
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombined chemoenzymatic strategy for sustainable continuous synthesis of the natural product hordenine†
Stefania
Gianolio
 ,
David
Roura Padrosa
* and
Francesca
Paradisi
*
,
David
Roura Padrosa
* and
Francesca
Paradisi
*
Department of Chemistry, Biochemistry and Pharmacology, University of Bern, Freistrasse 3, Bern, Switzerland. E-mail: Francesca.paradisi@unibe.ch
First published on 12th October 2022
Abstract
To improve sustainability, safety and cost-efficiency of synthetic methodologies, biocatalysis can be a helpful ally. In this work, a novel chemoenzymatic stategy ensures the rapid synthesis of hordenine, a valuable phenolic phytochemical under mild working conditions. In a two-step cascade, the immobilized tyrosine decarboxylase from Lactobacillus brevis (LbTDC) is here coupled with the chemical reductive amination of tyramine. Starting from the abundant and cost-effective amino acid L-tyrosine, the complete conversion to hordenine is achieved in less than 5 minutes residence time in a fully-automated continuous flow system. Compared to the metal-catalyzed N,N-dimethylation of tyramine, this biocatalytic approach reduces the process environmental impact and improves its STY to 2.68 g L−1 h−1.
Introduction
Hordenine (4-(2-dimethylaminoethyl)phenol) is a phenolic phytochemical named after the genus of malting Barley (Hordeum vulgare), where it is mainly present.1 This natural product, also known as anhaline, eremursine, peyocactine or N,N-dimethyltyramine (Fig. 1), is part of our diet through the consumption of edible plants, fruits and herbs.2–4 Significant effort has been dedicated to elucidate its biological activity and pharmacological potential.5–12 However, despite growing evidence of its beneficial role on human health, the exact mechanism of action is still unclear and research on human models is limited to investigating its biokinetics and metabolism.13 To date it is known that hordenine displays neurological activity since it can cross the blood–brain barrier (BBB), and acts as an agonist of the dopamine D2 receptor, with an influence on the central dopaminergic pathways linked to the reward system.14 In parallel, during the last 20 years, the nutraceutical industry has gone through an astonishing evolution broadening the plethora of molecules from natural origin on the market. While numerous compounds have an established place among the food ingredients with proven health benefits (antioxidants, flavonoids, vitamins, fish oil preparations etc.), many other molecules with interesting properties are not yet categorized as nutraceuticals. A limitation in exploring new dietary supplements is often the capacity to produce these compounds in an economically sustainable process matching the required strict regulations, with minimal environmental impact. In October 2021, Hordenine has been removed from the FDA “Ingredient Advisory List” and recognized as a new dietary ingredient (NDI), opening the possibility of incorporating this natural product in nutritional supplements. As a result, its application in the pharmacological and nutraceutical field is becoming highly relevant.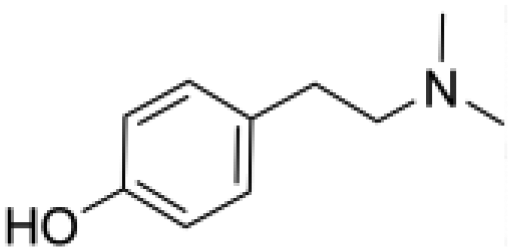 | ||
| Fig. 1 Chemical structure of hordenine. | ||
Hordenine production is achieved either by extraction from natural resources15 or via chemo catalytic methods.16–19 The extraction process requires a vast amount of chlorinated solvent20,21 and does not represent a practical and efficient approach due to the low yield and D value (distribution ratio, 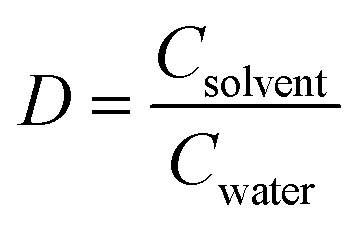 ). The described chemical syntheses typically start from the biogenic aromatic amine tyramine (which carries a number of biosafety hazards),22 and involve high temperature and pressure, as well as precious metal catalysts for the amine methylation step. More recently, an iron-based catalyst has been used which, while it eliminates the need of high hydrogen pressure, still requires temperatures above 100 °C and a reaction time of 24 h.17
). The described chemical syntheses typically start from the biogenic aromatic amine tyramine (which carries a number of biosafety hazards),22 and involve high temperature and pressure, as well as precious metal catalysts for the amine methylation step. More recently, an iron-based catalyst has been used which, while it eliminates the need of high hydrogen pressure, still requires temperatures above 100 °C and a reaction time of 24 h.17
