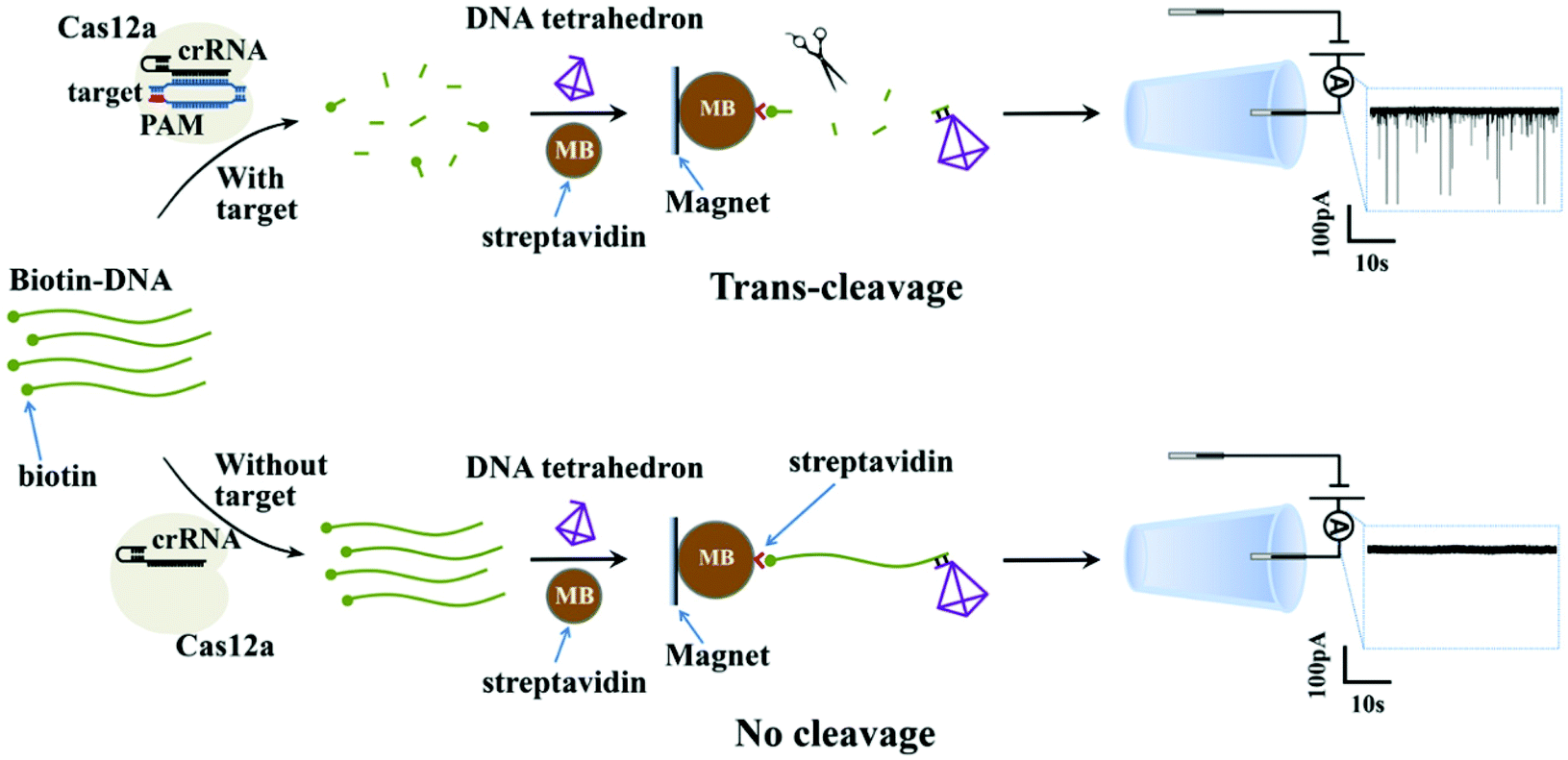 | ||
| Fig. 1 Schematic illustration of a solid-state nanopore sensor using a DNA tetrahedron as a signal transducer based on the CRISPR-cas12a conversion mechanism. With target molecules, the trans-cleavage activity of the Cas12a causes degradation of the linker ssDNA, resulting in the increased translocation rate of the DNA tetrahedron transducer through the nanopore. Without target molecules, the Cas12a is not activated and thus the DNA tetrahedron transducers are pulled-down by a magnet. No events can be observed. | ||
To further verify that the event was caused by the translocation of the DNA tetrahedron signal transducer rather than other components in the solution, we performed several experiments. As shown in Fig. 2a–c, the glass nanopore used in our experiment was typically about 20 nm in diameter and no translocation event was observed for the samples containing only the HPV18 target or Cas12a complex, indicating that the nanopore was inert to these small-sized biomolecules. In fact, as already reported,56 ssDNA that is less than 1 kbp in length does not generate a detectable signal in the glass nanopore. Instead, a distinct translocation event can be observed when the DNA tetrahedron was added. Then, if magnetic beads (MBs) and biotin-DNA (linker) were added, the event disappeared, suggesting that DNA tetrahedra were bound to the surface of the magnetic beads and fixed there. Then, we carried out the whole conversion method with or without HPV18 target DNA, and two totally different results were observed. Without a target, no translocation signal was seen, while with a target, translocation signals can be seen and the pattern of the signal was exactly the same as that of the DNA tetrahedron (Fig. 2e and f). This suggests that the translocation signal was actually caused by converting the target into a DNA tetrahedron that acts as a signal transducer. Thus, we have achieved a short-chain ssDNA or dsDNA nanopore sensor using a DNA tetrahedron as a signal transducer with the assistance of the CRISPR-Cas12a system, which can be applied in HPV18 detection.
