
Cell-SELEX-based selection of ssDNA aptamers for specifically targeting BRAF V600E-mutated melanoma
Wanming
Li
a,
Tao
Bing
 b,
Rui
Wang
c,
Sihan
Jin
a,
Dihua
Shangguan
*b and
Hang
Chen
*d
b,
Rui
Wang
c,
Sihan
Jin
a,
Dihua
Shangguan
*b and
Hang
Chen
*d
aDepartment of Cell Biology, Key Laboratory of Cell Biology, Ministry of Public Health, and Key Laboratory of Medical Cell Biology, Ministry of Education, China Medical University, Shenyang North New Area, Shenyang 110122, Liaoning, China
bBeijing National Laboratory for Molecular Sciences, Key Laboratory of Analytical Chemistry for Living Biosystems, CAS Research/Education Center for Excellence in Molecular Sciences, institute of chemistry, Chinese Academy of Sciences, 100190, Beijing, China. E-mail: sgdh@iccas.ac.cn
cDepartment of Stem Cells and Regenerative Medicine, Key Laboratory of Cell Biology, Ministry of Public Health of China, and Key Laboratory of Medical Cell Biology, Ministry of Education of China, China Medical University, Shenyang North New Area, Shenyang 110122, Liaoning, China
dExperiment Teaching Center of Functional Subjects, College of Basic Medicine, China Medical University, No.77 Puhe Road, Shenyang North New Area, Shenyang 110122, Liaoning, China. E-mail: lnfsch@hotmail.com; Tel: +86-024-31939079
First published on 16th November 2021
Abstract
Malignant melanoma is regarded as the most aggressive form of skin cancer, and is responsible for most death caused by skin cancer. BRAF mutations occur in approximately 40–50% of melanomas, with V600E being the most common mutation. Testing for BRAF mutations and targeted therapy have markedly improved long-term survival for patients with BRAF-mutated melanoma. Here, we report two aptamers, CH1 and CH5 generated by Cell-SELEX, against BRAF V600E-mutated human melanoma cells A375. The two aptamers exhibited high affinity to target cells with low dissociation constants (Kd) in the nanomolar range. Furthermore, the binding of two aptamers to target cells was independent of incubation temperature, and their molecular targets were demonstrated to be membrane proteins on the cell surface. We also demonstrated that aptamer CH1 was able to bind to metastatic colorectal cancer cells harboring BRAF V600E mutation, indicating a relationship between aptamer CH1 and BRAF V600E-related metastatic cancer. Owing to the structure stability and high selectivity to BRAF V600E-mutated targeting cells, aptamer CH1 holds great potential as a molecular probe for the detection of BRAF V600E-mutated metastatic melanoma.
Introduction
Malignant melanoma is a highly aggressive skin cancer, comprising 90% of all deaths from skin cancers.1 It is “curable” at the early stages by surgical resection.2,3 However, after metastases, prognosis is poor and the 5-year survival is less than 10%.4 Approximately 50% of melanoma patients harbor a BRAF mutation, with V600E being the most common mutation, and genetic abnormality has a strong correlation with the cancer development and prognosis.5,6BRAF-mutated melanoma tends to exhibit more likely remote metastases than BRAF wild-type (WT) melanoma, and has been linked to decreased overall survival.7 Targeted therapies utilizing small molecule drugs are rapid developing for precision treatment of cancer, and BRAF inhibitors were approved by the FDA (Food and Drug Administration) as first or second-line for the treatment of metastatic melanoma patients with BRAF V600E mutation.8,9 Although BRAF-targeted therapy produced marked tumor response in most patients, these responses were usually limited by drug resistance within 5–7 months.10,11 Given these features, it is crucial to early determine whether melanoma patients have tumors harboring BRAF mutations. Currently, BRAF mutational tests include DNA-based sequencing methods or antibody-based VE1 immunostaining.12,13 However, test results can be challenged due to discordance in the BRAF mutational status between primary and metastatic sites.14 Therefore, developing molecular biomarker-based techniques for the detection of BRAF-mutated melanoma cells is still needed.Aptamers are short single-stranded DNA (ssDNA) or RNA oligonucleotides, and generated from random oligonucleotide pools by a selection process known as Systematic Evolution of Ligands by Exponential Enrichment (SELEX) against various targets, such as small organic molecules, protein, and cells.15–18 Selection using living cells as targets (Cell-SELEX) is able to isolate aptamers that can bind specifically to molecules on cell surfaces in their native conformation.19,20 Furthermore, with the application of the additional subtractive step, a cancer cell line is used as the target, and other cancer or normal cell lines as the subtractive group, and as a result the selected aptamers can differentiate phenotypes between target cells and subtractive cells. A variety of molecular probes to detect tumor cells have been identified using this approach, including leukemia,21 ovarian cancer,22 gastric cancer,23 and glioblastoma.24 Moreover, these aptamers can be functionalized for various applications, including drug delivering,25 molecular imaging,26 and cell capture.27
To date, several aptamers have been reported against some proteins related to melanoma, such as aptamers against RAGE,28 CD20,29 and nucleolin.30 Though these aptamers can inhibit the proliferation of melanoma cells, their targeting proteins are expressed in many different types of cancer cells, and the specificity of aptamers was low. Recently, Liu et al. have reported an aptamer LL4A that could recognize vemurafenib-resistant melanoma, and CD63 was identified as its target protein.31 However, no aptamer was generated to specifically recognize melanoma harboring BRAF mutation by Cell-SELEX.
Here, we used the BRAF-mutated melanoma cell line A375 as the target cells and the BRAF wild-type squamous carcinoma cell line A431 as the negative cells to perform Cell-SELEX. After sequence optimization, two aptamers, CH1 and CH5 were identified that specifically bind to A375 cells with high specificity and affinity. Furthermore, the effects of proteinases and incubation temperature on binding of aptamers to target cells have also been investigated. Additionally, we demonstrated that one of the selected aptamers, CH1 could specifically target the tumors harboring BRAF V600E mutation, indicating that CH1 holds the potential as a molecular probe for the detection of BRAF V600E mutated cancer cells.
