
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFerrocene as a potential electrochemical reporting surrogate of abasic sites in DNA†
Chiara
Figazzolo
 ab,
Yifeng
Ma
c,
James H. R.
Tucker
*c and
Marcel
Hollenstein
*a
ab,
Yifeng
Ma
c,
James H. R.
Tucker
*c and
Marcel
Hollenstein
*a
aInstitut Pasteur, Université Paris Cité, Department of Structural Biology and Chemistry, Laboratory for Bioorganic Chemistry of Nucleic Acids, CNRS UMR3523, 28, rue du Docteur Roux, 75724 Paris Cedex 15, France. E-mail: marcel.hollenstein@pasteur.fr
bLearning Planet Institute, 8, rue Charles V, 75004 Paris, France
cSchool of Chemistry, University of Birmingham, Birmingham, B15 2TT, UK. E-mail: j.tucker@bham.ac.uk
First published on 5th October 2022
Abstract
Methods for the real-time monitoring of the substrate acceptance of modified nucleotides by DNA polymerases are in high demand. In a step towards this aim, we have incorporated ferrocene-based abasic nucleotides into DNA templates and evaluated their compatibility with enzymatic synthesis of unmodified and modified DNA. All canonical nucleotides can be incorporated opposite ferrocene sites with a strong preference for purines. DNA polymerases with lesion-bypass capacity such as Dpo4 allow DNA synthesis to be resumed beyond the site of incorporation. Modified purine nucleotides can readily be incorporated opposite ferrocene basic site analogs, while pyrimidine nucleotides decorated with simple side-chains are also readily tolerated. These findings open up directions for the design of electrochemical sensing devices for the monitoring of enzymatic synthesis of natural or modified DNA.
Introduction
Interest in chemical modification of nucleic acids stems from early developments of nucleoside and nucleotide-based antiviral drugs as well as of antisense oligonucleotide therapeutic agents.1–4 The need for synthesizing oligonucleotides equipped with chemical alterations has been spurred further by the recent advent of mRNA-based vaccines.5,6 In this context, besides solid-phase synthesis,7–9 modified oligonucleotides can be constructed using polymerase-catalysed incorporation of unnatural nucleoside triphosphates ((d)N*TPs).10 This method is particularly alluring since it is compatible with Darwinian evolution methods to identify functional nucleic acids and it is not limited in terms of size or nature of the functional groups that can be incorporated.11–20 In the latter, modified oligonucleotides can be accessed either by primer extension (PEX) reactions or under PCR amplification conditions by substituting one or multiple canonical nucleotides with the corresponding unnatural counterparts. Natural or engineered polymerases are particularly tolerant to nucleotides bearing modifications attached at position C5 of pyrimidine or N7 of 7-deazapurine nucleotides or with simple backbone modifications but can also be coerced to incorporate nucleotides with sugar or unnatural base surrogates.21–36In order to evaluate whether modified nucleotides are accepted as substrates by polymerases, the products stemming from PEX reactions or PCR are evaluated by gel electrophoresis and by MS techniques including MALDI, ESI-MS, and LC-MS. On the other hand, real-time monitoring of the incorporation of nucleoside triphosphates, particularly when equipped with chemical modifications, is a more difficult undertaking and can only be achieved by techniques such as nanopore sequencing37,38 fluorescence microscopy,39 or by the application of FRET-based methods.40,41 These techniques often require the positioning of one or multiple dyes on oligonucleotides or incoming nucleotides or necessitate the use of specialized equipment. Hence, simple methods that enable the real-time monitoring of incorporation of canonical and modified nucleotides into DNA and RNA by polymerases are in high demand since they would reduce the workload involved in the biochemical characterization of modified nucleotides. In this article, we suggest first steps towards the development of monitoring devices based on an electrochemical read-out mediated by a chemical analog of a THF-abasic site (Φ in Fig. 1). Apurinic/apyrimidinic (AP) sites or abasic sites are one of the most common DNA lesions and occur thousands of times in mammalian sites after hydrolysis of the glycosidic bond.42,43 Besides their biological and clinical relevance mainly caused by their inherent highly mutagenic nature, AP sites are also interesting modifications for the development of tools in chemical biology. For instance, the formation of AP sites combined after modification of DNA with 5-iodo-deoxyuridine was harnessed to explore the role of stochastic variations in gene expression.44 In addition, AP sites have also been employed to gauge the effect of nucleobase shape as well as their solvation energies, hydrogen bonding capacities, and π–electron interactions on the DNA polymerisation cycle.45–48
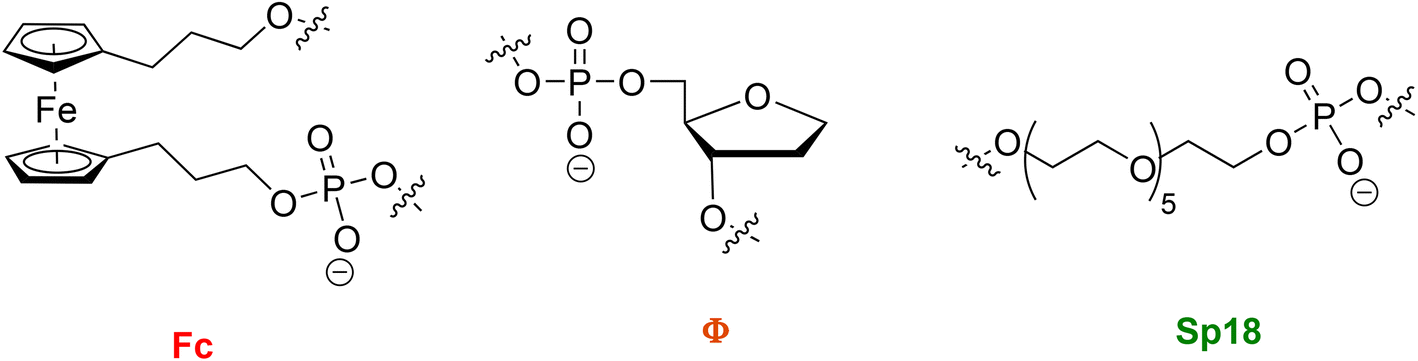 | ||
| Fig. 1 Chemical structures of abasic site analogs investigated in PEX reactions. | ||
Here, we have explored the possibility of substituting one or two tetrahydrofuran (THF in Fig. 1) deoxyribose sugar units with a ferrocene moiety (Fc in Fig. 1) which could concomitantly be used to electrochemically monitor the incorporation of nucleotides by polymerases. In a first step towards this aim, we provide here a thorough biochemical investigation of incorporation assays of canonical and modified nucleotides opposite ferrocene-modified abasic sites. This has been achieved by incorporating a ferrocene unit into the backbone of various DNA templates using solid-phase synthesis.49 These ferrocene-containing templates were then used in PEX reactions with natural and modified nucleotides. Overall, we demonstrate that ferrocene moieties incorporated into DNA display a similar behavior to AP sites during enzymatic DNA synthesis. We also show that modified nucleotides can be incorporated opposite such analogs and hence, ferrocene units embedded within DNA templates might be integrated in future electrochemical probes to monitor the efficiency of nucleotide incorporation.
