
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sizing individual dielectric nanoparticles with quantitative differential interference contrast microscopy†
Samuel
Hamilton
 a,
David
Regan
a,
Lukas
Payne
ab,
Wolfgang
Langbein
*b and
Paola
Borri
a
a,
David
Regan
a,
Lukas
Payne
ab,
Wolfgang
Langbein
*b and
Paola
Borri
a
aSchool of Biosciences, Cardiff University, Cardiff, UK
bSchool of Physics and Astronomy, Cardiff University, Cardiff, UK. E-mail: langbeinww@cardiff.ac.uk
First published on 18th March 2022
Abstract
We report a method to measure the size of single dielectric nanoparticles with high accuracy and precision using quantitative differential interference contrast (DIC) microscopy. Dielectric nanoparticles are detected optically by the conversion of the optical phase change into an intensity change using DIC. Phase images of individual nanoparticles were retrieved from DIC by Wiener filtering, and a quantitative methodology to extract nanoparticle sizes was developed. Using polystyrene beads of 100 nm radius as size standard, we show that the method determines this radius within a few nm accuracy. The smallest detectable polystyrene bead is limited by background and shot-noise, which depend on acquisition and analysis parameters, including the objective numerical aperture, the DIC phase offset, and the refractive index contrast between particles and their surrounding. Measurements on small beads of 15 nm nominal radius are shown, and a sensitivity limit potentially reaching down to 1.8 nm radius was inferred. As application example, individual nanodiamonds with nominal sizes below 50 nm were measured, and were found to have a nearly exponential size distribution with 28 nm mean value. Considering the importance of dielectric nanoparticles in many fields, from naturally occurring virions to polluting nanoplastics, the proposed method could offer a powerful quantitative tool for nanoparticle analysis, combining accuracy, sensitivity and high-throughput with widely available and easy-to-use DIC microscopy.
1. Introduction
Dielectric nanoparticles (NPs) exist in a multitude of forms and are ubiquitous in our world. They can be naturally occurring (e.g. virions and exosomes), synthetically fabricated (e.g. silica beads, nanodiamonds), or by-products of material degradation (e.g. nanoplastics). These NPs are widely utilised in research and industry, with applications ranging from drug delivery in biomedicine1,2 to the fabrication of advanced functional materials.3 A key requirement for all these applications is the knowledge of the NP size. For example, in cell biology it is well known that the uptake of a NP by the plasma membrane, and the subsequent intracellular trafficking route, tightly depends on the NP size, which in turn is acrucial parameter in the use of NPs as vehicles for drug delivery and therapeutics.1,2Different from metallic particles, dielectric NPs are typically not electron dense, hence their sizes are more challenging to measure with electron microscopy (EM), the industry-standard technique for NP characterisation. EM analysis is also expensive and typically “low-throughput”, since only a limited number of NPs can be examined in one field of view under vacuum (for a review on NP characterisation methods see ref. 4 and 5). To that end, the use of wide-field optical microscopy to determine the size of individual NPs offers many advantages, including simplicity, low cost, high speed and high throughput, with hundreds of individual NPs rapidly imaged in one field of view, under ambient conditions. However, the spatial resolution of an optical microscope is limited by light diffraction (usually to about 200 nm), typically larger than the size of the investigated NPs. In other words, differently from EM, optical microscopy methods cannot directly resolve NP dimensions. On the other hand, they can exploit the physical relationship between measurable optical properties and NP sizes to accurately determine the latter. Using this concept, we recently showed that NP sizes could be determined from the optical absorption (σabs) and scattering cross-section (σsca) of individual NPs measured by wide-field extinction microscopy, with uncertainties down to about 1 nm in diameter.6
While optical extinction microscopy is in principle applicable to any NP material, for particle sizes much smaller than the light wavelength (dipole limit) the technique is practically useful only when NPs exhibit significant optical absorption, such as gold6 and silver NPs.7 This is because σabs scales with the NP volume while σsca scales with the square of the NP volume, severely penalising small NPs which are not absorptive. In other words, by measuring the magnitude of σext = σabs + σsca one can be sensitive to small NP sizes only if particles are strongly absorbing such that σext ∼ σabs. For example, using gold NPs, we have demonstrated a sensitivity limit down to sizes of 2 nm diameter.8 For significantly larger diameters, quantitative extinction and scattering measurements can be used, together with simulations, to extract sizes of dielectric NPs, as shown for 100 nm polystyrene (PS) beads in ref. 9.
We note that light scattering can also be exploited to determine the hydrodynamic radius of NPs diffusing in a liquid of known viscosity.14 Commercial instruments are available, either as ensemble measurements techniques (using dynamic light scattering) or by tracking individual particles (nanoparticle tracking analysis). In addition to the mentioned limitation of light scattering methods penalizing small NPs, these techniques do not measure the geometrical size of a NP. Indeed, the hydrodynamic radius can overestimate the NP geometrical radius, depending e.g. on the particle surface charges. Moreover, nanoparticle tracking analysis methods have been shown to overestimate the size distribution.14
Small dielectric NPs exhibit a negligible absorption in the visible wavelength range and are weakly scattering. Hence, for such NPs optical sizing by extinction microscopy is less suited and a different optical contrast method is required. Notably, it is possible to achieve an image contrast proportional to the NP volume using interferometric approaches. For example, it has been recently shown that weakly scattering single dielectric nanoparticles (including biological macromolecules) can be detected with high sensitivity by means of reflection microscopy on a weakly reflecting interface, termed interferometric scattering microscopy (iSCAT).10–12 Alternatively, a transmission technique called COBRI,13 attenuating the un-scattered light similar to conventional phase contrast, has been used to track silica NPs of 50 nm and 100 nm size diffusing in water, with a size sensitivity limit of about 30 nm for these NPs.
One of the simplest interferometric techniques to generate an image contrast scaling with the NP volume exploits the conversion of the optical phase change of transmitted light introduced by the sample into an amplitude change, by means of differential interference contrast microscopy.15 Briefly, DIC uses a Nomarski prism to split the two linear light polarization components in direction in the condenser back focal plane (BFP), creating a shear distance in the image plane. The two components are recombined by a second prism in the objective BFP. By choosing a shear distance comparable to the spatial resolution of the system, an intensity changing linearly with the differential of the transmitted optical phase along the shear direction is created. Since the phase is proportional to the thickness of an object, the intensity is proportional to the thickness slope. This is akin to the brightness of a modulated surface height under oblique illumination, and thus provides an intuitively interpretable image. DIC also provides optical sectioning since the contrast of a particle decays with the third power of the defocus. Notably, DIC is widely available in most commercial optical microscopes and is commonly used.
To create quantitative phase information from DIC, various methods have been developed in terms of image acquisition and analysis, eventually resulting in a spatially-resolved map of the optical phase, integrated from the differential phase. For example, the acquisition of images for two orthogonal shear directions and four phase offsets, with subsequent Fourier-space phase integration, was simulated by Arnison et al.16 and later experimentally demonstrated by King et al.17 and Duncan et al.18 Alternatively, using only a focussed and a defocussed DIC image, phase retrieval was shown via the transport of intensity equation.19 To simplify the acquisition of two shear directions without sample rotation, two orthogonal Nomarski prisms and polarization control can be employed,20 and axially-offset circularly-polarized DIC was shown.21 Moreover, using only a single shear direction, Wiener filtering was demonstrated to be effective in extracting phase images,22 and iterative phase reconstruction23 can further improve the results. By exploiting quantitative DIC (qDIC) with Wiener filtering, we have previously shown that the thickness of lipid bilayers could be measured with a precision of 0.1 nm.24,25 Furthermore, by directly fitting the DIC contrast without phase integration, the lamellarity of giant lipid vesicles was quantified.26
The use of qDIC to measure the volume of individual dielectric NPs was proposed by us in an earlier work on single nanodiamonds.27 However, there the investigated nanoparticles were rather large (200–500 nm diameter) hence the challenge to measure small dielectric NPs with this method was not addressed, neither the detection sensitivity limit, nor the precision or accuracy of the technique was quantified. In this work, we have characterised the application of qDIC for sizing single dielectric NPs and determined the precision and accuracy of the method, depending on the acquisition parameters. As an application example, we show sizing of individual nanodiamonds with only 28 nm mean size.