Materials and methods
Cell lines and buffers
The human malignant melanoma cell line A375 (BRAF V600E) and human squamous carcinoma cell line A431 (BRAF WT) were used for the selection. The other cell lines used in this study include the human colorectal cancer cell lines HCT116, RKO, COLO 205, HCT-8 and CL187, the human melanoma cell lines SK-MEL-28 and SK-MEL-2, the Chinese hamster ovary cell line CHO, and the human embryonic kidney cell line HEK293. A375, A431, HCT116, RKO, COLO 205, HCT-8 and CHO were purchased from the Cell Bank of the Chinese Academy of Sciences. CCL187 was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). For cell culture, A375, A431 and HEK293 were cultured in a basic medium of high-glucose Dulbecco's Modified Eagle's Medium (DMEM) (GIBCO), and the other cells in PRMI1640 (GIBCO). All the basic media were supplemented with 10% fetal bovine serum (FBS, GIBCO) and 100 units per mL penicillin–streptomycin (GIBCO). All the cells were routinely cultured at 37 °C under a 5% CO2 atmosphere. Washing buffer (WB) was prepared from Dulbecco's phosphate buffered saline (PBS) with additional glucose (4.5 g L−1). The binding buffer (BB) was prepared by adding salmon sperm DNA (0.1 mg mL−1), BSA (1 mg mL−1) and MgCl2 (1 mM) to the washing buffer.ssDNA library and primers
An initial library containing two flanking regions of 20-nucleotide sequences for primer hybridization and 45-nucleotide randomized sequences (5′-ACGCTCGGATGCCACTACAG-N45-CTCATGGACGTGCTGGTGAC-3′) was synthesized by Sangon Biotechnology Co., Ltd (Shanghai, China), where N represents a randomized nucleotide of either A, G, C, or T. The fluorescein (FAM)-labeled forward primer (5′-FAM-ACGCTCGGATGCCACTACAG-3′) and the biotin-labeled reverse primer (5′-Bio-GTCACCAGCACGTCCATGAG-3′) were used for PCR amplification of the DNA library or to separate the ssDNA by streptavidin-coated Sepharose beads (GE Healthcare, USA). The forward primer was used to monitor the progress of selection by flow cytometry (BD, USA).Procedure of Cell-SELEX
The Cell-SELEX procedure was performed as previously described.32 An initial ssDNA (10 nmol) library was first dissolved in 1 mL of BB. After annealing, the library was incubated with target cells (1 × 107) at 4 °C for 60 min. After incubation, the cells were washed with WB to remove the unbound ssDNA sequences, scraped from cell dishes, and re-suspended in Dnase-free deionized water. The bound ssDNAs were eluted by heating at 95 °C for 10 min. After being eluted from target cells, the bound ssDNAs were amplified by PCR using biotin and FAM-labeled primers. Streptavidin-coated Sepharose beads (GE Healthcare, USA) were used to isolate FAM-labeled ssDNAs from PCR products. After desalting using a NAP-5 column (GE Healthcare, USA), the FAM-labeled ssDNAs were collected as the ssDNA library for the next round of selection.For the second and subsequent rounds, the annealed ssDNA pool was firstly incubated with negative control cells (A431 cells), and the A431-unbound ssDNAs were collected and incubated with the target-A375 cells for positive selection. The selection pressure was gradually increased by decreasing the number of A375 cells (from approximately 107 to 105), decreasing the amount of the ssDNA pool (from 10 nmol to 50 pmol), decreasing the incubation time with target cells (from 60 min to 20 min), increasing the proportion of FBS in BB (from 0% to 30%), and increasing the washing strength. After 11 rounds of selection, the enriched ssDNA pool was subjected to high-throughput sequencing using Illumina MiSeq by Sangon Biotech (Shanghai, China).
Flow cytometry analysis
Target and negative cells were cultured in 100 mm culture dishes to 80–90% confluence. Cells were washed with PBS and were collected and suspended in PBS. Then, A375 cells and negative A431 cells were respectively incubated with 250 nM FAM-labeled selected ssDNA pools in 200 μL of BB at 4 °C for 30 min to monitor the enrichment of Cell-SELEX. After incubation, the cells were washed and analyzed using a BD FACSCalibur flow cytometer by exciting the sample at 488 nm. The FAM-labeled initial library was used as the negative control. In the same way, each cell line was incubated with the FAM-labeled selected aptamers to analyze the binding specificity of aptamers using flow cytometry. All experiments were repeated three times independently.Affinity analysis
