
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSonochemistry in an organocatalytic domino reaction: an expedient multicomponent access to structurally functionalized dihydropyrano[3,2-b]pyrans, spiro-pyrano[3,2-b]pyrans, and spiro-indenoquinoxaline-pyranopyrans under ambient conditions†
Biplob Borah a,
Jahnu Boraa,
Pambala Rameshb and
L. Raju Chowhan*a
a,
Jahnu Boraa,
Pambala Rameshb and
L. Raju Chowhan*a
aSchool of Applied Material Sciences, Centre for Applied Chemistry, Central University of Gujarat, Sector-30, Gandhinagar-382030, Gujarat, India. E-mail: rchowhan@cug.ac.in
bCSIR-Indian Institute of Chemical Technology, Hyderabad-50007, India
First published on 28th April 2022
Abstract
A highly convenient and sustainable one-pot approach for the diversely-oriented synthesis of a variety of medicinally privileged amino-substituted 4,8-dihydropyrano[3,2-b]pyran-3-carbonitriles, and spiro[indoline-3,4′-pyrano[3,2-b]pyran]-3-carbonitrile/carboxylate derivatives on the basis of a domino three-component reaction of readily available carbonyl compounds including aryl aldehydes or isatins, active methylene compounds, and kojic acid as a Michael donor using secondary amine catalyst L-proline under ultrasound irradiation in aqueous ethanolic solution at ambient temperature has been developed. This methodology can involve the assembly of C–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–O, C–N bonds via a one-pot operation, and following this protocol, a series of novel amino-substituted spiro[indeno[1,2-b]quinoxaline-11,4-pyrano[3,2-b]pyran]-3-carbonitrile/carboxylates have been synthesized. The practical utility of this method was found to be very efficient for scale-up reaction and other useful transformations. The methodology provides significant advantages including mild reaction conditions, energy-efficiency, short reaction time, fast reaction, simple work-up procedure, broad functional group tolerances, utilization of reusable catalyst, green solvent system, being metal-free, ligand-free, waste-free, inexpensive, etc. Excellent chemical yields have been achieved without using column chromatography. To address the issues of green and more sustainable chemistry, several metrics including Atom Economy (AE), Reaction Mass Efficiency (RME), Atom efficiency, E-factor, Process Mass Intensity (PMI), and Carbon Efficiency (CE) have been quantified for the present methodology that indicates the greenness of the present protocol.
C, C–O, C–N bonds via a one-pot operation, and following this protocol, a series of novel amino-substituted spiro[indeno[1,2-b]quinoxaline-11,4-pyrano[3,2-b]pyran]-3-carbonitrile/carboxylates have been synthesized. The practical utility of this method was found to be very efficient for scale-up reaction and other useful transformations. The methodology provides significant advantages including mild reaction conditions, energy-efficiency, short reaction time, fast reaction, simple work-up procedure, broad functional group tolerances, utilization of reusable catalyst, green solvent system, being metal-free, ligand-free, waste-free, inexpensive, etc. Excellent chemical yields have been achieved without using column chromatography. To address the issues of green and more sustainable chemistry, several metrics including Atom Economy (AE), Reaction Mass Efficiency (RME), Atom efficiency, E-factor, Process Mass Intensity (PMI), and Carbon Efficiency (CE) have been quantified for the present methodology that indicates the greenness of the present protocol.
Introduction
Over 60% of organic compounds produced by diverse chemical industries in the form of fine chemicals, drugs, optoelectronic materials, and agrochemicals contain heterocycles as the key active ingredients. In particular, the synthesis of novel oxygen and nitrogen-containing heterocycles is of significant importance on account of their occurrences in natural compounds, medicinally active components, and optoelectronic materials.1 5-Hydroxy-2-(hydroxymethyl)-4H-pyran-4-one, commonly known as kojic acid is an oxygen-containing natural γ-pyrone generated by filamentous fungi and numerous kinds of bacteria including Aspergillus, Penicillium, and Acetobacter, etc.2 The kojic acid nucleus and its analogs have drawn immense significance in recent years due to their various application in the medicinal and pharmaceutical industry,3 food industry,4 cosmetic and personal care products,5 agriculture,6 and chemical industry.7 By taking into consideration kojic acid's structural characteristics and broad-spectrum activities, a substantial attempt was made not just towards its synthesis but also to using them as an essential constituent for construction and identification of potent structural fragments.8 In line with this, one of the most important fused heterocycles namely, pyrano[3,2-b]pyrans, was synthesized mostly in the last two decades based on kojic acid.9 Pyrano[3,2-b]pyrans are found to be the major structural constituent of many natural products (Fig. 1) and they are established as the most important target for total synthesis. Elatenyne (A), a dibrominated natural product extracted from Laurencia elata, belongs to a class of non-isoprenoid, halogenated ethers having an unbranched C15 backbone and a conjugated enyne terminal in their structure.10 The compound B, also a halogenated C15 natural product was derived from Laurencia majuscule, a marine red alga.11 Both compounds A and B have received a lot of attention as potential total synthesis targets due to the presence of the functionalized pyrano[3,2-b]pyran core in their skeleton. Alternatively, the presence of strategically situated halogen atoms on the tetrahydropyran rings of the natural products (3Z)-Dactomelynes (C) and (3E)-Dactomelynes (D) isolated from the digestive glands of the sea hare Aplysia dactylomela make them an important skeleton for laboratory synthesis to evaluate pharmacologically.12,13 Pyrano[3,2-b]pyrans are often found in the core structure of many non-natural products, synthetic drug candidates and holds a pivotal position in drug discovery and medicinal chemistry on account of their remarkable biological activities14 such as antioxidant, antifungal, antibacterial, anti-tumor activity [Fig. 1(E)–(I)].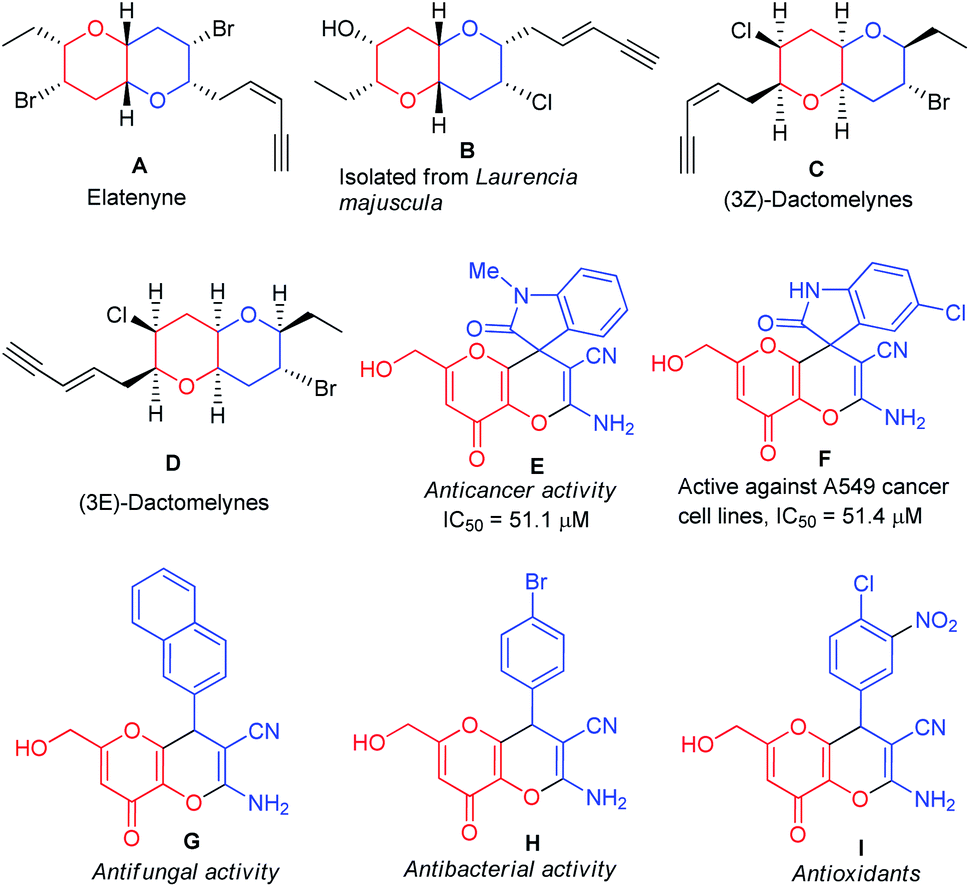 | ||
| Fig. 1 Naturally occurring molecules (A)–(D) and some biologically active compounds (E)–(I) containing the pyrano[3,2-b]pyran moiety. | ||
Furthermore, the synthesis of spiro-heterocyclic compounds especially spiro-oxindoles have drawn much more attention not only in synthetic chemistry but also in medicinal chemistry for their outstanding reactivity and prolific therapeutic potential along with their utilization as key intermediates for direct access to natural product types molecules and medicinally privileged heterocycles.15
Considering these prominent significances and the broad-spectrum pharmacological profile of pyrano[3,2-b]pyran derivatives, the synthesis of these structural motifs becomes extremely demanding. Several synthetic procedures for pyrano[3,2-b]pyrans have been established during the past decades.9 Although, the previously reported methods offer some advantages, many of them suffer drawbacks, including the use of volatile organic solvents, metal-catalyst, tedious work-up procedure, time-consuming, limited substrate scopes, harsh reaction conditions, and relatively low products yield. Consequently, developing an alternative approach for conducting organic synthesis that avoids or minimizes the use of volatile organic solvents and hazardous reagents by introducing environmentally benign conditions with the main focus to reduce the cost-effectiveness of the chemical transformation is highly desired.
With these perspectives and people's ever-increasing awareness about their living environment to protect from chemical pollution associated with a chemical process, sonochemistry appears to be a promising and alternative eco-friendly activation method for organic synthesis due to attractive features associated with it such as the improved reaction rates and product selectivity under mild condition, low energy requirements, along with an immediate reduction in reaction times.16 The ability to accomplish or promote chemical reactions by means of sound waves triggered by ultrasound can make the sonochemical-assisted synthetic method much more superior to the conventional method with respect to green and sustainable chemistry.17 When exposed to a reaction mixture, ultrasound irradiation caused a large number of cavitation bubbles to form once the pressure reached a specific level. These cavitation bubbles will rapidly growing up and then violently collapsed. This repeated dynamic process of oscillation of formation, growing up, and collapses generates the fine emulsion between the reactants and elevated the local temperature of the reaction mixture to cross the activation energy of the reaction.18
But, again the occurrence of transition-metal-catalyst(s) in chemical processes even at the lowest level often triggers an undesirable impact on the greenness or sustainability of the transformation.19 Notwithstanding, transition metal catalyst(s) has been successfully employed in the synthesis of valuable structural building blocks;20 their occurrences in a chemical process is associated with limitations because of their highly toxic nature, and high cost involved in the preparation of transition metal catalyst alongside the requirement of non-commercial supporting ligands for catalyst preparation. Besides these, the elimination of transition-metal-catalyst(s) from chemical transformation which is predominantly required in the pharmaceutical industry is extremely challenging and often results in contamination of the target compounds. Consequently, the recent years have witnessed tremendous progress in the use of small organic molecules known as organocatalyst(s) in organic transformations, which has resulted in a revolution in the synthesis of molecular diversity and complexity in asymmetric and non-asymmetric ways using a variety of activation modes and has emerged as a popular research topic on the context of synthetic effectiveness and also from a green and sustainability standpoint.21 Among other organocatalysts, secondary amine catalyst, L-proline has received immeasurable attention in synthetic chemistry on account of its easy availability, non-toxic, economical nature, and bifunctional mode of reactivity that could be operated simply in chemical transformation and recovered easily from the chemical process. The versatile structural features of L-proline have allowed it to act either as Brønsted acid or Brønsted base or show both the behaviours and it can undergo covalent organocatalysis by generating iminium or enamine intermediates.22 The effectiveness of L-proline was demonstrated by its applicability in various organic transformations23 for example Aldol reaction,23a Mannich reaction,23b Knoevenagel condensation reaction,23c Michael reaction,23d Diels–Alder,23e Biginelli reaction,23f and multicomponent reactions.23g,h
Multicomponent reactions (MCRs) have drastically received a spectacular application in organic synthesis owing to their prolific propensity for the synthesis of complex molecular structures in a single operation. The ability to accomplish reaction from simple, cheap, and readily available starting materials through the creation of several bonds in a one-pot with immediate prevention in the isolation and purification of product intermediates makes MCRs a powerful tool for green or sustainable synthesis as compared to the conventional stepwise method which avoids green chemistry principle.24
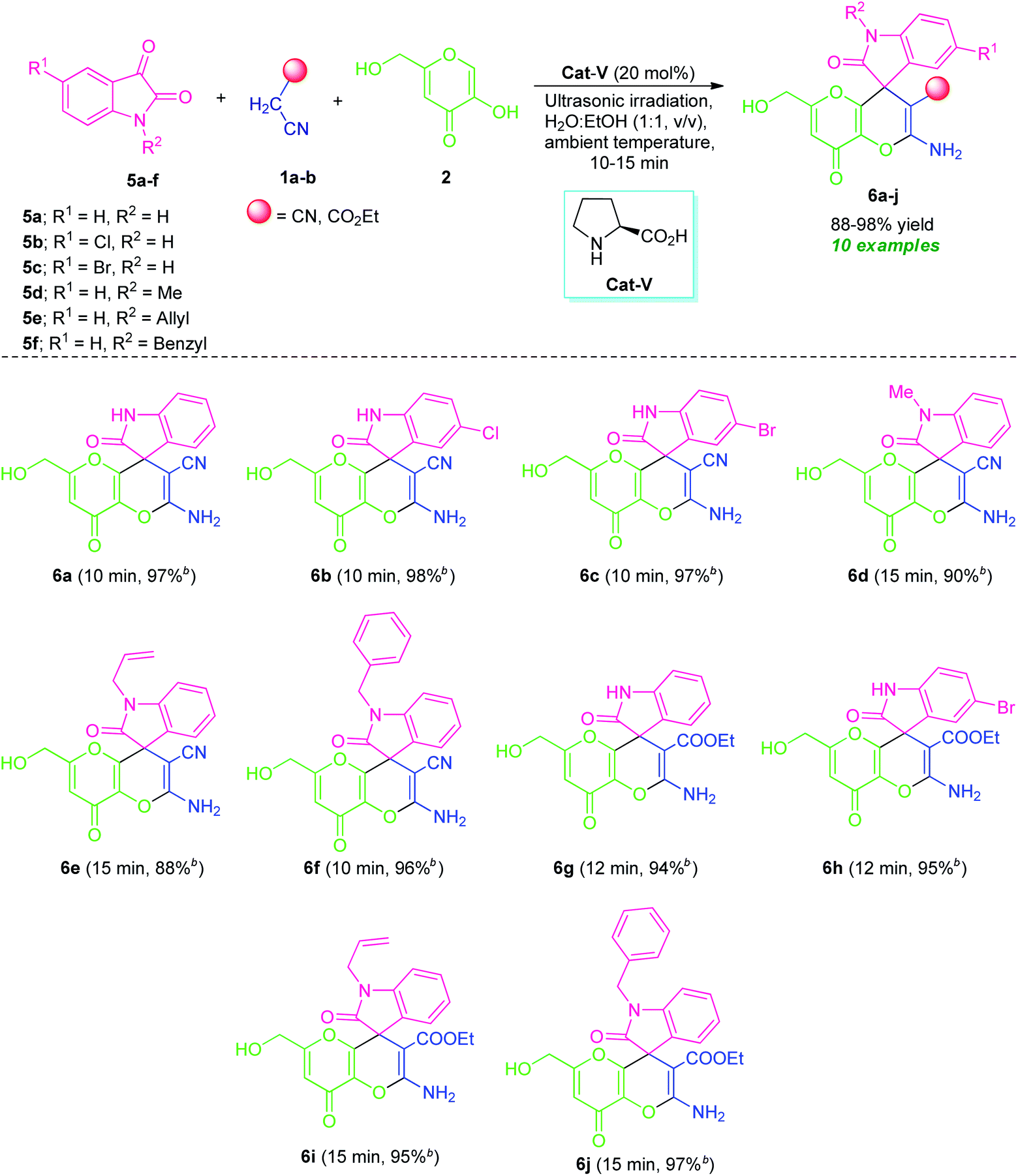
By considering these all aspects and in accordance with making a pollution-free environment, the present study reports an ultrasound-assisted superficial organocatalytic one-pot approach for the assembly of a variety of amino-substituted pyrano[3,2-b]pyrans such as dihydropyrano[3,2-b]pyran-3-carbonitriles (4a–n), and spiro[indoline-3,4′-pyrano[3,2-b]pyran]-3′-carbonitrile/carboxylate derivatives (6a–j) from a domino three-component reaction of active methylene compounds 1a–b, kojic acid 2, and substituted aldehydes 3a–n/isatins 5a–f in aqueous ethanolic solution by employing secondary amine, L-proline as the catalyst at ambient temperature (Scheme 1).