Here, we present the synthesis of hordenine starting from abundantly available natural amino acid L-tyrosine, achieved with an innovative combined chemoenzymatic system that simultaneously improves safety, sustainability and economic efficiency. The in situ (bio)generation of tyramine eliminates the risks associated with handling this reagent which is then directly chemically converted to the final product. The integration of biocatalytic steps within traditional synthetic pathways is essential to validate biocatalysis as a versatile and complementary chemical tool. The number of chemoenzymatic strategies in the fine chemical manufacturing has indeed increased steadily,23–29 and it is clear that when enzymatic steps can be smoothly integrated, the process sustainability greatly increases.30,31 However, compatibility between biocatalysis and organic chemistry is often a significant challenge and the implementation of hybrid cascades with enzymes and chemocatalysts is predominantly confined to batch processes.32–34 To tackle these problematics, the synergy between immobilized biocatalysts and flow chemistry offers the possibility to compartmentalize biocatalytic steps within complex synthetic planning.35–39 Flow bioreactors represent a prominent process innovation to intensify high-value compound production by exploiting their modularity, compartmentalization, reactor versatility, and customization. They can be equipped with real-time monitoring and in-line work-up systems, minimizing energy consumption and manual intervention as a result of process automation. Several examples have been reported on the synthesis of complex molecules which can be achieved with high efficiency and increased sustainability, successfully assembling fully enzymatic cascade reactions in continuous mode, mimicking biosynthetic pathways or, albeit yet less established, combining chemoenzymatic reactors in line.40,41 By compartmentalizing each step in separated flow reactors, we designed a simple chemoenzymatic cascade in continuous flow mode (Fig. 2), obtaining hordenine as a valuable natural product.
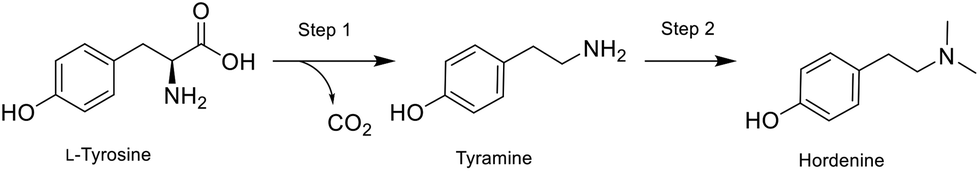 | ||
| Fig. 2 Hordenine production via biocatalytic decarboxylation (step 1) and reductive amination (step 2). | ||
The naturally abundant starting material L-tyrosine is here converted into tyramine by the immobilised tyrosine decarboxylase from Lactobacillus brevis (LbTDC),42 and then a chemical reductive amination follows in-line. In the second step, the application of both sodium triacetoxyborohydride (STAB) and picoline borane were trialled to provide a greener alternative to the classic use of cyanoborohydride as reducing agent.
Results and discussion
Immobilisation of LbTDC tyrosine decarboxylase from Lactobacillus brevis
The soluble expression of LbTDC was successfully achieved as previously reported,43 with a volumetric yield of 3.5 mg Lculture−1 (Fig. S1†). The enzyme showed very high activity and specificity for its natural substrate, L-tyrosine, with a specific activity of 43.8 U mg−1. To enable the reuse of the enzyme, extend its stability and increase the catalytic efficiency of the biotransformation via its integration in continuous reactors, a method for the immobilisation of the biocatalyst was designed. Following the screening of different methacrylic supports with epoxide handles, as well as a range of protein loadings (Tables S1 and S2†), the optimal conditions were found to be 5 mg of enzyme per g of methacrylic support ReliSorb™ 400(SS). In the selected preparation, the activity on the support reached up to 32.85 U g−1, with a 15% recovered activity. The stability of the immobilised biocatalyst was found to be excellent when stored at 4 °C (no loss of activity for 1 month) (Graph S1†).Production of tyramine
Following immobilisation, the biocatalyst could be reused in consecutive biotransformations, increasing its productivity in the conversion to tyramine (μmol tyramine per mg of biocatalyst). As the enzyme has high activity against L-tyrosine, a low concentration (0.006 mg mL−1) of the free biocatalyst was sufficient to achieve full conversion at 2.5 mM scale in 20 minutes. When bound to the support, LbTDC specific activity is reduced by approximately 5-fold. Nevertheless, in batch biotransformations at 5 mM scale, complete conversion was systematically obtained for up to five reuse cycles of 45 minutes, with 15 mg of LbTDC-EP400(SS) 5 mg g−1, in 2 mL reaction volume. Therefore, the enzyme productivity increased from 415 μmol mg−1 to 670 μmol mg−1 (see ESI†). After establishing standard working conditions of the decarboxylation reaction in batch, the immobilized LbTDC was tested in continuous flow mode (Table S3†). The higher local concentration and the enhanced efficiency of the biocatalytic system afforded the product quantitatively in just 2.5 minutes of contact time (Table 1).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