To assess the affinity of the aptamer to target A375 cells, we measured the Kd value of the selected aptamers. A375 cells were incubated with increasing concentrations of FAM-labeled aptamers in BB at 4 °C for 30 min, and the fluorescence intensity was then analyzed by flow cytometry. The ssDNA library was used as the negative control. The Kd value of each aptamer was obtained using Sigma plot 10.0 software, according to the following equation Y = Bmax × X/(Kd + X), where Bmax is the saturated binding, Y is the fluorescence intensity and X is the concentration of aptamers. All experiments were performed in triplicate.Effect of proteinase and temperature on aptamer binding
A375 cells (1 × 106) were dissociated with 0.02% ethylenediaminetetraacetic acid (EDTA), 0.25% trypsin, or 0.1 mg/Ml proteinase K at 37 °C for 10 min, respectively. After adding FBS to terminate the digestion, the treated cells were washed, and incubated with the FAM-labeled aptamer in BB at 4 °C for 30 min, and then analyzed by flow cytometry as described above. To investigate whether the temperature will affect the binding ability of aptamers, A375 cells were digested with 0.02% EDTA, and incubated with FAM-labeled aptamers in BB for 30 min at 4 °C, 25 °C or 37 °C, respectively. The fluorescence intensities of aptamers were measured using flow cytometry. All experiments were repeated three times independently.Fluorescence microscopy analysis
The cells were cultured in a chamber overnight. After washing twice with WB, the cells were incubated with 250 nM FAM-labeled aptamers or ssDNA library at 4 °C for 1 h. After washing, the cells were fixed with 4% formaldehyde for 15 min, and stained with DAPI for 5 min to counterstain the nucleus. Fluorescence imaging was performed using an Olympus IX51 fluorescence microscope (Olympus, Japan).Statistical analysis
All experiments were repeated at least three times, and data are presented as the mean ± standard deviation. Student's two-tailed t-test was used to analyse the results and a p value <0.05 was considered statistically significant. The statistical analyses were performed using GraphPad Prism software.Results and discussion
Selection and enrichment of aptamers against A375 cells
The BRAF V600E mutation is the most common genetic mutation in melanoma. In order to generate aptamers specifically targeting melanoma harboring BRAF V600E mutation, we performed Cell-SELEX using a human malignant melanoma cell line A375 carrying the BRAF V600E mutation as target cells. The human squamous carcinoma cell line A431 with BRAF WT was introduced for the negative selection from the 2nd round. The cell-SELEX process is illustrated in Scheme 1. The ssDNA pool was firstly incubated with A431 cells for a negative selection, and the unbound ssDNAs were then incubated with A375 cells. ssDNA sequences bound to A375 cells were isolated and PCR amplified for use as the ssDNA pool in the subsequent round. Flow cytometry was used to monitor the enrichment of the ssDNA pool after the 3th, 6th, 9th and 11th rounds. The fluorescence intensity curve reflects the amount of FAM-labeled aptamer bound to the cells. As shown in Fig. 1A, with the increasing rounds of selection, the fluorescence intensity of A375 cells gradually increased from the 3th round to 11th round, whereas there was no obvious change in the fluorescence intensity of the negative A431 cells, suggesting that enriched sequences specific for the target cells were obtained as the selection pressure increased (Fig. 1B). However, no further change in fluorescence intensity was observed from the 9th pool to 11th pool, implying that the enrichment of ssDNA sequences bound to target cells reach saturation. Fluorescence microscopy imaging was used to further confirm the specific binding of the 11th round pool for A375 cells. Compared with the ssDNA library, the fluorescence signal was observed mainly on the surface of A375 cells after the 11th round pool treatment, but not on A431 cells (Fig. 1C), confirming that the enriched ssDNA sequences specifically bound to the target A375 cells, but not to A431 cells. Thus, the ssDNA pool from the 11th round was further sequenced through high throughput sequencing.