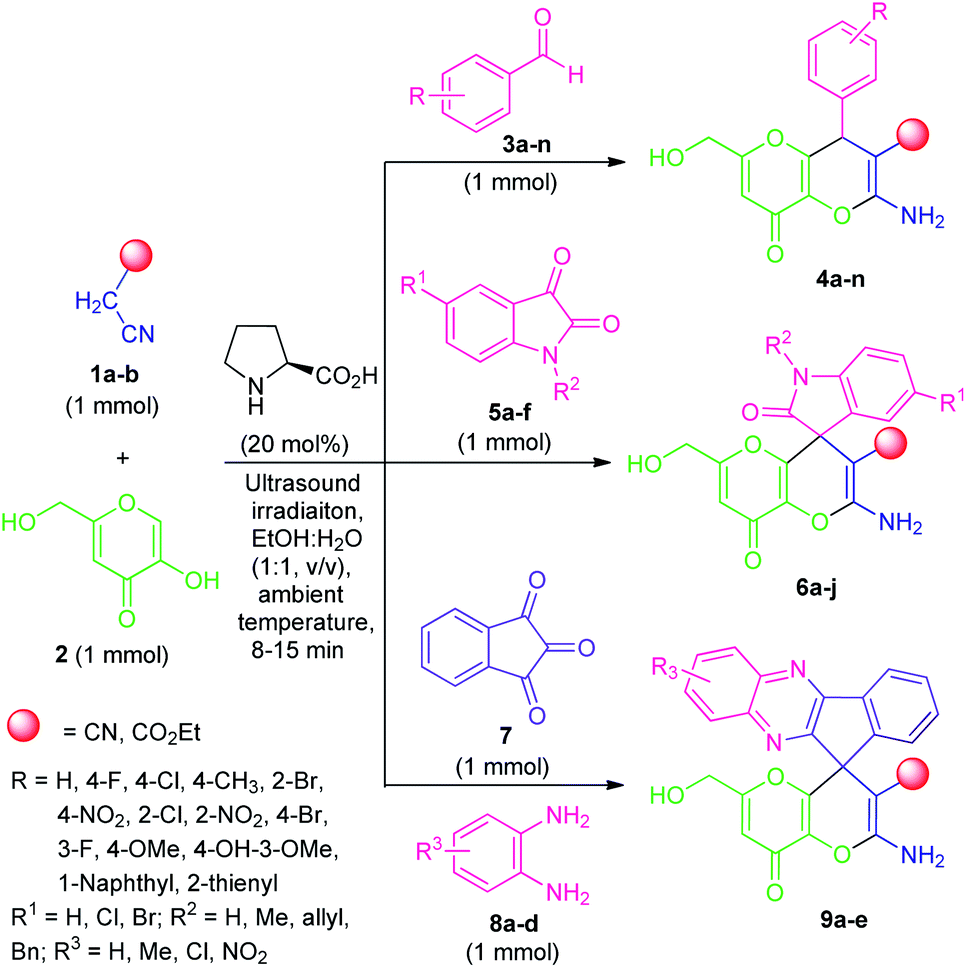 | ||
| Scheme 1 Ultrasound-assisted organocatalytic domino synthesis of dihydro-pyrano[3,2-b]pyran 4, spiro-pyrano[3,2-b]pyran 6 and spiro-[indeno[1,2-b]quinoxaline-11,4′-pyrano[3,2-b]pyran] 9. | ||
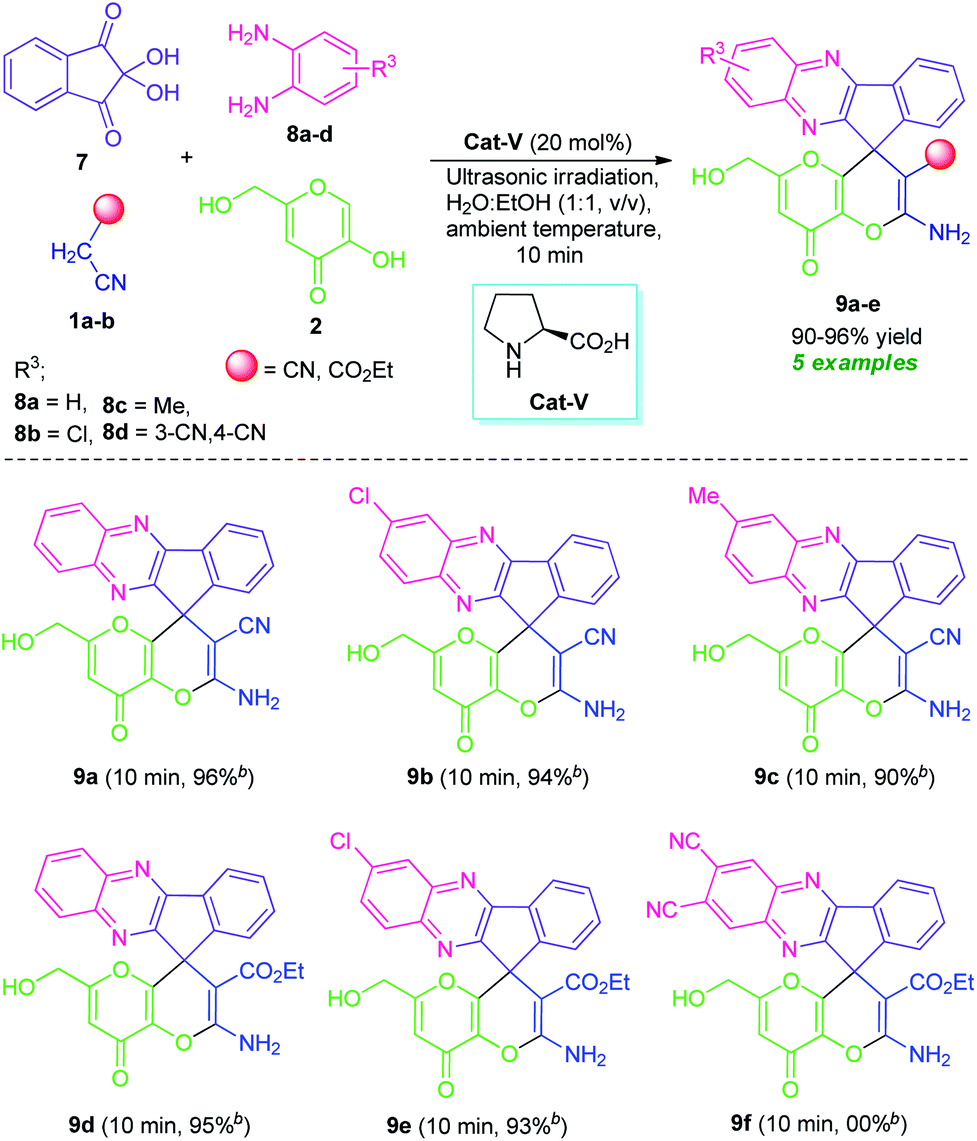
Quinoxaline, a nitrogen-containing bicyclic heterocycle, and its fused derivatives, on the other hand, have been shown to represent attractive structural features in synthetic and medicinal chemistry.19a They are commonly encountered in many natural products, agrochemicals, synthetic drug candidates and represent a wide variety of therapeutic potential such as antidiabetic, antibacterial, anticancer, anti-inflammatory, antituberculosis, anti-HIV, antiprotozoal, etc. These impressive properties of the privileged quinoxaline nucleus have prompted us to synthesize some novel hybrid molecules incorporating both quinoxaline and pyrano[3,2-b]pyran moiety by joining them through C-11 of the spiro-system. In this context, we reported here the synthesis of several novel amino-substituted spiro[indeno[1,2-b]quinoxaline-11,4′-pyrano[3,2-b]pyran] derivatives (9a–e) for the first time via ultrasound-assisted organocatalytic domino four-component reactions of active methylene compound 1, kojic acid 2, ninhydrin 7, and 1,2-diamine 8 under the influences of L-proline in aqueous ethanolic solution at ambient temperature (Scheme 1).
Result and discussion
We commence our studies with an equimolar amount of malononitrile 1a, kojic acid 2, benzaldehyde 3a, as our model substrates and the reaction was performed under the influences of various types of catalysts, solvents, along with catalyst-free and solvent-free conditions using ultrasonic irradiation as a green energy source at ambient temperature. Our preliminary investigation was aimed to authenticate the general feasibility of our approach for aromatic aldehydes to provide 2-amino-6-(hydroxymethyl)-8-oxo-4-aryl/heteroaryl-4,8-dihydropyrano[3,2-b]pyran-3-carbonitriles 4a–n. A quick investigation of the results outlined in Table 1 revealed that the reaction executed in absence of any catalyst failed to yield the intended product 3a even after 1 hour (entry 1, Table 1). Then, we turned our focus on examining the effects of different catalysts on the model reaction and the results were summarized in Table 1. Under the influences of bases such as Et3N, K2CO3, DBU, DABCO; the reaction can require a longer time to complete, and the yield of the product was found to be not satisfactory (entries 2–5, Table 1). Changing the reaction condition from base to acid catalysts such as p-TSA, trifluoroacetic acid (TFA), acetic acid, and benzoic acid, seemed to have no effect on the reaction rate or product yield (entries 6–9, Table 1). With the expectation to improve the yield of the product, next, we investigated the model reaction under the influence of different types of organocatalysts (Fig. 2) such as Cat-I, Cat-II, Cat-III, Cat-IV, Cat-V, and Cat-VI (entries 10–15, Table 1). The catalysts Cat-I, Cat-II, Cat-III, and Cat-VI are found to be slightly able to catalyze the reaction albeit with lower yields of the product, and catalyst Cat-IV gave good conversion of the starting materials to the final product with a slight increase in the yield. However, no significant improvement in the yield as compared to other acids and bases were achieved. But surprisingly, the high completion rate of the model reaction with an excellent yield (96% yields for 4a, entry 14, Table 1) of the product has been achieved under the influence of 20 mol% of Cat-V in only 10 minutes. As a consequence, the exploration of the high catalytic activity of L-proline (Cat-V) was demonstrated as the best optimum catalytic condition for the present study (entry 14, Table 1). To find out the appropriate reaction medium necessary for the formation of 4a, the representative reaction was then performed in solvent-free as well as in a different solvent system such as MeOH, EtOH, H2O, H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOH (v/v, 1:1), H2O:EtOH (v/v, 2:1), toluene, DMF, CH2Cl2, and CH3CN in presence of Cat-V in sonication at ambient temperature (entries 1–10, Table 2). Initial observation in solvent-free conditions realizes the unsuccessful completion of the reaction. While insufficient yields of the product were obtained in different solvent systems such as MeOH, toluene, DMF, CH2Cl2, and CH3CN (entry 2, 7, 8, 9, 10; Table 2). Conversely, the reaction performed in presence of ethanol was found to lead to the product in good yield (88% yields, entry 3, Table 2). Then we investigated the reaction using water as the solvent system and noticeably, the preferred product has been achieved in 90% yields (entry 4, Table 2). Furthermore, mixing the solvent system including water and ethanol in a 1:1 ratio was found to be very efficient to deliver the product in excellent yields (entry 5, Table 2). However, there was no change in product yield with changing the ratio of the mixing solvent system (entry 6, Table 2). Although the reaction was very compatible with water as the medium, the high yield of the product in aqueous ethanol (1:1, v/v) over pure water promoted us to consider L-proline (Cat-V) in aqueous ethanol by employing ultrasound irradiation as the optimum reaction condition for the synthesis of dihydropyrano[3,2-b]pyran-3-carbonitrile 4a.