| L-Tyrosine disodium salt (mM) | t (min) | M.c.b (%) | |
|---|---|---|---|
| a Reaction set up: 5 mM L-tyrosine disodium salt hydrate, 0.2 mM PLP, 200 mM sodium acetate buffer, pH 5. Batch conditions: 0.05 mg mL−1LbTDC in a 2 mL biotransformation. Flow conditions: 1 g LbTDC-EP400(SS), 1.3 mL PBR, 0.54 mL min−1 flow rate. b M.c.: molar conversion determined by HPLC. | |||
| Batch | 5 | 45 | >99 |
| Flow | 5 | 2.5 | >99 |
Complete conversion of the starting material using the immobilised enzyme in a packed bed reactor (PBR) was successfully maintained over 56 reaction cycles. After 103 reaction cycles, the conversion was still above 95% and it remained above 90% after 8 hours (corresponding to 192 reaction cycles).
Optimization studies: process intensification in batch for tyramine production
L-Tyrosine solubility in the optimal buffer for the enzyme is limited to 5 mM. In an attempt to increase the solubility, the addition of surfactants was explored.44 Jiang et al. screened nine different surfactants and co-solubilizers succeeding in improving the conversion to tyramine with LbTDC in batch. Although not yielding a limpid solution, Tween 20 (15% v/v) allowed to increase the solubility of L-tyrosine up to 30 mM, obtaining a micro-suspension and leading to full conversion to tyramine, without loss of activity by the immobilized LbTDC in batch (Tables S4 and S5†). Hence, a 6-fold increase in TON (Turnover Number) of the immobilized biocatalyst was recorded with this set up. However, while a fine dispersion in solution favours the enzymatic activity in batch, over time the cloudy micro-suspension of L-tyrosine caused significant back pressure in the flow instrumentation and the follow-on reactions were performed again at 5 mM.Reductive alkylation of tyramine
The second step in the synthesis of hordenine was then addressed. It was previously reported that the dimethylation of tyramine can be achieved in biological samples, exploiting formaldehyde and sodium cyanoborohydride (NaBH3CN) in ammonium acetate buffer.45 For laboratory scale reductive amination, NaBH3CN, among other hydrides, is the most employed because it is highly selective, soluble in many solvents, and stable in acid medium. Nevertheless, it generates toxic products such as HCN or NaCN during the reaction or work-up, and thus it is not recommended for medium or large-scale reactions. The efficiency of less hazardous reducing agents, such as sodium borohydride and sodium triacetoxyborohydride (STAB) was therefore tested in batch. STAB was effective but required the use of aprotic solvents, because it is unstable in aqueous systems. Therefore, acetonitrile had to be added to reduce the water effective concentration and suppress the hydrolysis of the reducing agent.We then sought to identify the minimum amount of reducing agent required for the complete conversion of tyramine to hordenine (Table S6†). Initially the reductive amination was performed in batch and then tested in the flow coil reactor (Table 2). This strategy aimed to minimise the reagents excess and to test the scalability of the reaction up to 40 mM tyramine. With STAB, we achieved full conversion in flow at 30 mM scale and 82% conversion at 40 mM scale, employing half of the equivalents of the reagents compared to batch mode (Table S8†). However, with the aim of coupling the two steps to establish a telescoped process, the working concentration of tyramine was reduced to 5 mM, as this is the solubility limit of the first module.
With 12.5 eq. formaldehyde and 12 eq. STAB, at room temperature, with 75% v/v MeCN which yielded almost complete conversion (96%) within a RT (residence time) of 4.62 min, without the requirement of back pressure regulator or temperature control in the flow set up (Table S9†). By virtue of the biocatalyst productivity, this system achieved a space–time-yield (STY) of 2.68 ghordenine L−1 h−1, an improvement of 50% compared to what previously reported.17
The strategy depicted in Scheme 1, allowed the processing of 75 mL of feedstock in just 2.5 hours. Following a liquid–liquid extraction, hordenine hydrochloride was then isolated by HCl titration (isolated yield of 90%) (Table 3).
