
Surface engineered CoP/Co3O4 heterojunction for high-performance bi-functional water splitting electro-catalysis†
Xintong
Li
 a,
Yizhe
Liu
a,
Qidi
Sun
a,
Wei-Hsiang
Huang
bc,
Zilong
Wang
d,
Chu-Chen
Chueh
e,
Chi-Liang
Chen
c and
Zonglong
Zhu
*a
a,
Yizhe
Liu
a,
Qidi
Sun
a,
Wei-Hsiang
Huang
bc,
Zilong
Wang
d,
Chu-Chen
Chueh
e,
Chi-Liang
Chen
c and
Zonglong
Zhu
*a
aDepartment of Chemistry, City University of Hong Kong, Kowloon, 999077, Hong Kong. E-mail: zonglzhu@cityu.edu.hk
bGraduate Institute of Applied Science and Technology, National Taiwan University of Science and Technology (NTUST), Taipei 10607, Taiwan
cNational Synchrotron Radiation Research Centre, Hsinchu 30076, Taiwan, Republic of China
dSiyuan Laboratory, Guangdong Provincial Engineering Technology Research Centre of Vacuum Coating Technologies and New Energy Materials, Department of Physics, Jinan University, Guangzhou, Guangdong 510632, P. R. China
eDepartment of Chemical Engineering, National Taiwan University, Taipei 10617, Taiwan
First published on 10th November 2021
Abstract
In the electrochemical water splitting process, integrating hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) in the same electrolyte with the same catalyst is highly beneficial for increasing the energy efficiency and reducing the fabrication cost. However, most OER catalysts are unstable in the acidic solution, while HER shows poor kinetics in the alkaline solution, which hinders the scale-up application of electro-catalytic water splitting. In this work, a CoP/Co3O4 heterostructure is firstly fabricated and then O and P defects are introduced via surface engineering (s-CoP/Co3O4). The as-prepared material was employed as the catalyst towards electrochemical water splitting in an alkaline environment. In alkaline HER, a current density of −10 mA cm−2 can be achieved at an overpotential of 106 mV vs. RHE. In the OER process, the overpotential of s-CoP/Co3O4 electrode is only 211 mV vs. RHE at 10 mA cm−2 in 1 M KOH, and the corresponding Tafel slope is only 58.4 mV dec−1 so that the s-CoP/Co3O4 electrode could be used as the bifunctional catalyst for alkaline water splitting. This work provides a simple and low-cost approach to fabricate a Co-based heterojunction electrode with unsaturated metal sites to improve the electro-catalytic activities towards water splitting.
 Zonglong Zhu | Since joining in CityU, Dr Zonglong Zhu has established optoelectronic materials and devices lab. Until now, more than 30 articles have been published as corresponding author, including Nat. Nano, Nat. Commun., J. Am. Chem. Soc., Adv. Mater., Adv. Energy Mater., Adv. Funct. Mater., Adv. Sci., Joule, and Small Methods, and one US patent has been filed. His research mainly focuses on the design inorganic/organic materials, as well connecting the materials synthesis, physical properties and device performance for optoelectronics application. The current application interests include solar cells, transistors, light-emitting diodes, and electrochemical devices (e.g. batteries, supercapacitors). |
Introduction
Renewable energy has been growing rapidly as over 80% of the global energy consumption relies on nonrenewable fossil fuels.1 As promising energy revolutions, wind and solar can generate electricity with zero pollutant emissions.2 However, how to utilize the engendered clean energy is limited by the sustainable energy storage and their scaled application.3 Compared with various energy storage technologies, electrocatalytic water splitting provides a possible strategy as an intermediate approach for energy storage and conversion.4 To efficiently operate the water splitting process, a large activation energy barrier for both hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) needs to be overcome.5,6 Noble metals based catalysts will benefit the water splitting kinetics for efficient HER and OER,7 but their widespread application has been limited by their high cost and scarcity in practical processes.8 Thus, it is necessary to develop high-performance catalysts for water splitting with earth-abundant materials.For practical applications, bifunctional HER and OER electro-catalysis can significantly increase energy utilization and reduce electrode costs.9 Although impressive progress has been made, only a few high-performance electrocatalysts with bifunctional HER and OER catalytic activity in the same electrolyte have been reported. As most of the catalysts have poor stability in acid solution, it is highly attractive to develop high-performance non-noble bifunctional electro-catalysts for overall water splitting in alkaline environment.10,11
Cobalt-based materials have been widely reported as the electro-catalysts for water splitting.12 Typically, cobalt oxides (CoOx) have performed efficiently during the OER process, thanks to accessible adsorption sites from electro-deficient Co 3d orbits.11 Their oxygen vacancies facilitate the pre-oxidation of the adjacent Co atoms and the formation of Co–OOH during the OER process, further boosting the catalytic activities of cobalt oxides towards oxygen evolution.13 Their oxygen vacancies facilitate the pre-oxidation of the adjacent Co atoms and the formation of Co–OOH during the OER process, further boosting the catalytic activities of cobalt oxides towards oxygen evolution.14 However, simple CoOx exhibited deficient catalytic activity under HER operation. On the contrary, CoP containing electronegative P atoms, which offer adsorption sites towards electropositive reactants and intermediate,6,15,16 is considered as a suitable catalytic candidate for HER.14 However, the H2 desorption process has been impeded due to the strong binding between CoP and H atoms, which further results in poor performance.