:EtOH (v/v, 1:1), H2O:EtOH (v/v, 2:1), toluene, DMF, CH2Cl2, and CH3CN in presence of Cat-V in sonication at ambient temperature (entries 1–10, Table 2). Initial observation in solvent-free conditions realizes the unsuccessful completion of the reaction. While insufficient yields of the product were obtained in different solvent systems such as MeOH, toluene, DMF, CH2Cl2, and CH3CN (entry 2, 7, 8, 9, 10; Table 2). Conversely, the reaction performed in presence of ethanol was found to lead to the product in good yield (88% yields, entry 3, Table 2). Then we investigated the reaction using water as the solvent system and noticeably, the preferred product has been achieved in 90% yields (entry 4, Table 2). Furthermore, mixing the solvent system including water and ethanol in a 1:1 ratio was found to be very efficient to deliver the product in excellent yields (entry 5, Table 2). However, there was no change in product yield with changing the ratio of the mixing solvent system (entry 6, Table 2). Although the reaction was very compatible with water as the medium, the high yield of the product in aqueous ethanol (1:1, v/v) over pure water promoted us to consider L-proline (Cat-V) in aqueous ethanol by employing ultrasound irradiation as the optimum reaction condition for the synthesis of dihydropyrano[3,2-b]pyran-3-carbonitrile 4a.
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Loading (mol%) | Timeb (min) | Yieldc (%) |
| a Reaction conditions: malononitrile 1a (1 mmol), kojic acid 2 (1 mmol), benzaldehyde 3a (1 mmol), in presence of different catalysts in H2O:EtOH (1:1, v/v) under ultrasonic irradiation at ambient temperature.b Reaction progress monitored by TLC.c Yields of the isolated products. NR-no reaction. |
||||
| 1 | None | — | 60 | NR |
| 2 | Et3N | 20 | 10 | 68 |
| 3 | K2CO3 | 20 | 10 | 70 |
| 4 | DBU | 20 | 10 | 60 |
| 5 | DABCO | 20 | 10 | 55 |
| 6 | p-TSA | 20 | 10 | 45 |
| 7 | TFA | 20 | 10 | 40 |
| 8 | AcOH | 20 | 10 | 52 |
| 9 | PhCO2H | 20 | 10 | 58 |
| 10 | Cat-I | 20 | 10 | 80 |
| 11 | Cat-II | 20 | 10 | 35 |
| 12 | Cat-III | 20 | 10 | 70 |
| 13 | Cat-IV | 20 | 10 | 75 |
| 14 | Cat-V | 20 | 10 | 96 |
| 15 | Cat-VI | 20 | 10 | 60 |
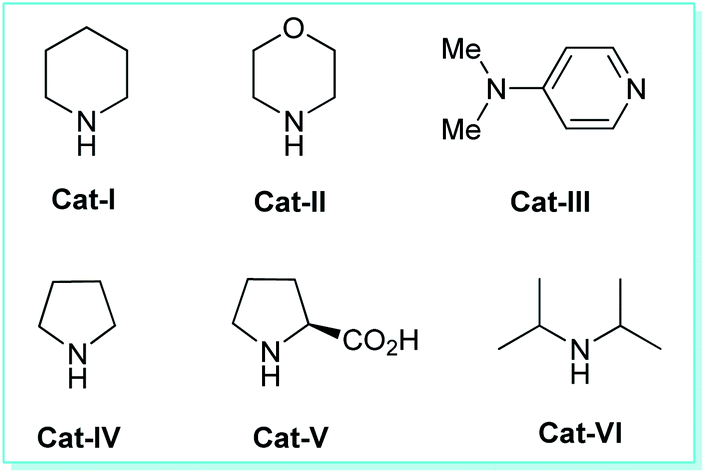 | ||
| Fig. 2 Organocatalysts used in this study. | ||
| Entry | Solvents | Timeb (min) | Yieldc (%) |
|---|---|---|---|
| a Reaction conditions: malononitrile 1a (1 mmol), kojic acid 2 (1 mmol), benzaldehyde 3a (1 mmol), solvent (3 mL), and Cat-V (20 mol%) under ultrasonic irradiation at ambient temperature.b Reaction progress monitored by TLC.c Yields of the isolated products. NR-no reaction. | |||
| 1 | Solvent-free | 10 | NR |
| 2 | MeOH | 10 | 45 |
| 3 | EtOH | 10 | 88 |
| 4 | H2O | 10 | 90 |
| 5 | H2O:EtOH (1:1, v/v) |
10 | 96 |
| 6 | H2O:EtOH (2:1, v/v) |
10 | 90 |
| 7 | Toluene | 10 | 55 |
| 8 | DMF | 10 | 42 |
| 9 | CH2Cl2 | 10 | 32 |
| 10 | CH3CN | 10 | 38 |
Moreover, the amounts of catalyst required for the conversion of reactant into the desired product were evaluated by varying the loading of catalyst (10 mol%, 15 mol%, 20 mol%, and 25 mol%) in the model reaction (Table 3).
| Entry | Catalyst V (mol%) | Timea,b (min) | Yieldc (%) |
|---|---|---|---|
| a Reaction conditions: malononitrile 1a (1 mmol), kojic acid 2 (1 mmol), benzaldehyde 3a (1 mmol), in Cat-V (10–25 mol%) in H2O:EtOH (1:1, v/v).b Reaction progress monitored by TLC.c Yields of the isolated products.d The reaction was performed in ultrasonic irradiation.e The reaction was carried out in reflux condition.f The reaction was carried out in stirred conditions. |
|||
| 1d | 10 | 10 | 80 |
| 2d | 15 | 10 | 90 |
| 3d | 20 | 10 | 96 |
| 4d | 25 | 10 | 96 |
| 4e | 20 | 180 | 82 |
| 5f | 20 | 360 | 85 |
A slight reduction in the rate of complete conversion of reactants was observed when the reaction was performed in presence of 10 mol% of catalyst. Pleasingly, increasing the amounts of catalyst from 10 mol% to 20 mol% leads to the corresponding products with excellent yield, and no further increase in the product yield was detected with increasing the catalyst loading. Therefore, 20 mol% of Cat-V is sufficient for the maximum conversion of the reactants to the final product. With this information in hand, then we investigate the model reaction by employing 20 mol% of Cat-V in aqueous ethanol under stirred conditions as well as reflux conditions. But no improvements in the reaction rate or product yields have been observed. Consequently, ultrasound irradiation was demonstrated as the efficient condition for this transformation as compared to both traditional conditions in terms of reaction time, energy, work-up procedure, and product yields.
After ascertaining the optimal conditions, the reaction was performed with various substituted aromatic aldehyde such as 4-flurobenzaldehyde (3b)/4-chlorobenzaldehyde (3c)/4-bromobenzaldehyde (3d)/4-methylbenzaldehyde (3e)/4-methoxybenzaldehyde (3f)/4-nitrobenzaldehyde (3g)/2-chlorobenzaldehyde (3h)/2-nitrobenzaldehyde (3i)/2-bromobenzaldehyde (3j)/3-fluorobenzaldehyde (3k)/4-hydroxy-3-methoxy-benzaldehyde (3m), active methylene compound 1a, and kojic acid 2 by using 20 mol% of L-proline (Cat-V) in H2O:EtOH (1:1, v/v) under ultrasound irradiation for 8–15 minutes at ambient temperature (Table 4). The electronic effect of substitution on the aryl ring of aldehydes undergoing this domino reaction was first examined. All halogenated substrates like fluoro, chloro, bromo located on C-2, C-3 and C-4 position of the aromatic ring of aldehydes (3b, 3c, 3d, 3h, 3j, and 3k) efficiently worked under this condition and afforded the amino substituted-4,8-dihydropyrano[3,2-b]pyran-3-carbonitrile derivatives 4b, 4c, 4d, 4h, 4j, and 4k with excellent yields without any side reaction. It is interesting to note that aldehyde possessing electron-donating groups such as methyl (3e), methoxy (3f) at C-4 and electron-withdrawing groups including nitro group (3g and 3i) at C-2, and C-4 positions successfully give the desired product 4e, 4f, 4g, and 4i in 90%, 92%, 94%, and 94% yields respectively for a time of 10–15 minutes. In addition, di-substituted groups like hydroxy and methoxy at different positions appeared to be very effective for this reaction (entry 3m). Concurrent to aryl aldehydes, heteroaryl aldehydes such as 2-naphthaldehyde (3l) and thiophene-2-carbaldehyde (3n) are also well suited for this transformation to deliver the desired products 4l and 4n, respectively in 94% and 97% yields at 13 and 10 minutes. The utilization of ultrasound as a green activation method for this reaction was found to increases the yield of the products in a very short reaction time as compared to the reaction performed in absence of ultrasonic irradiation. Furthermore, the temperature of the water bath was remained constant during the reaction and not increased even after the optimized reaction time, which indicates the acceleration of the reaction by irradiation as the energy source not for an increased in the temperature of the reaction. However, aliphatic aldehydes were unable to react under this condition.
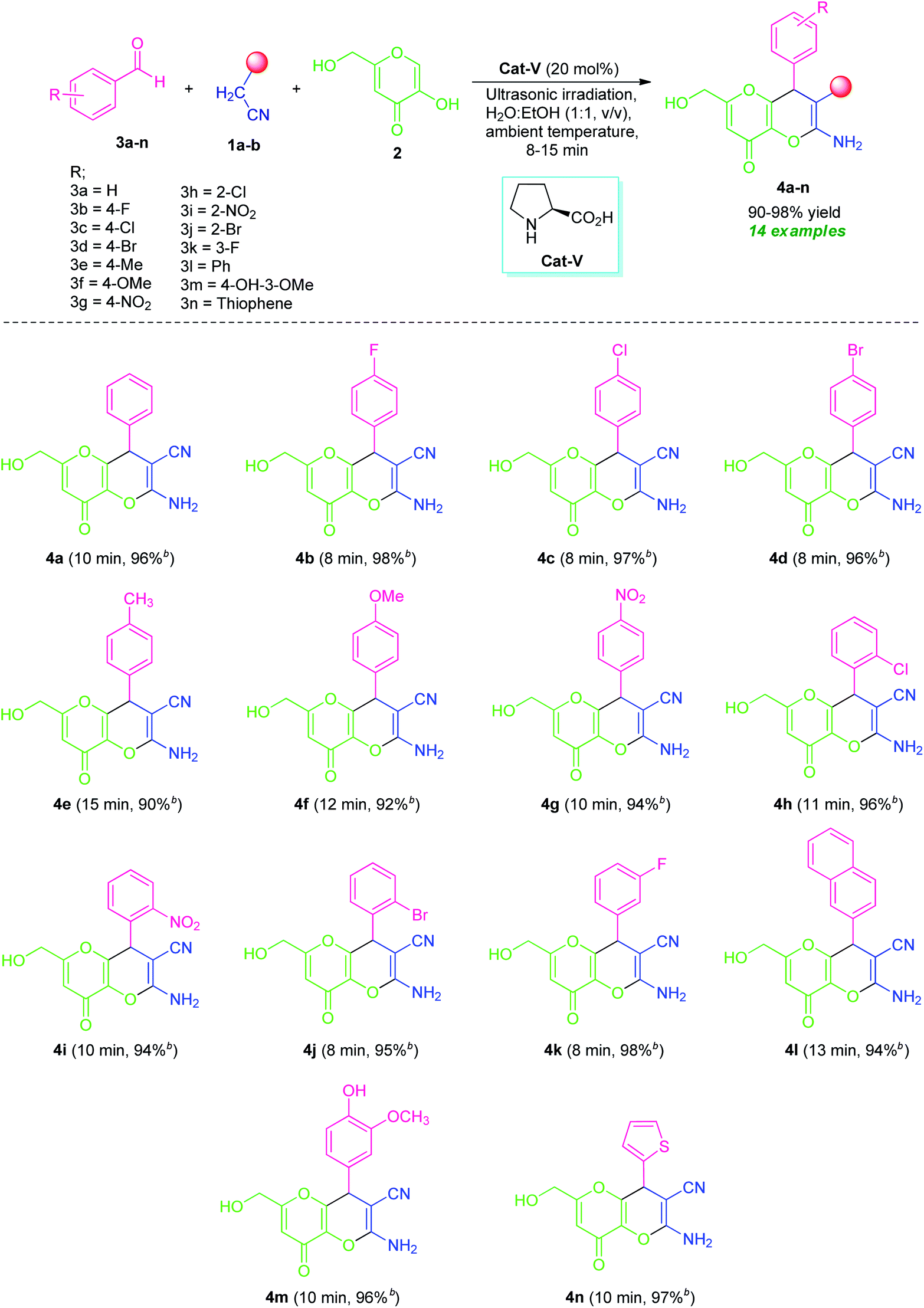
To further explore the synthetic potentiality of the current approach, we investigate this domino reaction with isatin 5a by replacing aldehyde 3a. To our delight, the reaction of active methylene compound 1a, kojic acid 2 and isatin 5a successfully worked under the similar reaction condition to deliver the corresponding product 2′-amino-6′-(hydroxymethyl)-2,8′-dioxo-8′H-spiro[indoline-3,4′-pyrano[3,2-b]pyran]-3′-carbonitrile (6a) in excellent yield (97%) within a very short reaction time. Encouraged by these results, we then attempted a series of reactions between several derivatives of isatin 5 such as 5-chloro isatin 5b/5-bromo isatin 5c/N-methyl isatin 5d/N-allyl isatin 5e/N-benzyl isatin 5f, active methylene compound 1 such as malononitrile 1a/ethyl cyanoacetate 1b, and kojic acid 2 that smoothly providing the corresponding amino-substituted spiro[indoline-3,4′-pyrano[3,2-b]pyran]-3′-carbonitrile/carboxylate derivatives 6a–j in good to excellent yields (Table 5). Variations in the yields of the obtained products were found to be not greatly influenced by the substitutions on the isatin ring. Halogen substituted isatins including chloro and bromo, are efficiently worked to provide the desired product 6b, 6c, and 6h in 98%, 97%, and 95% yield respectively within 10–12 minutes. The reaction was found to be very compatible with both N-substituted as well as N-unsubstituted isatins. For N-substituted isatin, the corresponding products 6d, 6e, 6f, 6i, and 6j derived from 5d, 5e, and 5f have been achieved in 88–97% yield at 10–15 minutes.