Results and discussion
Design and synthesis of phosphoramidite building block and modified oligonucleotides
Molecular dynamics simulations of duplexes containing a THF-AP site have revealed a large structural flexibility compared to unmodified DNA since the deoxyribose sugar is capable of adopting an unusual amount of sugar puckers.50 This flexibility was ascribed to the lack of base pairing and stacking interactions and was further confirmed by NMR structural investigations.51 AP-containing DNA duplexes mainly adopt a B-DNA structure and kinks of up to 30° caused by flipping out of the orphan nucleobase have been observed.50 We rationalized that introducing an aromatic and more rigid AP analog would avoid this kinking of the helical structure and facilitate enzymatic DNA synthesis and favor π-stacking interactions with the incoming nucleotide. Ferrocene (Fc) displays interesting electrochemical properties11 and is often used in drug discovery as a bioisostere for aromatic rings.52 In addition, we have previously demonstrated that ferrocene acts as a good surrogate for a dinucleotide motif due to the similar size (i.e. the distance between the cyclopentadienyl (Cp) rings is 3.3 Å compared to 3.4 Å for adjacent base pairs in B-DNA).53 Hence, we surmised that a ferrocene moiety could represent a valid surrogate for one or potentially two consecutive AP-sites. In order to verify this hypothesis, we prepared a suitable ferrocene phosphoramidite building block following literature protocols (see Scheme S1 in the ESI†).54–57 The resulting building block was then incorporated into DNA sequences using standard solid-phase synthesis (see Table 1 and the ESI†). The sequence composition of the resulting modified templates is based on similar oligonucleotides containing either abasic sites or imidazole-modified nucleotides which have recently been used to probe the incorporation of C-nucleotides into DNA48 or the enzymatic formation of metal base pairs, respectively.58 Two modified templates (M1 and M2) were designed so as to contain one and two Fc units respectively immediately following the 3′-terminus of the 19 nucleotide-long, 5′-FAM-labelled primer P1. We also synthesized a template (M3) where the primer end was three bases upstream of an Fc nucleotide and one template (M4) where the Fc nucleotide was located within the primer binding region. Lastly, we also synthesized templates containing one or two tetrahydrofuran-AP sites (AP1 and AP2, respectively) using the corresponding phosphoramidite building block48 as well as a template containing an 18-atom hexa-ethyleneglycol Sp18-spacer unit (M1-Sp18). All modified sequences were purified using HPLC and their chemical integrity verified by mass spectrometry (see ESI†).| Oligonucleotide sequence | |
|---|---|
| a Fc represents a ferrocene nucleotide; Φ represents a THF-abasic site; Sp18 represents an 18-atom hexa-ethyleneglycol chemical spacer. | |
| P1 | 5′ – TAC GAC TCA CTA TAG CCT C – 3′ |
| M1 | 5′ – GGA GFcG AGG CTA TAG TGA GTC GTA – 3′ |
| M2 | 5′ – G GAG FcFcG AGG CTA TAG TGA GTC GTA – 3′ |
| M3 | 5′ – GGFc GCG AGG CTA TAG TGA GTC GTA – 3′ |
| M4 | 5′ – GGA GTG AGG CTA TAG FcGA GTC GTA – 3′ |
| AP1 | 5′ – GGA GΦG AGG CTA TAG TGA GTC GTA – 3′ |
| AP2 | 5′ – GGA GΦΦ GAG GCT ATA GTG AGT CGT A – 3′ |
| M1-Sp18 | 5′ – GGA GSp18G AGG CTA TAG TGA GTC GTA – 3′ |
| P2 | 5′ – TAC GAC TCA CTA TAG CCT CA – 3′ |
| P3 | 5′ – TAC GAC TCA CTA TAG CCT CAC – 3′ |
Primer extension reactions with unmodified nucleotides
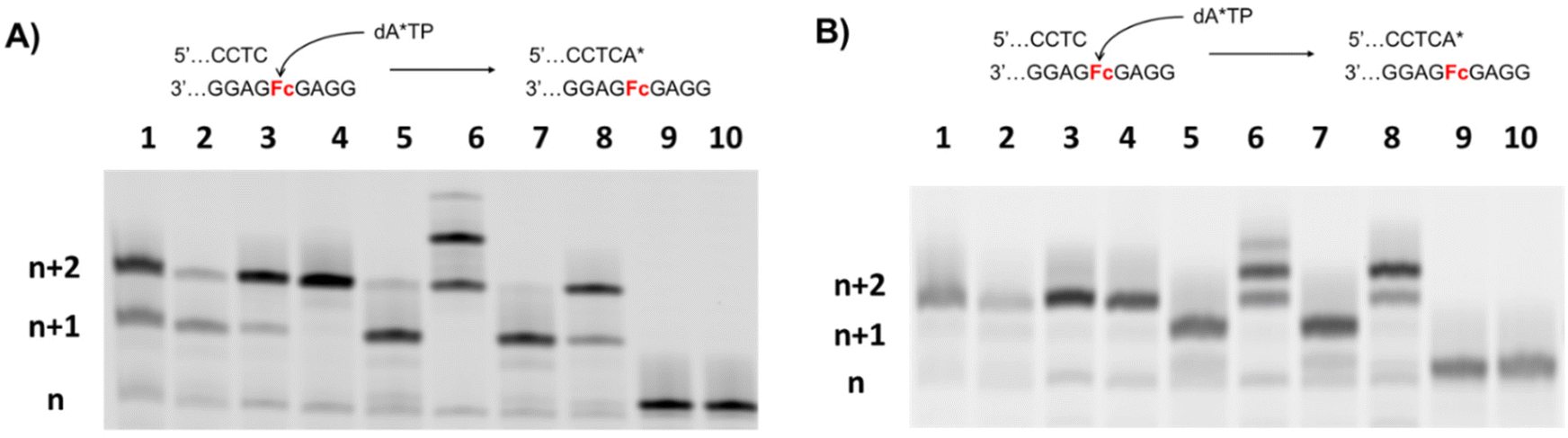
With the modified templates to hand, we first evaluated the possibility of incorporating canonical nucleotides opposite a Fc site. To do so, we carried out primer extension (PEX) reactions with different polymerases (A, B, and Y families) with each individual nucleoside triphosphate (dNTP) using the P1/M1 primer/template system (Fig. 2). Analysis of the reaction products by gel electrophoresis reveals that all natural nucleotides can be incorporated opposite an Fc abasic site analog with higher incorporation efficiencies for the larger purine nucleotides (Fig. 2A). Indeed, most polymerases incorporate one dA and one dG opposite a Fc site with full conversion of the primer to the corresponding n + 1 products. In the case of pyrimidines, yields of n + 1 product formation are significantly lower and reach only about 50%. The Sulfolobus DNA polymerase IV (Dpo4) is a Y family DNA polymerase that displays lesion-bypass capacity and it is therefore unsurprising that multiple incorporation events are observed with all nucleotides except with dGTP (lanes 7 in Fig. 2A). Reactions conducted with Deep Vent led to complete hydrolytic degradation of the primer (lanes 8 in Fig. 2). In order to further characterize the efficiency and specificity of incorporation of the different natural nucleotides, we reduced the reaction times for purine nucleotides while concomitantly increasing that for pyrimidine nucleotides (Fig. 2B). A decrease of reaction time had little effect on the outcome of reactions carried out with purine nucleotides, while an increase in reaction time served to improve n + 1 product formation with pyrimidine nucleotides (Fig. 2B). A similar outcome was observed when template M1 was substituted with template AP1 which contains a single THF-AP site instead of the Fc unit (Fig. S1†). | ||