In this work, we firstly designed and synthesized COP/Co3O4 heterostructure by an electro-deposition method. To improve the electro-catalytic properties both under the HER and OER processes, we introduced oxygen and phosphorus vacancies into the COP/Co3O4 heterostructure using a partial reduction process, respectively. The surface engineered COP/Co3O4 (s-COP/Co3O4) was then characterized using Raman, X-ray diffraction (XRD), transmission electron microscopy (TEM), and X-ray photoelectron spectroscopy (XPS). The results show that the CoP nanosheets grow vertically and uniformly on the surface of the Co3O4 layers, and the ratio of Co2+/Co3+ increased after the reduction process. s-COP/Co3O4 shows a much better catalytic performance than pristine COP/Co3O4, a current density of 10 mA cm−2 can be achieved at an overpotential of 211 mV vs. RHE in the OER process and the overpotential is only 106 mV vs. RHE at a current density of −10 mA cm−2 in HER, which surpasses most of the transition metal-based catalysts from the previously reported works. X-ray absorption spectroscopy (XAS) was also employed to further explain the elevated performances after the surface engineering process, the results show the successful formation of P vacancies in CoP. Different effects of the heterojunction structure in HER and OER are also presumed in the s-COP/Co3O4 system. This work provides a feasible approach to fabricate a low-cost, high-performance bi-functional electrocatalyst for HER and OER as well as verifying the positive effects of the heterojunction structure and surface engineering process.
Results and discussion
The fabrication process of the s-CoP/Co3O4 electrode is illustrated in Fig. 1a. Co(OH)2 was first obtained by precipitation and then thermally treated in the air at 250 °C to obtain Co3O4. The black Co3O4 powder was coated on the carbon paper and then electro-deposited in the electrolyte containing Co2+, H2PO42− and sequestrant (sodium citrate) at −1.0 V for 900 s to involve CoP. The CoP-coated Co3O4 electrode was soaked in a freshly-prepared NaBH4 solution (0.5 M) to obtain the final s-CoP/Co3O4 electrode.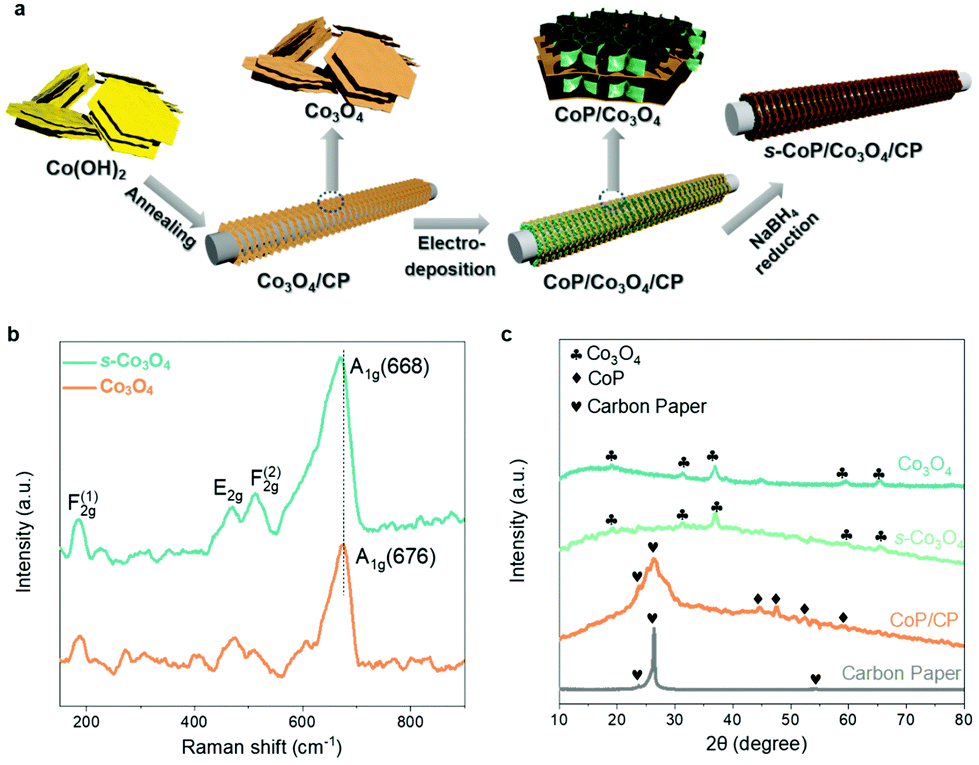 | ||
| Fig. 1 (a) Illustration of the preparation process of s-CoP/Co3O4. (b) Raman spectra at the wave length of 532 nm of Co3O4 and s-Co3O4. (c) XRD patterns of Co3O4, s-Co3O4, CoP/carbon paper, and carbon paper. | ||
Raman spectra of Co3O4 and s-Co3O4 were recorded to analyze the variation of crystal structure and increase of surface defects. As shown in Fig. 1b, the bands at 186, 470, 510 and 676 cm−1 correspond to the F(1)2g, E2g, F(2)2g and A1g symmetries, respectively, of Co3O4.17 The peak of A1g symmetry in the Raman spectrum of Co3O4 negatively shifts from 676 to 668 cm−1, which indicates that more defects are created during surface engineering of Co3O4, thus verifying the successful involvement of oxygen vacancies after NaBH4 reduction.18 The XRD patterns of Co3O4, s-Co3O4, CoP/carbon paper and bare carbon paper are shown in Fig. 1c. The diffraction peaks at 19.0°, 31.3°, 36.8°, 59.4° and 65.3° can be ascribed to (111), (220), (311), (400) and (440) planes of the cubic Co3O4, respectively. After the reduction treatment, the crystal structure remained in the s-Co3O4 structure, indicating that there was no crystal phase variation compared with Co3O4. The intense peaks at 23.8° and 26.4° of CoP/carbon paper electrode correspond to the peaks of the carbon paper at 46.2°, 48.1°, 52.3° and 56.7° correspond to (112), (211), (103) and (301) peaks of CoP. SEM images of Co3O4, s-Co3O4, CoP electrodeposited at different potentials (−0.8, −1.0 and −1.5 V vs. RHE) and s-CoP/Co3O4 are shown in Fig. S1.† The results show that Co3O4 is of the layered structure. After the NaBH4 reduction process, pores appeared on the surface of s-Co3O4. The electro-deposited CoP also had a nanolayered structure and the thickness of the CoP layer was the smallest when the deposition potential was −1.0 V vs. Ag/AgCl. The SEM image of s-CoP/Co3O4 showed that the CoP layers grew vertically on the surface of the Co3O4 layer, forming a three-dimensional connected structure, facilitating the transfer processes, and creating more exposed active sites.
The high-resolution transmission electron microscopy (HR-TEM) image of the s-CoP/Co3O4 electrode is shown in Fig. 2a and the interplanar distances of CoP and Co3O4 are demonstrated in Fig. 2b1–3. The lattice fringes of 0.283, 0.244 and 0.202 nm indicate the (011) facet of CoP and (113), (400) facets of Co3O4, respectively.19 The elemental mapping result of s-CoP/Co3O4 (Fig. 2c–f) shows that Co, O and P atoms are distributed uniformly on the surface of the electrode, suggesting that the CoP nanolayers grow homogeneously on the surface of Co3O4.
 | ||
| Fig. 2 (a) HR-TEM image of s-CoP/Co3O4, corresponding lattice fringe images of (b1) CoP and (b2–b3) Co3O4, (c–f) TEM elemental mapping of s-CoP/Co3O4 electrode. | ||
The elemental compositions and chemical states of the as-prepared Co3O4, s-Co3O4, CoP, CoP/Co3O4 and s-CoP/Co3O4 were further analyzed from XPS spectra (Fig. 3). High-resolution XPS spectra of Co 2p in Co3O4 and s-Co3O4 are shown in Fig. 3a. The major peaks at about 795 and 780 eV can be ascribed to Co 2p1/2 and Co 2p3/2, respectively, both of which could be deconvoluted to the peaks of Co2+ (782.2 and 796.9 eV) and Co3+ (779.88 and 794.9 eV), and the additional peaks at 788.5 and 804.2 eV are satellite peaks.20 The Co2+/Co3+ ratios are estimated to be 50.2% and 54.6% in Co3O4 and s-Co3O4, respectively, according to the relative area of the deconvoluted peaks corresponding to Co2+ and Co3+. The detailed calculation process is shown in the ESI.† It has been reported that NaBH4 will react with Co3O4 during the surface engineering process,15 making Co3O4 remaining at the oxygen-deficient states, thus the ratio of Co2+ will increase to achieve charge balance in s-Co3O4.22 Therefore, the increased ratio of Co2+/Co3+ indicates the successful reduction of Co3O4 by the NaBH4 treatment. The high-resolution XPS spectra of Co 2p of CoP/Co3O4 and s-CoP/Co3O4 from Fig. 3b show that the peak of Co0 appears at 777.5 eV after reduction, suggesting a small amount of CoP was reduced to Co by NaBH4.11 Schottky junction structures are formed at the interface of Co and CoP, facilitating electrons transfer from Co to CoP according to the Mott–Schottky effect, thus further facilitating the OER process.21
 | ||
| Fig. 3 High-resolution XPS spectra of Co 2p in (a) Co3O4, s-Co3O4, (b) CoP/Co3O4 and s-CoP/Co3O4. High-resolution XPS spectra of (c) O 1s in Co3O4, s-Co3O4, CoP/Co3O4 and s-CoP/Co3O4, and (d) P 2p in CoP and CoP/Co3O4. | ||
The O 1s high-resolution XPS spectra of the as-prepared materials could be deconvoluted to three peaks in Co3O4, s-Co3O4, CoP/Co3O4 and s-CoP/Co3O4 (Fig. 3c). The peaks at 529.9, 531.0 and 531.8 eV correspond to the O atoms coordinating with metal species (lattice O), surface oxygen defect species (OV) and O from adsorbed molecular water (absorbed O), respectively.22 The peaks corresponding to the adsorbed O exist in Co3O4 and s-Co3O4 because they absorbed water molecules from the air. However, the peak areas corresponding to the absorbed O in CoP/Co3O4 and s-CoP/Co3O4 decrease because they are characterized when coated on the carbon paper while the XPS signals of Co3O4 and s-Co3O4 were obtained from the powder sample. Moreover, the ratio of OV increases from 5.8% in Co3O4 to 12.3% in s-Co3O4, verifying the involvement of oxygen vacancies. With the protection of the electro-deposited CoP, the ratio of OV also increases from 9.8% to 19.6% after the reduction process. In Fig. 3d, the high-resolution XPS spectra of P 2p can be deconvoluted to three peaks. The binding energies of 129.6 and 130.6 eV can be attributed to 2p3/2 and 2p1/2 while the peak at 133.3 eV is ascribed to the surface oxidized P species, indicating the successful involvement of CoP.23 Moreover, the peak corresponding to P 2p3/2 was positively shifted by 0.20 eV after the reduction process, signifying fewer electrons occupied P atoms, which facilitates the H* desorption in the HER process.24 The ΔGH* value of CoP is much negative, in other words, H* adsorbs too strongly on the surface of CoP, so that the H* desorption is the rate-determining step of the HER process. Therefore, the catalytic activity of CoP towards HER will be enhanced after the partial reduction procedure.