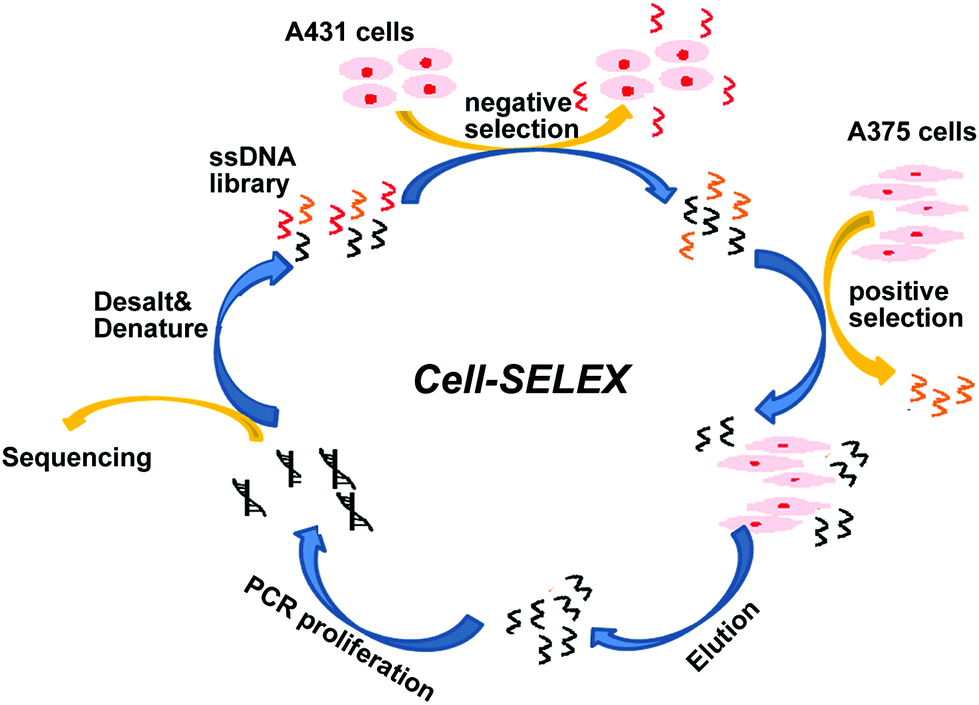 | ||
| Scheme 1 | ||
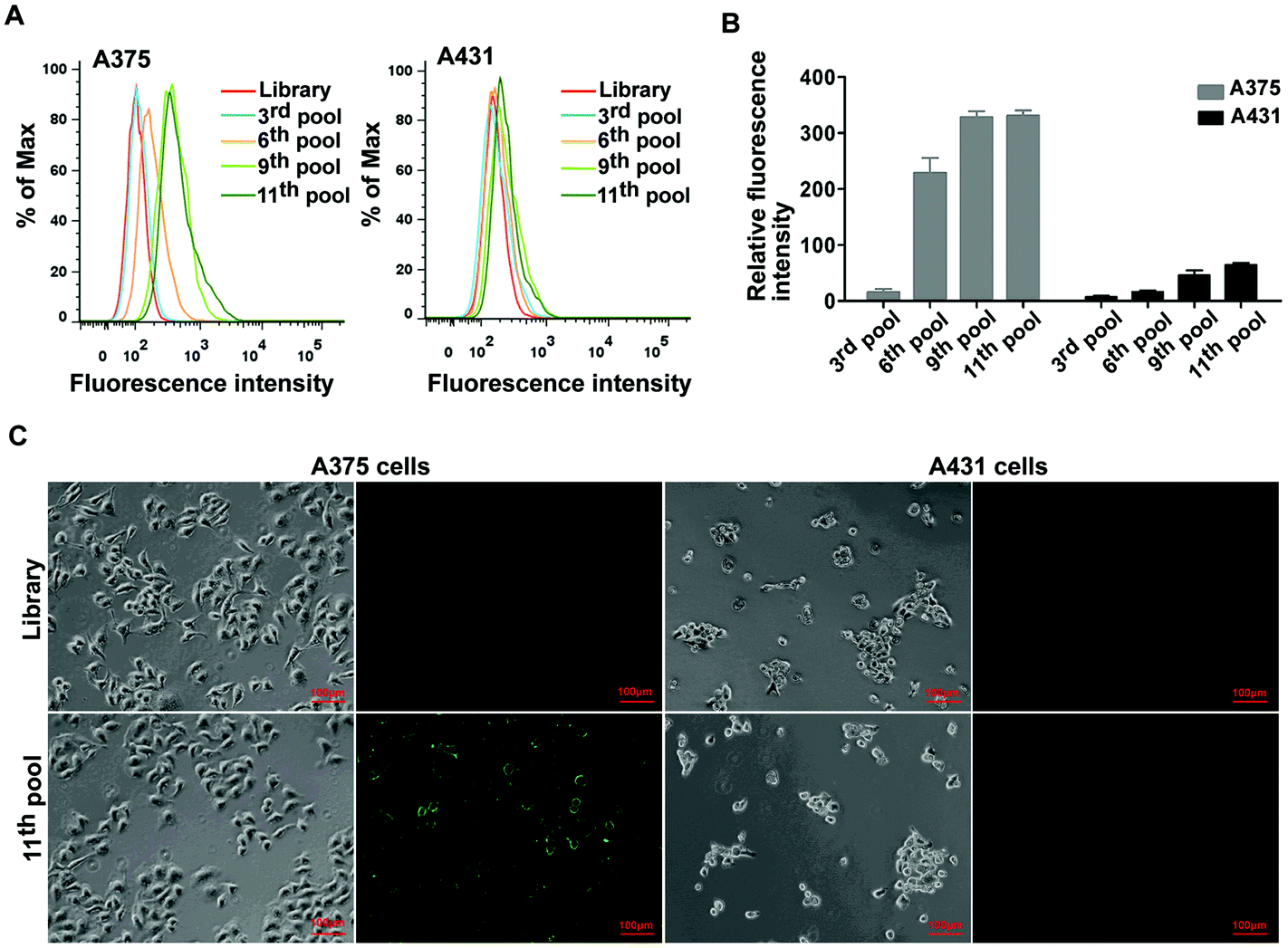 | ||
| Fig. 1 The binding affinity analyses of the ssDNA pools with cells. (A) Flow cytometry assay to monitor the binding of selected pools (3rd, 6th, 9th and 11th) with A375 cells (target cells) and 431 cells (negative cells). (B) Bar graph of the flow cytometry assay results. (C) The fluorescence imaging of the 11th pools bound to A375 and A431 cells using fluorescence microscopy. In each picture, left is the optical image and right is the fluorescence image. Scale bar was 100 μm. | ||
After sequencing, we first calculated the sequence homology in 15 most frequent sequences using program ClustalW2 (https://www.ebi.ac.uk/Tools/msa/clustalw2/). The result showed these sequences were aligned into three groups: sequences 2, 3, 5, 7, 14 and 15 (group-1); sequences 1, 9, 10, 11 and 12 (group-2); sequences 4, 6, 8 and 13(group-3) (Fig. 2A). Furthermore, the top five enriched sequences were selected and chemically synthesized for binding affinity analysis. Flow cytometry results demonstrated that two of these five sequences, sequences 1and 5, bound to 375 cells, resulting in an obvious fluorescence shift in comparison with the ssDNA library, but did not bind to A431 cells, indicating that these two sequences could specifically recognize the target cells (Fig. 2B and C). Three other sequences, sequences 2, 3, and 4, did not bind to the target cells (data not shown). Thus, sequences 1 and 5 were chosen for further optimization.
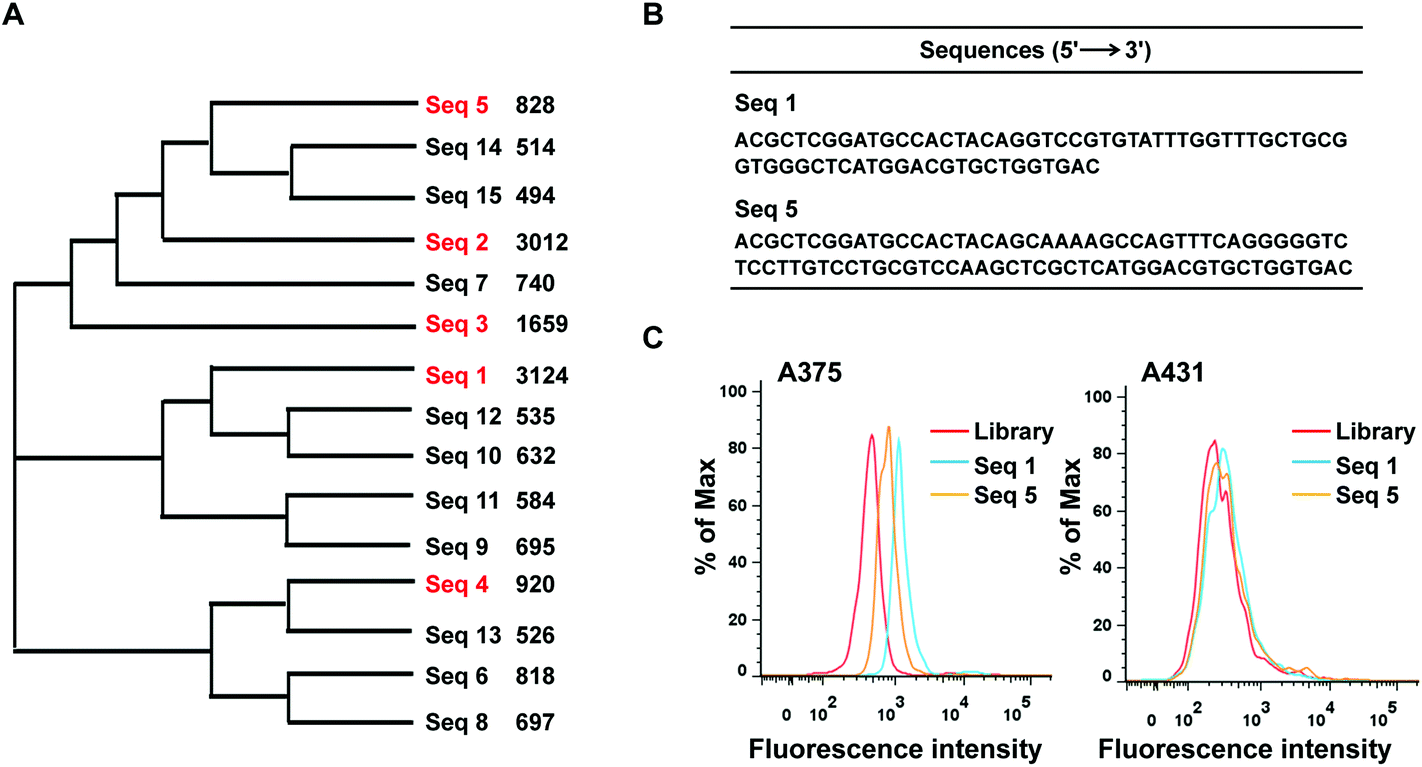 | ||
| Fig. 2 (A) The biological evolutionary tree analysis data of the top 15 sequences from high-throughput sequencing. (B) The list of two aptamer sequences (C) binding assays of two sequences, 1 and 5, with the target cells A375 and the negative cells A431. | ||
Identification of aptamers
It has been reported that not all the nucleotides of one aptamer are required for the targeted binding.33 In general, the stem-loop structures of aptamers are important for binding to the target molecules. Thus, for retaining loop related sequences, the full-length sequences 1 and 5 were optimized by mutation and removing nucleotides at the 5′ and 3′ ends. The acquired aptamers were termed CH1 and CH5, respectively. As shown in Fig. 3A, we first predicted the secondary structures of sequences 1 and 5 using the web-based IDT oligoAnalyzer 3.1 program. According to prediction, CH1 was obtained from sequence 1 by respectively truncating 15 nucleotides from its 5′ end and 8 nucleotides from its 3′ end, and mutating 1 nucleotide (indicated by an arrow). For CH5, the aptamer was acquired from sequence 5 by respectively truncating 13 nucleotides from its 5′ end and 2 nucleotides from its 3′ end, and mutating 3 nucleotides (indicated by an arrow). Next, we examined the binding specificity of the two aptamers to A375 and A431 cells by flow cytometry. As shown in Fig. 3B, compared to the ssDNA library, aptamers CH1 and CH5 exhibited their binding specificity with A375 cells, but they did not bind to A431 cells. Moreover, the results from fluorescence microscopy imaging were also similar to that of flow cytometry. There was no obvious fluorescence on A375 cells after treatment with the ssDNA library. However, green fluorescence signals were observed on the surface of A375 cells following treatment with CH1 or CH5 (Fig. 3C). These findings demonstrated that aptamers CH1 and CH5 could specifically bind to A375 cells and the central stem-loop structures are the key binding motif of aptamers. | ||