| Fig. 2 Gel images (PAGE 20%) of PEX reactions with primer P1 (10 pmol) and template M1 (15 pmol) in the presence of the individual natural dNTPs (100 μM) and different DNA polymerases. (A) Reactions carried out for 1 h; (B) reactions carried out for 30 min with dATP and dGTP and for 2 h with dCTP and dTTP. List of polymerases used: 1. Hemo Klem Taq (8 reactions), 2. Taq (5 U), 3. Bst (8 U), 4. Therminator (2 U), 5. Vent (exo−) (2 U), 6. Kf exo− (5 U), 7. Dpo4 (2 U), 8. Deep Vent (2 U). The negative controls are: 9. No polymerase, 10. No dNTP. The label n is connected to the length of the primer, while n + 1 is referred to the length of the primer with the addition of one dNMP. | ||
Surprisingly, when we introduced an 18-atom long hexa-ethyleneglycol spacer moiety (Sp18 in Fig. 1) into the template (template M1-Sp18, Table 1), at the same location as ferrocene or as THF-AP sites, we also observed n + 1 product formation rather than a halting of polymerase-mediated synthesis (Fig. S2†). We believe however that this effect is likely caused by non-templating addition (NTA) of nucleotides at the 3′-end of the primer rather than A rule type of incorporation. Indeed, when DNA or RNA polymerases reach the end of DNA templates, one or multiple, mainly adenosine, nucleotides are incorporated. NTA is known to plague enzymatic synthesis of DNA and RNA and various non-nucleosidic59 and nucleosidic60 inhibitors have been proposed to suppress NTA. In addition, NTA is probably assisted with a strand slippage mechanism in which misaligned primers cause polymerases to slip and produce indel mutations.61 Such a mechanism was recently described for modified and natural nucleotides on templates containing longer homoplymeric sequences as well as shorter polyethylene glycol spacers.62
Next, we performed a time-course experiment to evaluate the relative efficiency of incorporation of each individual nucleotide. To that effect, we performed PEX reactions with Vent (exo−) as polymerase and the P1/M1 primer/template system (Fig. 3). The relative efficiency of incorporation of canonical nucleotides opposite a templating Fc nucleotide was dA > dG ≫ dC ∼ dT which compares to that observed for the incorporation opposite THF-abasic sites.63 Moreover, the “A rule”, meaning a strong preference of polymerases to incorporate a dA nucleotide opposite an abasic site, appears to be obeyed in the context of an Fc nucleotide as well.
 | ||
| Fig. 3 Gel images (PAGE 20%) of PEX reactions with primer P1 (10 pmol) and template M1 (15 pmol) in the presence of the individual natural dNTPs (100 μM) and Vent (exo−) DNA polymerase (0.5 U). Time points were taken after 1 min (1), 5 min (2), 10 min (3), 20 min (4), 30 min (5), 60 min (6). | ||
Overall, these experiments allowed us to identify conditions permitting the efficient incorporation of all individual, canonical nucleotides opposite Fc sites and revealed that purine nucleotides are preferentially incorporated following the well-established A rule. Next, we evaluated the effect of adding two consecutive Fc nucleotides in the template strand on enzymatic DNA synthesis. To that effect, we performed PEX reactions using the P1/M2 primer/template system using various DNA polymerases and each individual nucleotide (Fig. 4A). When dATP was used as substrate, all polymerases except for Dpo4 incorporated at least one nucleotide opposite the two consecutive Fc nucleotides. Interestingly, Therminator and Deep Vent polymerases incorporated two while Kf exo− incorporated multiple dA nucleotides opposite the Fc sites. In the case of deoxyguanosine, most polymerases incorporated single nucleotides with complete conversion of the primer to n + 1 and n + 2 product formation was observed only when Therminator was used as polymerase. As observed with templates containing single abasic sites, reactions carried out with pyrimidine proceeded less efficiently. Interestingly, similar results were observed when a template (AP2) containing two consecutive THF-abasic sites was used in PEX reactions except that Dpo4 efficiently incorporated single dA and dG nucleotides opposite Φ but not opposite Fc (Fig. 4B).
