
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards polymerase-mediated synthesis of artificial RNA–DNA metal base pairs†
Fabienne
Levi-Acobas
a,
Luke K.
McKenzie
ab and
Marcel
Hollenstein
 *a
*a
aInstitut Pasteur, Université de Paris, CNRS UMR3523, Laboratory for Bioorganic Chemistry of Nucleic Acids, Department of Structural Biology and Chemistry, 28, rue du Docteur Roux, 75724 Paris Cedex 15, France. E-mail: marcel.hollenstein@pasteur.fr
bChimie ParisTech, PSL University, CNRS, Institute of Chemistry for Life and Health Sciences, Laboratory for Inorganic Chemical Biology, 75005 Paris, France
First published on 15th February 2022
Abstract
Metal-mediated base pairs are formed by the connection of two nucleobases via coordination to a metal cation. The resulting metal-containing duplexes have been used in a large variety of applications ranging from allosteric control of functional nucleic acids to the construction of nanowires. Recently, enzymatic approaches are being developed for the construction of metal-mediated base pairs. Here, we have studied the possibility of constructing HgII- and AgI-mediated DNA/RNA hetero base pairs using primer extension reactions. The high thermodynamic stabilities of metal base pairs can be harnessed to trigger the formation of two rU–HgII–dT base pairs. Bypass experiments demonstrate the possibility of constructing RNA containing DNA sequences which are used in multiple applications including DNAzyme selections. These findings will be useful for the enzymatic construction of xenonucleic acid (XNA) based metal pairs.
Introduction
Metal base pairs are structural analogs of canonical Watson–Crick pairs where the hydrogen pattern is substituted with the coordinative interaction of metal cations with the nucleobases.1,2 Metal base pairs can be constructed using the canonical nucleobases or synthetic nucleoside analogs with nucleobases that are specifically designed to serve as ligands for the specific binding of transition metal cations.3,4 These metal base pairs have advanced as promising candidates for a number of applications including the development of nanomolecular devices,5 ion sensors and biosensing devices6,7 and metal nanowires and nanodevices8–10 as well as for the allosteric control of functional nucleic acids.11–14The formation of metal base pairs mainly occurs by annealing short synthetic oligonucleotides together with specific metal cations. While this approach has allowed the identification of metal base pairs based on natural and modified nucleotides, it is restricted in oligonucleotide size and in terms of diversity of functional groups that can be explored due to the rather harsh conditions imposed by solid-phase synthesis.15 Alternatively, metal base pairs can be formed via enzymatic synthesis where polymerases incorporate modified or natural nucleotides into DNA in the strict presence of metal cations.16–20
So far, most synthetic efforts have been dedicated to the identification of modified nucleobases that can act as potent ligands for metal coordination. Surprisingly, very little attention has been devoted to combining sugar and/or backbone modifications and metal base pairs,21–24 especially in the context of enzymatic synthesis. This might be ascribed to the fact that DNA polymerases are finely tuned biological machineries that have the capacity of strongly distinguishing dNTPs from sugar modified nucleotides including NTPs.25,26 The strong discrimination of NTPs (by a factor of up to 105) stems from a steric exclusion caused by a clash between the 2′-hydroxyl moiety of the incoming rNTP and an active site residue, usually equipped with a bulky side chain such as tryptophan or phenylalanine.26–29 Hence, most naturally occurring DNA polymerases predominantly incorporate deoxynucleoside monophosphates (dNMPs) and depending on the polymerase, insert only one ribonucleoside monophosphate every hundred thousand correct nucleotides.30 This rather strong discrimination can be alleviated by using naturally occurring polymerases that lack such a steric gate,31,32 engineered polymerases equipped with an alternate, more permissive steric gate33,34 or by adding Mn2+ cofactors which also promote the incorporation of NTPs by DNA polymerases.26 Here, we demonstrate that the thermodynamic stabilities of metal base pairs35,36 can be harnessed to facilitate enzymatic RNA synthesis by DNA polymerases.
The dT–HgII–dT undoubtedly is the most prominent and best studied metal base pair35 and both the RNA equivalent, rU–HgII–rU,37 and the chimeric RNA–DNA variant, rU–HgII–dT,38,39 have been identified (Fig. 1A). The addition of mercury cations to these mismatches leads to large thermal stabilizations of duplexes (ΔTm ranging from +6 to +10 °C) driven by favorable enthalpy and entropy of formation.40 These favorable assets have allowed the enzymatic construction of single41 and multiple42 dT–HgII–dT base pairs under primer extension (PEX) reaction conditions. Similarly, silver cations have been shown to stabilize duplexes containing dC–dC and dC–dA (Fig. 1B) mismatches (ΔTm = +8.3 °C and +4.0 °C, respectively)43,44 which also could be produced enzymatically.45–47 However, the possibility of using these stable metal base pairs to form RNA–DNA heteroduplexes by enzymatic synthesis has never been investigated.
