
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotochemical transformation of chlorobenzenes and white phosphorus into arylphosphines and phosphonium salts†
Marion
Till
a,
Verena
Streitferdt
b,
Daniel J.
Scott
 a,
Michael
Mende
a,
Ruth M.
Gschwind
b and
Robert
Wolf
*a
a,
Michael
Mende
a,
Ruth M.
Gschwind
b and
Robert
Wolf
*a
aUniversität Regensburg, Institut für Anorganische Chemie, Regensburg 93040, Germany. E-mail: robert.wolf@ur.de
bUniversität Regensburg, Institut für Organische Chemie, Regensburg 93040, Germany
First published on 6th December 2021
Abstract
Chlorobenzenes are important starting materials for the preparation of commercially valuable triarylphosphines and tetraarylphosphonium salts, but their use for the direct arylation of elemental phosphorus has been elusive. Here we describe a simple photochemical route toward such products. UV-LED irradiation (365 nm) of chlorobenzenes, white phosphorus (P4) and the organic superphotoreductant tetrakis(dimethylamino)ethylene (TDAE) affords the desired arylphosphorus compounds in a single reaction step.
Commercially valuable and academically interesting organo-phosphorus compounds (OPCs) such as arylated phosphines (Ar3P) and phosphonium salts ([Ar4P]+) are currently prepared by a wasteful and inefficient multi-step procedure which relies on the initial oxidation of white phosphorus (P4) using toxic Cl2 gas to give highly reactive PCl3 which is further converted into the desired monophosphorus species (Fig. 1a).1,2 Given the numerous drawbacks of this method the direct transformation of P4 into OPCs and other P-containing compounds, avoiding potentially hazardous intermediates, has long been an exceedingly important yet challenging objective.3
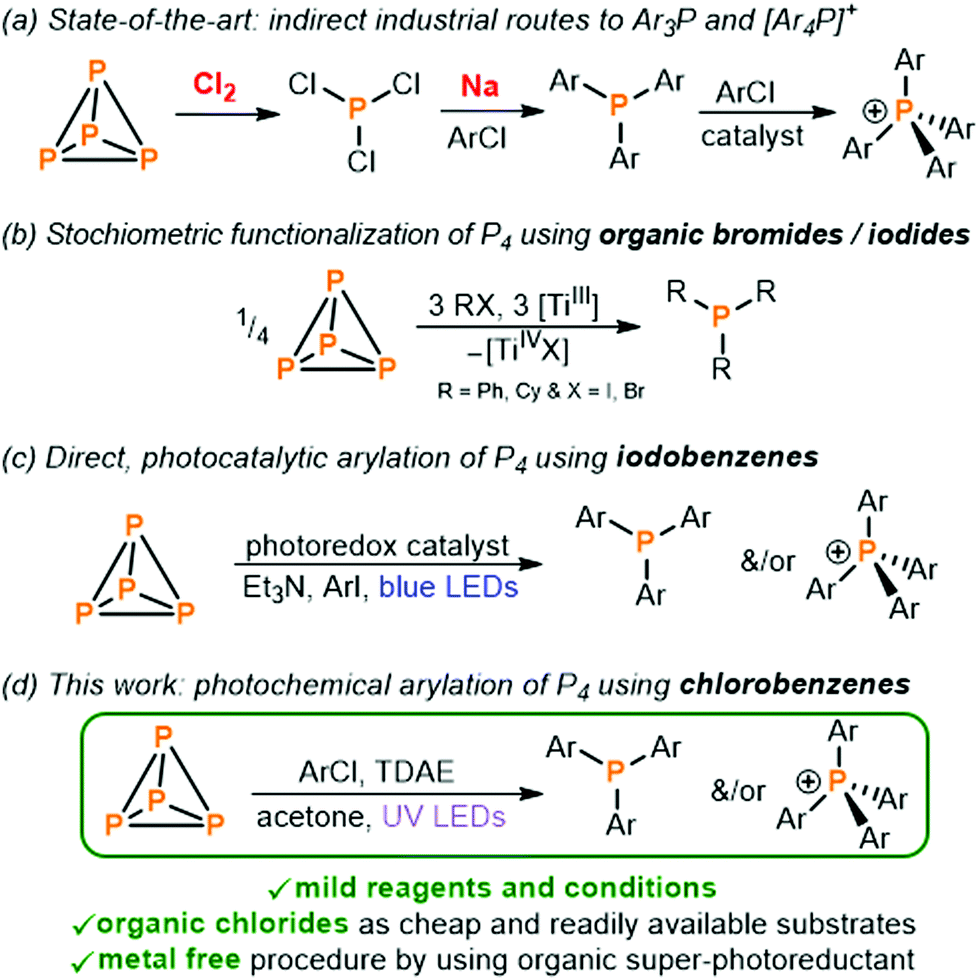 | ||
| Fig. 1 (a) Indirect methods for the synthesis of triarylphosphines Ar3P and tetraarylphosphonium salts [Ar4P]+ (Ar = aryl substituent) employed industrially,1,2 (b) stoichiometric transformation of P4 into tertiary phosphines R3P ([TiIII] = Ti(N[tBu]Ar)3),6 (c) recently reported direct photocatalytic transformation of P4 into Ar3P and [Ar4P]+,8–10 and (d) photochemical arylation of P4 using chlorobenzenes and an organic super-photoreductant (TDAE = tetrakis(dimethylamino)ethylene). | ||
