
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of N-glycan positional isomers by combining IMS and vibrational fingerprinting of structurally determinant CID fragments†
Priyanka
Bansal
 ,
Ahmed
Ben Faleh
,
Stephan
Warnke
and
Thomas R.
Rizzo
*
,
Ahmed
Ben Faleh
,
Stephan
Warnke
and
Thomas R.
Rizzo
*
Laboratoire de Chimie Physique Moléculaire, École Polytechnique Fédérale de Lausanne, EPFL SB ISIC LCPM, Station 6, CH-1015, Lausanne, Switzerland. E-mail: thomas.rizzo@epfl.ch
First published on 21st January 2022
Abstract
While glycans are present on the surface of cells in all living organisms and play key roles in most biological processes, their isomeric complexity makes their structural characterization challenging. Of particular importance are positional isomers, for which analytical standards are difficult to obtain. We combine ultrahigh-resolution ion-mobility spectrometry with collision-induced dissociation and cryogenic infrared spectroscopy to determine the structure of N-glycan positional isomers. This approach is based on first separating the parent molecules by SLIM-based IMS, producing diagnostic fragments specific to each positional isomer, separating the fragments by IMS, and identifying them by comparing their IR fingerprints to a previously recorded spectral database. We demonstrate this strategy using a bottom-up scheme to identify the positional isomers of the N-linked glycan G0-N, in which a terminal N-acetylglucosamine (GlcNAc) is attached to either the α-3 or α-6 branch of the common N-glycan pentasaccharide core. We then use IR fingerprints of these newly identified isomers to identify the positional isomers of G1 and G1F, which are biantennary complex-type N-glycans with a terminal galactose attached to either the α-3 or α-6 branch, and in the case of G1F a fucose attached to the reducing-end GlcNAc. Starting with just a few analytical standards, this fragment-based spectroscopy method allows us to develop a database which we can use to identify positional isomers. The generalization of this approach would greatly facilitate glycan analysis.
Introduction
Glycosylation is one of the most common post-translational modifications of proteins in eukaryotic cells.1 As such, glycans play diverse biological functions ranging from cell-to-cell recognition and cell adhesion, to inflammatory and immune response.2,3 However, the analysis of glycans represents a major challenge due to their inherent structural diversity and complex biosynthesis. The isomeric nature of the monosaccharide building blocks gives rise to a myriad of glycan isomers differing in sequence and linkage. Of particular importance are positional isomers resulting from the addition of a monosaccharide unit to different branches of a glycan. For example, galactose (Gal) is ubiquitously found at the non-reducing end of N-glycans and plays an important role as a biomarker for many diseases.4–6 The terminal Gal position of N-glycans can also affect the efficacy of biotherapeutics. For example, recent studies have shown that the position of the terminal Gal in the N-glycan G1F, in which Gal is either attached to the α-3 (G1F(3)) or the α-6 (G1F(6)) branch, influences the effector functions of therapeutic monoclonal antibodies.7 It is therefore important to be able to differentiate such structural isomers.The inherent complexity of glycan analysis has led to the use of various experimental approaches. Nuclear magnetic resonance (NMR) and X-ray crystallography can be extremely valuable for glycan structural determination, but they require large amounts of highly purified samples, which limits their application.8–10 Single-stage mass spectrometry (MS) is by its very nature blind to isomers and must be combined with other techniques to extract structural information from isomeric glycans. One approach is to selectively fragment isomers using collision-induced dissociation (CID) in MSn schemes to elucidate glycan sequence and branching patterns.11–13 However, interpretation of fragmentation data is not always straightforward, and MSn alone cannot distinguish among all isomeric forms.14 It is therefore often used in conjunction with separation methods such as capillary electrophoresis (CE) and liquid chromatography (LC).15–20 Despite its extensive use in glycan analysis, the identification of subtly different glycan isomers by LC-MS remains challenging.21
The use of various IMS technologies together with tandem MS has been reported for the analysis of isomeric glycans, including positional isomers.22–32 While ultrahigh-resolution IMS based on structures for lossless ion manipulation (SLIM)33,34 has been applied to achieve separation of different kinds of glycan isomers,35,36 the assignment of a drift peak to a precise isomeric form often remains elusive.35,37
We have previously demonstrated that cryogenic vibrational spectroscopy can be used for the identification of glycan isomers based on their unique infrared (IR) absorption spectrum.38–40 Such fingerprint spectra are robust identifiers, since they are inherent to a given molecule and largely insensitive to experimental conditions. An IR spectrum can thus positively identify a compound even when the spectra of other possible isomeric species are not known. The combination of IR fingerprinting with SLIM-based ultrahigh-resolution cyclic IMS was subsequently demonstrated to be a powerful tool for the analysis of glycan mixtures.41–44 However, any identification method that relies on a database, such as IR spectral fingerprinting, requires access to pure analytical standards. For many complex glycan structures, such standards are either expensive to synthesize or unavailable, which raises the need to create reference data from glycans in isomeric mixtures. Our recently developed IMS-IMS approach, where fragments of isomer-separated species can undergo further isomer separation before spectroscopic interrogation,45 poses an ideal starting point for the development of such methods.