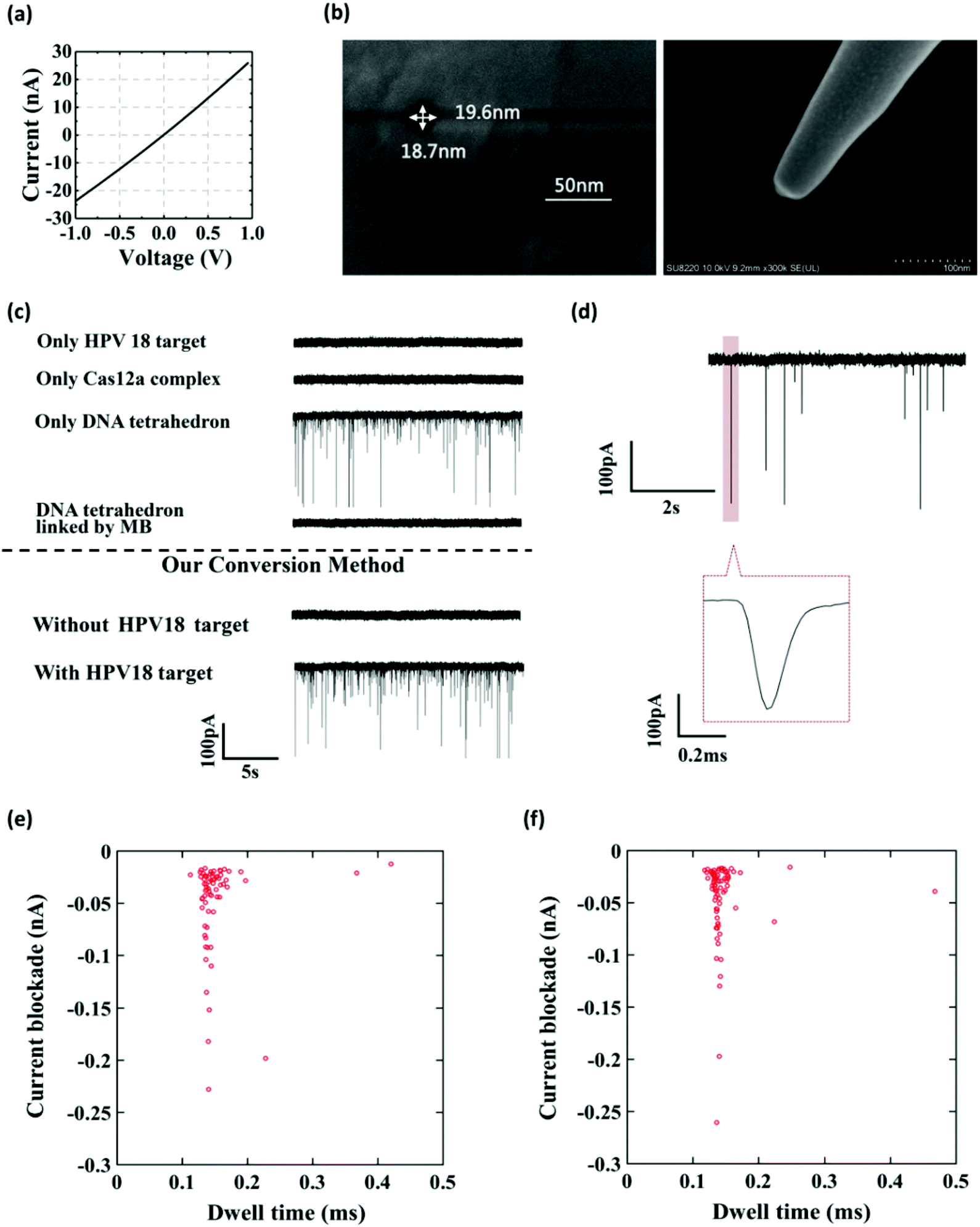 | ||
| Fig. 2 Verification of the current pulse in this experiment. (a) A typical I–V curve of our glassy nanopore in the test buffer solution. (b) Top and side views of the SEM image of the glassy nanopore. (c) Current traces of the glassy nanopore in the presence of different components with a bias of 400 mV. (d) Enlarged view of a typical event signal. (e) Scatter plot of the current blockade versus dwell time of the DNA tetrahedron measured at 400 mV applied bias. (f) Scatter plot of the current blockade versus dwell time of the DNA tetrahedron released in the solution with our conversion method measured at 400 mV applied bias. The threshold for event detection in all experiments was 3 times the standard deviation of the baseline. | ||
It is worth noting that we have achieved an indirect detection method by converting the small-sized target into a relatively big DNA nanostructure as a signal transducer. As a result, the time-consuming process of adjusting the pore size to match the small-sized target, which is necessary for the direct detection method, was avoided. Owing to their three-dimensional and adjustable conformation, DNA nanostructures can match the pore-size very well and distinguish them from interferences in the solution, so that the sensor could achieve a good translocation signal and low background noise. We also tried other DNA nanostructures (cube, bucket and triangular prism) as signal transducers, the results of which are shown in Fig. S2 and S3† and the sequences are presented in Table S1.† All DNA nanostructures can produce a detectable signal, but the performance of the DNA tetrahedron is the best over the other three DNA nanostructures, because of the easy synthesis and temperature stability. Owing to the good spatial structure of the DNA tetrahedron, the event rate was relatively stable with the fluctuation of the pore size, showing the good repeatability of the sensor which is vital for the practical use of the nanopore sensor (Fig. S4†). Furthermore, due to their flexible modification, DNA nanostructures can selectively interact with external triggers, such as proteins, enzymes, small molecules and so on, making this conversion method possible to be extended to other biomolecular detection methods.
Buffer optimization
The reaction buffer in the experiment is of significant importance because it can greatly influence the trans-cleavage activity of the CRISPR-Cas12a system and thus affect the performance of the glass nanopore sensor. NEBuffer 2.1 is commonly used as the reaction buffer. However, previous studies have shown that a high concentration (1.5 μM) of bovine serum albumin (BSA) in the buffer can severely deteriorate the performance of a glass nanopore sensor due to its strong adsorption on the surface of the glass nanopipette. Therefore, it is necessary to find out a BSA-free buffer with high catalytic activity. We then carried out a Φ174, a kind of natural long ssDNA, cleavage experiment with a Cas12a system in four buffer candidates: NEBuffer 2.1, NEBuffer 2.1 with BSA removed, NEBuffer 2.1 with BSA replaced with DTT or IDT buffer (see the ESI Table S2† for detailed compositions) to explore their catalytic activity. The gel analysis of the cleavage of Φ174 by the CRISPR-Cas12a system with a target HPV18 concentration of 50 nM is shown in Fig. 3. The commonly used NEBuffer 2.1 with BSA indeed showed good catalytic activity which can be used as a reference. For the NEBuffer 2.1 with BSA removed, the result showed that it did not activate the cleavage activity of Cas12a. Comparing the IDT buffer with NEBuffer 2.1 with BSA replaced with DTT, it is obvious that the IDT buffer has better catalytic activity. Therefore, the reaction buffer used in the following experiment is the IDT buffer.