
Tuning and understanding the electronic effect of Co–Mo–O sites in bifunctional electrocatalysts for ultralong-lasting rechargeable zinc–air batteries†
Yi Guan‡
 a,
Nan Li‡a,
Jiao Hea,
Yongliang Li*a,
Lei Zhanga,
Qianling Zhanga,
Xiangzhong Ren*a,
Chuanxin Hea,
LiRong Zhengb and
Xueliang Sunc
a,
Nan Li‡a,
Jiao Hea,
Yongliang Li*a,
Lei Zhanga,
Qianling Zhanga,
Xiangzhong Ren*a,
Chuanxin Hea,
LiRong Zhengb and
Xueliang Sunc
aCollege of Chemistry and Environmental Engineering, Shenzhen University, Shenzhen, Guangdong 518060, P. R. China. E-mail: renxz@szu.edu.cn; liyli@szu.edu.cn; Fax: +86-755-26558134; Fax: +86-755-26931162; Tel: +86-755-26558134 Tel: +86-755-26931162
bInstitute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, P. R. China
cDepartment of Mechanical and Materials Engineering, University of Western Ontario, London, Ontario N6A 6B9, Canada
First published on 19th July 2021
Abstract
Herein, we report a post-assembly strategy by growing bimetallic Co/Zn zeolitic imidazolate frameworks (BIMZIF) on the surface of customized Mo metal–organic frameworks (MOFs) (Mo-MOFs) to prepare core–shell structured Mo-MOF@BIMZIF, which results in a porous Co–MoO2 polyhedral nanocage (Co–MoO2-NC) structure and abundant Co–Mo–O active sites after high temperature pyrolysis. Co–MoO2-NC-900 which was obtained by pyrolysis at 900 °C displays a low overpotential (370 mV) for the oxygen evolution reaction (OER) and a half-wave potential (E1/2 ≈ 0.89 V) for the oxygen reduction reaction (ORR), exhibiting bifunctional features. A Zn–air battery with Co–MoO2-NC-900 shows a high power density of 176.5 mW cm−2 at 217.1 mA cm−2, which is better than that of reference Pt/C + Ir/C electrocatalysts, and is capable of exhibiting a smaller discharge–charge overpotential (0.85 V) and excellent stability (1145 h). Density functional theory calculations and experiments reveal that the structure and electron transfer between the Co and MoO2 components reduce the free energy of the intermediates, resulting in bifunctional electrocatalytic activity.
Introduction
In order to solve the problem of increasing energy shortage, sustainable and clean energy has been vigorously developed.1,2 Due to the high theoretical energy densities, rechargeable metal–air batteries present great potential to serve as next generation energy systems.3–7 The oxygen evolution reaction (OER) and oxygen reduction reaction (ORR) are two important reactions during the operation process of metal–air batteries, respectively.8–13 But sluggish kinetics for reactions mentioned above significantly limit the reaction efficiency. Although precious metal catalysts can effectively reduce the free energy of reaction and promote electron transport, they still have the disadvantages of high cost, easy poisoning, and short service life.14–20Improving the electrochemical activity of inexpensive catalysts to replace precious metal-based catalysts is still a harsh challenge. Notably, Mo-based compounds attract considerable attention in many fields like electrocatalysis, nitrogen reduction reaction and photocatalysis.21–23 Typically, MoO2 is regarded as a reasonable OER electrocatalyst with high chemical stability but its ORR activity is not sufficient.24,25 However, the catalytic performance of a single molybdenum-based metal oxide is insufficient to produce the dual-functional catalytic capacity required by zinc–air battery catalysts, and it also has the characteristics of poor conductivity and active sites that are prone to irreversible structural evolution in electrochemical reactions.
Optimizing the material structure and modulating the intrinsic electronic properties are feasible methods to increase the activity of catalysts.26–29 Zeolitic imidazolate frameworks (ZIFs) hold great potential to construct well-designed nanocarbon materials because of their high surface area, adjustable structures, and nitrogen self-doping.30,31 Besides, a hollow structure constructed from ZIFs can further reduce the ion migration resistance and ion diffusion path during the reaction by exposing active surface sites efficiently.32–34 Moreover, doping a secondary metal atom is capable of optimizing the electronic state of the as-prepared catalyst effectively to improve its intrinsic activity.35 Thus, a novel approach to construct MOF-derived core–shell structured carbon-coated double transition metal particle materials to investigate the influence of inserting a second metal atom on the electronic state and to explore the catalytic mechanism of the active site is extremely desirable and challenging.