In this work we describe a technique to identify positional isomers of underivatized N-glycans with a reduced need for pure analytical standards. To this end, we have developed an experimental protocol based on isomer separation using ultrahigh-resolution IMS followed by fragmentation and IR fingerprinting of structurally diagnostic fragments specific to each positional isomer. We then construct an IR spectral database for the parent glycans and their fragments, the latter of which we use to identify positional isomers from an isomeric mixture. We demonstrate the principle of this new approach by following a bottom-up scheme to identify the positional isomers of the N-glycans G0-N, G1 and G1F.
Experimental approach
Ion-mobility selective cryogenic IR spectroscopy
Experiments were performed on a previously described home-built instrument,44 which combines travelling wave ion mobility spectrometry ((TW)-IMS) using SLIM33,34 with messenger-tagging IR spectroscopy.46 We make use of new CID capabilities of our SLIM module, which allow us to perform IMS-IMS experiments followed by cryogenic IR spectroscopy of the fragments. Ions produced by a nano-electrospray (nESI) source are transferred into the instrument and guided toward the ion mobility region using a dual-stage ion funnel assembly. In the SLIM-IMS device, the travelling wave potentials are created between electrodes of two mirrored printed circuit boards (PCBs) that propel ions through the N2 drift gas with a pressure maintained at 2.2 mbar. Once transferred onto the SLIM module, ions are accumulated in a two meter storage section47 from which short packets are released into the separation region, where ions follow a serpentine path. With a single-pass pathlength of 10 meters, we achieve a mobility resolving power of ∼200.44 The design of the boards allows routing the ions back to the entrance of the separation region for additional cycles, increasing the pathlength and hence the resolving power.44 After mobility separation, ions are sent through differential pumping stages into a cryogenic ion trap maintained at 45 K. Prior to the arrival of the incoming ions, a He![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N2 mixture (80:20) is pulsed into the trap to help confine and cool them. During this process, the ions form weakly bound clusters with one or two N2 molecules, which are used to perform messenger-tagging IR spectroscopy.46,48 The tagged ions are irradiated for 50 ms with a continuous wave, mid-IR, fiber-pumped laser (IPG Photonics), operated at 1 W output power with a linewidth of ∼1 cm−1. Photons are absorbed by the N2-tagged ions when the frequency of the incident infrared light is resonant with a vibrational transition of the molecule, leading to redistribution of vibrational energy and causing the N2 tag to detach. The ions are then extracted towards a commercial time-of-flight mass spectrometer (Tofwerk) to measure their mass spectrum. We obtain an IR spectrum by monitoring the depletion of N2-tagged molecules as a function of the laser wavenumber. All ions were studied in their singly sodiated charge state, and our spectral database was constructed for the sodiated species. Since we are trying to determine the glycan primary structure and not 3-dimensional structure, we are free to choose whatever charge state is most convenient.
:N2 mixture (80:20) is pulsed into the trap to help confine and cool them. During this process, the ions form weakly bound clusters with one or two N2 molecules, which are used to perform messenger-tagging IR spectroscopy.46,48 The tagged ions are irradiated for 50 ms with a continuous wave, mid-IR, fiber-pumped laser (IPG Photonics), operated at 1 W output power with a linewidth of ∼1 cm−1. Photons are absorbed by the N2-tagged ions when the frequency of the incident infrared light is resonant with a vibrational transition of the molecule, leading to redistribution of vibrational energy and causing the N2 tag to detach. The ions are then extracted towards a commercial time-of-flight mass spectrometer (Tofwerk) to measure their mass spectrum. We obtain an IR spectrum by monitoring the depletion of N2-tagged molecules as a function of the laser wavenumber. All ions were studied in their singly sodiated charge state, and our spectral database was constructed for the sodiated species. Since we are trying to determine the glycan primary structure and not 3-dimensional structure, we are free to choose whatever charge state is most convenient.
(IMS)n experiments