| Fig. 3 Identification of potential aptamer candidates. (A) Predicted structures and structure optimization of CH1 and CH5. The structures above the red line represent the truncated forms. Mutating nucleotides were indicated by the arrow. (B) Binding assays of the selected aptamers, CH1 and CH5, with the target cells A375 and the negative cells A431. (C) A375 cells were incubated with FAM-labeled CH1, CH5 and initial ssDNA library, respectively. Fluorescence images revealed that CH1 and CH5 specifically bound to the surface of A375cells. Scale bar was 50 μm. | ||
Characterization of aptamers CH1 and CH5
To test the ability of aptamers to serve as a probe in clinical applications, we analyzed the binding affinity, the molecular types of targets and temperature adaptability. First, we quantitatively evaluated the binding affinity of aptamers CH1 and CH5. Various concentrations of aptamers were incubated with target A375 cells and the fluorescence intensity was measured to determine the Kd by flow cytometry. As shown in Fig. 4A, the Kd values of aptamers CH1 and CH5 were approximately 19.9 ± 4.2 nM and 65.6 ± 5.4 nM, respectively, indicating that these two aptamers could specifically recognize target A375 cells with high affinity. Furthermore, to investigate whether proteins on cell surface are recognized by the aptamers, binding of aptamers to proteinase-digested cells was measured. As shown in Fig. 4B, after being treated with trypsin or proteinase K, compared with the non-treatment groups, the binding abilities of A375 cells to CH1 and CH5 remarkably decreased, indicating that the targets of CH1 and CH5 on the cell surface are most likely proteins.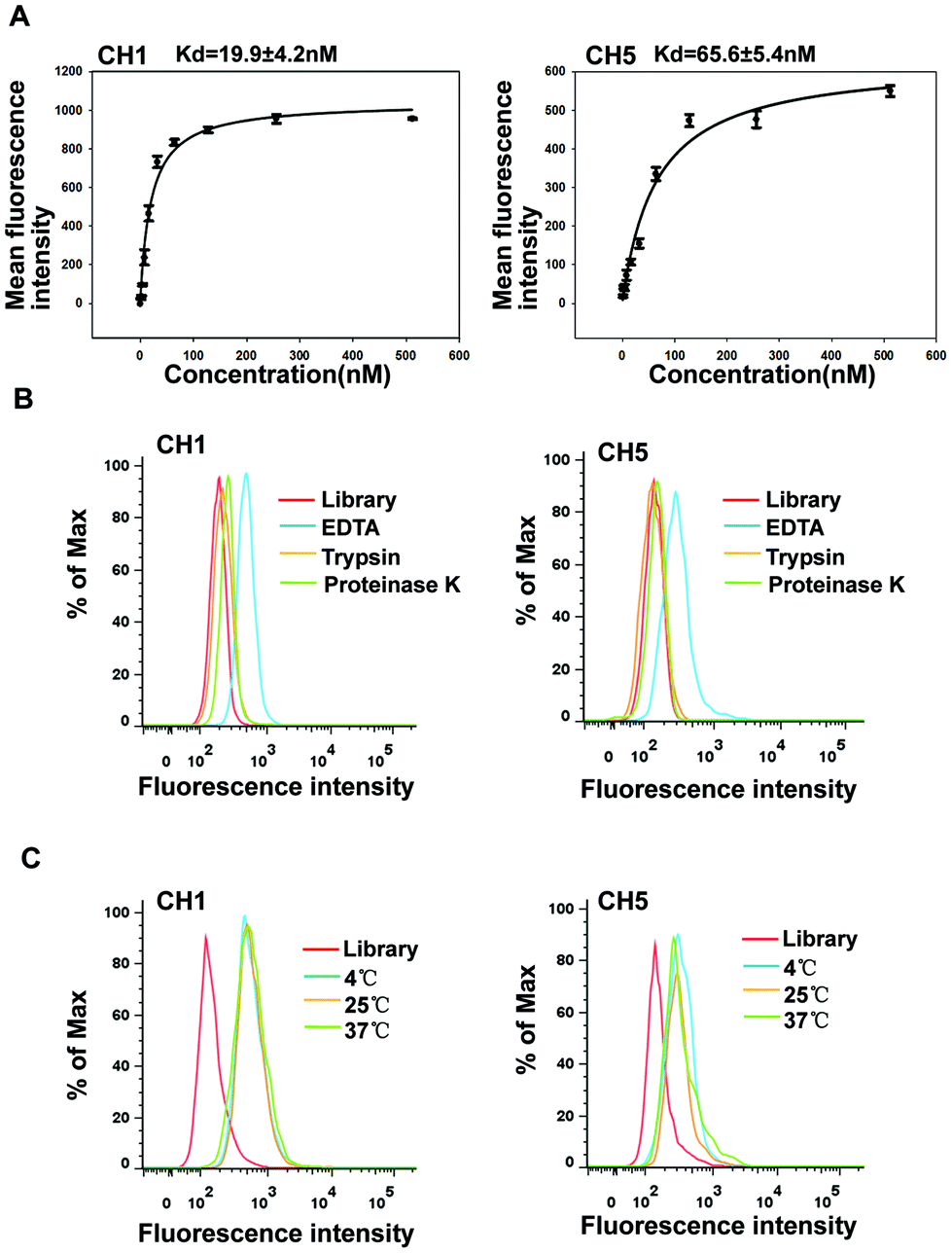 | ||
| Fig. 4 Binding properties of aptamers CH1 and CH5. (A) Binding curve of aptamers CH1 and CH5 with A375 cells. Cells were incubated with varying concentrations of FAM-labeled CH1 or CH5 in triplicate, respectively. (B) Cells were treated with trypsin or proteinase K for 10 min, respectively, and the binding of the FAM-labeled CH1 or CH5 to A375 cells was then assessed by flow cytometry. (C) Flow cytometry assays were performed to assess the binding capacity of CH1 or CH5 at 4 °C, 25 °C, and 37 °C, respectively. The red curve represents the background binding of the initial library. | ||
A temperature of 4 °C was used in the selection process for avoiding enriching nonspecific DNA sequences. Nevertheless, temperature change may influence the formation of nucleic acid secondary structures. Considering that aptamers as targeted probes may be used at 25 °C or 37 °C, we investigated the binding capacity of aptamers after temperature change. A375 cells were respectively incubated with CH1 and CH5 in binding buffer (BB) for 30 min at 4 °C, 25 °C or 37 °C, and then analyzed by flow cytometry. The results illustrated that A375 cells showed the same fluorescence intensity and patterns after incubation at all temperatures (Fig. 4C), indicating that increases in temperature did not influence the aptamers’ binding to cells, and aptamers can be used both in vitro and in vivo. Above all, these results showed that CH1 and CH5 were with good structure stability, and incubation temperatures did almost not influence the aptamers’ binding to cells. Compared with aptamer CH5, aptamer CH1 exhibited a better binding affinity, so we chose aptamer CH1 for further evaluation.
Selectivity of aptamer CH1