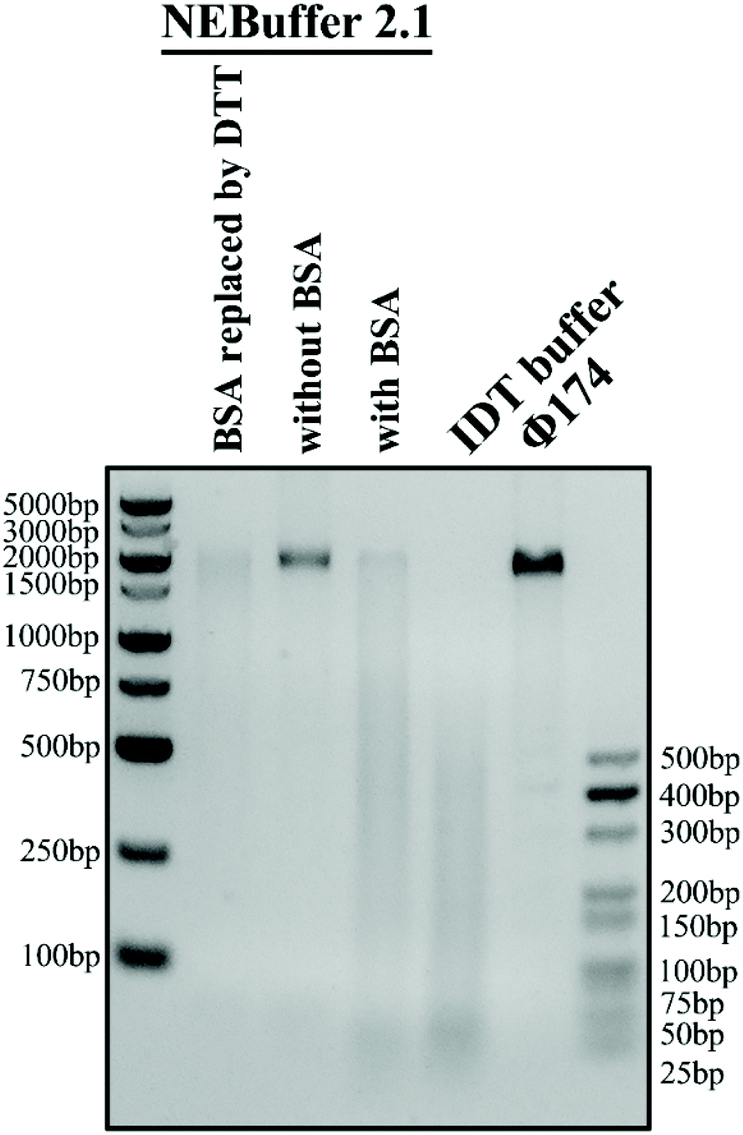 | ||
| Fig. 3 Buffer optimization for the CRISPR-Cas12a conversion system. Agarose gel electrophoresis analysis of the collateral cleavage activity of Φ174 in four buffer candidates was explored. NEBuffer 2.1 without BSA has the lowest catalytic activity while IDT buffer has the highest catalytic activity. | ||
Cleavage method optimization
As we know, the activated Cas12a could cleave only the surrounding ssDNAs. Since the ssDNA linker was modified on the surface of the magnetic beads rather than the direct dispersal in solution, the lack of good dispersion and the large space resistance will reduce the trans-cleavage efficiency of the CRISPR-Cas12a system. In order to get an efficient cleavage rate, we explored two cleavage methods: namely, Cleavage 1 and Cleavage 2 (Fig. 4a). The difference between the two strategies is whether the ssDNA substrate is first cleaved in the solution and then bound to the magnetic beads (Cleavage 1) or first bound to the magnetic beads and then cleaved (Cleavage 2). The cleavage efficiencies of the two cleavage methods with different lengths of the ssDNA linker (Bio-DNA (long and short) 25-mer and 95-mer) were compared using gel electrophoresis. As shown in Fig. 4b, the cleavage efficiency of Cleavage 1 is much higher than that of Cleavage 2 and the length of the ssDNA linker has no effect on the cleavage efficiency. This is easy to understand because in Cleavage 1 the ssDNA linker was evenly distributed in the solution during the cleavage process, and Cas12a easily reached the substrate no matter how long the ssDNA linker was, which resulted in a higher cleavage efficiency. In contrast, in Cleavage 2, the ssDNA substrate was first immobilized on the magnetic beads, and the poor dispersion and strong surface hindrance prevented Cas12a from approaching the substrate, which would result in a low cleavage efficiency. Moreover, since the length of the ssDNA linker in Cleavage 2 could strongly affect the steric hindrance, the use of the longer ssDNA linker evidently improved the cleavage efficiency. In this experiment, we chose Cleavage 1 to achieve a higher cleavage efficiency and higher sensitivity of the sensor. Moreover, we optimized the number of magnetic beads used before cleavage and the bias voltage used during the experiment (shown in Fig. S5 and S6†).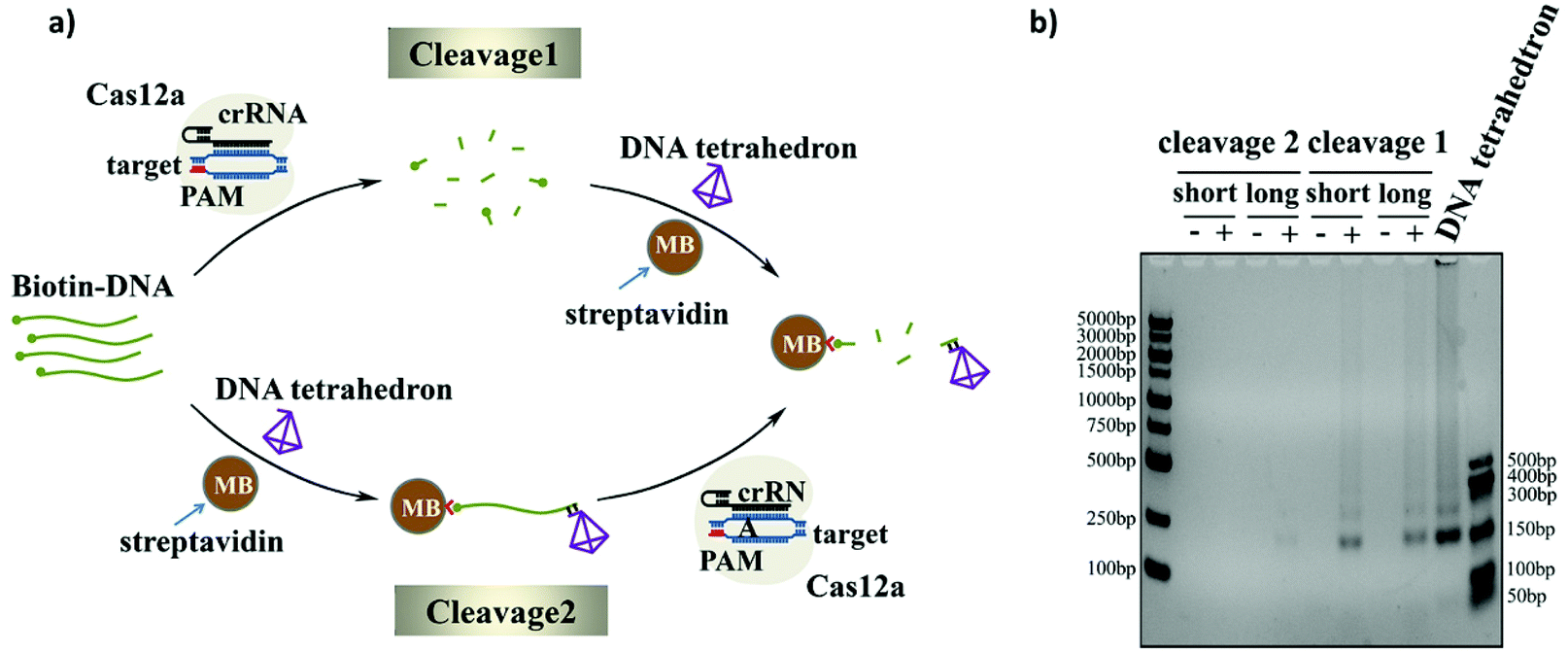 | ||
| Fig. 4 (a) Illustration of the two cleavage methods. Two methods go through the same steps, but in different order. (b) Agarose gel electrophoresis analysis to evaluate the cleavage efficiency of two cleavage methods with different lengths of the ssDNA linker. | ||
Nanopore event rate for DNA target quantification