We recently reported the use of a SLIM-IMS device to perform (IMS)n type experiments, where a difference in DC voltage bias of an on-board trapping region with respect to the bias of the separation region enabled to perform collision induced dissociation (CID).45 In the newly designed SLIM-IMS device used here, an assembly consisting of two metallic grids (90% transmission), separated by a distance of 0.8 mm, was placed at the entrance of the CID trap to define a homogenous electric field of up to 3300 V cm−1 for efficient CID. After the bias voltage of the trapping region is raised to the level of the separation region, the ions can exit the trap and follow the serpentine path to either exit the SLIM device or undergo additional mobility separation. The arrival-time distributions (ATDs) of fragments that we report represent the total amount of time that ions reside on the SLIM device, including the drift time of the parent molecules, the time that ions spend inside the CID trap, and the subsequent drift time used to separate the fragments. The setup thus enables (IMS)n-type experiments followed by the acquisition of cryogenic IR spectra of mobility-separated fragments. Exemplary fragment mass spectra of mobility-separated G0-N ions are shown in Fig. S1.†Sample preparation
Isomerically pure Man-2 standards, shown in Fig. 1a, were purchased from Creative Biolabs. The G0-N glycan sample, specified to be a mixture of two positional isomers, was purchased from TheraProteins. The glycans G1 and G1F, also specified to be a mixture of two positional isomers, and G0, which does not present any positional isomers, were purchased from Dextra Laboratories. All samples were prepared in a 30/70 solution of acetonitrile/H2O to yield a concentration of 5–10 μM. | ||
| Fig. 1 (a) Structure of the glycans used in this work, using CFG notation. (b) Glycan fragment notation according to Domon and Costello.49 | ||
We use glycan notation issued by the Consortium for Functional Glycomics (CFG). The nomenclature of glycan fragments follows that of Domon and Costello, which is summarized in Fig. 1b.49
Results and discussion
Isomer identification and database construction
A schematic describing the approach used to identify positional isomers and to successively populate a database with IR fingerprints from newly identified isomers is shown in Fig. 2, using N-glycans as an example. We illustrate this approach starting from the two possible N-glycan positional isomers of Man-2, where the terminal mannose building block is bound with either an α-3 or α-6 glycosidic linkage to Man-1.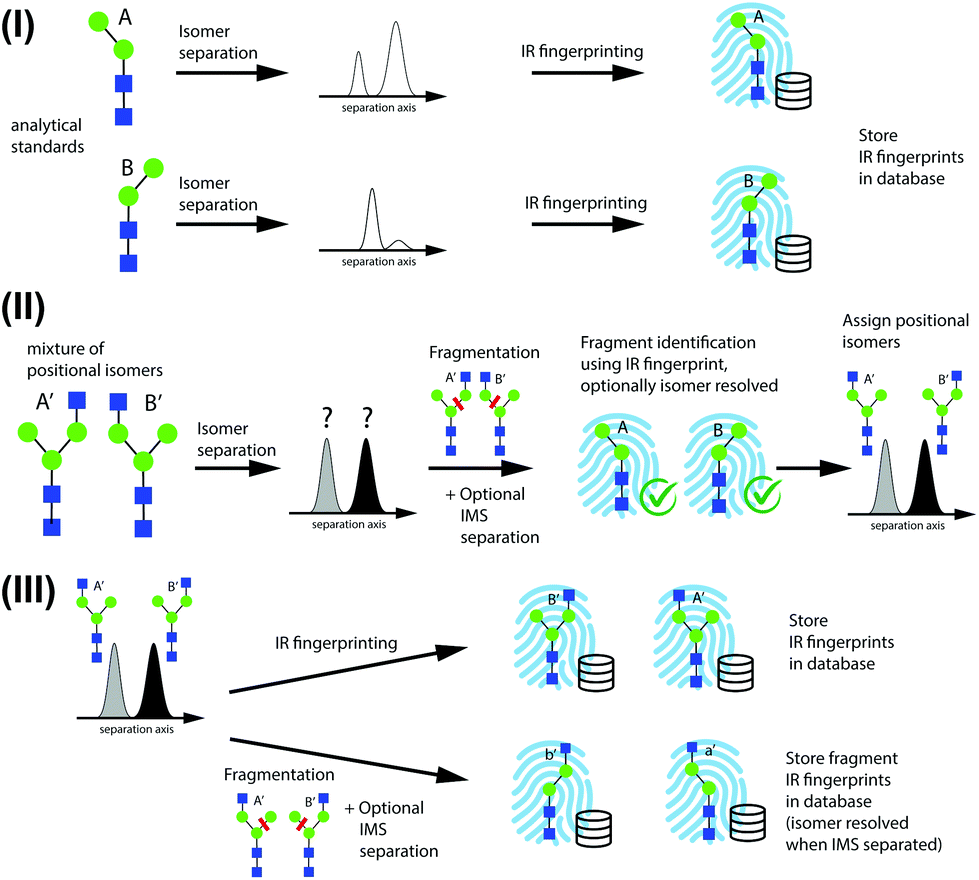 | ||
| Fig. 2 Scheme for identification of positional isomers and database construction, consisting of (I) initial database population using standards, (II) identification of positional isomers through identification of structurally diagnostic fragments, and (III) expansion of the database. | ||
In step I of Fig. 2, reference IR fingerprints of small analytical standards are recorded after IMS to separate α and β reducing-end anomers26,35,42 or coexisting gas-phase conformers. In step II, positional isomers are identified by first separating them by IMS and then subjecting them to CID to create structurally diagnostic fragments from each isomer peak. The diagnostic species for isomers A′ and B′ are labeled A and B, respectively. Fragments are then identified based on their IR fingerprint spectrum that has been recorded in step I, and the precursor positional isomers A′ and B′ can subsequently be assigned. Further IMS isomer separation of fragments might be necessary if the presence of positional fragment isomers cannot be excluded.
Once an isomer is identified based on the IR fingerprints of its fragments, its own IR fingerprint is recorded and stored in the database in step III. This serves two purposes: (1) in a subsequent encounter of this species, it can be directly identified by its IR fingerprint without the need to analyse its fragments; and (2) its fingerprint can potentially be used to identify structurally diagnostic fragments of larger positional isomers. Further in step III, isomers identified by the database can serve as precursor structures to create other fragments that were not accessible through fragmentation of smaller precursor species, such as species a′ and b′ in Fig. 2. It is important to note that these diagnostic fragments need not correspond to molecules that exist in solution. The proposed method can thus serve both to identify isomers of glycans and to construct an IR fingerprint database in a bottom-up fashion.
Identification of G0-N positional isomers
In the first part of this work, we aimed to identify two positional isomers of G0-N by applying steps I and II from the scheme presented in Fig. 2. The starting point was a commercially available glycan sample, labeled by the provider as a mixture of two positional isomers, G0-N(3) and G0-N(6), differing in the position of a terminal GlcNAc monosaccharide, which is either attached to the α-3 or the α-6 branch, respectively (see Fig. 1(a)). The isomeric fragments Y3α and Y3β (m/z 771) generated from G0-N upon CID are characteristic for the positional isomers G0-N(3) and G0-N(6), respectively, and correspond to the intact glycan Man-2, for which the two possible positional isomers Man-2(3) and Man-2(6) are readily available. Hence, IR fingerprints of these individual compounds were recorded after IMS separation to build the initial reference database (Fig. S2†). It is noteworthy that two IMS features could be separated for Man-2(3), while only one feature was observed for Man-2(6). The latter must therefore represent a mixture of the α and β reducing-end anomers, which coexist in solution.42An important assumption on which we base our identification scheme is that the dissociation of a single covalent bond is more likely to occur under the CID conditions applied here than the dissociation of two bonds. Therefore, only Y3α and Y3β fragments of G0-N were taken into consideration.
Figure 3(a) shows the arrival time distribution (ATD) of singly sodiated G0-N ions (m/z 1136) obtained from the commercial sample, where two distinct peaks were observed after one mobility-separation cycle (10 m drift path). While it might be tempting to assume that these two drift peaks correspond to the two positional isomers of G0-N, they might also be a result of the two reducing-end anomers for one of the species42 or different conformational states of the molecules.
 | ||
| Fig. 3 (a) ATD of G0-N after mobility separation (one cycle). (b) Cryogenic IR fingerprint spectra of the diagnostic fragments (m/z 771) generated from each peak in the ATD of G0-N (tan). Synthetic spectrum (orange) generated using 40% and 60% contribution of spectra from peak 1 and peak 2, respectively, compared with the database fingerprint spectrum of Man-2(6) (grey). (c) G0-N and the assigned fragment and parent structures. | ||
Structurally diagnostic fragment ions (m/z 771) corresponding to the Man-2 species in the initial database were generated upon CID of each mobility-selected drift peak, and their resulting IR fingerprints are depicted in Fig. 3(b) (tan). The two fingerprint spectra are strikingly similar in position and intensity of the resolved absorption bands, with the only difference being the precise position of three bands in a wavenumber region corresponding to free OH-stretching vibrations (3620 cm−1–3670 cm−1). While both spectra have similarities with the database spectrum of Man-2(6), neither of them poses a perfect match that would confirm its presence. On the other hand, the similarity of the fingerprint spectra can be an indicator that these species represent the two reducing-end anomers42,50 that were not separated for the Man-2(6) standard. Under this assumption, we prepared a synthetic mixture in a 40/60 ratio of the two fragment IR fingerprints, corresponding to the relative intensities of the drift peaks in the ATD in Fig. 3(a). The resulting spectrum represents an extremely good match to the database fingerprint of Man-2(6), depicted in orange and grey, respectively, in Fig. 3(b). By assigning fragments from both mobility features of G0-N to Man-2(6), the content of the analysed sample was determined to be constituted solely by one positional isomer of G0-N, namely G0-N(3), despite the specifications from the supplier. Consequently, the two drift peaks observed for G0-N are likely a result of the two reducing-end anomers.42Figure 3(c) summarizes the identification procedure.
In accordance with step III in the scheme of Fig. 2, we recorded the IR fingerprint spectra of both G0-N(3) species (Fig. S3†), which can then be used for its identification in complex samples without having to analyse its fragments again. One should also conclude from these results that sample description from suppliers for subtly different isomers should be cautiously evaluated.
To obtain an IR fingerprint of the other positional isomer, G0-N(6), we applied our CID scheme to the glycan G0, which should yield both positional isomers of G0-N as fragments (Fig. 4(a)). Lacking any positional isomers, singly sodiated G0 ions were fragmented without prior ion-mobility separation. Fragments with m/z 1136, generated upon the loss of a GlcNAc monosaccharide, can exist in three isomeric forms Y4α, Y4β, and C4, where Y4α and Y4β correspond to G0-N(6) and G0-N(3), respectively (Fig. 4(a)). Following fragmentation of G0, the m/z 1136 fragments underwent one additional cycle of mobility separation. The resulting ATD, which features three well-separated peaks, is displayed in Fig. 4(b). The IR spectra of all three peaks are shown in Fig. 4(c), where they are paired with their best matching database fingerprint (displayed in grey, where applicable). A simple visual comparison of spectra confirms the assignment of the first two peaks in the ATD to the two isomers of G0-N(3) (i.e. fragment Y4β) for which we previously obtained the reference spectrum shown in Fig. S3.† The glycan corresponding to the C4 fragment is available commercially, and its IR fingerprint was measured and added to the database after mobility separation, however it does not correspond to that of the third mobility species in the fragment ATD (compare the bottom two spectra in Fig. 4(c)). Moreover, the molecule corresponding to the C4 fragment has an arrival time in between that of the second and third drift peak in the fragment ATD of Fig. 4(b) (Fig. S4†). We can therefore, by exclusion, assign the third drift peak and its spectrum in Fig. 4(c) to the Y4α fragment, which corresponds to the glycan G0-N(6), and store its fingerprint in the database to be used either for its future identification or to identify a structurally diagnostic fragment of a larger glycan. It should be noted that the spectrum of this isomer represents a mixture of both reducing-end anomers.
 | ||
| Fig. 4 (a) Structure and possible structurally diagnostic fragments of G0. (b) ATD of m/z 1136 fragments with regions subjected to IR spectroscopy highlighted. (c) Cryogenic IR spectra of mobility-separated drift peaks according to the colour scheme in the ATD, paired with their best-matching database spectra (grey) where possible. | ||
Database extension through identification of G1 positional isomers
The terminal galactose in the N-glycan G1 can either be linked to the α-3 or the α-6 branch, leading to the positional isomers G1(3) and G1(6), respectively. The strategy for identification of these isomers follows step (B) of the scheme presented in Fig. 2: the fragments Y4α and Y4β (m/z 1136) represent structurally diagnostic species for the isomers G1(3) and G1(6), respectively, and correspond to glycans G0-N(6) and G0-N(3), for which database fingerprints were acquired as described above.Isomer separation of singly sodiated G1 ions was performed prior to fragmentation, and the m/z 1136 fragments were sent through one additional cycle of ion mobility separation. An ATD of G1 after one separation cycle is shown in Fig. 5(a), where the drift-time windows selected for CID are highlighted. The ATD of the fragments generated from the first mobility peak of the G1 ions (blue), exhibits two features, whereas the fragment ATD generated from the second mobility region (pink) shows a single peak at longer drift times.
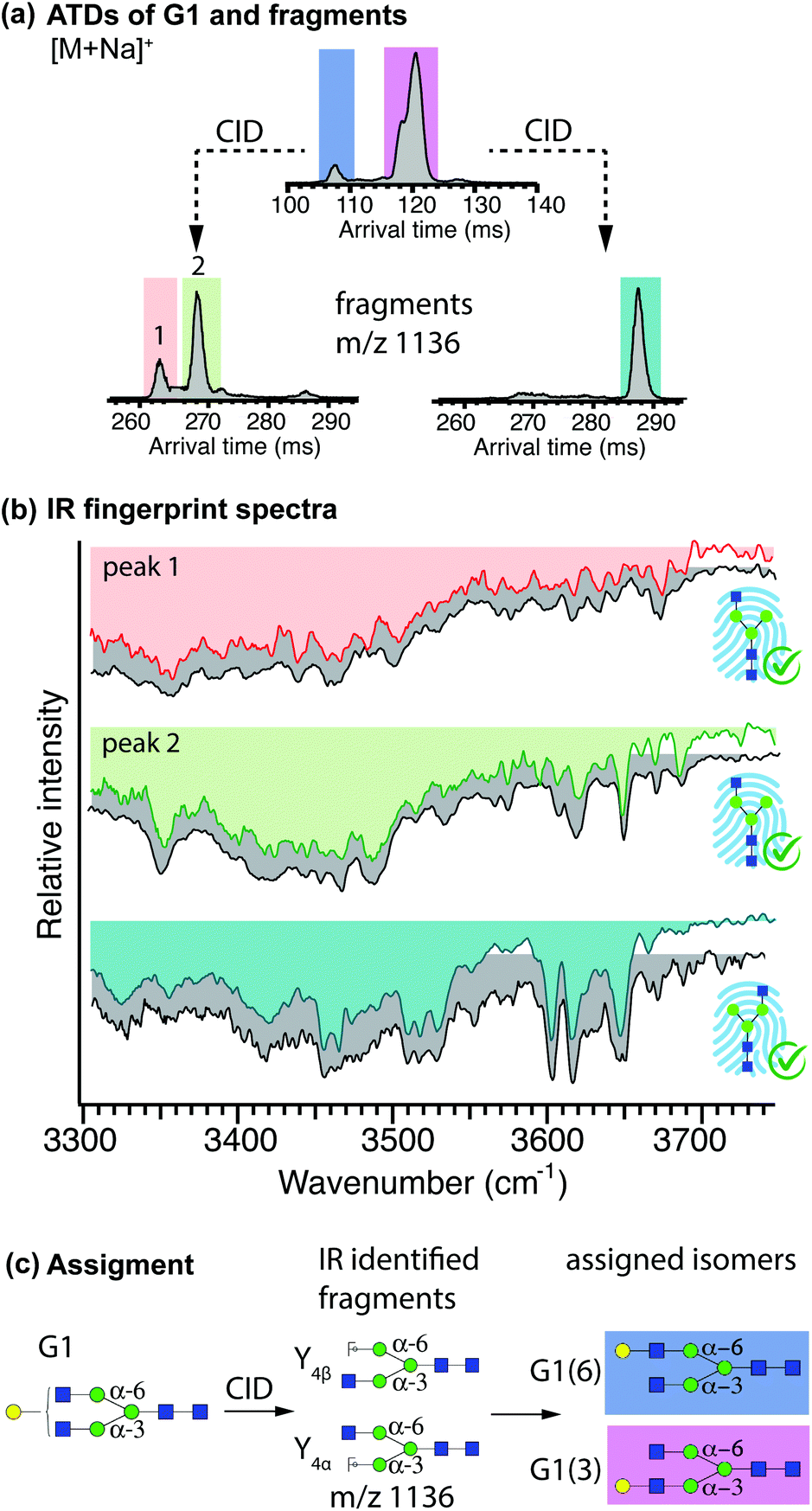 | ||
| Fig. 5 (a) ATD of G1 after one separation cycle. The regions that were subjected to CID followed by one more separation cycle are highlighted and the ATDs of the m/z 1136 fragments are shown below. (b) Cryogenic IR fingerprints of mobility-separated drift peaks of mobility-separated Y fragments of G1 (in colour), paired with the best-matching IR fingerprints from the database (grey). (c) Structure of G1, assigned fragments, and assigned parent structures (colour code as in ATDs). | ||
The IR spectra of all individual peaks in the fragment ATDs were recorded and compared to best-matching database IR fingerprints (Fig. 5(b)). Visual comparison confirms that both fragments generated from the first mobility peak (peach and green) correspond to the two species of G0-N(3) for which two isomers (presumably the reducing-end anomers) were added to the database, whereas fragments generated from the second mobility region (cyan) correspond to the database entry of G0-N(6). Based on the identification of these structurally diagnostic fragments, we can assign the first and second mobility region in the ATD of G1 to the positional isomers G1(6) and G1(3), respectively. The procedure is summarized in Fig. 5(c).
Spectra for both positional isomers G1(3) and G1(6) were recorded (Fig. S5†) and added to the IR fingerprint database and can be used for subsequent identification of G1 positional isomers or as structurally diagnostic fragments of yet larger glycans. It should be noted that under the separation conditions used here, the reducing-end anomers of G1 are completely unresolved for the positional isomers G1(6) and partially resolved for G1(3). However, given the partial separation observed for the latter, a few more cycles would be necessary to fully resolve these anomers, which might be useful if anomer-resolved reference spectra are required for fragment identification.
Further database extension through identification of G1F positional isomers
The terminal galactose in the N-glycan G1F can either be linked to the α-3 or the α-6 branch, leading to the positional isomers G1F(3) and G1F(6), respectively. The only possible isomeric fragments with m/z 1501, generated upon the loss of the fucose monosaccharide, corresponds to previously identified G1(3) and G1(6), which can be used as structurally diagnostic fragments for identification of G1F(3) and G1F(6), respectively.In accordance with the scheme described in Fig. 2, ion-mobility separation of G1F was performed prior to fragmentation. Figure 6(a) shows the ATD of singly sodiated G1F ions after four separation cycles and the drift-time windows selected for fragmentation are highlighted in different colours (red and purple). Following fragmentation, structurally diagnostic fragments (m/z 1501) underwent one additional cycle of mobility separation. The ATD of fragments generated from first two mobility peaks of G1F taken together (red), displays a single peak with a hint of an unresolved shoulder on its left. The ATDs obtained after fragmenting each of the parent drift peaks separately confirms the presence of two overlapping features (see Fig. S6†). The resulting fragment ATD from the second mobility region of G1F (purple) exhibits two partially resolved peaks.
 | ||
| Fig. 6 (a) ATD of G1F after four separation cycles. The regions that were subjected to CID followed by one more separation cycle are highlighted, and the ATDs of the m/z 1501 fragments are shown below. (b) Cryogenic IR spectra of the diagnostic fragments (m/z 1501) generated from the drift time windows highlighted in the ATD of G1F, paired with the best-matching IR fingerprints from the database (grey). (c) Structure of G1F, assigned fragments, and assigned parent structures (colour code as in ATDs). | ||
The IR spectra of the fragments are shown in Fig. 6(b), where they are paired with their best matching database fingerprint (displayed in grey), which we obtained as described in the previous section (see Fig. S5†). Visual comparison confirms that the spectrum of fragments (pink) generated from the first region in the ATD of G1F corresponds to G1(3) and the fragment (blue) generated from the second region in the ATD of G1F corresponds to G1(6). This identifies the first two peaks in the ATD of G1F as belonging to the G1F(3) positional isomer, and the broader ATD peak arriving at longer drift time to the G1F(6) positional isomer. The assignment procedure is summarized in Fig. 6(c).
We recorded the IR fingerprints of the newly identified positional isomers of G1F (Fig. S7†), which can now be used either for their identification in complex mixtures or as structurally diagnostic fragment for yet larger glycans. It is important to note that the spectra for first two peaks, assigned as G1F(3), are very similar in their band positions and intensities, with the only noticeable difference being the bands at 3660 cm−1 and 3655 cm−1. This furthermore supports the hypothesis that these two drift peaks likely correspond to the two reducing-end anomers of G1F(3). Under the separation conditions used here, the broad peak assigned to be the G1F(6) isomer could not be fully resolved, and therefore the resulting IR spectrum represents a mixture of both reducing-end anomers.
Conclusions
We present here a new approach for the identification of glycan positional isomers using a combination of high-resolution IMS-IMS and cryogenic IR fingerprint spectroscopy of structurally diagnostic fragments. Starting with only two pure analytical standards, Man-2(3) and Man-2(6), we were able to first identify and then record reference IR fingerprints of the positional isomers of the glycan G0-N. We subsequently used these newly obtained fingerprint spectra to identify positional isomers of the glycan G1, which we then used to identify the positional isomers of G1F. With this example, we demonstrate how the proposed workflow could be used to successively build a database including IR fingerprint spectra without the need for isomerically pure glycan standards for larger species, which are often expensive to produce or simply not available. We anticipate that this approach could be extended to different classes of glycans, such as human-milk oligosaccharides (HMOs) or O-glycans. It is important to note that this approach does not require complex fragment-based analysis to determine the sequence and branching of a glycan but simply a comparison of experimental IR fingerprints with those in a database, which can be implemented in an algorithm as we have recently demonstrated.44 In combination with a user-friendly software that facilitates automated IR comparison, and our latest advancements in acquisition speed and robustness,44 we believe that cryogenic IR fingerprinting and the identification workflow proposed here has the potential to become an important tool in the field of glycan analysis.Author contributions
PB, AB, SW and TRR designed the project. All authors were involved in editing/revising the manuscript. TRR supervised the project and acquired the funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the European Research Council (Grant 788697-GLYCANAL) and the Swiss National Science Foundation (Grants 200020_184838 and 206021_177004) for their generous support of this work.References
- A. Varki, Glycobiology, 2017, 27, 3–49 CrossRef CAS PubMed.
- R. D. Cummings and J. M. Pierce, Chem. Biol., 2014, 21, 1–15 CrossRef CAS PubMed.
- R. A. Dwek, Chem. Rev., 1996, 96, 683–720 CrossRef CAS PubMed.
- E. Walker-Nasir, A. Kaleem, D. C. Hoessli, A. Khurshid and Nasir-ud-Din, Curr. Org. Chem., 2008, 12, 940–956 CrossRef CAS.
- R. B. Parekh, R. A. Dwek, B. J. Sutton, D. L. Fernandes, A. Leung, D. Stanworth, T. W. Rademacher, T. Mizuochi, T. Taniguchi, K. Matsuta, F. Takeuchi, Y. Nagano, T. Miyamoto and A. Kobata, Nature, 1985, 316, 452–457 CrossRef CAS PubMed.
- R. Parekh, D. Isenberg, B. Ansell, I. Roitt, R. Dwek and T. Rademacher, Lancet, 1988, 331, 966–969 CrossRef.
- M. Aoyama, N. Hashii, W. Tsukimura, K. Osumi, A. Harazono, M. Tada, M. Kiyoshi, A. Matsuda and A. Ishii-Watabe, mAbs, 2019, 11, 826–836 CrossRef CAS PubMed.
- J. F. G. Vliegenthart, L. Dorland and H. V. Halbeek, Adv. Carbohydr. Chem. Biochem., 1983, 41, 209–374 CrossRef CAS.
- M. R. Wormald, A. J. Petrescu, Y.-L. Pao, A. Glithero, T. Elliott and R. A. Dwek, Chem. Rev., 2002, 102, 371–386 CrossRef CAS PubMed.
- M. D. Battistel, H. F. Azurmendi, B. Yu and D. I. Freedberg, Prog. Nucl. Magn. Reson. Spectrosc., 2014, 79, 48–68 CrossRef CAS PubMed.
- J. Zaia, Mass Spectrom. Rev., 2004, 23, 161–227 CrossRef CAS PubMed.
- D. J. Harvey, J. Am. Soc. Mass Spectrom., 2005, 16, 647–659 CrossRef CAS PubMed.
- V. N. Reinhold, B. B. Reinhold and C. E. Costello, Anal. Chem., 1995, 67, 1772–1784 CrossRef CAS PubMed.
- D. J. Harvey, Proteomics, 2005, 5, 1774–1786 CrossRef CAS PubMed.
- Y. Liu, O. Salas-Solano and L. A. Gennaro, Anal. Chem., 2009, 81, 6823–6829 CrossRef CAS PubMed.
- J. Zhao, S. Li, C. Li, S.-L. Wu, W. Xu, Y. Chen, M. Shameem, D. Richardson and H. Li, Anal. Chem., 2016, 88, 7049–7059 CrossRef CAS PubMed.
- C. Ashwood, C.-H. Lin, M. Thaysen-Andersen and N. H. Packer, J. Am. Soc. Mass Spectrom., 2018, 29, 1194–1209 CrossRef CAS PubMed.
- S. Zhou, Y. Huang, X. Dong, W. Peng, L. Veillon, D. A. S. Kitagawa, A. J. A. Aquino and Y. Mechref, Anal. Chem., 2017, 89, 6590–6597 CrossRef CAS PubMed.
- Y.-M. She, R. Y. Tam, X. Li, M. Rosu-Myles and S. Sauvé, Anal. Chem., 2020, 92, 14038–14046 CrossRef CAS PubMed.
- J. Wei, Y. Tang, Y. Bai, J. Zaia, C. E. Costello, P. Hong and C. Lin, Anal. Chem., 2020, 92, 782–791 CrossRef CAS PubMed.
- L. Veillon, Y. Huang, W. Peng, X. Dong, B. G. Cho and Y. Mechref, Electrophoresis, 2017, 38, 2100–2014 CrossRef CAS PubMed.
- P. Both, A. P. Green, C. J. Gray, R. Šardzík, J. Voglmeir, C. Fontana, M. Austeri, M. Rejzek, D. Richardson, R. A. Field, G. Widmalm, S. L. Flitsch and C. E. Eyers, Nat. Chem., 2014, 6, 65–74 CrossRef CAS PubMed.
- M. M. Gaye, R. Kurulugama and D. E. Clemmer, Analyst, 2015, 140, 6922–6932 RSC.
- E. G. Pallister, M. S. F. Choo, I. Walsh, J. N. Tai, S. J. Tay, Y. S. Yang, S. K. Ng, P. M. Rudd, S. L. Flitsch and T. Nguyen-Khuong, Anal. Chem., 2020, 92, 15323–15335 CrossRef CAS PubMed.
- J. Wei, Y. Tang, M. E. Ridgeway, M. A. Park, C. E. Costello and C. Lin, Anal. Chem., 2020, 92, 13211–13220 CrossRef CAS PubMed.
- J. Ujma, D. Ropartz, K. Giles, K. Richardson, D. Langridge, J. Wildgoose, M. Green and S. Pringle, J. Am. Soc. Mass Spectrom., 2019, 30, 1028–1037 CrossRef CAS PubMed.
- D. J. Harvey, C. A. Scarff, M. Edgeworth, K. Pagel, K. Thalassinos, W. B. Struwe, M. Crispin and J. H. Scrivens, J. Mass Spectrom., 2016, 51, 1064–1079 CrossRef CAS PubMed.
- D. J. Harvey, G. E. Seabright, S. Vasiljevic, M. Crispin and W. B. Struwe, J. Am. Soc. Mass Spectrom., 2018, 29, 972–988 CrossRef CAS PubMed.
- J. Hofmann and K. Pagel, Angew. Chem., Int. Ed., 2017, 56, 8342–8349 CrossRef CAS PubMed.
- Y. Pu, M. E. Ridgeway, R. S. Glaskin, M. A. Park, C. E. Costello and C. Lin, Anal. Chem., 2016, 88, 3440–3443 CrossRef CAS PubMed.
- C. J. Gray, B. Thomas, R. Upton, L. G. Migas, C. E. Eyers, P. E. Barran and S. L. Flitsch, Biochim. Biophys. Acta, Gen. Subj., 2016, 1860, 1688–1709 CrossRef CAS PubMed.
- T. L. Peterson and G. Nagy, Anal. Chem., 2021, 93, 9397–9407 CrossRef PubMed.
- A. M. Hamid, S. V. B. Garimella, Y. M. Ibrahim, L. Deng, X. Zheng, I. K. Webb, G. A. Anderson, S. A. Prost, R. V. Norheim, A. V. Tolmachev, E. S. Baker and R. D. Smith, Anal. Chem., 2016, 88, 8949–8956 CrossRef CAS PubMed.
- L. Deng, I. K. Webb, S. V. B. Garimella, A. M. Hamid, X. Zheng, R. V. Norheim, S. A. Prost, G. A. Anderson, J. A. Sandoval, E. S. Baker, Y. M. Ibrahim and R. D. Smith, Anal. Chem., 2017, 89, 4628–4634 CrossRef CAS PubMed.
- G. Nagy, I. K. Attah, S. V. B. Garimella, K. Tang, Y. M. Ibrahim, E. S. Baker and R. D. Smith, Chem. Commun., 2018, 54, 11701–11704 RSC.
- J. C. May, K. L. Leaptrot, B. S. Rose, K. L. W. Moser, L. Deng, L. Maxon, D. DeBord and J. A. McLean, J. Am. Soc. Mass Spectrom., 2021, 32, 1126–1137 CrossRef CAS PubMed.
- I. Dyukova, A. Ben Faleh, S. Warnke, N. Yalovenko, V. Yatsyna, P. Bansal and T. R. Rizzo, Analyst, 2021, 146, 4789–4795 RSC.
- C. Masellis, N. Khanal, M. Z. Kamrath, D. E. Clemmer and T. R. Rizzo, J. Am. Soc. Mass Spectrom., 2017, 28, 2217–2222 CrossRef CAS PubMed.
- N. Khanal, C. Masellis, M. Z. Kamrath, D. E. Clemmer and T. R. Rizzo, Analyst, 2018, 143, 1846–1852 RSC.
- N. Khanal, C. Masellis, M. Z. Kamrath, D. E. Clemmer and T. R. Rizzo, Anal. Chem., 2017, 89, 7601–7606 CrossRef CAS PubMed.
- A. Ben Faleh, S. Warnke and T. R. Rizzo, Anal. Chem., 2019, 91, 4876–4882 CrossRef CAS PubMed.
- S. Warnke, A. Ben Faleh, V. Scutelnic and T. R. Rizzo, J. Am. Soc. Mass Spectrom., 2019, 30, 2204–2211 CrossRef CAS PubMed.
- N. Yalovenko, V. Yatsyna, P. Bansal, A. H. AbiKhodr and T. R. Rizzo, Analyst, 2020, 145, 6493–6499 RSC.
- S. Warnke, A. Ben Faleh and T. R. Rizzo, ACS Meas. Sci. Au, 2021, 1, 157–164 CrossRef CAS PubMed.
- P. Bansal, V. Yatsyna, A. H. AbiKhodr, S. Warnke, A. Ben Faleh, N. Yalovenko, V. H. Wysocki and T. R. Rizzo, Anal. Chem., 2020, 92, 9079–9085 CrossRef CAS PubMed.
- A. B. Wolk, C. M. Leavitt, E. Garand and M. A. Johnson, Acc. Chem. Res., 2014, 47, 202–210 CrossRef CAS PubMed.
- A. Li, G. Nagy, C. R. Conant, R. V. Norheim, J. Y. Lee, C. Giberson, A. L. Hollerbach, V. Prabhakaran, I. K. Attah, C. D. Chouinard, A. Prabhakaran, R. D. Smith, Y. M. Ibrahim and S. V. B. Garimella, Anal. Chem., 2020, 92, 14930–14938 CrossRef CAS PubMed.
- M. Z. Kamrath, E. Garand, P. A. Jordan, C. M. Leavitt, A. B. Wolk, M. J. Van Stipdonk, S. J. Miller and M. A. Johnson, J. Am. Chem. Soc., 2011, 133, 6440–6448 CrossRef CAS PubMed.
- B. Domon and C. E. Costello, Glycoconjugate J., 1988, 5, 397–409 CrossRef CAS.
- S. Warnke, A. B. Faleh, R. P. Pellegrinelli, N. Yalovenko and T. R. Rizzo, Faraday Discuss., 2019, 217, 114–125 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1an01861b |
| This journal is © The Royal Society of Chemistry 2022 |