Herein, we propose a novel post-assembly strategy by growing bimetallic Co/Zn ZIFs (BIMZIF) on the external surface of a customized Mo-MOF to prepare core–shell structured Mo-MOF@BIMZIF, which is composed of porous Co–MoO2 polyhedral nanocages (Co–MoO2-NC-900) after pyrolysis. During the growth process, Co/Zn bimetallic ions not only linked with 2-methylimidazole (2-mIm) organic ligands to form Co/Zn-ZIF, but also doped into Mo-MOF because of the similarity of the two ligands, which could effectively enhance the combination of the two MOFs. After pyrolysis, the Mo-MOF derived MoO2-embedded N-doped carbon not only offered conductive MoO2 active sites to catalyze the OER process, but also served as a nanostructured hollow framework to reduce ion migration resistance. Besides, the even distribution of Co nanoparticles in the composite provides additional active sites, which further increase the electrochemical activity. In addition, the Zn species will evaporate if the temperature goes above 900 °C, which could promote the growth of carbon nanotubes (CNTs). Therefore, the well-designed structure of Co–MoO2-NC will effectively improve diffusion kinetics and increase the active site exposure area. The electrocatalyst presents a remarkable bifunctional activity (ΔE = 0.71 V). More importantly, a zinc–air battery (ZAB) with Co–MoO2-NC electrocatalysts was assembled and exhibited excellent long-term stability for 1145 h and high peak power density (176.5 mW cm−2). Our findings also show that the Co–Mo–O active site constructed after the successful introduction of Co atoms can efficiently control the electronic state of MoO2 and give full play to the synergistic effect of these two components.
Results and discussion
The schematical representation of the synthesis of Co–MnO2-NC is shown in Fig. 1. Firstly, the customized Mo-MOF was synthesized referring to a method reported before with modifications.36 Then, the core–shell Mo-MOF@BIMZIF was prepared by seed epitaxial growth because the organic ligands of the two MOFs are very similar. Finally, the porous Co–MoO2 polyhedral nanocages (Co–MoO2-NC-900) were obtained by a two-step pyrolysis process. The morphology of Mo-MOF, Co–MoO2-NC-900 and its precursor was observed by SEM and TEM. Fig. S1a and S1b in the ESI† demonstrate that the customized Mo-MOF displays an irregular cube-like structure and its pyrolysis products appear granular with a tendency to agglomerate. When BIMZIF is successfully grown on the surface of Mo-MOF, the as-synthesized product presents a rhombic dodecahedral appearance (Fig. 2a and S1c†). From Fig. S2 in the ESI,† we can confirm that the core–shell Mo-MOF@BIMZIF was prepared successfully because the TEM image reveals a hollow interior of polyhedral nanocages through contrast between the shell and internal void. After pyrolysis, the final product (Co–MoO2-NC-900) displays a porous polyhedral nanocage morphology with carbon nanotubes (CNTs) over its surface as shown in Fig. 2b and S1d.† By means of TEM, we further confirm that the Co nanoparticles (NPs) are crystalline and distributed in the tips of CNTs, which present a lattice fringe of about 2.05 Å, corresponding to β-Co (111). The lattice fringes of 3.7 Å around the Co nanoparticle correspond to C (002) facets, which matched well with the top growth mechanism for the formation of CNTs, as shown in Fig. 2c.37,38 Besides, the well-defined lattice fringes (3.42 Å) match well with the (011) crystal faces of MoO2 and there is an obvious interface between MoO2 and Co (Fig. 2d).39,40 In addition, by means of HAADF-STEM and the corresponding EDS mapping, we can find the Co, Mo and O species coexist with each other in both CNTs and the carbon layer (Fig. 2e and S3 in the ESI†). The above results suggested that abundant Co–Mo–O moieties as active sites have been prepared.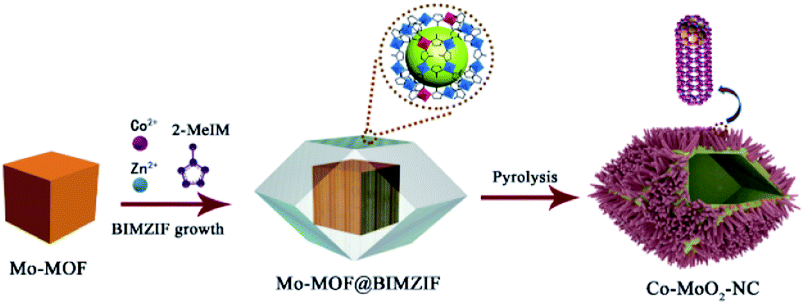 | ||
| Fig. 1 A schematic representation of the synthesis of Co–MoO2-NC products. | ||
 | ||
| Fig. 2 SEM images of (a) Mo-MOF@BIMZIF and its resultant (b) Co–MoO2-NC-900; TEM images of the (c) Co species that exists at the tip of the CNT and (d) MoO2 encapsulated in the carbon layer in Co–MoO2-NC-900; (e) EDS mapping of Co–MoO2-NC-900. | ||
To further study how the pyrolysis temperature affects the morphology and elemental form of the hybrids, Mo-MOF@BIMZIF was calcined at 800 and 1000 °C as the reference, and the products are donated as Co–MoO2-NC-800 and Co–MoO2-NC-1000, respectively. Because the zinc element will only evaporate when the pyrolysis temperature is above 800 °C, even Co–MoO2-NC-800 has a similar appearance to Co–MoO2-NC-900, and it presents a polyhedral nanocage structure but it is hard to find the CNTs on the surface of the composite, as shown in Fig. S4a in the ESI.† The huge agglomerated nanoparticle also demonstrates the importance of adequate evaporation of zinc at higher temperatures (Fig. S4b in the ESI†). When the temperature rises to 1000 °C, enormous nanospheres appeared, demonstrating that the Co NPs agglomerated severely (Fig. S5 in the ESI†).