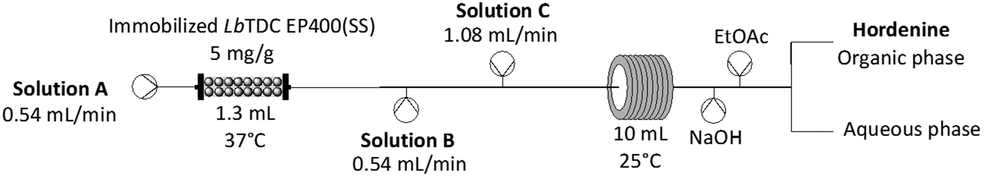 | ||
| Scheme 1 Scheme of the continuous flow chemoenzymatic system for hordenine production with STAB. Solution A: 5 mM L-tyrosine disodium salt hydrate, 0.2 mM PLP in 200 mM sodium acetate buffer. Solution B: 62.5 mM formaldehyde, 2.5% v/v AcOH in MeCN. Solution C: 60 mM sodium triacetoxyborohydride in MeCN. | ||
While this system worked very well, the requirement of MeCN negatively impacted the sustainability of the process. We therefore tested picoline borane (pic-BH3), as a different green reducing agent. This reagent is well established, non-toxic, and while it is easily dissolved in different solvents, it remains nearly insoluble in water.46 Exploiting this characteristic, working with pic-BH3 in aqueous phase as previously reported in the literature,47–50 we packed the reducing agent (300 mg bulked up with an equivalent amount of Celite) inside a PBR and used this heterogenous reducing module in continuous flow (Scheme 2). In batch, we previously observed that pic-BH3 is more efficient in reducing the imine intermediate at alkaline pH rather at acidic pH (Table 4 and Table S7†). Therefore, 250 mM sodium carbonate (pH 11.5) was used as medium for the formaldehyde feedstock solution, allowing the reductive amination reaction to happen at pH 9.
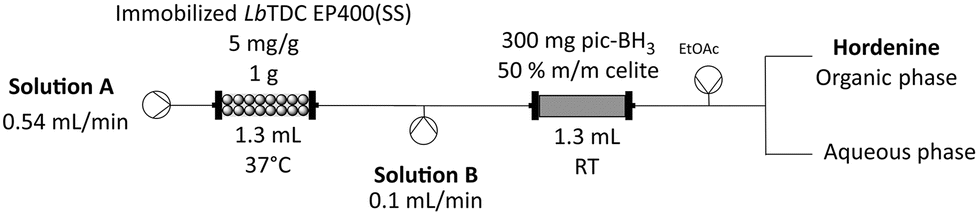 | ||
| Scheme 2 Scheme of the continuous flow chemoenzymatic system for hordenine production with pic-BH3. Solution A: 5 mM L-tyrosine disodium salt hydrate, 0.2 mM PLP in 200 mM sodium acetate buffer. Solution B: 202.5 mM formaldehyde, in 250 mM sodium carbonate (pH 11.5). | ||
With this alternative strategy 130 mL of 5 mM feedstock solution were processed within 4 hours, achieving almost complete conversion with only 7.5 eq. of formaldehyde and 2.5 min residence time (Table 5). The system achieved a higher STY of 11.4 ghordenine L−1 h−1, improving by 4-fold the result obtained with STAB (Table 3). Again, a liquid–liquid extraction, followed by HCl titration led to the isolation of hordenine hydrochloride (isolated yield of 77%).
Conclusions
In this work, we present an alternative method to synthesize the natural product hordenine. By implementing a new 2-step cascade in continuous flow system, L-tyrosine was biocatalytically decarboxylated to tyramine with immobilised LbTDC, and the following in-line reductive alkylation of the intermediate was successfully achieved under mild conditions. To replace the use of sodium cyanoborohydride, alternative reducing agents such as STAB and pic-BH3, afforded hordenine in very good isolated yield, with residence times as low as 2.5 min. The great efficiency of the initial biocatalytic step allows a straightforward and unambiguous process design, forestalling unreacted substrate and side reaction products. The rationale behind the selection of the reaction reagents was to achieve a balance of their availability, toxicity and effectiveness: the starting material, L-tyrosine is a natural amino acid, non-hazardous, and broadly available; despite the loss of chirality via decarboxylation in the first step, the fact that tyramine is synthesized directly in the reactor increases the safety of the process.22 The choice of STAB, in itself a very convenient, safe, and effective option, posed the challenge of having to use acetonitrile, compromising the process sustainability. Pic-BH3, despite its higher cost, was an excellent alternative and very easily integrated in the flow as solid reagent in an in-line PBR. The calculated E-factor (environmental factor = kg of waste per kg of desired product) for our system, with pic-BH3 suspended in Celite, is 36. This value fares extremely well when related to standard pharmaceutical processes at the same conceptual phase (E-factors around 25–100).51 Unveiling its potential, LbTDC showed excellent stability, leading to an efficient strategy capable of contributing to a more sustainable tyramine production.52 Hordenine is synthesized rapidly and efficiently, working under mild conditions, mitigating the need to use hazardous processes commonly associated with the currently reported catalytic methods described in the introduction. In conclusion, the resulting profitable process, where the starting material value raises by 200-fold (L-tyrosine disodium salt hydrate −1.72 € per g and hordenine −382 € per g), was designed to safeguard sustainability and safety, and can be taken as a successful example of the integration between biocatalysis and chemical synthesis in continuous manufacturing, representing a flexible and efficient hordenine production alternative.Experimental
Materials
All reagents and solvents were obtained from commercial suppliers and were used without further purification. The methacrylate support was purchased from ResindionS.R.L. In the immobilisation support screening, the methacrylic resins involved were RelizymeTM EP403(S), RelizymeTM HFA403(S), RelisorbTM EP400(SS).Analytical methods
HPLC analyses were carried out using a Liquid Chromatography UHPLC system, equipped with a binary pump and a diode array detector (Dionex UltiMate 3000, Thermo Scientific). All the samples were dissolved in 0.2% HCl and filtered with 0.45 μm nylon filters. See ESI† for detailed information.Expression, purification and Immobilisation of LbTDC
LbTDC was expressed and purified as previously reported.44 After dialysis against phosphate buffer (25 mM phosphate buffer, 150 mM NaCl, 0.2 mM PLP, pH 7.4), the protein was quantified by measuring the absorbance at 280 nm. The protein concentration was calculated using the Lambert–Beer law. Molar extinction coefficient was estimated by ProtPram (ε: 115630 M−1 cm−1). The absorbance measurements were carried out using a BioTek Take3 Microplate reader. The immobilisation of LbTDC was performed on the optimal methacrylate support, through first the coordination of the His-tag with immobilized metal in the resin and, after, promoting the covalent bonding between the epoxy groups on the resin and the ε-amino groups of the lysines in the protein sequence.53 In short, 1 g of resin was treated with 2 mL of modification buffer (sodium borate (0.1 M), iminodiacetic acid (2 M) in phosphate buffer (50 mM), pH 8.5) under gentle shaking for 2 h at room temperature. The resin was then filtered, washed three times with distilled water and mixed with 5 mL of the metal solution (1 M sodium chloride and 5 mg mL−1 of cobalt chloride in 50 mM phosphate buffer, pH 6) for another 2 h. Following the same procedure, the sample was washed 3 times with distilled water and then 4 mL of the protein solution were added. The sample was kept under agitation for 4 h. After determining the immobilization yield by measuring the concentration of the enzyme in the supernatant, the support was then filtered and washed thoroughly with desorption buffer (50 mM EDTA, 0.5 M NaCl in 20 mM phosphate buffer, pH 7.4) and washed with distilled water. Finally, 4 mL of blocking buffer (3 M glycine in 50 mM phosphate buffer, pH 8.5) were added and the suspension was left under agitation for 20 hours. In the end, the beads were washed, collected and stored at 4 °C.
Activity assay of LbTDC
The activity assay was performed using the HPLC, measuring the conversion of L-tyrosine disodium salt hydrate to tyramine at 280 nm. One unit (U) of activity is defined as the amount of enzyme which catalyses the consumption of 1 μmol of L-tyrosine per minute under reference conditions i.e., 2.5 mM L-tyrosine disodium salt hydrate, 0.2 mM PLP, in 200 mM sodium acetate buffer, pH 5.0. 50 μL aliquots were collected at different reaction times (0 min, 5 min, 10 min, 15 min, 20 min) and quenched by the addition of 0.2% HCl before the HPLC analysis.Small scale batch biotransformations
Batch reactions were performed in 2 mL Eppendorf tubes. The substrate was suspended in 200 mM or 500 mM sodium acetate buffer, pH 5, containing 0.2 mM PLP. If needed, sonication of the reaction solution was performed to dissolve L-tyrosine disodium salt hydrate. After adding the enzyme, the reaction mixture was left at 37 °C, 150 rpm. The final reaction volume was 2 mL. 50 μL aliquots were collected and added to 0.2% HCl before been submitted for the HPLC analysis.Small scale batch reductive amination reaction
Batch reactions were performed in 15 mL falcon tubes. 2 mL of 10 mM tyramine solution in 200 mM sodium acetate buffer was mixed with the desired volume of formaldehyde solution 37%. In the case of STAB, 200 μl of acetic acid and2 mL of MeCN were then mixed with the solution, before the final addition of the reducing agent. In the case of pic-BH3, reactions were performed adding the same volume of acetic acid, for the acid pH, or diluting formaldehyde in sodium carbonate 250 mM, for basic pH. The use of organic solvents was not necessary. The reaction mixture was left under agitation and 50 μL aliquots were collected and added to 0.2% HCl before been submitted for the HPLC analysis.Continuous flow bioconversion
The feedstock solutions were prepared in 50 mL falcon tubes. The tyrosine solution consisted of 5 mM L-tyrosine disodium salt hydrated and 0.2 mM PLP in 200 mM sodium acetate buffer, pH 5. The solution of formaldehyde (62.5 mM) in MeCN, with 2.5% v/v acetic acid was prepared by dilution of the commercial 37% formaldehyde solution. Similarly the solution of formaldehyde (202.5 mM) in sodium carbonate (250 mM, pH 11.5) was also prepared by dilution of the commercial 37% formaldehyde solution. The reducing agent STAB was dissolved in MeCN, in concentration of 60 mM and the suspension was kept homogeneous with magnetic stirring during the experiment (1500 rpm).The 1.3 mL PBR with the immobilised biocatalyst was coupled with two T-tubes (Scheme 1), corresponding to the inlets for formaldehyde (62.5 mM formaldehyde, 2.5% v/v acetic acid in MeCN) and reducing agent (60 mM STAB in MeCN). The feeding flow rate for the PBR was 0.54 mL min−1, the same as for the formaldehyde inlet and exactly half of that for the reducing agent feed. In line, a 10 mL flow coil reactor was connected and fed, at the resulting flow rate of 2.16 mL min−1, with the mixed solution.
The same 1.3 mL PBR with the immobilised biocatalyst was coupled with one T-tubes (Scheme 2), corresponding to the inlet for formaldehyde (202.5 mM formaldehyde, 250 mM sodium carbonate). The feeding flow rate for the immobilized enzyme PBR was kept 0.54 mL min−1, while for the formaldehyde solution it was set at 0.1 mL min−1. In line, we connected the PRB with 300 mg pic-BH3 mixed homogeneously in 300 mg Celite.
Product isolation and purification
300 mL of the final product solution obtained with the STAB system were extracted one time with 300 mL ethyl acetate after adjusting the pH to 9.6. The water phase was washed twice with its same volume of ethyl acetate and the organic phase was collected in one flask to be mixed with sodium sulfate anhydrous. After filtration, the ethyl acetate was evaporated and the crude product was resuspended in 5 mL of MeCN. To the suspension, an equimolar amount of 37% HCl solution was added for the formation of hordenine hydrochloride and the precipitate was dried to be submitted for NMR analysis.From the pic-BH3 alternative process, 130 mL of the collected hordenine solution were extracted with ethyl acetate (5 × 30 mL organic phase). After drying with sodium sulfate anhydrous, the solution was filtered and the hordenine hydrochloride precipitation was performed in ethyl acetate as previously described. The precipitate was dried to be submitted for NMR analysis.
Author contributions
F. P. and D. R. P. conceptualized the idea and supervised the project. S. G. performed the experiments and wrote the initial draft. All the authors discussed the results and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported the Swiss National Science Foundation (SNSF) (grantnumber200021_192274). Open access funding provided by Universität Bern.References
- X. Gong, J. Tao, Y. Wang, J. Wu, J. An, J. Meng, X. Wang, Y. Chen and J. Zou, J. Ethnopharmacol., 2021, 273, 113994 CrossRef CAS PubMed.
- T. da Silveira Agostini-Costa, Food Chem., 2020, 327, 126961 CrossRef CAS PubMed.
- B. Avula, V. C. Joshi, A. Weerasooriya and I. A. Khan, Chromatographia, 2005, 62, 379–383 CrossRef CAS.
- L. Servillo, D. Castaldo, A. Giovane, R. Casale, N. D'Onofrio, D. Cautela and M. L. Balestrieri, J. Agric. Food Chem., 2017, 65, 892–899 CrossRef CAS PubMed.
- J. Ma, S. Wang, X. Huang, P. Geng, C. Wen, Y. Zhou, L. Yu and X. Wang, J. Pharm. Biomed. Anal., 2015, 111, 131–137 CrossRef CAS PubMed.
- H. J. Hapke and W. Strathmann, Dtsch. Tierarztl. Wochenschr., 1995, 102, 228–232 CAS.
- X. Zhang, L. Du, J. Zhang, C. Li, J. Zhang and X. Lv, Front. Pharmacol., 2021, 12, 712232 CrossRef CAS PubMed.
- S.-C. Kim, J.-H. Lee, M.-H. Kim, J.-A. Lee, Y. B. Kim, E. Jung, Y.-S. Kim, J. Lee and D. Park, Food Chem., 2013, 141, 174–181 CrossRef CAS PubMed.
- C. J. Barwell, A. N. Basma, M. A. K. Lafi and L. D. Leake, J. Pharm. Pharmacol., 2011, 41, 421–423 CrossRef PubMed.
- H. Ohta, Y. Murakami, Y. Takebe, K. Murasaki, K. Oshima, H. Yoshihara and S. Morimura, Food Sci. Technol. Res., 2020, 26, 313–317 CrossRef CAS.
- A. A. E. Said, T. F. S. Ali, E. Z. Attia, A.-S. F. Ahmed, A. H. Shehata, U. R. Abdelmohsen and M. A. Fouad, Nat. Prod. Res., 2021, 35, 5493–5497 CrossRef CAS PubMed.
- S. Anwar, T. Mohammad, A. Shamsi, A. Queen, S. Parveen, S. Luqman, G. M. Hasan, K. A. Alamry, N. Azum, A. M. Asiri and M. I. Hassan, Biomedicines, 2020, 8, 32–228 CrossRef PubMed.
- C. G. Viejo, R. Villarreal-Lara, D. D. Torrico, Y. G. Rodríguez-Velazco, Z. Escobedo-Avellaneda, P. A. Ramos-Parra, R. Mandal, A. P. Singh, C. Hernández-Brenes and S. Fuentes, Foods, 2020, 9, 821 CrossRef CAS PubMed.
- T. Sommer, T. Göen, N. Budnik and M. Pischetsrieder, J. Agric. Food Chem., 2020, 68, 1998–2006 CrossRef CAS PubMed.
- Nanjing Zelang, Medical Technology Co Ltd, CN101948392A, 2011.
- Shanxi Jiahe, Plant Chemical CO Ltd, CN103483209A, 2014.
- K. Natte, H. Neumann, R. V. Jagadeesh and M. Beller, Nat. Commun., 2017, 8, 1344 CrossRef PubMed.
- V. Goyal, J. Gahtori, A. Narani, P. Gupta, A. Bordoloi and K. Natte, J. Org. Chem., 2019, 84, 15389–15398 CrossRef CAS PubMed.
- N. Sarki, V. Goyal, N. K. Tyagi, Puttaswamy, A. Narani, A. Ray and K. Natte, ChemCatChem, 2021, 13, 1722–1729 CrossRef CAS.
- A. Percot, A. Yalçin, H. Erduğan and K. C. Güven, Acta Pharm. Sci., 2007, 49, 127–132 CAS.
- S. Berkov, J. Bastida, R. Tsvetkova, F. Viladomat and C. Codina, Z. Naturforsch., C: J. Biosci., 2009, 64, 311–316 CrossRef CAS PubMed.
- Tyramine, Generic EU MSDS - no country specific data, Sigma-Aldrich, 2022, 1–9 Search PubMed.
- F. Annunziata, M. Letizia Contente, D. Betti, C. Pinna, F. Molinari, L. Tamborini and A. Pinto, Catalysts, 2020, 10, 939 CrossRef CAS.
- J. H. Schrittwieser, S. Velikogne, M. Hall and W. Kroutil, Chem. Rev., 2018, 118, 270–348 CrossRef CAS PubMed.
- F. F. Özgen, M. E. Runda and S. Schmidt, ChemBioChem, 2021, 22, 790–806 CrossRef PubMed.
- P. Luan, Y. Liu, Y. Li, R. Chen, C. Huang, J. Gao, F. Hollmann and Y. Jiang, Green Chem., 2021, 23, 1960–1964 RSC.
- Y. Liu, P. Liu, S. Gao, Z. Wang, P. Luan, J. González-Sabín and Y. Jiang, Chem. Eng. J., 2021, 420, 127659 CrossRef CAS.
- L. Bering, E. J. Craven, S. A. Sowerby Thomas, S. A. Shepherd and J. Micklefield, Nat. Commun., 2022, 13, 380 CrossRef CAS PubMed.
- E. J. Craven, J. Latham, S. A. Shepherd, I. Khan, A. Diaz-Rodriguez, M. F. Greaney and J. Micklefield, Nat. Catal., 2021, 4, 385–394 CrossRef CAS.
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2018, 118, 801–838 CrossRef CAS PubMed.
- R. A. Sheldon and D. Brady, ChemSusChem, 2019, 12, 2859–2881 CrossRef CAS PubMed.
- L. Pan, Q. Li, Y. Tao, C. Ma, H. Chai, Y. Ai and Y.-C. He, Ind. Crops Prod., 2022, 186, 115203 CrossRef CAS.
- A. Romero and C.-H. Wong, J. Org. Chem., 2000, 65, 8264–8268 CrossRef CAS PubMed.
- J. Zheng, H. Xu, J. Fang and X. Zhang, Carbohydr. Polym., 2022, 291, 119564 CrossRef CAS PubMed.
- M. Dias Gomes and J. M. Woodley, Molecules, 2019, 24, 3573 CrossRef PubMed.
- L. Tamborini, P. Fernandes, F. Paradisi and F. Molinari, Trends Biotechnol., 2018, 36, 73–88 CrossRef CAS PubMed.
- M. Santi, L. Sancineto, V. Nascimento, J. Braun Azeredo, E. V. M. Orozco, L. H. Andrade, H. Gröger and C. Santi, Int. J. Mol. Sci., 2021, 22, 990 CrossRef CAS PubMed.
- M. P. Thompson, I. Peñafiel, S. C. Cosgrove and N. J. Turner, Org. Process Res. Dev., 2019, 23, 9–18 CrossRef CAS.
- M. Heidlindemann, G. Rulli, A. Berkessel, W. Hummel and H. Gröger, ACS Catal., 2014, 4, 1099–1103 CrossRef CAS.
- E. T. Hwang and S. Lee, ACS Catal., 2019, 9, 4402–4425 CrossRef CAS.
- A. I. Benítez-Mateos, D. Roura Padrosa and F. Paradisi, Nat. Chem., 2022, 14, 489–499 CrossRef PubMed.
- H. Zhu, G. Xu, K. Zhang, X. Kong, R. Han, J. Zhou and Y. Ni, Sci. Rep., 2016, 6, 27779 CrossRef CAS PubMed.
- K. Zhang and Y. Ni, Protein Expression Purif., 2014, 94, 33–39 CrossRef CAS PubMed.
- M. Jiang, G. Xu, J. Ni, K. Zhang, J. Dong, R. Han and Y. Ni, Appl. Biochem. Biotechnol., 2019, 188, 436–449 CrossRef CAS PubMed.
- K. Guo, C. Ji and L. Li, Anal. Chem., 2007, 79, 8631–8638 CrossRef CAS PubMed.
- S. Sato, T. Sakamoto, E. Miyazawa and Y. Kikugawa, Tetrahedron, 2004, 60, 7899–7906 CrossRef CAS.
- X. Li, K. S. Iyer, R. R. Thakore, D. K. Leahy, J. D. Bailey and B. H. Lipshutz, Org. Lett., 2021, 23, 7205–7208 CrossRef CAS PubMed.
- V. A. Cosenza, D. A. Navarro and C. A. Stortz, ARKIVOC, 2011, 2011, 182–194 Search PubMed.
- A. H. Orrego, M. Romero-Fernández, M. Millán-Linares, M. Yust, J. Guisán and J. Rocha-Martin, Catalysts, 2018, 8, 333 Search PubMed.
- A. Ambrogelly, C. Cutler and B. Paporello, Protein J., 2013, 32, 337–342 CrossRef CAS PubMed.
- F. Roschangar, R. A. Sheldon and C. H. Senanayake, Green Chem., 2015, 17, 752–768 RSC.
- Z. Guo, N. Yan and A. A. Lapkin, Curr. Opin. Chem. Eng., 2019, 26, 148–156 CrossRef.
- C. Mateo, V. Grazú, B. C. C. Pessela, T. Montes, J. M. Palomo, R. Torres, F. López-Gallego, R. Fernández-Lafuente and J. M. Guisán, Biochem. Soc. Trans., 2007, 35, 1593–1601 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc02767d |
| This journal is © The Royal Society of Chemistry 2022 |