To evaluate the sensitivity of the glassy nanopore sensor using a DNA tetrahedron as a signal transducer based on the CRISPR-Cas12a conversion mechanism, we first calibrated our sensor with a known concentration of the DNA tetrahedron. As shown in Fig. S7,† in the range of 0 to 100 nM, the event rate increased linearly with the increase of the tetrahedral concentration. According to this calibration curve, the event rate would be about 5 s−1 if all DNA tetrahedra were released into the solution from the magnetic beads (making the concentration about 33.3 nM). Then the event rate of the sensor with different concentrations of the HPV18 target (ranging from 0.5 nM to 50 nM) was tested, and each concentration was tested three times. As shown in Fig. 5a and b, the event rate is proportional to the HPV18 concentration from 0.5 nM to 50 nM. A clear linear relationship between the event rate and the HPV18 concentration was observed ranging from 0.5 nM to 10 nM (R = 0.988), which validates that the abundance of HPV18 can be quantified by nanopore counting. The detection limit of the sensor is 3 nM (event rate was higher than 1 s−1) which is low compared with other nanopore sensors for the detection of short-chain DNA and very close to the lower limit of the activation concentration of Cas12a. Compared with the latest nanopore technology using the tetrahedron (Jin's work38) or CRISPR technology (Guan's work56) to detect short-strand DNAs, the data analysis used in our method is the most intuitive which only needs the calculation of the number of peaks and a much better detection limit was also achieved compared to them (both of them were over 10 nM). These results suggest that the glass nanopore sensor using a DNA tetrahedron as a signal transducer based on the CRISPR-cas12a conversion method is a good candidate for short-strand DNA detection.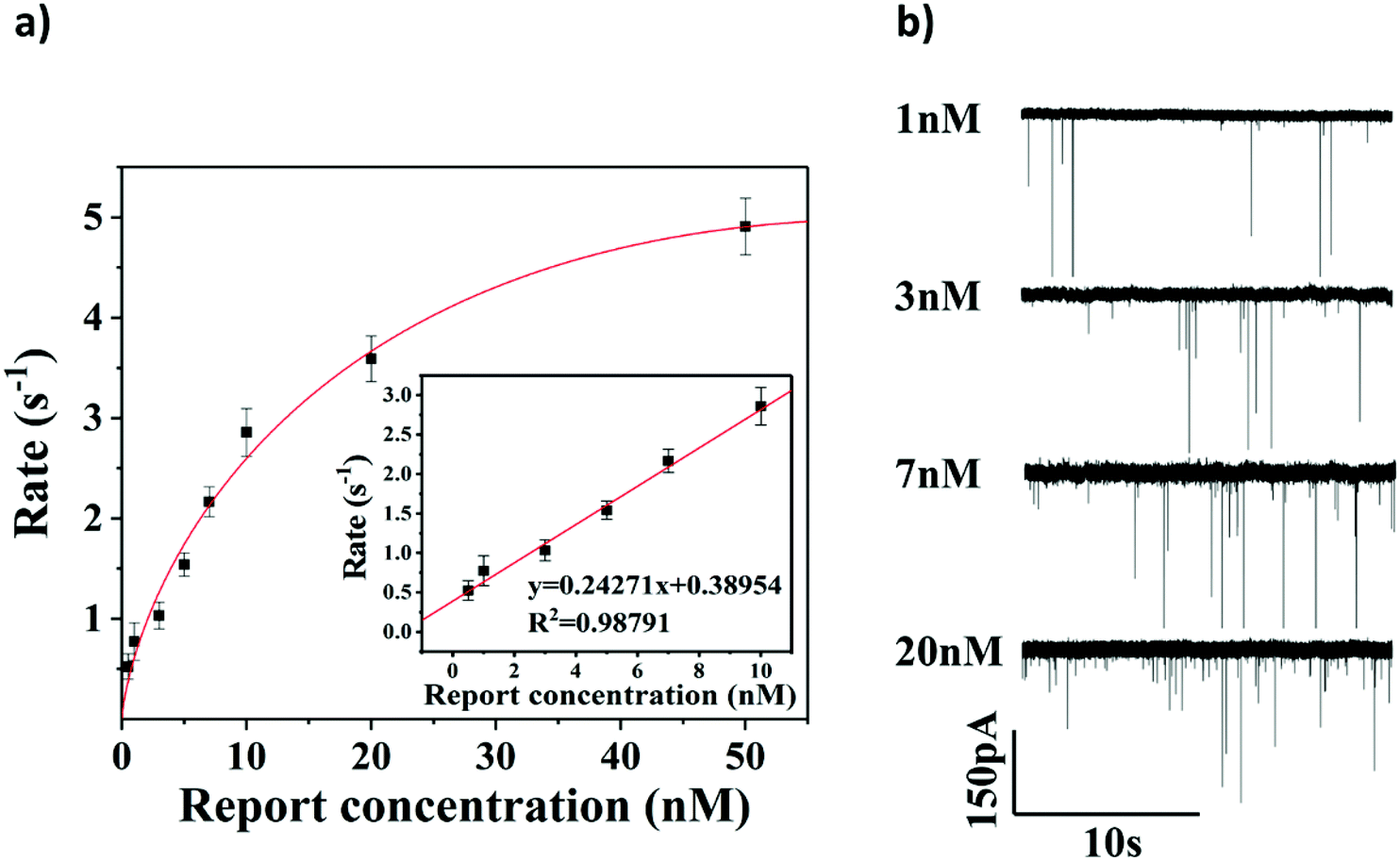 | ||
| Fig. 5 Quantification test of the sensor. (a) Calibration curve of the sensor with the HPV 18 concentration ranging from 0.5 nM to 50 nM. The inset indicates the linear zone of the sensor. (b) Translocation recording of the sensor with different concentrations of HPV18 under 400 mV bias. | ||
Sequence-specific test
To further verify the selectivity of the sensor, two other targets HPV16 (another subtype of HPV virus) and HIV (another completely different virus) were used as interferences. 50 nM HPV18, HPV16 and HIV were added, respectively, to the reaction solution to test if they could activate Cas12a to cleave the ssDNA linker. Both agarose gel analysis and nanopore signal analysis were carried out and are shown in Fig. 6. It is clearly shown in Fig. 6a that cleavage of the ssDNA linker happens only when the HPV18 target was added and hardly any cleavage was observed with the other nonspecific target. For HPV18 DNA targets, the translocation event rate is 4.908 ± 0.281 s−1, whereas for HPV16 DNA targets and HIV DNA targets, the event rate is relatively low (1.125 ± 0.172 s−1 and 1.166 ± 0.160 s−1). The selectivity in our experiment is about 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. However, it was not as high as the sensing technologies based on the CRISPR-Cas12a system reported in other studies. The reason may be that these short ssDNAs occupy some binding sites on the magnetic beads, resulting in a slight release of the DNA tetrahedra and thus an increase in the background noise.