The XRD pattern illustrates the crystalline nature of Co–MoO2-NC-900 (Fig. S6a in the ESI†). The sharp peaks at 26° corresponds to the (011) of monoclinic MoO2 (PDF #32-0671).41 Besides, the three sharp peaks located at 76.1°, 51.5°, and 44.3° match well with the (220), (200), and (111) facets of Co (PDF #15-0806).42,43 Broadened Bragg peaks located at 25.8° match the (002) lattice planes of graphitic carbon. The XRD pattern also illustrates that there is no obvious diffraction peak of MoO2 for Co–MoO2-NC-800, possibly because the Zn species cannot be completely removed at 900 °C, so BIMZIF at the shell completely covers the MoO2 as the core.44 When the pyrolysis temperature rises, the pattern of Co–MoO2-NC-1000 displays a more obvious Co characteristic peak than Co–MoO2-NC-900, inferring that a large amount of cobalt is aggregated, which is in great agreement with TEM results. The electronic states of Mo, Co and O elements in the Co–MoO2-NC-900 sample are investigated by XPS. The six main peaks at 232.6, 235.9, 229.5, 231.9, 235.0 and 232.9 eV of Mo 3d spectra match well with the Mo4+ 3d3/2, Mo6+ 3d3/2, Mo4+ 3d5/2, Moδ+ 3d5/2, Moδ+ 3d3/2 and Mo6+ 3d5/2 corresponding to MoO2 (Fig. S6b in the ESI†).39 Moreover, the Co 2p peak of the three samples is deconvoluted into six peaks of Co3+ 3d3/2, Co2+ 3d1/2, Co2+ 3d3/2, Coδ+ 3d1/2, Coδ+ 3d3/2 and Co3+ 3d3/2 at approximately 780.8, 799.1, 782.97, 803.4, 787.1 and 796.7 eV, respectively (Fig. S6c in the ESI†).45–47 Furthermore, there are five types of N in N 1s spectra: oxidized N (403 eV), graphitic N (400.9 eV), pyrrolic N (399.8 eV), M–N (398.9 eV) (M = Co, Mo), and pyridinic N (398.3 eV) (Fig. S6d in the ESI†).9,48–51 In order to prove that there is electron transfer between MoO2 and Co, Co–MoO2-NC-900 and MoO2-NC-900 samples have been synthesized as references. As shown in Fig. S6e in the ESI,† the peak of Co in Co–MoO2-NC-900 shows a 0.4 eV positive shift compared to the as-prepared Co-NC-900 samples. When compared to the as-prepared MoO2-NC-900 samples, the Mo peak in Co–MoO2-NC-900 displays a 0.4 eV negative shift (Fig. S6f in the ESI†). The above results demonstrate that the surface charge and composition of MoO2 are regulated by the doped Co, and the electrons are transferred from the MoO2 surface to the Co cluster. Furthermore, Zn species are favourable for the formation of Co NPs because they can increase the space between Co atoms, and accelerate the growth of CNTs.52 Fig. S7a in the ESI† shows that there are no signal peaks in the Zn 2p spectra of Co–MoO2-NC-900 and Co–MoO2-NC-1000, which indicates that the Zn species has completely evaporated. However, as for Co–MoO2-NC-800, a significant Zn signal peak can be observed because the temperature of vaporization of metallic Zn is over 900 °C. Besides, from Fig. S7b in the ESI,† at a temperature of 900 °C, it can be seen that Co–MoO2-NC-900 has good structural integrity so that it can retain more active sites during the pyrolysis process. It is interesting to find that the change of the Co and Zn ion ratio will greatly affect the morphology and structure of Mo-MOF@BIMZIF and its pyrolysis products. As shown in Fig. S8 in the ESI,† the pyrolysis products obtained have a large number of carbon nanotubes and fully dispersed metal active sites only when the precursor has a cobalt–zinc ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
:1.
To further characterize the coordination properties and electronic structure of the hybrids, especially the existence form of Co–Mo–O active sites, EXAFS and XANES spectroscopy technologies have been employed. According to Fig. 3a, the Co K-edge EXAFS Fourier transform for Co–MoO2-NC-900 reveals a dominated peak centered at 2.1 Å and 2.5 Å corresponding to Co–Co coordination.53 Further EXAFS fitting parameters at the Co K-edge demonstrate that the coordination spheres of Co species in Co–MoO2-NC-900 are similar to those in Co foil but have a lower coordination numbers (NCo–MoO2a-NC-900Co–MoO2-NC-900 = 10.1, NCo foila = 12), which indicates that the Co species are in clusters rather than alloys (Fig. 3b and c and Table S1†). Furthermore, XANES is employed to further characterize the local environment of Co with a higher sensitivity. As shown in Fig. 3d, there is an obvious difference between the Co foil and Co–MoO2-NC-900_Co, which reveals an obvious electron transfer between the Co and Mo–O components in the Co–MoO2-NC-900 hybrid. The white-line peak of Co–MoO2-NC-900_Co is obviously higher than that of the Co foil, which further proved the electron transfer phenomenon between the two components. Besides, the high intensity of Co–MoO2-NC-900_Co in XANES indicates an oxidized electronic structure of Co, indicating electron transfers between Co and Mo via O atoms.34 In Fig. 3e and f, wavelet transform (WT) EXAFS was carried out for Co foil and Co–MoO2-NC-900_Co in K space. Two WT intensity maxima near 4 and 8 Å−1 match well with Co–N and Co–Co coordination, indicating that the Co coordination environment of Co–MoO2-NC-900_Co was different from that of Co foil.53,54 The electronic structure of Mo in the Co–MoO2-NC-900 sample was also investigated. The Mo K-edge EXAFS Fourier transform for Co–MoO2-NC-900_Mo reveals two dominant peaks centered at 2.54 and 1.75 Å, which match well with Mo–Co and Mo–O coordination, respectively, which reveals that Co–Mo–O motives exist (Fig. S9 and Table S1 in the ESI†).