Inspired by these elegant examples, as well as our ongoing interest in the multicomponent synthesis of bioactive heterocycles,25 we investigated a domino four-component reaction between ninhydrin 7, benzene-1,2-diamine (8a), malononitrile 1a, and kojic acid 2 by employing the optimized reaction conditions. Pleasingly, the reaction was found to proceeded efficiently to deliver the corresponding product 2′-amino-6′-(hydroxymethyl)-8′-oxo-8′H-spiro[indeno[1,2-b]quinoxaline-11,4′-pyrano[3,2-b]pyran]-3′-carbonitrile 9a in 96% yield at only 10 minutes. To study the scope and limitation of this ultrasound-assisted organocatalytic domino four-component reaction, a series of substituted 1,2-diamine 8 such as 4-chlorobenzene-1,2-diamine (8b)/4-methylbenzene-1,2-diamine (8c)/4,5-diaminophthalonitrile (8d), and active methylene 1 including malononitrile 1a/ethyl cyanoacetate 1b were employed in the reaction. For 1,2-diamine, the aromatic ring bearing halogen substituent (chloro, entry 8b) showed to be particularly compatible for this reaction, affording the desired product 2′-amino-7-chloro-6′-(hydroxymethyl)-8′-oxo-8′H-spiro[indeno[1,2-b]quinoxaline-11,4′-pyrano[3,2-b]pyran]-3′-carbonitrile 9b and ethyl 2′-amino-7-chloro-6′-(hydroxymethyl)-8′-oxo-8′H-spiro[indeno[1,2-b]quinoxaline-11,4′-pyrano[3,2-b]pyran]-3′-carboxylate 9e in excellent yield (94% and 93% yield respectively, Table 6). Electron-donating substituents (4-Me, entry 8c) present on the phenyl ring was also amenable to this domino reaction and had no greater influences on the rate of the reaction, delivering the desired product 2′-amino-6′-(hydroxymethyl)-7-methyl-8′-oxo-8′H-spiro[indeno[1,2-b]quinoxaline-11,4′-pyrano[3,2-b]pyran]-3′-carbonitrile 9c in good yield. The reaction was limited to 4,5-diaminophthalonitrile 8d and failed to yield any product by this transformation. From our knowledge, we disclosed herein the first report of the synthesis of fused quinoxalines and pyrano[3,2-b]pyran together in a single molecule, a hybrid molecule linked through the spiro carbon at C-11 position and we hope these structural features may hold future application to medicinal and synthetic organic chemistry as well as in material science by the evaluation of their prolific pharmacological profile and optoelectronic properties.
| a Reaction condition: ninhydrin 7 (1 mmol), o-phenylenediamine 8a–d (1 mmol), active methylene 1a–b (1 mmol), and kojic acid 2 (1 mmol), with the help of 20 mol% of Cat-V in H2O:EtOH (1:1, v/v; 3 mL) under ultrasound irradiation (40 kHz, 180 W) at ambient temperature.b Isolated yield of the products. |
|---|
 |
All of the synthesized compounds were isolated pure just by filtering off the precipitate along with continuous washing by water followed by recrystallization from ethanol. Notably, the approach does not necessitate the use of time-consuming column chromatographic techniques. The structure of the synthesized compounds has been determined based on spectroscopic analysis such as FT-IR, 1H NMR, 13C NMR, and HRMS (see ESI†).
To further established the effectiveness of this ultrasound-assisted domino ‘one-pot’ reaction system, we performed a gram-scale reaction for the preparation of pyrano[3,2-b]pyran derivatives 4a, 6a, and 9a on a 10 mmol scale (Scheme 2). With the help of 20 mol% of Cat-V, the reaction between benzaldehyde (3a)/isatin (5a) with malononitrile 1a and kojic acid 2, smoothly furnished 95% and 96% yield of the respective product 4a and 6a at 15 minutes. Similarly, the gram-scale synthesis of 9a was also performed using the standard condition and the corresponding product 9a was achieved in 93% yield. Gratifyingly, the gram-scale reaction is found to be almost similar to the 1 mmol scale protocol in terms of product yield albeit with a slightly longer reaction time.
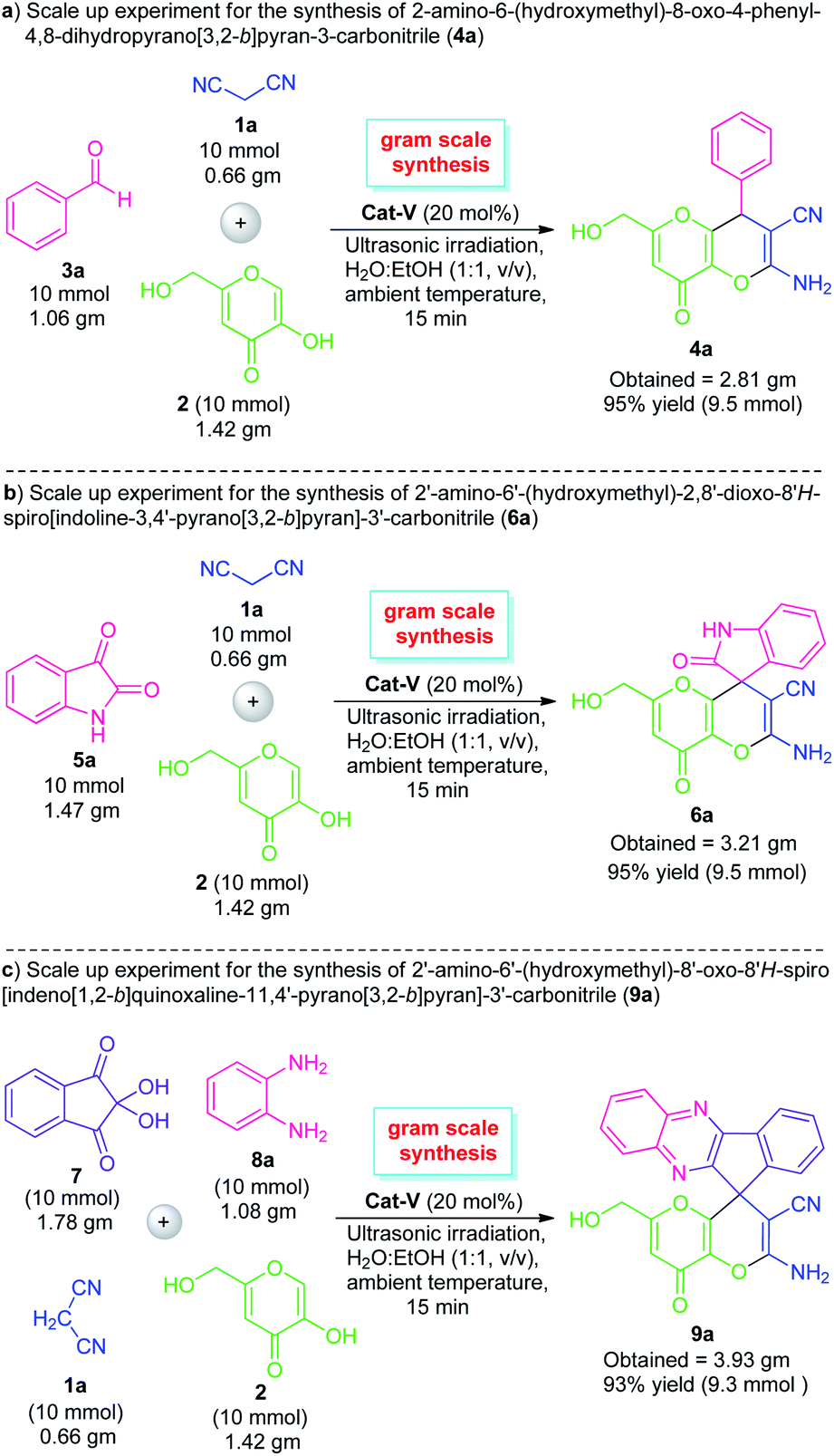 | ||
| Scheme 2 Preparative scale-up synthesis of 4a, 6a, and 9a. | ||
The synthetic applicability of the present approach was also established by the convergent synthesis of a fused amino-substituted pyrimidine derivative 11 in 68% yield over a one-step procedure from dihydropyrano[3,2-b]pyran 4h by treating it with formamide 10 (Scheme 3). The successful construction of quinoxaline, as well as pyrimidine derivatives fused with the pyrano[3,2-b]pyran core, marks the most notable aspects of this approach.
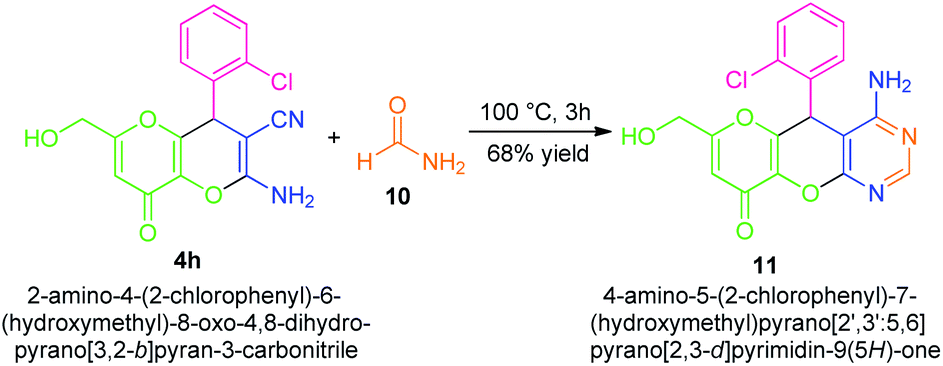 | ||
| Scheme 3 Further synthetic applications of the synthesized compounds to more complex-fused compounds 11. | ||
Recyclability experiments
Apart from this, the possibility of recyclability and sustainability of the catalyst for the present protocol was examined by employing 2-bromobenzaldehyde 3j, malononitrile 1a, and kojic acid 2 as the reaction partners (Fig. 3). After that, the solid reaction mixture was filtered off once the reaction was completed, and the filtrate containing Cat-V was recovered and successfully utilized for further subsequent reactions. From the observations, it has been revealed that there was no considerable change in the yields of the product (95–90% yields for 4j) at the end of the 4th catalytic cycle which demonstrated the effectiveness and efficiency of the catalyst for the present work.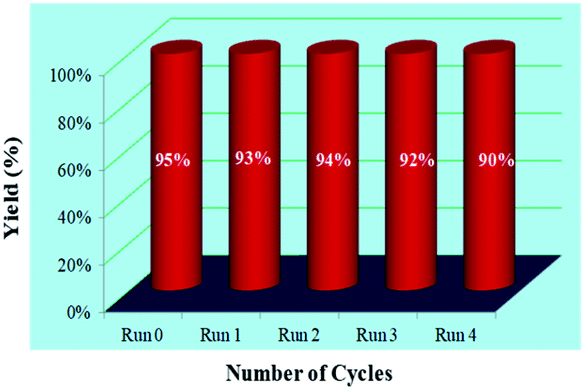 | ||
| Fig. 3 Recyclability experiment with malononitrile 1a, kojic acid 2, and 2-bromobenzaldehyde 3j for the synthesis of 4j. | ||
Plausible mechanism
To demonstrate the mechanism of this domino reaction, we executed some control experiments as shown in Scheme 4. Initially, when ninhydrin 7 was subjected to malononitrile 1a under the standard reaction condition, the reaction was found to be incomplete and failed to yield any product. Treatments of kojic acid 2 with ninhydrin 7 were also unable to form any product. These two results indicated that ninhydrin 7 was not likely to undergo either Knoevenagel condensation or Michael addition in this domino reaction. Meanwhile, a relatively accessible key intermediate 11H-indeno[1,2-b]quinoxalin-11-one 12 was successfully synthesized in 98% yield under the standard condition. The reaction of 12 with kojic acid 2 was again proved to be not compatible under the standard reaction condition. But surprisingly, the reaction of 2-(11H-indeno[1,2-b]quinoxalin-11-ylidene)malononitrile 13 with kojic acid 2 proceeded efficiently under the identical reaction condition and deliver the expected product 9a in 97% yield which indicates the possibilities of being having this intermediate in this transformation.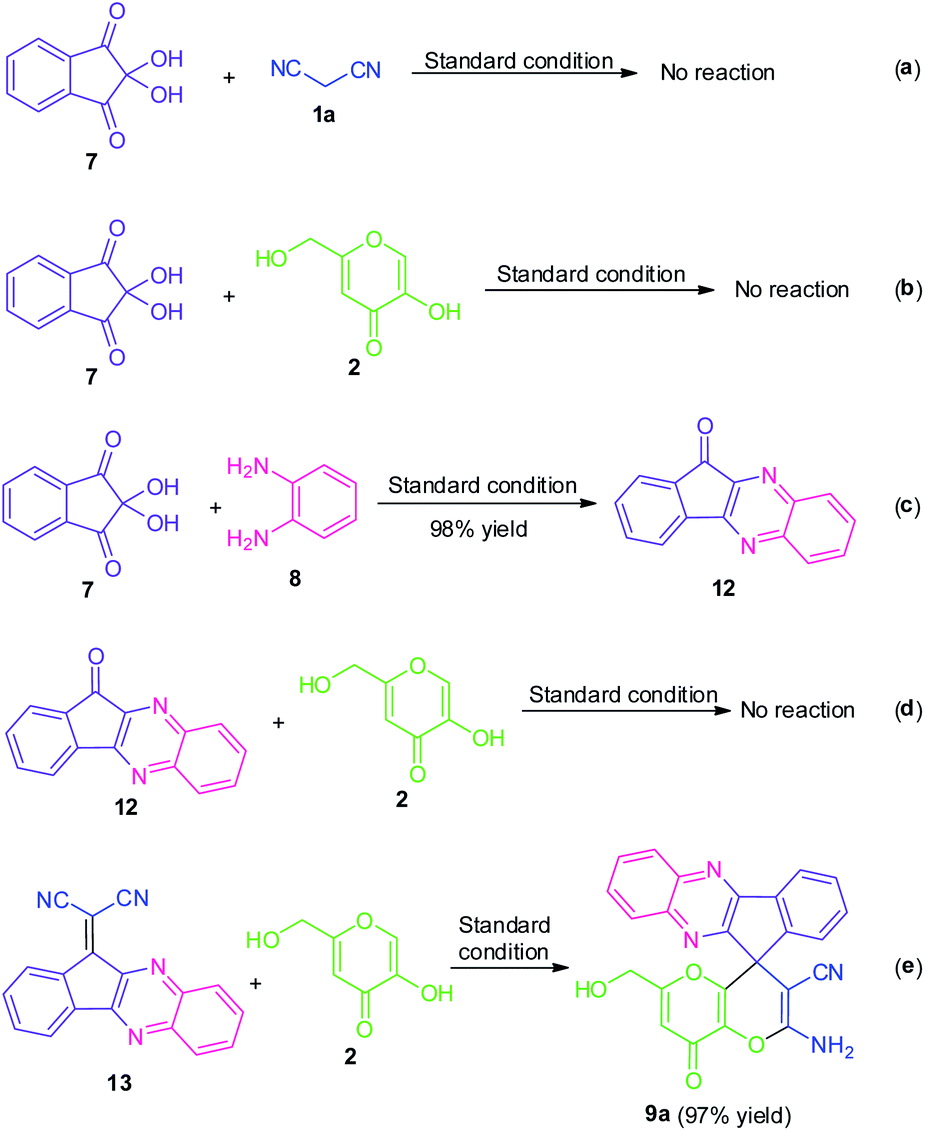 | ||
| Scheme 4 Control experiments. | ||
Based on these experimental results, we suggest the following plausible mechanistic pathway (Scheme 5) for the ultrasound-assisted synthesis of amino-substituted 8H′-spiro[indeno[1,2-b]quinoxaline-11,4′-pyrano[3,2-b]pyran]-3′-carbonitrile/carboxylate derivatives 9 from the domino four-component reaction of ninhydrin 7 with 1,2-diamine 8, active methylene 1 and kojic acid 2 under the influences of secondary amine catalyst, L-proline (Cat-V) in aqueous ethanolic solution at ambient temperature. We assume that initially, the reversible reaction of intermediate Int-1 (generated in situ from ninhydrin 7 and 1,2-diamine 8) with L-proline formed an iminium ion intermediate Int-2 that could facilitate the Knoevenagel condensation with active methylene compound 1 to form the Knoevenagel adduct Int-4 via subsequent elimination of L-proline from Int-3. Thereafter, a Michael addition between the adduct Int-4 and kojic acid 2 took place to form an intermediate Int-5 which is followed by a 6-exo-dig cyclization of –OH group to the –CN moiety to afford the corresponding products 9, after an imine–enamine tautomerization.