:1. However, it was not as high as the sensing technologies based on the CRISPR-Cas12a system reported in other studies. The reason may be that these short ssDNAs occupy some binding sites on the magnetic beads, resulting in a slight release of the DNA tetrahedra and thus an increase in the background noise.
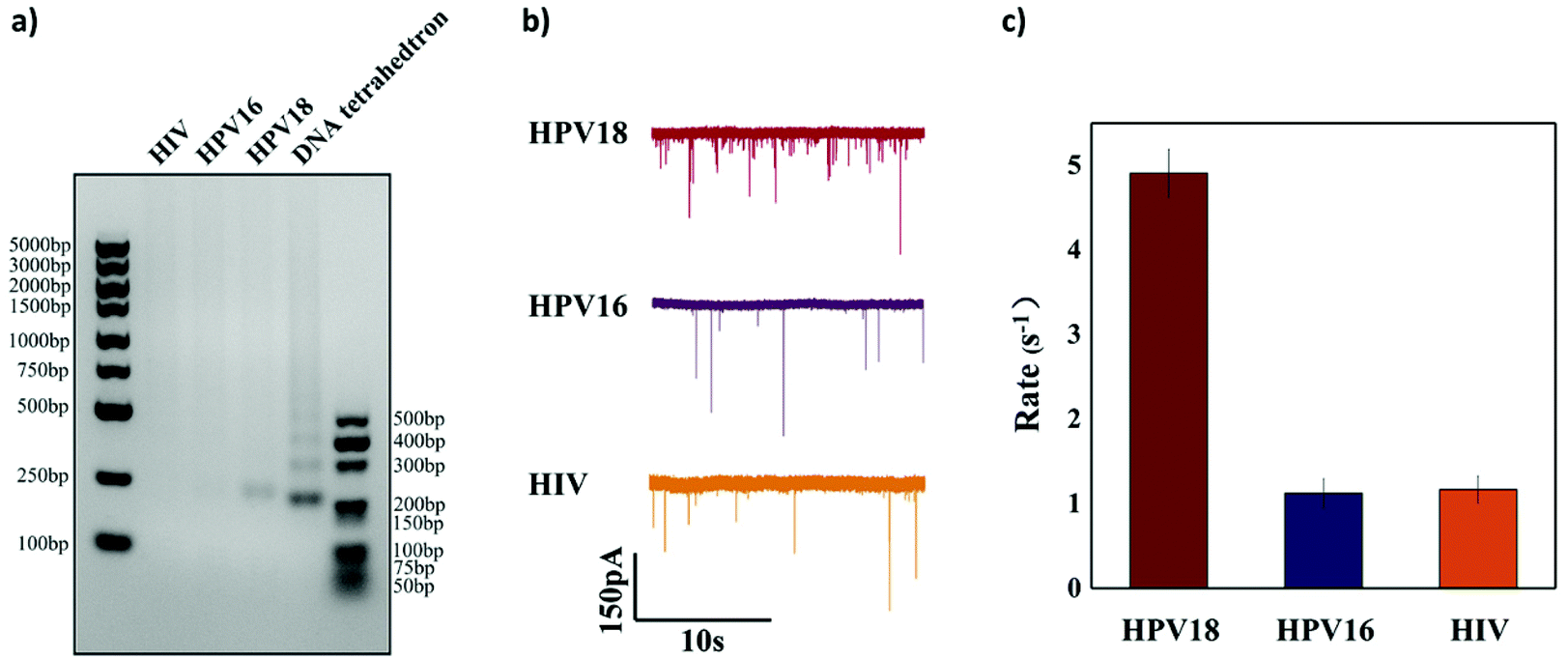 | ||
| Fig. 6 Specificity test of the sensor. (a) Agarose gel electrophoresis images of the designed specificity test. Only the HPV18 target leads to the release of the DNA tetrahedron. (b) Translocation recording of the sensor with HPV18, HPV16 and HIV. A significant increase in the number of translocation events can be seen only with the HPV18 target. (c) The event rate of the sensor with 50 nM of HPV18, HPV16 and HIV. | ||
But still, it is clear that in our experiment only the matched Cas12a assay and its target can significantly increase the number of translocation events after the reaction. Moreover, due to the compatibility of this detection method, the high specificity of the DNA amplification step (PCR, LAMP, RPA, etc.) before the detection of clinical samples will compensate for the selectivity. The integration of the CRISPR-Cas12a and nucleic acid amplification techniques has been proved by several works.22,60,63–65 These results demonstrated that our nanopore sensor using a DNA tetrahedron as a signal transducer based on the CRISPR-Cas12a conversion mechanism could detect targets specifically, which we believe could become a promising alternative to biomolecular detection.
Experimental
Materials and reagents
EnGen®Lba Cas12a (Cpf1) and Φ174 were purchased from NEW ENGLAND Biolabs Inc. (NEB). All DNA and RNA strands were synthesized, labeled, and purified with HPLC by Sangon Biotechnology Co., Ltd (Shanghai, China), the sequences of which are listed in Table S1 (ESI).† Streptavidin coated magnetic beads were purchased from Sigma-Aldrich (Shanghai) Trading Co., Ltd and were used due to their good quality and easy adsorption by magnets. All solutions in the experiment were prepared using ultrapure water (>18.25 MΩ cm−1) obtained from a Millipore Milli-Q water purification system. NEBuffer 2.1 was purchased from NEW ENGLAND Biolabs Inc. (NEB). 1× IDT reaction buffer (20 mM HEPES, 100 mM NaCl, 5 mM MgCl2 and 0.1 mM EDTA, pH 6.5 at 25 °C), TM buffer (50 mM MgCl2 and 20 mM Tris, pH 8.0 at 25 °C) and test buffer solutions (1 M KCl, 10 mM Tris and 0.1 mM EDTA) were made at the lab. DNA marker and 4S GelRed (10000×) were purchased from Sangon Biotechnology Co., Ltd. 6× Loading buffer was from Takara Bio Inc., and agarose was from Aladdin Chemistry Co. Ltd (Shanghai, China). Piranha solution (mixing sulfuric acid (H2SO4) with hydrogen peroxide (H2O2) at a ratio of 7:3) was freshly made in the lab. Quartz capillaries (O.D: 1 mm; I.D: 0.5 mm; QF100-50-10) were purchased from Sutter Instrument Co.