 | ||
| Fig. 3 (a) K-Edge and best-fitted EXAFS spectra of Co–MoO2-NC-900_Co and Co foil. (b) R space. (c) K space. (d) Experimental XANES spectra. WT for the k3-weighted EXAFS signals for (e) Co foil and (f) Co–MoO2-NC-900_Co. | ||
The pore structure information and BET surface area were characterized by N2 adsorption/desorption. Fig. S10a in the ESI† illustrates that Co–MoO2-NC-900 has a porous structure with obvious hysteresis loops. Besides, Co–MoO2-NC-900 presents a 336 m2 g−1 specific surface area, larger than that of other products (131 m2 g−1 for Co–MoO2-NC-800 and 192 m2 g−1 for Co–MoO2-NC-1000). Furthermore, pore size distribution curves (Fig. S10b in the ESI†) indicate that Co–MoO2-NC-900 also exhibits more mesopores and the size is concentrated in the 1–10 nm range, which is beneficial for charge and mass transport.55
The ORR performance of electrocatalysts was investigated by LSV, CV and RRDE tests. Co–MoO2-NC-900 presents a more positive half-wave potential (E1/2 ≈ 0.89 V) and onset potential (Eonset ≈ 0.99 V) than the other Co–MoO2-NC samples, which are comparable to those of the commercial Pt/C electrocatalyst (E1/2 ≈ 0.87 V and Eonset ≈ 1.00 V) (Fig. 4a and S11 in the ESI†). According to Fig. 4b, Co–MoO2-NC-900 displays a lower Tafel slope (74.8 mV dec−1) than the other Co–MoO2-NC samples and the reference Pt/C (88.8 mV dec−1). The Koutecky–Levich (K–L) equation was applied to investigate the electron transfer number (n) by fitting the linear regions from 0.5 to 0.7 V. Besides, the n of Co–MoO2-NC-900 is near 4 and produces a low ring current density of 0.01084 mA cm−2 during the ORR procedure, comparable to commercial Pt/C (Fig. 4c and S12 in the ESI†). Furthermore, the high kinetic current density (Jk) at 0.85 V of Co–MoO2-NC-900 is 26.0 mA cm−2, which is higher than that of the other Co–MoO2-NC samples and Pt/C electrocatalyst (Jk = 7.7 mA cm−2). Besides, the ORR activity of Co–MoO2-NC with different Co/Zn ratios has also been evaluated as shown in Fig. S13 in the ESI.† When there is only zinc in the precursor, the prepared Co–MoO2-NC material has the worst ORR/OER performance. When the cobalt–zinc ratio is 1:2, the performance of the prepared Co–MoO2-NC-1:2 is significantly improved, and when the ratio is 1:1, the optimal performance is reached, and then the activity decreases as the ratio increases. When only cobalt is present, Co–MoO2-NC-3:0 shows poor performance due to a large amount of cobalt agglomeration.
 | ||
| Fig. 4 Electrochemical performance of Co–MoO2-NC products, Pt/C and Ir/C. (a) ORR curves. (b) Tafel plots for the ORR; (c) ring current densities (×10) and current densities of Co–MoO2-NC-900 and Pt/C; (d) OER curves and (e) Tafel plots for the OER; (f) i–t cures at 0.5 V for 25000 s; (g) LSV curves before and after 1000 potential cycles for the OER and (h) LSV curves of different catalysts for the ORR and OER. | ||
Co–MoO2-NC-900 shows the lowest overpotential of 370 mV at 10 mA cm−2 compared with Co–MoO2-NC-800 (Ej=10 = 434 mV), Co–MoO2-NC-1000 (Ej=10 = 561 mV) and the reference Ir/C (Ej=10 = 388 mV) (Fig. 4d). Furthermore, the Tafel slope of Co–MoO2-NC-900 (142.1 mV dec−1) is also lower than that of Co–MoO2-NC-800 (285.3 mV dec−1), Co–MoO2-NC-1000 (218.5 mV dec−1) and the reference Ir/C (186.9 mV dec−1) (Fig. 4e). Fig. S14 in the ESI† also demonstrates that when the ratio of cobalt to zinc is 1:1, the prepared product has the best OER performance and the lowest Tafel slope. The electrochemical properties of all samples have been summarized in Table S2 in the ESI.† To further investigate the electrochemical properties of the electrocatalysts, their effective electrochemically active surface area (ECSA) was evaluated from the double layer capacitance (Cdl) based on the CV results at different scan rates from 2 to 10 mV s−1. Fig. S15 in the ESI† illustrates that Co–MoO2-NC-900 shows a larger Cdl value of 28.92 mF cm−2 than the other Co–MoO2-NC products (26.32 mF cm−2 for Co–MoO2-NC-800 and 17.32 mF cm−2 for Co–MoO2-NC-1000).