The electro-catalytic activities of the as-prepared materials towards water splitting (OER and HER) are tested in the alkaline electrolyte (1 M KOH) with a conventional three-electrode system. The polarization curves were recorded using linear sweep voltammetry (LSV) at a scan rate of 5 mV s−1. As shown in Fig. 4a, the Co3O4/carbon paper (Co3O4/CP) electrode carries a limited catalytic efficiency towards OER, while s-Co3O4/CP performs better activity owing to the larger number of oxygen vacancies. The dosage of NaBH4 was adjusted to optimize the reduction conditions of Co3O4 and the result is shown in Fig. S2.† The catalytic performance of s-Co3O4 improves when the concentration of NaBH4 increases from 0.3 M to 0.5 M yet it gets poorer when the concentration of NaBH4 further increases to 0.7 M, owing to the impaired conductivity of Co3O4 caused by excessive oxygen vacancies. This conclusion can be further verified by the increased Tafel slope (from 63.8 mV dec−1 of 0.5-Co3O4 to 82.1 mV dec−1 of 0.7-Co3O4, Fig. S3†). The involvement of CoP enhances the catalytic performance of Co3O4 to a large extent, the overpotential of CoP/Co3O4 was decreased to 298 mV at a current density of 10 mA cm−2. The deposition time of CoP was also adjusted to optimize the catalytic efficiency of CoP/Co3O4, which is shown in Fig. S4,† and the electro-catalytic performance of CoP/Co3O4 was the best when the deposition time was 900 s. The electro-catalytic efficiency could be further enhanced while the CoP was coated on the surface engineered Co3O4, obtaining an overpotential of 265 mV at 10 mA cm−2. s-CoP/Co3O4 realizes the earliest overpotential of 211 mV at 10 mA cm−2 and the smallest Tafel slope (58.4 mV dec−1) (Fig. 4b), surpassing the performance of IrO2/carbon paper (281 mV, Fig. S5†). The electrochemical impedance spectroscopy (EIS) spectra of the electrodes were recorded at −0.2 V vs. RHE and 1.6 V vs. RHE and the results are shown in Fig. 4c and Fig. S10.† The impedance of Co3O4 is slightly enlarged after the reduction process, indicating an impaired electron transfer property though more exposed active sites are created after the reduction process. The impedances of CoP, CoP/Co3O4 and s-CoP/Co3O4 are much smaller than that of Co3O4 and s-Co3O4, further verifying that the involvement of CoP layers improves the electrons transfer properties to a large extent.
 | ||
| Fig. 4 (a) OER polarization curves, (b) corresponding Tafel plots (c) EIS (@−0.2 V vs. RHE) spectra, (d) HER polarization curves and (e) corresponding Tafel plots of Co3O4, s-Co3O4, CoP, CoP/Co3O4, CoP/s-Co3O4 and s-CoP/Co3O4 in 1 M KOH, (f) water splitting polarization curves of s-CoP/Co3O4 in 1 M KOH. | ||
The HER performance of samples were also investigated under alkaline conditions (1 M KOH). As shown in Fig. 4d, Co3O4 and s-Co3O4 electrodes showed poor catalytic activities towards HER as expected. With the involvement of the CoP layer, the overpotentials of CoP/Co3O4 and s-CoP/Co3O4 towards HER get much earlier, because the involved CoP offers the effective adsorption sites towards electropositive reactants and intermediates. Moreover, these two materials also perform better than the CoP, which is attributed to the water dissociation effects of Co3O4. Similar to OER systems, the earliest overpotential at 10 mA cm−2 was obtained for s-CoP/Co3O4 (106 mV), and the corresponding Tafel slope was only 51.2 mV dec−1 (Fig. 4e), which is close to the performance of Pt/C/carbon paper (56 mV, Fig. S6†). The much-elevated performance can mainly be attributed to two aspects, (1) P defects further optimize the electronegativity of CoP and (2) s-Co3O4 catalyzes the water dissociation efficiently. Due to the outstanding electrocatalytic activity of the s-CoP/Co3O4 electrode towards both HER and OER, it was adopted as the bifunctional catalyst in the water splitting cell. The water spitting performance of s-CoP/Co3O4 was recorded in a two-electrode system. As presented, to offer the current densities of 10 and 50 mA cm−2, the cell potentials were only 1.529 and 1.605 V, respectively (Fig. 4f), which surpass those reported for most of the electrodes in the latest journals (Table S3† and Fig. 5e).
 | ||
| Fig. 5 (a and b) Polarization curves of s-CoP/Co3O4 recorded before and after 5000 CV cycles, (c) Cdl values of Co3O4, CoP, CoP/Co3O4 and s-CoP/Co3O4 in 1 M KOH, comparison of overpotentials of s-CoP/Co3O4 and other catalysts reported in the latest literature reports towards (d) OER and (e) OER and HER at η10 in 1 M KOH. | ||
The durability of the s-CoP/Co3O4 electrode is verified by the LSV polarization before/after 5000 CV cycles at a scan rate of 50 mV s−1 and current–time (i–t) curves conducted at 211 mV (overpotential of OER at 10 mA cm−2) and −106 mV (overpotential of HER at −10 mA cm−2), respectively. As shown in Fig. 5a and b, no obvious deactivation could be observed after 5000 CV cycles in both OER and HER systems. Moreover, the current density barely shows any attenuation after operating at fixed overpotentials for 24 h. The electrochemical double-layer capacitances (Cdls, Fig. 5c) of the Co3O4, CoP, CoP/Co3O4 and s-CoP/Co3O4 electrodes were calculated according to the cyclic voltammograms (CVs, Fig. S7†) at different scan rates (10 to 100 mV s−1). The results show that the Cdl of s-CoP/Co3O4 (28.5 mF cm−2) is much larger than that of Co3O4 (1.02 mF cm−2), CoP (1.13 mF cm−2) and CoP/Co3O4 (1.99 mF cm−2). As well established, Cdl of the material is proportional to its electrochemically active surface areas (ECSA).25 Therefore, the Cdl results indicate that the coated CoP layer increases the ECSA of Co3O4 and the reduction process further enlarges the ECSA of CoP/Co3O4 to a larger extent, which can be attributed to the involvement of unsaturated cationic sites, they act as active sites and facilitate the pre-oxidation of the low valence Co and initiate OER at a lower applied potential. Therefore, the reduction process increases the density of active sites thus increasing the ECSA of CoP/Co3O4. The largest ECSA of s-CoP/Co3O4 further explains its best electro-catalytic activity towards water splitting. The electrochemically active surface areas (ECSA, @50 mV s−1, Fig. S11†) normalized LSV curves of Co3O4, CoP, CoP/Co3O4, and s-CoP/Co3O4 are summarized in Fig. S12,† the results show that the intrinsic activity of active sites in s-CoP/Co3O4 is similar to that of Co3O4, CoP and CoP/Co3O4, indicating that the CoP coating and surface engineering process improve the catalytic performances of the electrode by creating new active sites towards OER rather than enhancing the intrinsic catalytic activity of active sites in Co3O4. Notably, the comparisons of overpotentials towards OER and HER of the as-prepared s-CoP/Co3O4 and transition metal-based catalysts in the latest literature reports are shown in Fig. 5d and e and Tables S1–S3,† which indicate that the s-CoP/Co3O4 performs competitive catalytic activities for both HER and OER.
To further characterize the effect of the NaBH4 reduction process, X-ray absorption spectroscopy (XAS) was employed to characterize the Co K-edge in CoP and s-CoP. The X-ray absorption near-edge spectroscopy (XANES) spectra in Fig. 6a show that the Co K-edge adsorption energy of CoP is lower than that of CoO, yet higher than that of the Co foil, indicating that the average valence in CoP is lower than +2. Moreover, the absorption energy of Co K-edge in s-CoP gets lower relative to CoP, thus verifying the successful reduction of Co species, which does well to the OER process. This conclusion can be further verified by the fitting curve (Fig. S8†), the absorption energy of Co K-edge in s-CoP is lower than that of the fit curve. The Fourier-transformed (FT) k2-weighted extended X-ray absorption fine structure (EXAFS) spectra of CoP and s-CoP at R space are shown in Fig. S9,† the main peak at 1.6 can be corresponded to Co–P coordination shells.26 The peak corresponding to Co–P shifts from 1.62 to 1.65 in reduced CoP, which indicates the prolonged bond length and impaired interaction between Co and P atoms. Therefore, the reduction process decreases the chemical environment of P atoms, thus decreasing the adsorption energy of H to CoP, facilitating the H* desorption process in HER. Moreover, the peak intensity of Co–P decreases obviously in s-CoP, further indicating the increased degree of disorder and the formation of P vacancies.
 | ||
| Fig. 6 (a) X-ray absorption near-edge spectroscopy (XANES) spectra of Co K-edge of CoP, r-CoP and reference materials (Co foil, CoO, Co2O3 and Co3O4), (b) promotion mechanism of unsaturated Co sites towards OER, (c) illustration of the HER process in the alkaline environment on the CoP/Co3O4 electrode. | ||
The effects of s-CoP in HER and OER can be presumed as shown in Fig. 6b and c according to the electro-catalytic performances of the as-prepared materials and previous research projects. In the oxygen evolution process, electron-deficient Co atoms in CoP act as the adsorption sites towards electro-negative reactants and intermediates, thus providing dual active sites towards OER. It has been reported that anionic vacancies will decrease the activation energy of OER,27 thus the P vacancies also act as the active sites towards OER. In the HER process, the catalytic reaction takes place in a tandem pathway,28 in which Co3O4 catalyzes the water dissociation process while the CoP catalyzes the hydrogen evolution process.29 The normalized turnover frequency (TOF) calculations have been involved to verify the speculation. The detailed calculation process has been added to the revised ESI,† where the number of active sites are estimate by ECSA of the as-prepared materials (Fig. S11,† @50 mV s−1). The results in Fig. S13 and S14† show that the normalized TOF of s-CoP/Co3O4 is similar to that of CoP and CoP/Co3O4 while slightly better Co3O4 in OER, indicating that the s-CoP enhances the catalytic performance of the electrode mainly by increasing the density of active sites in OER. Conversely, the intrinsic catalytic activity of active sites in s-CoP/Co3O4 is obviously better than other materials in HER process, suggesting that the involvement of Co3O4 and surface engineering process enhances the catalytic performance of CoP not only by enlarging the density of active sites. Therefore, the s-CoP/Co3O4 shows excellent catalytic activity towards water splitting in alkaline environment.
Conclusions
In this work, layered Co3O4 was synthesized by thermal treatment of Co(OH)2, then the CoP nanolayer was coated on the surface of Co3O4 by electrodeposition, forming a heterojunction structure. The CoP/Co3O4 is further reduced by NaBH4 solution to obtain anionic vacancies at the surface of s-CoP/Co3O4, which was used as the electrode towards water splitting in an alkaline environment. The vacancies cause fewer electro-positive Co atoms, promoting oxygen evolution. It can be presumed that in the HER process, the s-CoP/Co3O4 electrode catalyzes reaction via a tandem reaction pathway and the s-CoP/Co3O4 offers dual active sites in the OER process. The current density of 10 mA cm−2 can be obtained at only 211 and −106 mV vs. RHE for OER and HER, respectively. This study provides a novel strategy to fabricate high-performance noble metal-free electrodes by involving efficient active sites for both OER and HER, which provide remarkable performance for water splitting in an alkaline environment.Experiment section
Chemicals and materials
Cobalt nitrate hexahydrate (Co(NO3)2·6H2O, 98%), potassium hydroxide (KOH, 85%), sodium borohydride (NaBH4, 98%), sodium hypophosphite monohydrate (NaH2PO2·H2O, 98%), sodium citrate (Na3C6H5O7, 98%), potassium chloride (KCl, 98%), ethanol (C2H5OH, 95%) hydrochloric acid (HCl, 35%–37%), nitric acid (HNO3, 98%) potassium hydroxide (KOH, 95%) were obtained from Aladdin Reagents Ltd (China) and used as received without further purification.Materials preparation
Layered Co3O4 was synthesized by thermal treatment of Co(OH)2. Specifically, Co(NO3)2 solution (0.01 g mL−1, 150 mL) was firstly heated to 80 °C under vigorous stirring. Then, 150 mL KOH solution (20 mM) was added dropwise to the Co(NO3)2 solution and kept for 1 h. The solution was filtered and washed using DI water three times after cooling to room temperature. The sediment was dried in a vacuum at 50 °C overnight to obtain Co(OH)2. Finally, the Co(OH)2 powder was transferred to the muffle furnace and annealed for 2 h at 250 °C to obtain Co3O4. The s-Co3O4 was obtained by the surface engineering of Co3O4. Typically, the as-prepared Co3O4 powder (50 mg) was dispersed in 20 mL freshly prepared NaBH4 solution (0.3, 0.5, 0.7 mM) for 1 h and then filtered, washed with DI water three times. The as-prepared powder was dried in vacuum at 50 °C for 24 h to obtain s-Co3O4. The working electrodes were fabricated by supporting the catalysts on the prepared carbon paper (the commercial carbon paper was first cut into pieces of 0.5 × 1 cm2 and washed by sonication for 15 minutes in water, ethanol, and hydrochloric acid consecutively. After that, the carbon paper was pre-functionalized with HNO3). Typically, 4 mg Co3O4 powder and 40 μL of 5 wt% Nafion solution were dispersed in 500 μL water and ethanol mixture solution (Vwater![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Vethanol = 3:2). Then, 20 μL catalyst ink (containing 100 μg catalysts) was loaded onto a carbon paper and dried in the vacuum at 50 °C to obtain the Co3O4/CP electrode. The CoP layer was electro-deposited on Co3O4/CP in 50 mL electrolyte containing 0.8 g Co(NO3)2·6H2O, 0.3 g sodium citrate and 1 g NaH2PO2·H2O at a potential of −1.2 V (vs. Ag/AgCl) for 900 s at room temperature to obtain the CoP/Co3O4/CP electrode. During the electrodeposition process, the Ag/AgCl (3.5 M KCl) was employed as the reference electrode and the graphite rod was the counter electrode. The CoP/Co3O4 electrode was finally sunk in 20 mL freshly prepared NaBH4 solution (0.3, 0.5, 0.7 mM) for 1 h and washed with DI water several times to obtain the s-CoP/Co3O4 working electrode.
:Vethanol = 3:2). Then, 20 μL catalyst ink (containing 100 μg catalysts) was loaded onto a carbon paper and dried in the vacuum at 50 °C to obtain the Co3O4/CP electrode. The CoP layer was electro-deposited on Co3O4/CP in 50 mL electrolyte containing 0.8 g Co(NO3)2·6H2O, 0.3 g sodium citrate and 1 g NaH2PO2·H2O at a potential of −1.2 V (vs. Ag/AgCl) for 900 s at room temperature to obtain the CoP/Co3O4/CP electrode. During the electrodeposition process, the Ag/AgCl (3.5 M KCl) was employed as the reference electrode and the graphite rod was the counter electrode. The CoP/Co3O4 electrode was finally sunk in 20 mL freshly prepared NaBH4 solution (0.3, 0.5, 0.7 mM) for 1 h and washed with DI water several times to obtain the s-CoP/Co3O4 working electrode.
Materials and electrochemical characterization
Raman spectra (Renishaw inVia confocal microscopy), X-ray powder diffraction (XRD, Bruker D2 Phaser), X-ray photoelectron spectroscopy (XPS, K-ALPHA), scan electron microscopy (SEM, QUATTRO S) and transmission electron microscopy (TEM, Philips Tecnai G2F20) were used for the characterization of the obtained materials. X-ray absorption spectroscopy (XAS, NSRRC BL17C1, following a previously published literature procedure30). Electrochemical measurements were performed on a CHI760E, workstation using a conventional three-electrode system with the sample loading on a carbon paper as the working electrode, Hg/HgO (1 M KOH solution) as the reference electrode and graphite rod as the counter electrode. The alkaline electrolyte was a 1 M KOH aqueous solution. All the measured potentials were calibrated to RHE by using the following equation: E(RHE) = E(Hg/HgO) + 0.098 V (25 °C) + 0.059 × pH.Author contributions
Xintong Li and Zonglong Zhu developed the research idea. Xintong Li synthesized the materials, recorded the electrocatalytic performances, analyzed the characterization results and wrote the original version of the manuscript under the supervision of Zonglong Zhu. Yizhe Liu and Qidi Sun assisted with the experimental design and revision of the drafts while Zilong Wang helped to characterize the materials. Wei-Hsiang Huang characterized the as-prepared materials by XAS. Chu-Chen Chueh and Chi-Liang Chen helped to revise and edit the manuscript. Zonglong Zhu offered funding, revised and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the New Faculty Start-up Grant of the City University of Hong Kong (9380086, 9610421), Innovation and Technology Fund (ITS/095/20), the ECS grant (21301319) and GRF grant (11306521) from the Research Grants Council of Hong Kong, Natural Science Foundation of Guangdong Province (2019A1515010761).References
- V. H. Hoa, D. T. Tran and D. C. Nguyen, et al. , Adv. Funct. Mater., 2020, 30, 2002533 CrossRef CAS; L. Zhuang, Y. Jia and H. Liu, et al. , Angew. Chem., Int. Ed., 2020, 59, 14664–14670 CrossRef PubMed; T. Ling, D.-Y. Yan and Y. Jiao, et al. , Nat. Commun., 2016, 7, 12876 CrossRef PubMed.
- Z. Li, J.-Y. Fu and Y. Feng, et al. , Nat. Catal., 2019, 2, 1107–1114 CrossRef CAS; H. Sun, C. Tian and G. Fan, et al. , Adv. Funct. Mater., 2020, 30, 2004375 CrossRef.
- Y. Pi, Y. Xu and L. Li, et al. , Adv. Funct. Mater., 2020, 30, 2004375 CrossRef CAS.
- C. Liang, P. Zou and A. Nairan, et al. , Energy Environ. Sci., 2020, 13, 86–95 RSC; B. Shan, M. K. Brenneman and L. Troian-Gautier, et al. , J. Am. Chem. Soc., 2019, 141, 10390–10398 CrossRef CAS PubMed.
- M. A. Ahsan, A. R. P. Santiago and Y. Hong, et al. , J. Am. Chem. Soc., 2020, 142, 14688–14701 CrossRef CAS PubMed.
- H. Huang, A. Cho and S. Kim, et al. , Adv. Funct. Mater., 2020, 30, 2003889 CrossRef CAS.
- J. Yin, J. Jin and M. Lu, et al. , J. Am. Chem. Soc., 2020, 142, 18378–18386 CrossRef CAS PubMed.
- K. Yuan, D. Luetzenkirchen-Hecht and L. Li, et al. , J. Am. Chem. Soc., 2020, 142, 2404–2412 CrossRef CAS PubMed.
- H. Huang, S. Zhou and C. Yu, et al. , Energy Environ. Sci., 2020, 13, 545–553 RSC; Y. Guo, J. Tang and J. Henzie, et al. , ACS Nano, 2020, 14, 4141–4152 CrossRef CAS PubMed; Q. Lian, L. Zhong and C. Du, et al. , ACS Nano, 2019, 13, 7975–7984 CrossRef PubMed.
- Q. Shi, Q. Liu and Y. Ma, et al. , Adv. Energy Mater., 2020, 10, 2002896 CrossRef CAS; L. L. Ji, J. Y. Wang and X. Teng, et al. , ACS Catal., 2020, 10, 412–419 CrossRef; Z. Y. Zhang, X. X. Zhao and S. B. Xi, et al. , Adv. Energy Mater., 2020, 10, 1902854 Search PubMed; Y. Liu, X. Wu and X. Guo, et al. , Mater. Today Energy, 2021, 19, 100610 CrossRef.
- H. W. Huang, S. Zhou and C. Yu, et al. , Energy Environ. Sci., 2020, 13, 545–553 RSC.
- A. Moysiadou, S. Lee and C. S. Hsu, et al. , J. Am. Chem. Soc., 2020, 142, 11901–11914 CrossRef CAS PubMed; J. Liu, Y. Ji and J. Nai, et al. , Energy Environ. Sci., 2018, 11, 1736–1741 RSC; M. Kuang, J. Zhang and D. Liu, et al. , Adv. Energy Mater., 2020, 10, 2002215 CrossRef.
- B. Zhang, L. Wang and Z. Cao, et al. , Nat. Catal., 2020, 3, 985–992 CrossRef CAS; P. W. Menezes, C. Panda and C. Walter, et al. , Adv. Funct. Mater., 2019, 29, 1808632 CrossRef.
- R. Zhang, Y.-C. Zhang and L. Pan, et al. , ACS Catal., 2018, 8, 3803–3811 CrossRef CAS; Y. Wang, T. Zhou and K. Jiang, et al. , Adv. Energy Mater., 2014, 4, 1400696 CrossRef; K.-Y. Niu, F. Lin and S. Jung, et al. , Nano Lett., 2015, 15, 2498–2503 CrossRef PubMed; L. Xu, Q. Jiang and Z. Xiao, et al. , Angew. Chem., Int. Ed., 2016, 55, 5277–5281 CrossRef PubMed.
- G. Zhou, M. Li and Y. Li, et al. , Adv. Funct. Mater., 2020, 30, 1905252 CrossRef CAS.
- L. Ji, J. Wang and X. Teng, et al. , ACS Catal., 2020, 10, 412–419 CrossRef CAS.
- X.-W. Lv, Y. Liu and R. Hao, et al. , ACS Appl. Mater. Interfaces, 2020, 12, 17502–17508 CrossRef CAS PubMed.
- X. Wang, X. Li and J. Mu, et al. , ACS Appl. Mater. Interfaces, 2019, 11, 41988–41999 CrossRef CAS PubMed.
- C. Tang, R. Zhang and W. B. Lu, et al. , Adv. Mater., 2017, 29, 1602441 CrossRef CAS PubMed; Z. Xiao, Y. Wang and Y.-C. Huang, et al. , Energy Environ. Sci., 2017, 10, 2563–2569 RSC.
- G. Dong, H. Hu and X. Huang, et al. , J. Mater. Chem. A, 2018, 6, 21003–21009 RSC; Q. Wang, X. Xue and Y. Lei, et al. , Small, 2020, 16, 2001571 CrossRef CAS PubMed.
- Z.-H. Xue, H. Su and Q.-Y. Yu, et al. , Adv. Energy Mater., 2017, 7, 1602355 CrossRef.
- Z. Li, Y. Zhang and Y. Feng, et al. , Adv. Funct. Mater., 2019, 29, 2000364 Search PubMed; Y. Liu, C. Ma and Q. Zhang, et al. , Adv. Mater., 2019, 31, 1900062 CrossRef PubMed.
- R. Boppella, J. Tan and W. Yang, et al. , Adv. Funct. Mater., 2019, 29, 1807976 CrossRef.
- X. Zhou, H. Gao and Y. Wang, et al. , J. Mater. Chem. A, 2018, 6, 14939–14948 RSC.
- Z. Chen, Y. Ha and H. Jia, et al. , Adv. Energy Mater., 2019, 9, 1807976 Search PubMed; M. Li, Y. Zhu and H. Wang, et al. , Adv. Energy Mater., 2019, 9, 1803918 CrossRef.
- E. Cao, Z. Chen and H. Wu, et al. , Angew. Chem., Int. Ed., 2020, 59, 4154–4160 CrossRef CAS PubMed.
- Z. Xiao, Y.-C. Huang and C.-L. Dong, et al. , J. Am. Chem. Soc., 2020, 142, 12087–12095 CrossRef CAS PubMed.
- C. Tang, Y. Zheng and M. Jaroniec, et al. , Angew. Chem., Int. Ed., 2021, 60, 2–21 CrossRef.
- S. Anantharaj, S. Noda and V. R. Jothi, et al. , Angew. Chem., Int. Ed, 2021, 60, 2–28 CrossRef.
- W.-H. Huang, W.-N. Su and C.-L. Chen, et al. , Appl. Surf. Sci., 2021, 562, 1873–5584 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr06044a |
| This journal is © The Royal Society of Chemistry 2021 |