2. Methods
2.1. Samples
For calibration of the qDIC method, PS beads, having a nominal radius of 100 nm with less than 3% coefficient of variance (cv), were purchased (Alpha Nanotech Colloidal PS Beads NP-PA07CPSX78). These PS beads were dispersed in water and drop cast onto (24 × 24) mm2 #1.5 coverslips (Menzel Gläser). After drop casting and drying, the beads were immersed in oil by pipetting 20 μl onto the coverslip. To avoid the formation of air bubbles, the samples were then degassed in a vacuum for 10 minutes immediately before adding a microscope slide and sealing the borders using clear nail varnish. Two types of oil were used, namely water immersion oil (Zeiss, Immersol W 2010) of refractive index nwo = 1.334, and silicone oil (Sigma Aldrich, AP 150 Wacker) of index nso = 1.518. Prior to use, all glass slides and coverslips were cleaned as follows. First, coverslips and slides were immersed in toluene and sonicated for 20 minutes, followed by being immersed in acetone and sonicated for 20 minutes. Next, they were immersed in deionized (DI) water which was then boiled for 3 minutes. Finally, slides and coverslips were immersed in a 30% hydrogen peroxide solution, and again sonicated for 20 minutes. After cleaning, slides and coverslips were kept in a refrigerator in the hydrogen peroxide solution, until needed. To demonstrate the sensitivity of the method, fluorescently labelled PS beads of 15 nm nominal radius were purchased (Sigma Aldrich, L5155) and samples were prepared in the same way. Nanodiamonds (NDs) were purchased from Microdiamant with nominal sizes (0–50) nm (MSY 0–0.05 micron), (0–150) nm, (MSY 0–0.15 micron), and (0–250) nm (MSY (0–0.25) micron). Purchased NDs were purified in-house to remove sp2 graphitic bonds from the surface, by immersion in sulfuric acid for 2 hours, followed by air annealing at 600 °C for 5 hours. Nanodiamonds deposited onto glass were prepared in the same way as described above for PS beads, using silicone oil as surrounding medium.2.2. Optical setup
DIC images were obtained using an inverted Nikon Ti-U microscope. Samples were illuminated using a 100 W halogen lamp (Nikon V2-A LL 100 W) followed by a Schott BG40 filter to remove wavelengths above 650 nm (for which the DIC polarisers are not suited) and a Nikon green interference filter (Nikon GIF), to define the wavelength range centred at 550 nm and having a full-width at half maximum (FWHM) of 53 nm. Limiting the bandwidth to about 10% of the central wavelength provides a defined average wavelength for the later analysis and limits chromatic errors of the optics. This illumination was then passed through a de-Sénarmont compensator (a rotatable linear polariser and quarter-wave plate, Nikon T-P2 DIC Polariser HT MEN51941) and a Nomarski prism (Nikon N2 DIC module MEH52400 or MEH52500) and focused onto the sample by a condenser of 0.72 numerical aperture (NA) or 1.34 NA (part number MEL56100 or MEL41410, respectively). The shear of the Nikon N2 DIC was measured to be (238 ± 10) nm by removing the condenser DIC module and measuring the image shift for linearly polarized illumination along and across the shear. The objectives used were a 20 × 0.75 NA planapochromat (MRD00205) in conjunction with the 0.72 NA condenser and a 1.5× tube lens, and a 60 × 1.27 NA water immersion planapochromat (MRD70650) or 100 × 1.45 NA oil immersion planapochromat (MRD01905), in conjunction with the 1.34 NA oil immersion condenser and a 1× tube lens. After the objectives, light passes through a suited Nomarski prism (DIC sliders MBH76220, MBH76264, and MBH76190, respectively) and a linear polariser (Nikon Ti-A-E DIC analyser block MEN 51980). Images were detected by a Hamamatsu Orca 285 CCD camera (18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 electrons full well capacity, 7 electrons read noise, and 4.6 electrons per count, 12 bit digitizer, 1344 × 1024 pixels, pixel size 6.45 μm, 192 counts offset).
000 electrons full well capacity, 7 electrons read noise, and 4.6 electrons per count, 12 bit digitizer, 1344 × 1024 pixels, pixel size 6.45 μm, 192 counts offset).
The NA of the condenser lens was matched to that of the chosen objective, with the maximum NA of 1.34 used for the 1.45 NA objective, and the maximum of 0.72 for the 0.75 NA objective. The microscope was adjusted for Köhler illumination, and the field aperture was set to be slightly larger than the imaged sample region.
A sequence of Na frames (up to 256) were acquired, with 120 ms exposure time per frame (given by highest stably achievable frame rate), with the lamp intensity adjusted to provide a mean intensity of about 3000 counts (13000 photoelectrons) per pixel. Data for de-Sénarmont polarizer angles ±θ as well as zero were taken to enable qDIC analysis, for θ of 15, 30, and 45 degrees. Images with opposite angles were taken in close temporal sequence to minimize drift between both data. The rotation of the de-Sénarmont polarizer was motorized (by a home-built modification) to improve positioning speed and reproducibility.
Wide-field epi-fluorescence of the labelled PS beads was excited by a metal–halide lamp (Prior Scientific, Lumen L200/D) set at 10% of the maximum power. A suitable exciter/emitter/dichroic filter cube (Semrock, GFP-A-Basic) was used (see also ESI section S3 ii†), resulting in an illumination intensity of about 4 W cm−2 at the sample.
2.3. qDIC analysis
In order to obtain quantitative phase information, we follow the analysis described in ref. 24 and 27, briefly summarised here for clarity. The transmitted intensity image in DIC can be expressed as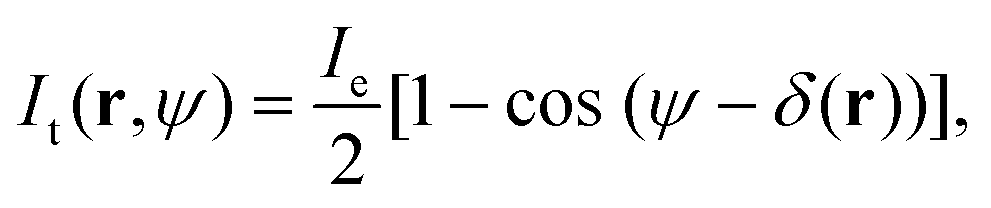 | (1) |
| δ(r) = ϕ(r + s/2) − ϕ(r − s/2). | (2) |
To reduce the influence of a residual spatial dependence of Ie, which includes inhomogeneities in illumination and detection, we acquire two images at opposite angles θ of the de-Sénarmont polarizer, providing the intensities I± = It(r,±ψ) where ψ = 2θ. The contrast image is then defined as
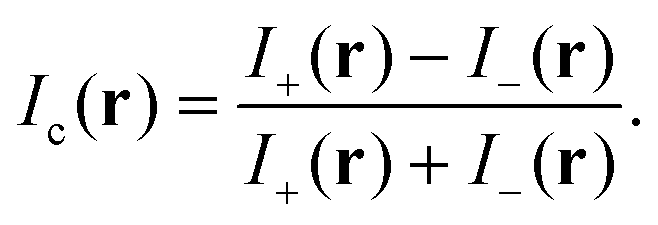 | (3) |
By combining eqn (1) and (3), we obtain
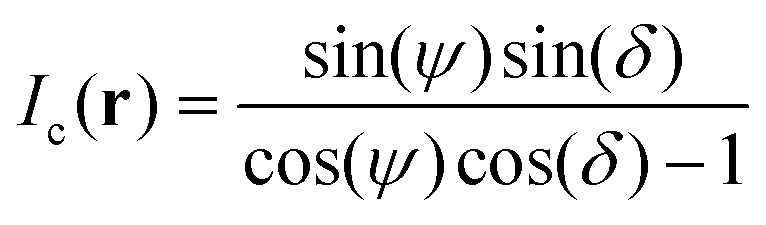 | (4) |
 | (5) |
To extract the phase ϕ from δ, a Wiener deconvolution in the Fourier domain of wave vector k is used. Eqn (2) is written in the Fourier domain as
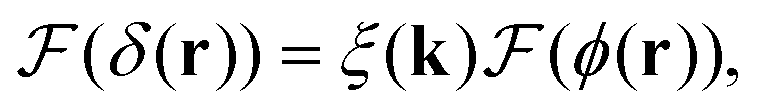 | (6) |
sin(k·s/2) and 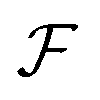 denoting the Fourier transform. Using Wiener deconvolution with a signal to noise parameter κ, we retrieve the phase using
denoting the Fourier transform. Using Wiener deconvolution with a signal to noise parameter κ, we retrieve the phase using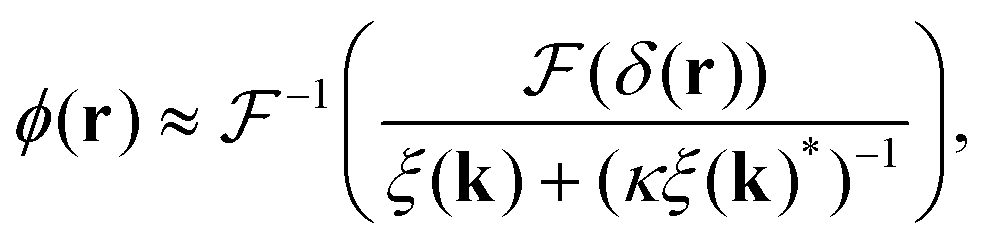 | (7) |
To analyze particle volumes, the phase ϕ(r) is then spatially integrated over a circular area centred at the NP (as previously applied to extinction images28,29) using a dual radius method, as follows. Firstly, particle positions are determined by maxima of ϕ. The background phase for a given particle is determined as the mean phase over the area Ab within the distance ri and 2ri from the particle position, namely
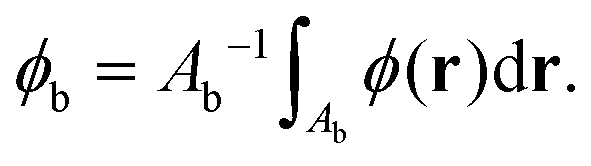 | (8) |
The measured integrated phase Amϕ over the particle is then calculated over an area Ai with a distance below ri from the particle position, using
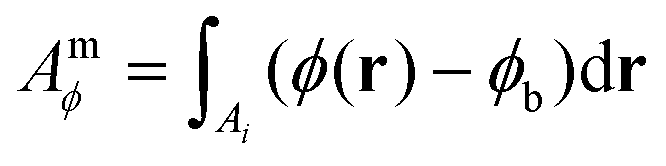 | (9) |
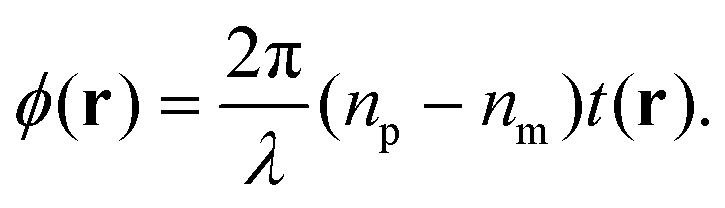 | (10) |
Evaluating eqn (9) for this phase difference, we find
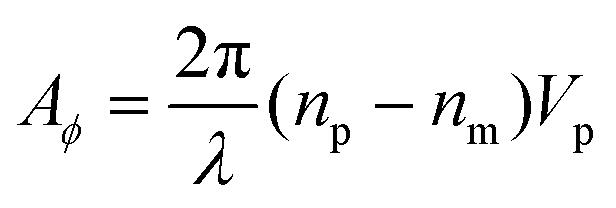 | (11) |
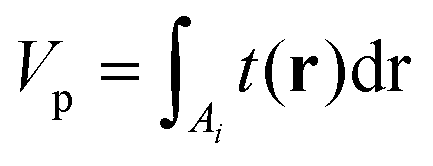 | (12) |
 | (13) |
Importantly, the measured phase area Amϕ is affected by the finite spatial resolution and the finite κ in eqn (7), and has to be corrected to obtain Aϕ, as we detail later. The software and parameters used for the analysis are described in the ESI section S8.†