The stability test of the Co–MoO2-NC-900 electrocatalyst was performed. Compared to commercial Pt/C (53% of the initial ORR current), Co–MoO2-NC-900 could keep 93% of the initial ORR current at 0.5 V for 25000 s, demonstrating its excellent durability (Fig. 4f). LSV of Co–MoO2-NC-900 shows a negligible change (ΔE1/2 ≈ 11 mV) after 1000 CV cycles (0.165 to 1.165 V), which is superior to that of Pt/C (ΔE1/2 ≈ 27 mV) (Fig. S16a in the ESI†). As for the OER performance, the Co–MoO2-NC-900 electrocatalyst can hold 93% of the initial current at 1.5 V for 60000 s (Fig. S16b in the ESI†), which is much better than that of the reference Ir/C (only 30% of the initial OER current for 36000 s). Besides, as Fig. 4e illustrates, there is no obvious attenuation for Co–MoO2-NC-900 after 1000 cycles, superior to commercial Ir/C (ΔEj=10 ≈ 40 mV). Furthermore, Co–MoO2-NC-900 also exhibited better resistance to the methanol crossover effect than Pt/C (Fig. S17 in the ESI†). Additionally, as an efficient bifunctional catalyst, the variance of ORR E1/2 and OER potential at 10 mA cm−2 (ΔE = Ej=10 − E1/2) could be used to evaluate its electrochemical performance. Herein, we can find Co–MoO2-NC-900 (ΔE = 0.71 V) presents better performance than the reference Pt/C + Ir/C (ΔE = 0.748 V) and other Co–MoO2-NC products, which is also comparable to that of other state of the art bifunctional electrocatalysts (Fig. 4h and Table S3 in the ESI†).
By utilizing as-synthesized Co–MoO2-NC-900 as an electrocatalyst for Zn–air batteries, the performance in practical applications was also investigated. Fig. 5a shows that the Zn–air battery constructed by using Co–MoO2-NC-900 presents a high open circuit voltage (1.6 V), superior to the commercial reference (1.41 V), as shown in Fig. S18a.† And Co–MoO2-NC-900 also shows a high-power density of 176.5 mW cm−2 at a current density of ≈217.1 mA cm−2, while for the battery based on Pt/C + Ir/C it is only 106.2 mW cm−2 at a current density of ≈166.5 mA cm−2. Moreover, the photograph in Fig. 5b shows that a LED can be powered by two batteries with Co–MoO2-NC-900. Fig. 5c and d show that the Zn–air battery with Co–MoO2-NC-900 displays a smaller discharge–charge overpotential of 0.85 V at a current density of 5 mA cm−2 smaller than that of the Pt/C + Ir/C cathode (0.9 V). Furthermore, Co–MoO2-NC-900 also shows a better durability and little voltage changes over 3400 charge/discharge cycles (for nearly 1145 h) compared to the reference Pt/C + Ir/C (highly significant polarization after 160 charge–discharge cycles for 52 h). Furthermore, as shown in Fig. S18b in the ESI,† the specific discharge capacity was found to be 819.08 mA h gZn−1 at 5 mA cm−2 for the Co–MoO2-NC-900–C based Zn–air battery (normalization by the mass of the consumed Zn in the Zn anode), corresponding to a high energy density of 1048.42 W h kgZn−1, which is superior to the respective values obtained in the Pt/C + Ir/C-based Zn–air battery (775.19 mA h gZn−1, corresponding to 930.23 W h kgZn−1).Additionally, Table S4† illustrates that when Co–MoO2-NC-900 is applied as a zinc–air battery catalyst, it can make the assembled battery device comparable to the most stable zinc–air batteries recently reported (Table S4 in the ESI†).
 | ||
| Fig. 5 (a) Discharge polarization curves and the corresponding power density plots; (b) photograph of a lit light emitting diode (LED, 3.0 V) powered by Zn–air batteries; (c) galvanostatic cycling stability of the Zn–air battery with coupled noble-metal Pt/C + Ir/C and (d) Co–MoO2-NC-900 electrocatalysts at a current density of 5 mA cm−2. | ||
To further theoretically elucidate the catalytic mechanism of Co–MoO2-NC-900 during ORR/OER processes, density functional theory (DFT) simulations were performed on the electrocatalysts based on the model of which Co clusters are on the (001) surface of MoO2. The total density of states (TDOS) demonstrates that the electronic state of the material changes due to the transfer of electrons between the two components after the introduction of Co into MoO2 hybrids (Fig. 6a).56,57 Fig. S19 in the ESI† reveals that the Mo atom in Co–MoO2-NC-900 holds a more filled d eg state compare to that in MoO2. The above results show that the introduction of Co allows electrons to transfer between the two components through the oxygen bridge and can effectively control the electronic state of the material, and it is also consistent with the EXAFS analysis.
 | ||
| Fig. 6 (a) TDOS for Co–MoO2-NC-900 and MoO2; (b) free energy pathways of the ORR/OER processes for Co–MoO2-NC-900 and MoO2. | ||
The free energies of the intermediates have been calculated to investigate the ORR/OER processes (Fig. 6b and S20 in the ESI†). The OER is considered as the reverse reaction of the ORR. It is interesting to find that the free energy values (overpotential) of three intermediate states, *O, *OH, and *OOH, on the as-prepared Co–MoO2-NC-900 are much lower than those of pure MoO2, indicating that the Co–Mo–O structure as the main motive site plays a key role in both ORR and OER processes (Table S5 in the ESI†). Besides, the DFT results demonstrate that the rate-limiting steps are totally different from those of Co–MoO2-NC-900 and pure MoO2 during ORR/OER processes. For Co–MoO2-NC-900, the adsorption of O2 (Step 1, 0.36 V) and transition from *OH to *O (Step 2, 0.47 V) are the rate-limiting steps. For pure MoO2, the formation of O2 (Step 2, −0.97 V) and transition from *OH to OH− (Step 3, −0.81 V) are the most sluggish steps. The above results demonstrate that after successfully doping Co sites to adjust the electronic state of the material, the eg orbital of the Mo sites in MoO2 has been optimized, making it easier to combine with oxygen species to reduce reaction barriers.
Conclusions
In conclusion, we report a novel post-assembly method by growing bimetallic Co/Zn ZIFs on the external surface of a customized Mo-MOF to prepare core–shell structured Mo-MOF@BIMZIF, which displays a hollow porous Co–MoO2 polyhedral nanocage (Co–MoO2-NC) structure after high temperature pyrolysis. The well-designed structure enables the active site to be covered by a carbon layer of appropriate thickness to avoid direct contact with the electrolyte while enhancing conductivity. Besides, the hollow porous nanocage also makes the material present a large specific surface area, high electrochemical surface area and abundant pores, which makes a great contribution to the electrochemical activity. As an efficient bifunctional electrocatalyst, Co–MoO2-NC-900 presents a low overpotential (370 mV) for the OER and a half-wave potential (E1/2 ≈ 0.89 V) for the ORR. The Zn–air battery with Co–MoO2-NC-900 shows a high-power density of 176.5 mW cm−2, which is superior to that of the battery based on Pt/C + Ir/C, and it can display a smaller discharge–charge overpotential of 0.85 V at a current density of 5 mA cm−2 for 1145 h. EXAFS and XANES results demonstrate the coordination geometry and electron coupling of the hybrid. Besides, DFT results show that when Co is successfully introduced into the MoO2 hybrid, the hybrid electronic state generated by the electron transfer between the two components will let the electrocatalysts show high activity during OER and ORR processes. This work gives a guideline to prepare hybrid nanostructures using MoO2 and M–Nx–C materials as bifunctional electrocatalysts with durable, efficient, and novel core–shell structures for broad application.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21671136 and 21878189), Guangdong Basic and Applied Basic Research Foundation (2020A1515010379), Project of Educational Commission of Guangdong Province of China (2020ZDZX2011), Shenzhen Science and Technology Project Program (JCYJ20190808144413257 and JCYJ20190808145203535), and Shenzhen Key Projects of Technological Research (JSGG20200925145800001) and Instrumental Analysis Center of Shenzhen University.References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed
.