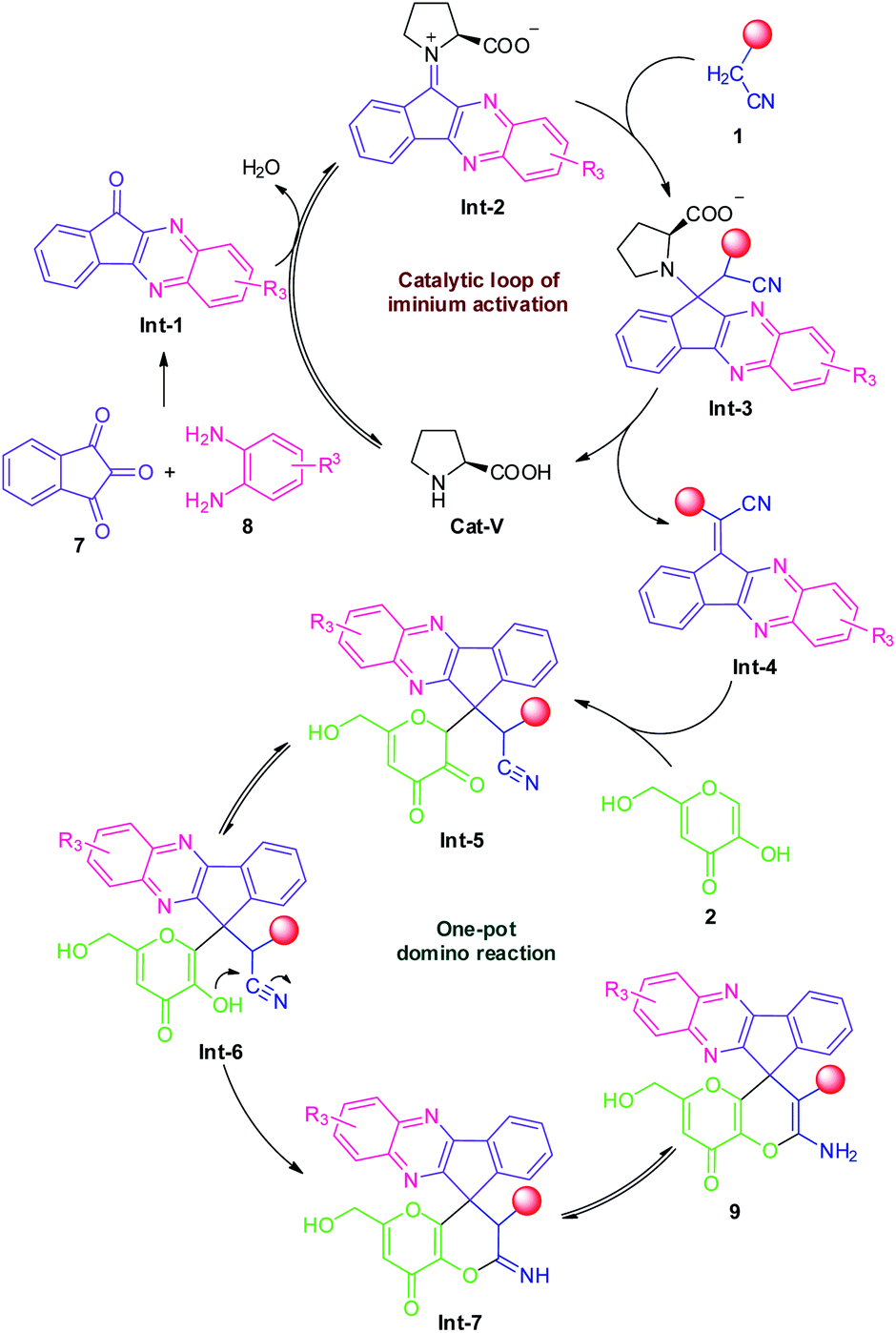 | ||
| Scheme 5 The plausible mechanism for the L-proline catalyzed synthesis of 9 via iminium activation strategy. | ||
Green chemistry metrics calculation
In order to formulate the actual progress of chemical syntheses and chemical processes in both industry and academia in the move toward more sustainable practices, the assessment of the greenness of different chemical processes is crucial.26 The quantification of sustainable practices has to lead to the development of a series of metrics to support and reinforce the behavior change of chemical technology with the aim to addressed green and sustainable chemistry.27 Most importantly used green metrics including Atom Economy (AE), atom efficiency, E-factor, Reaction Mass Efficiency (RME), Process Mass Intensity (PMI), Carbon Efficiency (CE) are calculated for our newly developed methodology towards the synthesis of pyrano[3,2-b]pyran derivatives among which we examined the value of the metrics for compound 4b, 6b and 9b.| AE = molecular weight of product ÷ Σ (molecular weight of stoichiometric reactants) × 100% |
| AE = 314.267 g mol−1 ÷ (124.112 g mol−1 + 66.061 g mol−1 + 142.109 g mol−1) × 100% = 94.57% |
For compound 6b
| AE = 371.731 g mol−1 ÷ (181.575 g mol−1 + 66.061 g mol−1 + 142.109 g mol−1) × 100% = 95.38% |
For compound 9b
| AE = 456.837 g mol−1 ÷ (178.142 g mol−1 + 142.586 g mol−1 + 66.061 g mol−1 + 142.109 g mol−1) × 100% = 86.38% |
| Atom efficiency = (yield × AE)/100 |
| Atom efficiency = (98% × 94.57%)/100 = 92.68% |
For compound 6b
| Atom efficiency = (98% × 95.38%)/100 = 93.47% |
For compound 9b
| Atom efficiency = (94% × 86.38%)/100 = 81.19% |
| E-factor = [total mass of wastes]/mass of product |
| Total mass of waste = total mass of raw materials − total mass of product |
For compound 4b
| E-factor = [(0.124 + 0.066 + 0.142) − 0.308]/0.308 = 0.07 |
For compound 6b
| E-factor = [(0.181 + 0.066 + 0.142)] − 0.364/0.364 = 0.06 |
For compound 9b
| E-factor = [(0.178 + 0.142 + 0.066 + 0.142) − 0.429]/0.429 = 0.23 |
| RME = mass of isolated product/Σ (mass of stoichiometric reactants) × 100% |
| RME = 0.308 ÷ (0.124 + 0.066 + 0.142) × 100% = 92.77% |
For compound 6b
| RME = 0.364 ÷ (0.181 + 0.066 + 0.142) × 100% = 93.58% |
For compound 9b
| RME = 0.429 ÷ (0.178 + 0.142 + 0.066 + 0.142) × 100% = 81.25% |
| PMI = Σ (mass of stoichiometric reactants + solvent)/mass of product |
| PMI = (0.124 + 0.066 + 0.142 + 0.018 + 0.046) ÷ 0.308 = 1.28 |
For compound 6b
| PMI = (0.181 + 0.066 + 0.142 + 0.018 + 0.046) ÷ 0.364 = 1.24 |
For compound 9b
| PMI = (0.178 + 0.142 + 0.066 + 0.142 + 0.018 + 0.046) ÷ 0.429 = 1.37 |
| CE = (amount of carbon in the product/total carbon present in reactant) × 100% |
| CE = [0.98 × 16/(1 × 7 + 1 × 3 + 1 × 6)] × 100% = 98% |
For compound 6b
| CE = [0.98 × 17/(1 × 8 + 1 × 3 + 1 × 6)] × 100% = 98% |
For compound 9b
| CE = [0.94 × 24/(1 × 9 + 1 × 6 + 1 × 3 + 1 × 6)] × 100% = 94% |
From Table 7, it was observed that the calculated value of atom economy and atom efficiency for compounds 4b, 6b, and 9b were much closer to the ideal value of atom economy which indicates the presence of all starting material in the final product. The ideal value of the E-factor is considered zero for a chemical process and the calculated E-factor for compounds 4b, 6b, and 9b was found to be almost similar to the ideal value that points out the avoidance of waste products from the reaction. Similarly, the value of RME ranges from 0–100%, and the greater amount of RME provides the “cleanness” of a chemical reaction. The calculated value of RME for compounds 4b, 6b, and 9b resemble the ideal value which confirms the cleanness of the presented protocol. Also, the value of PMI and CE is matched with the calculated value for compounds 4b, 6b, and 9b. The above results point towards the greenness and sustainability of the present approach.
| Entry | Green metrics | Ideal value | Compound 4b | Compound 6b | Compound 9b |
|---|---|---|---|---|---|
| 1 | Atom economy (AE) (%) | 100 | 94.57 | 95.38 | 86.38 |
| 2 | Atom efficiency (%) | 100 | 92.68 | 93.47 | 81.19 |
| 3 | E-factor | 0 | 0.07 | 0.06 | 0.23 |
| 4 | Reaction mass efficiency (RME) (%) | 100 | 92.77 | 93.58 | 81.25 |
| 5 | Process mass intensity (PMI) | 1 | 1.28 | 1.24 | 1.37 |
| 6 | Carbon efficiency (CE) (%) | 100 | 98 | 98 | 94 |
Further, we had investigated the effectiveness and efficiency of the present protocol by comparing it with the previously reported procedure for the assembly of amino-substituted dihydro-pyrano[3,2-b]pyran derivatives 4 and spiro[indoline-3,4′-pyrano[3,2-b]pyran] derivatives 6. First, we compared the previously published method (entries 1–5) for the synthesis of 4a with the present method (entry 6) and then we compared the previously published method (entries 7–10) for the synthesis of 6a with our method (entry 11). From the results depicted in Table 8, it was observed that the present protocol is undeniably more efficient in terms of reaction time, reaction temperature, energy source, solvent, product yield, and substrate scopes.