 | ||
| Fig. 1 Chemical structures of (A) HgII-mediated and (B) AgI-mediated metal base pairs with canonical nucleotides. | ||
Results and discussion
In a first step towards this aim, we wanted to evaluate the possibility of forming RNA–DNA mixed metal base pairs on short synthetic oligonucleotides using UV melting experiments. To do so, we designed 4 different duplexes containing central rU–dT, rA–dA, rC–dA, and rC–dC mismatches using 13 nucleotide long sequences (Table 1).48,49 The thermal stability of these duplexes in the presence of AgI, HgII, and a combination of HgII and MnII was investigated by temperature-dependent UV spectroscopy (Table 1 and ESI†). We have also determined the Tm values of a fully matched dsDNA duplex (duplex 1) and a duplex containing a central dT–dT mismatch to compare with the stability of the duplexes containing RNA–DNA mismatches. Expectedly, this analysis revealed that AgI and HgII had little incidence on the thermal stability of a fully matched dsDNA duplex (duplex 1) and that HgII could specifically stabilize a dT–dT mismatch by formation of a metal base pair (ΔTm = +9.8 °C; dsDNA duplex 2). The insertion of an rU–dT mismatch (RNA–DNA hetereo-duplex 3) led to a further decrease in duplex stability compared to the system containing a dT–dT mismatch (ΔTm = −3.0 °C) and the presence of HgII could substantially compensate for this loss of thermal stability (ΔTm = +8.3 °C). Hence, the dT–HgII–dT and rU–HgII–dT base pairs stabilize mismatched duplexes with comparable efficiencies (ΔTm = +9.8 and +8.3 °C, respectively) in this sequence context. The introduction of rA–dA, rC–dA, and rC–dC mismatches leads to a similar destabilization as observed between duplexes 2 and 3 (ΔTm ranging from −3.0 to −4.0 °C). The addition of AgI leads to marked increases in Tm values of the systems containing rC–dA (RNA–DNA hetereo-duplex 5) and rC–dC (RNA–DNA hetereo-duplex 6) mismatches (ΔTm = +3.8 and +4.3 °C, respectively) but appears to be thermoneutral in the case of RNA–DNA hetereo-duplex 4 that contains an rA–dA mismatch (ΔTm = 1.0 °C). Hence, the gain in duplex stabilization generated by the formation of an rC–AgI–dC pair appears to be reduced compared to that of the corresponding dC–AgI–dC (Fig. 1B) base pair (ΔTm of +4.3 °C and +8.3 °C,43 respectively) and compares to the stabilization imparted by an all-RNA rC–AgI–rC pair (ΔTm = +4.0 °C).39 On the other hand, the rC–AgI–dA and dC–AgI–dA pairs (Fig. 1B) display similar increases in thermal stabilities (ΔTm of +3.8 °C and +3.5 °C,50 respectively). Based on this UV-melting analysis, it appears unlikely that a silver mediated rA–AgI–dA base pair can be formed under these conditions due to the rather modest increase in Tm value observed upon addition of AgI to RNA–DNA hetereo-duplex 4.| Duplex | Sequences | Metal cation | T m (°C) | ΔTmb (°C) |
|---|---|---|---|---|
| a Standard deviations are given in parenthesis. b ΔTm is calculated for each duplex system as the difference in Tm between a measurement performed in the presence and one in the absence of metal cations. | ||||
| Duplex 1 | 5′-d(GAGGGTATGAAAG) | — | 50.6(4) | — |
| 3′-d(CTCCCATACTTTC) | ||||
| Duplex 1 | 5′-d(GAGGGTATGAAAG) | AgI | 51.4(1) | +0.8(2) |
| 3′-d(CTCCCATACTTTC) | ||||
| Duplex 1 | 5′-d(GAGGGTATGAAAG) | HgII | 50.3(5) | −0.3(3) |
| 3′-d(CTCCCATACTTTC) | ||||
| Duplex 2 | 5′-d(GAGGGTTTGAAAG) | — | 41.8(2) | — |
| 3′-d(CTCCCATACTTTC) | ||||
| Duplex 2 | 5′-d(GAGGGTTTGAAAG) | HgII | 51.6(1) | +9.8(2) |
| 3′-d(CTCCCATACTTTC) | ||||
| Duplex 2 | 5′-d(GAGGGTTTGAAAG) | MnII | 41.4(2) | −0.4(2) |
| 3′-d(CTCCCATACTTTC) | ||||