 | ||
| Fig. 4 Gel images (PAGE 20%) of PEX reactions with primer P1 (10 pmol) and (A) template M2 (15 pmol) and (B) template AP2 (15 pmol) with each individual dNTP (100 μM for A) and 20 μM for (B). List of polymerases used: 1. Hemo Klem Taq (8 reactions), 2. Taq (5 U), 3. Bst (8 U), 4. Therminator (2 U), 5. Vent (exo−) (2 U), 6. Kf exo− (5 U), 7. Dpo4 (2 U), 8. Deep Vent (2 U). The negative controls are: 9. No polymerase, 10. No dNTP. The label n is connected to the length of the primer, while n + 1 is referred to the length of the primer with the addition of one dNMP while n + 2 is referred to the length of the primer with the addition of two dNMPs. | ||
We have also investigated the possibility of inserting nucleotides opposite Fc sites under running start rather than standing start conditions since this allows to evaluate the incorporation efficiency of nucleotides at a site more remote from the end of the primer (Fig. S3†). When either two (dGTP, dCTP) or three (dGTP, dCTP, dATP) nucleotides were used in PEX reactions with the P1/M3 primer/template system, the expected n + 3 product predominantly formed under all experimental conditions. This is consistent with extensive kinetic studies which suggest higher misincorporation rates of polymerases under running start conditions.64 Lastly, we examined the effect of an Fc nucleotide located in the primer binding region of the template on DNA synthesis (Fig. S4†). In this context, we designed template M4 where the ferrocene abasic site is located opposite a dA nucleotide in primer P1 due to the favorable incorporation capacity of dAMP nucleotides as demonstrated in Fig. 1. When PEX reactions were carried out, all polymerases extended primer P1 with six nucleotides corresponding to the addition of five complementary nucleotides and one additional (presumably dA) nucleotide by NTA. Hence, these experiments reveal that the presence of a single Fc nucleotide located within the primer binding region does not negatively impact enzymatic synthesis and appears to be rather well accommodated in the B-DNA structure. In addition, Fc represents a chemically stable analog of an abasic site since it is resistant to the β- and δ-elimination process leading to DNA strand sission.65
Bypass of Fc and Φ sites
After demonstrating that Fc and Φ sites adopted a similar behavior when assayed under PEX reaction conditions and establishing conditions that permitted the incorporation of one or multiple canonical nucleotides opposite Fc sites, we investigated the possibility of extending DNA synthesis beyond such a dN–Fc pair. Bypass of a dN–Fc is of crucial importance to develop an electrochemical sensing platform for the incorporation of modified/natural nucleotides. Hence, we first installed one of each canonical nucleotide opposite Fc by PEX reactions as described previously and incubated the resulting, extended primer with dCTP in the presence of various polymerases (Fig. S5†). When dA was first installed on the primer opposite Fc, Kf exo− efficiently incorporated a dCMP moiety in high (∼80%) yields (see lane 8 of Fig. S5†). In addition, Dpo4 due to its lesion-bypass capacity was capable of adding one or two dCMPs after a dN–Fc pair when dN was dA, dC, or dT but not dG (lanes 6 in Fig. S5†). To shed some light into this discrepancy we further investigated the capacity of Dpo4 at bypassing a dN–Fc pair (Fig. S6A†). We first installed a dG or a dA nucleotide by a first Dpo4-catalyzed PEX reaction and then further extended the resulting product by incubation with dCTP alone or a combination of dCTP and dTTP. These experiments confirm that Dpo4 can resume DNA synthesis beyond a dA–Fc pair. On the contrary, even though n + 5 full length product (i.e. full length product with an additional nucleotide stemming from NTA) can be achieved in the presence of a dG–Fc pair, the efficiency is impaired in terms of yields and additional bands are observed that originate from yet unidentified side-products. In addition, the formation of the n + 6 product observed after the addition of both dCTP and dTTP (lane 3 in Fig. S6A†) might arise via NTA of two nucleotides or if the Fc site acts as a dinucleotide surrogate (Fig. S6B†).In order to confirm these observations, we carried out PEX reactions with template M1 and primer P2 that is equipped with an additional terminal 3′-dA nucleotide compared to P1. Primer P2 thus represents a synthetic analog of enzymatically constructed dA–Fc pairs. When PEX reactions were carried out with this system in the sole presence of dCTP, Kf exo− incorporated one nucleotide at the end of the primer in ∼50% yield, while the reaction catalyzed by Dpo4 led to a distribution of products with complete conversion of the primer (Fig. 5). When both dCTP and dTTP were added to the reaction mixtures, n + 3 and n + 4 products were observed in equal quantities with Dpo4 as polymerase and low yields of n + 1 product with Kf exo− (we believe the double banding observed in this case to correspond to products resulting from single dC and dT nucleotide incorporation). When similar PEX reactions were carried out with template AP1 containing a THF rather than an Fc site, reactions catalyzed by the Vent (exo−), Dpo4, and Kf exo− polymerases led to full conversion of the primer to the n + 4 product when both dCTP and dTTP were present (Fig. S7A†). On the other hand, polymerases struggled markedly to extend primer P2 when template M1-Sp18 was used (Fig. S7B†). Lastly, we carried out PEX reactions with all three modified templates (M1, AP1, and M1-Sp18) along with primer P3 that contains additional dA and dC nucleotides thus mimicking an efficient bypass reaction (Fig. S8†). Under these conditions, most polymerases were capable of adding a single dT unit at the end of primer P3 when template AP1 was used, either in moderate yields (40–60%) or with full conversion to n + 1 product (lanes 4–6 in Fig. S8B†). With template M1 containing an Fc site, only Dpo4 led to the expected n + 1 product while Kf exo− struggled to produce n + 1 extended product (∼30% conversion of primer) and all other polymerases did not incorporate any nucleotides (Fig. S8A†). When template containing the C18 spacer was assayed under these conditions, Dpo4 led to a clean conversion to n + 1 product and yielded the product with an additional dT nucleotide in ∼40% yield while all other polymerases failed to incorporate a nucleotide (Fig. S8C†). A similar trend was observed when both dCTP and dTTP were included in the reaction mixture, except that full length products were observed rather than single incorporation events.
 | ||
| Fig. 5 Gel images (PAGE 20%) of PEX reactions with primer P2 (10 pmol) and template M1 (15 pmol) with dCTP alone (dC, 100 μM) or with dCTP and dTTP (dC + dT, 100 μM) for 1 h. List of polymerases used: 1. Hemo Klem Taq (8 reactions), 2. Taq (5 U), 3. Bst (8 U), 4. Vent (exo−) (2 U), 5. Dpo4 (2 U), 6. Kf exo− (5 U). The negative controls are: 7. No polymerase, 8. No dNTP. The label n is connected to the length of the primer, while n + 1 is referred to the length of the primer with the addition of one dNMP while n + 2 is referred to the length of the primer with the addition of two dNMPs, n + 3 and n + 4 to three and four added nucleotides. | ||
Evaluation of incorporation of modified nucleotides opposite ferrocene nucleotides
Our biochemical characterization of templates containing ferrocene analogs of abasic sites revealed that (i) canonical nucleotides can be incorporated opposite Fc sites and as for THF-AP sites, the A rule appears to be obeyed; (ii) conditions could be met to support the incorporation of less favored pyrimidine nucleotides; (iii) polymerases such as Dpo4 do not stall after the installation of a dN–Fc pair and permit bypass synthesis; (iv) a two-step protocol method could be envisioned by coupling Dpo4 to other polymerases that stall after the installation of a dN–Fc pair.21,48,66–68 Since our aim is to use Fc sites to monitor the incorporation of modified nucleotides, we next investigated the compatibility of such ferrocene abasic sites with nucleotides containing different nucleobase modification patterns (Fig. 6). | ||
| Fig. 6 Chemical structures of modified nucleotides evaluated in PEX reactions. | ||
First, we evaluated the compatibility of nucleotides that are either characterized by the absence of a base (nucleotide 1) or with an unnatural base surrogate (nucleotide 2) with enzymatic synthesis on templates containing one or multiple Fc sites. To do so, we carried out PEX reactions with the P1/M1 and P1/M2 systems in the presence of various DNA polymerases (Fig. 7).