Pioneering studies by Barton and co-workers demonstrated the excellent ability of P4 to trap carbon-centered radicals, and that this can provide the basis for subsequent transformation of white phosphorus into valuable OPCs.4,5 Subsequently, stoichiometric functionalization reactions were reported by Cummins and co-workers in which the generation of reactive organoradicals by reacting organic bromides and iodides with a titanium(III) complex (or more recently samarium(II) halides) is again the fundamental first step in converting P4 into the corresponding tertiary phosphines (Fig. 1b).6,7 Expanding on these stoichiometric methodologies in which organic halides are reduced by metal-containing reductants in the presence of P4, recently, we reported the first catalytic procedure to obtain OPCs directly from P4 (Fig. 1c).8–10 The reduced form of a photoexcited iridium photocatalyst or, alternatively, an organophotocatalyst is able to reduce aryl iodides (ArI) to generate carbon-centered aryl radicals which are again trapped by P4 to directly form arylated phosphines and phosphonium salts.
All these efforts to prepare OPCs directly from P4 through the use of carbon-centered radicals were realized only by using organic bromides and iodides.11–13 However, from an economic point of view the use of bromo- and iodobenzenes for the preparation of tertiary phosphines is infeasible, with these substrates often even being more expensive than the target phosphines. In contrast, organic chlorides are cheap and abundant substrates which comprise over two-thirds of commercially available aryl halides,14,15 but their use as substrates and radical precursors in photocatalysis and photochemistry is challenging because of their chemical inertness.16–19 Herein, we describe a simple photochemical method for the preparation of various valuable triarylphosphines and tetraarylphosphonium salts directly from P4 by using only readily and cheaply commercially available aryl chlorides.
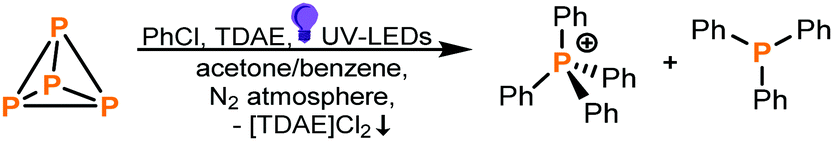
Recently, Wenger and co-workers reported on a remarkably simple light-driven reductive hydrodehalogenation of organic chlorides via aryl radicals mediated by tetrakis(dimethylamino)ethylene (TDAE) as a photoreductant.20 We anticipated that radicals generated in the same manner could be trapped to functionalize P4. And, indeed, UV light irradiation (365 nm) of a solution of P4 (added as a stock solution in benzene), PhCl (as a simple model substrate) and TDAE in acetone for 20 hours yielded the phosphonium salt [Ph4P]Cl (53% yield determined by NMR) and tertiary phosphine Ph3P (9% NMR yield) (Table 1, entry 1). To our knowledge, this represents the first example of an aryl chloride successfully being reacted with P4 to provide such products directly. Further variations of solvent, light source, temperature, stoichiometry and reaction time (see ESI,† S3 and Tables 1–7) indicated that the reaction parameters given in Table 1 (entry 1) are optimal.