| Duplex 2 | 5′-d(GAGGGTTTGAAAG) | HgII, MnII | 50.8(1) | +9.0(2) |
| 3′-d(CTCCCATACTTTC) | ||||
| Duplex 3 | 5′-d(GAGGGT)rUd(TGAAAG) | — | 38.8(2) | — |
| 3′-d(CTCCCATACTTTC) | ||||
| Duplex 3 | 5′-d(GAGGGT)rUd(TGAAAG) | HgII | 47.1(2) | +8.3(2) |
| 3′-d(CTCCCATACTTTC) | ||||
| Duplex 3 | 5′-d(GAGGGT)rUd(TGAAAG) | MnII | 39.0(1) | +0.2(2) |
| 3′-d(CTCCCATACTTTC) | ||||
| Duplex 3 | 5′-d(GAGGGT)rUd(TGAAAG) | HgII, MnII | 46.4(1) | +7.6(2) |
| 3′-d(CTCCCATACTTTC) | ||||
| Duplex 4 | 5′-d(GAGGG)rAd(ATGAAAG) | — | 39.2(1) | — |
| 3′-d(CTCCCATACTTTC) | ||||
| Duplex 4 | 5′-d(GAGGG)rAd(ATGAAAG) | AgI | 40.2(4) | +1.0(2) |
| 3′-d(CTCCCATACTTTC) | ||||
| Duplex 5 | 5′-d(GAGGG)rCd(ATGAAAG) | — | 39.8(1) | — |
| 3′-d(CTCCCATACTTTA) | ||||
| Duplex 5 | 5′-d(GAGGG)rCd(ATGAAAG) | AgI | 43.6(1) | +3.8(1) |
| 3′-d(CTCCCATACTTTA) | ||||
| Duplex 6 | 5′-d(GAGGG)rCd(ATGAAAG) | — | 38.1(2) | — |
| 3′-d(CTCCCCTACTTTA) | ||||
| Duplex 6 | 5′-d(GAGGG)rCd(ATGAAAG) | AgI | 42.4(4) | +4.3(3) |
| 3′-d(CTCCCCTACTTTA) | ||||
Having established the possibility of constructing RNA–DNA chimeric metal base pairs with synthetic oligonucleotides, we next sought to evaluate whether this could be translated to enzymatic synthesis. To do so, we carried out primer extension (PEX) reactions with templates containing an overhang of seven consecutive dT (T1), dC (T2), dA (T3), or dG (T4) nucleotides immediately following the 3′-terminus of the FAM-labelled primer P1 (Table 2).51 Initial PEX reactions were carried out with five different commercially available DNA polymerases (Taq, Bst, Vent (exo−), Sulfolobus DNA Polymerase IV (Dpo4), and the Klenow fragment of DNA polymerase I exo− (Kf exo−)) and in the presence or absence of either HgII or AgI. All the PEX reaction products were analyzed by 20% denaturing gel electrophoresis and visualised using fluorescence imaging.
| Name | Sequence |
|---|---|
| P1 | 5′-FAM-d(CAT GGG CGG CAT GGG) |
| T1 | 5′-d(TTT TTT TCC CAT GCC GCC CAT G) |
| T2 | 5′-d(CCC CCC CCC CAT GCC GCC CAT G) |
| T3 | 5′-d(AAA AAA ACC CAT GCC GCC CAT G) |
| T4 | 5′-d(GGG GGG GCC CAT GCC GCC CAT G) |
Expectedly, all DNA polymerases investigated were capable of incorporating one (for Bst) or multiple deoxythymidine monophosphate (dTMP) units opposite templating dT nucleotides in the presence of HgII when the primer/template P1/T1 system and dTTP were used (Fig. S1, ESI†). Moreover, when PEX reactions were conducted in the presence of both rUTP and HgII, partial incorporation of a single uridine monophosphate (UMP) moiety could be observed when Kf exo− was used as a polymerase, albeit in modest yields (∼20%). The control reaction performed in absence of the metal cation did not yield any extended primer product. When the reaction mixtures were supplemented with the MnII cofactor which is known to relax polymerase fidelity52 and favor the formation of mercury metal mediated base pairs,42 the yield of n + 1 product formation significantly increased to ∼50% (Fig. 2A and Fig. S2, ESI†). In order to confirm that MnII was not implicated in metal base pair formation but served to improve the substrate tolerance of the polymerase, we recorded UV-melting experiments with duplexes containing homo- and hetero-mismatches (dT–dT and rU–dT) in the presence of MnII. The addition of this metal cation had little effect on the Tm values of duplexes 2 and 3, confirming that MnII does not trigger metal base pair formation in duplex DNA. This is further confirmed by the small decrease in Tm observed when dsDNA duplex 2 and RNA–DNA hetereo-duplex 3 were supplemented both with MnII and HgII instead of HgII alone (−0.8 and −0.7 °C, respectively).
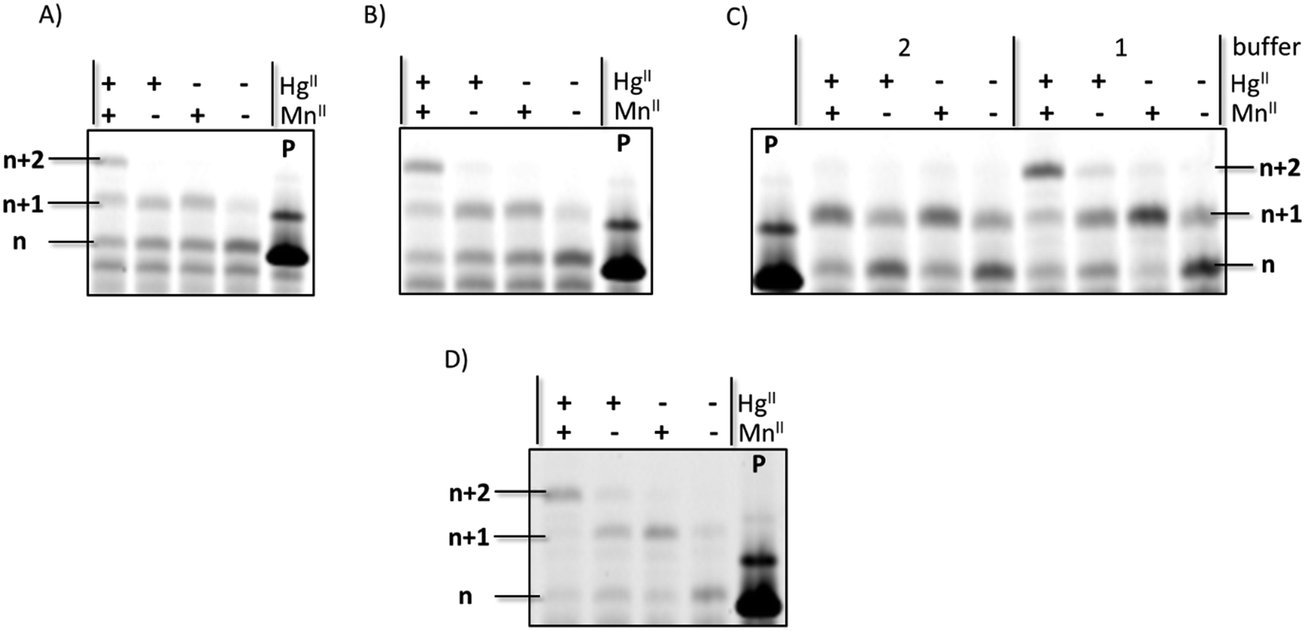 | ||
| Fig. 2 Gel images (PAGE 20%) of PEX reactions carried out with primer P1 and template T1 using different UTP concentrations, reaction times, and metal cations. All reactions were performed with 5 U of Kf exo− at 37 °C. (A) Reactions with 200 μM UTP and 3 h reaction time in buffer 1; (B) reactions with 400 μM UTP and 3 h reaction time in buffer 1; (C) reactions with 200 μM UTP and 12 h reaction time; (D) reactions in buffer 1 with 400 μM UTP and 12 h reaction time. 5 U of Kf exo− were used in all reactions. Buffer 1 did not contain any source of Cl− and buffer 2 is the supplied buffer. In all cases, Hg(ClO4)2 was used as source of mercury. P indicates unreacted primer. | ||
Based on these observations, we next sought to fine tune the experimental conditions to improve both yields and number of incorporation events (Fig. 2). When both the reaction time (from 3 h to 12 h; Fig. 2C) and the UTP concentration (from 200 to 400 μM; Fig. 2B) were increased, full conversion of the primer to the n + 1 (∼10%) and n + 2 (∼90%) products could be achieved when HgII and MnII were present (Fig. 2D). In addition, no incorporation of a uridine moiety into DNA could be observed in the absence of HgII, underscoring the need for metal base-pair formation for the incorporation of this mismatched RNA nucleotide into DNA. We have also carried out PEX reactions with templates T1–T4 and the corresponding, complementary, NTPs (Fig. S3, ESI†) under similar experimental conditions. This analysis reveals that in PEX reactions with template T3, which contains seven dA units, two UMPs are appended on the 3′-end of primer P1 (Fig. S3, ESI†) with a comparable efficiency to that of the misincoporation of UMPs opposite templating dT units triggered by mercury cations shown in Fig. 2C and D. Surprisingly, Kf exo− was capable of appending up to 7 rNMP units onto primer P1 when templates T1, T2, and T4 were used in PEX reactions in conjunction with the corresponding nucleoside triphosphate (Fig. S3, ESI†), confirming earlier findings that Pol I can incorporate matched ribonucleotides in vitro.28
Having established conditions that enable the enzymatic formation of an rU–HgII–dT base pair, we next questioned whether this metal base pair could be bypassed once installed so that DNA synthesis could resume.53 To do so, we carried out PEX reactions with the P1/T1 system and UTP to install rU–HgII–dT base pairs (Fig. 3A). The resulting products were then incubated with dTTP and HgII (Fig. S5, ESI†) or dATP (Fig. 3B–F) following an experimental protocol established for silver-mediated artificial base pairs (see ESI†).53 Full length products (n + 7) could be observed upon the addition of dATP and Kf exo− albeit in low yields (Fig. 3B). Interestingly, when Kf exo− was substituted by other polymerases including Bst, Vent (exo−), and Dpo4, the expected full lengths products could be formed in high yields (Fig. 3C, E, and F, respectively), while reactions conducted with Therminator produced slower running products that stem from untemplated addition of dAMP residues (Fig. 3D). These results suggest that the rU–HgII–dT base pairs did not induce termination of DNA synthesis and can be bypassed. On the other hand, excess HgII used for the installation of the rU–HgII–dT base pairs appears to inhibit the extension reactions with dATP and need to be removed with EDTA prior to the bypass reactions (Fig. S5C and ESI†). Lastly, formation of dT–HgII–dT base pair after the installation of the rU–HgII–dT hetero-metal base pairs did not proceed very well and was observed in low yields only (Fig. S5B, ESI†).
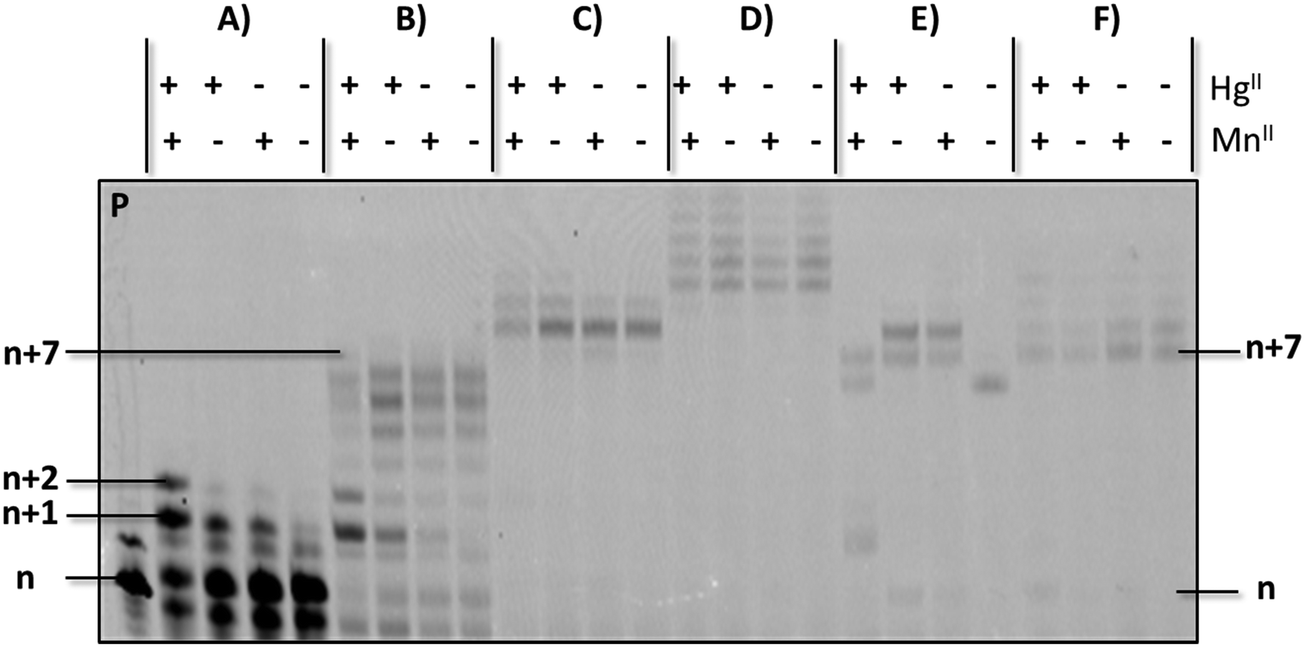 | ||
| Fig. 3 Gel analysis (PAGE 20%) of products from the bypass experiments carried out with the primer P1/template T1 system. (A) Reactions with UTP (400 μM) and Kf exo− (5 U) at 37 °C; (B) reaction with UTP (400 μM) and Kf exo− (5 U) at 37 °C for 3 h followed by removal of excess HgII, addition of dATP (200 μM), and reaction for 1 h; (C) as in (B) but with Bst (8 U) instead of Kf exo−; (D) as in (B) but with Therminator (2 U) instead of Kf exo−; (E) as in (B) but with Vent (exo−) (2 U) instead of Kf exo−; (F) as in (B) but with Dpo4 (2 U) instead of Kf exo−. P indicates unreacted primer. | ||
Having established the possibility of forming rU–HgII–dT base pairs, we next considered the possibility of using AgI to trigger the formation of DNA/RNA hetero base pairs. UV melting experiments revealed that the rC–dA and rC–dC mismatches could be stabilized by the addition of AgI, albeit to a lesser extent than rU–dT mismatches with HgII. Nonetheless, we attempted to use CTP as a substrate for polymerases to enzymatically construct rC–AgI–dA and rC–AgI–dC base pairs since similar syntheses have been reported for the corresponding DNA base pairs.45–47 We thus carried out PEX reactions with the primer/template systems P1/T2 (Fig. S4, ESI†) and P1/T3 (Fig. S6, ESI†) in the presence of different DNA polymerases and AgI. Even by changing multiple reaction parameters (CTP and polymerase concentration, reaction times) we could not observe any difference with the control reactions carried out in absence of metal cofactor and the incorporation of the RNA nucleotide remained modest. These results suggest that the strength of the rC–AgI–dA and rC–AgI–dC base pairs might not be sufficient to coerce the introduction of mismatched RNA nucleotides into DNA duplexes under enzymatic conditions. Finally, when PEX reactions were conducted with ATP with primer P1 and template T3, no extended primer products could be observed (data not shown) which confirms the absence of enzymatic rA–AgI–dA metal base pair formation observed in UV melting experiments (RNA–DNA hetereo-duplex 4 in Table 1).
Conclusions
In conclusion, we have investigated the possibility of using the favorable thermodynamic parameters of metal base pair formation to insert RNA nucleotides into an all-DNA setting using DNA polymerases. In a first step towards this aim, we have used UV melting experiments on short synthetic duplexes to confirm the possibility of forming rU–HgII–dT base pairs. These mercury-mediated base pairs stabilized mismatched duplexes albeit to a slightly lesser extent than the parent dT–HgII–dT base pair. A similar trend was observed with a duplex containing rC–AgI–dA base pair but not with rC–AgI–dC which is less stable than in the all-DNA case. These trends are reflected in PEX reactions using natural RNA nucleotides since efficient rU–HgII–dT base pair formation could be observed. On the other hand, the stability of silver-mediated pairs appeared to be insufficient to coerce DNA polymerases to misincorporate RNA nucleotides even in the presence of metal cations. Taken together, the favorable thermodynamic parameters of mercury-mediated base pairs can be hijacked to generate RNA–DNA oligonucleotides using polymerase-assisted synthesis. Such an approach alleviates the synthetic efforts required for the generation of such chimeric oligonucleotides which are used in numerous applications such as substrates for DNAzyme selections.54,55 Finally, we are currently investigating the compatibility of other sugar chemistries with the enzymatic synthesis of artificial metal base pairs in order to eventually achieving our long standing aim of creating orthogonal xenonucleic acids based on metal base pairs.56 The use of engineered polymerases20 with a broader substrate tolerance will certainly advance as important tools in this context since they will permit the efficient construction of additional RNA–DNA hetero-metal base pairs and expand this concept to XNAs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Institut Pasteur (starting funds to M. H.) and The Joe W. and Dorothy Doresett Brown Foundation (research grants to M. H.) are acknowledged for funding. L. K. M. thanks the ARC Foundation for cancer research for a postdoctoral Research Fellowship (grant number: S-FB18006).Notes and references
- J. Müller, Coord. Chem. Rev., 2019, 393, 37–47 CrossRef.
- Y. Tanaka, J. Kondo, V. Sychrovský, J. Šebera, T. Dairaku, H. Saneyoshi, H. Urata, H. Torigoe and A. Ono, Chem. Commun., 2015, 51, 17343–17360 RSC.
- K. Kowalski, Coord. Chem. Rev., 2021, 432, 213705 CrossRef CAS.
- G. H. Clever, C. Kaul and T. Carell, Angew. Chem., Int. Ed., 2007, 46, 6226–6236 CrossRef CAS PubMed.
- A. Perez-Romero, A. Dominguez-Martin, S. Galli, N. Santamaria-Diaz, O. Palacios, J. A. Dobado, M. Nyman and M. A. Galindo, Angew. Chem., Int. Ed., 2021, 60, 10089–10094 CrossRef CAS PubMed.
- A. Gonzalez-Rosell, C. Cerretani, P. Mastracco, T. Vosch and S. M. Copp, Nanoscale Adv., 2021, 3, 32 RSC.
- A. Aro-Heinila, T. Lönnberg and P. Virta, ChemBioChem, 2021, 22, 354–358 CrossRef PubMed.
- J. Kondo, Y. Tada, T. Dairaku, Y. Hattori, H. Saneyoshi, A. Ono and Y. Tanaka, Nat. Chem., 2017, 9, 956–960 CrossRef CAS PubMed.
- A. Ono, H. Kanazawa, H. Ito, M. Goto, K. Nakamura, H. Saneyoshi and J. Kondo, Angew. Chem., Int. Ed., 2019, 58, 16835–16838 CrossRef CAS PubMed.
- X. L. Zhou, D. Kondhare, P. Leonard and F. Seela, Chem. – Eur. J., 2019, 25, 10408–10419 CrossRef CAS PubMed.
- Y. Takezawa, A. Suzuki, M. Nakaya, K. Nishiyama and M. Shionoya, J. Am. Chem. Soc., 2020, 142, 21640–21644 CrossRef CAS PubMed.
- M. H. Heddinga and J. Müller, Beilstein J. Org. Chem., 2020, 16, 2870–2879 CrossRef CAS PubMed.
- Y. Takezawa, L. Y. Hu, T. Nakama and M. Shionoya, Angew. Chem., Int. Ed., 2020, 59, 21488–21492 CrossRef CAS PubMed.
- T. Nakama, Y. Takezawa, D. Sasaki and M. Shionoya, J. Am. Chem. Soc., 2020, 142, 10153–10162 CrossRef CAS PubMed.
- L. K. McKenzie, R. El-Khoury, J. D. Thorpe, M. J. Damha and M. Hollenstein, Chem. Soc. Rev., 2021, 50, 5126–5164 RSC.
- M. Flamme, C. Figazzolo, G. Gasser and M. Hollenstein, Metallomics, 2021, 13, mfab016 CrossRef PubMed.
- T. Kobayashi, Y. Takezawa, A. Sakamoto and M. Shionoya, Chem. Commun., 2016, 52, 3762–3765 RSC.
- C. Kaul, M. Müller, M. Wagner, S. Schneider and T. Carell, Nat. Chem., 2011, 3, 794–800 CrossRef CAS PubMed.
- E.-K. Kim and C. Switzer, ChemBioChem, 2013, 14, 2403–2407 CrossRef CAS PubMed.
- N. Freund, M. J. L. J. Fürst and P. Holliger, Curr. Opin. Biotechnol, 2022, 74, 129–136 CrossRef CAS PubMed.
- B. Jash, P. Scharf, N. Sandmann, C. Fonseca Guerra, D. A. Megger and J. Müller, Chem. Sci., 2017, 8, 1337–1343 RSC.
- L. Zhang and E. Meggers, J. Am. Chem. Soc., 2005, 127, 74–75 CrossRef CAS PubMed.
- Z. Ma, F. Olechnowicz, Y. A. Skorik and C. Achim, Inorg. Chem., 2011, 50, 6083–6092 CrossRef CAS PubMed.
- O. Nakagawa, H. Aoyama, A. Fujii, Y. Kishimoto and S. Obika, Chem. – Eur. J., 2021, 27, 3842–3848 CrossRef CAS PubMed.
- E. Medina, E. J. Yik, P. Herdewijn and J. C. Chaput, ACS Synth. Biol., 2021, 10, 1429–1437 CrossRef CAS PubMed.
- W.-J. Wu, W. Yang and M.-D. Tsai, Nat. Rev. Chem., 2017, 1, 0068 CrossRef CAS.
- C. M. Joyce, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 1619–1622 CrossRef CAS PubMed.
- M. Astatke, K. M. Ng, N. D. F. Grindley and C. M. Joyce, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 3402–3407 CrossRef CAS PubMed.
- J. A. Brown and Z. C. Suo, Biochemistry, 2011, 50, 1135–1142 CrossRef CAS PubMed.
- Z. X. Zhou, J. S. Williams, S. A. Lujan and T. A. Kunkel, Crit. Rev. Biochem. Mol. Biol., 2021, 56, 109–124 CrossRef CAS PubMed.
- S. A. N. McElhinny, B. E. Watts, D. Kumar, D. L. Watt, E. B. Lundstrom, P. M. J. Burgers, E. Johansson, A. Chabes and T. A. Kunkel, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4949–4954 CrossRef PubMed.
- S. Ohashi, F. Hashiya and H. Abe, ChemBioChem, 2021, 22, 2398–2406 CrossRef CAS PubMed.
- A. F. Gardner, K. M. Jackson, M. M. Boyle, J. A. Buss, V. Potapov, A. M. Gehring, K. M. Zatopek, I. R. Correa, J. L. Ong and W. E. Jack, Front. Mol. Biosci., 2019, 6, 9 CrossRef PubMed.