 | ||
| Fig. 7 Gel images (PAGE 20%) of PEX reactions with primer P1 (10 pmol) and templates M1 and M2 (15 pmol) with nucleotides 1 (abasic) or 2 (biphenyl). List of polymerases used: 1. Hemo Klem Taq (8 reactions), 2. Taq (5 U), 3. Bst (8 U), 4. Therminator (2 U), 5. Vent (exo−) (2 U), 6. Kf exo− (5 U), 7. Dpo4 (2 U), 8. Deep Vent (2 U). The negative control is reaction without polymerase (lanes 9). All reactions were carried out with 200 μM modified nucleotides for 1 h. The label n is connected to the length of the primer, while n + 1 is referred to the length of the primer with the addition of one dNMP. | ||
The abasic site nucleotide 1 (dΦTP) was previously shown to be tolerated by engineered DNA polymerases and incorporation of single and even multiple abasic site nucleotides could be observed opposite templating dA nucleotides as well as THF-AP sites.69 In stark contrast, no dΦMP units were incorporated in primer P1 when one or two Fc sites were present in the template. The C-nucleotide analog 2 is readily incorporated opposite Fc sites by Therminator, Kf exo−, and Deep Vent polymerases but the presence of a second ferrocene moiety on template M2 causes polymerases to halt and only n + 1 products can be observed (Fig. 7). Similar results were obtained previously with nucleotide 2 and templates containing THF-abasic sites.48 Next, we turned out attention to nucleobase-modified nucleotides due to their relevance in various fields such as aptamer and DNAzyme selection experiments18,70 or in the context of epigenetic modifications.71 Since 2′-deoxyadenine is preferentially incorporated opposite Fc and THF-abasic sites, we evaluated whether the presence of functional groups interfered with n + 1 product formation. We performed PEX reactions with the P1/M1 (Fig. 8) and P1/AP1 (Fig. S9†) systems in the presence of dATP analogs modified at two particular position of the nucleobase. Nucleotide 3 bears an iodine atom at position N7 (which is a popular site for the introduction of side-chains), while nucleotide 4 is equipped with an aliphatic linker connected to an amino functional group on the exocyclic amine of the purine nucleobase (which are commonly introduced into DNA via nick translation reactions72). Nucleotide 3 is readily incorporated into DNA opposite Fc sites by all polymerases that were tested (Fig. 8A) and most reactions even lead to NTA and thus to n + 2 product formation. Surprisingly, nucleotide 4 bearing a modification at position N6 which is expected to be less well tolerated by polymerases was incorporated with a comparable efficiency to that of nucleotide 3 (Fig. 8B). Similar results were obtained when both nucleotides were incorporated opposite single THF-abasic site analogs (Fig. S9†).
 | ||
| Fig. 8 Gel images (PAGE 20%) of PEX reactions with primer P1 (10 pmol) and template M1 (15 pmol) with modified dATP nucleotides (A) 3 or (B) 4. List of polymerases used: 1. Hemo Klem Taq (8 reactions), 2. Taq (5 U), 3. Bst (8 U), 4. Therminator (2 U), 5. Vent (exo−) (2 U), 6. Kf exo− (5 U), 7. Dpo4 (2 U), 8. Deep Vent (2 U). The negative controls are: 9. No polymerase, 10. No dNTP. All reactions were carried out with 200 μM modified nucleotides for 1 h. The label n is connected to the length of the primer, while n + 1 and n + 2 refer to the length of the primer with the addition of one or two dNMPs, respectively. | ||
Lastly, we evaluated the possibility of incorporating modified dUTP analogs bearing various side-chains and functional groups at position C5 of the pyrimidine which is readily tolerated by numerous polymerases.10,18,67,70,71,73,74 Therminator, Hemo Klem Taq, and Dpo4 efficiently incorporated the commercially available dU*TP analog 5 opposite an Fc site (Fig. S10A†), while additional polymerases also appended this nucleotide on primer P1 opposite a THF-abasic site (Fig. S10B†). A slightly lower incorporation efficiency was observed when M1-Sp18 was used as template (Fig. S10C†). Surprisingly, dU*TP analog 6, decorated with a cubane moiety and which has been successfully used for the identification of specific modified aptamers, was not readily incorporated opposite any of the abasic sites (i.e. Fc, Φ, or C18) that were evaluated and only Therminator displayed some capacity at producing extended primers (Fig. 9). Cubane is an isostere of benzene75 and therefore we synthesized nucleotide 7 to probe whether the structure and chemical nature of the substituent had an effect on the incorporation efficiency. Under PEX reaction conditions with templates M1, AP1, and M1-Sp18, a similar product distribution and incorporation efficiency was observed with nucleotide 7 as with the corresponding isostere in nucleotide 6 (Fig. S11†). We also synthesized nucleotide 8 which is modified with a ferrocene which is also a good analog and isostere of benzene.52 The ferrocene-modified dU*TP acted as a better substrate for polymerases with the modified templates (Fig. S12†), however, PEX reactions led to multiple products as observed by gel electrophoresis and yields remained modest. Taken together, these results indicate that base-modified nucleotides can be incorporated opposite Fc sites. However, the incorporation efficiency strongly depends on the nature and the size of the chemical modifications.
 | ||
| Fig. 9 Gel images (PAGE 20%) of PEX reactions with primer P1 (10 pmol) and 5-cubane-modified dUTP 6 and templates (all 15 pmol) (A) M1, (B) AP1, and (C) M1-Sp18. List of polymerases used: 1. Hemo Klem Taq (8 reactions), 2. Taq (5 U), 3. Bst (8 U), 4. Therminator (2 U), 5. Vent (exo−) (2 U), 6. Kf exo− (5 U), 7. Dpo4 (2 U), 8. Deep Vent (2 U). The negative controls are: 9. No polymerase, 10. No dNTP. All reactions were carried out with 200 μM modified nucleotides for 1 h. The label n is connected to the length of the primer, while n + 1, n + 2, and n + 3 refer to the length of the primer with the addition of one, two, or three dNMPs, respectively. | ||
Conclusions
Chemical modifications can help nucleic acids to bind to targets, catalyze chemical transformations, and enhance their resistance against nuclease-mediated degradation. Functional groups can be included either via chemical means or by the enzymatic polymerization of modified nucleoside triphosphates. Enzymatic synthesis is particularly attractive since it is directly amenable to SELEX and related methods of in vitro selection and it is devoid of any size limitations. Methods for the real-time monitoring of incorporation of chemically modified nucleoside triphosphates are in dire need since these would enable to rapidly evaluate whether these analogs are tolerated as substrates by polymerases. In a first step towards to realizing this goal through the use of electrochemistry, we have explored the possibility of using a ferrocene analogue of abasic site in templates during enzymatic DNA synthesis. We demonstrate that canonical nucleotides can readily be incorporated by PEX reactions opposite such a chemical surrogate and as for naturally occurring abasic sites, the A-rule appears to be obeyed. Ferrocene also represents a chemically stable abasic site analogue since it not subjected to the elimination followed by strand cleavage process. Under more forcing conditions, all canonical nucleotides can be incorporated into DNA. We have also demonstrated that DNA polymerases with lesion-bypass capacity can be used to extend DNA synthesis beyond dN–Fc pairs. Lastly, we have evaluated the possibility of incorporating modified nucleotides in conjunction with templates containing Fc sites. Modified dATPs are readily incorporated opposite Fc sites, while dUTP analogs bearing small side-chains such as propargylamine are also well-tolerated. On the other hand, nucleotides bearing more complex modification patterns such as C-glycosidic aromatic nucleobases or cubane residues are only incorporated opposite Fc analogs by specific polymerases in moderate yields. Collectively, these experiments demonstrate the compatibility of ferrocene moieties integrated within DNA templates with enzymatic synthesis of natural and modified DNA. Such an approach could also be extended to nucleotides bearing different modification patterns such as xenonucleic acids (XNAs).76 The next imminent steps towards the development of an electrochemical monitoring device will include square-wave voltammetry measurements on surface-immobilized oligonucleotides to examine the redox behavior of Fc-sites in the presence of natural and modified nucleotides.77–79Experimental section
Modified nucleotides and oligonucleotides
Nucleotides 1,692,48 and 614 were synthesized according to literature protocols, nucleotides 3–5 were purchased from Jena Bioscience. Synthesis of nucleotides 7 and 8 will be reported elsewhere. Details for the synthesis of Fc abasic phosphoramidite and oligonucleotide synthesis can be found in the ESI.†General protocol for PEX reactions
A 5′-FAM-labelled primer (10 pmol) was annealed to the appropriate template (15 pmol) in MilliQ H2O by heating to 95 °C and gradually cooling to room temperature over 1 hour. The appropriate DNA polymerase, the relative buffer (1 μL of 10×), and the dNTP(s) were then added to the reaction mixture for a total reaction volume of 10 μL. The reaction mixture was incubated for the appropriate time at the optimal temperature for the polymerase. The reaction was stopped by adding 10 μL of the quenching solution (formamide (70%), ethylenediaminetetraacetic acid (EDTA, 50 mM), bromophenol (0.1%), xylene cyanol (0.1%)). The reaction mixtures were subjected to gel electrophoresis in denaturing polyacrylamide gel (20%) containing Trisborate–EDTA (TBE) 1× buffer (pH 8) and urea (7 M). Visualization was performed by fluorescence imaging by using a Typhoon Trio phosphorimager.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
C. F. gratefully acknowledges the Pasteur–Paris University (PPU) International Ph.D. Program that has received funding from INCEPTION project (PIA/ANR-16-CONV-0005), the “Ecole Doctorale Frontières de l'Innovation en Recherche et Education—Programme Bettencourt” for financial support. The authors gratefully acknowledge financial support from Institut Pasteur. The Centre for Chemical and Materials Analysis in the School of Chemistry at the University of Birmingham is acknowledged for technical support.References
- M. A. Maverick, M. Gaillard, J.-J. Vasseur, F. Debart and M. Smietana, Eur. J. Org. Chem., 2022, e202101061 CAS.
- L. K. McKenzie, R. El-Khoury, J. D. Thorpe, M. J. Damha and M. Hollenstein, Chem. Soc. Rev., 2021, 50, 5126–5164 RSC.
- T. C. Roberts, R. Langer and M. J. A. Wood, Nat. Rev. Drug Discovery, 2020, 19, 673–694 CrossRef CAS.
- K. L. Seley-Radtke and M. K. Yates, Antiviral Res., 2018, 154, 66–86 CrossRef CAS PubMed.
- M. J. Hogan and N. Pardi, Annu. Rev. Med., 2022, 73, 17–39 CrossRef PubMed.
- Y. Xiao, Z. Tang, X. Huang, W. Chen, J. Zhou, H. Liu, C. Liu, N. Kong and W. Tao, Chem. Soc. Rev., 2022, 51, 3828–3845 RSC.
- M. H. Caruthers, Science, 1985, 230, 281–285 CrossRef CAS.
- P. F. Conlon, O. Eguaogie, J. J. Wilson, J. S. T. Sweet, J. Steinhoegl, K. Englert, O. G. A. Hancox, C. J. Law, S. A. Allman, J. H. R. Tucker, J. P. Hall and J. S. Vyle, Chem. Sci., 2019, 10, 10948–10957 RSC.
- A. F. Sandahl, T. J. D. Nguyen, R. A. Hansen, M. B. Johansen, T. Skrydstrup and K. V. Gothelf, Nat. Commun., 2021, 12, 2760 CrossRef CAS PubMed.
- M. Hocek, Acc. Chem. Res., 2019, 52, 1730–1737 CrossRef CAS PubMed.
- D. Kodr, C. P. Yenice, A. Simonova, D. P. Saftić, R. Pohl, V. Sýkorová, M. Ortiz, L. Havran, M. Fojta, Z. J. Lesnikowski, C. K. O'Sullivan and M. Hocek, J. Am. Chem. Soc., 2021, 143, 7124–7134 CrossRef CAS PubMed.
- A. Chakrapani, O. Ruiz-Larrabeiti, R. Pohl, M. Svoboda, L. Krásný and M. Hocek, Chem. – Eur. J., 2022, 28, e202200911 CrossRef CAS.
- L. Appy, S. Peyrottes and B. Roy, Eur. J. Org. Chem., 2022, e202100544 CAS.
- Y.-W. Cheung, P. Röthlisberger, A. E. Mechaly, P. Weber, F. Levi-Acobas, Y. Lo, A. W. C. Wong, A. B. Kinghorn, A. Haouz, G. P. Savage, M. Hollenstein and J. A. Tanner, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 16790–16798 CrossRef CAS PubMed.
- Q. Li, V. A. Maola, N. Chim, J. Hussain, A. Lozoya-Colinas and J. C. Chaput, J. Am. Chem. Soc., 2021, 143, 17761–17768 CrossRef CAS PubMed.
- C. M. McCloskey, Q. Li, E. J. Yik, N. Chim, A. K. Ngor, E. Medina, I. Grubisic, L. Co Ting Keh, R. Poplin and J. C. Chaput, ACS Synth. Biol., 2021, 10, 3190–3199 CrossRef CAS PubMed.
- J. Siegl, O. Plückthun and G. Mayer, RSC Chem. Biol., 2022, 3, 288–294 RSC.
- S. Paul, A. A. W. L. Wong, L. T. Liu and D. M. Perrin, ChemBioChem, 2022, 23, e202100600 CrossRef CAS.
- M. Hollenstein, C. J. Hipolito, C. H. Lam and D. M. Perrin, Nucleic Acids Res., 2009, 37, 1638–1649 CrossRef CAS PubMed.
- B. McGorman, N. Z. Fantoni, S. O'Carroll, A. Ziemele, A. H. El-Sagheer, T. Brown and A. Kellett, Nucleic Acids Res., 2022, 50, 5467–5481 CrossRef CAS.
- M. Flamme, P. Röthlisberger, F. Levi-Acobas, M. Chawla, R. Oliva, L. Cavallo, G. Gasser, P. Marlière, P. Herdewijn and M. Hollenstein, ACS Chem. Biol., 2020, 15, 2872–2884 CrossRef CAS.
- G. S. Ravi Kumara and Y. J. Seo, Org. Biomol. Chem., 2021, 19, 5788–5793 RSC.
- D. Ji, M. G. Mohsen, E. M. Harcourt and E. T. Kool, Angew. Chem., Int. Ed., 2016, 55, 2087–2091 CrossRef CAS PubMed.
- K. Sefah, Z. Yang, K. M. Bradley, S. Hoshika, E. Jiménez, L. Zhang, G. Zhu, S. Shanker, F. Yu, D. Turek, W. Tan and S. A. Benner, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1449–1454 CrossRef CAS PubMed.
- S. Hoshika, N. A. Leal, M.-J. Kim, M.-S. Kim, N. B. Karalkar, H.-J. Kim, A. M. Bates, N. E. Watkins, H. A. SantaLucia, A. J. Meyer, S. DasGupta, J. A. Piccirilli, A. D. Ellington, J. SantaLucia, M. M. Georgiadis and S. A. Benner, Science, 2019, 363, 884–887 CrossRef CAS PubMed.
- A. X. Z. Zhou, X. Dong and F. E. Romesberg, J. Am. Chem. Soc., 2020, 142, 19029–19032 CrossRef CAS PubMed.
- M. Kimoto and I. Hirao, Chem. Soc. Rev., 2020, 49, 7602–7626 RSC.
- M. Luo, E. Groaz, M. Froeyen, V. Pezo, F. Jaziri, P. Leonczak, G. Schepers, J. Rozenski, P. Marliere and P. Herdewijn, J. Am. Chem. Soc., 2019, 141, 10844–10851 CrossRef CAS PubMed.
- H. Nguyen, M. Abramov, J. Rozenski, E. Eremeeva and P. Herdewijn, ChemBioChem, 2022, 23, e202200060 CrossRef CAS PubMed.
- S. Arangundy-Franklin, A. I. Taylor, B. T. Porebski, V. Genna, S. Peak-Chew, A. Vaisman, R. Woodgate, M. Orozco and P. Holliger, Nat. Chem., 2019, 11, 533–542 CrossRef CAS.
- H. Cahová, A. Panattoni, P. Kielkowski, J. Fanfrlík and M. Hocek, ACS Chem. Biol., 2016, 11, 3165–3171 CrossRef PubMed.
- J. Balintova, M. Welter and A. Marx, Chem. Sci., 2018, 9, 7122–7125 RSC.
- L. Bornewasser, C. Domnick and S. Kath-Schorr, Chem. Sci., 2022, 13, 4753–4761 RSC.
- F. Levi-Acobas, L. K. McKenzie and M. Hollenstein, New J. Chem., 2022, 46, 4871–4876 RSC.
- A. S. Schröder, E. Parsa, K. Iwan, F. R. Traube, M. Wallner, S. Serdjukow and T. Carell, Chem. Commun., 2016, 52, 14361–14364 RSC.
- B. Hu, Y. Wang, S. Sun, W. Yan, C. Zhang, D. Luo, H. Deng, L. R. Hu and Z. Huang, Angew. Chem., Int. Ed., 2019, 58, 7835–7839 CrossRef CAS.
- J. Chu, M. González-López, S. L. Cockroft, M. Amorin and M. R. Ghadiri, Angew. Chem., Int. Ed., 2010, 49, 10106–10109 CrossRef CAS.
- C. W. Fuller, S. Kumar, M. Porel, M. C. Chien, A. Bibillo, P. B. Stranges, M. Dorwart, C. J. Tao, Z. M. Li, W. J. Guo, S. D. Shi, D. Korenblum, A. Trans, A. Aguirre, E. Liu, E. T. Harada, J. Pollard, A. Bhat, C. Cech, A. Yang, C. Arnold, M. Palla, J. Hovis, R. Chen, I. Morozova, S. Kalachikov, J. J. Russo, J. J. Kasianowicz, R. Davis, S. Roever, G. M. Church and J. Y. Ju, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5233–5238 CrossRef CAS PubMed.
- M. T. Walsh and X. H. Huang, Nucleic Acids Res., 2017, 45, e175 CrossRef CAS.
- A. Krieg, S. Laib, T. Ruckstuhl and S. Seeger, ChemBioChem, 2003, 4, 589–592 CrossRef CAS.
- C. Fijen, A. Montón Silva, A. Hochkoeppler and J. Hohlbein, Phys. Chem. Chem. Phys., 2017, 19, 4222–4230 RSC.
- S. Boiteux and M. Guillet, DNA Repair, 2004, 3, 1–12 CrossRef CAS PubMed.
- Z. J. Liu, S. Martínez Cuesta, P. van Delft and S. Balasubramanian, Nat. Chem., 2019, 11, 629–637 CrossRef CAS.
- R. V. Desai, X. Y. Chen, B. Martin, S. Chaturvedi, D. W. Hwang, W. H. Li, C. Yu, S. Ding, M. Thomson, R. H. Singer, R. A. Coleman, M. M. K. Hansen and L. S. Weinberger, Science, 2021, 373, eabc6506 CrossRef CAS PubMed.
- E. A. Motea, I. Lee and A. J. Berdis, ChemBioChem, 2013, 14, 489–498 CrossRef CAS.
- A. Dasari, T. Deodhar and A. J. Berdis, J. Mol. Biol., 2017, 429, 2308–2323 CrossRef CAS.
- T. J. Matray and E. T. Kool, Nature, 1999, 399, 704–708 CrossRef CAS PubMed.
- P. Röthlisberger, F. Levi-Acobas, C. J. Leumann and M. Hollenstein, Bioorg. Med. Chem., 2020, 28, 115487 CrossRef.
- J.-L. H. A. Duprey and J. H. R. Tucker, Chem. Lett., 2014, 43, 157–163 CrossRef CAS.
- D. Barsky, N. Foloppe, S. Ahmadia, D. M. Wilson and A. D. MacKerell, Nucleic Acids Res., 2000, 28, 2613–2626 CrossRef CAS.
- R. D. Hazel, K. Tian and C. de los Santos, Biochemistry, 2008, 47, 11909–11919 CrossRef CAS.
- M. Patra and G. Gasser, Nat. Rev. Chem., 2017, 1, 0066 CrossRef CAS.
- H. V. Nguyen, Z.-y. Zhao, A. Sallustrau, S. L. Horswell, L. Male, A. Mulas and J. H. R. Tucker, Chem. Commun., 2012, 48, 12165–12167 RSC.
- A.-E. Navarro, N. Spinelli, C. Moustrou, C. Chaix, B. Mandrand and H. Brisset, Nucleic Acids Res., 2004, 32, 5310–5319 CrossRef CAS.
- G. Chatelain, H. Brisset and C. Chaix, New J. Chem., 2009, 33, 1139–1147 RSC.
- N. K. Gupta, E. A. Wilkinson, S. K. Karuppannan, L. Bailey, A. Vilan, Z. Zhang, D.-C. Qi, A. Tadich, E. M. Tuite, A. R. Pike, J. H. R. Tucker and C. A. Nijhuis, J. Am. Chem. Soc., 2021, 143, 20309–20319 CrossRef CAS.
- T. Ihara, D. Sasahara, M. Shimizu and A. Jyo, Supramol. Chem., 2009, 21, 207–217 CrossRef CAS.
- P. Röthlisberger, F. Levi-Acobas, I. Sarac, P. Marlière, P. Herdewijn and M. Hollenstein, Org. Biomol. Chem., 2017, 15, 4449–4455 RSC.
- P. Güixens-Gallardo, M. Hocek and P. Perlíková, Bioorg. Med. Chem. Lett., 2016, 26, 288–291 CrossRef PubMed.
- S. Moran, R. X.-F. Ren, C. J. Sheils, S. Rumney, IV and E. T. Kool, Nucleic Acids Res., 1996, 24, 2044–2052 CrossRef CAS PubMed.
- M. Garcia-Diaz and T. A. Kunkel, Trends Biochem. Sci., 2006, 31, 206–214 CrossRef CAS PubMed.
- A. V. Chudinov, V. A. Vasiliskov, V. E. Kuznetsova, S. A. Lapa, N. A. Kolganova and E. N. Timofeev, Sci. Rep., 2021, 11, 2423 CrossRef CAS PubMed.
- S. Obeid, N. Blatter, R. Kranaster, A. Schnur, K. Diederichs, W. Welte and A. Marx, EMBO J., 2010, 29, 1738–1747 CrossRef CAS.
- J. G. Bertram, K. Oertell, J. Petruska and M. F. Goodman, Biochemistry, 2010, 49, 20–28 CrossRef CAS PubMed.
- M. Mosimann, P. A. Küpfer and C. J. Leumann, Org. Lett., 2005, 7, 5211–5214 CrossRef CAS PubMed.
- Y. Takezawa, T. Nakama and M. Shionoya, J. Am. Chem. Soc., 2019, 141, 19342–19350 CrossRef CAS PubMed.
- P. Ghosh, H. M. Kropp, K. Betz, S. Ludmann, K. Diederichs, A. Marx and S. G. Srivatsan, J. Am. Chem. Soc., 2022, 144, 10556–10569 CrossRef CAS.
- P. Ménová, H. Cahová, M. Plucnara, L. Havran, M. Fojta and M. Hocek, Chem. Commun., 2013, 49, 4652–4654 RSC.
- P. Röthlisberger, F. Levi-Acobas, I. Sarac, R. Ricoux, J. P. Mahy, P. Herdewijn, P. Marliere and M. Hollenstein, Tetrahedron Lett., 2018, 59, 4241–4244 CrossRef.
- K. Y. Chan, A. B. Kinghorn, M. Hollenstein and J. A. Tanner, ChemBioChem, 2022, 23, e202200006 CAS.
- F. Gracias, O. Ruiz-Larrabeiti, V. Vaňková Hausnerová, R. Pohl, B. Klepetářová, V. Sýkorová, L. Krásný and M. Hocek, RSC Chem. Biol., 2022, 3, 1069–1075 RSC.
- G. Gebeyehu, P. Y. Rao, P. Soochan, D. A. Simms and L. Klevan, Nucleic Acids Res., 1987, 15, 4513–4534 CrossRef CAS PubMed.
- M. Kuwahara, J.-i. Nagashima, M. Hasegawa, T. Tamura, R. Kitagata, K. Hanawa, S.-i. Hososhima, T. Kasamatsu, H. Ozaki and H. Sawai, Nucleic Acids Res., 2006, 34, 5383–5394 CrossRef CAS PubMed.
- M. Hollenstein, Chem. – Eur. J., 2012, 18, 13320–13330 CrossRef CAS PubMed.
- K. F. Biegasiewicz, J. R. Griffiths, G. P. Savage, J. Tsanaktsidis and R. Priefer, Chem. Rev., 2015, 115, 6719–6745 CrossRef CAS.
- T. N. Malik and J. C. Chaput, Curr. Res. Chem. Biol., 2021, 1, 100012 CrossRef CAS.
- A. Simonova, I. Magriñá, V. Sýkorová, R. Pohl, M. Ortiz, L. Havran, M. Fojta, C. K. O'Sullivan and M. Hocek, Chem. – Eur. J., 2020, 26, 1286–1291 CrossRef CAS PubMed.
- M. Ortiz, M. Jauset-Rubio, V. Skouridou, D. Machado, M. Viveiros, T. G. Clark, A. Simonova, D. Kodr, M. Hocek and C. K. O'Sullivan, ACS Sens., 2021, 6, 4398–4407 CrossRef CAS PubMed.
- M. Ortiz, M. Jauset-Rubio, D. Kodr, A. Simonova, M. Hocek and C. K. O'Sullivan, Biosens. Bioelectron., 2022, 198, 113825 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob01540d |
| This journal is © The Royal Society of Chemistry 2022 |