|
|
||||
|---|---|---|---|---|
| Entry | Conditions | Full conv. of P4 | Conv. to [Ph4P]Cl/% | Conv. to Ph3P/% |
| a Reactions were performed with 40.7 µL chlorobenzene (10 equiv. based on phosphorus atoms, 40 equiv. based on P4), 46.5 µL TDAE (5 equiv. based on phosphorus atoms) and P4 (0.01 mmol, as a stock solution in 71.3 µL benzene) in 0.5 mL acetone as solvent. Samples were prepared under N2 atmosphere in a sealed tube and placed in a water-cooled block during irradiation (20 h) with UV light (365 nm). Conversions were determined by quantitative 31P{1H} NMR experiments with subsequently added Ph3PO as an internal standard. | ||||
| 1 | Standarda | ✓ | 53 | 9 |
| 2 | No light | ✗ | 0 | 0 |
| 3 | No TDAE | ✗ | 0 | 0 |
| 4 | No PhCl | ✗ | 0 | 0 |
| 5 | PhF instead of PhCl | ✗ | 0 | 0 |
| 6 | PhBr instead of PhCl | ✓ | 43 | 10 |
| 7 | PhI instead of PhCl | ✓ | 14 | 10 |
| 8 | Pred instead of P4 | — | 0 | 0 |
For the transformation of one P4 molecule into four [Ph4P]Cl at least 16 PhCl have to be reduced to form the needed 16 P–C bonds. For a clean stoichiometric functionalization, a stoichiometric ratio of TDAE to PhCl of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :16 (equiv. based on P4) should suffice to efficiently generate the final phosphonium salt (assuming TDAE acts as a two-electron donor; vide infra). Nevertheless, adapting the reaction stoichiometry to this level was found to result in appreciably reduced conversion (see ESI,† S3 and Table S5). In part this is likely due to side reactions such as forming benzene from an aryl radical/anion or generating biphenyl by recombination of two radicals (confirmed by GC-MS). Although all P4 is consumed quantitative product formation is not observed, even after an increase of the reaction time (40 h) as shown by quantitative 31P{1H} NMR spectroscopy (see ESI,† S3 and Table S6). The loss of phosphorus intensity is possibly caused by formation of polymerized phosphorus species, which was also a limiting factor in our recently reported photocatalytic procedure.8
:16 (equiv. based on P4) should suffice to efficiently generate the final phosphonium salt (assuming TDAE acts as a two-electron donor; vide infra). Nevertheless, adapting the reaction stoichiometry to this level was found to result in appreciably reduced conversion (see ESI,† S3 and Table S5). In part this is likely due to side reactions such as forming benzene from an aryl radical/anion or generating biphenyl by recombination of two radicals (confirmed by GC-MS). Although all P4 is consumed quantitative product formation is not observed, even after an increase of the reaction time (40 h) as shown by quantitative 31P{1H} NMR spectroscopy (see ESI,† S3 and Table S6). The loss of phosphorus intensity is possibly caused by formation of polymerized phosphorus species, which was also a limiting factor in our recently reported photocatalytic procedure.8
Control experiments confirmed that all reaction components (P4, PhCl, TDAE and UV light) are necessary to observe product formation (Table 1, entries 2–4). Switching from PhCl to the less reactive PhF or the more easily reducible substrates PhBr or PhI was found to be detrimental (Table 1, entries 5–7). In the case of PhF no reaction occurred at all. Also, replacing P4 with red phosphorus yielded no product (Table 1, entry 8).
Various other substituted aryl chlorides could also successfully be employed under identical reaction conditions, albeit with somewhat lower conversions than for chlorobenzene (see ESI,† Table S8 and Fig. S22). The investigated substrate scope shows that the method is more suitable for substrates bearing electron-donating (Me, OMe) groups than for chloroarenes with electron-withdrawing groups (EWGs, such as CF3, COOMe), which generally gave poor conversions. Gratifyingly, not only aryl radicals led to product formation. For benzyl chloride 27% conversion to tribenzylphosphine was achieved. Even more impressively, employing (2-chloroethyl)benzene as substrate the product tris(2-phenylethyl)phosphine was formed selectively in 75% NMR yield. Selected products (tetraphenylphosphonium chloride and tris(2-phenylethyl)phosphine) were synthesized by performing reactions on a preparative (0.8 mmol) scale and isolated in modest yields by precipitation from dichloromethane/n-hexane (for the phosphonium salt) or fractional distillation (for the phosphine). See the ESI† for details of the purification and isolation of these products.
The chemical inertness of these chlorobenzenes (e.g. PhCl; −2.78 V vs. SCE in DMF,21Fig. 2) is due to the high energetic barrier for C–Cl bond activation. To overcome this a strong (photo)reductant is needed. TDAE as a single-electron reducing agent with an oxidation potential of −0.78 V vs. SCE (CH3CN)22 is not strong enough to split off a chloride anion to generate aryl radicals. By contrast the photoactive excited state of TDAE (*TDAE) offers an extraordinary reducing power of −3.4 V vs. SCE20 (Fig. 2) which makes it a super-photoreductant. Luminescence lifetime measurements showed that the excited state of TDAE is quenched by acetone.20 Thus, instead of a direct single electron transfer (SET) from photoexcited TDAE to the substrate, acetone (−2.84 V vs. SCE)23 is reduced and forms an acetone radical anion which subsequently undergoes SET to the organic chlorides and generates carbon-centered radicals. Using a solvent which cannot be reduced by photoexcited TDAE such as acetonitrile or benzene (−3.42 V vs. SCE),24 or employing the substrate PhCl as solvent decreased the yield of phosphonium salt and phosphine (see ESI,† S3, Table S4).
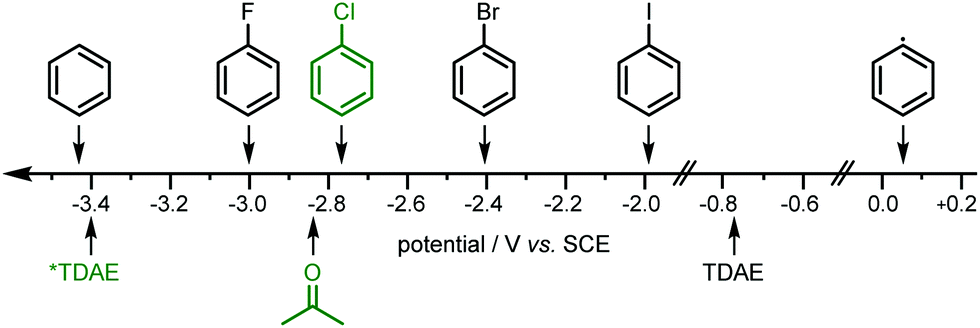 | ||
| Fig. 2 Reducing power of ground-state and exited-state organic photoreductant TDAE20 and reduction potentials of used solvents (acetone and benzene),23,24 aryl halides (fluoro-, chloro-,21 bromo- and iodobenzene)25 and the corresponding aryl radical.26 | ||
Usually, organic chlorides are more inactive substrates than the corresponding bromides and iodides because of their high bond dissociation energy (PhCl 327 kJ mol−1).16 As mentioned above, however, using substrates such as bromobenzene or iodobenzene (PhBr −2.44 V vs. SCE, PhI −1.93 V vs. SCE)25 for this photochemical procedure reduced the product formation. While the reasons for this are not entirely clear, it is presumably related to the fact that the bromide and iodide can more easily be reduced than aryl chlorides, leading to more rapid accumulation of aryl radicals, that may then be more prone to unwanted side-reactions such as dimerization or over-reduction (+0.05 V vs. SCE).26,27 In contrast, using aryl fluorides as radical source yielded no product at all, presumably because PhF is outcompeted by the solvent as an oxidative quencher of *TDAE, and the resulting acetone radical anion is incapable of reducing PhF (see Fig. 2).
Time-resolved 31P{1H} NMR spectroscopic investigation of the model reaction using chlorobenzene showed rapid consumption of P4 (Fig. 3, completely consumed within one hour under in situ NMR conditions, see also ESI,† S3 and Table S6) and the formation of Ph3P, [Ph4P]Cl and another intermediate (Int1). The structure of Int1 could be assigned based on a combination of its 31P chemical shift, 1H–31P HMQC experiments and comparison with literature data,28 which suggest a TDAE skeleton that is coordinated to a Ph2P moiety via a methylene group (Fig. 3 and see also ESI,† S6 and Fig. S36).
 | ||
| Fig. 3 In situ 31P{1H} NMR monitoring of phosphorus speciation during photochemical functionalization of white phosphorus using chlorobenzene and TDAE (see ESI,† S6 for full reaction details and qualitative kinetics). The diagram is cut off at the x (30 h) and y axis (400). Overall the reaction was monitored for 90 h and the initial P4 intensity is defined as 1000. The relatively long reaction time can be attributed to the reduced irradiation power of the in situ light source and lack of stirring. | ||
On the basis of the results mentioned above, the light-driven functionalization of P4 in the presence of chlorobenzene and TDAE in acetone is proposed to proceed by the mechanism summarized in Fig. 4. First, irradiation of TDAE with UV light provides a super-photoreductant (TDAE*) (see Fig. 4(i)) which is then able to reduce the solvent acetone by a SET generating TDAE˙+ and acetone˙− (Fig. 4(ii)). Next, the acetone radical anion reduces the substrate PhCl (Fig. 4(iii)) which can then provide phenyl radicals after elimination of Cl− (Fig. 4(iv)). We denote steps (ii–iv) as the solvent radical mechanism. The aryl radicals thus generated then sequentially functionalize P4 by controlled cleavage of all six P–P bonds of the P4 tetrahedron (Fig. 4(v)), producing a sequence of different arylated intermediates analogous to that observed in our previous photocatalytic method,8–10 such as the primary phosphine (PhPH2), the secondary phosphine (Ph2PH), the diphosphine (Ph4P2), Ph3P and finally [Ph4P]Cl. The first arylation step to the primary phosphine (PhPH2) is presumably too rapid to be tracked by 31P{1H} NMR, but small amounts of Ph2PH were observable during NMR monitoring (ESI,† S6.1 and Fig. S32). For the formation of the intermediates PhPH2 and Ph2PH the required H-atoms could potentially be transferred from the oxidized radical cation TDAE˙+ (see Fig. 4) which is an effective H-atom donor.29,30 Nevertheless, mechanistic investigations and in situ NMR experiments31,32 (see ESI,† S6) using deuterated acetone-d6 showed clear deuterium incorporation into these intermediates, suggesting that acetone is involved in the mechanism. It should be noted, however, that P4 arylation also proceeds in the absence of acetone with 30–40% total conversion to a mixture of [Ph4P]Cl and Ph3P (31P NMR spectroscopic monitoring, see the ESI,† Table S4). Hence, the direct, non-acetone mediated reduction may be an additional, significant pathway.
 | ||
| Fig. 4 Proposed mechanism for the light-driven photochemical arylation of P4 in the presence of aryl chloride and TDAE. | ||
To validate their intermediacy, the photochemical phenylation was also analyzed starting from the different arylated phosphorus compounds PhPH2, Ph2PH, Ph4P2 and Ph3P (see ESI,† S7). By using Ph2PH as a starting material a mixture of [Ph4P]Cl (76%) and PhP3 (15%) could be obtained in good yields. PhPH2 also showed good conversion to the phosphonium salt (68%) and using Ph4P2 as the P-containing species yielded a mixture of 55% [Ph4P]Cl and 28% Ph3P. Interestingly, further investigations of the stability of the phosphonium salt under the applied photochemical conditions confirmed some decomposition of the product (loss of [Ph4P]Cl intensity dependent on amount of TDAE, see ESI,† S7 and Table S13) which is probably a factor behind the consistently sub-quantitative yields of the P4 arylation reaction.
Finally, it was noted that during the photochemical reaction [TDAE]Cl233 is formed as a side product (see ESI,† S8) which is almost completely insoluble in acetone and precipitates as a white solid over the course of the reaction and can be recovered by simple filtration. Since it is known that [TDAE]Cl2 can be used to regenerate TDAE using simple reductants such as Zn,34 this recovery suggests the possibility of using the organic photoreductant as part of a closed loop, in which a much cheaper reagent acts as the effective terminal reductant. Synthetic efforts in this direction are currently underway.
In conclusion, we have described herein the direct photochemical arylation of white phosphorus using inexpensive, commercially available aryl chlorides for the first time, which has long been an important yet challenging objective. The simple light-driven procedure is mediated by TDAE and acetone under irradiation with UV light. The remarkable reducing power of photoexcited TDAE enables the reduction of different substituted organic chlorides to generate the corresponding aryl radicals which can be trapped by P4 to form new P–C bonds and ultimately valuable aryl phosphines and phosphonium salts. While these observations represent an important proof-of-principle, the present method still suffers from several practical disadvantages, e.g. the need for an excess of aryl chloride and TDAE, poor selectivities toward the formation of either phosphonium salt or phosphine and our current inability to recycle the TDAE photoreductant effectively. Solutions to these limitations are the subject of ongoing investigations, alongside further studies into the activation of chloro- and bromobenzenes for (catalytic) P4 functionalization.
This project received funding from the European Research Council (ERC CoG 772299) and the Deutsche Forschungsgemeinschaft (DFG, CRC 325 Assembly Controlled Chemical Photocatalysis – 444632635 – and RTG 2620 Ion Pair Effects in Molecular Reactivity).
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. Bettermann, W. Krause, G. Riess and T. Hofmann, Ullmann's Encyclopedia of Industrial Chemistry, Wiley, 2000 Search PubMed.
- D. E. C. Corbridge, Chemistry, Biochemistry and Technology, Elsevier, 2000 Search PubMed.
- C. M. Hoidn, D. J. Scott and R. Wolf, Chem. – Eur. J., 2021, 27, 1886–1902 CrossRef CAS.
- D. H. R. Barton and J. Zhu, J. Am. Chem. Soc., 1993, 115, 2071–2072 CrossRef CAS.
- D. H. R. Barton and R. A. Vonder Embse, Tetrahedron, 1998, 54, 12475–12496 CrossRef CAS.
- B. M. Cossairt and C. C. Cummins, New J. Chem., 2010, 34, 1533–1536 RSC.
- S. K. Ghosh, C. C. Cummins and J. A. Gladysz, Org. Chem. Front., 2018, 5, 3421–3429 RSC.
- U. Lennert, P. B. Arockiam, V. Streitferdt, D. J. Scott, C. Rödl, R. M. Gschwind and R. Wolf, Nat. Catal., 2019, 2, 1101–1106 CrossRef CAS PubMed.
- P. B. Arockiam, U. Lennert, C. Graf, R. Rothfelder, D. J. Scott, T. G. Fischer, K. Zeitler and R. Wolf, Chem. – Eur. J., 2020, 26, 16374–16382 CrossRef CAS.
- R. Rothfelder, V. Streitferdt, U. Lennert, J. Cammarata, D. J. Scott, K. Zeitler, R. M. Gschwind and R. Wolf, Angew. Chem., Int. Ed., 2021, 60, 24650–24658 CrossRef CAS PubMed.
- N. G. W. Cowper, C. P. Chernowsky, O. P. Williams and Z. K. Wickens, J. Am. Chem. Soc., 2020, 142, 2093–2099 CrossRef CAS PubMed.
- For direct organofunctionalization of P4 using Sn-based radicals: D. J. Scott, J. Cammarata, M. Schimpf and R. Wolf, Nat. Chem., 2021, 13, 458–464 CrossRef CAS.
- (a) D. G. Yakhvarov, E. V. Gorbachuk, R. M. Kagirov and O. G. Sinyashin, Russ. Chem. Bull., 2012, 61, 1300–1312 CrossRef CAS; (b) D. G. Yakhvarov, E. V. Gorbachuk and O. G. Sinyashin, Eur. J. Inorg. Chem., 2013, 4709–4726 CrossRef CAS; (c) Y. H. Budnikova, T. V. Gryaznova, V. V. Grinenko, Y. B. Dudkina and M. N. Khrizanforov, Pure Appl. Chem., 2017, 89, 311–330 CAS.
- L. Huang, L. K. G. Ackerman, K. Kang, A. M. Parsons and D. J. Weix, J. Am. Chem. Soc., 2019, 141, 10978–10983 CrossRef CAS.
- A. F. Chmiel, O. P. Williams, C. P. Chernowsky, C. S. Yeung and Z. K. Wickens, J. Am. Chem. Soc., 2021, 143, 10882–10889 CrossRef CAS.
- M. Cybularczyk-Cecotka, J. Szczepanik and M. Giedyk, Nat. Catal., 2020, 3, 872–886 CrossRef CAS.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS.
- H. Kim, H. Kim, T. H. Lambert and S. Lin, J. Am. Chem. Soc., 2020, 142, 2087–2092 CrossRef CAS.
- N. Kvasovs and V. Gevorgyan, Chem. Soc. Rev., 2021, 50, 2244–2259 RSC.
- F. Glaser, C. B. Larsen, C. Kerzig and O. S. Wenger, Photochem. Photobiol. Sci., 2020, 19, 1035–1041 CrossRef CAS.
- L. Pause, M. Robert and J. M. Savéant, J. Am. Chem. Soc., 1999, 121, 7158–7159 CrossRef CAS.
- C. Burkholder, W. R. Dolbier Jr. and M. Médebielle, J. Org. Chem., 1998, 63, 5385–5394 CrossRef CAS.
- T. Fuchigami, M. Atobe and S. Inagi, Fundamentals and Applications of Organic Electrochemistry: Synthesis, Materials, Devices, John Wiley & Sons, Chichester, West Sussex, UK, 2015 Search PubMed.
- J. Mortensen and J. Heinze, Angew. Chem., Int. Ed. Engl., 1984, 23, 84–85 CrossRef.
- C. J. Zeman, IV, S. Kim, F. Zhang and K. S. Schanze, J. Am. Chem. Soc., 2020, 142, 2204–2207 CrossRef.
- C. P. Andrieux and J. Pinson, J. Am. Chem. Soc., 2003, 125, 14801–14806 CrossRef CAS.
- M. Mahesh, J. A. Murphy, F. LeStrat and H. P. Wessel, Beilstein J. Org. Chem., 2009, 5, 1 Search PubMed.
- E. G. Bowes, D. D. Beattie and J. A. Love, Inorg. Chem., 2019, 58(5), 2925–2929 CrossRef CAS.
- P. J. Delaive, T. K. Foreman, C. Giannotti and D. G. Whitten, J. Am. Chem. Soc., 1980, 102, 5627–5631 CrossRef CAS.
- Y. Pellegrin and F. Odobel, C. R. Chim, 2017, 20, 283–295 CrossRef CAS.
- C. Feldmeier, H. Bartling, E. Riedle and R. M. Gschwind, J. Magn. Reson., 2013, 232, 39–44 CrossRef CAS PubMed.
- A. Seegerer, P. Nitschke and R. M. Gschwind, Angew. Chem., Int. Ed., 2018, 57, 7493–7497 CrossRef CAS PubMed.
- C. Knopf, U. Herzog, G. Roewer, E. Brendler., G. Rheinwald and H. Lang, J. Organomet. Chem., 2002, 662, 14–22 CrossRef CAS.
- N. Wiberg and J. W. Buchler, Chem. Ber., 1963, 96, 3223–3229 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc05691c |
| This journal is © The Royal Society of Chemistry 2022 |