3. Results and discussion
3.1. PS beads
PS beads of known radius and refractive index were used as a reference standard, to test the accuracy of NP sizing by qDIC. A representative differential phase image δ(r) for nominally 100 nm radius PS beads deposited onto glass and embedded in silicon oil is shown in Fig. 1a using the 1.45 NA objective and a phase offset of ψ = 30° (see Methods for details of the sample and optical set-up). The corresponding images for the 0.75 NA and 1.27 NA objectives are shown in the ESI Fig. S5 and S6,† respectively. The typical shadow-cast appearance of the individual beads is observed. Note the remarkable absence of blemishes or vignetting on a scale of only ± 50 mrad, which is a result of using the DIC contrast eqn (3), as compared to individual DIC images (for illustration the I+ image corresponding to Fig. 1a shown in the ESI Fig. S7†). | ||
| Fig. 1 qDIC microscopy on individual PS beads of nominal 100 nm radius, drop cast onto glass and surrounded by silicon oil, imaged with a 1.45 NA objective and a phase offset of ψ = 30°. (a) δ(r) on a grey scale as shown, from m = −50 mrad to M = 50 mrad. The shadow cast impression is evident, with the shear s = 0.16(1,1) μm in the (x,y) coordinates (x is the horizontal axis and y the vertical in the image). Optical phase maps ϕ(r) showing a region of (2.71 × 2.07) μm2 around a selected bead indicated by the dashed circle, for κ = 1 (b, m = −15 mrad to M = 30 mrad), κ = 200 (c, m = −20 mrad to M = 40 mrad), and κ = 1000 (d, m = −30 mrad to M = 30 mrad). The red and blue circles have the radii ri and 2ri, respectively, with ri = 2.5, 4, 8 pixels in (b), (c), (d), respectively, representing different integration areas Ai and Ab used in the analysis for eqn (8) and (9). | ||
3.2. qDIC optimization and calibration
The qDIC analysis discussed in section 2.3 uses as parameters the signal to noise ratio κ in the Wiener deconvolution, and the area radius ri to evaluate the integrals. To choose the parameter values for best precision and accuracy, the dependence of the measured integrated phase Amϕ and its noise is evaluated as function of these parameters. For the discussion, let us consider here data taken on PS beads mounted in silicone oil, for the 1.45 NA microscope objective, at a phase offset of ψ = 30°. The data were analysed for κ ranging from 0.5 to 105, and ri from 0.5 to 9 pixels. Representative images of the optical phase ϕ(r) for κ = 1, 200, and 1000 are shown in Fig. 1b–d around a single bead.As κ is increased, the extension of spatial features along the shear direction is increasing proportional to  . This is the result of the spatial high-pass filter along the shear resulting from the Wiener filter of qDIC, eqn (7). Its cut-off wave vector kc is given by the condition κ|ξ(kc)|2 = 1, which for small |ξ| is approximated by |kc·s|
. This is the result of the spatial high-pass filter along the shear resulting from the Wiener filter of qDIC, eqn (7). Its cut-off wave vector kc is given by the condition κ|ξ(kc)|2 = 1, which for small |ξ| is approximated by |kc·s| = 1, so that |kc| is proportional to 1/
= 1, so that |kc| is proportional to 1/ . While allowing for longer range features to be retrieved, increasing κ also increases the noise due to the larger amplification of the data by the filter function for small |ξ|.
. While allowing for longer range features to be retrieved, increasing κ also increases the noise due to the larger amplification of the data by the filter function for small |ξ|.
Notably, the stripes show here a triplet structure, which is attributed to the asymmetry of the point-spread function for linearly polarised light. As the two sheared components have linear polarization along and across the shear, their spatial elongation is oriented also in this way, resulting in accordingly different shapes of the probed regions. For smaller NA, this asymmetry is reduced, and with it the triplet structure, as can be seen in the results for the 0.75 NA objective in Fig. S5.†
The radius ri instead determines the size of the circular areas Ai and Ab over which the integrals of the optical phase are calculated (eqn (8) and (9)), as shown by the red and blue circles in Fig. 1b–d. For ri larger than the cut-off of the Wiener filter, Ai contains also regions of inverted (negative) contrast (see dark tails in Fig. 1b–d), reducing the resulting Amϕ. On the other hand, for ri smaller than the spatial resolution, Ai will not contain the full response and again Amϕ will be reduced. Furthermore, the areas scale with ri2, so that the shot-noise in Amϕ will scale with ri, favouring small ri for high signal to noise ratio (SNR).
The evaluated Amϕ as function of κ and ri is given in Fig. 2a for the bead selected in Fig. 1. We find, in accordance with the above qualitative arguments, that Amϕ is increasing steeply with ri up to about 4 pixels, which is the size of the point-spread function (PSF) (see red circle in Fig. 1c). For larger ri, Amϕ reduces for small values of κ, and increases for large κ, converging to a stable value for κ > 500 and ri > 7.
 | ||
| Fig. 2 Phase area Amϕ (a, m = 0 to M = 2950 nm2) and SNR Amϕ/σ (b, from m = 0 to M = 550) as function of κ and ri for a PS bead in silicon oil imaged with the 1.45 NA objective and a phase offset of ψ = 30° as in Fig. 1. The chosen (κ, ri) pairs SN, SE, and C are indicated. | ||
We define three (κ,ri) pairs according to the following criteria: the pair that provides the highest SNR (called SN pair), the one for which Amϕ converges to the highest value (called C pair), and a compromise choice which still gives a good SNR but has a reduced systematic error due to a lower sensitivity to the specific shape of the PSF (called SE pair). Based on Fig. 2a, as C pair we use (κ, ri) = (1000, 8), where the units of ri are pixels (one pixel has a size of 65 nm on the sample for these data). To choose the SN pair, we determine the SNR by evaluating Amϕ at positions not showing a visible particle in the image, and fit its histogram with a Gaussian to determine its standard deviation σ, as can be seen in the ESI Fig. S1.† The ri dependencies of Amϕ and σ are shown in Fig. S4† for κ = 1000. For small ri, σ scales with ri, as expected from shot noise, while for larger ri, we find that σ scales with ri2, indicating the dominance of background structure. The resulting SNR Amϕ/σ corresponding to Fig. 2a is given in Fig. 2b. We find that the SNR is increasing with ri up to about 2 pixels. This can be understood considering that for small ri, Amϕ is scaling with ri2, while σ scales only with ri. For larger ri instead, Amϕ is saturating or even decreasing, as seen in Fig. 2a, so that the SNR decreases, due to the increasing σ. Moreover, for κ above 2, for which the Wiener filter cut-off is larger than the PSF, the SNR is decreasing as expected from the qualitative arguments mentioned previously. The highest SNR is obtained for the SN pair (1, 2.5). Finally, for the SE pair we chose a larger ri corresponding to the PSF size, and accordingly the κ giving the highest SNR, which is (200, 4). This choice reduces systematic errors observed for lower ri, as will be shown later.
Note that the values of κ and ri for the SN, SE, and C pairs depend on the objective and tube lens used, which determine the optical resolution. We report in the ESI†Amϕ as function of κ and ri for the 0.75 NA and 1.27 NA objectives, see Fig. S2a and S3a,† with the corresponding SNR Amϕ/σ, see Fig. S2b and S3b,† and the resulting parameters for the SN, SE, and C pairs.
3.3. Correction factors and polystyrene bead radii
Since the SN and SE pairs provide a phase area Amϕ which is lower than the converged value given by the C pair, we determine correction factors ρ for these pairs to scale Amϕ to the converged value representing Aϕ (see also eqn (9) and (11)). To do this, each particle's Amϕ for the C pair was divided by the value for the SN or SE pair, and the histogram of the resulting ratios was fitted with a Gaussian distribution, to determine center and standard deviation. The distribution obtained for the PS beads mounted in silicone oil imaged using the 1.45 NA objective is shown in Fig. 3 for both the SN and SE pair, resulting in correction factors of ρ = 3.48 ± 0.27 and ρ = 1.25 ± 0.07, respectively. The correction factors found for the different optics and corresponding SE and SN pairs are given in Table 1. Notably, the relative standard deviation of correction factors is generally larger for the SN than the SE pair, showing a reduced systematic error for the SE pair. We emphasize that the correction factors for a given pair (κ, ri) depend only on the optical setup, and not on the particles investigated, and are therefore valid for any dielectric particle which is smaller than the PSF. | ||
| Fig. 3 Analysis of PS beads in silicon oil measured using the 1.45 NA objective and a phase offset of ψ = 30° as in Fig. 1. (a and c) Histogram of the ratio of Amϕ for the (κ,ri) pair C to Amϕ for pair SN (a) or SE (c), with Gaussian fits yielding the mean correction factor ρ. (b and d) Histograms of the resulting bead radii R, for the SN (b) and SE (d) pair. | ||
| ψ | NA | Pair | κ | r i | ρ | A ϕ (nm2) | R (nm) | σ s (nm2) | σ b (nm2) | Radius limit (nm) |
|---|---|---|---|---|---|---|---|---|---|---|
| Silicone oil nso = 1.518 | ||||||||||
| 30 | 0.75 | SN | 1 | 1 | 7.07 | 3193.4 | 97.5 ± 4.30 | 62.0 ± 1.3 | 2.05 ± 0.32 | 16 |
| SE | 100 | 2 | 1.27 | 3164 | 97.2 ± 4.5 | 634.7 ± 4.7 | 50.39 ± 0.85 | 27 | ||
| 1.27 | SN | 1 | 1.5 | 3.76 | 3466.1 | 100.2 ± 4.4 | 13.61 ± 0.41 | 0.62 ± 0.09 | 8.8 | |
| SE | 100 | 2 | 1.63 | 3539.3 | 100.9 ± 3.5 | 110.7 ± 2.6 | 9.36 ± 0.44 | 16 | ||
| 1.45 | SN | 1 | 2.5 | 3.48 | 3570.9 | 101.2 ± 2.2 | 17.9 ± 1.0 | 1.94 ± 0.17 | 13 | |
| SE | 200 | 4 | 1.25 | 3497.3 | 100.5 ± 3.0 | 131.3 ± 5.1 | 16.66 ± 0.87 | 18 | ||
| 60 | 0.75 | SN | 1 | 1 | 7.07 | 4045.7 | 105.5 ± 5.7 | 121.5 ± 3.9 | 3.9 ± 1.0 | 20 |
| SE | 100 | 2 | 1.27 | 3518.2 | 100.7 ± 3.4 | 1250.6 ± 10.5 | 58.6 ± 2.1 | 28 | ||
| 1.27 | SN | 1 | 1.5 | 3.76 | 2907.6 | 94.5 ± 4.1 | 28.11 ± 0.55 | 0.46 ± 0.25 | 7.9 | |
| SE | 100 | 2 | 1.63 | 3105.8 | 96.6 ± 4.2 | 235.3 ± 6.5 | 10.1 ± 1.4 | 17 | ||
| 1.45 | SN | 1 | 2.5 | 3.48 | 3549.8 | 101.0 ± 2.3 | 31.21 ± 0.50 | 1.53 ± 0.10 | 12 | |
| SE | 200 | 4 | 1.25 | 3486.9 | 100.4 ± 2.5 | 215.1 ± 8.8 | 18.4 ± 1.5 | 19 | ||
| 90 | 0.75 | SN | 1 | 1 | 7.07 | 4011.3 | 105.2 ± 5.4 | 212 ± 13 | 0.0 ± 5.4 | 22 |
| SE | 100 | 2 | 1.27 | 3507.8 | 100.6 ± 4.4 | 2222 ± 30 | 77.2 ± 7.3 | 31 | ||
| 1.27 | SN | 1 | 1.5 | 3.76 | 2639 | 91.5 ± 4.2 | 49.17 ± 0.55 | 1.17 ± 0.18 | 11 | |
| SE | 100 | 2 | 1.63 | 2735.7 | 92.6 ± 4.7 | 375.4 ± 3.9 | 14.14 ± 0.91 | 19 | ||
| 1.45 | SN | 1 | 2.5 | 3.48 | 3645.5 | 101.9 ± 2.1 | 51.1 ± 1.8 | 2.27 ± 0.38 | 13 | |
| SE | 200 | 4 | 1.25 | 3602.8 | 101.5 ± 2.5 | 361 ± 13 | 24 ± 24 | 21 | ||
| Water immersion oil nwo = 1.334 | ||||||||||
| 30 | 0.75 | SN | 1 | 1 | 6.75 | 13000.1 |
102.0 ± 4.3 | 61.8 ± 1.4 | 4.00 ± 0.24 | 13 |
| SE | 100 | 2 | 1.23 | 12434.9 |
100.5 ± 2.5 | 644 ± 17 | 91.9 ± 3.0 | 21 | ||
| 1.27 | SN | 1 | 1.5 | 5.26 | 12103.9 |
99.6 ± 4.2 | 24.03 ± 0.54 | 1.56 ± 0.09 | 8.7 | |
| SE | 100 | 2 | 2.12 | 11922.5 |
99.1 ± 3.7 | 106.94 ± 0.86 | 10.52 ± 0.14 | 12 | ||
| 1.45 | SN | 1 | 2.5 | 4.12 | 12472.1 |
100.6 ± 5.5 | 15.98 ± 0.37 | 1.45 ± 0.06 | 7.9 | |
| SE | 200 | 4 | 1.38 | 12711.7 |
101.2 ± 4.3 | 139 ± 45 | 17.7 ± 7.2 | 13 | ||
| 60 | 0.75 | SN | 1 | 1 | 6.75 | 12031.1 |
99.4 ± 3.6 | 122.3 ± 3.8 | 4.66 ± 0.88 | 14 |
| SE | 100 | 2 | 1.23 | 10940.2 |
96.3 ± 2.8 | 1276 ± 14 | 100.0 ± 2.4 | 22 | ||
| 1.27 | SN | 1 | 1.5 | 5.26 | 10838.3 |
96.0 ± 5.1 | 50.6 ± 1.4 | 2.07 ± 0.29 | 9.6 | |
| SE | 100 | 2 | 2.12 | 11008.5 |
96.5 ± 4.5 | 227.7 ± 3.4 | 11.83 ± 0.65 | 13 | ||
| 1.45 | SN | 1 | 2.5 | 4.12 | 14019.8 |
104.6 ± 5.6 | 32.43 ± 0.80 | 1.56 ± 0.16 | 8.1 | |
| SE | 200 | 4 | 1.38 | 15431.8 |
108.0 ± 4.2 | 233 ± 12 | 18.7 ± 2.0 | 13 | ||
| 90 | 0.75 | SN | 1 | 1 | 6.75 | 11145.9 |
96.9 ± 4.2 | 197.2 ± 6.5 | 3.1 ± 3.1 | 12 |
| SE | 100 | 2 | 1.23 | 10272.6 |
94.3 ± 2.9 | 2108 ± 61 | 141 ± 11 | 24 | ||
| 1.27 | SN | 1 | 1.5 | 5.26 | 11249.8 |
97.2 ± 5.4 | 86.8 ± 3.6 | 2.5 ± 1.1 | 10 | |
| SE | 100 | 2 | 2.12 | 11284.6 |
97.3 ± 4.6 | 395 ± 13 | 11.8 ± 3.5 | 13 | ||
| 1.45 | SN | 1 | 2.5 | 4.12 | 14923.1 |
106.8 ± 4.4 | 53.8 ± 1.7 | 2.39 ± 0.35 | 9.3 | |
| SE | 200 | 4 | 1.38 | 14425.7 |
105.6 ± 5.7 | 370 ± 13 | 27.2 ± 2.2 | 15 | ||
The mean correction factor was then used to define the phase area Aϕ = ρAmϕ, which in turn determines the particle volume and corresponding radius from the measurements. Histograms of the resulting radii for the SE and SN pairs are shown in Fig. 3b and d. Note that the C pair, while not requiring a correction factor, corresponds to the lowest SNR of the three pairs, about 20 for the 1.45 NA objective. The noise in the C pair is the dominant contribution to the standard deviation of the measured correction factor for the SE pair. The error of the mean correction factor, considering the roughly 100 particles analysed, is roughly 10 times smaller than the standard deviation, resulting in a relative error below 1%.
To find the mean particle size and standard deviation for each measured radius distribution, the following fit function was used, given by a sum of Gaussian distributions to account for particle aggregates:
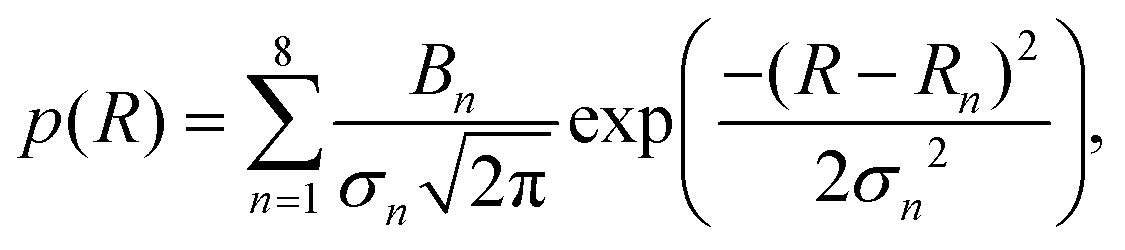 | (14) |
 | (15) |
3.4. Background and shot noise
To characterise the precision of the method, we evaluated the error derived from the noise in the measurements. The noise in the qDIC δ images in the absence of strong contrast, that is for Ic ≈ 0, consists of two components. Firstly, the photon shot noise in the measured images I±, which depends on the average number of detected photoelectrons (phe) per pixel Ne. For an acquisition consisting of Na frames which are averaged, the shot noise is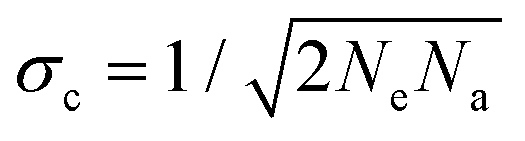 in the DIC contrast Ic (where the factor 1/
in the DIC contrast Ic (where the factor 1/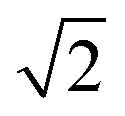 accounts for the use of two images in Ic, see eqn (3)). For typical values used in our work, Ne = 104 and Na = 256, we find σc = 0.04%, and for a single frame σc = 0.7%. To evaluate the corresponding noise σδ of δ, which is related to Ic by eqn (4), we find
accounts for the use of two images in Ic, see eqn (3)). For typical values used in our work, Ne = 104 and Na = 256, we find σc = 0.04%, and for a single frame σc = 0.7%. To evaluate the corresponding noise σδ of δ, which is related to Ic by eqn (4), we find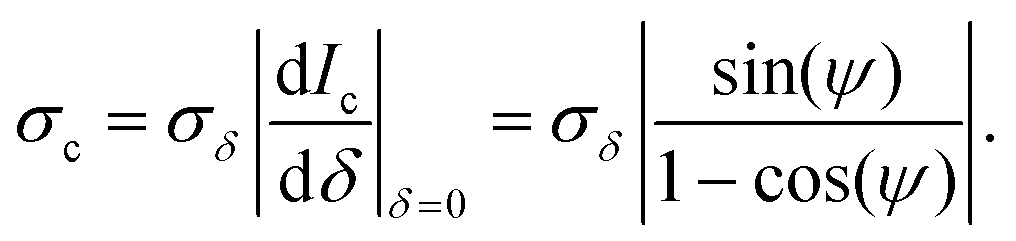 | (16) |
We can see that for small offset angles 0 < ψ ≪ 1, σδ is reduced, by a factor of about ψ/2 compared to the noise for ψ = 90°, as discussed previously.26 However, smaller ψ also reduces the range which can be retrieved, and the transmitted intensity (eqn (1)) is reduced, requiring longer measurement times or stronger illumination. Furthermore, the non-ideal optical elements used (finite extinction of the polarizers, non-perfect matching of the DIC prisms, birefringence of the objective due to residual strain and oblique transmission) results in deviations of the measured data from the ideal behaviour given by eqn (1). Most notably, in high-quality objectives as used here, light rays incident at large oblique angles, collected and collimated by the objectives, are also at oblique incidence on the lens surfaces of the objectives. The resulting polarization dependent transmission of s and p polarized waves provides a variation of the polarization of the collimated ray after the objective, which depends on its incident direction. As a consequence, the rays are not completely blocked by the polarizer, and a significant transmission at ψ = 0 can be observed also without sample.
This issue has been identified early on in polarization microscopy,30 and a recent calculation of the diattenuation and phase shift for a 1.27 NA objective can be found in ref. 31. It has also been shown that the polarization mixing can be reduced by a polarization rectifier, and a reduction of the background from 0.2% to 0.01% for a 1.25 NA objective was shown.32 A more recent discussion including results from a liquid crystal rectifier is given in ref. 33. However, presently such rectifiers are not offered by manufacturers of microscope objectives. We note that the objectives used in our work have an apochromatic and aplanatic performance and high transmission across a wide wavelength range (435–850 nm), constraining the lens design including the anti-reflex coatings. We can thus assume that a given value for the upper limit of the polarization mixing is one of the lens design goals. When limiting the detection and excitation to a single optical mode in confocal microscopy, a complete extinction can in principle be achieved using polarization compensation.34 Notably, 10−8 extinction has been shown35 and a discussion of remaining limits in a model setup reaching 10−9 is given in ref. 36. We emphasize that in DIC additionally a mismatch of their shear of the two Nomarski prisms and a mismatch of their axial position from the conjugated planes can contribute to the finite background.
We have quantified the background transmission as a fraction η of Ie, which was found to be η = 0.80%, 0.64%, and 0.86% for the 1.45 NA, 1.27 NA, and 0.75 NA objectives, respectively. Notably, for the smallest ψ used in this work, i.e. 30°, the transmission (eqn (1)) is only 6.7% of Ie, so that the background constitutes a significant fraction of the ideal transmission without sample. To correct for this residual transmission (i.e. non perfect extinction) in the analysis, we have subtracted this background from the measured intensities Im± to determine I±. This equates to using I± = Im± − 〈Im±〉2η/(1 − cos(ψ)) in eqn (3), where 〈.〉 denotes the spatial average.
Other than shot-noise, we have random structures in our samples unrelated to the particles of interest (POI). Notably, the samples that we study consist of a glass coverslip with attached particles, embedded in an immersion oil. We are imaging the glass-immersion oil interface, while other interfaces are out of focus by at least 10 μm, making them essentially invisible in DIC. Therefore, the background in the absence of POIs originates from unwanted structures at the glass – immersion oil interface. It is thus paramount to use high-quality coverslips and clean them properly before attaching the POIs (see sample preparation protocol in Methods section). Even after cleaning, however, there is a remaining surface roughness of a few nanometers which is an intrinsic feature of glass surfaces fabricated by float techniques, due to the thermally excited surface waves at the glass transition during cooling. Since in DIC the contrast at the interface scales with the refractive index difference between glass and immersion oil, optically clearing the interface by matching the refractive indices is an effective way to suppress background from surface roughness. The two immersion oils used in the present work have an index difference to glass of about 0.2 (water oil) and <0.002 (silicone oil). Thus, when using silicon oil, surface roughness is not relevant.
To unpick the background and shot noise contributions, we determine the noise in Amϕ using 1000 points in regions without evident PS beads, which were then analysed with the SN and SE pairs. A Gaussian function was fitted to the resulting integrated phase area distribution to determine its standard deviation σ. An example of this histogram is shown in the inset of Fig. 4, and the resulting σ is shown in Fig. 4 as function of Na for the 1.45 NA objective on the sample in silicone oil and phase offsets of ψ = 30, 60, and 90°. We find a decreasing σ with increasing Na, as expected for shot noise, which tends to saturate for Na > 100, indicating the background noise limit. We fit this dependence as
 | (17) |
 | ||
| Fig. 4 Standard deviation σ, from the distribution of Amϕ in regions of the sample without PS beads, versus number of averages Na, for PS beads mounted in silicone oil imaged using the 1.45 NA objective and phase offsets ψ of 30° (black), 60° (blue), and 90° (red). The inset shows the histogram of Amϕ for Na = 100, analysed using the SE pair imaged at ψ = 30°, and the fitted Gaussian distribution (black line). | ||
Recalling the scaling of the noise given in eqn (16), we fitted the dependence of the resulting σs for the SN pair with the phase offset ψ (see Fig. 5) using
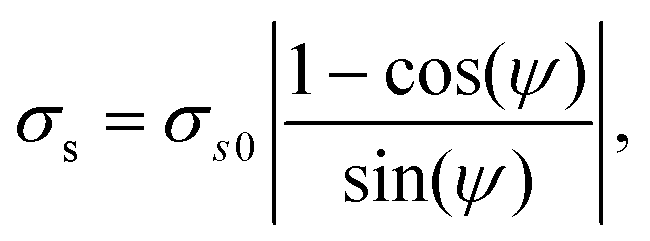 | (18) |
 | ||
| Fig. 5 Single frame shot-noise σs of Amϕ for the different objectives and phase offset angles, ψ, when analysed using the corresponding SN pair. The lines are fits using eqn (18). | ||
As can be seen in Table 1, the smallest single frame shot noise σs is found for the 1.27 NA objective and SN pair for beads in silicon oil, yielding about 14 nm2. Using eqn (11) (and taking into account the correction factor ρ to scale the phase area), this corresponds to a PS bead of 25 nm radius. For samples in water oil, σs is 16 nm2 using the 1.45 NA objective, corresponding to a PS bead radius of 17.5 nm as sensitivity limit from shot noise with a single frame acquisition. Generally, σs decreases with (i) increasing NA due to the improving spatial resolution, (ii) going from the SE to the SN pair due to the better SNR (Fig. 2), (iii) decreasing phase offset due to the increased contrast (eqn (16)). The size limit scales with the third root of the noise, and decreases going from silicone to water oil due to the increased refractive index difference (eqn (11)).
Ultimately, for a sufficient number of frames Na, the shot-noise can be decreased to a point where the background noise σb limits the sensitivity. An overview over the measured σb is given in Fig. 6. On some occasions, and specifically for the 0.75 NA having a larger σs, an increase of σb with ψ is seen, which is not expected, and is attributed to a systematic error; see also the fits in Fig. 4. We note the reduction of σb going from the SE to the SN pair by a factor of 10 to 20. Interestingly, σb for the two immersion oils is generally quite similar, even though the water oil has much larger refractive index difference to the glass substrate than silicone oil. This indicates that glass substrate roughness is not the dominant background, suggesting that it is instead due to other impurities, e.g. remaining debris upon sample preparation.
 | ||
| Fig. 6 Overview of the background noise σb for different phase angles ψ, objectives 0.75, 1.27, and 1.45 NA, water oil (WO) and silicone oil (SO) immersion, and analysis pairs SE and SN. | ||
For ψ = 30°, the SN pair, and the 1.27 NA objective, σb = (0.62 ± 0.06) nm2 for silicone oil, corresponding to a smallest detectable bead radius of 8.8 nm. For samples immersed in water oil instead, we find σb = (1.56 ± 0.06) nm2 and a smallest detectable bead radius of 8.7 nm. Such similar radii would be expected if σb would be caused by dielectric debris on the surface of a refractive index similar to the one of PS.
Other quantitative phase contrast techniques have a reported uncertainty of 0.3 to 2 nm in optical path length.20 Using a wavelength of 550 nm and a point-spread function size of 300 nm, this corresponds to an uncertainty in Aϕ of 308–2056 nm2, and thus a radius limit for PS beads in water of 32 to 60 nm. Notably, the radius limit in the present work is about 5 times lower.
It should be noted that the iSCAT technique11 avoids the static background noise σb by analyzing particles which attach and/or detach during measurements, so that the difference can be detected. Such a method can also be applied to qDIC, resulting in a sensitivity only limited by the shot-noise. For example, using the 1.45 NA objective, samples in water oil, ψ = 30°, the SN pair, and Na = 1000 acquisitions, the radius limit (scaling with Na−1/6) is down to 3.8 nm, which can be achieved within 1 s with a modern camera. We also note that by reducing the phase offset, the sensitivity can be increased, see eqn (16). Assuming ideal optics and η = 0, we find that for ψ = 1° the limit for Na = 1000 is a radius of 1.8 nm.
3.5. PS bead sizes
Samples with PS beads as described in section 1 were measured and the resulting Aϕ was converted into a PS volume, using the refractive index of np = 1.59. Note that this index can vary depending on the packing density of the PS. Hence, rather than assuming a nominal value, we have determined the refractive index for the PS beads used here, by considering the measured change of the DIC contrast versus immersion medium index, as discussed in the ESI section S2.†The retrieved particle radii, using PS beads in silicone oil and the 1.45 NA objective, are shown in Fig. 3 for the SE and SN pairs. The histograms were fitted with eqn (14) yielding (R1 ± σ1) = (101.2 ± 2.2) nm and (100.5 ± 3.0) nm, respectively, as mentioned previously and summarised in Table 1. Results for other objectives and immersion oils are also given in Table 1, with the histograms shown in the ESI Fig. S15 to S19.† Importantly, we find a quantitative agreement of the measured radii for all immersion oils, objectives, and phase offset combinations within a few %.
The smallest σ1 is found using the 1.45 NA objective with beads in silicone oil and for the SN pair, which gives a relative variation of σ1/R1 = 2.2%, consistent with the cv below 3% specified by the manufacturer. This suggests that the contribution of the measurement noise to the uncertainty in the size distribution is negligible. The influence of the measurement noise σ to the size distribution can also be calculated, and in turn removed, resulting in a corrected σc1 given by
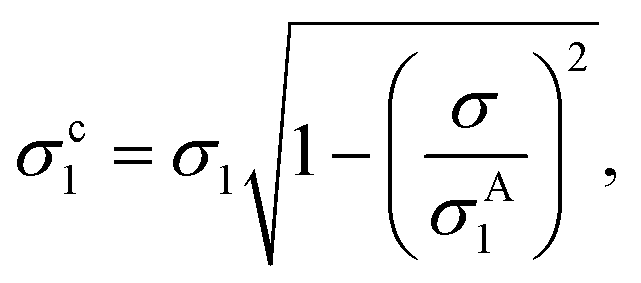 | (19) |
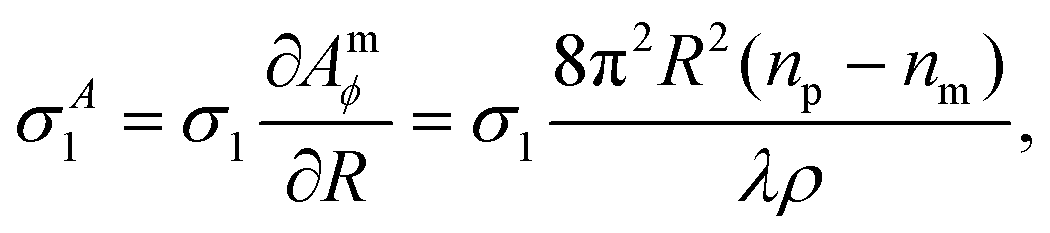 | (20) |
As detailed in the ESI section S3 i,† the bead diameter of the batch used was measured with transmission electron microscopy (TEM) to be 187 nm, with a cv of 2.1%. Scanning electron microscopy (SEM) gave a diameter of 190 nm, dynamic light scattering (DLS) resulted in 183 nm diameter, and atomic force microscopy (AFM) gave a height of 179 nm with a cv of 3.7%. The most reliable result is SEM, since measurements were performed over a large number of beads (see ESI Fig. S9†), giving 95 nm radius, which is consistent with the qDIC results (see Table 1). The about 5% difference in radius found in qDIC compared to SEM corresponds to a 15% difference in the qDIC phase area. This is consistent with the accuracy of determining the saturated Amϕ for the C pair seen in Fig. S4a.† A systematic variation of about 5% is also seen between the different objectives and pairs, which each have their own correction factor.
3.6. Small PS beads
To demonstrate the capability of the technique to measure small PS particles, fluorescent PS beads of nominally 15 nm radius were examined. Here a 100 × 1.49 NA apochromat objective (part number MRY10059) was used, keeping the SN pair and correction factor of the 1.45 NA objective. Its background transmission η has been determined to 0.96% (see Table S1†). A phase offset of ψ = 20° was used, providing a low σs.The measured phase ϕ(r) of a representative region of a sample using silicone oil is shown in Fig. 7a. Analysis of this data with the SN pair shows a phase area Amϕ with a noise of σ = 1.6 nm2, and the dependence on Na is described by eqn (17) with σs = 13.3 ± 0.8 nm2 and σb = 1.4 ± 0.1 nm2. The inset shows a bead clearly visible above background, having a phase area corresponding to 24 nm radius.
 | ||
| Fig. 7 (a) qDIC phase ϕ(r) on fluorescent PS beads of nominally 15 nm radius, drop cast onto glass and surrounded by silicone oil, imaged with a 1.49 NA objective at a phase offset of ψ = 20° and analyzed using κ = 1 and Na = 256. Grey scale from m = −1 mrad to M = 1 mrad. The inset shows a region of (2.07 × 1.55) μm2 around a bead highlighted by the yellow dashed circle, on a greyscale from m = −0.4 mrad to M = 0.4 mrad. This bead has an Amϕ corresponding to a radius of 24 nm. (b) epi-Fluorescence intensity Ifl (average of 5 frames with 3 s exposure time each) of the same sample region, on a greyscale from m = 37 to M = 4177 phe. The excitation area was limited to the discernible disk region by a field aperture. Inset as in (a), greyscale m = 41 to M = 1647 phe. | ||
The histogram of background and particle Amϕ is shown in Fig. 8a. To reliably distinguish particles above background, we require that Amϕ>4σ (see grey shaded region in Fig. 8a), which in the absence of particles occurs with a probability of p = 3 × 10−5 for a Gaussian background distribution. Considering a spatial resolution of about s = 300 nm, this probability corresponds to an average distance of s/ = 53 μm between noise-induced particle detections, or about two detections over the region shown in Fig. 7a. The retrieved particle radius distribution is given in Fig. 8b. The 4σ limit corresponds to a radius of 18 nm. The distribution is decaying above this radius, as expected from the nominally 15 nm radius beads.
= 53 μm between noise-induced particle detections, or about two detections over the region shown in Fig. 7a. The retrieved particle radius distribution is given in Fig. 8b. The 4σ limit corresponds to a radius of 18 nm. The distribution is decaying above this radius, as expected from the nominally 15 nm radius beads.
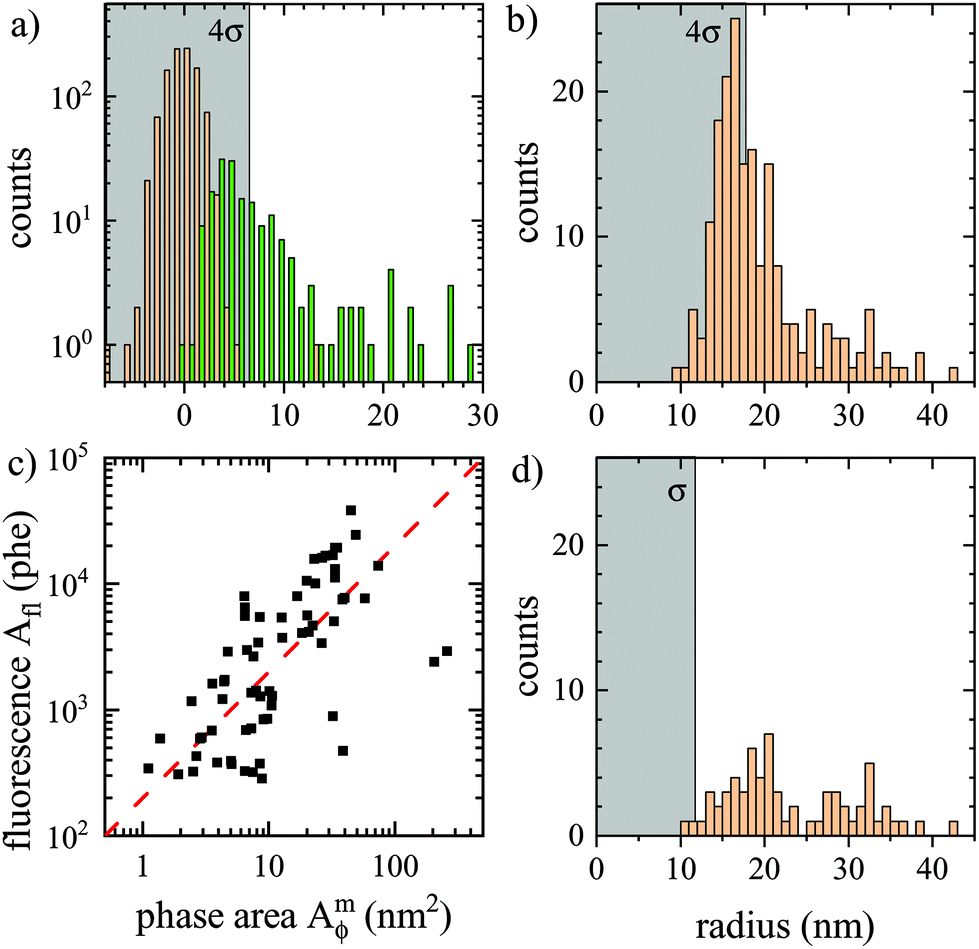 | ||
| Fig. 8 Analysis of data shown in Fig. 7. (a) histogram of the phase area Amϕ for background (orange, σ = 1.6 nm2), and particles (green) located as maxima in ϕ(r) above 0.13 mrad. The region below 4σ is indicated in gray. (b) resulting histogram of particle radius. (c) fluorescence photoelectrons Aflversus Amϕ, with a proportionality indicated as dashed line. (d) Histogram of particle radius with corresponding Afl above 274 phe. | ||
Importantly, the fluorescence of the beads provides an independent reporter of the bead position, and even an estimate of bead size assuming that the fluorescence intensity Ifl is proportional to the bead volume. We have therefore imaged the fluorescence of the same region, as shown in Fig. 7b. A good correlation between the particles visible in ϕ(r) and in Ifl(r) can be seen, including the particle in the inset. For each particle identified in ϕ(r), the integrated fluorescence Afl was determined using the same double radius method as for Amϕ, but in area units of pixels, and counts converted to photoelectrons, and is shown in Fig. 8a. We can see a linear correlation of Afl and Amϕ, consistent with the fluorescence intensity Ifl being proportional to the particle volume. However, a significant spread of the proportionality is observed, which could be an intrinsic property of the PS beads. Notably we found that the beads lose their fluorophores over a timescale of a few hours when immersed in oil (possibly due to the missing surface tension of water allowing for swelling and solubility of the fluorophores in oil). The histogram of particles having a corresponding Afl above a threshold of four standard deviations of the background in Afl is given in Fig. 7d. It shows a similar distribution as the original one, but fewer particles, specifically with radii below 20 nm. This is attributed to the limitation of the analysed region to the fluorescence excitation area, and the loss of fluorophores reducing the fluorescence of small beads to below the threshold.
We have performed a similar analysis using water oil immersion, with results shown in the ESI Fig. S25 and Fig. S26.† Interestingly, the detection limit is similar to the one seen for silicone oil immersion, even though the index contrast to PS has increased by a factor of 3.56. However, the index contrast to glass has increased by a factor of about 100 and the glass roughness is clearly visible in the background structure, indicating it to be the limiting factor in this case.
As detailed in the ESI section S3 ii,† the bead diameter distribution of the batch used was measured with DLS to have a z-mean of 24 nm and a cv of 52%, while the height distribution was measured by AFM to have a mean of 16 nm with a cv of 42%. This shows that these beads have a wide distribution of sizes, consistent with the above qDIC results. Notably, the large number of particles below 20 nm diameter likely contribute to σb in the measured samples, while particles above 36 nm diameter can be reliably identified and measured using only qDIC.
3.7. Nanodiamond volume measurement
To showcase the method with an application example, we measured the size of individual nanodiamonds, specified by the manufacturer to have a broad distribution of sizes below 50 nm, 150 nm, and 250 nm (see also Methods section). NDs were embedded in silicon oil. Particles with nominal diameter ranges (0–250) nm and (0–150) nm were measured using the 0.75 NA objective, while those with (0–50) nm diameters were imaged with the 1.27 NA objective (examples of the δ(r) and ϕ(r) images for each are shown in the ESI Fig. S27 to S29†). Stacks of Na = 256 images were obtained at ψ = 30° for the (0–250) nm and (0–150) nm NDs, and at ψ = 60° for the (0–50) nm NDs. Analysis was carried out using the SE pairs. The resulting particle volume histograms using a ND refractive index37 of n = 2.42 show a nearly exponential decay, and thus were fitted with an exponential distribution p0exp(−Vp/![[V with combining overline]](https://www.rsc.org/images/entities/i_char_0056_0305.gif) ), with the mean volume . NDs are typically brick shaped, hence we defined a characteristic particle size using a cube geometry as
), with the mean volume . NDs are typically brick shaped, hence we defined a characteristic particle size using a cube geometry as 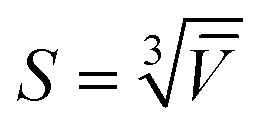 . Fig. 9 shows the volume distributions for each ND sample and the exponential fits. The mean volumes were found to be 2.1 × 104 nm3, 2.4 × 105 nm3 and 4.1 × 105 nm3, for the (0–50) nm, (0–150) nm and (0–250) nm NDs, respectively, yielding characteristic sizes S of 27.6 nm, 62.1 nm, and 74.3 nm.
. Fig. 9 shows the volume distributions for each ND sample and the exponential fits. The mean volumes were found to be 2.1 × 104 nm3, 2.4 × 105 nm3 and 4.1 × 105 nm3, for the (0–50) nm, (0–150) nm and (0–250) nm NDs, respectively, yielding characteristic sizes S of 27.6 nm, 62.1 nm, and 74.3 nm.
 | ||
| Fig. 9 Size histograms measured with qDIC on nanodiamonds in silicon oil. Particles with nominal diameter ranges (0–250) nm and (0–150) nm were measured using the 0.75 NA objective, while those with (0–50) nm diameters were imaged with the 1.27 NA objective. The SE pair was used for the analysis. Solid lines are the exponential fits. | ||
3.8. Limitations and refinements
Let us review the assumptions of the method as presented, the resulting limitations, and further refinements possible.(i) NPs are assumed to be smaller than the optical resolution of the microscope, in order to use the microscope PSF independent of the particle size in the analysis. This is relevant when using the SN pair and corresponding correction factors ρ, to minimize the noise in the retrieval of Aϕ. For larger objects, one can use the C pair, and retrieve the integrated phase area similar to other quantitative phase imaging (QPI) techniques.38 However, long-range phase gradients are difficult to determine with a DIC based approach, which measures only the spatial differential of the phase, while other QPI methods such as off-axis holography38 are advantageous when used for measuring extended structured objects such as whole cells.
(ii) The refractive index of the NPs is assumed to be constant over the probed wavelength range. Since the wavelength range has a relative bandwidth of only 10% and the NPs are assumed to be dielectric, this is not a limiting factor for the NPs investigated here. NPs showing a strong dispersion of the refractive index typically also show significant absorption, e.g. plasmonic NPs close to the plasmon resonances. They are less suited for the present method and can be better measured using their extinction cross-section.8
(iii) NPs are assumed to be non-birefringent. This is specifically relevant for the DIC method used in the present work, which is based on two sheared beams linearly polarized along and orthogonal to the shear. A birefringence aligned with the shear would thus be measured as a phase difference. Notably, such a signal, which is a single peak in δ, leads to a differential shape in the phase ϕ(r), which in first-order would not affect the integrated phase area Aϕ. An instrumentation refinement suppressing this effect would be to add quarter wave plates on both sides of the sample to convert the orthogonal linear polarizations into circular polarizations of opposite helicity, probing the circular birefringence (optical activity), which is weak in natural materials. Notably, this would also suppress the triple pattern seen in the integrated phase for the high NA objectives (Fig. 1d) as a result of the elongated PSFs of linearly polarized light. However, a change of the polarization state of the transmitted light of the beams by a birefringence phase shift would still affect their recombination by the objective DIC prism, which would be relevant for strongly birefringent NPs.
(iv) NPs are assumed to have an isotropic response. An anisotropic response due to the NP shape would affect Aϕ similar to the birefringence discussed in (iii). Elongated shapes result in a field-orientation dependent NP polarisability, which for smallelliptical NPs is given by the components39
 | (21) |
η) + O(η3), showing that the orientation dependence is given by the factor 1 − Ljη. The depolarization factors Lj are 1/3 for a sphere, and with increasing aspect ratio decrease towards zero along the long axis, and increase towards 1 along the short axis. Importantly, they are bounded between zero and one, so that for small η the anisotropy is small independent of shape, for NPs described by the dipole limit eqn (21). For typical organic particles in water, η is about 0.2, so that even for shapes with large aspect ratios the resulting differences between the αj are below 10%.
For the ND samples investigated in the present work, the index contrast is higher, yielding η ≈ 1.5, so that the effect can be more pronounced. The NDs have a range of brick-like shapes,27 with aspect ratios of 1–1.5. In the data we find examples of the resulting asymmetric response in ϕ(r), showing up as bipolar stripes tailing away from the ND, as is exemplified in the inset of Fig. S28.† Since the size distribution in the case of the NDs is wide, the resulting volume error is not important for the conclusions drawn.
(v) The permittivity contrast η is assumed to be small in order to neglect variations in beam propagation direction in the ray picture, or a significantly screened local field inside the particle in the wave-picture. Layered structures such as lipid bilayers24 will be less affected by the screening due to the Maxwell's boundary conditions conserving the dominant in-plane field. For a higher index contrast, the above analytical expressions for elliptical particles could be a starting point to provide corrections.6 For more complex shapes, full numerical simulations would be required to provide calibration-free quantitative sizing.7 To avoid the complications of large |η|, one can choose a suited immersion medium, which is commercially available40 with refractive indexes between 1.3 and 1.8.
(vi) Typical biological NPs such as endosomes, liposomes, or viruses, are not homogeneous in material and shape, so that the size and the composition cannot be easily separated. However, the phase area Aϕ can still be used to determine the organic mass in the NP in a similar way as it is done in quantitative phase imaging of cells.41 Combining this analysis with nanoparticle tracking to determine the NP hydrodynamic radius from the spatial diffusion would then allow to determine both size and mass of the particle.
Furthermore, for particles with unknown refractive index, a correlative measurement of the same particle in two different immersion media7 can be used to determine the refractive index of each particle, analogous to the determination of the PS index using ensemble averaged phase areas in the ESI section S2†.
Finally, we note that apart from using calculated responses for sizing, there is the possibility to calibrate the method for a specific particle type by measuring particles of a range of known sizes.
4. Conclusion
In conclusion, we have investigated the application of quantitative DIC microscopy with Wiener filtering for sizing individual dielectric NPs, and determined the precision and accuracy of the method. Using polystyrene beads of 100 nm radius as size standard, we found that the accuracy in determining their radius was within few nm, corresponding to a relative accuracy of only a few percent. In terms of precision, we found the smallest detectable PS bead radius to be about 10 nm, limited by background structure at the glass interface onto which the nanoparticles were deposited. For reliable identification of NPs, a signal of at least four standard deviations above the background is required, corresponding to a radius of 16 nm. This has been verified experimentally using small fluorescent PS beads. Notably, this limit can be overcome when observing particles which attach and/or detach from a glass surface during measurements, eventually reaching a sensitivity only limited by shot noise. The latter was found to equate to 4 nm PS bead radius when averaging over 1000 frames, which can be achieved within 1 s total acquisition time with modern cameras. Such sensitivity could be further increased by using small phase offsets in the DIC acquisition, potentially reaching a size limit down to only 2 nm radius. As application example, we demonstrated sizing of individual nanodiamonds having poly-disperse distributions. Small nanodiamonds with nominal sizes below 50 nm were well above the detection limit, and were found to have a nearly exponential size distribution with 28 nm mean size.Considering the importance of dielectric nanoparticles in many fields, from naturally occurring virions and exosomes to polluting nanoplastics, the proposed method could offer a powerful tool for nanoparticle analysis, combining accuracy, sensitivity and high-throughput with widely available and easy-to-use DIC microscopy.
Author contributions
W. L. and P. B. conceived the work. S. H. prepared the samples for the optical measurements and performed the related measurements and data analysis. D. R. and L. P. supported the data analysis. S. H., P. B. and W. L. wrote the manuscript. All authors contributed to the data interpretation and manuscript review.CRediT: Conceptualization WL, PB; data curation SH, DR, LP, WL; formal analysis SH, WL, DR; funding acquisition WL, PB; investigation SH, DR; methodology WL, PB, SH, DR; project administration PB, WL; resources PB, WL; software WL, DR, LP; supervision WL, PB; validation SH; visualization SH, PB, WL; writing – original draft SH, WL, PB; writing – review & editing PB, WL, SH, DR, LP.
Data availability
Information on the data underpinning the results presented here, including how to access them, can be found in the Cardiff University data catalogue at http://doi.org/10.17035/d.2022.0153720825.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. H. acknowledges support for his PhD studies by the EPSRC Diamond Science and Technology CDT [grant no. EP/L015315/1] and Cardiff University. The microscope equipment was supported by the EPSRC grant no. EP/M028313/1. We acknowledge Joseph Bleddyn Williams for contributing to the development of the qDIC analysis software, Iestyn Pope for support of the microscope instrumentation, Martina Recchia and Ozan Aksakal for measuring the bead fluorescence spectra, and Vikramdeep Singh for help with the atomic force microscopy measurements.References
- T. Sun, Y. S. Zhang, B. Pang, D. C. Hyun, M. Yang and Y. Xia, Angew. Chem., Int. Ed., 2014, 53, 12320–12364 CAS.
- J. Liu, R. Zhang and Z. P. Xu, Small, 2019, 15, 1900262 CrossRef PubMed.
- Z.-M. Dang, J.-K. Yuan, S.-H. Yao and R.-J. Liao, Adv. Mater., 2013, 25, 6334–6365 CrossRef CAS PubMed.
- W. Anderson, D. Kozak, V. A. Coleman, Å. K. Jämting and M. Trau, J. Colloid Interface Sci., 2013, 405, 322–330 CrossRef CAS PubMed.
- S. Mourdikoudis, R. M. Pallares and N. T. K. Thanh, Nanoscale, 2018, 10, 12871–12934 RSC.
- L. M. Payne, W. Albrecht, W. Langbein and P. Borri, Nanoscale, 2020, 12, 16215–16228 RSC.
- Y. Wang, A. Zilli, Z. Sztranyovszky, W. Langbein and P. Borri, Nanoscale Adv., 2020, 2, 2485–2496 RSC.
- L. M. Payne, W. Langbein and P. Borri, Phys. Rev. Appl., 2018, 9, 034006 CrossRef CAS.
- A. Zilli, W. Langbein and P. Borri, ACS Photonics, 2019, 6, 2149–2160 CrossRef CAS PubMed.
- M. Piliarik and V. Sandoghdar, Nat. Commun., 2014, 5, 4495 CrossRef CAS PubMed.
- G. Young, N. Hundt, D. Cole, A. Fineberg, J. Andrecka, A. Tyler, A. Olerinyova, A. Ansari, E. G. Marklund, M. P. Collier, S. A. Chandler, O. Tkachenko, J. Allen, M. Crispin, N. Billington, Y. Takagi, J. R. Sellers, C. Eichmann, P. Selenko, L. Frey, R. Riek, M. R. Galpin, W. B. Struwe, J. L. P. Benesch and P. Kukura, Science, 2018, 360, 423–427 CrossRef CAS PubMed.
- R. W. Taylor and V. Sandoghdar, Nano Lett., 2019, 19, 4827–4835 CrossRef CAS PubMed.
- C.-Y. Cheng, Y.-H. Liao and C.-L. Hsieh, Nanoscale, 2019, 11, 568–577 RSC.
- R. D. Boyd, S. K. Pichaimuthu and A. Cuenat, Colloids Surf., A, 2011, 387, 35–42 CrossRef CAS.
- G. M. Nomarski, J. Phys. Radium, 1955, 16, 9S Search PubMed.
- M. R. Arnison, K. G. Larkin, C. J. R. Sheppard, N. I. Smith and C. J. Cogswell, J. Microsc., 2004, 214, 7–12 CrossRef CAS PubMed.
- S. V. King, A. Libertun, R. Piestun, C. J. Cogswell and C. Preza, J. Biomed. Opt., 2008, 13, 024020 CrossRef PubMed.
- D. D. Duncan, D. G. Fischer, A. Dayton and S. A. Prahl, J. Opt. Soc. Am. A, 2011, 28, 1297 CrossRef PubMed.
- S. S. Kou and C. Sheppard, International Conference on Advanced Phase Measurements Methods in Optics and Imaging, 2010, pp. 301–306.
- M. Shribak, K. G. Larkin and D. Biggs, J. Biomed. Opt., 2017, 22, 016006 CrossRef PubMed.
- C. Ding, C. Li, F. Deng and G. J. Simpson, Opt. Express, 2019, 27, 3837 CrossRef CAS PubMed.
- E. B. van Munster, L. J. van Vliet and J. A. Aten, J. Microsc., 1997, 188, 149–157 CrossRef CAS PubMed.
- K. Koos, J. Molnár, L. Kelemen, G. Tamás and P. Horvath, Sci. Rep., 2016, 6, 30420 CrossRef CAS PubMed.
- D. Regan, J. Williams, P. Borri and W. Langbein, Langmuir, 2019, 35, 13805–13814 CrossRef CAS PubMed.
- D. Regan, J. Williams, F. Masia, P. Borri and W. Langbein, Quantitative Phase Imaging V, 2019 Search PubMed.
- C. I. McPhee, G. Zoriniants, W. Langbein and P. Borri, Biophys. J., 2013, 105, 1414–1420 CrossRef CAS PubMed.
- I. Pope, L. Payne, G. Zoriniants, E. Thomas, O. Williams, P. Watson, W. Langbein and P. Borri, Nat. Nanotechnol., 2014, 9, 940–946 CrossRef CAS PubMed.
- L. M. Payne, W. Langbein and P. Borri, Appl. Phys. Lett., 2013, 102, 131107 CrossRef.
- L. Payne, G. Zoriniants, F. Masia, K. P. Arkill, P. Verkade, D. Rowles, W. Langbein and P. Borri, Faraday Discuss., 2015, 184, 305–320 RSC.
- S. Inoué, Exp. Cell Res., 1952, 3, 199–208 CrossRef.
- X. Xu, W. Huang and M. Xu, Opt. Express, 2015, 23, 27911 CrossRef CAS PubMed.
- S. Inoué and W. L. Hyde, J. Biophys. Biochem. Cytol., 1957, 3, 831–838 CrossRef PubMed.
- M. I. Shribak, S. Inoue and R. Oldenbourg, SPIE Proceedings, 2002.
- P. Higdon, R. Juškaitis and T. Wilson, J. Microsc., 1997, 187, 8–11 CrossRef.
- A. V. Kuhlmann, J. Houel, D. Brunner, A. Ludwig, D. Reuter, A. D. Wieck and R. J. Warburton, Rev. Sci. Instrum., 2013, 84, 073905 CrossRef PubMed.
- M. Benelajla, E. Kammann, B. Urbaszek and K. Karrai, Phys. Rev. X, 2021, 11, 021007 CAS.
- G. Turri, S. Webster, Y. Chen, B. Wickham, A. Bennett and M. Bass, Opt. Mater. Express, 2017, 7, 855–859 CrossRef CAS.
- Y. Park, C. Depeursinge and G. Popescu, Nat. Photonics, 2018, 12, 578–589 CrossRef CAS.
- C. F. Bohren and D. R. Huffman, Absorption and scattering of light by small particles, John Wiley & Sons, New York, 1983 Search PubMed.
- https://www.cargille.com/refractive-index-liquids/ .
- T. A. Zangle and M. A. Teitell, Nat. Methods, 2014, 11, 1221–1228 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1an02009a |
| This journal is © The Royal Society of Chemistry 2022 |