
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible single-crystal to single-crystal phase transformation between a new Werner clathrate and its apohost†
Catiúcia R. M. O.
Matos
 ab,
Rana
Sanii
a,
Shi-Qiang
Wang
a,
Célia M.
Ronconi
b and
Michael J.
Zaworotko
*a
ab,
Rana
Sanii
a,
Shi-Qiang
Wang
a,
Célia M.
Ronconi
b and
Michael J.
Zaworotko
*a
aDepartment of Chemical Sciences, Bernal Institute, University of Limerick, Limerick V94 T9PX, Republic of Ireland. E-mail: Michael.Zaworotko@ul.ie
bDepartment of Inorganic Chemistry, Universidade Federal Fluminense (UFF), Outeiro de São João Batista, s/n, Campus do Valonguinho, Centro, 24020-141, Niterói, RJ, Brazil
First published on 23rd August 2021
Abstract
In this work, we report the synthesis and structural characterisation of the ligand 2-(pyridin-3-yl)-benzo[de]isoquinoline-1,3(2H)-dione, 5, its isostructural Werner complexes ML4(NCS)2 (L = 5; M = Co(II) and Ni(II)), and five clathrates with three aromatic guests, ML4(NCS)2·2G (M = Co(II) and Ni(II), G = nitrobenzene (NB); M = Co, G = 1,2-dichlorobenzene (1,2-DCB); M = Co(II) and Ni(II), G = o-xylene (OX)). 5 was prepared in high yield by condensation in the solid-state (C3S3, Cocrystal Controlled Solid-State Synthesis). The Werner complexes ML4(NCS)2 (M = Co(II) and Ni(II)) (apohosts) were prepared by reacting M(NCS)2 (M = Co(II) and Ni(II)) and 5 in 1-butanol at 60 °C for 24 h. The Werner clathrates were prepared by reacting M(NCS)2 (M = Co(II) and Ni(II)), G and 5 in 1-butanol at 60 °C for 48–96 h. The clathrates were observed to transform to the apohost ML4(NCS)2 upon heating. CoL4(NCS)2·2NB was subsequently regenerated by exposing CoL4(NCS)2 to liquid NB at 60 °C for 48 h. This phase change occurred as a single-crystal to single-crystal phase transformation and was studied by single crystal X-ray diffraction, powder X-ray diffraction and thermal analyses. Structural analyses of the apohost CoL4(NCS)2 and its Werner clathrate CoL4(NCS)2·2NB indicated that rotational freedom of the Co-N bonds together with torsional flexibility of the ligand between the imide bond and the pyridine moiety are key to enabling the structural switching induced by exposure to NB or its removal.
Introduction
Werner complexes of general formula ML4X2 are historically important in the context of coordination chemistry1 and are exemplified by octahedral transition metal (M) complexes that involve coordination of four equatorial aromatic N-donor ligands (L) and two axial anionic ligands (X). Powell introduced the concept of “clathrate” in the 1940s to classify compounds in which guest molecules are trapped within a void present in the structure of host compound.2,3 By the late 1950s it had been recognised that Werner complexes can form clathrates of general formula ML4X2·nG (G = guest).4–6 Schaeffer reported that the Werner complex Ni(4-MePy)4(NCS)2, (4-MePy = 4-methylpyridine) forms clathrates with xylenes, cymenes and methylnaphthalenes and that selective clathration enables separation of aromatic hydrocarbons.4 Several other groups subsequently studied the inclusion chemistry of Werner complexes. Radzitzk investigated Werner complexes containing a variety of primary substituted benzylamine ligands, e.g. Ni(α-propylbenzylamine)4(NCS)2, and their selective clathration of mono- and poly-substituted benzenes and naphthalenes.5 Williams studied the clathrates of M(4-MePy)4(NCS)2 (M = Fe, Co, Ni) formed in the presence of mixtures of dichlorobenzenes or methylstyrenes and observed selectivity towards the respective para isomers.6 In the 1980s, Lipkowski detailed how Ni(4-MePy)4(NCS)2 forms clathrates with 1-bromonaphthalene and azulene.7 Lipkowski's group also studied clathrate formation in terms of kinetics and thermodynamic stability for Ni(4-MePy)4(NCS)2.8 The term “Werner clathrate” was coined by Nassimbeni and co-workers in the 1980s,9–12 who explored the versatility of [Ni(NCS)2(4-MePy)4] complexes through substitution of the pyridine ligands and/or counterions and their clathrates with xylenes and other guests.13,14The porous (open) phases of the Werner clathrates are typically crystallised by forming a Werner complex in the presence of guest molecules or through exposure of an as-synthesised non-porous (closed) Werner complex to solid–liquid and/or solid–vapour reactions with guests.15 The inclusion of guest molecules within the structures of Werner clathrates can be classified into four categories according to their crystal packing motifs: (i) densely packed non-porous, α-phase; (ii) porous β-phase, a cage-type clathrate; (iii) porous δ-phase, a channel-type clathrate and (iv) porous γ-phase, a layer-type clathrate.10,16,17
An important feature of Werner complexes is the rotational freedom of metal-N bonds.11 For substituted pyridine ligands there can be additional rotational freedom for the Werner complex thanks to torsional flexibility at the substituent.13,18 In essence, this is what enables a Werner complex to adjust its shape to accommodate a variety of guests of different sizes and shapes11 while also facilitating the reverse phase transformation through removal of guest molecules. The potential utility of Werner complexes in separations is critically dependent upon both the selectivity of Werner clathrates towards the components of a mixture and whether or not guest uptake/removal is reversible.19–27
A survey of the literature has revealed that some Werner complexes can exhibit reversible sorption isotherms between their non-porous and porous phases. We recently termed such complexes Switching Adsorbent Molecular Materials (SAMMs).15,16,28,29 Structural switching between non-porous and porous phases in SAMMs is demonstrated by a single-step or “Type F-IV”30 sorption isotherms. Such isotherms are of particular interest because which can offer higher working capacities than rigid porous materials, which tend to exhibit Type I isotherms.31,32 To our knowledge, the first reversible sorption isotherm for a Werner complex was reported in 1969 by Allison and Barrer,31 who investigated the sorption properties of Co(4-ethylpyridine)4(NCS)2, SAMM-1-Co-NCS, when exposed to benzene, toluene or C8 isomer.31SAMM-2-Cu-PF6 (2 = 4-methylpyridine) was studied by Nakamura et al.28 and found to form clathrates with guests such as acetone, butanone and benzene. Sorption isotherms were reported for benzene and CO2. Nakamura et al. also reported on SAMM-4-Cu-PF6 and SAMM-4-Cu-BF4 (4 = pyridine) and their acetone and acetonitrile vapour sorption isotherms.29 Barbour et al. studied SAMM-3-Ni-NCS (3 = 4-phenylpyridine) and its clathrates with o-xylene (OX), p-xylene (PX) and m-xylene (MX). SAMM-3-Ni-NCS was found to exhibit the highest selectivity observed for the three xylene isomers at that time.16 Sorption isotherms of benzene and toluene were also reported.16 Recently, we reported that SAMM-3-Cu-OTf switches between non-porous and porous phases upon exposure to OX but not when exposed to other C8 isomers at high partial pressure.33
That Werner complexes and related materials, such as flexible porous coordination networks, can exhibit selective capture of organic compounds34,35 is a topical subject thanks to the lack of scientific understanding about what drives switching phenomena and the potential utility for separations. With respect to coordination networks, we and others36–40 have demonstrated that square lattice topology coordination networks based upon Werner complexes connected by linker ligands41 can exhibit reversible phase transformations when exposed to a range of guests. In addition, metal complexes that undergo structural switching and reversible isotherms triggered by a guest molecule remain quite rare. A survey of the Cambridge Structural Database (CSD, ConQuest 2020 2.0, CSD v5.41 + August 2020 update)42 revealed that 345 octahedral metal complexes containing pyridine-based terminal ligands are archived but only six are SAMMs that are known to exhibit reversible clathrate formation (CSD database search details, ESI†). SAMMs based upon Werner complexes therefore remain an understudied class of compounds and represent an attractive platform for structure–function studies thanks in part to the ready availability of many N-donor ligands that in turn allow for creation of families of closely related complexes. In addition, we note that Werner complexes offer synthetic simplicity and structural diversity in comparison to most coordination networks. Our specific interest in the study of Werner complexes lies with properties, especially the study of Werner complexes that exhibit pore adaptive behaviour that can result in selectivity towards a specific guest.32,33 Such behaviour opens the opportunity to generate high working capacity sorbents for selective separation by enclathration.
In this contribution, we report the solid-state synthesis 2-(pyridin-3-yl)-benzo[de]isoquinoline-1,3(2H)-dione, 5, and its use as a Werner complex ligand. The formation of imides from Cocrystal Controlled Solid-State Synthesis (C3S3) has been previously exploited by our group as a mechanochemical approach to prepare ligands43–45 and typically involves solvent-drop grinding (SDG)46–48 followed by heating. Advantages of C3S3 include the following: (i) little solvent is required, especially compared to solution-based synthesis methods; (ii) yields are typically high, often approaching quantitative; (iii) new condensation products can be readily accessible from simple organic building blocks.49
Results and discussion
Synthesis of ligand 5
(2-(Pyridin-3-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione), 5, was previously reported as an intermediate of an N-oxide organic ligand obtained by microwave-assisted synthesis50 and has been used to form Eu(III), Gd(III)50 and Ru(II) complexes51 but is otherwise unstudied. We prepared 5 from condensation of 1,8-naphthalene anhydride (1,8-NA) and the 3-amino-pyridine (3APy) using C3S3 (Fig. 1a).43,44 A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ground mixture of 1,8-NA and 3APy formed an intermediate via SDG when using 120 μL of EtOH. The imide was obtained by heating the intermediate at 185 °C for 5 h, affording 5 in 93% yield (Fig. 1a). In the synthesis of 5, the selection of EtOH was guided by solvent selection guidelines developed by and for industry.52 To our knowledge, the synthesis of 5 through mechanochemistry has not been previously reported.
:1 ground mixture of 1,8-NA and 3APy formed an intermediate via SDG when using 120 μL of EtOH. The imide was obtained by heating the intermediate at 185 °C for 5 h, affording 5 in 93% yield (Fig. 1a). In the synthesis of 5, the selection of EtOH was guided by solvent selection guidelines developed by and for industry.52 To our knowledge, the synthesis of 5 through mechanochemistry has not been previously reported.
 | ||
| Fig. 1 (a) Synthesis of 5 was accomplished using Cocrystal Controlled Solid-State Synthesis (C3S3) approach via solvent-drop grinding (SDG). (b) Preparation of SAMM-5-Co-NCS and its Werner clathrates with nitrobenzene, NB, SAMM-5-Co-NCS·NB; o-xylene, OX, SAMM-5-Co-NCS·OX and 1,2-dichlorobenzene, 1,2-DCB, SAMM-5-Co-NCS·(1,2-DCB). | ||
Synthesis of Werner complexes and clathrates
5 and M(NCS)2 (M = Co(II) and Ni(II)) were dissolved by heating in 1-butanol or 1-butanol/MeOH and layered to form the Werner complexes reported herein. Reaction of 5 with Co(NCS)2 in 1-butanol at 60 °C afforded pink crystals of the apohost, SAMM-5-Co-NCS (Fig. 1b, 3a, b and Scheme S1†), whereas reaction with Ni(NCS)2 in 1-butanol/MeOH afforded green crystals of SAMM-5-Ni-NCS (Scheme S1†). SAMM-5-Ni-NCS was also obtained from heating the Werner clathrates of Ni(II) with OX and NB.(Fig. S17a and b†) Reaction of 5 with M(NCS)2 (M = Co(II) or Ni(II)) in NB with 1-butanol or 1-butanol/MeOH, afforded the isostructural Werner clathrates SAMM-5-Co-NCS·NB and SAMM-5-Ni-NCS·NB as pink and green crystals, respectively (Fig. 1b, 3c, d, S17c, d and Scheme S1†). A detailed description of the structure of SAMM-5-Co-NCS·NB is presented below.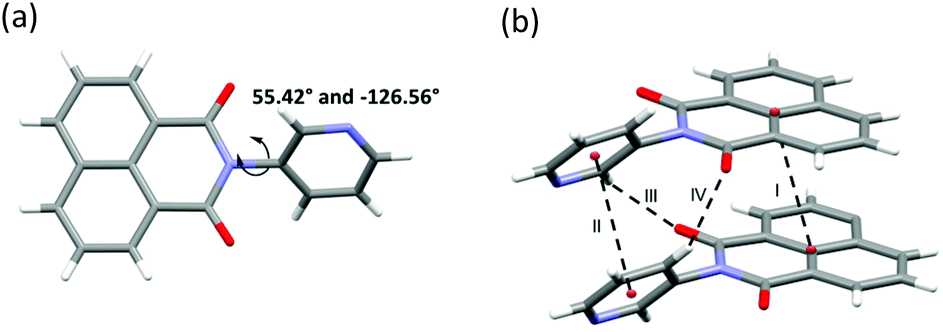 | ||
| Fig. 2 (a) Asymmetric unit of 5 and its torsion angles. (b) Supramolecular interactions I, II, III and IV. | ||
 | ||
| Fig. 3 Crystal structure of the: α-phase of SAMM-5-Co-NCS representing (a) the coordination environment around Co2+ cations; (b) crystal packing of SAMM-5-Co-NCS molecules viewed along the a-axis; δ-phase of SAMM-5-Co-NCS·NB representing (c) the coordination environment around Co2+ cations; (d) crystal packing viewed along the a-axis; δ-phase of SAMM-5-Co-NCS·(1,2-DCB) representing (e) the coordination environment around Co2+ centre; (f) view of the crystal packing through c-axis; δ-phase of SAMM-5-Co-NCS·OX representing (g) the coordination environment around Co2+ centre; (h) view of the crystal packing through a-axis. | ||
Reaction of 5 and Co(NCS)2 in a mixture of 1-butanol and 1,2-DCB formed SAMM-5-Co-NCS·(1,2-DCB) as pink crystals suitable for SCXRD (Fig. 1b, 3e, f and Scheme S1†). Reaction of 5 with M(NCS)2 (M = Co(II) or Ni(II)) in a mixture of OX, 1-butanol or 1-butanol/MeOH afforded the isostructural Werner clathrates SAMM-5-Co-NCS·OX and SAMM-5-Ni-NCS·OX (Fig. 1b, 3g, h and Scheme S1†). Only the structural details of SAMM-5-Co-NCS·OX are discussed in detail herein. Our initial attempts to obtain Werner clathrates using OX at 60 °C for 48 h afforded SAMM-5-Co-NCS and SAMM-5-Ni-NCS with some crystals of SAMM-5-Co-NCS·OX and SAMM-5-Ni-NCS·OX, respectively (Fig. S9a†). However, reactions at room temperature for 48 h resulted in the formation of the clathrate phases SAMM-5-Co-NCS·OX and SAMM-5-Ni-NCS·OX as bulk products (Fig. S9b†). Attempts to obtain Werner clathrates at 60 °C using ethylbenzene (EB), methylnaphthalene (MN), meta-xylene (MX) and para-xylene (PX) were unsuccessful and resulted instead in isolation of SAMM-5-Co-NCS. SAMM-5-Co-NCS·EB was obtained at room temperature and found to be isostructural with SAMM-5-Co-NCS·OX. Full characterisation details are provided in ESI.†
Structural analyses
SCXRD studies revealed that 5 crystallised as an anhydrate in the orthorhombic space group P212121 with one molecule of 5 comprising the asymmetric unit (Fig. 2a and Table S2, ESI†). The torsional flexibility of the imide bond allowed for the pyridyl ring to be twisted by 55.42° and −126.56° (torsion angles) with respect to the imide moiety. A number of CH⋯π, CH⋯N and CH⋯O contacts, π⋯π stacking directed the crystal packing (Fig. 2b).SAMM-5-Co-NCS crystallised in the monoclinic space group P21/n (Table S3 and Fig. S17a and b, ESI†) with two crystallographically independent molecules of 5, one Co2+ cation on a special position, and one independent NCS− anion comprising the asymmetric unit (Fig. S13a, ESI†). The crystal structure of SAMM-5-Co-NCS revealed that each Co2+ cation adopts octahedral coordination geometry with nitrogen atoms from four different ligands forming the equatorial plane and Co–N bond distances ranging from 2.218(6) to 2.293(7) Å. Octahedral coordination is completed by two NCS− anions at the axial positions of the Co2+ with Co–N bond distances of 2.055(9) Å (Fig. 3a). These distances are in good agreement with related coordination compounds.53–55 As mentioned earlier, Werner complexes and clathrates can be classified into four categories, α, β, δ or γ, according to the type of crystal packing.10,16,17SAMM-5-Co-NCS and SAMM-5-Ni-NCS are therefore classified as α-phases since they are densely-packed and non-porous (Fig. 3a, b and Fig. S17a, b†).10 Intermolecular interactions include CH⋯π, CH⋯N and CH⋯O contacts along with π⋯π stacking (Table S5, ESI†).
SAMM-5-Co-NCS·NB crystallised in the monoclinic space group P21/n, whereas SAMM-5-Co-NCS·OX adopted the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Fig. 3c, g, S17, S18, Tables S3 and S6, ESI†). The asymmetric unit of each Werner clatharate contains two crystallographically independent molecules of 5, one Co2+ cation at a special position, one coordinated NCS— anion and one guest molecule (NB or OX) (Fig. S13, ESI†). The octahedral geometries around Co2+ are retained in SAMM-5-Co-NCS·NB and SAMM-5-Co-NCS·OX, with Co–N bond distances ranging from 2.055 (18) to 2.213(19) Å. The structural analysis revealed that NB or OX molecules lie in channels of SAMM-5-Co-NCS·NB and SAMM-5-Co-NCS·OX, respectively, classifying them as porous δ-phases (Fig. 3d and h). The rotational freedom about Co–N bonds, as well as the flexibility between the imide bond and the pyridine moiety of 5, provided two distinct conformations and crystal packing for each δ-phase (Fig. 3c and g). Intermolecular interactions in both δ-phases are governed by CH⋯π and CH⋯O contacts as well as π⋯π stacking enabled by the structural flexibility of 5 (ESI†). When single crystals of SAMM-5-Co-NCS·NB and SAMM-5-Ni-NCS·NB were stored at room temperature for two days they underwent polymorphic transformations from P21/n to C2/m (Tables S3 and S4, ESI†). Both polymorphs can also be classified as δ-phases. In the P21/n phases, NB molecules lie within 1D channels (Fig. 3c, d and S17a, b†) whereas in the C2/m phases NB molecules are disordered inside the channels. SAMM-5-Co-NCS·(1,2-DCB) was found to be isostructural to the C2/m phases of NB clathrates with disordered 1,2-DCB molecules inside the channels (Fig. 3e, f and Table S4, ESI†).
(Fig. 3c, g, S17, S18, Tables S3 and S6, ESI†). The asymmetric unit of each Werner clatharate contains two crystallographically independent molecules of 5, one Co2+ cation at a special position, one coordinated NCS— anion and one guest molecule (NB or OX) (Fig. S13, ESI†). The octahedral geometries around Co2+ are retained in SAMM-5-Co-NCS·NB and SAMM-5-Co-NCS·OX, with Co–N bond distances ranging from 2.055 (18) to 2.213(19) Å. The structural analysis revealed that NB or OX molecules lie in channels of SAMM-5-Co-NCS·NB and SAMM-5-Co-NCS·OX, respectively, classifying them as porous δ-phases (Fig. 3d and h). The rotational freedom about Co–N bonds, as well as the flexibility between the imide bond and the pyridine moiety of 5, provided two distinct conformations and crystal packing for each δ-phase (Fig. 3c and g). Intermolecular interactions in both δ-phases are governed by CH⋯π and CH⋯O contacts as well as π⋯π stacking enabled by the structural flexibility of 5 (ESI†). When single crystals of SAMM-5-Co-NCS·NB and SAMM-5-Ni-NCS·NB were stored at room temperature for two days they underwent polymorphic transformations from P21/n to C2/m (Tables S3 and S4, ESI†). Both polymorphs can also be classified as δ-phases. In the P21/n phases, NB molecules lie within 1D channels (Fig. 3c, d and S17a, b†) whereas in the C2/m phases NB molecules are disordered inside the channels. SAMM-5-Co-NCS·(1,2-DCB) was found to be isostructural to the C2/m phases of NB clathrates with disordered 1,2-DCB molecules inside the channels (Fig. 3e, f and Table S4, ESI†).
Investigation of the reversibility of phase transformations
We next investigated whether or not the δ-phase (SAMM-5-Co-NCS·NB) can revert back to the α-phase (SAMM-5-Co-NCS) in a SC to SC phase transformation. Thermogravimetric Analysis (TGA) of the as-synthesised crystals of SAMM-5-Co-NCS·NB revealed a weight loss of 15% between 152 and 188 °C consistent with the loss of two NB molecules per formula unit (Fig. 6). A single crystal of as-synthesised SAMM-5-Co-NCS·NB was heated at 185 °C for 1 h to remove NB (process I, Fig. 4a and b). PXRD and SCXRD data from the single crystal obtained after heating were consistent with as-synthesised SAMM-5-Co-NCS (Fig. 5 and Table S7, ESI†). Single crystals of SAMM-5-Co-NCS as obtained from process I were then soaked in NB at 60 °C for 48 h (process II, Fig. 4a and b). The PXRD patterns of the single crystals obtained after soaking in NB matched well with the calculated and experimental PXRD patterns of as-synthesised SAMM-5-Co-NCS·NB (Fig. 5). TGA (Fig. 6) and SCXRD results also support the reversibility of process II (Fig. 4a and b and Table S7, ESI†). A video recording of a single crystal of SAMM-5-Co-NCS in contact with liquid NB revealed evolution of bubbles from the crystal surface, which we attribute to the release of gases trapped in cavities during transformation from non-porous to porous phases (ESI†). SAMM-5-Co-NCS prepared from 5 and Co(NCS)2 in 1-butanol also formed SAMM-5-Co-NCS·NB when soaked in NB at 60 °C for 48 h (process II) (Fig. S20, ESI†). However, no phase transformation occurred following exposure of SAMM-5-Co-NCS to NB vapour at 60 °C for 48 h or after soaking in OX, MX, PX or EB at 60 °C for 48 h (Fig. S21a, ESI†). As-synthesised SAMM-5-Ni-NCS did not undergo phase transformation when soaked in NB at 60 °C for 96 h or after soaking in OX, MX, PX or EB for 48 h. PXRD revealed that SAMM-5-Ni-NCS remains after soaking in NB. (Fig. S21b and S22a, ESI†). The δ-phase SAMM-5-Ni-NCS·NB transformed to the α-phase of SAMM-5-Ni-NCS upon heating at 185 °C for 1 h (Fig. S21b and S22b, ESI†). SAMM-5-Ni-NCS did not undergo a reversible phase transformation upon soaking in liquid NB at 60 °C for 72 h as indicated by the TGA curve of a sample that had been soaked in NB (Fig. S21b and S22b, ESI†). Single crystals of the δ-phase SAMM-5-Co-NCS·1,2DCB prepared from 5, Co(NCS)2 and 1,2-DCB transformed to SAMM-5-Co-NCS upon heating at 200 °C for 30 min (Fig. S23a ESI†). The reverse structural transformation did not occur following soaking in 1,2-DCB at 60 °C for 48 h according to TGA and PXRD data (Fig. S23a and b, ESI†). | ||
| Fig. 4 (a) Reversible SC-SC phase transformation occurs upon heating single crystals of the δ-phase SAMM-5-Co-NCS·NB at 185 °C to remove NB (process I, removal) followed by soaking crystals of the α-phase SAMM-5-Co-NCS in NB to regenerate SAMM-5-Co-NCS·NB (process II, uptake). (b) Images of a single crystal subjected to processes I and II. | ||
 | ||
| Fig. 5 PXRD pattern of (a) calculated SAMM-5-Co-NCS generated from SCXRD; (b) experimental SAMM-5-Co-NCS·NB after heating at 185 °C resulting in α-phase SAMM-5-Co-NCS; (c) calculated SAMM-5-Co-NCS·NB generated from SCXRD; (d) experimental SAMM-5-Co-NCS·NB as-synthesised; and (e) SAMM-Co-NCS·NB obtained after soaking α-phase SAMM-5-Co-NCS in NB at 60 °C resulting in δ-phase. | ||
 | ||
| Fig. 6 TGA of the (a) as-synthesised δ-phase SAMM-5-Co-NCS·NB; (b) α-phase SAMM-5-Co-NCS obtained by heating δ-phase SAMM-5-Co-NCS·NB at 185 °C 1 h−1 and (c) δ-phase SAMM-5-Co-NCS·NB obtained after soaking α-phase SAMM-5-Co-NCS in NB at 60 °C 48 h−1. | ||
A single crystal of the δ-phase SAMM-5-Co-NCS·OX prepared from 5, Co(NCS)2 and OX transformed to SAMM-5-Co-NCS upon exposure to ambient conditions for 12 h (Table S7, ESI†). This phase transformation can also be induced by heating crystals of SAMM-5-Co-NCS·OX and SAMM-5-Ni-NCS·OX at 200 °C for 1 h, resulting in the formation of SAMM-5-Co-NCS and SAMM-5-Ni-NCS, respectively (Scheme S1, Fig. S9b, ESI†). After heating, SAMM-5-Ni-NCS was washed with MeOH to remove residual 5, while SAMM-5-Co-NCS required no additional purification (Fig. S24, ESI†). The reverse transformation did not occur following soaking of the regenerated phases of SAMM-5-Co-NCS and SAMM-5-Ni-NCS in OX at 60 °C for 48 h. Rather, SAMM-5-Co-NCS and SAMM-5-Ni-NCS were isolated, respectively (Fig. S24, ESI†). Therefore, among the Werner complexes reported herein, only the NB clathrate of SAMM-5-Co-NCS exhibited reversible transformations analogous to those seen in other Werner complexes and related coordination networks.56,57
Structural flexibility of Werner complexes and the enclathration process
The rotational freedom of the Co–N bonds and the additional rotational freedom around the imide moiety is a feature of SAMM-5-Co-NCS that is evident from the structural results and could be key to enabling reversible transformations.11 Fig. S25† reveals the torsion angles between the nitrogen atom from the pyridyl group and the Co2+ cation (Co–N bonds). For the α-phase of SAMM-5-Co-NCS, there are two sets of torsion angles ranging from 8.27° to 9.16° and from −80.13° to 99.87°. In the δ-phase SAMM-5-Co-NCS·NB, torsion angles about the Co–N bond were found to range from 63.57° to −116.46° and from 64.64° to 115.36°. The δ-phase of SAMM-5-Co-NCS·OX exhibits two sets of torsion angles ranging from 81.80° to 98.20° and from 57.69° to −122.31° (Fig. S25, ESI†). For the δ-phase SAMM-5-Co-NCS·OX, one set of torsion angles about Co-N bonds (−81.80° to 98.20°) are close to those encountered in the corresponding α-phase SAMM-5-Co-NCS (−80.13° to 99.87°) (Fig. S25, ESI†). Fig. S26† reveals the torsion angles about the C–N bond between pyridyl and imide group for each ligand of the Werner complex. In the α-phase SAMM-5-Co-NCS, the torsion angles about C–N bonds are ±51.93°/129.89° and ±70.60°/109.80°. In the δ-phase SAMM-5-Co-NCS·NB, the torsion angles about C–N bonds are ±60.74°/119.18° and ±63.27°/116.82°, while in the δ-phase SAMM-5-Co-NCS·OX the torsion angles are twisted by ±64.18°/114.50° and ±76.59°/101.74°.Conclusions
In conclusion, ligand 5 was synthesised in high yield by mechanochemistry and formed new Werner complexes that can exist as δ-phase Werner clathrates with channels that contain NB, 1,2-DCB or OX. Regarding the ligand, its potential for widespread use is good given that it is facile to prepare using simple and low-cost starting materials. Likewise, both the Werner complexes and the clathrates reported herein can be synthesised under mild conditions. A reversible transformation from the porous δ-phase SAMM-5-Co-NCS·NB to its non-porous α-phase SAMM-5-Co-NCS occurred via a SC-SC process, making it a rare example of a Werner clathrate that undergoes switching between non-porous and porous phases. Whereas earlier examples are known to readily undergo phase transformation via liquid or vapour contact with multiple guests, SAMM-5-Co-NCS transformed only in the presence of liquid NB. The isostructural analogue SAMM-5-Ni-NCS did not transform to its NB porous δ-phase in the same manner. The torsion angles about Co-N bonds or the C–N bonds from the imide moiety in the structures of SAMM-5-Co-NCS·NB, SAMM-5-Co-NCS·OX and SAMM-5-Co-NCS can help to explain why SAMM-5-Co-NCS reversibly switches from porous to non-porous phase in the presence of NB, although it did not undergo any structural switching in the presence of the other guests. In the case of SAMM-5-Co-NCS·NB, enclathrated NB molecules induced higher structural distortion in the host compared to SAMM-5-Co-NCS·OX. The OX Werner clathrates exhibited one set of torsion angles analogous to the guest-free SAMM-5-Co-NCS, which means that the OX molecules induced smaller structural distortion, resulting in an irreversible adsorption process in SAMM-5-Co-NCS. We consider that the inherent torsional flexibility of the Werner complex with respect to its Co-N bonds and the ligand with respect to its imide moiety play important roles in enabling the observed reversible phase transformation.Experimental section
General aspects
Reagents and solvents were purchased from Sigma-Aldrich, Fluorochem, Alpha or TCL and used without further purification. C3S3 reaction was performed using agate mortar and pestle. 1H and 13C Nuclear Magnetic Resonance (NMR) spectra of 5 were recorded on a Jeol ECX400 at a frequency of 400 and 100 MHz, respectively. PXRD of the Werner complexes, clathrates and the reversible phase transformation in SAMM-5-Co-NCS·NB to SAMM-5-Co-NCS were acquired on Empyrean diffractometer (PAN-analytical, Philips) using CuKα (λ = 1.54178 Å) source. PXRD data of 5, (SAMM-5-Co-NCS and SAMM-5-Ni-NCS after soaking in OX) and (SAMM-5-Co-NCS after removal and soaking in 1,2-DCB) were acquired on Proto AXRD Benchtop Powder Diffractometer. TGA curves were performed on a TA Instrument Q50 TG under flow of N2 with heating rate of 10 °C min−1. The balance purge was 40 mL min−1 and the sample purge was 60 mL min−1 of N2. Differential scanning calorimetry (DSC) analyses were carried out on a TA Instrument DSC Q20 under a sample purge of 50 mL min−1 of N2 with the heating rate of 10 °C min−1 for all compounds. Fourier Transform Infrared (FTIR) spectra of 5, Werner complexes and clathrates were collected on a PerkinElmer Spectrum 100 spectrometer with ATR accessory.X-ray crystallography
Crystal structures of all compounds were determined by SCXRD using either MoKα or CuKα radiation in either Bruker D8 Quest fixed-chi diffractometer equipped with Photon II or Photon 100 detector, respectively. The unit-cells, data reduction and absorption correction (multi-scan method) were conducted using APEX358 suit (Bruker) including SADABS software.59 Space groups were determined using XPREP60 implemented in APEX3. Structures were solved using intrinsic phasing method (SHELXT)61 and refined on F2 using nonlinear least-squares techniques with SHELXL62 contained in OLEX2 v1.2.8 programs packages.63 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were added to the structure in idealised positions and were further refined according to the riding model. All crystal structures have been deposited with the Cambridge Crystallographic Data Centre (CCDC 1995785–1995796 and 2065192).:1 mixture of CH2Cl2 (1.0 mL) and MeOH (1.0 mL). The final solution was sonicated for one min. Instantly suitable needles precipitated from the solution.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge Science Foundation Ireland (16/IA/B2549, MJZ), the European Research Council (ADG 885695, MJZ), the Brazilian Federal Agency for Support and Evaluation of Graduate Education (CAPES) for a fellowship to Catiúcia R. M. O. Matos, the Brazilian agency National Council for Scientific and Technological Development (CMR, CNPq research fellowship grant number 313799/2017-2) and the Rio de Janeiro Research Foundation (FAPERJ, Cientistas do Nosso Estado grant number E-26/202.629/2019, CMR).Notes and references
- J. Lipkowski, in Inclusion Compounds: Structural Aspects of Inclusion Compounds Formed by Inorganic and Organometallic Host Lattices, ed. J. L. Atwood, J. E. D. Davies and D. D. MacNicol, Academic Press: London, 1984, vol. 1 Search PubMed.
- H. M. Powell, J. Chem. Soc., 1948, 61–73 RSC.
- H. M. Powell and J. H. Rayner, Nature, 1949, 163, 566–567 CrossRef CAS.
- W. D. Schaeffer, W. S. Dorsey, D. A. Skinner and C. G. Christian, J. Am. Chem. Soc., 1957, 79, 5870–5876 CrossRef CAS.
- P. de Radzitzky and J. Hanotier, Ind. Eng. Chem. Process Des. Dev., 1962, 1, 10–14 CrossRef.
- F. V. Williams, J. Am. Chem. Soc., 1957, 79, 5876–5877 CrossRef CAS.
- J. Lipkowski, L. Gluzinski, K. Suwinska and G. D. Andreetti, J. Inclusion Phenom., 1984, 2, 327–332 CrossRef CAS.
- P. Starzewski, W. Zielenkiewicz and J. Lipkowski, J. Inclusion Phenom., 1984, 1, 223–232 CrossRef CAS.
- D. R. Bond, G. E. Jackson and L. R. Nassimbeni, S. Afr. J. Chem., 1983, 36, 19–26 CAS.
- M. H. Moore, L. R. Nassimbeni and M. L. Niven, J. Chem. Soc., Dalton Trans., 1987, 2125–2140 RSC.
- L. R. Nassimbeni, M. L. Niven and M. W. Taylor, J. Coord. Chem., 1989, 19, 339–348 CrossRef CAS.
- L. Lavelle and L. R. Nassimbeni, J. Inclusion Phenom. Mol. Recognit. Chem., 1993, 16, 25–54 CrossRef CAS.
- L. R. Nassimbeni, M. L. Niven and M. W. Taylor, Inorg. Chim. Acta, 1987, 132, 67–73 CrossRef CAS.
- F. M. A. Noa, S. A. Bourne, H. Su and L. R. Nassimbeni, Cryst. Growth Des., 2017, 17, 1876–1883 CrossRef.
- M. Lusi and L. J. Barbour, Chem. Commun., 2013, 49, 2634–2636 RSC.
- M. Lusi and L. J. Barbour, Angew. Chem., Int. Ed., 2012, 51, 3928–3931 CrossRef CAS PubMed.
- J. Lipkowski, in Comprehensive Supramolecular Chemistry, ed. J. L. Atwood, J. E. D. Davies, D. D. MacNicol and F. Vogtle, Elsevier, Oxford, 1999, vol. 6 Search PubMed.
- M. M. Wicht, N. B. Báthori and L. R. Nassimbeni, Dalton Trans., 2015, 44, 6863–6870 RSC.
- A. Y. Manakov, J. Lipkowski, K. Suwinska and M. Kitamura, J. Inclusion Phenom. Mol. Recognit. Chem., 1996, 26, 1–20 CrossRef CAS.
- J. Lipkowski and D. V. Soldatov, J. Inclusion Phenom. Mol. Recognit. Chem., 1994, 18, 317–329 CrossRef CAS.
- D. V. Soldatov and J. Lipkowski, J. Inclusion Phenom. Mol. Recognit. Chem., 1998, 30, 99–109 CrossRef CAS.
- D. V. Soldatov, V. A. Logvinenko, Y. A. Dyadin, J. Lipkowski and K. Suwinska, J. Struct. Chem., 1999, 40, 757–771 CrossRef CAS.
- D. V. Soldatov, G. D. Enright and J. A. Ripmeester, Cryst. Growth Des., 2004, 4, 1185–1194 CrossRef CAS.
- E. A. Ukraintseva, D. V. Soldatov and Y. U. A. Dyadin, J. Inclusion Phenom., 2004, 48, 19–23 CrossRef CAS.
- M. M. Wicht, H. Su, N. B. Báthori and L. R. Nassimbeni, CrystEngComm, 2016, 18, 2509–2516 RSC.
- M. M. Wicht, L. R. Nassimbeni and N. B. Báthori, Polyhedron, 2019, 163, 7–19 CrossRef CAS.
- N. M. Sykes, H. Su, S. A. Bourne and L. R. Nassimbeni, Cryst. Growth Des., 2020, 20, 274–280 CrossRef CAS.
- S. Noro, T. Ohba, K. Fukuhara, Y. Takahashi, T. Akutagawa and T. Nakamura, Dalton Trans., 2011, 40, 2268–2274 RSC.
- S. I. Noro, K. Fukuhara, K. Sugimoto, Y. Hijikata, K. Kubo and T. Nakamura, Dalton Trans., 2013, 42, 11100–11110 RSC.
- Q. Y. Yang, P. Lama, S. Sen, M. Lusi, K. J. Chen, W. Y. Gao, M. Shivanna, T. Pham, N. Hosono, S. Kusaka, J. J. Perry, S. Ma, B. Space, L. J. Barbour, S. Kitagawa and M. J. Zaworotko, Angew. Chem., Int. Ed., 2018, 57, 5684–5689 CrossRef CAS PubMed.
- S. A. Allison and R. M. Barrer, J. Chem. Soc., 1969, 1717–1723 RSC.
- S.-Q. Wang, S. Mukherjeea and M. J. Zaworotko, Faraday Discuss., 2021 10.1039/D1FD00037C.
- A. M. Kałuża, S. Mukherjee, S.-Q. Wang, D. J. O'Hearn and M. J. Zaworotko, Chem. Commun., 2020, 56, 1940–1943 RSC.
- M. Shivanna, K. Otake, J. Zheng, S. Sakaki and S. Kitagawa, Chem. Commun., 2020, 56, 9632–9635 RSC.
- X. Tian, H. Zhou, X. Zhang, C. Wang, Z. Qiu, D. Zhou and J. Zhang, Inorg. Chem., 2020, 59, 6047–6052 CrossRef CAS PubMed.
- S.-Q. Wang, S. Mukherjee, E. Patyk-Kaźmierczak, S. Darwish, A. Bajpai, Q. Y. Yang and M. J. Zaworotko, Angew. Chem., Int. Ed., 2019, 58, 6630–6634 CrossRef CAS PubMed.
- S.-Q. Wang, Q. Y. Yang, S. Mukherjee, D. O'Nolan, E. Patyk-Kaźmierczak, K. J. Chen, M. Shivanna, C. Murray, C. C. Tang and M. J. Zaworotko, Chem. Commun., 2018, 54, 7042–7045 RSC.
- N. Kumar, S.-Q. Wang, S. Mukherjee, A. A. Bezrukov, E. Patyk-Kaźmierczak, D. O'Nolan, A. Kumar, M.-H. Yu, Z. Chang, X.-H. Bu and M. J. Zaworotko, Chem. Sci., 2020, 11, 6889–6895 RSC.
- H. Kanoh, A. Kondo, H. Noguchi, H. Kajiro, A. Tohdoh, Y. Hattori, W. C. Xu, M. Inoue, T. Sugiura, K. Morita, H. Tanaka, T. Ohba and K. Kaneko, J. Colloid Interface Sci., 2009, 334, 1–7 CrossRef CAS PubMed.
- H. Kajiro, A. Kondo, K. Kaneko and H. Kanoh, Int. J. Mol. Sci., 2010, 11, 3803–3845 CrossRef CAS PubMed.
- G. B. Gardner, D. Venkataraman, J. S. Moore and S. Lee, Nature, 1995, 374, 792–795 CrossRef CAS.
- P. Willett, J. C. Cole and I. J. Bruno, CrystEngComm, 2020, 22, 7233–7241 RSC.
- M. L. Cheney, G. J. McManus, J. A. Perman, W. Zhenqiang and M. J. Zaworotko, Cryst. Growth Des., 2007, 7, 616–617 CrossRef CAS.
- R. Sanii, A. Bajpai, E. Patyk-Kaźmierczak and M. J. Zaworotko, ACS Sustainable Chem. Eng., 2018, 6, 14589–14598 CrossRef CAS.
- R. Sanii, C. Hua, E. Patyk-Kaźmierczak and M. J. Zaworotko, Chem. Commun., 2019, 55, 1454–1457 RSC.
- N. Shan, F. Toda and W. Jones, Chem. Commun., 2002, 2, 2372–2373 RSC.
- T. Friščić, A. V. Trask, W. Jones and W. D. S. Motherwell, Angew. Chem., Int. Ed., 2006, 45, 7546–7550 CrossRef PubMed.
- G. A. Bowmaker, Chem. Commun., 2013, 49, 334–348 RSC.
- M. L. Cheney, M. J. Zaworotko, S. Beaton and R. D. Singer, J. Chem. Educ., 2008, 85, 1649–1651 CrossRef CAS.
- Z. Wang, N. Liu, H. Li, P. Chen and P. Yan, Eur. J. Inorg. Chem., 2017, 2017, 2211–2219 CrossRef CAS.
- A. Bacchi, D. Capucci, A. Gatti, C. Loffi, M. Pioli, D. Rogolino, F. Terenziani and P. Pelagatti, ChemistrySelect, 2017, 2, 7000–7007 CrossRef CAS.
- D. Prat, O. Pardigon, H.-W. Flemming, S. Letestu, V. Ducandas, P. Isnard, E. Guntrum, T. Senac, S. Ruisseau, P. Cruciani and P. Hosek, Org. Process Res. Dev., 2013, 17, 1517–1525 CrossRef CAS.
- S. Wöhlert, T. Fic, Z. Tomkowicz, S. G. Ebbinghaus, M. Rams, W. Haase and C. Näther, Inorg. Chem., 2013, 52, 12947–12957 CrossRef PubMed.
- E. Loukopoulos, B. Berkoff, K. Griffiths, V. Keeble, V. N. Dokorou, A. C. Tsipis, A. Escuer and G. E. Kostakis, CrystEngComm, 2015, 17, 6753–6764 RSC.
- S. Suckert, L. S. Germann, R. E. Dinnebier, J. Werner and C. Näther, Crystals, 2016, 6, 1–17 CrossRef.
- Y. Inubushi, S. Horike, T. Fukushima, G. Akiyama, R. Matsuda and S. Kitagawa, Chem. Commun., 2010, 46, 9229–9231 RSC.
- R. Kitaura, K. Seki, G. Akiyama and S. Kitagawa, Angew. Chem., Int. Ed., 2003, 42, 428–431 CrossRef CAS PubMed.
- APEX3. Ver. 2017.3-0, Bruker AXS Inc., Madison, Wisconsin, USA, 2017 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- XPREP. Ver. 2014/2, Bruker AXS Inc., Madison, Wisconsin, USA, 2014 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, C71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, C71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Complete synthetic protocol, compound characterisation and crystallographic details. CCDC 1995785–1995796 and 2065192. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt01839f |
| This journal is © The Royal Society of Chemistry 2021 |