We performed subtractive Cell-SELEX using A375 cells as the target, and generated malignant melanoma related aptamers. A375 cells were reported to harbor BRAF V600E mutations. BRAF V600E mutations also occur in 10–15% of colorectal cancer (CRC) and are frequently associated with poor prognosis.34 To further verify whether aptamer CH1 can specifically bind tumor cells harboring BRAF V600E mutations, we examined the binding selectivity of CH1 against two normal cell lines, two melanoma cell lines and five CRC cell lines, and three of these cell lines, SK-MEL-28, RKO and COLO 205, harbor BRAF V600E mutations. As shown in Fig. 5, aptamer CH1 only bound to SK-MEL-28 cells but not SK-MEL-2 cells, validating that aptamer CH1 specifically binds BRAF V600E-mutated melanoma cells, as SK-MEL-28 is a BRAF V600E-mutated melanoma cell line and SK-MEL-2 is a BRAF WT cell line. Moreover, this phenomenon also occurred in CRC cells. CH1 was shown to recognize CRC cells harboring BRAF V600E mutations, but not three other BRAF WT CRC cells, including HCT116, CL187, and HCT-8, which might suggest that molecular targets of CH1 highly expressed on cancer cells harboring BRAF V600E mutation. In addition, there was no obvious fluorescence shift when two normal cell lines were tested, including the human embryonic kidney cell line HEK293 and the Chinese hamster ovary cell line CHO, revealing the targeted tumor specificity of CH1. We also measured the Kd value to quantitatively evaluate the binding affinity of CH1 to COLO 205 cells. Fig. 6A shows that the Kd of CH1 for COLO 205 cells was approximately 34.9 ± 7.1 nM, indicating that aptamer CH1 could specifically recognize COLO 205 cells with high affinity. The binding specificity of CH1 to COLO 205 cells was further confirmed by fluorescence microscopy. As illustrated in Fig. 6B, there was no obvious fluorescence on COLO 205 cells following treatment with the ssDNA library. However, after incubation with CH1, the fluorescence signal was observed mainly on the surface of COLO 205 cells, suggesting CH1 recognized molecular targets on the cell surface, which is consistent with the flow cytometry results mentioned above. The poorly differentiated COLO 205 cells represent highly metastatic cancer cells with an endogenous BRAF V600E mutation. Aptamer CH1 could specifically bind to the high-metastatic cells with BRAF V600E mutations, including COLO 205 and A375 cells, indicating a relationship between molecular targets of CH1 and BRAF V600E-related metastasis. BRAF mutations are associated with less-differentiated and metastatic colorectal cancer. Studies demonstrated that BRAF V600E mutation induced promigratory molecules like β1 and β4-integrin in CRC cells. Conversely, BRAF inhibition suppressed several metastasis-associated transcripts that have not been implicated as biomarkers of BRAF signaling.35,36 Huang et al. also reported that BRAF mutations were significantly related to CRC metastasis including lymphatic or distant metastases in an Asian population.37 Therefore, aptamer CH1 has the potential as a promising probe for specifically targeting metastatic tumor harboring BRAF V600E mutations. | ||
| Fig. 5 Cell-binding selectivity of aptamer CH1. FAM-labeled CH1 was incubated with SK-MEL-28, SK-MEL-2, PKO, COLO 205, HCT116, CL187, HCT-8, CHO and HEK293 cells, respectively. Binding ability was analyzed by flow cytometry. | ||
 | ||
| Fig. 6 Binding property of aptamer CH1 to COLO 205 cells. (A) Binding curve of aptamer CH1 to COLO 205 cells. (B) COLO 205 cells were incubated with FAM-labeled CH1, FAM-labeled library, respectively. Fluorescence images revealed that CH1 specifically bound to the surface of COLO 205 cells. Scale bar was 100 μm. | ||
Conclusion
Using Cell-SELEX technology, aptamers against malignant melanoma cells A375 were successfully obtained after 11 rounds of selection. After truncation and mutation, two DNA aptamers, CH1 and CH5, were identified to specifically recognize targeting melanoma cells with the low nanomolar Kd values of 19.9 ± 4.2 nM and 65.6 M ± 5.4 nM, respectively. Furthermore, we discovered that the target type of aptamer CH1 or CH5 was the membrane protein on the cell surface, and the binding of both aptamers to A375 cells was almost not influenced by incubation temperatures (4 °C, 25 °C and 37 °C), suggesting that the two aptamers could be applied in complex circumstances. In addition, aptamer CH1 not only specifically bound to malignant A375 cells but also bound to other highly metastatic cancer cell lines harboring BRAF V600E mutations, indicating a relationship between aptamer CH1 and BRAF V600E-related metastasis. Given the good binding selectivity and affinity, aptamer CH1 could be developed as a promising molecular probe for the diagnosis and therapy of malignant melanoma. Additionally, further identification of targets of aptamer CH1 will provide novel insights into BRAF mutated cancers.Author contributions