| Entry | Catalyst | Solvent | Condition | Time (min) | Yield (%) | Ref. |
|---|---|---|---|---|---|---|
| 1 | Imidazole | EtOH | Reflux | 90 | 88 | 28 |
| 2 | Nanozeolite clinoptilolite | H2O | Reflux | 25 | 95 | 29 |
| 3 | β-cyclodextrin | H2O | 70 °C | 60 | 90 | 30 |
| 4 | SnCl4/SiO2 NPs | Solvent-free | 60–70 °C | 14 | 595 | 31 |
| 5 | Fe3O4@SiO2-s-triazinium chloride MNPs | H2O | 100 °C | 40 | 94 | 14b |
| 6 | L-Proline | H2O:EtOH (1:1, v/v) |
Sonication, r.t | 10 | 96 | Present work |
| 7 | DABCO | MeOH | Reflux | 720 | 95 | 32 |
| 8 | Cu(OTf)2 | (CH2)2Cl2 | Reflux | 30 | 93 | 14d |
| 9 | SBA-15-DABCO | H2O | Reflux | 240 | 95 | 33 |
| 10 | [DABCO-H]Cl | CH3CN | 50 °C | 90 | 92 | 34 |
| 11 | L-Proline | H2O:EtOH (1:1, v/v) |
Sonication, r.t | 10 | 97 | Present work |
Conclusion
In conclusion, we have demonstrated a energy-efficient, environmentally benign, and eco-compatible one-pot domino organocatalytic methodology for the expedient access to a series of medicinally privileged amino-substituted dihydro-pyrano[3,2-b]pyrans 4a–n, spiro[indoline-3,4′-pyrano[3,2-b]pyran] (6a–j) and novel spiro[indeno[1,2-b]quinoxaline-11,4-pyrano[3,2-b]pyran]-3-carbonitrile/carboxylates (9a–e) by employing the reactivity of 5-hydroxy-2-(hydroxymethyl)-4H-pyran-4-one in aqueous ethanolic solution under ultrasonic irradiation at ambient temperature. Good to excellent yields were obtained and the product could be isolated without using column chromatography techniques. Highly economical bifunctional natural amino acid L-proline was used as a recyclable catalyst with low catalytic loading. The methodology provides a vast array of incredible advantages including utilization of readily available starting material, mild reaction condition, fast reaction rate, cost-effectiveness, easy work-up procedure, short reaction time, metal-free, ligand-free, waste-free, work in an open vessel, high conversion rate, a wide range of substrate scope that makes it more useful synthetic protocol from the perspective of green and sustainable chemistry. The ability to accomplish multiple C–C, CC, C–N, C–O bonds in a one-pot operation under the metal-free organocatalytic condition in presence of a green solvent system by employing sonochemistry as green energy input features the significant advantages of this protocol. The synthetic application of the product to more complex-fused compounds points toward another advantage of the present study (Scheme 3). The effectiveness of the methodology was also established by a scale-up reaction and it was observed to be equally efficient in all three cases. Sustainability of the present protocol was studied by using several green metrics such as Atom Economy (AE), Atom efficiency, E-factor, Reaction Mass Efficiency (RME), Process Mass Intensity (PMI), Carbon Efficiency (CE), and the entire three compounds examined were found to be very close with the ideal value that indicates the greenness of the present protocol.
Experimental section
General experimental detail
All commercially available chemicals were used without further purification. Thin Layer Chromatography (TLC) was executed utilizing silica gel 60 F254 (Merck) plates. Proton nuclear magnetic resonance spectra (1H NMR spectra) were obtained on Bruker 500 MHz FT-NMR spectrometers and 400 MHz FT-NMR in CDCl3 and DMSO solvents. 13C NMR spectra were recorded at 125 MHz and 100 MHz. Chemical shifts are reported in parts per million (ppm) relative to the TMS signal. Multiplicity is indicated as follows: s (singlet); bs (broad singlet); d (doublet); t (triplet); q (quartet); m (multiplet); dd (doublet of doublets), etc. Sonication was performed using an ultrasonic bath cleaner (model: SB-3200DT) with an operating frequency of 40 kHz and nominal power of 180 W. TOF and quadrupole mass analyzer types are used for the HRMS measurements The IR spectra were recorded using an FT-IR spectrometer (Shimadzu) in the range of 400–4000 cm−1.:1, v/v; 3 mL) was subjected to ultrasonic irradiation (40 kHz, 180 W) at ambient temperature for the indicated time. The progress of the reaction was monitored by TLC (thin layer chromatography); however, the same can be inferred by visualizing the disappearance of the colour of the starting material. After the complete consumption of starting material, as indicated by the TLC, the reaction mixture was filtered off by using filter paper and washed with dilute water, and the crude solid product was recrystallized from ethanol to give analytically pure products 4, and 6. All the compounds were fully characterized based on analytical data and detailed spectral studies including FT-IR, 1H NMR, 13C NMR, and HRMS.
2-Amino-6(hydroxymethyl)-8-oxo-4-phenyl-4,8-dihydropyrano[3,2-b]pyran-3-carbonitrile, 4a. 95% yield, white solid. Rf = 0.45 (40% EtOAc/hexane); mp 221–224 °C. IR (KBr) νmax (cm−1): 3361, 3351, 3340, 3280, 3064, 2218, 1646, 985. 1H NMR (500 MHz, CDCl3 + DMSO-d6) δ 7.38 (t, J = 7.1 Hz, 2H), 7.30 (t, J = 9.9 Hz, 3H), 7.10 (s, 2H), 6.34 (s, 1H), 5.64 (t, J = 5.9 Hz, 1H), 4.69 (s, 1H), 4.25 (dd, J = 15.9, 5.8 Hz, 1H), 4.15 (dd, J = 15.8, 5.8 Hz, 1H). 13C NMR (126 MHz, CDCl3 + DMSO-d6) δ 169.83, 168.15, 159.41, 148.61, 140.19, 136.29, 128.62, 127.68, 127.50, 119.07, 111.12, 59.19, 55.56, 40.62. HRMS (ESI+): m/z calculated for [C16H12N2O4 + H+]: 297.0875; found 297.0888.
2-Amino-4-(-fluorophenyl)-6-(hydroxymethyl)-8-oxo-4,8-dihydropyrano[3,2-b]pyran-3-carbonitrile, 4b. 98% yield, white solid. Rf = 0.4 (40% EtOAc/hexane); mp 247–249 °C. IR (KBr) νmax (cm−1): 3330, 3253, 3204, 2286, 1654, 980. 1H NMR (500 MHz, CDCl3 + DMSO-d6) δ 7.55 (s, 1H), 7.38–7.22 (m, 2H), 7.07 (t, J = 8.3 Hz, 2H), 6.43 (d, J = 27.0 Hz, 2H), 5.52 (t, J = 6.0 Hz, 1H), 4.67 (s, 1H), 4.32 (dd, J = 15.7, 6.0 Hz, 1H), 4.23 (dd, J = 16.0, 5.9 Hz, 1H). 13C NMR (126 MHz, CDCl3 + DMSO-d6) δ 169.95, 168.20, 162.78, 160.83, 159.40, 148.30, 136.20, 135.92, 129.38, 118.93, 115.51, 115.34, 111.12, 59.23, 55.52, 40.02. HRMS (ESI+): m/z calculated for [C16H11FN2O4 + H+]: 315.0781; found 315.0786.
2-Amino-4-(4-chlorophenyl)-6-(hydroxymethyl)-8-oxo-4,8-dihydropyrano[3,2-b]pyran-3-carbonitrile, 4c. 97% yield, white solid. Rf = 0.4 (40% EtOAc/hexane); mp 199–202 °C. IR (KBr) νmax (cm−1): 3357, 3290, 3152, 2215, 1676, 1590, 876. 1H NMR (500 MHz, CDCl3 + DMSO-d6) δ 7.37 (d, J = 8.4 Hz, 2H), 7.29 (d, J = 8.4 Hz, 2H), 6.96 (s, 2H), 6.38 (s, 1H), 5.64 (s, 1H), 4.70 (s, 1H), 4.28 (d, J = 18.3 Hz, 1H), 4.18 (d, J = 15.7 Hz, 1H). 13C NMR (126 MHz, CDCl3 + DMSO-d6) δ 170.39, 170.06, 168.45, 159.52, 148.21, 138.85, 136.37, 133.18, 129.31, 128.77, 119.02, 111.22, 59.84, 59.29, 55.25, 13.98. HRMS (ESI+): m/z calculated for [C16H11ClN2O4 + H+]: 331.0486; found 331.0468.
2-Amino-4-(4-bromophenyl)-6-(hydroxymethyl)-8-oxo-4,8-dihydropyrano[3,2-b]pyran-3-carbonitrile, 4d. 96% yield, white solid. Rf = 0.4 (40% EtOAc/hexane); mp 223–225 °C. IR (KBr) νmax (cm−1): 3365, 3280, 3145, 2220, 1652, 802. 1H NMR (500 MHz, CDCl3 + DMSO-d6) δ 7.52 (d, J = 8.2 Hz, 2H), 7.24 (d, J = 8.1 Hz, 2H), 7.06 (s, 2H), 6.36 (s, 1H), 5.63 (t, J = 5.8 Hz, 1H), 4.70 (s, 1H), 4.26 (dd, J = 15.7, 5.6 Hz, 1H), 4.17 (dd, J = 16.2, 5.8 Hz, 1H). 13C NMR (126 MHz, CDCl3 + DMSO-d6) δ 169.80, 168.32, 159.47, 148.17, 139.73, 136.41, 131.75, 129.83, 121.37, 119.09, 111.28, 59.21, 55.02, 48.87. HRMS (ESI+): m/z calculated for [C16H11BrN2O4 + H+]: 374.9980; found 374.9969.
2-Amino-6-(hydroxymethyl)-4-(4-nitrophenyl)-8-oxo-4,8-dihydropyrano[3,2-b]pyran-3-carbonitrile, 4g. 94% yield, light pink solid. Rf = 0.4 (40% EtOAc/hexane); mp 230–232 °C. IR (KBr) νmax (cm−1): 3538, 3454, 3329, 3176, 2194, 1670, 1649, 1520, 1352. 1H NMR (500 MHz, CDCl3 + DMSO-d6) δ 8.25 (d, J = 8.2 Hz, 2H), 7.56 (d, J = 8.2 Hz, 2H), 6.99 (s, 2H), 6.42 (s, 1H), 5.63 (d, J = 5.7 Hz, 1H), 4.88 (s, 1H), 4.29 (dd, J = 16.1, 5.4 Hz, 1H), 4.19 (dd, J = 16.2, 5.4 Hz, 1H). 13C NMR (126 MHz, DMSO-d6) δ 169.68, 168.48, 159.46, 148.09, 147.84, 147.24, 136.79, 129.42, 124.33, 119.13, 111.58, 59.14, 54.74, 30.79. HRMS (ESI+): m/z calculated for [C16H11N3O6 + H+]: 342.0726; found 342.0714.
2-Amino-4-(3-flurophenyl)-6-(hydroxymethyl)-8-oxo-4,8-dihydropyrano[3,2-b]pyran-3-carbonitrile, 4k. 98% yield, white solid. Rf = 0.42 (40% EtOAc/hexane); mp 220–222 °C. IR (KBr) νmax (cm−1): 3463, 3425, 3325, 3220, 2214, 1642. 1H NMR (500 MHz, CDCl3 + DMSO-d6) δ 7.35 (d, J = 16.9 Hz, 3H), 7.10 (d, J = 7.4 Hz, 1H), 6.51 (s, 1H), 5.82 (s, 2H), 5.15 (s, 1H), 4.66 (s, 1H), 4.38–4.31 (m, 1H), 4.30–4.23 (m, 1H). 13C NMR (126 MHz, CDCl3 + DMSO-d6) δ 170.37, 168.47, 163.60, 161.64, 159.59, 148.21, 142.34, 136.46, 130.37, 123.49, 118.89, 114.79, 111.38, 59.47, 55.66, 40.44. HRMS (ESI+): m/z calculated for [C16H11FN2O4 + H+]: 315.0781; found 315.0767.
2-Amino-4-(4-hydroxy-3-methoxyphenyl)-6-(hydroxymethyl)-8-oxo-4,8-dihydropyrano[3,2-b]pyran-3-carbonitrile, 4m. 96% yield, white solid. Rf = 0.4 (40% EtOAc/hexane); mp 218–220 °C. IR (KBr) νmax (cm−1): 3380, 3320, 3185, 2848, 2228, 1610, 975. 1H NMR (500 MHz, CDCl3 + DMSO-d6) δ 8.91 (s, 1H), 6.95 (s, 2H), 6.86–6.76 (m, 2H), 6.69 (s, 1H), 6.35 (s, 1H), 5.65 (s, 1H), 4.58 (s, 1H), 4.24 (d, J = 19.8 Hz, 1H), 4.19 (d, J = 16.1 Hz, 1H), 3.83 (s, 3H). 13C NMR (126 MHz, CDCl3 + DMSO-d6) δ 170.16, 168.30, 159.51, 149.34, 147.75, 146.32, 136.22, 131.47, 120.22, 119.49, 115.72, 111.46, 111.34, 59.47, 56.09, 55.74, 40.35. HRMS (ESI+): m/z calculated for [C17H14N2O6 + H+]: 343.0930; found 343.0914.
2′-Amino-6′-(hydroxymethyl)-2,8′-dioxo-8′H-spiro[indoline-3,4′-pyrano[3,2-b]pyran]-3′-carbonitrile, 6a. 97% yield, white solid. Rf = 0.25 (90% EtOAc/hexane); mp 235–237 °C. IR (KBr) νmax (cm−1): 3436, 3298, 3173, 2204, 1716, 1634, 1597, 1475, 1226, 1051. 1H NMR (500 MHz, CDCl3 + DMSO-d6) δ 10.66 (s, 1H), 7.78 (s, 2H), 7.28 (t, J = 7.5 Hz, 1H), 7.18 (d, J = 7.4 Hz, 1H), 7.07–7.01 (m, 1H), 6.94 (d, J = 7.4 Hz, 1H), 6.43 (s, 1H), 5.56 (t, J = 5.8 Hz, 1H), 4.17 (dd, J = 16.5, 5.9 Hz, 1H), 4.08 (dd, J = 15.4, 6.0 Hz, 1H). 13C NMR (126 MHz, CDCl3 + DMSO-d6) δ 175.39, 169.47, 168.31, 159.94, 145.59, 141.59, 137.74, 130.18, 129.56, 124.49, 122.52, 117.05, 111.27, 110.21, 59.02, 54.97, 51.13. HRMS (ESI+): m/z calculated for [C17H11N3O5 + H+]: 338.0777; found 338.0763.
2′-Amino-5-chloro-6′-(hydroxymethyl)-2,8′-dioxo-8′H-spiro[indoline-3,4′-pyrano[3,2-b]pyran]-3′-carbonitrile, 6b. 95% yield, white solid, Rf = 0.3 (90% EtOAc/hexane); mp 296–298 °C. IR (KBr) νmax (cm−1): 3310, 3190, 2204, 1738, 1636, 1590, 1480, 1444, 1215, 1060, 1031. 1H NMR (500 MHz, CDCl3 + DMSO-d6) δ 10.71 (s, 1H), 7.54 (s, 1H), 7.26 (d, J = 8.3 Hz, 1H), 7.16 (s, 1H), 6.91 (d, J = 8.3 Hz, 1H), 6.74 (s, 2H), 6.49 (s, 1H), 5.49 (t, J = 6.1 Hz, 1H), 4.21 (dd, J = 16.2, 6.0 Hz, 1H), 4.14 (dd, J = 16.4, 5.9 Hz, 1H). 13C NMR (126 MHz, CDCl3 + DMSO-d6) δ 175.17, 169.74, 168.57, 160.05, 144.90, 140.26, 137.86, 131.63, 129.63, 127.39, 124.70, 116.82, 111.60, 111.36, 59.18, 54.86, 51.31. HRMS (ESI+): m/z calculated for [C17H10ClN3O5 + H+]: 372.0387; found 372.0372.
2′-Amino-5-bromo-6′-(hydroxymethyl)-2,8′-dioxo-8′H-spiro[indoline-3,4′-pyrano[3,2-b]pyran]-3′-carbonitrile, 6c. 97% yield, white solid. Rf = 0.3 (90% EtOAc/hexane); mp 220–222 °C. IR (KBr) νmax (cm−1): 3350, 3195, 2205, 1732, 1646, 1471, 1440, 1297, 1215, 1072, 1033. 1H NMR (500 MHz, CDCl3 + DMSO-d6) δ 10.80 (s, 1H), 7.69 (d, J = 6.4 Hz, 2H), 7.41 (d, J = 6.6 Hz, 1H), 7.04 (s, 1H), 6.87 (d, J = 7.9 Hz, 1H), 6.46 (s, 1H), 5.55 (d, J = 5.4 Hz, 1H), 4.19 (dd, J = 16.2 Hz, 1H), 4.14 (dd, J = 6.3 Hz, 1H). 13C NMR (126 MHz, CDCl3 + DMSO-d6) δ 180.18, 174.97, 173.78, 165.22, 150.08, 145.87, 143.04, 137.69, 137.15, 132.60, 121.95, 119.94, 117.28, 116.56, 64.40, 60.25, 56.40. HRMS (ESI+): m/z calculated for [C17H10BrN3O5 + H+]: 415.9882; found 415.9870.
2′-Amino-6′-(hydroxymethyl)-1-methyl-2,8′-dioxo-8′H-spiro[indoline-3,4′-pyrano[3,2-b]pyran]-3′-carbonitrile, 6d. 90% yield, white solid. Rf = 0.2 (90% EtOAc/hexane); mp 270–272 °C. IR (KBr) νmax (cm−1): 3340, 3190, 3070, 2987, 2205, 1720, 1645, 1612, 1490, 1228, 1035. 1H NMR (500 MHz, CDCl3 + DMSO-d6) δ 7.50 (d, J = 7.8 Hz, 2H), 7.31 (t, J = 7.6 Hz, 1H), 7.09 (d, J = 7.1 Hz, 1H), 7.03 (t, J = 7.4 Hz, 1H), 6.89 (d, J = 7.7 Hz, 1H), 6.45 (s, 1H), 5.40–5.34 (m, 1H), 4.17 (dd, J = 16.0, 5.7 Hz, 1H), 4.05 (dd, J = 16.3, 5.7 Hz, 1H), 2.60 (s, 3H). 13C NMR (101 MHz, CDCl3 + DMSO-d6) δ 175.74, 169.72, 168.28, 167.04, 159.91, 147.23, 143.29, 136.56, 132.44, 128.68, 122.81, 122.49, 110.93, 107.70, 73.55, 59.01, 50.39, 26.43. HRMS (ESI+): m/z calculated for [C18H13N3O5 + H+]: 352.0933; found 325.0915.
2′-Amino-1-benzyl-6′-(hydroxymethyl)-2,8′-dioxo-8′H-spiro[indoline-3,4′-pyrano[3,2-b]pyran]-3′-carbonitrile, 6f. 96% yield, white solid. Rf = 0.21 (90% EtOAc/hexane); mp 234–236 °C. IR (KBr) νmax (cm−1): 3428, 3326, 2931, 2190, 1705, 1635, 1610, 1486, 1468, 1207. 1H NMR (500 MHz, CDCl3 + DMSO-d6) δ 7.57 (s, 2H), 7.34 (s, 4H), 7.26 (d, J = 7.5 Hz, 2H), 7.12 (d, J = 7.1 Hz, 1H), 6.80 (s, 2H), 6.49 (s, 1H), 5.50 (s, 1H), 5.12 (d, J = 15.8 Hz, 1H), 4.88 (d, J = 16.2 Hz, 1H), 4.15 (s, 1H), 4.10 (s, 1H). 13C NMR (126 MHz, CDCl3 + DMSO-d6) δ 174.10, 169.58, 168.49, 160.22, 145.42, 142.05, 137.78, 135.03, 129.92, 129.54, 128.50, 127.41, 126.72, 124.65, 123.55, 117.11, 111.47, 109.78, 59.05, 54.61, 50.97, 43.53. HRMS (ESI+): m/z calculated for [C24H17N3O5 + H+]: 428.1246; found 428.1220.
Ethyl 2′-amino-6′-(hydroxymethyl)-2,8′-dioxo-8′H-spiro[indoline-3,4′-pyrano[3,2-b]pyran]-3′-carboxylate, 6g. 94% yield, white solid. Rf = 0.2 (90% EtOAc/hexane); mp 283–285 °C. IR (KBr) νmax (cm−1): 3438, 3290, 2916, 1720, 1688, 1656, 1640, 1619, 1522, 1474, 1295, 1210. 1H NMR (500 MHz, CDCl3 + DMSO-d6) δ 10.22 (s, 1H), 7.55 (s, 2H), 7.19 (t, J = 7.3 Hz, 1H), 7.03 (d, J = 6.4 Hz, 1H), 6.94 (t, J = 7.4 Hz, 1H), 6.89 (t, J = 6.8 Hz, 1H), 6.44 (s, 1H), 5.42 (t, J = 5.8 Hz, 1H), 4.24–4.17 (m, 1H), 4.09 (d, J = 15.9 Hz, 1H), 3.91–3.79 (m, 2H), 0.85 (t, J = 6.4 Hz, 3H). 13C NMR (126 MHz, CDCl3 + DMSO-d6) δ 177.69, 169.93, 168.35, 167.05, 159.94, 147.71, 142.08, 136.53, 133.50, 128.38, 123.01, 121.74, 110.98, 109.37, 73.50, 59.13, 51.16, 50.27, 12.93. HRMS (ESI+): m/z calculated for [C19H16N2O7 + H+]: 358.1036; found 358.1052.
Ethyl 2′-amino-1-benzyl-6′-(hydroxymethyl)-2,8′-dioxo-8′H-spiro[indoline-3,4′-pyrano[3,2-b]pyran]-3′-carboxylate, 6j. 97% yield, white solid. Rf = 0.21 (90% EtOAc/hexane); mp 240–242 °C. IR (KBr) νmax (cm−1): 3384, 3286, 3070, 2986, 1692, 1676, 1650, 1624, 1610, 1520, 1225, 1092. 1H NMR (500 MHz, CDCl3 + DMSO-d6) δ 7.96 (s, 2H), 7.45–7.34 (m, 2H), 7.34–7.27 (m, 1H), 7.26–7.18 (m, 1H), 7.17–7.09 (m, 1H), 7.05–6.97 (m, 1H), 6.80 (dd, J = 7.7, 2.8 Hz, 1H), 6.42 (s, 1H), 5.61 (dd, J = 7.6, 4.5 Hz, 1H), 4.58 (dd, J = 15.7, 2.7 Hz, 1H), 4.17–4.07 (m, 1H), 4.07–3.96 (m, 1H), 3.86–3.77 (m, 1H), 3.76–3.67 (m, 1H), 0.79 (t, 2H). 13C NMR (101 MHz, CDCl3 + DMSO-d6) δ 175.65, 169.39, 168.24, 166.73, 160.01, 147.25, 142.60, 136.48, 135.79, 132.72, 128.59, 128.29, 127.20, 126.88, 123.08, 122.57, 111.13, 108.39, 73.12, 58.92, 58.76, 50.59, 43.39, 13.27. HRMS (ESI+): m/z calculated for [C26H22N2O7 + H+]: 475.1505; found 475.1530.
:1, v/v; 3 mL) was irradiated under ultrasound (40 kHz, 180 W) at ambient temperature for the required time period. After the complete consumption of starting material as indicated by TLC, the residue was poured on ice water and the solid was filtered. The crude product was further washed with cold ethanol to afford analytically pure products 9.
2′-Amino-6′-(hydroxymethyl)-8′-oxo-8′H-spiro[indeno[1,2-b]quinoxaline-11,4′-pyrano[3,2-b]pyran]-3′-carbonitrile, 9a. 96% yield, white solid. Rf = 0.3 (90% EtOAc/hexane); mp 278–280 °C. IR (KBr) νmax (cm−1): 3467, 3277, 3233, 3152, 2194, 1679, 1661, 1633, 1597, 1426, 1339, 1228, 1208, 1062. 1H NMR (500 MHz, CDCl3 + DMSO-d6) δ 8.20 (t, J = 7.4 Hz, 2H), 8.14 (d, J = 7.9 Hz, 1H), 8.05 (s, 1H), 7.86 (t, J = 7.4 Hz, 1H), 7.80 (t, J = 7.3 Hz, 1H), 7.70 (s, 3H), 7.47 (s, 2H), 6.35 (s, 1H), 5.42 (t, J = 5.8 Hz, 1H), 3.81 (d, J = 5.8 Hz, 2H). 13C NMR (126 MHz, CDCl3 + DMSO-d6) δ 168.99, 167.79, 161.09, 159.59, 152.32, 147.45, 146.36, 141.93, 140.59, 137.50, 135.81, 131.97, 129.50, 128.33, 121.47, 116.63, 110.72, 58.27, 55.59, 50.06. HRMS (ESI+): m/z calculated for [C24H14N4O4 + H+]: 423.1093; found 423.1093.
2′-Amino-6′-(hydroxymethyl)-7-methyl-8′-oxo-8′H-spiro[indeno[1,2-b]quinoxaline-11-4′-pyrano[3,2-b]pyran]-3′-carbonitrile, 9c. 90% yield, pale yellow solid. Rf = 0.2 (90% EtOAc/hexane); mp 260–262 °C. IR (KBr) νmax (cm−1): 3397, 3310, 3250, 2951, 2833, 2190, 1710, 1670, 1646, 1621, 1579, 1453, 1409, 1353, 1208, 1136, 1062. 1H NMR (500 MHz, CDCl3 + DMSO-d6) 8.20 (s, 1H), 8.02–7.94 (m, 1H), 7.69–7.56 (m, 5H), 6.72 (s, 2H), 6.43 (s, 1H), 5.30 (s, 1H), 3.85 (s, 2H), 2.64 (s, 3H). 13C NMR (126 MHz, CDCl3 + DMSO-d6) δ 170.00, 168.43, 160.23, 160.16, 152.78, 147.74, 147.22, 142.58, 140.71, 139.61, 138.04, 136.48, 132.10, 131.34, 129.87, 128.49, 127.72, 125.13, 121.94, 117.03, 111.17, 58.94, 57.03, 50.32, 21.40. HRMS (ESI+): m/z calculated for [C25H16N4O4 + H+]: 437.1250; found 437.1265.
Ethyl 2′-amino-6′-(hydroxymethyl)-8′-oxo-8′H-spiro[indeno[1,2-b]quinoxaline-11,4′-pyrano[3,2-b]pyran]-3′-carboxylate, 9d. 95% yield, white solid. Rf = 0.25 (90% EtOAc/hexane); M. P. 296–298 °C. IR (KBr) νmax (cm−1): 3435, 3390, 3290, 2961, 2914, 2713, 1981, 1720, 1688, 1656, 1640, 1522, 1474, 1295, 1210. 1H NMR (400 MHz, CDCl3 + DMSO-d6) δ 8.22–8.10 (m, 2H), 8.05 (d, J = 8.1 Hz, 1H), 7.81–7.67 (m, 4H), 7.55 (ddd, J = 19.9, 6.6, 3.3 Hz, 3H), 6.34 (d, J = 1.0 Hz, 1H), 5.25 (d, J = 5.6 Hz, 1H), 3.76 (dd, J = 14.2, 8.3 Hz, 2H), 3.46–3.31 (m, 2H), 0.25 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3 + DMSO-d6) δ 169.27, 167.50, 166.12, 159.61, 153.71, 150.16, 148.23, 141.03, 139.94, 136.22, 135.96, 135.13, 131.02, 128.55, 128.01, 127.88, 123.36, 120.76, 112.43, 110.16, 73.92, 58.14, 57.79, 49.94, 49.17, 11.90. HRMS (ESI+): m/z calculated for [C26H19N3O6 + H+]: 470.1352; found 470.1375.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
BB thanks UGC-India for the fellowship. BB thanks Central Instrumentation Facility (CIF), Central University of Gujarat. Author thanks the Central University of Gujarat for the infrastructure to carry out the work.Notes and references
- (a) A. R. Katritzky, Chem. Heterocycl. Compd., 1992, 28, 241–259 CrossRef; (b) J. A. Joule, Adv. Heterocycl. Chem., 2016, 119, 81–106 CrossRef CAS; (c) A. P. Taylor, R. P. Robinson, Y. M. Fobian, D. C. Blakemore, L. H. Jones and O. Fadeyi, Org. Biomol. Chem., 2016, 14, 6611–6637 RSC; (d) B. Borah, K. D. Dwivedi and L. R. Chowhan, Arkivoc, 2021,(part i), 273–278 Search PubMed; (e) K. D. Dwivedi, B. Borah and L. R. Chowhan, Front. Chem., 2020, 7, 944 Search PubMed; (f) B. Kumar, M. S. Reddy, K. D. Dwivedi, A. Dahiya, J. N. Babu and L. R. Chowhan, Appl. Organomet. Chem., 2022, 36, e6455 CrossRef CAS.