- T. Chen, N. Hongdilokkul, Z. Liu, R. Adhikary, S. S. Tsuen and F. E. Romesberg, Nat. Chem., 2016, 8, 556–562 CrossRef CAS PubMed.
- Y. Miyake, H. Togashi, M. Tashiro, H. Yamaguchi, S. Oda, M. Kudo, Y. Tanaka, Y. Kondo, R. Sawa, T. Fujimoto, T. Machinami and A. Ono, J. Am. Chem. Soc., 2006, 128, 2172–2173 CrossRef CAS PubMed.
- O. P. Schmidt, G. Mata and N. W. Luedtke, J. Am. Chem. Soc., 2016, 138, 14733–14739 CrossRef CAS PubMed.
- S. Johannsen, S. Paulus, N. Düpre, J. Müller and R. K. O. Sigel, J. Inorg. Biochem., 2008, 102, 1141–1151 CrossRef CAS PubMed.
- S. Manna and S. G. Srivatsan, Org. Lett., 2019, 21, 4646–4650 CrossRef CAS PubMed.
- T. Kozasa, Y. Miyakawa, A. Ono and H. Torigoe, Nucleic Acids Symp. Ser., 2008, 52, 197–198 CrossRef CAS PubMed.
- H. Torigoe, Y. Miyakawa, A. Ono and T. Kozasa, Thermochim. Acta, 2012, 532, 28–35 CrossRef CAS.
- H. Urata, E. Yamaguchi, T. Funai, Y. Matsumura and S. Wada, Angew. Chem., Int. Ed., 2010, 49, 6516–6519 CrossRef CAS PubMed.
- T. Funai, C. Tagawa, O. Nakagawa, S. Wada, A. Ono and H. Urata, Chem. Commun., 2020, 56, 12025–12028 RSC.
- A. Ono, S. Cao, H. Togashi, M. Tashiro, T. Fujimoto, T. Machinami, S. Oda, Y. Miyake, I. Okamoto and Y. Tanaka, Chem. Commun., 2008, 4825–4827 RSC.
- J. Kondo, Y. Tada, T. Dairaku, H. Saneyoshi, I. Okamoto, Y. Tanaka and A. Ono, Angew. Chem., Int. Ed., 2015, 54, 13323–13326 CrossRef CAS.
- T. Funai, Y. Miyazaki, M. Aotani, E. Yamaguchi, O. Nakagawa, S. Wada, H. Torigoe, A. Ono and H. Urata, Angew. Chem., Int. Ed., 2012, 51, 6464–6466 CrossRef CAS PubMed.
- T. Funai, J. Nakamura, Y. Miyazaki, R. Kiriu, O. Nakagawa, S. Wada, A. Ono and H. Urata, Angew. Chem., Int. Ed., 2014, 53, 6624–6627 CrossRef CAS.
- T. Funai, M. Aotani, R. Kiriu, J. Nakamura, Y. Miyazaki, O. Nakagawa, S. Wada, H. Torigoe, A. Ono and H. Urata, ChemBioChem, 2020, 21, 517–522 CrossRef CAS.
- B. Jash, J. Neugebauer and J. Müller, Inorg. Chim. Acta, 2016, 452, 181–187 CrossRef CAS.
- S. Naskar and J. Müller, Chem. – Eur. J., 2019, 25, 16214–16218 CrossRef CAS PubMed.
- H. Torigoe, I. Okamoto, T. Dairaku, Y. Tanaka, A. Ono and T. Kozasa, Biochimie, 2012, 94, 2431–2440 CrossRef CAS PubMed.
- P. Röthlisberger, F. Levi-Acobas, I. Sarac, R. Ricoux, J. P. Mahy, P. Herdewijn, P. Marliere and M. Hollenstein, Tetrahedron Lett., 2018, 59, 4241–4244 CrossRef.
- P. Röthlisberger, F. Levi-Acobas, I. Sarac, P. Marliere, P. Herdewijn and M. Hollenstein, Org. Biomol. Chem., 2017, 15, 4449–4455 RSC.
- M. Flamme, P. Röthlisberger, F. Levi-Acobas, M. Chawla, R. Oliva, L. Cavallo, G. Gasser, P. Marliere, P. Herdewijn and M. Hollenstein, ACS Chem. Biol., 2020, 15, 2872–2884 CrossRef CAS PubMed.
- M. Hollenstein, Curr. Opin. Chem. Biol., 2019, 52, 93–101 CrossRef CAS PubMed.
- Y. J. Wang, K. Nguyen, R. C. Spitale and J. C. Chaput, Nat. Chem., 2021, 13, 13 Search PubMed.
- J. C. Chaput, P. Herdewijn and M. Hollenstein, ChemBioChem, 2020, 21, 1408–1411 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2nj00427e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2022 |