- L. Wei, H. E. Karahan, S. Zhai, H. Liu, X. Chen, Z. Zhou, Y. Lei, Z. Liu and Y. Chen, Adv. Mater., 2017, 29, 1701410 CrossRef PubMed
- Q. Hu, X. Liu, B. Zhu, L. Fan, X. Chai, Q. Zhang, J. Liu, C. He and Z. Lin, Nano Energy, 2018, 50, 212–219 CrossRef CAS
- W. Zang, A. Sumboja, Y. Ma, H. Zhang, Y. Wu, S. Wu, H. Wu, Z. Liu, C. Guan, J. Wang and S. J. Pennycook, ACS Catal., 2018, 8, 8961–8969 CrossRef CAS
- X. F. Lu, S. L. Zhang, E. Shangguan, P. Zhang, S. Gao and X. W. D. Lou, Adv. Sci., 2020, 7, 2001178 CrossRef CAS PubMed
- J. Lin, C. Zeng, X. Lin, C. Xu, X. Xu and Y. Luo, ACS Nano, 2021, 15, 4594–4607 CrossRef CAS PubMed
- X. F. Lu, Y. Fang, D. Luan and X. W. D. Lou, Nano Lett., 2021, 21, 1555–1565 CrossRef CAS PubMed
- H. J. Qiu, P. Du, K. Hu, J. Gao, H. Li, P. Liu, T. Ina, K. Ohara, Y. Ito and M. Chen, Adv. Mater., 2019, 31, 1900843 CrossRef PubMed
- Q. Hu, G. M. Li, G. D. Li, X. F. Liu, B. Zhu, X. Y. Chai, Q. L. Zhang, J. Liu and C. He, Adv. Energy Mater., 2019, 9, 1803867 CrossRef
- D. Ji, L. Fan, L. Li, S. Peng, D. Yu, J. Song, S. Ramakrishna and S. Guo, Adv. Mater., 2019, 31, 1808267 CrossRef PubMed
- Y. Wu, J. Cai, Y. Xie, S. Niu, Y. Zang, S. Wu, Y. Liu, Z. Lu, Y. Fang, Y. Guan, X. Zheng, J. Zhu, X. Liu, G. Wang and Y. Qian, Adv. Mater., 2020, 32, 1904346 CrossRef CAS PubMed
- J. Yin, Y. Li, F. Lv, M. Lu, K. Sun, W. Wang, L. Wang, F. Cheng, Y. Li, P. Xi and S. Guo, Adv. Mater., 2017, 29, 1704681 CrossRef PubMed
- D. Ji, J. Sun, L. Tian, A. Chinnappan, T. Zhang, W. A. D. M. Jayathilaka, R. Gosh, C. Baskar, Q. Zhang and S. Ramakrishna, Adv. Funct. Mater., 2020, 30, 1910568 CrossRef CAS
- T. Zhou, N. Zhang, C. Wu and Y. Xie, Energy Environ. Sci., 2020, 13, 1132–1153 RSC
- L. An, Z. Zhang, J. Feng, F. Lv, Y. Li, R. Wang, M. Lu, R. B. Gupta, P. Xi and S. Zhang, J. Am. Chem. Soc., 2018, 140, 17624–17631 CrossRef CAS PubMed
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed
- B.-C. Hu, Z.-Y. Wu, S.-Q. Chu, H.-W. Zhu, H.-W. Liang, J. Zhang and S.-H. Yu, Energy Environ. Sci., 2018, 11, 2208–2215 RSC
- S. Chen, X. Shu, H. Wang and J. Zhang, J. Mater. Chem. A, 2019, 7, 19719–19727 RSC
- X. F. Lu, Y. Chen, S. Wang, S. Gao and X. W. Lou, Adv. Mater., 2019, 31, 1902339 CrossRef PubMed
- Q. Cui, G. Qin, W. Wang, K. R. Geethalakshmi, A. Du and Q. Sun, J. Mater. Chem. A, 2019, 7, 14510–14518 RSC
- Q. Yue, Y. Wan, Z. Sun, X. Wu, Y. Yuan and P. Du, J. Mater. Chem. A, 2015, 3, 16941–16947 RSC
- A. Wu, Y. Xie, H. Ma, C. Tian, Y. Gu, H. Yan, X. Zhang, G. Yang and H. Fu, Nano Energy, 2018, 44, 353–363 CrossRef CAS
- Z. Yan, J. Xie, J. Jing, M. Zhang, W. Wei and S. Yin, Int. J. Hydrogen Energy, 2012, 37, 15948–15955 CrossRef CAS
- Y. Jin and P. K. Shen, J. Mater. Chem. A, 2015, 3, 20080–20085 RSC
- C.-L. Zhang, B.-R. Lu, F.-H. Cao, Z.-Y. Wu, W. Zhang, H.-P. Cong and S.-H. Yu, Nano Energy, 2019, 55, 226–233 CrossRef CAS
- G. Zhang, R. Chenitz, M. Lefèvre, S. Sun and J.-P. Dodelet, Nano Energy, 2016, 29, 111–125 CrossRef CAS
- A. Morozan and F. Jaouen, Energy Environ. Sci., 2012, 5, 9269–9290 RSC
- Y. Guan, Y. Li, S. Luo, X. Ren, L. Deng, L. Sun, H. Mi, P. Zhang and J. Liu, Appl. Catal., B, 2019, 256, 117871 CrossRef CAS
- J. Wang, D. Gao, G. Wang, S. Miao, H. Wu, J. Li and X. Bao, J. Mater. Chem. A, 2014, 2, 20067–20074 RSC
- Z. Chen, R. Wu, Y. Liu, Y. Ha, Y. Guo, D. Sun, M. Liu and F. Fang, Adv. Mater., 2018, 30, 1802011 CrossRef PubMed