- (a) A. Beélik, Kojic acid, in Advances in Carbohydrate Chemistry, Academic Press, 1956, vol. 11, pp. 145–183 Search PubMed; (b) H. E. Morton, W. Kocholaty, R. Junowicz-Kocholaty and A. Kelner, J. Bacteriol., 1945, 50, 579–584 CrossRef CAS PubMed; (c) T. Yabuta, J. Electrochem. Soc. Jpn., 1916, 37, 1185–1233 CAS.
- (a) G. Karakaya, A. Türe, A. Ercan, S. Öncül and M. D. Aytemir, Bioorg. Chem., 2019, 88, 102950 CrossRef PubMed; (b) M. D. Aytemir, B. Özçelik and G. Karakaya, Bioorg. Med. Chem. Lett., 2013, 23, 3646–3649 CrossRef CAS PubMed; (c) M. D. Aytemir and B. Özçelik, Eur. J. Med. Chem., 2010, 45, 4089–4095 CrossRef CAS PubMed; (d) G. Karakaya, A. Ercan, S. Oncul and M. D. Aytemir, Anti-Cancer Agents Med. Chem., 2018, 18, 2137–2148 CrossRef CAS PubMed; (e) S. Oncul, G. Karakaya, M. Dilsiz Aytemir and A. Ercan, Chem. Biol. Drug Des., 2019, 94, 2084–2093 CrossRef CAS PubMed.
- (a) G. A. Burdock, M. G. Soni and I. G. Carabin, Regul. Toxicol. Pharmacol., 2001, 33, 80–101 CrossRef CAS PubMed; (b) K. Uchino, M. Nagawa, Y. Tonosaki, M. Oda and A. Fukuchi, Agric. Biol. Chem., 1988, 52, 2609–2610 CAS.
- (a) M. Uher, J. Brtko, O. Rajniakova, M. Kovac and E. Novotana, Parfumerie und Kosmetik, 1993, 74, 554–556 CAS; (b) A. K. Gupta, M. D. Gover, K. Nouri and S. Taylor, J. Am. Acad. Dermatol., 2006, 55, 1048–1065 CrossRef PubMed; (c) M. Saeedi, M. Eslamifar and K. Khezri, Biomed. Pharmacother., 2019, 110, 582–593 CrossRef CAS PubMed.
- (a) J. Marui, N. Yamane, S. Ohashi-Kunihiro, T. Ando, Y. Terabayashi, M. Sano and M. Machida, J. Biosci. Bioeng., 2011, 112, 40–43 CrossRef CAS PubMed; (b) M. Uher, V. Konecny and O. Rajniakova, Chem. Pap., 1994, 48, 282–284 CAS; (c) R. M. Saleh, S. A. Kabli, S. M. Al-Garni and S. A. Mohamed, Afr. J. Microbiol. Res., 2011, 5, 1619–1628 Search PubMed.
- (a) R. Mohamad, M. S. Mohamed, N. Suhaili, M. M. Salleh and A. B. Ariff, Biotechnol. Mol. Biol. Rev., 2010, 5, 24–37 CAS; (b) S. Katoh, J. Toyama, I. Kodama, K. Kamiya, T. Akita and T. Abe, Eur. Surg. Res., 1992, 24, 349–355 CrossRef CAS PubMed; (c) B. Ochiai, M. Kamiya and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3493–3498 CrossRef CAS.
- (a) C. Balakrishna, N. Payili, S. Yennam, P. U. Devi and M. Behera, Bioorg. Med. Chem. Lett., 2015, 25, 4753–4756 CrossRef CAS PubMed; (b) K. Morisaki and S. Ozaki, Chem. Pharm. Bull., 1996, 44, 1647–1655 CrossRef CAS PubMed; (c) Y. Wei, C. Zhang, P. Zhao, X. Yang and K. Wang, J. Inorg. Biochem., 2011, 105, 1081–1085 CrossRef CAS PubMed.
- B. Borah, K. D. Dwivedi and L. R. Chowhan, Polycyclic Aromat. Compd., 2021, 1–45, DOI:10.1080/10406638.2021.1962923.
- J. G. Hall and J. A. Reiss, Aust. J. Chem., 1986, 39, 1401–1409 CrossRef CAS.
- A. D. Wright, G. M. König, R. de Nys and O. Sticher, J. Nat. Prod., 1993, 56, 394–401 CrossRef CAS.
- A. P. Kozikowski and J. Lee, J. Org. Chem., 1990, 55, 863–870 CrossRef CAS.
- E. Lee, C. M. Park and J. S. Yun, J. Am. Chem. Soc., 1995, 117, 8017–8018 CrossRef CAS.
- (a) F. O. Memar, L. Khazdooz, A. Zarei and A. Abbaspourrad, Med. Chem. Res., 2020, 29, 1792–1803 CrossRef; (b) D. Azarifar, H. Ebrahimiasl, R. Karamian and M. Ahmadi-Khoei, J. Iran. Chem. Soc., 2019, 16, 341–354 CrossRef CAS; (c) S. Asghari, R. Baharfar, M. Alimi, M. Ahmadipour and M. Mohseni, Monatsh. Chem., 2014, 145, 1337–1342 CrossRef CAS; (d) K. Parthasarathy, C. Praveen, C. Balachandran, S. Ignacimuthu and P. T. Perumal, Bioorg. Med. Chem. Lett., 2013, 23, 2708–2713 CrossRef CAS PubMed.
- (a) P. Saraswat, G. Jeyabalan, M. Z. Hassan, M. U. Rahman and N. K. Nyola, Synth. Commun., 2016, 46, 1643–1664 CrossRef CAS; (b) G. S. Singh and Z. Y. Desta, Chem. Rev., 2012, 112, 6104–6155 CrossRef CAS PubMed; (c) A. Ding, M. Meazza, H. Guo, J. W. Yang and R. Rios, Chem. Soc. Rev., 2018, 47, 5946–5996 RSC.
- (a) B. Banerjee, Ultrason. Sonochem., 2017, 35, 15–35 CrossRef CAS PubMed; (b) G. Brahmachari, M. Mandal, I. Karmakar, K. Nurjamal and B. Mandal, ACS Sustainable Chem. Eng., 2019, 7, 6369–6380 CrossRef CAS; (c) T. J. Mason, Chem. Soc. Rev., 1997, 26, 443–451 RSC; (d) R. B. Nasir Baig and R. S. Varma, Chem. Soc. Rev., 2012, 41, 1559–1584 RSC.
- (a) G. Chatel, Ultrason. Sonochem., 2018, 40, 117–122 CrossRef CAS PubMed; (b) P. Cintas and J. L. Luche, Green Chem., 1999, 1, 115–125 RSC; (c) B. Borah, K. D. Dwivedi, B. Kumar and L. R. Chowhan, Arabian J. Chem., 2022, 15, 103654, DOI:10.1016/j.arabjc.2021.103654.
- (a) G. Chatel and R. S. Varma, Green Chem., 2019, 21, 6043–6050 RSC; (b) C. Einhorn, J. Einhorn and J. L. Luche, Synthesis, 1989, 1989, 787–813 CrossRef; (c) M. Vinatoru and T. J. Mason, Ultrason. Sonochem., 2019, 52, 2–5 CrossRef CAS PubMed.
- (a) B. Borah and L. R. Chowhan, RSC Adv., 2021, 11, 37325–37353 RSC; (b) B. Borah, K. D. Dwivedi and L. R. Chowhan, Asian J. Org. Chem., 2021, 10, 3101–3126 CrossRef CAS.
- (a) I. Nakamura and Y. Yamamoto, Chem. Rev., 2004, 104, 2127–2198 CrossRef CAS PubMed; (b) D. M. D'Souza and T. J. Mueller, Chem. Soc. Rev., 2007, 36, 1095–1108 RSC.
- (a) S. Bertelsen and K. A. Jørgensen, Chem. Soc. Rev., 2009, 38, 2178–2189 RSC; (b) C. F. Barbas III, Angew. Chem., Int. Ed., 2008, 47, 42–47 CrossRef PubMed; (c) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2001, 40, 3726–3748 CrossRef CAS; (d) J. Seayad and B. List, Org. Biomol. Chem., 2005, 3, 719–724 RSC; (e) H. Pellissier, Adv. Synth. Catal., 2012, 354, 237–294 CrossRef CAS; (f) P. Melchiorre, M. Marigo, A. Carlone and G. Bartoli, Angew. Chem., Int. Ed., 2008, 47, 6138–6171 CrossRef CAS PubMed; (g) B. Borah, K. D. Dwivedi and L. R. Chowhan, RSC Adv., 2021, 11, 13585–13601 RSC; (h) B. Borah, K. D. Dwivedi and L. R. Chowhan, Asian J. Org. Chem., 2021, 10, 2709–2762 CrossRef CAS; (i) P. Renzi and M. Bella, Chem. Commun., 2012, 48, 6881–6896 RSC; (j) Q. Ren and J. Wang, Asian J. Org. Chem., 2013, 2, 542–557 CrossRef CAS.
- (a) B. S. Vachan, M. Karuppasamy, P. Vinoth, S. Vivek Kumar, S. Perumal, V. Sridharan and J. C. Menéndez, Adv. Synth. Catal., 2020, 362, 87–110 CrossRef CAS; (b) M. Gruttadauria, F. Giacalone and R. Noto, Chem. Soc. Rev., 2008, 37, 1666–1688 RSC.
- (a) B. Alcaide, P. Almendros, A. Luna and M. R. Torres, J. Org. Chem., 2006, 71, 4818–4822 CrossRef CAS PubMed; (b) J. M. Janey, Y. Hsiao and J. D. Armstrong, J. Org. Chem., 2006, 71, 390–392 CrossRef CAS PubMed; (c) M. Venkatanarayana and P. K. Dubey, Synth. Commun., 2012, 42, 1746–1759 CrossRef CAS; (d) B. M. Choudary, C. V. Rajasekhar, G. Gopi Krishna and K. Rajender Reddy, Synth. Commun., 2007, 37, 91–98 CrossRef CAS; (e) W. Notz, F. Tanaka and C. F. Barbas, Acc. Chem. Res., 2004, 37, 580–591 CrossRef CAS PubMed; (f) J. S. Yadav, S. P. Kumar, G. Kondaji, R. S. Rao and K. Nagaiah, Chem. Lett., 2004, 33, 1168–1169 CrossRef CAS; (g) S. M. Rajesh, B. D. Bala, S. Perumal and J. C. Menendez, Green Chem., 2011, 13, 3248–3254 RSC; (h) A. Kumar, M. K. Gupta and M. Kumar, Green Chem., 2012, 14, 290–295 RSC.
- (a) B. B. Toure and D. G. Hall, Chem. Rev., 2009, 109, 4439–4486 CrossRef CAS PubMed; (b) C. de Graaff, E. Ruijter and R. V. Orru, Chem. Soc. Rev., 2012, 41, 3969–4009 RSC; (c) B. H. Rotstein, S. Zaretsky, V. Rai and A. K. Yudin, Chem. Rev., 2014, 114, 8323–8359 CrossRef CAS PubMed.
- (a) M. S. Reddy, L. R. Chowhan, N. S. Kumar, P. Ramesh and S. B. Mukkamala, Tetrahedron Lett., 2018, 59, 1366–1371 CrossRef CAS; (b) M. S. Reddy, N. S. Kumar and L. R. Chowhan, RSC Adv., 2018, 8, 35587–35593 RSC; (c) V. Singh, S. R. Lakshmi and L. R. Chowhan, Front. Chem., 2021, 9, 759436 CrossRef CAS PubMed; (d) P. Ramesh, M. S. Reddy, N. S. Kumar, B. Rathod, R. S. Prakasham and L. R. Chowan, ChemistrySelect, 2018, 3, 9096–9101 CrossRef CAS.
- C. Jiménez-González, D. J. Constable and C. S. Ponder, Chem. Soc. Rev., 2012, 41, 1485–1498 RSC.
- (a) R. A. Sheldon, ACS Sustainable Chem. Eng., 2018, 6, 32–48 CrossRef CAS; (b) C. Jimenez-Gonzalez, C. S. Ponder, Q. B. Broxterman and J. B. Manley, Org. Process Res. Dev., 2011, 15, 912–917 CrossRef CAS; (c) J. Augé, Green Chem., 2008, 10, 225–231 RSC; (d) D. J. Constable, A. D. Curzons and V. L. Cunningham, Metrics to ‘green’chemistry—which are the best?, Green Chem., 2002, 4, 521–527 RSC.
- M. N. Khan, S. Pal, S. Karamthulla and L. H. Choudhury, RSC Adv., 2014, 4, 3732–3741 RSC.
- S. M. Baghbanian, N. Rezaei and H. Tashakkorian, Green Chem., 2013, 15, 3446–3458 RSC.
- E. A. Kataev, M. R. Reddy, G. N. Reddy, V. H. Reddy, C. S. Reddy and B. V. S. Reddy, New J. Chem., 2016, 40, 1693–1697 RSC.
- H. R. Molaei, B. Sadeghi and M. H. Moslemin, Bulg. Chem. Commun., 2017, 41, 308–314 RSC.
- A. Rahmati, Z. Khalesi and T. Kenarkoohi, Comb. Chem. High Throughput Screening, 2014, 17, 132–140 CrossRef CAS PubMed.
- R. Azimi and R. Baharfar, Can. J. Chem., 2014, 92, 1163–1168 CrossRef CAS.
- M. M. Li, C. S. Duan, Y. Q. Yu and D. Z. Xu, Dyes Pigm., 2018, 150, 202–206 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01917e |
| This journal is © The Royal Society of Chemistry 2022 |