Apparatus
Glassy nanopores were fabricated with a CO2-laser-actuated pipette puller (model P2000, Sutter Instrument Co.). The concentrations of DNA solutions were determined using a UV-1800 spectrophotometer (Shimadzu, Kyoto, Japan) at 260 nm. A Bio-Rad T100 thermal cycler (Bio-Rad, USA) was used with temperature gradients for the formation of DNA tetrahedra. The gel electrophoresis analysis was carried out using an Automatic Gel Imaging Analysis System (Peiqing Science and Technology Co., Ltd).The fabrication of the glass nanopore
Before fabrication, all glass capillaries should be thoroughly cleaned. They were immersed in freshly prepared piranha solution for two hours to remove organic impurities and then washed with deionized water and dried at 80 °C for 20 minutes in a vacuum drying oven. The capillaries used in most of the experiments were pulled using the pipette puller with a two-line program including the following settings: (1) heat 750, filament 5, velocity 50, delay 140, and pull 50 and (2) heat 710, filament 4, velocity 30, delay 155, and pull 250. Moreover, to verify the reproducibility of our method, we fabricated capillaries with larger pore sizes using the following program: (1) 30 nm. Line 1, Heat 750, Filament 5, Velocity 50, Delay140, and Pull 50; Line 2, Heat 710, Filament 4, Velocity 30, Delay 155, and Pull 220; (2) 50 nm. Line 1, Heat 650, Fil 3, Vel 35, Del 145, Pul 75; Line 2, Heat 900, Fil 2, Vel 15, Del 128, Pul 200;66 (3) 65 nm. Line 1, Heat 575, Fil 3, Vel 35, Del 145, Pul 75; Line 2, Heat 900, Fil 2, Vel 15, Del 128, Pul 20028.Self-assembly of the DNA tetrahedra
DNA tetrahedra with a sticky end were assembled according to a previous protocol.40 Briefly, equimolar quantities of four different DNA strands were heated to 95 °C for 5 minutes and rapidly cooled to 4 °C within 30 s in a TM buffer. Then the DNA tetrahedra were stored at 4 °C for more than 6 hours before use. The sequences of the four DNAs are shown in ESI Table S1.†Gel electrophoresis
The self-assembly of DNA tetrahedra was identified by 12% native PAGE in a 1× TBE running buffer (89 mM Tris, 89 mM boric acid, and 2 mM EDTA) at 120 V constant voltages for 40 min (ESI Fig. S1†). The buffer optimization, cleavage method optimization, quantitative test and specificity test were carried out in 2% agarose gels containing 1× TAE buffer (40 mM Tris, 20 mM glacial acetic acid, and 2 mM Na2·EDTA·2H2O) at 120 V for 30 minutes. The gels were visualized under UV light and finally photographed with an Automatic Gel Imaging Analysis System (Peiqing Science and Technology Co., Ltd).Experimental procedures of HPV18 detection based on the CRISPR-Cas12a converter system
Here, we tried two cleavage strategies. Briefly, the difference between the two strategies is whether the tetrahedron is first cleaved and then bound to the magnetic beads (Cleavage 1) or first bound to the magnetic beads and then cleaved (Cleavage 2).Experimental procedures of cleavage 1
First, a total of 20 μL of CRISPR/Cas12a pre-assembled solution including 35 nM Cas12a, 36 nM crRNA and different concentrations of the target DNA in a 1× IDT buffer was incubated in a BioRad T100 thermal cycler at 37 °C for 10 min to activate. Then, 1.2 μl of 10 μM biotin-DNA was added and incubated at 37 °C for 30 min for cleavage and inactivated at 65 °C for 10 min. 1 μl of 10 μM DNA tetrahedron was added into the solution and incubated at room temperature for 30 min to form a biotin-DNA-tetrahedron structure. After that, a 30 μL suspension of magnetic beads washed with the TM buffer five times was added to the solution and held for another 30 min while gently shaking the tube to form a sandwich hybrid structure. Finally, the PCR tube was placed on a magnetic rack and 10 μL of the supernatant was pipetted out and added into the test buffer for nanopore analysis.Experimental procedures of cleavage 2
First, 1.2 μl of 10 μM biotin-DNA and 1 μl of 10 μM DNA tetrahedron were hybridized to form a biotin-DNA-tetrahedron structure in TM buffer by incubating at room temperature for 30 min. A 30 μL suspension of magnetic beads was transferred to a clean PCR tube and washed with 160 μl of TM buffer five times. Subsequently, all of the above biotin-DNA-tetrahedron system was added to the beads and incubated at room temperature for 30 min while gently shaking the tube to form a sandwich hybrid structure. After the reaction was complete, the whole solution was magnetically separated to remove the excess unbound DNA sequences and washed three times with TM buffer. A total of 20 μL of CRISPR/Cas12a pre-assembled solution including 35 nM Cas12a, 36 nM crRNA and different concentrations of the target DNA in a 1× IDT buffer was incubated in a BioRad T100 thermal cycler at 37 °C for 10 min for activation. After that, all the above CRISPR/Cas12a pre-assembled solution was added to the PCR tube and incubated at 37 °C for 30 min and inactivated at 65 °C for 10 min. Finally, the PCR tube was placed on a magnetic rack and 10 μL of the supernatant was pipetted out and added into the test buffer for nanopore analysis.Nanopore sensing and data analysis
The ionic current change of DNA tetrahedron translocation through the nanopore was studied using a HEKA system. Briefly, a constant voltage of 400 mV was applied across the glass nanopore in a homemade electrolyte cell filled with the test buffer. One Ag/AgCl electrode was inserted into the quartz nanopipette filled with the solution, which serves as the working electrode while the other Ag/AgCl electrode was immersed in the solution outside the glass nanopore which serves as the reference electrode. During the experiment, the ionic currents were amplified, digitized and recorded using a HEKA EPC 10 system with a sampling rate of 50 kHz and a bandwidth of 2.9 kHz. All the experiments were carried out at room temperature. Transalyzer, a Matlab GUI based package for nanopore signal analysis, was used to analyze the current time trace and extract the single molecule translocation information. Nonlinear fitting results are obtained using Origin.Conclusions
The low-cost and reproducible detection of small-sized molecules by solid-state nanopores remains a significant challenge due to the difficulty in fabricating nanopores with matched small pore sizes and good reproducibility. In this work, we develop a glass nanopore sensor for small-sized DNA fragments using a DNA tetrahedron as a signal transducer in combination with the utilization of a CRISPR-Cas12a system as a converter. Since the well-designed DNA nanostructure matched the pore size and the great specificity of the CRISPR-Cas12a system, the sensor achieves a good signal to noise ratio, and exhibits good repeatability and selectivity. The sensor can detect the test target molecule, HPV18 DNA, within the concentration range of 0.5–50 nM with a linear range from 0.5 nM to 10 nM and a detection limit of 3 nM. Although the selectivity towards HPV18 was evident, the detection rates were 5:1 towards HPV18 and HIV, respectively. We expect that a better optimization of the system can provide higher selectivity, to be verified also towards additional interferents (DNA molecules or small entities).
The conversion method demonstrated in this paper represents a promising tool for sensitive DNA detection, combining the CRISPR-Cas12a conversion mechanism with the superior single molecule sensitivity of the nanopore sensor. It shows great potential to achieve extremely high sensitivity when combined with other existing nucleic acid amplification technologies (PCR, LAMP, RPA, etc.). Moreover, applying the indirect measurement approach using a DNA nanostructure as a signal transducer has great advantages for their flexibly programmable shape and volume and easy interaction with external triggers such as ligands, DNAs, RNAs, proteins and small molecules, which will greatly broaden the applications of this method but avoid the tedious process of adjusting the pore size to match different target molecules. Our demonstration of the glass nanopore sensor using a DNA tetrahedron as a signal transducer based on the CRISPR-cas12a conversion mechanism can be easily applied to other types of solid-state nanopores and could be a good candidate for practical diagnostic applications.
Author contributions
Shumin Zhang carried out most of the experiment, analyzed the data in this work and wrote the experimental part of the article. Minyi Liu helped to obtain the data in the cleavage method optimization part and target quantification part. Haofa Cui helped to obtain the data in the cleavage method optimization part. Muhammad Asad Ziaee helped to link the DNA tetrahedron with the magnetic beads and cut the linker using a CRISPR/Cas system. Rongwei Sun and Liting Chen helped to obtain the data in the specific test part. Daqi Chen designed the research concept and the experiment, helped to collect and analyze the data, and wrote most of the article. Denis Garoli put forward some valuable suggestions to the article and wrote the critical revision of the article. Jiahai Wang designed the experiment and put forward some valuable suggestions to the data analysis, wrote the introduction part of the article and gave the final approval to the article.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 62003105 and 22074025) and the Open Research Project of the State Key Laboratory of Industrial Control Technology, Zhejiang University, China (no. ICT20049). D. D. G. acknowledges funding from the European Union under the H2020 Programme (no. FETOPEN-01-2018-2019-2020, grant no. 964995 ‘DNAFAIRYLIGHT’).References
- N. A. W. Bell and U. F. Keyser, Nat. Nanotechnol., 2016, 11, 645–652 CrossRef CAS PubMed.
- R. D. Bulushev, S. Marion and A. Radenovic, Nano Lett., 2015, 15, 7118–7125 CrossRef CAS PubMed.
- A. Arima, I. H. Harlisa, T. Yoshida, M. Tsutsui, M. Tanaka, K. Yokota, W. Tonomura, J. Yasuda, M. Taniguchi, T. Washio, M. Okochi and T. Kawai, J. Am. Chem. Soc., 2018, 140, 16834–16841 CrossRef CAS PubMed.
- Y. L. Ying, Y. X. Hu, R. Gao, R. J. Yu, Z. Gu, L. P. Lee and Y. T. Long, J. Am. Chem. Soc., 2018, 140, 5385–5392 CrossRef CAS PubMed.
- K. Zhang, X. He, Y. Liu, P. Yu, J. Fei and L. Mao, Anal. Chem., 2017, 89, 6794–6799 CrossRef CAS PubMed.
- H. Liu, Q. Jiang, J. Pang, Z. Jiang, J. Cao, L. Ji, X. Xia and K. Wang, Adv. Funct. Mater., 2018, 28 Search PubMed.
- A. K. Thakur and L. Movileanu, Nat. Biotechnol., 2018, 37, 96–102 CrossRef.
- Y. Wang, Y. Zhang, X. Chen, X. Guan and L. Wang, Talanta, 2021, 223 Search PubMed.
- A. Singh, Nat. Methods, 2020, 17, 874–874 CrossRef CAS PubMed.
- Z.-L. Hu, M.-Z. Huo, Y.-L. Ying and Y.-T. Long, Angew. Chem., Int. Ed., 2021, 60, 14738–14749 CrossRef CAS PubMed.
- Y.-L. Ying, C. Cao and Y.-T. Long, Analyst, 2014, 139, 3826–3835 RSC.
- A. Crnkovic, M. Srnko and G. Anderluh, Life, 2021, 11, 27 CrossRef CAS PubMed.
- A. Biesemans, M. Soskine and G. Maglia, Nano Lett., 2015, 15, 6076–6081 CrossRef CAS PubMed.
- Z. Xie, J. Lei, M. Yang, Y. Li, X. Geng, S. Liu and J. Wang, Biosens. Bioelectron., 2019, 127, 200–206 CrossRef CAS PubMed.
- L. Xue, H. Yamazaki, R. Ren, M. Wanunu, A. P. Ivanov and J. B. Edel, Nat. Rev. Mater., 2020, 5, 931–951 CrossRef CAS.
- Z. Yuan, C. Wang, X. Yi, Z. Ni, Y. Chen and T. Li, Nanoscale Res. Lett., 2018, 13, 56 CrossRef PubMed.
- Q. Chen and Z. Liu, Sensors, 2019, 19, 1886 CrossRef CAS PubMed.
- A. Fragasso, S. Schmid and C. Dekker, ACS Nano, 2020, 14, 1338–1349 CrossRef CAS PubMed.
- Z. Tang, D. Zhang, W. Cui, H. Zhang, W. Pang and X. Duan, Nanomater. Nanotechnol., 2016, 6, 35 CrossRef.
- D. Garoli, H. Yamazaki, N. Maccaferri and M. Wanunu, Nano Lett., 2019, 19, 7553–7562 CrossRef CAS PubMed.
- Z. Zhu, R. Wu and B. Li, Chem. Sci., 2019, 10, 1953–1961 RSC.
- Z. Tang, G. Choi, R. Nouri and W. Guan, Nano Lett., 2019, 19, 7927–7934 CrossRef CAS PubMed.
- M. A. Alibakhshi, J. R. Halman, J. Wilson, A. Aksimentiev, K. A. Afonin and M. Wanunu, ACS Nano, 2017, 11, 9701–9710 CrossRef CAS.
- N. Farajpour, L. S. Lastra, V. Sharma and K. J. Freedman, Nanoscale, 2021, 13, 5780–5790 RSC.
- R. Ren, X. Wang, S. Cai, Y. Zhang, Y. Korchev, A. P. Ivanov and J. B. Edel, Small Methods, 2020, 4, 2000356 CrossRef CAS.
- R.-J. Yu, S.-M. Lu, S.-W. Xu, Y.-J. Li, Q. Xu, Y.-L. Ying and Y.-T. Long, Chem. Sci., 2019, 10, 10728–10732 RSC.
- S. Chen, Y. Tang, K. Zhan, D. Sun and X. Hou, Nano Today, 2018, 20, 84–100 CrossRef CAS.
- K. J. Freedman, L. M. Otto, A. P. Ivanov, A. Barik, S. H. Oh and J. B. Edel, Nat. Commun., 2016, 7, 10217 CrossRef CAS PubMed.
- J. A. Bafna and G. V. Soni, PLoS One, 2016, 11, e0157399 CrossRef PubMed.
- W. Li, N. A. W. Bell, S. Hernandez-Ainsa, V. V. Thacker, A. M. Thackray, R. Bujdoso and U. F. Keyser, ACS Nano, 2013, 7, 4129–4134 CrossRef CAS PubMed.
- X. Xu, C. Li, Y. Zhou and Y. Jin, ACS Sens., 2017, 2, 1452–1457 CrossRef CAS PubMed.
- R. Jia and M. V. Mirkin, Chem. Sci., 2020, 11, 9056–9066 RSC.
- P. Cadinu, G. Campolo, S. Pud, W. Yang, J. B. Edel, C. Dekker and A. P. Ivanov, Nano Lett., 2018, 18, 2738–2745 CrossRef CAS PubMed.
- R. Gao, Y.-L. Ying, Y.-J. Li, Y.-X. Hu, R.-J. Yu, Y. Lin and Y.-T. Long, Angew. Chem., Int. Ed., 2018, 57, 1011–1015 CrossRef CAS PubMed.
- P. Pandey, N. Panday, S. Chang, P. Pang, J. Garcia, X. Wang, Q. Fu and J. He, ChemElectroChem, 2018, 5, 3102–3112 CrossRef CAS.
- R. Wu, Y. Wang, Z. Zhu, C. Yu, H. Li, B. Li and S. Dong, ACS Appl. Mater. Interfaces, 2021, 13, 9482–9490 CrossRef CAS.
- S. Zhang, G. Liu, H. Chai, Y.-D. Zhao, L. Yu and W. Chen, Electrochem. Commun., 2019, 99, 71–74 CrossRef CAS.
- D. Wang, X. Xu, Y. Zhou, H. Li, G. Qi, P. Hu and Y. Jin, Anal. Chim. Acta, 2019, 1063, 57–63 CrossRef CAS PubMed.
- S. Cai, J. Y. Y. Sze, A. P. Ivanov and J. B. Edel, Nat. Commun., 2019, 10, 1797 CrossRef PubMed.
- M. Lin, P. Song, G. Zhou, X. Zuo, A. Aldalbahi, X. Lou, J. Shi and C. Fan, Nat. Protoc., 2016, 11, 1244–1263 CrossRef CAS PubMed.
- Q. Hu, H. Li, L. Wang, H. Gu and C. Fan, Chem. Rev., 2019, 119, 6459–6506 CrossRef CAS PubMed.
- R. Duan, X. Lou and F. Xia, Chem. Soc. Rev., 2016, 45, 1738–1749 RSC.
- F. Yang, Q. Li, L. Wang, G.-J. Zhang and C. Fan, ACS Sens., 2018, 3, 903–919 CrossRef CAS PubMed.
- J. Y. Y. Sze, A. P. Ivanov, A. E. G. Cass and J. B. Edel, Nat. Commun., 2017, 8, 1552 CrossRef PubMed.
- L. Reynaud, A. Bouchet-Spinelli, C. Raillon and A. Buhot, Sensors, 2020, 20, 4495 CrossRef CAS.
- S. Feng, C. Chen, W. Wang and L. Que, Biosens. Bioelectron., 2018, 105, 36–41 CrossRef CAS PubMed.
- M. Liang, Z. Li, W. Wang, J. Liu, L. Liu, G. Zhu, L. Karthik, M. Wang, K.-F. Wang, Z. Wang, J. Yu, Y. Shuai, J. Yu, L. Zhang, Z. Yang, C. Li, Q. Zhang, T. Shi, L. Zhou, F. Xie, H. Dai, X. Liu, J. Zhang, G. Liu, Y. Zhuo, B. Zhang, C. Liu, S. Li, X. Xia, Y. Tong, Y. Liu, G. Alterovitz, G.-Y. Tan and L.-X. Zhang, Nat. Commun., 2019, 10, 3672 CrossRef PubMed.
- B. Paul and G. Montoya, Biomed. J., 2020, 43, 8–17 CrossRef PubMed.
- J. P. Broughton, X. Deng, G. Yu, C. L. Fasching, V. Servellita, J. Singh, X. Miao, J. A. Streithorst, A. Granados, A. Sotomayor-Gonzalez, K. Zorn, A. Gopez, E. Hsu, W. Gu, S. Miller, C.-Y. Pan, H. Guevara, D. A. Wadford, J. S. Chen and C. Y. Chiu, Nat. Biotechnol., 2020, 38, 870–874 CrossRef CAS PubMed.
- R. K.-K. Leung, Q.-X. Cheng, Z.-L. Wu, G. Khan, Y. Liu, H.-Y. Xia and J. Wang, Methods, 2021 DOI:10.1016/j.ymeth.2021.02.018.
- F. Yan, W. Wang and J. Zhang, Cell Biol. Toxicol., 2019, 35, 489–492 CrossRef PubMed.
- O. Mukama, J. Wu, Z. Li, Q. Liang, Z. Yi, X. Lu, Y. Liu, Y. Liu, M. Hussain, G. G. Makafe, J. Liu, N. Xu and L. Zeng, Biosens. Bioelectron., 2020, 159, 112143 CrossRef CAS PubMed.
- A. Bonini, N. Poma, F. Vivaldi, A. Kirchhain, P. Salvo, D. Bottai, A. Tavanti and F. Di Francesco, J. Pharm. Biomed. Anal., 2021, 192, 113645 CrossRef CAS PubMed.
- J. S. Chen, E. Ma, L. B. Harrington, M. Da Costa, X. Tian, J. M. Palefsky and J. A. Doudna, Science, 2018, 360, 436–439 CrossRef CAS PubMed.
- M. J. Kellner, J. G. Koob, J. S. Gootenberg, O. O. Abudayyeh and F. Zhang, Nat. Protoc., 2019, 14, 2986–3012 CrossRef CAS PubMed.
- R. Nouri, Y. Jiang, X. L. Lian and W. Guan, ACS Sens., 2020, 5, 1273–1280 CrossRef CAS PubMed.
- M. Hu, C. Yuan, T. Tian, X. Wang, J. Sun, E. Xiong and X. Zhou, J. Am. Chem. Soc., 2020, 142, 7506–7513 CrossRef CAS PubMed.
- N. Shao, X. Han, Y. Song, P. Zhang and L. Qin, Anal. Chem., 2019, 91, 12384–12391 CrossRef CAS PubMed.
- W. Yang, L. Restrepo-Perez, M. Bengtson, S. J. Heerema, A. Birnie, J. van der Torre and C. Dekker, Nano Lett., 2018, 18, 6469–6474 CrossRef CAS PubMed.
- R. Nouri, Y. Jiang, Z. Tang, X. L. Lian and W. Guan, Nano Lett., 2021, 21, 8393–8400 CrossRef CAS PubMed.
- R. Ghittoni, R. Accardi, S. Chiocca and M. Tommasino, Ecancermedicalscience, 2015, 9, 526 CrossRef PubMed.
- K. Munger, A. Baldwin, K. M. Edwards, H. Hayakawa, C. L. Nguyen, M. Owens, M. Grace and K. Huh, J. Virol., 2004, 78, 11451–11460 CrossRef PubMed.
- K. Yao, D. L. Peng, C. Jiang, W. Zhao, G. K. Li, W. K. Huang, L. A. Kong, H. F. Gao, J. W. Zheng and H. Peng, Int. J. Mol. Sci., 2021, 22, 12577 CrossRef CAS PubMed.
- Z. Tang, R. Nouri, M. Dong, J. Yang, W. Greene, Y. Zhu, M. Yon, M. S. Nair, S. V. Kuchipudi and W. Guan, Biosens. Bioelectron., 2022, 197, 113759 CrossRef CAS PubMed.
- X. Wang, S. Zhou, C. Chu, M. Yang, D. Huo and C. Hou, ACS Sens., 2021, 6, 2419–2428 CrossRef CAS PubMed.
- Y. X. Hu, Y. L. Ying, R. Gao, R. J. Yu and Y. T. Long, Anal. Chem., 2018, 90, 12352–12355 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1an02313f |
| This journal is © The Royal Society of Chemistry 2022 |