- H. Zhang, P. An, W. Zhou, B. Y. Guan, P. Zhang, J. Dong and X. W. Lou, Sci. Adv., 2018, 4, eaao6657 CrossRef PubMed
- P. Zhang, X. F. Lu, J. Nai, S.-Q. Zang and X. W. Lou, Adv. Sci., 2019, 6, 1900576 CrossRef PubMed
- J. Chen, J. Liu, J.-Q. Xie, H. Ye, X.-Z. Fu, R. Sun and C.-P. Wong, Nano Energy, 2019, 56, 225–233 CrossRef CAS
- E. Hu, Y. Feng, J. Nai, D. Zhao, Y. Hu and X. W. Lou, Energy Environ. Sci., 2018, 11, 872–880 RSC
- X. Shi, A. Wu, H. Yan, L. Zhang, C. Tian, L. Wang and H. Fu, J. Mater. Chem. A, 2018, 6, 20100–20109 RSC
- M. Zeng, Y. Liu, F. Zhao, K. Nie, N. Han, X. Wang, W. Huang, X. Song, J. Zhong and Y. Li, Adv. Funct. Mater., 2016, 26, 4397–4404 CrossRef CAS
- H. Guo, Q. Feng, J. Zhu, J. Xu, Q. Li, S. Liu, K. Xu, C. Zhang and T. Liu, J. Mater. Chem. A, 2019, 7, 3664–3672 RSC
- L. Yang, J. Yu, Z. Wei, G. Li, L. Cao, W. Zhou and S. Chen, Nano Energy, 2017, 41, 772–779 CrossRef CAS
- Y. Jin, H. Wang, J. Li, X. Yue, Y. Han, P. K. Shen and Y. Cui, Adv. Mater., 2016, 28, 3785–3790 CrossRef CAS PubMed
- X. Liu, D. Wu, W. Ji and W. Hou, J. Mater. Chem. A, 2015, 3, 968–972 RSC
- I. S. Amiinu, X. Liu, Z. Pu, W. Li, Q. Li, J. Zhang, H. Tang, H. Zhang and S. Mu, Adv. Funct. Mater., 2018, 28, 1704638 CrossRef
- P. Sabhapathy, I. Shown, A. Sabbah, P. Raghunath, J.-L. Chen, W.-F. Chen, M.-C. Lin, K.-H. Chen and L.-C. Chen, Nano Energy, 2021, 80, 105544 CrossRef CAS
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed
- J. Wu, L. Hu, N. Wang, Y. Li, D. Zhao, L. Li, X. Peng, Z. Cui, L.-J. Ma, Y. Tian and X. Wang, Appl. Catal., B, 2019, 254, 55–65 CrossRef CAS
- Z. Lei, Y. Tan, Z. Zhang, W. Wu, N. Cheng, R. Chen, S. Mu and X. Sun, Nano Res., 2021, 14, 868–878 CrossRef CAS
- B. K. Kang, S. Y. Im, J. Lee, S. H. Kwag, S. B. Kwon, S. Tiruneh, M.-J. Kim, J. H. Kim, W. S. Yang, B. Lim and D. H. Yoon, Nano Res., 2019, 12, 1605–1611 CrossRef CAS
- A. C. Mtukula, X. Bo and L. Guo, J. Alloys Compd., 2017, 692, 614–621 CrossRef CAS
- Y.-Y. Chen, Y. Zhang, W.-J. Jiang, X. Zhang, Z. Dai, L.-J. Wan and J.-S. Hu, ACS Nano, 2016, 10, 8851–8860 CrossRef CAS PubMed
- T. Wang, Z. Kou, S. Mu, J. Liu, D. He, I. S. Amiinu, W. Meng, K. Zhou, Z. Luo, S. Chaemchuen and F. Verpoort, Adv. Funct. Mater., 2018, 28, 1705048 CrossRef
- L.-Y. Zhang, M.-R. Wang, Y.-Q. Lai and X.-Y. Li, J. Power Sources, 2017, 359, 71–79 CrossRef CAS
- Y. Pan, K. Sun, S. Liu, X. Cao, K. Wu, W. C. Cheong, Z. Chen, Y. Wang, Y. Li, Y. Liu, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 2610–2618 CrossRef CAS PubMed
- P. Yu, L. Wang, F. Sun, Y. Xie, X. Liu, J. Ma, X. Wang, C. Tian, J. Li and H. Fu, Adv. Mater., 2019, 31, 1901666 CrossRef PubMed
- Y. Liu, F. Chen, W. Ye, M. Zeng, N. Han, F. Zhao, X. Wang and Y. Li, Adv. Funct. Mater., 2017, 27, 1606034 CrossRef
- Q. Liu, J. Tian, W. Cui, P. Jiang, N. Cheng, A. M. Asiri and X. Sun, Angew. Chem., Int. Ed., 2014, 53, 6710–6714 CrossRef CAS PubMed
- S. Liu, Z. Wang, S. Zhou, F. Yu, M. Yu, C.-Y. Chiang, W. Zhou, J. Zhao and J. Qiu, Adv. Mater., 2017, 29, 1700874 CrossRef PubMed
- S. Zhao, Y. Wang, J. Dong, C.-T. He, H. Yin, P. An, K. Zhao, X. Zhang, C. Gao, L. Zhang, J. Lv, J. Wang, J. Zhang, A. M. Khattak, N. A. Khan, Z. Wei, J. Zhang, S. Liu, H. Zhao and Z. Tang, Nat. Energy, 2016, 1, 16184 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: SEM and TEM images, XPS spectra, CV curves, simulation module, etc. See DOI: 10.1039/d1ta04408g |
| ‡ Yi Guan and Nan Li contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |