
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAllylic alcohol synthesis by Ni-catalyzed direct and selective coupling of alkynes and methanol†
Herong
Chen
,
Zhijun
Zhou
and
Wangqing
Kong
 *
*
The Institute for Advanced Studies (IAS), Wuhan University, Wuhan, Hubei 430072, P. R. China. E-mail: wqkong@whu.edu.cn
First published on 7th June 2021
Abstract
Methanol is an abundant and renewable chemical raw material, but its use as a C1 source in C–C bond coupling reactions still constitutes a big challenge, and the known methods are limited to the use of expensive and noble metal catalysts such as Ru, Rh and Ir. We herein report nickel-catalyzed direct coupling of alkynes and methanol, providing direct access to valuable allylic alcohols in good yields and excellent chemo- and regioselectivity. The approach features a broad substrate scope and high atom-, step- and redox-economy. Moreover, this method was successfully extended to the synthesis of [5,6]-bicyclic hemiacetals through a cascade cyclization reaction of alkynones and methanol.
To address the sustainability issues in the production of new chemicals, the development of new catalytic processes that are free of by-products and using abundant renewable feedstocks is one of the most important challenges facing chemists today. The simplest alcohol, methanol, is very abundant, with a total annual production capacity of approximately 110 million metric tons per year,1 and is an important C1-feedstock in the chemical industry. Beller2 and Milstein3 made fundamental developments in catalytic dehydrogenation reactions of methanol.4 Krische and coworkers pioneered the study of Ir-catalyzed direct C–C coupling of methanol with reactive π-unsaturated reactants (1,3-dienes, 1,3-enynes and allenes).5 The groups of Glorius,6 Donohoe,7 Obora,8 Andersson9 and others10 demonstrated the direct methylation of ketones or amines using methanol. Despite these achievements, the catalytic C–C bond coupling reactions with methanol are still extremely rare and are limited to the use of precious and noble metal-catalysts such as Ru, Rh or Ir.11 The development and use of cheap and abundant metal catalysts for methanol activation is uphill and remains an important field that urgently needs to be developed.
On the other hand, allylic alcohols are highly versatile building blocks in organic synthesis and the pharmaceutical industry, and much effort has been devoted to their synthesis. Among them, nickel-catalyzed reductive coupling of alkynes and aldehydes represents an effective and powerful method. However, this method generally requires the use of stoichiometric reducing reagents that are air-sensitive, metallic or pyrophoric (e.g. ZnR2, BEt3, and R3SiH, Scheme 1a).12 The direct cross-coupling of alcohols and alkynes to synthesize allylic alcohols without the use of any reductant or oxidant represents a significant advancement (Scheme 1b).13 However, this approach still poses many limitations that will require considerable effort to overcome. (1) Alkynes are limited to dialkyl alkynes, and poor regioselectivities were observed for unsymmetrical alkynes, which greatly limits the scope of application of the reaction. (2) Alcohols are restricted to active benzyl alcohols and higher alcohols. The direct cross-coupling of alkynes with methanol has not yet been reported.
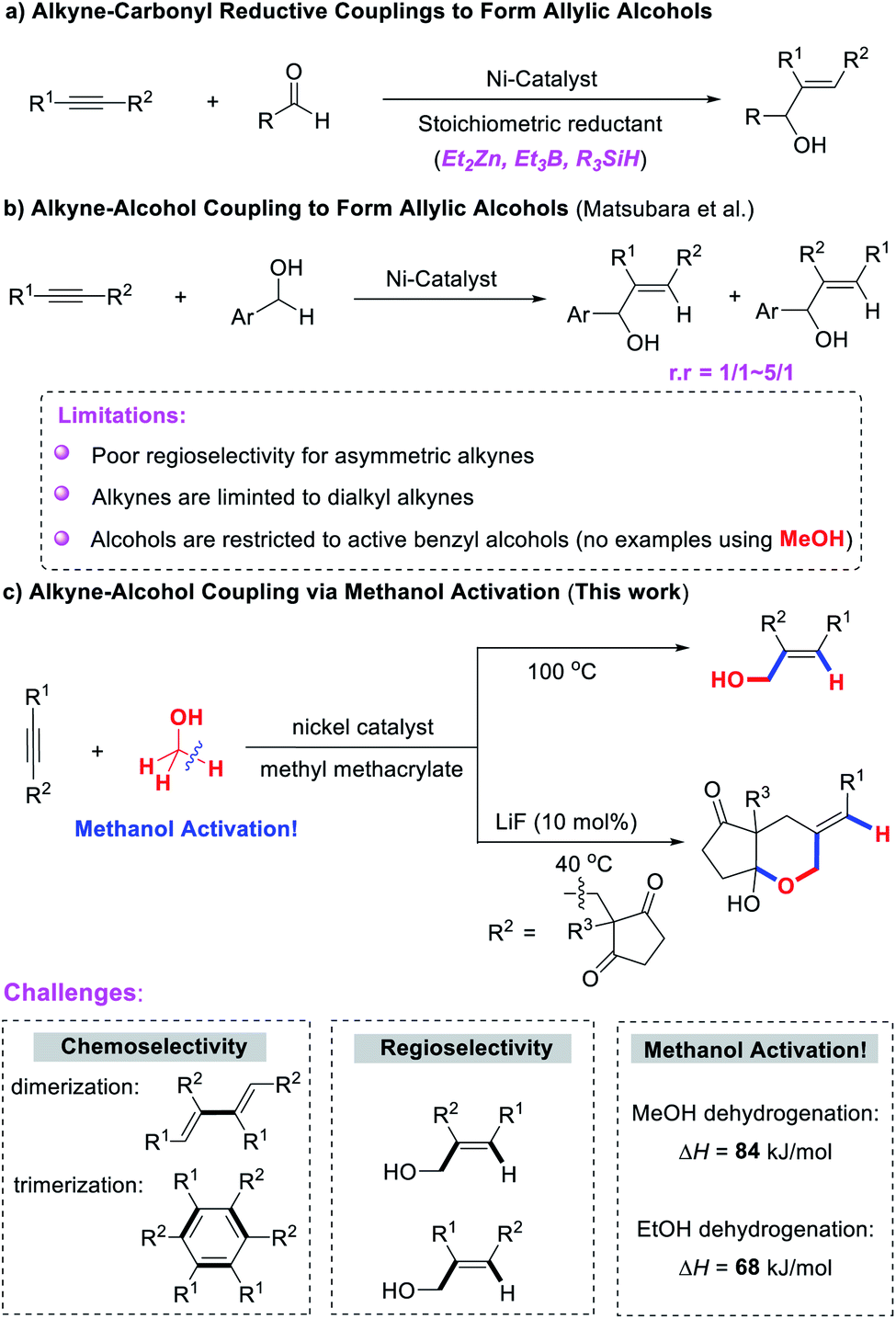 | ||
| Scheme 1 Synthesis of allylic alcohols by Ni-catalyzed coupling reaction with alkynes. | ||
Although the alkyne–paraformaldehyde reductive coupling has been developed,14 the paraformaldehyde was itself prepared from synthesis gas (through methanol). Therefore, the development of a new strategy for the direct coupling of alkynes and methanol without the use of any reductant or oxidant is still of great value, but also extremely challenging: (1) alkynes are reactive and could rapidly dimerize to 1,3-dienes15 or cyclotrimerize to aromatic ring derivatives in the interaction with nickel.16 (2) Unsymmetric alkynes could result in a mixture of regioisomers that are difficult to separate. (3) The activation energy of methanol in the dehydrogenation process (ΔH = +84 kJ mol−1) is significantly higher than that of higher alcohols or even ethanol (ΔH = +68 kJ mol−1).17
Herein we report the nickel-catalyzed direct and regioselective hydrohydroxymethylation of alkynes for the first time using methanol as a C1-feedstock, providing a broad and efficient approach for the synthesis of high added-value allylic alcohols in a high atom-, step- and redox-economic manner. In addition, a cascade cyclization reaction of alkynones and methanol has also been developed for the synthesis of [5,6]-bicyclic hemiacetals in good yields and excellent regio- and diastereoselectivity (Scheme 1c).
In our initial experiments, we chose unsymmetrical internal alkyne 1a as a model substrate to optimize the reaction conditions (Table 1). As expected, no reaction occurred under the previously reported reaction conditions using Ni(COD)2/L1(IPr) as the catalyst (entry 1).13a Even if the reaction temperature was increased to 100 °C, only a trace amount of allylic alcohol product 2a was observed (entry 2), which indicates that the use of methanol in the catalytic C–C coupling reactions is indeed a big challenge. Various N-heterocyclic carbene ligands (L2–L6) were investigated (entries 3–9). We found that the selectivity of allylic alcohol 2a is challenged by a number of side reactions, such as the hydrogenation (3a), dimerization (4a) and trimerization (5a) of alkyne 1a. L4 is the most effective, providing 2a with the highest yield (40%) and excellent regioselectivity (14/1), but an appreciable quantity of dimerization and trimerization by-products 4a and 5a was still obtained (entry 5). Krische14 reported that PCy3 could promote the reductive coupling of alkynes and paraformaldehyde, but we found that it is not effective for alkyne–methanol coupling (entry 8).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ligand | Additive | Yieldb (2, %) | Yieldb (3, %) | Yieldb (4, %) | Yieldb (5, %) |
| a Reactions conditions: 1a (0.2 mmol), Ni(COD)2 (10 mol%), ligand (20 mol%), tBuOK (12 mol%), additive (1 equiv.) in toluene (1 mL) and MeOH (3 mL) in a sealed tube at 100 °C. b Determined by GC analysis using adamantane as the internal standard. c Without tBuOK. d Room temperature. e Regioselectivity (2a/2a′). f Isolated yield. g 0.2 equivalent. | ||||||
| 1c,d | L1 | — | No reaction | |||
| 2c | L1 | — | 6 | 6 | <2 | 20 |
| 3 | L2 | — | 30 (14/1)e | 15 | 13 | 20 |
| 4 | L3 | — | 3 | 5 | <2 | 43 |
| 5 | L4 | — | 40 (14/1)e | 9 | 22 | 23 |
| 6 | L5 | — | 30 (7/1)e | 6 | 18 | 27 |
| 7 | L6 | — | No reaction | |||
| 8 | Cy3P | — | Complicated | |||
| 9 | PPh3 | — | 10 | 4 | <2 | 59 |
| 10 | L4 | A1 | 10 | <2 | <2 | <2 |
| 11 | L4 | A2 | 38 (14/1)e | <2 | <2 | <2 |
| 12 | L4 | A3 | 60f (14/1)e | 6 | <2 | <2 |
| 13 | L4 | A4 | No reaction | |||
| 14 | L4 |
A3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) g g |
54f (14/1)e | 8 | 6 | 7 |
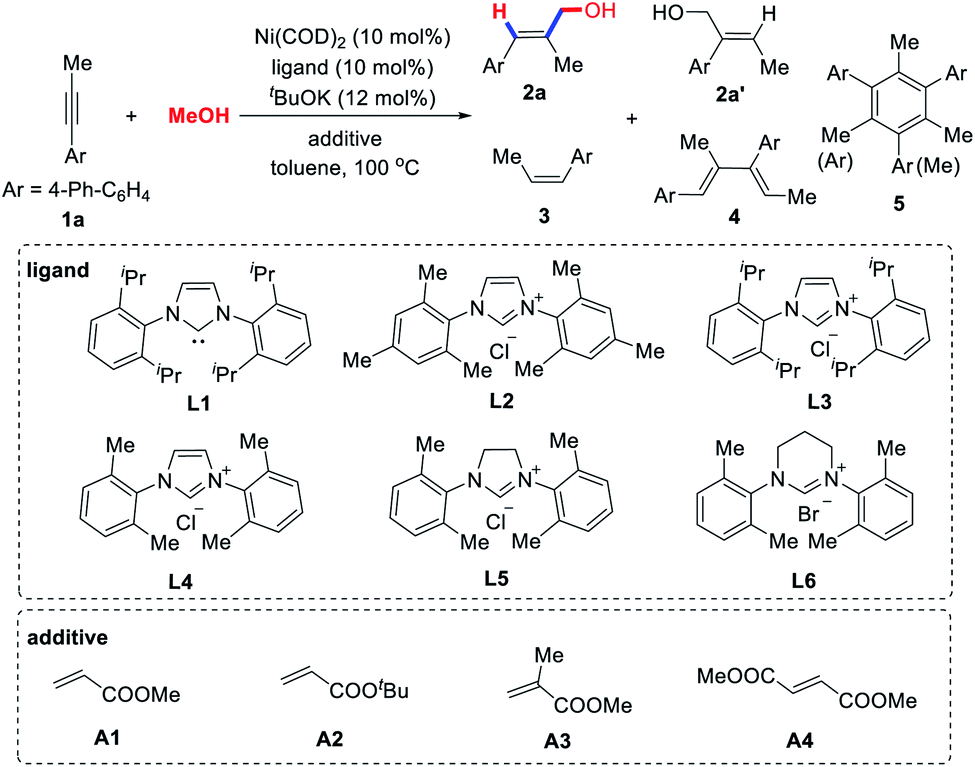
Many examples have reported that olefins can affect the outcomes of transition metal-catalyzed cross coupling reactions through increased activity, stability, or selectivity.18 More recently, Montgomery et al.19 found that adding electron-deficient olefins to NHC–Ni(0) complexes can improve their catalytic performance. Inspired by this discovery, we examined various acrylates A1–A4 (entries 10–13). Excitingly, the addition of methyl methacrylate (A3) can indeed significantly improve the chemoselectivity of the reaction, providing the allylic alcohol 2a in 60% isolated yield and a more than 14/1 ratio of regioisomers (entry 12). The structure of acrylates has a great influence on the reaction outcome, indicating that they may act as additional ligands to coordinate with the nickel catalyst, thereby suppressing these undesired dimerization or cyclotrimerization side reactions. However, by-products formed by the reductive coupling of acrylates and alkynes have also been observed (see Section 3 in the ESI†).20 It is worth mentioning that stoichiometric acrylate additives are not necessary. As shown in entry 14, even if 0.2 equivalent of A3 was used, 54% of the target product 2a can be obtained, thus showing the subtleties of our catalytic system.
With the optimized reaction conditions in hand, we turned our attention to explore the substrate scope of alkynes (Scheme 2). We were pleased to find that various unsymmetrical aryl–alkyl alkynes were coupled with methanol to provide the corresponding allylic alcohols 2a–2q in moderate to good yields and high regioselectivities. Various functional groups, such as fluorine (2b), trifluoromethyl (2c), chlorine (2e), allyl (2f), bromine (2g), amine (2h–2j) and amide (2k and 2l) could all be well-tolerated. Heteroaromatic ring-substituted alkynes, such as 5-indole,21 2-dibenzothiophene and 2-dibenzofuran could also proceed smoothly to furnish allylic alcohols 2n–2p in 40–63% yield. It is worth mentioning that complex biologically active molecules such as estrone derivatives, could also be successfully incorporated into the desired product 2q in 62%, thus demonstrating the robustness and generality of this methodology for late-stage modification of complex biologically active molecules. Terminal alkynes were also found to be compatible with the reaction conditions, providing the corresponding products 2r–2s in moderate yields and excellent regioselectivity (>20/1). Symmetric diarylalkynes bearing electron-donating or electron-withdrawing groups were applicable to the reaction (2t–2y). Strikingly, both 1,2-di(furan-2-yl)ethyne and 1,2-di(thiophen-2-yl)ethyne were competent substrates and furnished the desired allylic alcohols 2x–2y in good yields.
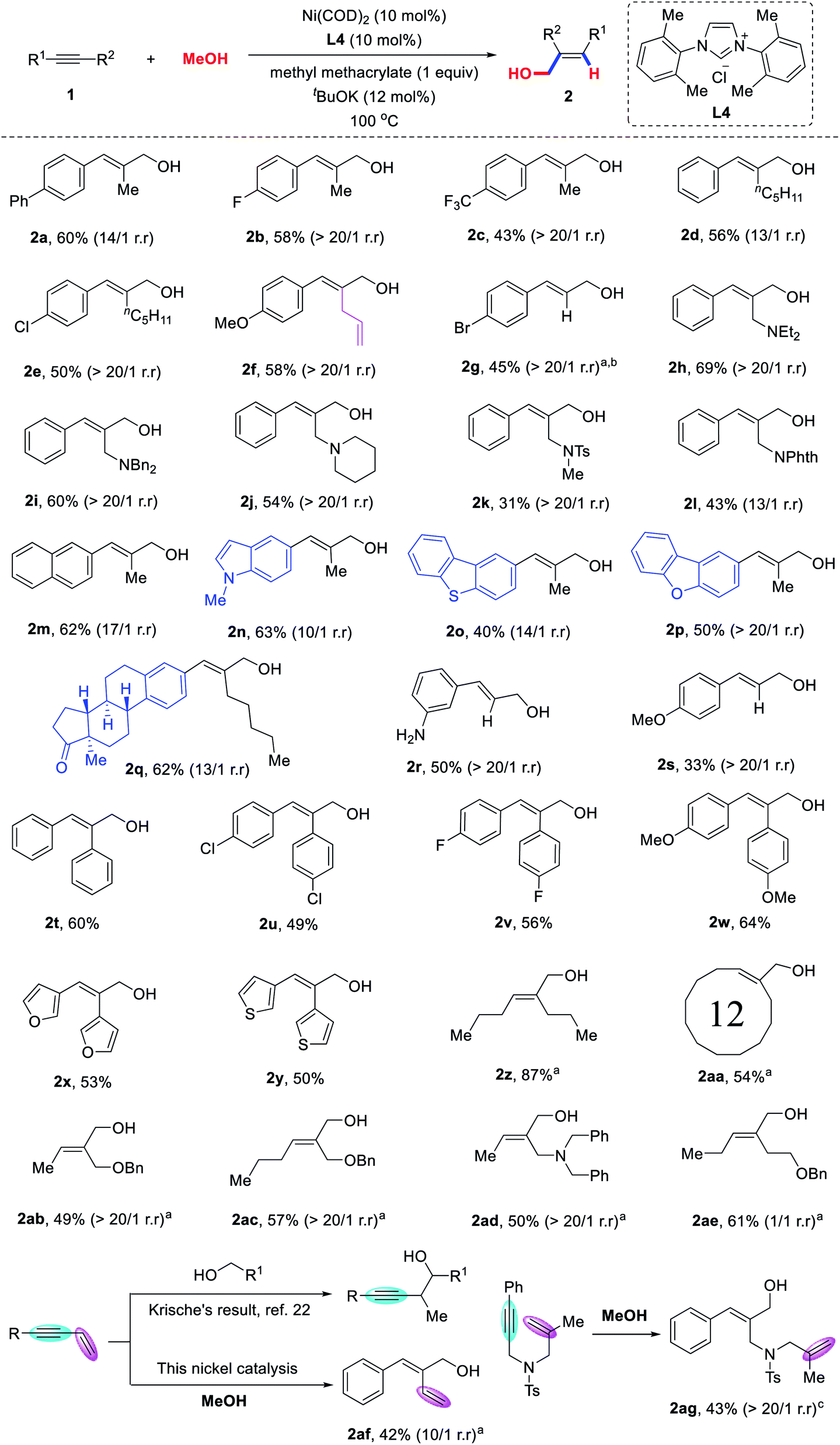 | ||
| Scheme 2 Substrate scope of alkynes for the synthesis of allylic alcohols. Reactions were carried out with 1 (0.2 mmol), Ni(COD)2 (10 mol%), L4 (10 mol%), tBuOK (12 mol%), and methyl methacrylate (0.2 mmol) in toluene (1.0 mL) and MeOH (3.0 mL) in a sealed tube at 100 °C. Isolated yields are given. a The reaction was conducted with Ni(COD)2 (15 mol%), L4 (15 mol%), and tBuOK (18 mol%). b ((4-Bromophenyl)ethynyl)trimethylsilane was used. c The reaction was conducted with toluene (0.5 mL) and MeOH (1.5 mL). | ||
In addition, this transformation is not restricted to aryl-substituted alkynes. As shown in Scheme 2, oct-4-yne and cyclododecane were coupled with methanol to produce allylic alcohols 2z and 2aa in 87% and 54% yields, respectively. To further evaluate the influence of the electronic properties of the substituents on the regioselectivity, we tested the hydrohydroxymethylation reaction of unsymmetrical dialkyl-substituted alkynes bearing benzyloxy or dibenzylamino groups at the propargylic position. To our delight, the corresponding allylic alcohols 2ab–2ad were obtained in moderate yields, with remarkably high regioselectivity (>20/1). However, the regioselectivity of this reaction was decreased by the alkyne bearing a benzyloxy group at the homopropargylic position.
To expand the potential synthetic applications of the transformation, we investigated the hydrohydroxymethylation of 1,3-enynes. The corresponding dienol 2af was obtained, which was selectively hydrohydroxymethylated on the alkyne but not on the alkene moiety. 1,6-Enyne was also compatible to give the corresponding allylic alcohol 2ag in 42% yield with >20/1 regioselectivity. This strategy can serve as a powerful supplement to the previous method reported by Krische et al.,22 in which alcohols were reacted with alkenes to obtain the corresponding homopropargylic alcohols.23
Alkynone substrates were also tested, but the expected product was not detected due to their sensitivity to base. After slightly modifying the reaction conditions, we were pleased to find that various [5,6]-bicyclic hemiacetals 7 could be obtained in good yields with excellent regio- and diastereoselectivities through the cascade cyclization reaction of alkynones 6 with methanol (Scheme 3). We first explored the influence of the substituents (R1) at the terminus of the triple bond. A variety of para-substituted aromatic rings at the alkyne terminus could undergo tandem cyclization to provide the target hemiacetals 7b–7g in 54–78% yields. The structure of 7a was confirmed by an X-ray crystal diffraction study. The aryl groups with substituents at the meta and ortho position were also found to be compatible, leading to the corresponding products 7h–7j in 56–74% yields. Moreover, various (hetero)aryl rings such as naphthalene (7k), benzodioxan (7l), 3,4-dihydrobenzodioxine (7m), thiophene (7n), dibenzofuran (7o), dibenzothiophene (7p), indole (7q) and pyridine (7r) at the terminal of the triple bond could be successfully incorporated into the desired products in good yields. Strikingly, estrone was also compatible with this transformation to afford the desired product 7s in 65% yield. However, no desired product was observed when the methyl substituted alkynone substrate was used. We then investigated the influence of the substituents (R2) at the 2-position of the cyclopentane-1,3-diones. Ethyl, benzyl, and allyl were all well tolerated leading to the corresponding [5,6]-bicyclic hemiacetals 7t–7w in moderate yields.
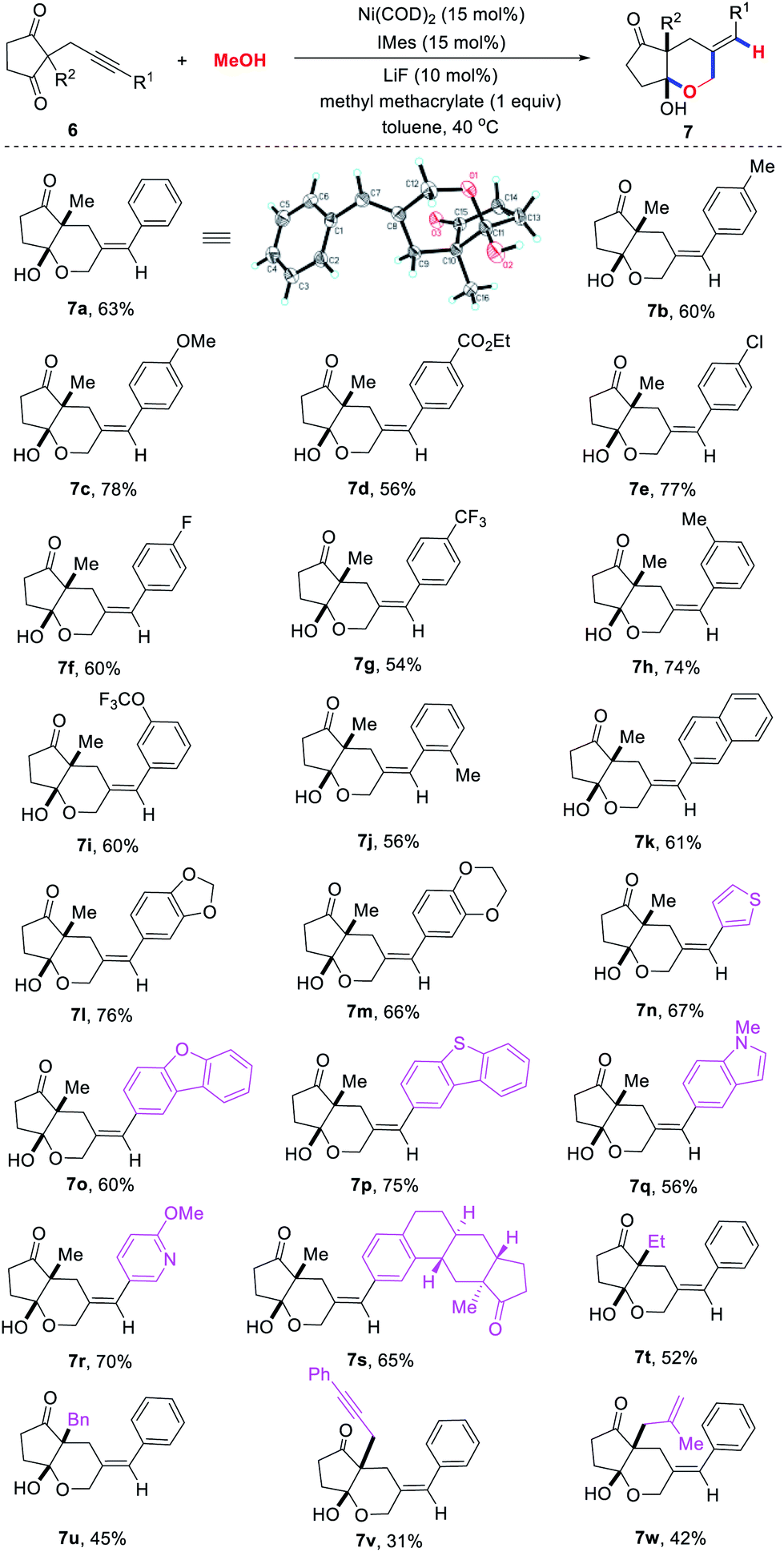 | ||
| Scheme 3 Substrate scope of alkynones for the synthesis of [5, 6]-bicyclic hemiacetals. Reactions were carried out with 6 (0.2 mmol), Ni(COD)2 (15 mol%), IMes (15 mol%), LiF (10 mol%), and methyl methacrylate (0.2 mmol) in toluene (1.5 mL) and MeOH (0.5 mL) in a sealed tube at 40 °C. Isolated yields are given. | ||
To provide a deeper insight into the reaction mechanism, deuterium-labelling experiments were performed. 1a was reacted with CH3OD under our standard reaction conditions; however, no incorporation of deuterium was detected in product 2a (Scheme 4a), revealing that the hydroxyl of methanol is not the proton source. This result is different from the previous report by Zhou et al.,24 in which the Ni(0) catalyst underwent oxidative addition to the O–H bond of methanol to form methoxyl nickel hydride species and then migratory insertion into unsaturated bonds. Further investigation using CD3OD as solvent provided 2a-D in 41% yield, in which 99% of the deuterium was incorporated into the olefinic position, but the reaction rate is obviously slowed down (Scheme 4b). We also conducted the kinetic isotope effect (KIE) experiment. The intermolecular competition reaction between 1a and CD3OD or CH3OH under standard reaction conditions provided a KIE (kH/kD) value of 6.1 (Scheme 4c). Taken together, these results may indicate that the dehydrogenation of methanol to form the key formaldehyde intermediate is the rate-determining step of this transformation.
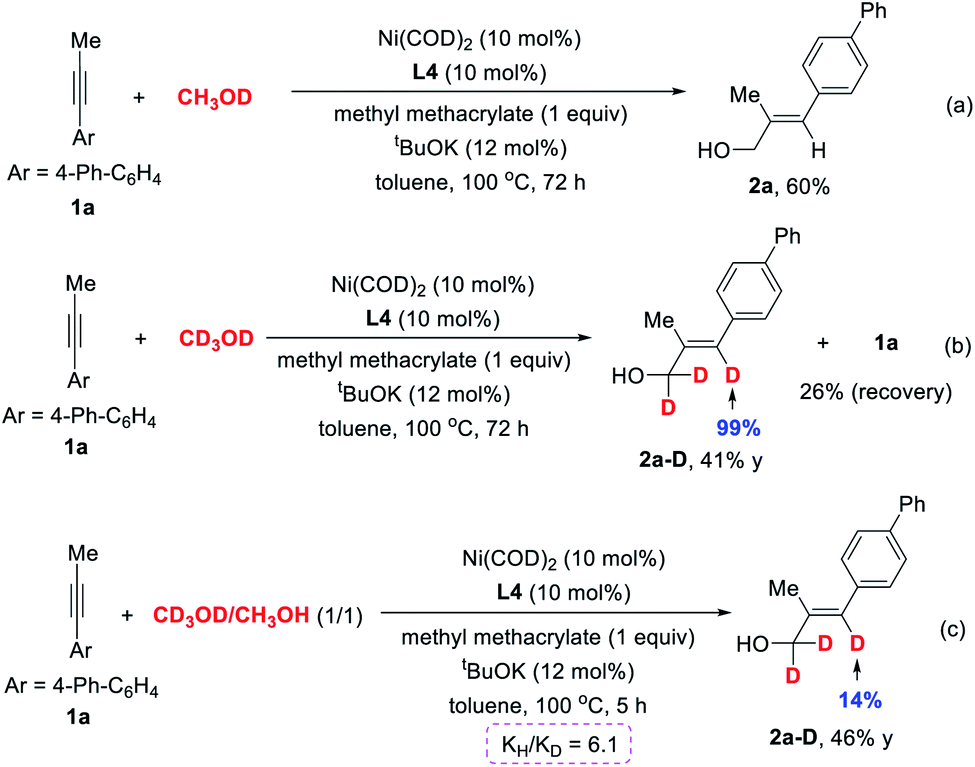 | ||
| Scheme 4 Deuterium-labelling experiments. | ||
On the basis of these experimental results and previous observations, a possible reaction mechanism is proposed in Scheme 5. The reaction is initiated by reducing alkyne to alkene and simultaneously oxidizing methanol to formaldehyde, as evidenced by the detection of catalytic amounts of alkene 3. Oxidative cyclization of acrylate-coordinated NHC–Ni(0) A17 with alkyne and formaldehyde gives oxa-nickelacycle intermediate B. Subsequent protonation of nickelacycle species B with methanol affords the vinylnickel intermediate C, which can undergo β-H elimination to generate vinyl nickel hydride species D and formaldehyde.25 Reductive elimination of D will furnish allylic alcohol 2 and the catalytically active Ni(0) catalyst A. Further nucleophilic addition of the hydroxyl group to one of the ketone carbonyl groups will produce [5,6]-bicyclic hemiacetal 7. We speculate that the acrylate is used as an additional ligand, thereby inhibiting the alkyne dimerization to 1,3-dienes or cyclotrimerization to aromatic ring derivatives.
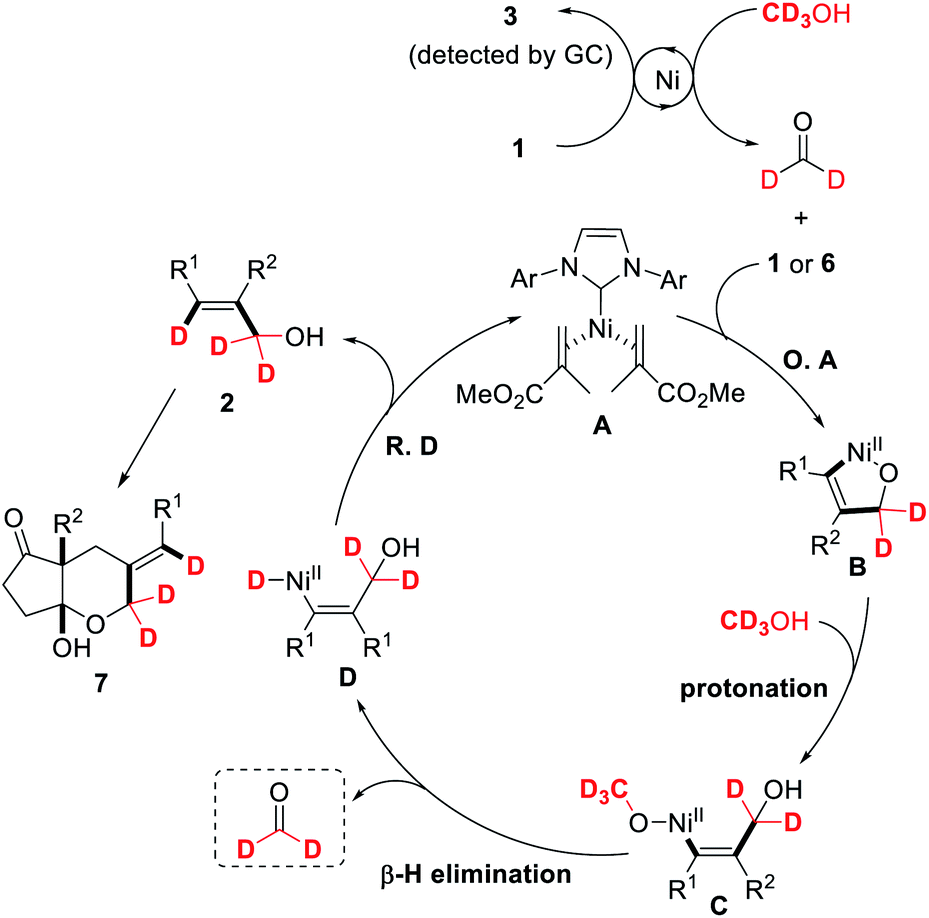 | ||
| Scheme 5 Proposed reaction mechanism. | ||
Conclusions
In summary, a nickel-catalyzed direct coupling of alkynes and methanol is developed for the first time, providing direct access to high added-value allylic alcohols in good yields and excellent chemo- and regioselectivity. This transformation features a wide substrate scope and high atom-, step- and redox-economy. In addition, a cascade cyclization reaction of alkynones and methanol has also been developed for the synthesis of [5,6]-bicyclic hemiacetals.Data availability
The ESI include experimental detail, NMR data and HRMS data.Author contributions
W. K. conceived and designed the experiments. H. C. and Z. Z. performed the experiments and prepared the ESI. W. K. directed the project and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support from Wuhan University, the “1000-Youth Talents Plan”, and the National Natural Science Foundation of China (No. 21702149).Notes and references
- (a) P. G. Cifre and O. Badr, Energy Convers. Manage., 2007, 48, 519 Search PubMed; (b) http://www.methanol.org/Methanol-Basics.aspx .
- M. Nielsen, E. Alberico, W. Baumann, H. J. Drexler, H. Junge, S. Gladiali and M. Beller, Nature, 2013, 495, 85 Search PubMed.
- (a) R. Langer, I. Fuchs, M. Vogt, E. Balaraman, Y. D. Posner, L. J. W. Shimon, Y. B. David and D. Milstein, Chem.–Eur. J., 2013, 19, 3407 Search PubMed; (b) C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588 Search PubMed.
- R. E. R. Lugo, M. Trincado, M. Vogt, F. Tewes, G. S. Quinones and H. Grützmacher, Nat. Chem., 2013, 5, 342 Search PubMed.
- (a) J. Moran, A. Preetz, R. A. Mesch and M. J. Krische, Nat. Chem., 2011, 3, 287 Search PubMed; (b) K. D. Nguyen, D. Herkommer and M. J. Krische, J. Am. Chem. Soc., 2016, 138, 14210 Search PubMed; (c) M. Holmes, K. D. Nguyen, L. A. Schwartz, T. Luong and M. J. Krische, J. Am. Chem. Soc., 2017, 139, 8114 Search PubMed.
- N. Ortega, C. Richter and F. Glorius, Org. Lett., 2013, 15, 1776 Search PubMed.
- (a) L. K. M. Chan, D. L. Poole, D. Shen, M. P. Healy and T. J. Donohoe, Angew. Chem., Int. Ed., 2014, 53, 761 Search PubMed; (b) D. Shen, D. L. Poole, C. C. Shotton, A. F. Kornahrens, M. P. Healy and T. J. Donohoe, Angew. Chem., Int. Ed., 2015, 54, 1642 Search PubMed.
- S. Ogawa and Y. Obora, Chem. Commun., 2014, 50, 2491 Search PubMed.
- X. Quan, S. Kerdphon and P. G. Andersson, Chem.–Eur. J., 2015, 21, 3576 Search PubMed.
- (a) Y. Li, H. Li, H. Junge and M. Beller, Chem. Commun., 2014, 50, 14991 Search PubMed; (b) E. A. Jo, J. H. Lee and C. H. Jun, Chem. Commun., 2008, 5779 Search PubMed; (c) F. Li, J. Xie, H. Shan, C. Sun and L. Chen, RSC Adv., 2012, 2, 8645 Search PubMed; (d) C. Sun, X. Zou and F. Li, Chem.–Eur. J., 2013, 19, 14030 Search PubMed.
- (a) A. Quintard and J. A. Rodriguez, ChemSusChem, 2016, 9, 28 Search PubMed; (b) B. G. R. Berendt, K. Polidano and L. C. Morrill, Org. Biomol. Chem., 2019, 17, 1595 Search PubMed; (c) T. Irrgang and R. Kempe, Chem. Rev., 2019, 119, 2524 Search PubMed.
- For reviews on Ni-catalyzed alkyne–carbonyl reductive couplings, see: (a) S. Saito and Y. Yamamoto, Chem. Rev., 2000, 100, 2901 Search PubMed; (b) J. Montgomery, Acc. Chem. Res., 2000, 33, 467 Search PubMed; (c) J. Montgomery, Angew. Chem., Int. Ed., 2004, 43, 3890 Search PubMed; (d) R. M. Moslin, K. Miller-Moslin and T. F. Jamison, Chem. Commun., 2007, 4441 Search PubMed; (e) Y. Nakao and T. Hiyama, Pure Appl. Chem., 2008, 80, 1097 Search PubMed; (f) M. Jeganmohan and C. H. Cheng, Chem.–Eur. J., 2008, 14, 10876 Search PubMed; (g) H. A. Malik, R. D. Baxter and J. Montgomery, Nickel-Catalyzed Reductive Couplings and Cyclizations, in Catalysis without Precious Metals, ed. R. M. Bullock, Wiley-VCH, Weinheim, 1st edn, 2010, pp. 181–210 Search PubMed; (h) Formation of C–C bonds via catalytic hydrogenation and transfer hydrogenation. J. Moran and M. J. Krische, in Sustainable Catalysis, ed. K. K. Hii, M. T. Williams, P. J. Dunn and M. J. Krische, John Wiley and Sons, New York, 2012, pp. 363–408 Search PubMed; (i) J. M. Ketcham, I. Shin, T. P. Montgomery and M. J. Krische, Angew. Chem., Int. Ed., 2014, 53, 9142 Search PubMed; (j) B. Sam, B. Breit and M. J. Krische, Angew. Chem., Int. Ed., 2015, 54, 3267 Search PubMed; (k) E. P. Jackson, H. A. Malik, G. J. Sormunen, R. D. Baxter, P. Liu, H. Wang, A. R. Shareef and J. Montgomery, Acc. Chem. Res., 2015, 48, 1736 Search PubMed; (l) E. A. Standley, S. Z. Tasker, K. L. Jensen and T. F. Jamison, Acc. Chem. Res., 2015, 48, 1503 Search PubMed.
- (a) K. Nakai, Y. Yoshida, T. Kurahashi and S. Matsubara, J. Am. Chem. Soc., 2014, 136, 7797 Search PubMed; (b) E. L. McInturff, K. D. Nguyen and M. J. Krische, Angew. Chem., Int. Ed., 2014, 53, 3232 Search PubMed; (c) Y. Cai, J. Zhang, F. Li, J. Liu and S. L. Shi, ACS Catal., 2019, 9, 1 Search PubMed.
- C. C. Bausch, R. L. Patman, B. Breit and M. J. Krische, Angew. Chem., Int. Ed., 2011, 50, 5687 Search PubMed.
- (a) T. Wu, J. Chen and Y. Wu, Org. Lett., 2011, 13, 4794 Search PubMed; (b) G. Zhang, Y. Xie, Z. Wang, Y. Liu and H. Huang, Chem. Commun., 2015, 51, 1850 Search PubMed; (c) Y. Liu, G. Zhang and H. Huang, Org. Lett., 2017, 19, 6674 Search PubMed; (d) S. Cañellas, J. Montgomery and M. À. Pericàs, J. Am. Chem. Soc., 2018, 140, 17349 Search PubMed; (e) Q. Liang, K. Hayashi and D. Song, ACS Catal., 2020, 10, 4895 Search PubMed; (f) H. Olivier-Bourbigou, P. A. R. Breuil, L. Magna, T. Michel, M. F. E. Pastor and D. Delcroix, Chem. Rev., 2020, 120, 7919 Search PubMed.
- For selected reviews, see: (a) S. Saito and Y. Yamamoto, Chem. Rev., 2000, 100, 2901 Search PubMed; (b) K. P. C. Vollhardt, Angew. Chem., Int. Ed., 1984, 23, 539 Search PubMed; (c) J. A. Varela and C. Saá, Chem. Rev., 2003, 103, 3787 Search PubMed; (d) Y. Yamamoto, Curr. Org. Chem., 2005, 9, 503 Search PubMed; (e) A. F. Orsino, M. Gutiérrez del Campo, M. Lutz and M. E. Moret, ACS Catal., 2019, 9, 2458 Search PubMed.
- (a) M. Qian, M. A. Liauw and G. Emig, Appl. Catal., A, 2003, 238, 211 Search PubMed; (b) W. H. Lin and H. F. Chang, Catal. Today, 2004, 97, 181 Search PubMed.
- J. B. Johnson and T. Rovis, Angew. Chem., Int. Ed., 2008, 47, 840 Search PubMed.
- (a) D. P. Todd, B. B. Thompson, A. J. Nett and J. Montgomery, J. Am. Chem. Soc., 2015, 137, 12788 Search PubMed; (b) A. J. Nett, S. Cañellas, Y. Higuchi, M. T. Robo, J. M. Kochkodan, M. T. Haynes, J. W. Kampf and J. Montgomery, ACS Catal., 2018, 8, 6606 Search PubMed; (c) Z. Zhou, J. Chen, H. Chen and W. Kong, Chem. Sci., 2020, 11, 10204 Search PubMed.
- C. Wang, P. Lin and C.-H. Cheng, J. Am. Chem. Soc., 2002, 124, 9696 Search PubMed.
- Trace amount of the methylated product at the C3-position of indole was observed, see: S. Chen, G. Lu and C. Cai, RSC Adv., 2015, 5, 70329 Search PubMed.
- (a) R. L. Patman, V. M. Williams, J. F. Bower and M. J. Krische, Angew. Chem., Int. Ed., 2008, 47, 5220 Search PubMed; (b) L. M. Geary, J. C. Leung and M. J. Krische, Chem.–Eur. J., 2012, 18, 16823 Search PubMed; (c) K. D. Nguyen, D. Herkommer and M. J. Krische, J. Am. Chem. Soc., 2016, 138(16), 5238 Search PubMed; (d) L. M. Geary, S. K. Woo, J. C. Leung and M. J. Krische, Angew. Chem., Int. Ed., 2018, 57, 461 Search PubMed.
- (a) J. R. Kong, M. Y. Ngai and M. J. Krische, J. Am. Chem. Soc., 2006, 128, 718 Search PubMed; (b) Y. T. Hong, C. W. Cho, E. Skucas and M. J. Krische, Org. Lett., 2007, 9, 3745 Search PubMed.
- L. Xiao, L. Cheng, W. Feng, M. Li, J. Xie and Q. Zhou, Angew. Chem., Int. Ed., 2018, 57, 461 Search PubMed.
- A. Herath, W. Li and J. Montgomery, J. Am. Chem. Soc., 2008, 130, 469 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectroscopic data, and NMR spectra. CCDC 2058189. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02625a |
| This journal is © The Royal Society of Chemistry 2021 |