Conceptualization: H.C. (Hang Chen) and D.S.; formal analysis: W.L., T.B. and H.C. (Hang Chen); funding acquisition: W.L., H.C. (Hang Chen) and D.S.; investigation: W.L., T.B., S.J. and H.C. (Hang Chen); methodology: W.L., T.B., R.W. and D.S.; project administration: W.L. and H.C. (Hang Chen); resources: W.L., D.S. and H.C. (Hang Chen); supervision: H.C. (Hang Chen); writing-original draft preparation: W.L. and H.C. (Hang Chen); writing-review and editing: W.L., H.C. (Hang Chen) and D.S.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This research work was financially supported by the Natural Science Foundation of Liaoning Province, China (No. 2020-MS-174 and 2020-MS-164) and the Shengyang Youth Science and Technology Project (No. RC190261).References
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2020, 70, 7–30 CrossRef PubMed.
- C. M. Balch, J. E. Gershenwald, S. J. Soong, J. F. Thompson, M. B. Atkins, D. R. Byrd, A. C. Buzaid, A. J. Cochran, D. G. Coit, S. Ding, A. M. Eggermont, K. T. Flaherty, P. A. Gimotty, J. M. Kirkwood, K. M. McMasters, M. C. Mihm Jr., D. L. Morton, M. I. Ross, A. J. Sober and V. K. Sondak, J. Clin. Oncol., 2009, 27, 6199–6206 CrossRef.
- T. Knackstedt, R. W. Knackstedt, R. Couto and B. Gastman, Plast. Reconstr. Surg., 2018, 142, 202–216 CrossRef.
- C. Lee, F. Collichio, D. Ollila and S. Moschos, Clin. Dermatol., 2013, 31, 141–147 CrossRef.
- P. B. Chapman, A. Hauschild, C. Robert, J. B. Haanen, P. Ascierto, J. Larkin, R. Dummer, C. Garbe, A. Testori, M. Maio, D. Hogg, P. Lorigan, C. Lebbe, T. Jouary, D. Schadendorf, A. Ribas, S. J. O'Day, J. A. Sosman, J. M. Kirkwood, A. M. M. Eggermont, B. Dreno, K. Nolop, J. Li, B. Nelson, J. Hou, R. J. Lee, K. T. Flaherty, G. A. McArthur and BRIM-3 Study Group, N. Engl. J. Med., 2011, 364, 2507–2516 CrossRef CAS.
- C. R. Schmidt, N. J. Achille, A. Kuntimaddi, A. M. Boulton, B. I. Leach, S. Zhang, N. J. Zeleznik-Le and J. H. Bushweller, BCOR Binding to MLL-AF9 Is Essential for Leukemia via Altered EYA1, SIX, and MYC Activity, Blood Cancer Discov., 2020, 1, 162–177 CrossRef.
- A. Ribas and K. T. Flaherty, Nat. Rev. Clin. Oncol., 2011, 8, 426–433 CrossRef CAS PubMed.
- A. Hauschild, J. J. Grob, L. V. Demidov, T. Jouary, R. Gutzmer, M. Millward, P. Rutkowski, C. U. Blank, W. H. M. Jr., E. Kaempgen, S. Martín-Algarra, B. Karaszewska, C. Mauch, V. Chiarion-Sileni, A. M. Martin, S. Swann, P. Haney, B. Mirakhur, M. E. Guckert, V. Goodman and P. B. Chapman, Lancet, 2012, 380, 358–365 CrossRef CAS.
- C. Jamieson, G. Martinelli, C. Papayannidis and J. E. Cortes, Hedgehog Pathway Inhibitors: A New Therapeutic Class for the Treatment of Acute Myeloid Leukemia., Blood Cancer Discov, 2020, 1, 134–145 CrossRef PubMed.
- K. K. Broman, L. A. Dossett, J. Sun, Z. Eroglu and J. S. Zager, Expert Opin. Drug Saf., 2019, 18, 381–392 CrossRef PubMed.
- G. A. McArthur, P. B. Chapman, C. Robert, J. Larkin, J. B. Haanen, R. Dummer, A. Ribas, D. Hogg, O. Hamid, P. A. Ascierto, C. Garbe, A. Testori, M. Maio, P. Lorigan, C. Lebbé, T. Jouary, D. Schadendorf, S. J. O'Day, J. M. Kirkwood, A. M. Eggermont, B. Dréno, J. A. Sosman, K. T. Flaherty, M. Yin, I. Caro, S. Cheng, K. Trunzer and A. Hauschild, Lancet Oncol., 2014, 15, 323–332 CrossRef CAS.
- L. M. Sholl, A. Andea, J. A. Bridge, L. Cheng, M. A. Davies, M. Ehteshami, T. C. Gangadhar, S. Kamel-Reid, A. Lazar, K. Raparia, A. Siroy, K. L. Watson and Members of Cancer Biomarker Reporting Committee and College of American Pathologists, Arch. Pathol. Lab. Med., 2016, 140, 355–357 CrossRef PubMed.
- M. A. F. Anwar, F. Murad, E. Dawson, Z. Y. A. Elmageed, K. Tsumagari and E. Kandil, J. Surg. Res., 2016, 203, 407–415 CrossRef CAS.
- L. Heinzerling, M. Baiter, S. Kühnapfel, G. Schuler, P. Keikavoussi, A. Agaimy, F. Kiesewetter, A. Hartmann and R. Schneider-Stock, Br. J. Cancer, 2013, 109, 2833–2841 CrossRef CAS.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS.
- Y. L. Song, Z. Zhu, Y. An, W. T. Zhang, H. M. Zhang, D. Liu, C. D. Yu, W. Duan and C. Y. J. Yang, Anal. Chem., 2013, 85, 4141–4149 CrossRef CAS PubMed.
- D. R. Bell, J. K. Weber, W. Yin, T. Huynh, W. Duan and R. H. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 8486–8493 CrossRef CAS PubMed.
- T. Bing, X. J. Liu, X. H. Cheng, Z. H. Cao and D. H. Shangguan, Biosens. Bioelectron., 2010, 25, 1487–1492 CrossRef CAS.
- A. Drabik, J. Ner-Kluza, K. Hartman, G. Mayer and J. Silberring, Proteomics: Clin. Appl., 2020, 14, e1800186 Search PubMed.
- Y. Zhong, J. Y. Zhao, J. Z. Li, X. Liao and F. L. Chen, Anal. Biochem., 2020, 598, 113620 CrossRef CAS PubMed.
- K. Sefah, Z. W. Tang, D. H. Shangguan, H. Chen, D. Lopez-Colon, Y. Li, P. Parekh, J. Martin, L. Meng, J. A. Phillips, Y. M. Kim and W. H. Tan, Leukemia, 2009, 23, 235–244 CrossRef CAS PubMed.
- J. Q. He, J. Y. Wang, N. Zhang, L. Y. Shen, L. L. Wang, X. Xiao, Y. Wang, T. Bing, X. J. Liu, S. Q. Li and D. H. Shangguan, Talanta, 2019, 194, 437–445 CrossRef CAS.
- W. M. Li, S. Wang, L. L. Zhou, Y. J. Cheng and J. Fang, Talanta, 2019, 199, 634–642 CrossRef CAS PubMed.
- N. Q. Lin, L. Wu, X. Xu, Q. Y. Wu, Y. Z. Wang, H. C. Shen, Y. L. Song, H. Y. Wang, Z. Zhu, D. Z. Kang and C. Y. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 9306–9315 CrossRef CAS PubMed.
- L. Yang, H. Sun, Y. Liu, W. J. Hou, Y. Yang, R. Cai, C. Cui, P. H. Zhang, X. S. Pan, X. W. Li, L. Li, B. S. Sumerlin and W. H. Tan, Angew. Chem., Int. Ed., 2018, 57, 17048–17052 CrossRef CAS PubMed.
- P. Gong, B. H. Shi, M. B. Zheng, B. Wang, P. F. Zhang, D. H. Hu, D. Y. Gao, Z. H. Sheng, C. F. Zheng, Y. F. Ma and L. T. Cai, Biomaterials, 2012, 33, 7810–7817 CrossRef CAS.
- W. M. Li, L. L. Zhou, M. Zheng and J. Fang, Mol. Ther.–Nucleic Acids, 2018, 12, 707–717 CrossRef CAS PubMed.
- N. Nakamura, T. Matsui, Y. J. Ishibashi, A. Sotokawauchi, K. Fukami, Y. Higashimoto and S. I. Yamagishi, Mol. Med., 2017, 23, 295–306 Search PubMed.
- Y. B. Zeng, Z. C. Yu, Y. N. He, T. Zhang, L. B. Du, Y. M. Dong, H. W. Chen, Y. Y. Zhang and W. Q. Wang, Acta Pharmacol. Sin., 2018, 39, 261–274 CrossRef CAS PubMed.
- J. Lopes-Nunes, J. Lifante, Y. L. Shen, E. C. Ximendes, D. Jaque, M. C. I. la Cruz and C. Cruz, Eur. J. Pharm. Biopharm., 2020, 154, 228–235 CrossRef CAS PubMed.
- H. Li, J. Liu, X. J. Xiao, S. M. Sun, H. Zhang, Y. B. Zhang, W. H. Zhou, B. Zhang, M. Roy, H. Liu, M. Ye, Z. Wang, F. Liu-Smith and J. Liu, Mol. Ther.–Nucleic Acids, 2019, 18, 727–738 CrossRef CAS PubMed.
- W. M. Li, T. Bing, J. Y. Wei, Z. Z. Chen, D. H. Shangguan and J. Fang, Biomaterials, 2014, 35, 6998–7007 CrossRef CAS PubMed.
- S. X. Gao, X. Zheng, B. H. Jiao and L. H. Wang, Anal. Bioanal. Chem., 2016, 408, 4567–4573 CrossRef CAS PubMed.
- A. I. Phipps, P. J. Limburg, J. A. Baron, A. N. Burnett-Hartman, D. J. Weisenberger, P. W. Laird, F. A. Sinicrope, C. Rosty, D. D. Buchanan, J. D. Potter and P. A. Newcomb, Gastroenterology, 2015, 148, 77–87 CrossRef CAS PubMed.
- R. Herr, M. Köhler, H. Andrlová, F. Weinberg, Y. Möller, S. Halbach, L. Lutz, J. Mastroianni, M. Klose, N. Bittermann, S. Kowar, R. Zeiser, M. A. Olayioye, S. Lassmann, H. Busch, M. Boerries and T. Brummer, Cancer Res., 2015, 75, 216–229 CrossRef CAS PubMed.
- N. Reischmann, G. Andrieux, R. Griffin, T. Reinheckel, M. Boerries and T. Brummer, Oncogene, 2020, 39, 6053–6070 CrossRef CAS.
- D. Huang, W. Sun, Y. Zhou, P. Li, F. Chen, H. Chen, D. Xia, E. Xu, M. Lai, Y. Wu and H. Zhang, Cancer Metastasis Rev., 2018, 37, 173–187 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |