
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Calcium carbonate: controlled synthesis, surface functionalization, and nanostructured materials
Yu-Qin
Niu
ac,
Jia-Hui
Liu
ac,
Cyril
Aymonier
 f,
Simona
Fermani
be,
Damir
Kralj
*d,
Giuseppe
Falini
*b and
Chun-Hui
Zhou
*ac
f,
Simona
Fermani
be,
Damir
Kralj
*d,
Giuseppe
Falini
*b and
Chun-Hui
Zhou
*ac
aResearch Group for Advanced Materials & Sustainable Catalysis (AMSC), State Key Laboratory Breeding Base of Green Chemistry-Synthesis Technology, College of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310032, China. E-mail: clay@zjut.edu.cn
bDepartment of Chemistry “Giacomo Ciamician”, University of Bologna, Via Selmi 2, I-40126 Bologna, Italy. E-mail: giuseppe.falini@unibo.it
cQing Yang Institute for Industrial Minerals, You Hua, Qing Yang, Chi Zhou 242804, China
dLaboratory for Precipitation Processes, Ruđer Bošković Institute, P. O. Box 1016, HR-10001 Zagreb, Croatia
eInterdepartmental Centre for Industrial Research Health Sciences & Technologies, University of Bologna, 40064 Bologna, Italy
fUniv Bordeaux, ICMCB, Bordeaux INP, UMR 5026, CNRS, F-33600 Pessac, France
First published on 22nd August 2022
Abstract
Calcium carbonate (CaCO3) is an important inorganic mineral in biological and geological systems. Traditionally, it is widely used in plastics, papermaking, ink, building materials, textiles, cosmetics, and food. Over the last decade, there has been rapid development in the controlled synthesis and surface modification of CaCO3, the stabilization of amorphous CaCO3 (ACC), and CaCO3-based nanostructured materials. In this review, the controlled synthesis of CaCO3 is first examined, including Ca2+–CO32− systems, solid–liquid–gas carbonation, water-in-oil reverse emulsions, and biomineralization. Advancing insights into the nucleation and crystallization of CaCO3 have led to the development of efficient routes towards the controlled synthesis of CaCO3 with specific sizes, morphologies, and polymorphs. Recently-developed surface modification methods of CaCO3 include organic and inorganic modifications, as well as intensified surface reactions. The resultant CaCO3 can then be further engineered via template-induced biomineralization and layer-by-layer assembly into porous, hollow, or core–shell organic–inorganic nanocomposites. The introduction of CaCO3 into nanostructured materials has led to a significant improvement in the mechanical, optical, magnetic, and catalytic properties of such materials, with the resultant CaCO3-based nanostructured materials showing great potential for use in biomaterials and biomedicine, environmental remediation, and energy production and storage. The influences that the preparation conditions and additives have on ACC preparation and stabilization are also discussed. Studies indicate that ACC can be used to construct environmentally-friendly hybrid films, supramolecular hydrogels, and drug vehicles. Finally, the existing challenges and future directions of the controlled synthesis and functionalization of CaCO3 and its expanding applications are highlighted.
 Yu-Qin Niu | Yu Qin Niu is currently a postgraduate at Research Group for Advanced Materials & Sustainable Catalysis (AMSC), College of Chemical Engineering, Zhejiang University of Technology (ZJUT), Hangzhou, China. She received her Bachelor's Degree in Chemical Engineering and Technology from Qingdao University of Science and Technology. Her research presently focuses on organic-inorganic mineral bio-composites under the supervision of Prof. Chun Hui ZHOU. She has coauthored scientific papers in respected peer-reviewed international journals. |
 Jia-Hui Liu | Jia Hui Liu is currently a PhD candidate at Research Group for Advanced Materials & Sustainable Catalysis (AMSC), College of Chemical Engineering, Zhejiang University of Technology (ZJUT), Hangzhou, China and under the supervision of Prof. Chun Hui Zhou. She received her Bachelor's Degree in Applied Chemistry from Anhui Jianzhu University. Her research presently focuses on colloid and surface chemistry of carbonate and clay minerals, and related hydrogels and nanostructured functional composites and their applications in healthcare, adsorption and catalysis. She has authored and coauthored several scientific papers in respected peer-reviewed international journals. |
 Damir Kralj | Damir Kralj is a senior scientist and a head of the Laboratory for Precipitation Processes, Ruder Boskovic Institute, Zagreb, Croatia. He studied Chemical Engineering and Chemistry at the University of Zagreb and completed his PhD in 1990. He held a research fellowship at the University of Copenhagen (Arne E. Nielsen) and a postdoc fellowship at the TU Delft (Gerda van Rosmalen). His research focus is on the kinetics and mechanisms of precipitation of slightly soluble ionic salts (calcium carbonates, oxalates, phosphates), metastable and precursor phases, interfacial interactions between mineral surfaces and dissolved species, biomineralization, pathological mineralization and industrial crystallization. |
 Giuseppe Falini | Prof. Giuseppe FALINI, PhD in Chemistry, is full professor in chemistry at the University of Bologna. Currently, his research activities are addressed to the design and preparation of innovative materials from waste marine biominerals and biopolymers and to the study of the biomineralization process in corals and echinoderms and under environmental stresses. He is co-author of about 230 scientific publications in international journals (two in Science). He also wrote 3 book chapters and is co-inventor of 3 patents. He has been awarded of grants from national institutions, companies and European Community (ERC Adv). |
 Chun-Hui Zhou | Dr Chun Hui ZHOU, born and brought up in Miaoqian, Qingyang, Anhui, is Professor of Chemical Engineering and Leader of Research Group for Advanced Materials and Sustainable Catalysis (AMSC), Zhejiang University of Technology. He is Director of Qing Yang Institute for Industrial Minerals. He acts as AIPEA Councilor (2017-) and Vice President (2022-). He also serves as Principal Editor of Clay Minerals, Associate Editor of Clays and Clay Minerals, and Editorial Member of Applied Clay Science, Journal of Porous Materials and Journal of Inclusion Phenomena and Macrocyclic Chemistry. He worked as a visiting academic at the University of Queensland (2006-2007) and as a visiting professor at the University of Western Australia (2010). His R&D centers on clay minerals, limestone and dolomite and related functional materials as well as catalysts for converting biochemicals and biomass, He teaches Catalysis, Materials Science and Engineering, and Scientific Literacy. |
1. Introduction
Calcium carbonate (CaCO3) ubiquitously exists in sedimentary rocks and minerals in the form of marble, limestone, and chalk, and can also be found in marine sediments.1,2 In addition, CaCO3 is present in many living organisms, functioning either as a structural support (e.g., in algae,3 sponges,4,5 corals6), a form of protection (e.g., shells),7 a hard buoyancy tank (e.g., cuttlebone),8 or as a component in photoreceptor systems (e.g., light-focusing eye lenses of chitons and brittlestars).9 CaCO3 is also synthesized by bacteria,10 even in extreme biomineralization conditions,11 and is an essential component of mineralized tissues as in the apatitic whale bone.12,13 Crystalline CaCO3 exhibits three polymorphs: hexagonal vaterite, orthorhombic aragonite, and rhombohedral calcite, in order of increasing thermodynamic stability.14 Two hydrated crystal phases of CaCO3, monohydrocalcite (CaCO3·H2O) and ikaite (CaCO3·6H2O), have been known for more than a century, while recently, hemihydrate CaCO3·½H2O with a monoclinic structure has been discovered.14–16 An unstable amorphous CaCO3 (ACC) phase can be found in Stylophora pistillata corals,17 crayfish gastroliths,18 sea urchin spicules,19 gastropods,20 earthworms,21 plant cystoliths, and other such organisms.22 The diversity of the origins, composition, morphologies, and polymorphs of CaCO3 makes it an extremely significant material for use in both scientific research and technological applications.CaCO3 is widely used as a filler material in paper, plastics, rubber, paints, and foodstuffs,23,24 yet its new applications, particularly as a functional nano-CaCO3 material, have driven the extensive research on synthesizing CaCO3 with a specific size, morphology, polymorph, or surface property.18,23 To improve the processes used to synthesize these specific materials and the properties of the products, liquid–liquid or solid–liquid–gas routes can be tailored by adding judiciously-chosen organic compounds or polymers, which act as templates or modifiers for the nucleation and growth of CaCO3.25,26 Moreover, water-in-oil reverse emulsion methods and ultrasonic intensification processes have also been introduced to control CaCO3 synthesis, while biomimetic approaches have been developed to produce CaCO3 with specific structures under mild conditions.27–29 Using these innovative strategies, a variety of CaCO3 particles with different sizes, polymorphs, and morphologies (e.g., spheres, hollow spheres, rods, and flower-like) have been successfully synthesized.24,30 Meanwhile, more in-depth knowledge of the scientific understanding of the mechanisms that underpin the nucleation and growth of CaCO3 under different conditions and environments and their effects on pH, temperature, supersaturation, organic modifiers, or templates, has been gained.31,32
In addition to the control of the dimensions, polymorphs, and morphologies of synthetic CaCO3 micro-/nanoparticles (MNPs), their surface functionalities are also crucial for their new applications. To this aim, the last decade or so has witnessed many types of in situ or post-modification methods having been developed to tune the surface polarity, hydrophilicity, oleophobicity, stability, and reactivity of CaCO3 MNPs.33,34 CaCO3 MNPs with distinct surface properties can thus be obtained and used to produce CaCO3-based or CaCO3-incorporated structured materials that have growing potential and applications in biomaterials and biomedicines, and environmental applications.35,36 It has been well established that CaCO3 particles are stable at pH 7, whereas they dissolve and release carbon dioxide (CO2) gas under acidic conditions,37 allowing them to be functionalized with targeting molecules or polymers for targeted pH-responsive drug/gene/protein delivery.38–40 The pH sensitivity of CaCO3 also makes it a useful self-sacrificing template for producing porous or hollow organic–inorganic biomaterials.41–43 Moreover, major advances have recently been made in combining CaCO3 with contrast agents to achieve various molecular imaging modalities for diagnosis;40,44 with different polymeric molecules to form porous biodegradable scaffolds for use in bone tissue engineering;45 and in integrating it with biomolecules to fabricate bioceramics,46 bone cement,47 and hydrogels.48 To date, CaCO3 has been used in many therapeutic and theranostic applications in chemotherapy, photothermal therapy (PTT), or photodynamic therapy (PDT);49,50 wound healing and blood clotting;51,52 in ultrasound (US),53,54 fluorescence,44 or magnetic resonance imaging (MRI);55 and multimodal imaging and therapy.56,59,60 In addition to the above-mentioned aspects of crystalline CaCO3, the preparation and stabilization of ACC have also presented a challenge. Although several synthetic routes to ACC inspired by and based on biomineralization have been developed,57–59 those processes are sophisticated. The preparation of ACC with distinct properties is affected by the mixing of reactants and additives,60,61 the control of emulsion formation,62 precipitation, and other parameters such as humidity, temperature, and pressure.63 Therein, much attention has been paid to understanding the role of additives, such as magnesium,64 phosphorus ions,65 polymeric compounds,66 and, in particular, the inclusion of carboxyl-containing compounds67–69 in the stabilization of ACC. Meanwhile, the latest studies have indicated that ACC can be used to prepare organic–inorganic hydrogels70 and films.71 Moreover, ACC hybrid NPs loaded with antitumor drug and coated with phospholipid/polyethylene glycol (PEG)/folic acid (FA) have been successfully prepared and used in enzyme-abundant and acidic tumor environments, exhibiting good drug release and antitumor effects, both in vitro and in vivo.72,73
In this review, the novel synthetic strategies for controlling the sizes, morphologies, and polymorphs of CaCO3 are first critically surveyed, with discussion on models describing the mechanisms of CaCO3 nucleation and growth. Newly-developed surface modification methods, particularly those that involve surface adsorption, surface grafting, and the encapsulation of CaCO3, are highlighted and discussed in detail. Then, the state-of-the-art in terms of engineering nanostructured materials with incorporated CaCO3 toward advanced applications is covered. Next, the major progress and challenges in producing and stabilizing ACC, which is an essential pre-requisite for the further development and utilization of ACC-based organic/inorganic composites, are examined. Finally, the existing issues and future direction of the controlled synthesis, surface modification of CaCO3, and the stabilization of ACC and CaCO3-based nanostructured materials are highlighted.
2. Controlled synthesis of CaCO3 MNPs and formation mechanisms
Although the control of the size distribution, morphologies, and polymorphs of CaCO3 MNPs have been the focus of many academic studies (Fig. 1),24,74 the theoretical understanding of the crystallization/precipitation of CaCO3 MNPs remains challenging toward practical technology applications. Although CaCO3 can be easily prepared via conventional precipitation reactions by adjusting the solution pH, ion concentration, solvent species, reaction temperature, time, or the presence of additives, there is still a need to develop sophisticated strategies and innovative methods using both chemistry and chemical engineering principles to produce CaCO3-based materials that have desirable properties and are suitable for use in advanced applications.75 The controlled synthesis of CaCO3 MNPs is associated with several significant shortcomings. First, CaCO3 can adopt chain-, needle-, flake-, cubic-, spherical-, spindle-like, and many other morphologies, with sizes ranging from MPs to NPs (Fig. 1B). In addition, the simultaneous initial formation of several polymorphs or/and hydrated forms, which is observed for the majority of CaCO3 precipitation systems, presents challenges in controlled synthesis when the aim is to isolate a pure sample of a specific polymorph of CaCO3 (Fig. 1A). In such systems, the kinetic or thermodynamic stabilization of metastable phases is crucial to enable control over the polymorphism and morphology of the resultant solid phase. To this aim, state-of-the-art techniques and synthetic platforms including a Ca2+–CO32− reaction system,76 solid–liquid–gas carbonation,77 and a water-in-oil reverse emulsion26 have been developed to synthesize specific types of CaCO3 MNPs and polymorphs of different sizes and/or with unusual morphologies (Table 1).78 The critical parameters that affect the size distribution, morphology, and polymorphism of CaCO3 are the temperature, solvent, pH, and presence of specific additives.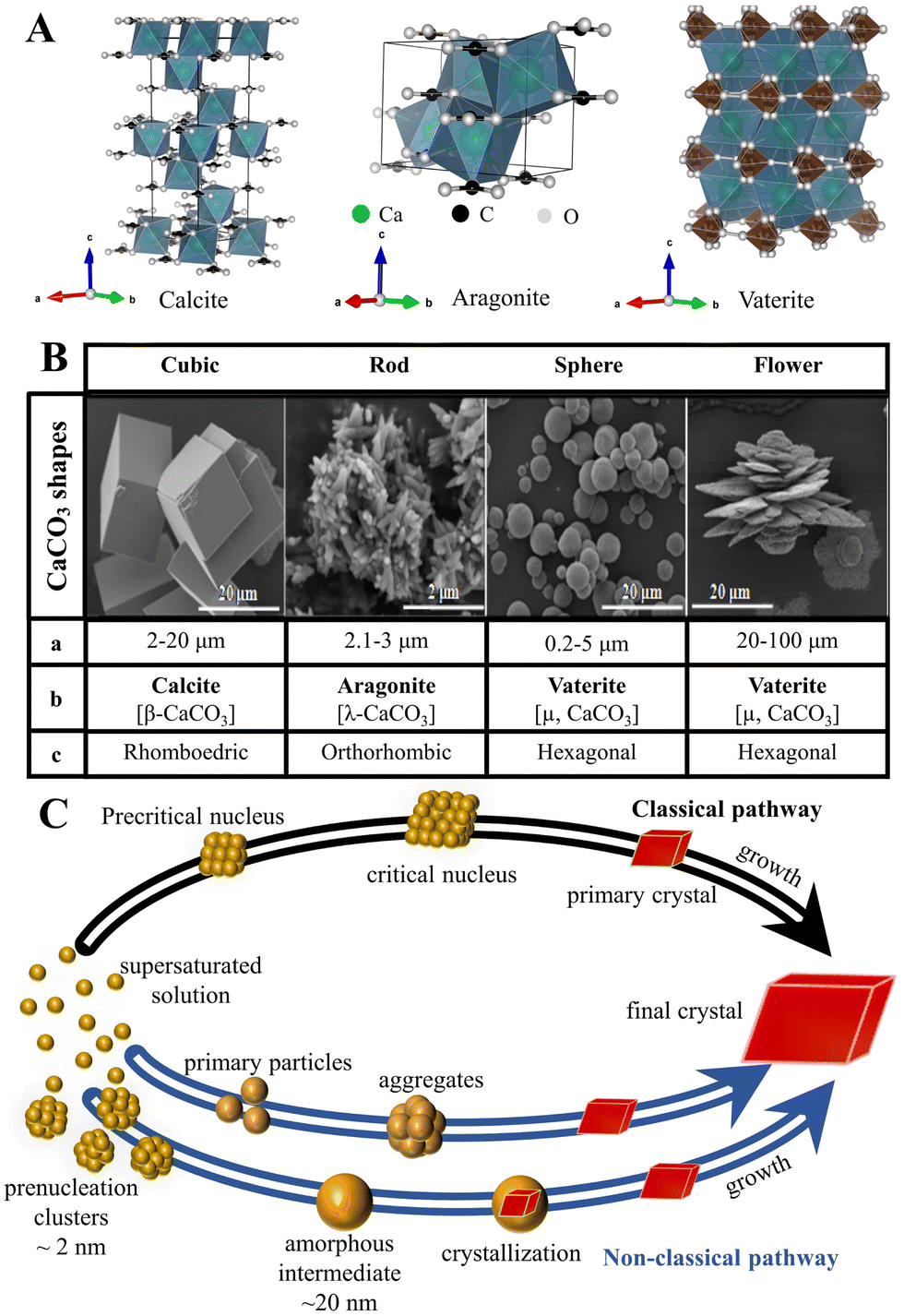 | ||
| Fig. 1 Structure, size and morphology, and polymorphism of CaCO3 micro-/nanoparticles and the associated formation pathways. (A) The polymorphs and crystal structure of CaCO3 (the crystal structure image was generated using the software VESTA, where the green, black, and white balls represent Ca, C, and O atoms, respectively). (B) Typical shapes of CaCO3 particles (a: average diameter; b: crystalline phase; and c: crystalline system). Adapted and reprinted with permission from ref. 79. Copyright 2017, the MedCrave Group under Creative Commons Non-Commercial Attribution License (CC-BY-NC 4.0). (C) Classical and non-classical pathways to CaCO3. Adapted and reprinted from ref. 80 and 81. Copyright (2008), with permission from AAAS. | ||
| Strategies | Conditions and additives | Characteristics | Advantages | Disadvantages | Ref. | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Additive(s) | pH | Temp. (°C) | Other | Morphology | Polymorph | Size (μm) | ||||
| a N′-Dodecyl-N,N-dimethyl acetamidine bicarbonate; AOT: sodium bis(2-ethylhexyl) sulfosuccinate; BDA: N-butydimethylamine; DBAE: 2-(dibutylamino) ethanol; DBU: 1,8-diazabicycloundec-7-ene; DMCHA: N,N-dimethylcyclohexylamine; DSS: dextran sodium sulfate; EDTA-2Na: disodium salt of ethylenediaminetetraacetic acid; EG: ethylene glycol; EP: N-Ethylpiperidine; PAA: poly(acrylic acid); Phe: L-phenylalanine; PSSS: poly(styrene sulfonate) sodium; SDS: sodium dodecyl sulfonate; US: ultrasonication; VHVEVS: Ac-Val-His-Val-Glu-Val-Ser-CONH2. | ||||||||||
| Ca2+–CO32-reaction system | DSS, PSSS | — | — | [CaCl2] = [Na2CO3] = 5 × 10−4–5 × 10−2 M | Spherical, toroidal, ellipsoidal | Vaterite, calcite | 0.5–1.8 | MPs to NPs; various morphologies; various polymorphs; (valcite, vaterite, aragonite); convenient doping with other ions; functionalized with molecules in situ | Poor crystallinity; difficult to control particles’ size | 25 |
| EG | 9.0 | 25 | [CaCl2] = [Na2CO3] = 0.05, 0.1, 0.33 M; EG/H2O ratios: 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 4:1, 6:1 :1, 4:1, 6:1 |
Spherical, ellipsoidal | Vaterite | 0.4–2.7 | 76 | |||
| EG | 8.0–10.0; 11.0–12.0 | 23 | [Ca2+]:[CO32−] ratios: 5:1, 2:1, 1:1, 1:2, 1:3 |
Ellipsoidal, spherical, spheroidal, rhombohedral, flower-like, irregular | Vaterite, calcite | — | 109 | |||
| SDS; PAA | — | 80 | PAA (Mw = 8000 g mol−1), [PAA] = 0.5 g L−1, [SDS] = 10 mM | Hollow microspheres | Calcite | 4–7 | 100 | |||
| Solid–liquid–gas carbonation | Urea | — | 150 | High-pressure CO2:12 MPa, mass ratios: Ca(OH)2/urea = 1:6 |
Spherical | 94.2% vaterite | — | Mainly NPs; mainly cubic or rhombohedral morphology; environment preservation; effective use of mineral resources; industrially | Difficult to control the crystal shape; mainly calcite; polymorph in absence of additives; low CO2 dissolution | 110 |
| DMCHA, DBU, DBAE, BDA, EP | 8.0–13.0 | 40–90 | [Ca(CH3COO)2] = 1.0 M | Rhombohedral, spherical, rod-like, shuttle-like | Calcite, vaterite, aragonite | — | 78 | |||
| EDTA-2Na | — | 120 | Pressurized-CO2 | Hierarchical hollow microspheres | Calcite | 4–6 | 74 | |||
| Water-in-oil reverse emulsion | AOT | — | 25 | AOT/isooctane/water reverse microemulsion, [CaCl2] = [Na2CO3] = 0.05 M | Rod-like, irregular, spherical | Calcite, vaterite | 1–3 | Size can be well controlled; soft template effect; reproducibility; simple maneuverability; low degree of agglomeration; high purity | Required a certain amount of an oil phase and surfactant | 111 |
| — | — | 65 | CO2/N2 switchable surfactant reverse micellesa | Spherical, rhombohedral, dendrite-like | Vaterite, calcite | – | 112 | |||
| Tween-80, Span-80 | — | 22 | US | Spherical | Vaterite | 0.02–0.03 | 113 | |||
| Biomineralization | Phe | 9.3–12.7 | 25 | [Phe]: 0 g L−1–16 g L−1; CO2 flow rate: 10–50 mL min−1 | Rhombohedral, spherical | Calcite, vaterite | 2–5 | MPs to NPs; controlled morphologies and polymorphs; good crystallinity; hierarchical structure | Complex mechanism | 24 |
| VHVEVS peptide | 7.2 | 20 | [Ca2+] = 22 mM; CO32−: hydrolyzed urea | Fiber-like, nanocubic | Aragonite, calcite | — | 114 | |||
In addition, the controlled synthesis of CaCO3 by biomineralization has drawn much attention because of its simplicity in controlling polymorphism, morphology, and sizes of the particles, via the reaction of CO32− and Ca2+ in the presence of an organic matrix.78 It is hypothesized that the organic matrix acts either as a substrate for heterogeneous nucleation or as an inhibitor of nucleation or crystal growth via adsorption.82 Besides the above-mentioned methods, some other synthesis strategies that have varying levels of efficiency, such as the decomposition of Ca(HCO3)2,83 spray drying techniques,84 electric field-controlled crystallization,85 ultrasonic irradiation,86–88 inorganic ion polymerization reactions,89 and the fusion of amorphous precursors under pressure,90 have also recently been developed.
To control the synthesis of CaCO3 and subsequently its respective physical and chemical properties, knowledge of the nucleation and growth mechanism of the material is fundamental. As such, models of CaCO3 nucleation have been of experimental or computational focus (Fig. 1C).91 Such processes can be predominantly explained using classical nucleation theory (CNT), which is based on the assumption that there is simple association between Ca2+ and CO32−, the formation of a supersaturated solution, and stable nuclei.92 However, CNT does not account for unexpected nucleation, proceeding via metastable/stable precursor phases, such as clusters at the pre-nucleation stage,92 liquid-like precursors,93 amorphous phases,94 and even oligomers,89 which may play important roles in the nucleation and subsequent structure control of CaCO3 (Fig. 1C). Such intermediate-based nucleation theory is referred to as a non-classical nucleation pathway (NCNT) to distinguish it from classical nucleation.
2.1 Controlled synthesis of CaCO3 MNPs
(1) Concentration of reactants
The initial concentrations of the reactant salts (Ca2+ and/or CO32−) affect the polymorphic selection, size, shape, surface charge, and hydrophilicity of the resultant CaCO3 particles, either via their instantaneous mixing or via the controlled addition of one component.76,97,101 For example, submicron vaterite particles have been synthesized from saturated Na2CO3 and CaCl2 solutions in the presence of ethylene glycol (EG) via dropwise precipitation, which allows variation in the Ca2+ concentration of the solution at each moment of the reaction.76 These techniques affect the crystallization process of CaCO3 by promoting the formation of new nucleation centers instead of providing the conditions for crystal growth. Such a reduced growth rate leads to a decrease in the rate of vaterite recrystallization, resulting in the formation of vaterite particles with spherical or ellipsoidal morphology. Moreover, in the presence of EG and with a controlled Ca2+ ion addition rate, the size of such vaterite particles can be controlled. A high concentration of CO32− leads to the formation of anisotropic rhomboidal and ellipsoidal morphology, while a low concentration results in the formation of isotropic spheroids.115 Furthermore, an excess of Ca2+ or CO32− in the system has multiple impacts on the reaction. An excess of CO32− accelerates the reaction and results in CaCO3 particle formation in the early stages, while an excess of Ca2+ slows down the process of particle formation and promotes the growth of spheroidal CaCO3 particles.25(2) pH
The precipitation and dissolution of CaCO3 in aqueous solution is a process in which CO32− reacts with Ca2+ (eqn (1)):| Ca2+(aq) + CO32−(aq) ↔ CaCO3(s) | (1) |
| H+ + OH− ↔ H2O (Kw) | (2) |
| CO2(g) ↔ CO2(aq) ↔ H2CO3(aq) (KH) | (3) |
| H+ + CO32− ↔ HCO3− (K1) | (4) |
| H+ + HCO3− ↔ H2CO30 (K2) | (5) |
| Ca2+ + CO32− ↔ CaCO30 (KCaCO3) | (6) |
| Ca2+ + HCO3− ↔ CaHCO3+ (KCaHCO3+) | (7) |
| Ca2+ + OH− ↔ CaOH+ (KCaOH+) | (8) |
| S = [(a(Ca2+)·a(CO32−))/(K0sp)]1/2 | (9) |
a(H+)), while from these data and known initial experimental conditions, the kinetics and the mechanisms of the process can be determined with relative precision at any given moment.118,119
It is obvious from the above-mentioned explanation that the initial pH can be clearly correlated with the composition and stability of the solution (supersaturation or undersaturation), which is, in turn, a key parameter for the determination of physical, chemical, structural, and/or morphological properties of the CaCO3 precipitate. However, to draw consistent conclusions about the role of the initial pH, other relevant parameters (concentrations and the ratio of reactants, hydrodynamics, temperature, presence of additives, and aging time, etc.) should be considered.88,120–122 To this aim, Trofimov et al.95 suggested that an increase in pH from 7 to 11 may lead to the precipitation of vaterite, which was explained by the observations of alterable supersaturation and increased CO32− content, but that the addition of negatively-charged inorganic or organic substances appeared to have the same effect. Moreover, Džakula et al.122 showed that in systems in which the supersaturation, ionic strength, ratio of the activity of the constituent ions (a(Ca2+)/a(CO32−)), and the type of stirring were identical that the calcite content increased with an increase in the pH from 8.5 to 10.5, while in the same systems that were magnetically stirred with the pH controlled at 8.5 and 9.0, the precipitated product was almost entirely vaterite. The morphology of the resultant vaterite was observed to continuously change with increasing pH. Similarly, in precipitation systems designed to produce vaterite, CaCO3 formation is accelerated in the presence of increased concentrations of CO32− ions, in particular, at pH >10.5, and it was demonstrated that spheroids, ellipsoids, or toroids can be specifically prepared via careful control of such parameters as the concentrations of the initial reagents, their ratio, reaction time, and organic additives.25 Transformation of vaterite morphology, from an almost spherical assemblage of primary NPs (5–10 nm) to hexagonal platelets (1–2 μm) and single crystals has been observed in an ammonia diffusion method.127 During this process, the pH, and consequently the supersaturation, continuously increased as a result of the decomposition of NH4HCO3 and thus led to the diffusion of NH3 into a CaCl2–NaHCO3 salt solution. It was also deduced that the existence of NH3 significantly affected the initial polymorph composition of the CaCO3 precipitates: high percentages of calcite were formed at low NH3 diffusion rates, whereas vaterite became the major phase when the NH3 diffusion was rapid and NH3 reached a concentration of >0.02 mol L−1.
(3) Reaction temperature
Temperature affects any precipitation, including the precipitation of CaCO3, in two ways.1 First, the rate constants of the nucleation and growth of a specific polymorph are influenced as indicated by the Arrhenius equation, which assumes their exponential relationship to temperature. Second, it has been observed that the solubility of different CaCO3 polymorphs and hydrated forms in water varies as a function of temperature as reflected by the changes of their solubility products (Ksp) within a defined temperature range (Fig. 2 and Table 2).97,102,123 Except for ikaite, all Ksp values continuously decrease with increasing temperature. The only difference is that the Ksp for ikaite has an opposite trend and changes more remarkable upon the changes of the temperature, primarily as a consequence of a large number of hydration water molecules in the crystal lattice.124 The calculated solubilities of polymorphs and hydrates (cs), in water and in closed precipitation systems, at 25 °C, are directly compared in Table 2. It is worth noting that although the effect of temperature on the solubility and subsequently on the formation of the polymorphic phases of CaCO3 cannot be simply evaluated without considering other parameters, a general tendency can still be observed. In addition, precipitation diagrams of CaCO3 polymorphs, which cover a broad span of initial concentrations and temperatures, ranging from the freezing to boiling points of water, have been also constructed.128,129 Such diagrams provide a basis for explaining the nature of the polymorphs precipitated in both inorganic and biological environments, and help to predict, design, and control the synthesis of CaCO3 particles.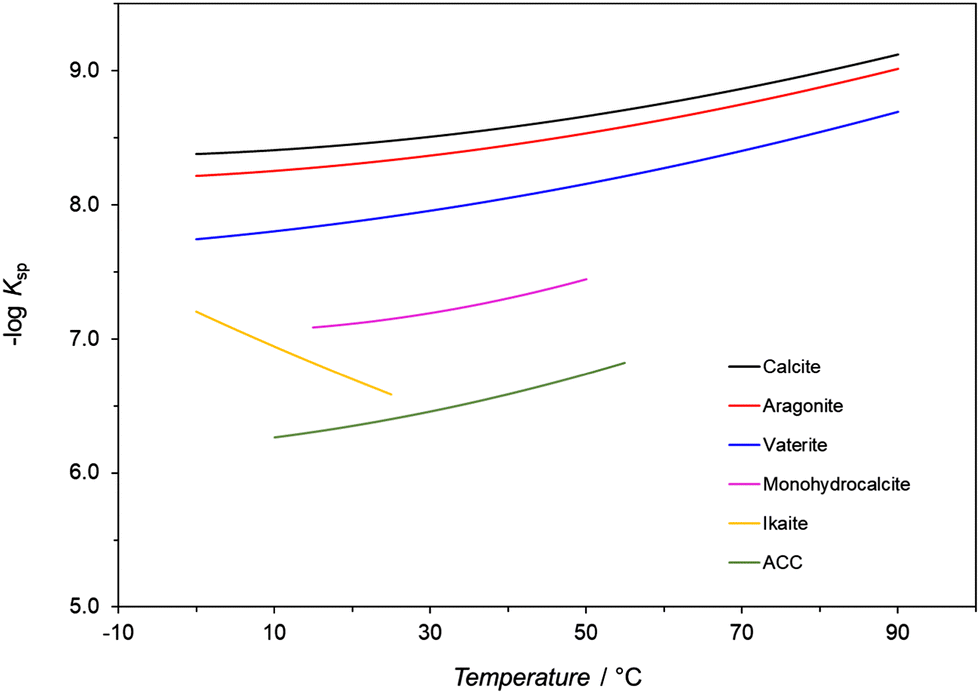 | ||
| Fig. 2 Minus log of values of thermodynamic solubility products (Ksp) of CaCO3 polymorphs and hydrates as a function of temperature. The data in the figure comes from ref. 123–126. | ||
| CaCO3 modification | Expression | Temperature range/°C | c s (25 °C)/mg dm−3 | Ref. |
|---|---|---|---|---|
| ACC – amorphous CaCO3; cs – solubility; Ksp – thermodynamic solubility product of respective modification; T – absolute temperature/K; t – temperature/°C. | ||||
| Calcite | logKsp = −171.9065 − 0.077993T + 2839.319/T + 71.595logT |
0–90 | 12.29 | 117 |
| Aragonite | logKsp = −171.9773 − 0.077993T + 2903.293/T + 71.595logT |
0–90 | 14.20 | 117 |
| Vaterite | logKsp = −172.1295 − 0.0779933T + 3074.688/T + 71.595logT |
0–90 | 22.22 | 117 |
| Ikaite | logKsp = 0.15981 − 2011.1/T |
0–25 | 125.97 | 124 |
| Monohydrocalcite | −logKsp = 7.050 + 0.000159·t2 |
15–50 | 56.06 | 125 |
| ACC | −logKsp = 6.1987 + 0.005336·t + 0.0001096·t2 |
10–55 | 168.73 | 126 |
Recently, aragonite has been found to be the dominant polymorph obtained at temperatures of >75 °C and a wide range of supersaturations.129 At moderate temperatures (50 °C) and low supersaturation, vaterite, aragonite, and calcite phases were observed to precipitate, while at high supersaturation, aragonite and calcite were the dominant phases. At low temperatures (25 °C) at Ca2+ concentrations ranging from 0.002 M to saturation, vaterite and calcite phases precipitated, while near to the freezing point of water, no precipitation occurred at 0.002 M and only the calcite phase precipitated at high concentrations. A similar tendency has been observed for temperature combined with other experimental parameters: at low temperatures and high ionic strengths, the precipitation of metastable ikaite and vaterite is favored, whereas at near-freezing temperature under highly alkaline conditions (pH 13.4), only ikaite is formed.15,130 Moreover, it is known that aragonite, which is slightly less stable than calcite, precipitates at a higher temperature.23 Since calcite is the only thermodynamic phase formed at ambient temperatures and pressures, the range of temperatures at which it forms in combination with many other parameters is relatively wide because all metastable phases finally transform into calcite, as predicted by Ostwald's law of stages.98
However, deviations from the above-mentioned empirical rules have been observed. In a double injection system in which CaCl2 and NH4HCO3 solutions were mixed at a range of temperatures from 30 °C to 80 °C, vaterite and aragonite phases with various structures and morphologies were predominantly synthesized.131 In particular, lamellar vaterite particles; a mixture of vaterite, aragonite, and traces of calcite; and aragonite whiskers were formed at 30–40 °C, 50–70 °C, and 80 °C, respectively. The formation of aragonite in the form of elongated needle-/rod-like or whisker-like morphologies with high surface energy, at high temperatures, was attributed to the increased energy of the reactive environment. However, the predominant formation of lamellar vaterite at temperatures up to 60 °C was explained as being due to a decrease in [CO32−]/[Ca2+] in line with an increase in the temperature. However, pure vaterite (≥99 wt%) was prepared at high temperatures (up to 60 °C) in systems in which the reactants were slowly mixed and then intensively stirred (600 rpm).132 The synthesis of pure vaterite under such circumstances was attributed to hydrodynamic conditions, under which increased local supersaturation effects and the formation of the precursor phase (ACC) were avoided.
Besides polymorphic selection, the temperature may also significantly affect the morphology and particle size distribution of precipitates. For example, according to a recent study from Sovova et al.,133 when concentrated aqueous solutions of 0.33 M CaCl2·2H2O and 0.33 M Na2CO3 were mixed in a vessel from 10 °C to 50 °C, the particle size of the vaterite increased linearly with the increasing temperature. A significant change in shape was observed over a temperature range of 20–45 °C; spherical vaterite particles were prepared, which then transformed from spheres to cauliflower-like shapes and then to croissant-like shapes. This change of vaterite particle shape was attributed to the increased diffusion of the Ca2+ and CO32− ions with increasing temperature and the decreased solubility of CaCO3, leading to an acceleration in the CaCO3 crystallization.133 The shape of vaterite particles, for example, changes from smooth spheres at 25 °C to cauliflower-like particles at 40 °C or 50 °C, in the systems in which the initial concentrations of the reactants (0.1 M Ca(NO3)2 and Na2CO3) and the stirring speed (1500 rpm) were kept the same.134,135 Upon an increase in the temperature, the overall transformation of vaterite to calcite is reduced, due to an increase in the growth rate of vaterite. However, in this case, the particle size distribution at 40 °C was found to be similar to that at 25 °C, indicating that the nucleation rate is virtually unaffected by temperature. In contrast, in a study121 in which an ethanol/water mixture was used with fixed initial concentrations of the reactants, only vaterite precipitated in the range of 0 °C to 100 °C and an increase in reaction temperature evidently led to a decrease in the particle size distribution and changes in morphology. This effect was attributed to the difference in the nucleation rate of the CaCO3 particles and the evaporation rate of ethanol at different temperatures, which is a trend that was the opposite to previously drawn conclusions about the role that temperature plays in CaCO3 precipitation.
In a precipitation system in which the temperature was varied together with the use of different organic additives, Altay et al.136 found that rhombohedral calcite particles were formed within a temperature range of 30–50 °C. At temperatures of >50 °C, calcite particles tended to agglomerate, while their surfaces and edges showed serious defects. At 80 °C and 90 °C, the formation of branch-like aragonite, with different aspect ratios, was observed. The importance of the role played by the organic additive in combination with temperature was also demonstrated in a system in which CaCO3 was precipitated as a result of the slow addition of Na2CO3 into a solution of CaCl2 containing diethylenetriaminepentaacetic acid (DTPA) over a temperature range of 60–230 °C.137 The results of product analyses were seemingly surprising, since the presence of DTPA in solution promoted the formation of pure aragonite at <100 °C, while pure calcite formed at 100 °C and 130 °C, and pure vaterite formed at 230 °C.
The interactions between the dominant experimental parameters, such as the temperature and presence of an inorganic additive, have been described in a publication by Fermani and coworkers,138 in which the influence of Mg2+ (additive concentration) on the aggregation and morphology of precipitated aragonite crystals at different temperatures (40 °C, 60 °C or 80 °C) was investigated. Different from some earlier investigations, aragonite precipitated in a chemical system in which Mg2+ acted specifically as a crystal growth modifier, rather than polymorph selector. Indeed, the results showed that an increase in Mg2+ concentration favored the aggregation of aragonite crystals in spherulites, while by increasing the temperature, the crystals showed a more regular morphology and rough spindle-like appearance (at 40 °C), later converting into needle-like structures (at 60 °C and 80 °C). In addition, the increase in the Mg2+ concentration favored the sharing of (110) faces among the crystals and the appearance of (001) faces.
(4) Additives
Since the properties of dispersed solid phases, particularly the particle size distribution, morphology, and polymorphism are determined during the early stages of CaCO3 formation, the critical question is how the additives control the nucleation process of CaCO3.91,139 CNT assumes that stochastic collisions of constituent ions take place in supersaturated solutions, and that thermodynamically unstable pre-critical nuclei are formed, which spontaneously grow into a crystalline phase after reaching a post-critical size. The size of the critical nuclei is dependent on supersaturation. A substantial point of CNT is the assumption that the nuclei exhibit all of the macroscopic and interfacial properties of the respective solid phases. However, the so-called pre-nucleation cluster (PNC) pathway assumes that the constituent ions form stable PNCs, which undergo aggregation and formation of larger liquid intermediate phases, with their final dehydration and solidification into amorphous phases and/or crystals.80 The PNCs are thermodynamically stable solutes with no phase interface and form independent of the level of supersaturation. Therefore, to discern the mechanism of action of different classes of additive molecules with CaCO3 minerals, as well as their interactions with precursors or intermediates that are postulated to exist during the early stages of mineralization, different experimental and/or computer modelling techniques, have been used and described in the literature.81,92,140However, the interaction of additives with the solid phase may take place during the crystal growth process, in which already-formed CaCO3 particles come into contact with the supersaturated solution. The additives thus may either inhibit or promote the growth and change in the morphology of the CaCO3 precipitate.28 Indeed, the additive molecules can interact with the CaCO3 surface via different mechanisms, changing the growth kinetics of the respective faces, or the surface energy.141 The additive molecules can strongly bind to the surface of the material or its crystal edges, and depending on the extent of the interactions and their surface concentration, growth can be slowed down or even completely disrupted at a given supersaturation level. When the additive molecules are adsorbed at growth sites for a short residence time, the growth is reduced. The additive molecules can also be incorporated into the crystal lattice during growth, distort the CaCO3 crystal structure, and consequently increase the free energy, which is manifested as an increase in solubility.141 Finally, certain types of additive molecules, typically surfactants, can lower the interfacial or step edge energy by adsorbing to surfaces or step edges.
Attention in recent studies has been given to controlling the growth rate, crystalline nature, stability, particle size, and surface morphology of CaCO3 in the presence of low and high molecular weight additives containing carboxyl, hydroxyl, sulfonate, and amino groups.95 In particular, additives such as DTPA,137 para-aminobenzoic acid (PABA),142 sodium dodecyl sulfate (SDS),143 sodium dodecyl sulfonate (SDSN),144 poly(acrylic acid) (PAA),145 PEG,143 poly(sodium-4-styrenesulfonate) (PSSS),25,145 poly (allylamine hydrochloride) (PAH),146 poly(vinylsulfonic acid) (PVSA),107 and polyvinyl pyrrolidone (PVP)144 have been used to control the synthesis of CaCO3. Typically, polyelectrolytes interact with Ca2+ ions and provide active sites for the nucleation of CaCO3 and subsequent agglomeration into MNPs. They can also stabilize nonequilibrium morphologies by changing the relative growth rates of different crystal faces via molecular interactions with specific crystallographic planes, resulting in the modification of the surface energy and/or the growth of CaCO3 crystals.115,144
Clearly, the temperature and initial concentration of reactants and additive are relevant precipitation parameters that may cause the formation of different precursor phases. However, it is worth noting that in complex CaCO3 precipitation systems, the subtle interplay of kinetics of nucleation, crystal growth, and dissolution also have a significant influence on the CaCO3 precipitates. In addition, the overall aging time of the systems should be considered. To obtain an in-depth understanding of such complicated precipitation and related influential parameters, consistent conditions including those mentioned above and hydrodynamic conditions need be experimentally used, which remains a tough task. Using a high concentration of reactants, high temperature and highly-charged additives, the understanding of the role of the transformation/stabilization of unstable polymorphs and the formation of specific morphologies has been to some degree revealed. For example, CaCO3 was precipitated from CaCl2 and Na2CO3 solutions in the presence of DTPA over a temperature range of 60–230 °C (Fig. 3).137 Structural analysis of the final precipitate showed that pure aragonite was formed at <100 °C (Fig. 3 and 4A, B), while calcite was formed in the range of 100–130 °C (Fig. 3 and 4C, D). Vaterite nucleation was observed to commence at 150 °C, with a steady increase in its mole fraction observed and this was in line with an increase in temperature (Fig. 4E and F), while pure vaterite was formed only at 230 °C (Fig. 4G). Such experimental results of a DTPA system highlighted the possible tendency for vaterite to be kinetically stabilized by the presence of additives in the range of 200–230 °C, whereas in the absence of DTPA, below 150 °C, ACC was converted into vaterite and rapidly transformed into calcite through dissolution–recrystallization (Fig. 3). The vaterite observed in the presence of DTPA in the range of 150–230 °C was due to the formation of precursor of metastable vaterite and the transformation of precursor of stable calcite (Fig. 3). It can be assumed that the formation of vaterite was attributed to the transformation of the existing polymorph of calcite. However, it is worth noting that, for revealing the exact transformation mechanisms in such complex CaCO3 precipitation systems, the appropriate sampling and structural analyses of the solid phases need be performed during the nucleation, crystallization and transformation. In addition, the consistent hydrodynamic conditions need be applied in experiments at low and high temperatures, and the aging times should be identical, in order to make the general conclusions about the formation mechanism. These remain a challenging task. However, the structural analysis of solid phases during precipitation in similar systems showed that, initially, the amorphous phase precipitated, which subsequently transformed into the most stable calcite phase via an intermediate vaterite phase (Fig. 3).146,147 Such experimental results of a DTPA system highlighted the possible tendency for calcite to transform into vaterite in the range of 200–230 °C (Fig. 3).
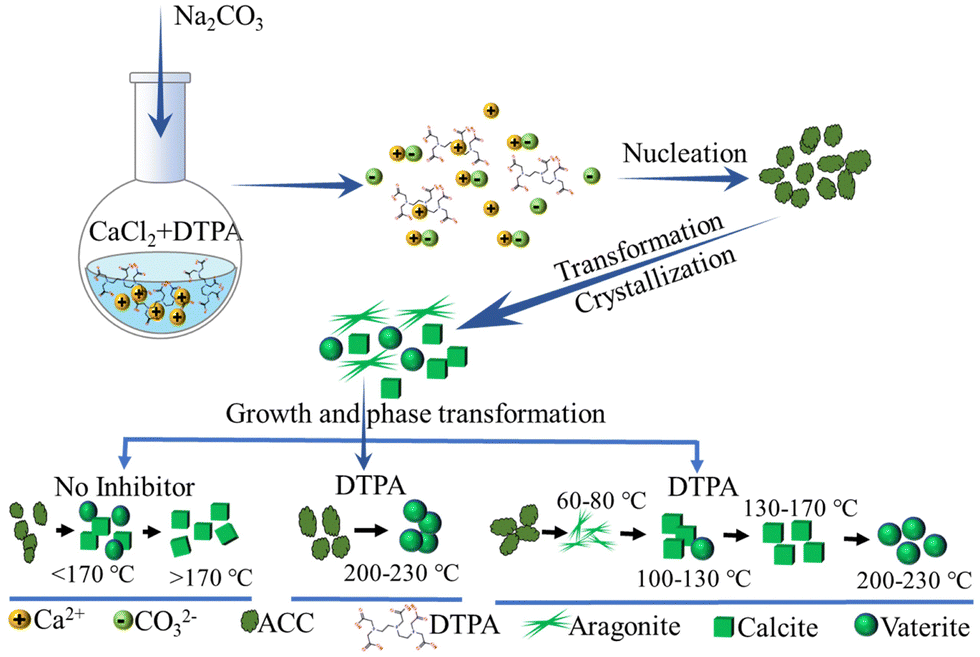 | ||
| Fig. 3 Schematic diagram showing the processes of CaCO3 precipitation, crystallization, and phase transformation in the presence of DTPA over a temperature range of 60–230 °C (designed and illustrated by the authors of the present Review based on the study reported in ref. 137). DTPA: diethylenetriaminepentaacetic acid. | ||
 | ||
| Fig. 4 SEM images of CaCO3 synthesized at different temperatures in the presence of DTPA. (A) 60 °C, (B) 80 °C, (C) 100 °C, (D) 130 °C, (E) 170 °C, (F) 200 °C, and (G) 230 °C. Reprinted from ref. 137. Copyright (2013), with permission from Elsevier. | ||
Additionally, CaCO3 polymorphs were precipitated over a range of temperatures in a precipitation system in which the pH and solvent were altered in the presence of PABA as a structure-directing agent.142 PABA features a benzene ring on which carboxylate (–COOH) and amino groups (–NH2) are attached, which makes the overall charge of this molecule susceptible to pH. At pH <7, PABA is positively charged due to the protonation of an amino group, and the formation of calcite was observed. At pH >7, the carboxylate groups were deprotonated and PABA competed with CO32− to bind Ca2+ ions. Under such conditions, the precipitate consists of calcite as the minor phase and vaterite as the major one. Pure vaterite was obtained when a water–methanol mixture was used as a solvent at pH = 8. To synthesize predominantly nanosized metastable vaterite particles, Nagaraja and co-workers introduced the negatively-charged polymeric additive PVSA into a CaCl2–Na2CO3 system.107 Besides the reaction temperature and the order of reagent addition, the study emphasized the decisive effect that the PVSA concentration has on the size and stability of vaterite. In this process, PVSA plays a dual role: it interacts with Ca2+ ions via ionic interactions, thus slowing down the nucleation rate, as well as preventing their aggregation into MPs. In addition, the transformation of vaterite into calcite was prevented by PVSA.
Furthermore, through the regulation of organic additives or templates, porous CaCO3 microspheres with various crystal polymorphs and morphologies can be obtained. In this aspect, recently, negatively- and positively-charged biodegradable and non-biodegradable polymers have been used to control the synthesis of CaCO3.145 The polymers are initially incorporated during the fabrication of the CaCO3 crystal matrix and then entrapped inside microcapsules. For example, negatively-charged PSSS having long hydrophilic was adsorbed on the surface of CaCO3 MPs and was observed to prevent re-crystallization in a similar manner to that of positively charged PVSA.107,145 Consequently, the shape of porous CaCO3 MPs of vaterite and calcite phases were shown to remain unchanged after six months of storage in water. This indicates that the CaCO3 MPs coating was stable, independently from the charge of the polymer. Such high stability in water of the CaCO3 MPs with different surface charge is very important for layer-by-layer assembly processes. In contrast, spherical vaterite MPs prepared and stabilized in the presence of positively-charged PAH transformed into rhombohedral calcite microcrystals after several weeks of storage in water at room temperature due to the occurrence of recrystallization.145 In addition, it was found that negatively-charged PAA has a strong effect on the electrostatic stability of CaCO3 particles by preventing their growth, resulting in the formed CaCO3 particles being smaller (400–600 nm) than particles produced using PSSS (600–1.1 μm).145
An additional strategy for the preparation of CaCO3 with specific properties is to use different classes of organic additives in the Ca2+–CO32− system. For example, Ji et al.143 investigated polymer–surfactant mixtures of PEG and SDS as a template for the controlled formation of hollow CaCO3 microspheres, wherein it was found that PEG strongly interacts with anionic SDS and forms complex micelles, which then act as a chemical microenvironment for the nucleation and growth of CaCO3. SDS provides a nucleation site for the crystallization of the solid phase, due to its interactions with Ca2+ in solution. Specifically, no hollow microspheres were obtained when SDS or PEG2000/10000 were used alone, while the addition of a PEG2000–SDS or PEG10000–SDS mixture into the Ca2+–CO32− system resulted in the formation of hollow calcite and vaterite crystals or hollow rhombohedral calcite crystals, respectively. The variation in molecular weight of the polymer also drastically changes the morphology and polymorph composition of the precipitate. Similarly, when using a mixture of PVP and SDSN as a template in an Na2CO3/CaCl2 system at 50 °C, hollow microspheres consisting of calcite and vaterite were prepared,144 whereas the combination of PAA/SDSN and Na2CO3/Ca(NO3)2 at 80 °C resulted in the formation of calcite hollow microspheres.100
Typically, in a solid–liquid–gas reaction system, the reaction temperature influences the solubility, ionic diffusion, and supersaturation of Ca(OH)2 and CO2. Upon an increase in temperature, the dissolution of solid Ca(OH)2 is increased, while that of CO2 is decreased.151 Recently, a new low-temperature dry ice carbonation approach was proposed for the preparation of calcite NP and porous vaterite microspheres, wherein the dry ice acts as both a source of CO2 and a coolant.152 Since the formation of CaCO3 is an exothermic process, the decrease in temperature in this process shifts the equilibrium toward the products.152,153 At low temperature, the nucleation dominates over the crystal growth, which enables the formation of more and considerably smaller CaCO3 NPs than during high-temperature synthesis.
Generally speaking, a key to preparing CaCO3 NPs is high supersaturation, which is beneficial for the nucleation process, while low supersaturation promotes crystal growth.154 In addition, intensification of mixing and mass transfer processes are appropriate for increasing supersaturation in a solid–liquid–gas carbonation system.33 The polymorph distribution of the precipitate can be controlled through the selection of the solvent of the system. Typically, calcite NPs have been obtained in water, while vaterite microspheres have been reported to form in a 75% methanol–25% water mixture.152
Since organic additives promote the absorption of CO2 gas into aqueous solution, a variety of compounds have been used to control the polymorphism of crystal phases and morphologies of CaCO3.155 The majority of the studies conducted found that the interactions of the functional moieties of the additives, such as amine, carboxyl, hydroxyl or ether groups, play significant roles in the precipitation/crystallization processes. These functional groups not only may interact with OH−, H3O+, CO32−, HCO3− or Ca2+ ions present in aqueous solution, but also with the surfaces of the solid phases during the precipitation process, which in turn affects the reaction dynamics and overall crystallization process of the system.155 Specifically, CaCO3 crystals have been found to precipitate from Ca(OH)2 suspensions and CO2 in the presence of aliphatic organic additives such as amines, diamines, and amino acids, which was attributed to be due to the alkyl chain length in the aliphatic part of the additive molecules.155 Consequently, the successive dissolution/recrystallization process is slowed in aqueous systems due to the adsorption of these organic additives on crystal surfaces, but also on reactants and intermediates. Not only polar interactions from hydrophilic functional groups, but also van der Waals interactions from hydrophobic alkyl groups, play important roles in the above-mentioned phase transformation.
In the presence of select organic additives, the formation of CaCO3 NPs with enhanced hydrophobicity typically proceeds in several steps: the nucleation of the NPs, their aggregation, and their final templated crystallization. For example, mixed polyethylene glycol phosphate (PGP) and stearic acid (SA) in a Ca(OH)2 slurry could play a synergistic role in this process.156 During the nucleation process, SA and PGP promote a certain preferential orientation at the Ca(OH)2–CaCO3–CO2 interfaces. Specifically, SA molecules are adsorbed on surfaces, while soluble PGP exists in the form of micelles and free PGP molecules. The free PGP molecules act as templates for the subsequent crystallization process. During PGP aggregation, hydrogen bonding form between the O of PGP and surface –OH groups of the CaCO3 crystals, resulting in their accretion and the formation of well-oriented polycrystalline aggregates. CaCO3 particles crystallize at the nucleation sites provided by SA and grow along the PGP chains, thereby leading to the formation of chain-like or rod-like CaCO3. To synthesize hydrophobic CaCO3 NPs with different morphologies via the carbonation of a suspension of Ca(OH)2 with a CO2/N2 gas mixture and by varying the octadecyl dihydrogen phosphate (C18H37OPO3H2) concentration, Wang et al.149 proposed that Ca2+ in (001) stereochemical (spindle-like) calcite layers might specifically interact with C18H37OPO3H2 to form P–O–Ca bonds. In this way, a number of calcium phosphate (CaP) precursor sites create favorable conditions for the nucleation and growth of CaCO3, as Ca2+ is located almost in the same lattice positions in the (001) layers, alternating with layers of CO32− in calcite. The pseudohexagonal net of C18H37OPO3H2 has a suitable inter-headgroup space, which matches the distance between the coplanar Ca2+ ions on the (001) face in calcite.157 In addition to geometric matching, the stereochemical arrangement of phosphate functional groups induces the nucleation of calcite due to the tridentate arrangement of P–O simulating the oxygen positions of CO32− lying parallel to the crystal surface.
Metastable CaCO3 polymorphs have also been obtained in Ca(OH)2–CO2 carbonation systems. Glycine (Gly) has been found to react with Ca(OH)2 to form calcium glycinate ((H2NCH2COO)2Ca).158 In this way, monodisperse spherical vaterite was prepared using Gly as an additive at room temperature and atmospheric pressure. Specifically, almost 100% spherical vaterite was obtained by adjusting the molar ratio of Gly:Ca2+. Initially, when CO2 was dissolved into a solution of (H2NCH2COO)2Ca, the concentration of CO32− increased above the precipitation limit, resulting in the nucleation of a mixture of CaCO3 polymorphs. On continuation of CO2 bubbling, the concentration of Gly liberated from (H2NCH2COO)2Ca gradually increased. This liberated Gly predominantly adsorbed onto the crystal faces of the growing calcite, thus inhibiting its further nucleation and growth, finally resulting in the formation of spherical vaterite. Such promotion of vaterite formation in the presence of Gly can also be extended to other systems such as CaCl2–NH3·H2O–Gly–CO2 or CaCl2–Gly–Na2CO3 reactions.158 In these systems, the resulting precipitate retains the polymorphic composition (vaterite), morphological characteristics (spherical particles), and size distribution, which are preserved even after thermal treatment and conversion into calcite. A model precipitation system was devised for vaterite nucleation, using an aqueous solution containing Ca(OH)2, NaCl, Gly, hyaluronic acid (HA) as a template, and supercritical CO2 as both as a reactant and a continuous (or external) phase.161 As a result of fast CO2 diffusion and dissolution in such a basic aqueous solution, spherical and hollow vaterite particles with an average diameter of 5 μm can be obtained. Recently, a novel free-solvent system was proposed for preparing spherical vaterite.110 In the high-pressure CO2 carbonation of Ca(OH)2 using urea as an additive, the urea entrapped Ca(OH)2via its melt to stabilize the formation of vaterite with its –NH2 functional group and triggered the formation of water by its decomposition to initiate the carbonation process. In this process, carbonation was initiated by the formation of Ca2+ and HCO3− upon pressurizing CO2 and the urea-entrapped Ca(OH)2, followed by the further formation of complexes between Ca2+ and the –NH2 of urea. HCO3− can be easily trapped by –NH2 to form an intermediate carbamate (–NHCOO–), which acts as a nucleation site for vaterite particles.162 In this solvent-free process, Ca(OH)2 completely transforms into spherical vaterite as the predominant phase without generating inorganic salts as by-products.
Furthermore, CaCO3 hollow or porous microspheres can be fabricated in the presence of additives. For example, high-purity hollow calcite microspheres with a micro–nano hierarchical structure have been prepared via a carbonation process at 120 °C using pressurized CO2 and the disodium salt of ethylenediaminetetraacetic acid (EDTA–2Na) as a crystal growth control agent.74 During the pressurized-CO2 carbonization reaction, EDTA complexed the Ca2+, which become the main nucleation site. In this way, CaCO3 clusters were trapped in the structure and automatically self-assembled into uniform hollow spheres. Therein, the introduction of pressurized CO2 gas increases the solubility of CO2 in the liquid phase, thus improving the driving force of the carbonization reaction. However, CO2 bubbles can also act as a template for fabricating a specific type of CaCO3. In a new one-step bubble-templating process to prepare hollow calcite rhombohedral NPs, no removal or decomposition of the template was unnecessary. Similarly, by bubbling CO2 gas through an aqueous Ca(OH)2 suspension, Kontrec et al.163 successfully prepared hollow rhombohedral calcite NPs with a mean particle size of around 100 nm. The dissolution of the precursor ACC particles that existed in close contact with the nucleation of calcite during the early stages of the process, accompanied by simultaneous release of the trapped amount of Ca(OH)2, resulted in the formation of holes with a diameter of around 50 nm at the calcite surface.
To elucidate the role of the micellar interface and to demonstrate that CaCO3 crystallization can be triggered “on-demand” by removal of the organic–inorganic interface, Stawski et al.26 investigated a highly supersaturated precipitation system comprising two aqueous phases (Na2CO3 and CaCl2) and the surfactant dioctyl sodium sulfosuccinate dissolved in oily 2,2,4-trimethylpentane. After mixing two microemulsions, the inter-droplet interactions enabled the exchange of Ca2+ and CO32− ions between the individual reverse micelles, leading to supersaturation with respect to CaCO3 inside the water nanoreactors, thus triggering the formation of Ca–CO3 clusters and the liquid-like CaCO3 phase, which may be akin to the concept of prenucleation clusters or ion pairs, and the precipitation of a solid phase.26 The instant formation of any solid CaCO3 phase could not be obtained in spite of the high initial supersaturation (c(Ca2+) = c(CO32−) = 0.075 mol dm−3) because each aqueous pool contained only approximately 1–3 Ca2+ or CO32− ions. However, the solute CaCO3 phase inside the water cores decreased the rigidity of the micellar surfactant/water interface, which promoted the aggregation of micelles and the formation of large globules. The subsequent precipitation of solid CaCO3 could be initiated by the addition of ethanol, which destabilized the globules containing the Ca–CO3 clusters. Within the clusters of the reverse micelles, the interactions between the organic interface and inorganic species compete with the purely inorganic drivers, thus changing the thermodynamics of the system and stabilizing the Ca–CO3 clusters.26 This observation is in contrast to fully inorganic aqueous systems, in which solids form instantaneously under the same chemical conditions. A different approach to initiate precipitation was proposed by Walsh et al.159 Instead of mixing two microemulsion systems, they prepared only one aqueous saturated Ca(HCO3)2 solution and subsequently subjected it to outgassing and evaporation to remove CO2 and consequently increase the supersaturation for CaCO3 precipitation. The organic phase used was SDS in octane, and in some additionally stabilized systems dodecanol co-surfactant was added. In water/SDS/oil microemulsions, discrete spheroidal vaterite MPs with a sponge-like microarchitecture and internal patterning were obtained. The spheroids are formed via the initial nucleation of vaterite at the oil–water droplet interface and patterned by microbubbles of CO2 entrapped at the surface of the water droplets (Fig. 5A).159 The precipitation of vaterite then proceeded inwardly as the droplets progressively decreased in volume as a result of the evaporation of water and oil. However, the sulfate head groups of the applied SDS most likely specifically interacted with dissolved Ca2+ and promoted the vaterite formation, as confirmed by control precipitation experiments in aqueous systems.159
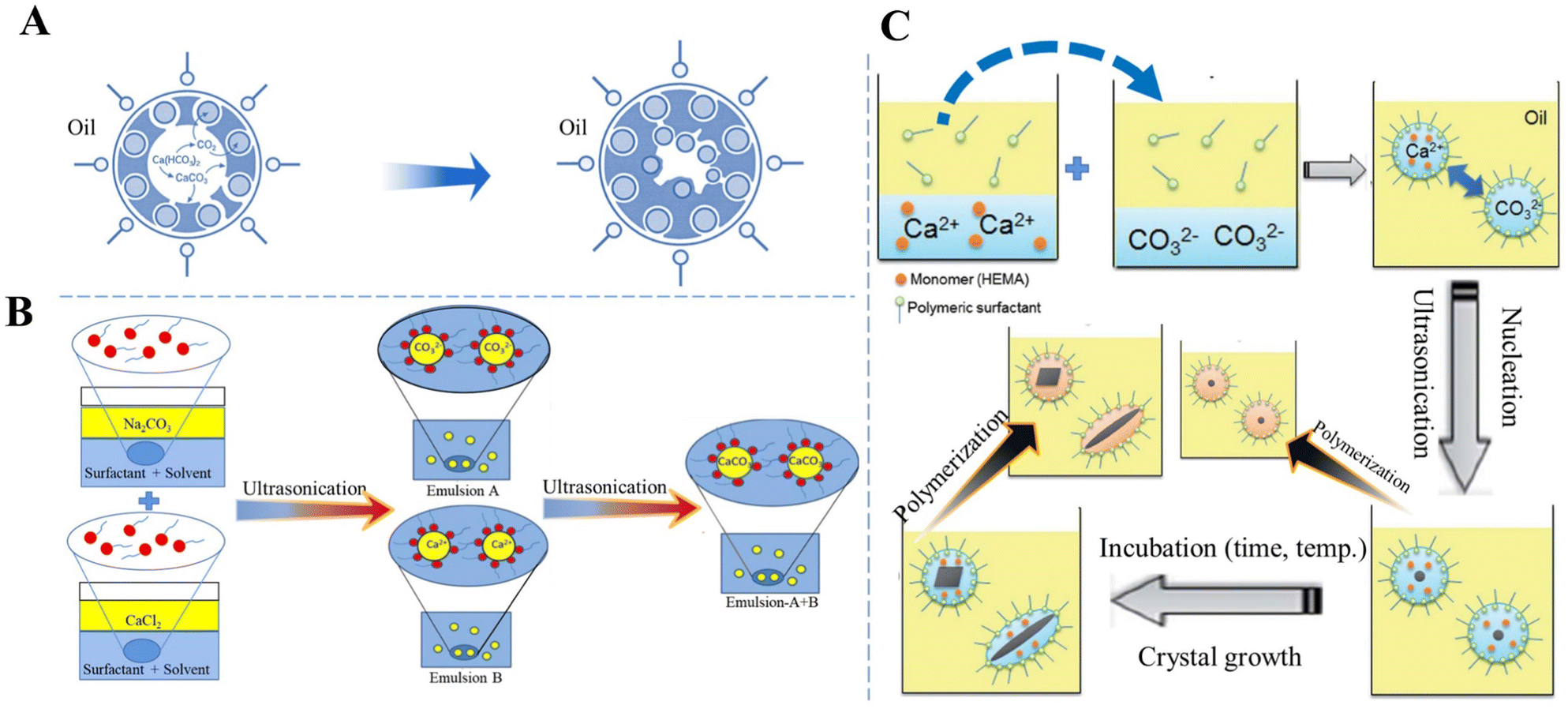 | ||
| Fig. 5 Schematic diagram showing the preparation of CaCO3 in W/O systems. (A) The formation of vaterite in water/SDS/oil microemulsions. Adapted and reprinted from ref. 159. Copyright (1999), with permission from John Wiley and Sons under the terms of the Creative Commons Attribution Non-Commercial (CC BY-NC 3.0) License. (B) Sonochemical synthesis of CaCO3via a miniemulsion technique using US. Reproduced from ref. 113. Copyright (2015), with permission from Elsevier. (C) Preparation of CaCO3 NPs with a variety of crystal shapes and structures by incubation with HEMA as a monomer and subsequent polymerization inside a nanoreactor of a miniemulsion system. Reprinted from ref. 160. Copyright (2012), with permission from the Royal Society of Chemistry. | ||
The size distribution, polymorphism, and morphology of CaCO3 precipitated in reverse emulsion systems can be controlled by changing the concentration of both the reactants and surfactant, and the water: surfactant molar ratio (ω).112 A CO2/N2 switchable surfactant/soft template (N′-dodecyl-N,N-dimethylethyl amidine bicarbonate) dissolved in either CaCl2 or Na2CO3 solution has been used to form reverse micellar solutions. At high reactant concentrations, a mixture of vaterite and calcite precipitated, whereas at intermediate or low concentrations (under 0.3 M) only calcite was produced. The size of the CaCO3 particles was increased as a result of the destabilization of the reverse micelles and their coalescence. The CaCO3 particles show a mixed crystal type of vaterite and calcite (ω = 3.52). As ω increases, the strength of the reverse micelle interfacial membrane decreases. However, with this increase in ω, the CaCO3 size becomes large and difficult to control due to the interface membrane being easily broken as a result of the collision between the reverse micelles.
To improve CaCO3 precipitation in W/O emulsion processes, Badnore et al.113 used US to prepare an emulsion and showed that in a model system containing aqueous Na2CO3 (emulsion A) and CaCl2 (emulsion B) solutions and toluene stabilized by biodegradable surfactants (Tween-80 and Span-80) as a continuous phase, CaCO3 NPs were synthesized with a diameter in the range of 20–30 nm (Fig. 5B).113 Particle size distribution was more uniform in the US CaCO3 system than that of CaCO3 prepared via conventional methods of synthesis. In addition, in the US system, porous spherical vaterite was the dominant solid phase.
A nanoreactor has also been developed based on a W/O emulsion system to carry out polymerization and mineralization simultaneously, in which the crystal structure, such as the shape and polymorph, of CaCO3 at the nanoscale can be tailored by changing the conditions (e.g., temperature, time, and ion concentration) for crystallization and the ones (e.g., initiation time and initiator concentration) for polymerization. For instance, two separate types of nanodroplets Ca(NO3)2/monomer and Na2CO3 were prepared and suspended in an cyclohexane/dimethicone oil phase, mixed via fusion and fission processes triggered by US to precipitate CaCO3 only inside the nanodroplets (Fig. 5C).160 In the presence of a monomer, 2-hydroxyethyl methacrylate (HEMA), spherical, rod-like, or ACC can be produced. The incubation period for nucleation and growth of CaCO3 with HEMA before the polymerization of HEMA inside the nonodroplets, as well as the polymerization rate, were recognized as critical factors which manipulated the structures and polymorphs of CaCO3 NPs. The crystal shape of CaCO3 was found to be controllable from rod-like to spherical forms upon increasing the rate of polymerization. Enhancement of the polymerization rate by increasing the concentration of a lipophilic initiator, AIBN, allowed HEMA to rapidly inhibit crystal growth to give rise to the entrapment of small and spherical CaCO3 particles.
Amino acids with different surface charges, polarity, and side chains can regulate the nucleation, growth, and crystal structure of CaCO3 (Fig. 6). It has been established earlier that the intraskeletal glycoproteins associated with CaCO3 biominerals are rich in side chains of carboxylated residues (i.e., aspartate and glutamate) and Gly.168 From this key information, in a pioneering study, Addadi and co-workers169 showed that the beta-sheet conformation of the adsorbed poly-aspartate macromolecules was responsible for the epitaxial oriented growth of calcite. In another milestone work, Gower and Tirrell170 revealed that the addition of poly(aspartate) to supersaturated solutions of CaCO3 led to unusual vaterite aggregates with helical extensions, as well as distorted calcite crystals that contained spiral pits. A reductionist approach, based on high-resolution synchrotron powder diffraction and analytical chemistry, was utilized to screen the 20 most common amino acids in terms of their incorporation into calcite. This research showed that the important factors were amino-acid charge, size, rigidity, and the relative pKa of the carboxyl and amino terminal groups. It was also demonstrated that cysteine, surprisingly, interacted very strongly with the mineral phase and therefore, like acidic amino acids, became richly incorporated.171 By in situ atomic force microscope observations and molecular modelling studies of calcite growth in the presence of chiral amino acids, it was found that the enantiomer-specific binding of the amino acids to calcite surface-step edges that offered the best geometric and chemical fit changed the step-edge free energies, which in turn resulted in modifications of macroscopic crystal shape.172 These results emphasize that the mechanism underlying crystal modifications through organic molecules will be better understood by considering both stereochemical recognition and the effects of binding on the interfacial energies of the growing crystal. In the presence of different concentrations of highly nonpolar hydrophobic L-valine (Val), positively-charged L-arginine (Arg) (Fig. 6A), and the less polar uncharged L-serine (Ser), CaCO3 with different morphologies and polymorphs were synthesized via the diffusion of NH3 into a saturated aqueous solution of Ca(HCO3)2 (Fig. 6B).173
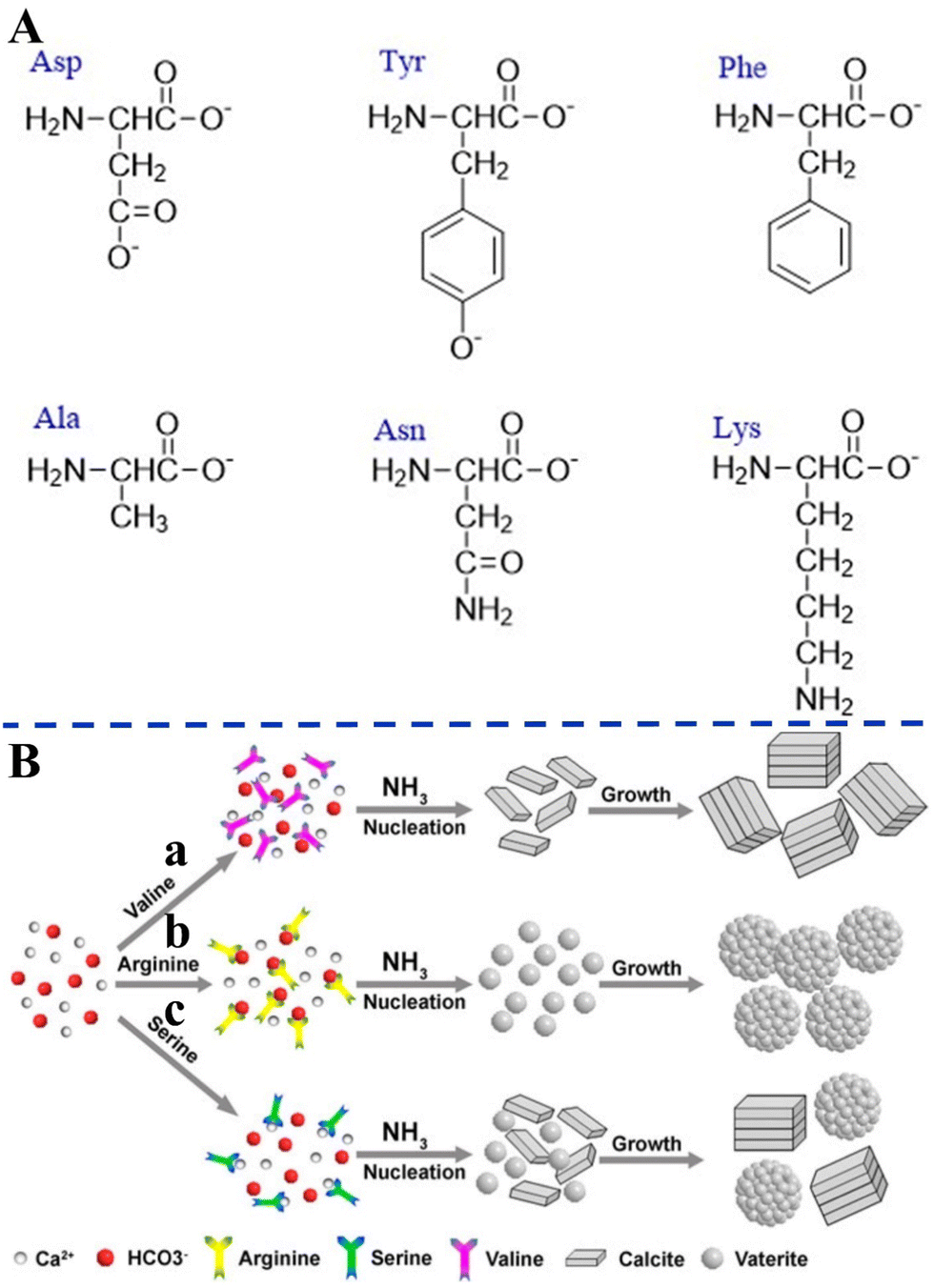 | ||
| Fig. 6 Schematic diagram illustrating the effects of different amino acids on biomimetic CaCO3 precipitation. (A) Amino acids with different surface charges, polarity, and side chains. (B) Schematic diagram illustrating the formation mechanisms of CaCO3 in the presence of different amino acids. (a) L-Valine; (b) L-arginine; and (c) L-serine. Reprinted by permission from [Springer Nature Customer Service Centre GmbH]: [Springer Nature] [ref. 173], [Copyright] (2013). | ||
As a result of the coordination interactions formed between the amino acids and Ca2+, Ca2+–amino acid complexes were first formed. Val did not affect nucleation at neutral pH, but merely changed the growth/assembly of calcite during the growth process at increasing values of pH (Fig. 6B-a). It can be speculated that this effect can be related to a change of the ζ-potential of the calcite crystal surface, which unfortunately was not reported in the paper. Without the addition of amino acids, rhombohedral calcite was predominantly produced. The effects on the nucleation were attributed to the charge of the side chains and their possible interactions with the constituent ions (Ca2+ or HCO3−) present in solution, while the effects on the crystal growth were attributed to the gradual increase in pH and deprotonation of –NH3 and –OH. The strong electrostatic interactions between the positively-charged side chain of the Arg and the HCO3− result in the formation of a stable complex, which affects the nucleation of the CaCO3 to form vaterite (Fig. 6B-b). However, with the less polar uncharged Ser, some of the Ca2+ form a complex with Ser, while the rest remained in the form of free ions, resulting in the simultaneous nucleation of calcite and vaterite to form the two polymorphs (Fig. 6B-c). Similarly, amino acids that have side chains with different charge and polarity under experimental conditions, such as L-aspartic acid (Asp), L-tyrosine (Tyr), Ser, L-asparagine (Asn), L-lysine (Lys), L-phenylalanine (Phe), and L-alanine (Ala), were found to significantly change the morphology and polymorph distribution of CaCO3 precipitates (Fig. 6A).30 Without the addition of amino acids, a mixture of typical calcite rhombohedral crystals and vaterite spherulites were observed. Asp contains two deprotonated –COOH groups that also cause relatively strong distortions of the calcite crystal lattice in the c-axis direction, which indicates Asp progressive incorporation into structure (Fig. 6A). The influences of nonpolar amino acids (Phe, Ala) on the structural and morphological properties of the CaCO3 precipitates were less pronounced. The strong effect observed for polar, particularly negatively-charged amino acids (Ser, Asn, Lys), may indicate that besides the strong impact of negatively-charged side groups on the precipitation of CaCO3, the hydrogen-bonding donor side-chain groups (–OH, –NH2 or –CO–NH–) could also influence the interactions of the amino acids with the calcite surface (Fig. 6A).
Although the majority of studies on the role of amino acids on biomimetic CaCO3 precipitation have focused on Asp and Gly, Phe, which makes up around 5% of the acid-soluble organic matrix of biomineralized structures, was also used as an organic template with the aim to induce the nucleation and growth of CaCO3.24 High concentrations of Phe have been shown to inhibit the nucleation and growth of calcite, and promote the formation of vaterite crystals with solid or hollow structures.174 Phe is an ampholyte that can release protons as an acid and accepts proton as a base. The structure of the anionic form of Phe is generated in solution due to deprotonation at high pH. This anionic form of Phe captures Ca2+via electrostatic interactions with –COO– and –NH2 groups to form a Ca2+–Phe complex. At low Phe concentration (<6 g L−1), a large amount of Ca2+ ions promote the generation of calcite and suppress the formation of vaterite. However, as the concentration of Phe increases, a large amount of Ca2+–Phe complex promotes the generation of vaterite and suppresses the formation of calcite.
Moreover, the results of the analysis of the crystal growth kinetics of a calcite seed in contact with zwitterionic model molecules with an acidic side chain, i.e., Asp derivatives, ((L-Asp)1, (L-Asp)2 and (L-Asp)3), which mimic the macromolecules found in biominerals, are a somewhat surprising and not intuitive.167 Most binding modes between dissolved molecules and calcite surface involve a positively-charged ammonium group, although attachment via negatively-charged side-chain carboxylate groups has also been frequently observed. The experimentally observed values of adsorption constants and binding free energies are in good agreement with free energy profiles determined from fully atomistic molecular dynamics simulations. As these features are also precisely the active sites for crystal growth, the growth inhibition mechanism relies primarily on the blocking of these sites, preventing further incorporation of dissolved ions and thus halting further growth. Montanari et al.175 concluded that Asp and its polymers (Asp5 and Aspn) inhibit growth, with a decreasing rate of calcite growth in line with an increase in the chain length of the amino acid.
The work on aragonite mineralization has also been inspired by the nacreous layer of marine organisms, which is the inner part of the shells of some mollusks, gastropods or cephalopods. Murai et al.114 used a synthetic multifunctional β-sheet Ac–VHVEVS–CONH2 peptide to act as a template for the in vitro biomimetic mineralization of aragonite via a “self-supplied mineralization” experimental setup. These synthetic β-sheet peptides formed three-dimensional (3D) nanofiber networks comprising assembled bilayer β-sheets, with His and Ser residues acting as the CaCO3 source supplier to hydrolyze urea to generate CO32− due to a charge relay effect between the His and the Ser residues. CaCO3 was selectively mineralized on the peptide assembly using the generated CO32−, thus leading to a fiber-like structure being obtained. However, in nature, biomineralization occurs in the presence of complex polypeptides, which interact with mineral surfaces electrostatically, but also stereochemical interactions and geometrical matching play critical roles in this process.28,176 The use of poly(amino acids) as model molecules for investigating organic/inorganic interactions in CaCO3 biomineralization is more appropriate than small monomeric molecular systems. A series of typical polypeptide molecule systems, including polyaspartic acid (PAsp), polyglutamic acid (PGlu), and polylysine (PLys), have been to precipitate CaCO3 crystals. Specifically, the crystallization of calcite on nonpolarized and polarized calcite single crystal substrates in the presence of PLys has a significant effect on the morphologies of the precipitate. At low PLys concentrations (<0.5 mg 10 mL−1), rhombohedral calcite crystals form and aggregate with an island structure, while at high PLys concentrations (1.0–3.0 mg 10 mL−1) calcite aggregates elongated in the direction perpendicular to the substrate were obtained.174 When CaCO3 was precipitated in a double-diffusion setup in agar hydrogel in the presence of PLys and PAsp, nucleation was found to occur heterogeneously on the polypeptide assemblies.82 In the presence of PLys alone, calcite and vaterite adopted dendritic and spherulitic morphologies, respectively, and calcite was the major component of the mixture. In the presence of PAsp alone or a mixture of PLys and PAsp, the precipitate comprised only hollow calcite spherulites, the cores of which might contain polypeptide assemblies and CaCO3 of poor crystallinity. It is interesting to note that the crystal growth of a calcite seed in the presence of PLys, PAsp, and PGlu suggests a dual action of PLys in its interaction with calcite.177 PLys interacts non selectively, electrostatically adsorbing at the crystal surface, thus increasing the rate of calcite growth at low concentrations and inhibiting it at high concentrations. Strong interactions between the crystal surfaces and PAsp are thought to be coordination between the carboxylic groups of the side chain of the PAsp (β-pleated sheet) and the Ca2+ cations of the calcite surface. Kim et al.166 investigated the interactions between CaCO3 and low-charge hydroxyl-rich macromolecules, by adsorbing proteins and homopolymers onto gold NPs, and concluded that the observed strong interactions may be similar to those observed in living organisms. Such complex systems also hold potential for synthesizing a class of unique single-crystal nanocomposites, which may be used as thermoelectrics, optoelectronics, catalysts, paints, and coatings or as drug delivery systems.
In addition to peptides, polypeptides, and proteins, other macromolecules have also been found to play a role in directing biomineralization and affect growth habits, phase selection, and precipitation kinetics. For example, Smeets et al.178 showed that polystyrene sulphonate (PSS) may mimic sulfated carbohydrates, which have been found to be responsible for the nucleation of aragonite in the nacreous layer of mollusk shells. The proposed mechanism of CaCO3 mineralization assumes that free Ca2+ is attracted to the SO3− group of PSS via electrostatic attraction, which leads to a locally high Ca2+ concentration in immobilized Ca–PSS globules (Fig. 7A). After the diffusion of CO32− into Ca–PSS globules, it binds with Ca2+ and replaces the weaker SO3−/Ca2+ interaction (Fig. 7B), therefore meaning that supersaturation increases and reaches the critical value for the nucleation of ACC (Fig. 7C). When the free Ca2+ in the globules is depleted, ACC stops growing, but due to the continuous generation of CO32− the supersaturation in the solution continues to rise to the level required for the nucleation of vaterite (Fig. 7D). The vaterite continues to grow until the free Ca2+ in the solution is depleted below the solubility values typical for a given set of conditions (Fig. 7E). In this model, the strong Ca binding capacity of SO3− generates the high local supersaturation required for nucleation of CaCO3.
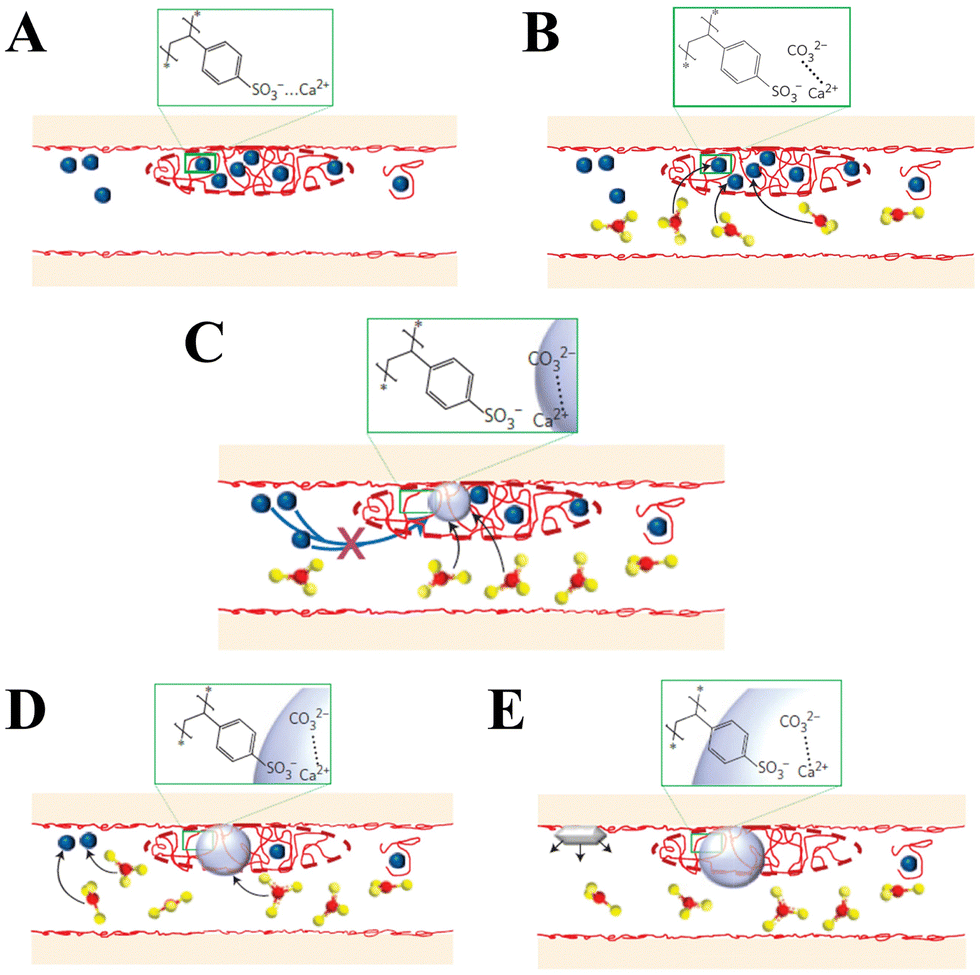 | ||
| Fig. 7 Schematic diagram illustrating the mechanism of CaCO3 mineral formation in the biomimetic matrix (blue dots: Ca2+; red dots: SO3−; red and yellow dots: CO32−). Reprinted by permission from [Springer Nature Customer Service Centre GmbH]: [Springer Nature] [ref. 178], [Copyright] (2015). | ||
As for the role played by common functional groups in CaCO3 biomineralization, Deng et al.179 proposed that nucleation of CaCO3 occurs mainly via an ion aggregation mechanism at the –COOH groups of self-assembled monolayer surfaces, resulting in the direct formation of calcite. At the surfaces of –OH and –NH2 groups of self-assembled monolayers, the synthesis of the CaCO3 phase proceeds via the formation of CaCO3 clusters, with their aggregation in solution and final adsorption onto the surface. It was also found that the surfaces of the –OH and –NH2 groups of self-assembled monolayers promote the formation of vaterite with preferred crystalline orientations, while neither amorphous nor crystalline CaCO3 modification has been observed on the –CH3 surface of groups. However, the interactions between the organic interfaces and CaCO3 surface and their effects on CaCO3 nucleation and growth could be highly complex. The polymorph distribution and precipitation rate are not equally affected by the selection of organic matrix. There are competitive or synergistic effects on nucleation and various precipitation pathways. Specifically, an organic matrix rich in ternary amines has been shown to strongly promote the nucleation of vaterite, while a carboxyl-enriched polyelectrolyte film has been found to significantly stabilize ACC in the near-surface region and equally promote the nucleation of both vaterite and calcite.31
2.2 Nucleation and growth of CaCO3
Regardless of the methods used for producing CaCO3 in a controllable way, understanding and utilizing the nucleation of CaCO3 polymorphs and their hydrated phases are crucial, yet challenging. The nucleation of CaCO3 is a process of forming a new phase in the form of small embryos from a homogeneous system (solution), the free energy of which is higher than that of the emerging new phase.141 Superficially, the driving force is expressed as the supersaturation of the solution, i.e., the ratio of the actual concentration of the solute and its equilibrium solubility at a given temperature. Nucleation is not only the initial stage of CaCO3 crystallization, it consequently determines some important properties of the obtained crystal phases, including the polymorph selection or particle size distribution.180 Following nucleation, the growth of crystals occurs via the adsorption of constituent units onto the existing crystal face, with their two-dimensional migration toward the growing steps, and final integration into the kink positions. The nucleation is an elemental process and must be controlled in to produce crystals with desired shapes, sizes and textures.22,176The mechanisms of the nucleation and growth of CaCO3 polymorphs, with and without additives, remain debatable.28,80 A basic theoretical model, also known as CNT, describes the formation of CaCO3 nuclei as being via the association of constituent units (ions) that overcome a free-energy barrier at a critical nucleus size (Fig. 8A-a).91,181 However, recent experimental evidence from advanced experimental techniques (cryo-transmission electron microscopy) and molecular simulation, have suggested that the crystallization process may proceed by pathways not as those predicted by CNT.92,140 The crystallization process of CaCO3 crystals is based on NPs rather than constituent units (ions) as in the CNT, a particle-based theoretical model referred to as NCNT. NCNT assumes that the nucleation and growth of CaCO3 are mediated by NPs, clusters, liquid-like precursors or oligomers (Fig. 8). After nucleation, the primary CaCO3 NPs assemble into iso-oriented crystals (Fig. 8A-b). When polymers or other additives are present, the primary CaCO3 NPs transform into mesocrystals (Fig. 8A-c). However, CaCO3 biominerals can be formed via stable PNCs, with aggregation into the ACC phase occurring via collision and coalescence, before transformation into a crystal phase (Fig. 8A-d).80 These above-mentioned crystallization pathways can only be interpreted using NCNT.
 | ||
| Fig. 8 Schematic diagram of the crystallization pathway of CaCO3. (A) The classical and nonclassical crystallization pathways of CaCO3. (a) Classical crystallization pathway involving layer-by-layer growth via ion (Ca2+/CO32−) addition. (b) Oriented aggregation of primary NPs forming iso-oriented crystals. (c) Mesocrystal formation via the self-assembly of primary NPs covered with organics. (d) Crystallization via liquid droplets or amorphous precursor phases. Adapted and reprinted with permission from ref. 181 and 182. Copyright (2003), with permission from John Wiley and Sons under the terms of the Creative Commons Attribution Non-Commercial (CC BY-NC 3.0) License. (B) The mechanism of nucleation according to the PNC pathway with the effects of PILP and nano-droplet adsorption by an additive (red ellipsoid). Reprinted from ref. 139. Copyright (2018), with permission from MDPI under the terms of the Creative Commons Attribution License. | ||
Stable prenucleation ionic clusters form even in undersaturated solution.80,183 For example, in a study by Gebauer,80 clusters formed at pH = 9.00 were found to have an average size of ∼70 ions, which is larger than predicted by CNT. Nevertheless, the structure of the PNCs is pH-dependent, with their precise structure remaining unknown, although it is apparent that they exhibit “solute character”. Such assumption means that hydration energy taking solvent effects into account can be ascribed to clusters, but not the surface tension, as predicted by CNT. Consequently, PNCs are formed with a negligible activation barrier, which can be matched with thermal energy. The activation barrier of PNCs is much lower than the critical nucleation enthalpy predicted by CNT for the formation of metastable clusters. However, Demichelis et al.184 concluded, using computer modelling with potential measurements, titration, and speciation calculations, as well as after structural analyses of quenched precursors, that the PNCs of CaCO3 are ionic polymers comprising Ca2+ and CO32− ions held together only via ionic interactions, with a dynamic topology consisting of chains, branches, and rings. The structural forms of the prenucleation clusters can be affected by the concentration of ions, as it results in a change in the frequency of collisions between the ions/ion pairs and clusters. In terms of a liquid-like precursor in the nucleation and growth of CaCO3, a polymer-induced liquid precursor (PILP) is a typical case (Fig. 8B).28 The additives incorporated in the PNCs or nano-droplets kinetically stabilize the PNCs against dehydration or the nano-droplets against aggregation and/or coalescence so that they grow into macroscopic PILPs. For instance, it has been found that the PILP of CaCO3 forms in the presence of charged polymers, then coalesces into thin films on a substrate (Fig. 8B), finally transforming into calcite or vaterite.93 In most cases, the formation of CaCO3 involves multiple nucleation and growth pathways. Various distinct precrystalline entities, liquid droplets, amorphous precursors and mesocrystals are present during the crystallization of CaCO3.182,185 For example, for sea urchin larval spicules growth, the transformation of a granular phase of ACC into calcite single crystals via a complex and tortuous propagation pathway by secondary nucleation. The crystallization of ACC leads to the final mesocrystalline structure.187,188
As discussed above, both the PNC- and PILP-mediated nucleation and growth pathways of CaCO3 involve an amorphous phase, followed by the formation of CaCO3 polymorph crystals. An as-yet unanswered question is how ACC transforms into crystalline CaCO3. According to a study by Bots et al.,186 the ACC crystallizes to vaterite in three stages (Fig. 9). During the first stage, hydrated and disordered ACC forms, which then rapidly transforms into a more ordered and dehydrated form of ACC. Meanwhile, vaterite grows via a spherulitic mechanism,189 followed by an intermediate stage during which the vaterite continues to form from the dissolving ACC. The third stage is controlled by the Ostwald ripening of the vaterite particles, via a dissolution reprecipitation mechanism.190 The further ripening of the vaterite is easily displaced by a dissolution–reprecipitation transformation and finally, calcite is formed. Such a pathway provides a comprehensive description of the abiotic mechanism of the crystallization of pure ACC.
 | ||
| Fig. 9 Schematic representation of the proposed multistage ACC → vaterite → calcite crystallization pathway (top) with the underlying combined reaction progress (the green triangles and full black squares represent ACC and vaterite, and the open squares and red triangles represent vaterite and calcite). Reprinted with the permission from ref. 186. Copyright (2012) American Chemical Society. | ||
When monomer and oligomer molecules are involved in CaCO3 nucleation processes as important precursors in polymers, these oligomer structures can produce CaCO3 with a specific structure. For example, an ionic polymer-like structure of CaCO3 can be generated by bubbling CO2 into an ethanol solution containing CaCl2·2H2O and triethylamine (TEA, (C2H5)3N).89 In this reaction, ethanol as the solvent favors the a formation of a hydrogen bonding between the nitrogen of TEA and the protonated carbonate (Fig. 10A). The capping with TEA stabilizes the precursors, namely ionic (CaCO3)n oligomers, in which n represents the number of Ca2+:CO32− units (Fig. 10A). The value of n within the ionic oligomers can be tuned to between 3 and 11 by altering the molar ratio of Ca2+ to TEA (Fig. 10B), where the (CaCO3)n oligomers possess a rod-like structure with a length of 1.2 nm (Fig. 10C). After removing the TEA, the oligomers change their shape from chain-like to branch-like structures (Fig. 10D). Pure monolithic ACC can be rapidly constructed via the crosslinking of the ionic oligomers with the increased density of the oligomers (Fig. 10E), which is then subsequently transformed into bulk crystals with a continuous and oriented internal structure (Fig. 10F). The oligomeric precursors can be molded into shapes to enable the construction of calcite rod arrays, which show the advantage of integrating classic inorganic and polymer chemistry in their crystallization.
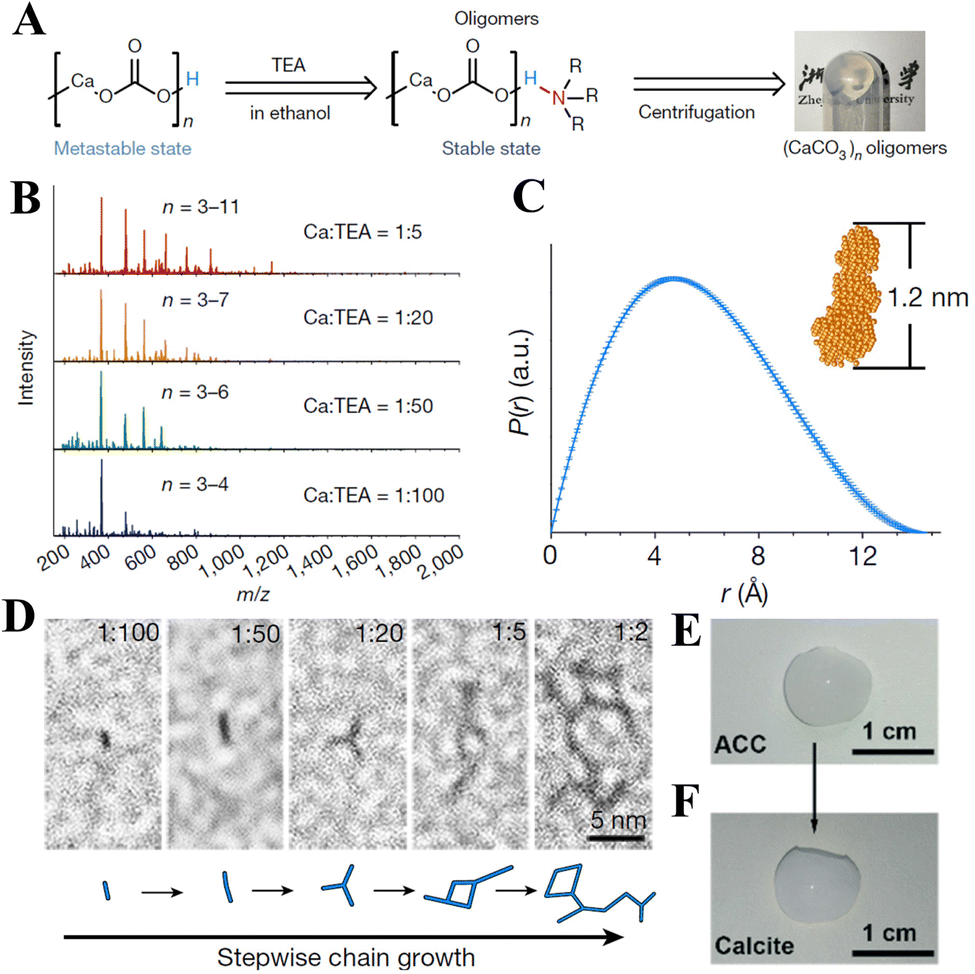 | ||
| Fig. 10 Schematic diagram of the oligomers involved in the crystallization of CaCO3. (A) Scheme of the capping strategy and reaction conditions for producing (CaCO3)n oligomers (left) and a photograph of the gel-like oligomers (right). (B) Mass spectra of (CaCO3)n oligomers with different Ca:TEA molar ratios. (C) Pair-distance distribution function (PIJr) of the (CaCO3)n oligomers. The inset shows the shape simulation of the oligomer. (D) High-resolution TEM images of (CaCO3)n oligomers grown at different Ca:TEA ratios from 1:100 to 1:2. (E) Photographs of monolithic ACC prepared from (CaCO3)n oligomers. (F) Photographs of monolithic calcite prepared from monolithic ACC. Reprinted by permission from [Springer Nature Customer Service Centre GmbH]: [Springer Nature] [ref. 89], [Copyright] (2020). | ||
3. Surface modification of CaCO3 NPs
CaCO3 NPs possess polar hydroxyl groups (–OH) on their surface, which endow them with hydrophilic and oleophobic properties.27 In addition, owing to their high surface energy, CaCO3 NPs are more prone to agglomerate via intermolecular forces and electrostatic interactions.191 For the same reason, the CaCO3 NPs are also not easy to uniformly disperse in either hydrophobic polymer or organic polymer matrices.192,193 Surface modification can be undertaken to overcome these issues and enhance the affinity and interfacial incompatibility of CaCO3 with organic molecules.194 The surface modification of CaCO3 NPs can be achieved by surface grafting and surface coating.27 Surface modifiers such as coupling agents, organic acids, anionic and cationic surfactants, and inorganic SiO2 have also been used to modify the surface of CaCO3 NPs.195 Furthermore, the surface modification process can be intensified by ball milling. Such auxiliary mechanochemical treatments not only help to break up the CaCO3 aggregates, but also enhance surface reactivity of the CaCO3 NPs toward the modifiers.3.1 Organic modification of CaCO3 NPs
It has now been found that the –OH groups on the surface of CaCO3 NPs can react with many types of organic modifiers, such as silane coupling agents, titanate coupling agents,196 aluminate coupling agents,197 unsaturated organic acids, and anionic and cationic surfactants. Silane coupling agents such as vinyltrimethoxysilane, triethoxyvinylsilane, γ-aminopropyl triethoxysilane, and methyltrimethoxysilane and γ-methacryloxypropyl trimethoxysilane have been used to modify the surface of CaCO3 NPs. Surface modification of CaCO3 NPs with these silane coupling agents occurs via condensation-like polymerization, in which the hydrolysis product of CaCO3 is used as monomer and a silane coupling agent as a chain terminator,198 where the [–Si–OR] group (where R is the methyl or ethyl of the molecular end of the coupling agent) of the silane coupling agent reacts with the –OH group on the surface of the CaCO3 NPs. Consequently, the silane coupling agents are grafted onto the surface of the CaCO3 NPs at one or several sites. The bulky organic moiety grafted onto the surface of the CaCO3 NPs lowers the surface-free energy of the CaCO3 NPs and generates surface exclusion and steric hindrance effects, thereby preventing the CaCO3 NPs from aggregating. For example, the silane coupling agent methyltrimethoxysilane (CH3Si(OCH)3) hydrolyzed in water to form CH3Si(OH)3, which then reacted with the –OH groups on the surface of the CaCO3 NPs to form Ca–O–Si bonds (Fig. 11).199 With an increase in the methyltrimethoxysilane concentration, a greater number of methyltrimethoxysilane molecules was then grafted onto the surface of the CaCO3 NPs. As a consequence, the grafted CaCO3 NPs exhibited a higher hydrophobicity than unmodified CaCO3 NPs and have potential applicability as a stabilizer for foams and emulsions. The use of US irradiation to uniformly disperse the agglomerated CaCO3 NPs into the reaction mixture reduces their surface energy, along with reducing the electrostatic forces of attraction in the material. When treated with triethoxyvinylsilane using a US-assisted technique, the surface modification of the CaCO3 NPs proceeds via the reaction of each Si–OC2H5 group with the –OH groups on the surface of the CaCO3 NPs.200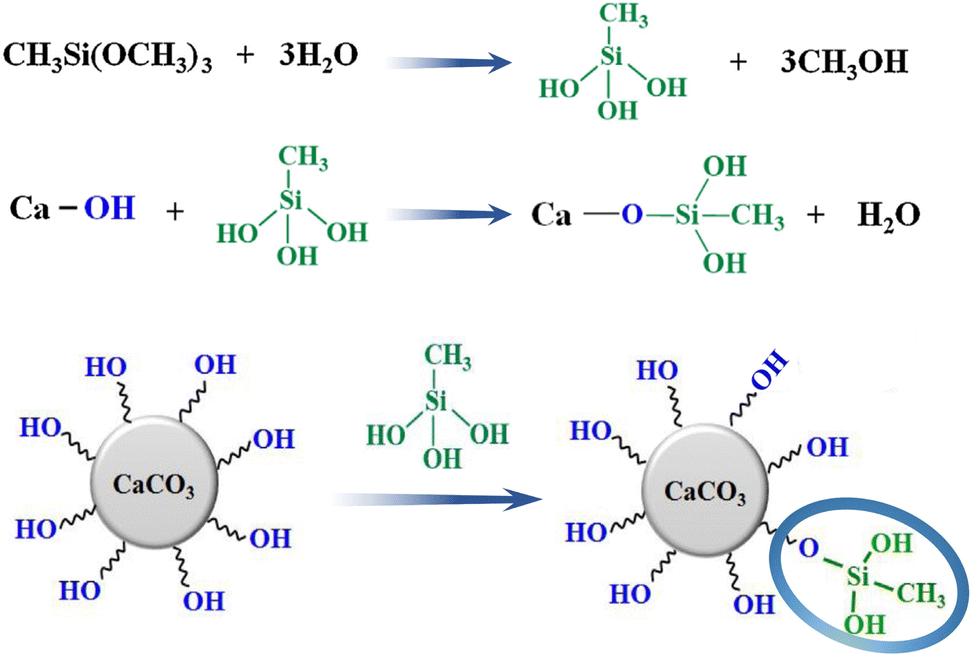 | ||
| Fig. 11 Schematic diagram showing surface modification of CaCO3 NPs by reaction with methyltrimethoxysilane. Reprinted from ref. 199. Copyright (2019), with permission from Elsevier. | ||
The surface of CaCO3 NPs can be modified with methacrylic,201 myristic,202 oleic acids,203 and SA.204 These acids are made up of two parts: a hydrophobic tail and a hydrophilic head. The carboxyl groups of these acids interact with Ca2+via an electrostatic attraction, creating a hydrophobic monolayer over the CaCO3 NPs.204 Modifications to the CaCO3 NPs can be made by so-called “dry” or “wet” methods.204 In the “dry” method, the SA is added to the filler while it is maintained in a dispersed state, usually by high shear mixing at a melting temperature that matches or exceeds that of SA. In this process, it is assumed that surface dissociation of molten SA occurs, where the H+ cations go to a surface CO32− to form surface HCO3−, and the stearate anions are chemisorbed by surface Ca2+ cations (Fig. 12A).205 Due to the steric effects of hydrocarbon chains of chemisorbed stearate (their oblique conformation), part of the Ca2+ surface centers are blocked, affecting the degree of surface dissociation of molten SA, or the amount of the chemisorbed SA. SA molecules can be adsorbed or bonded to the surface of calcite via hydrogen bonding or as dimers due to the interaction of hydrocarbon chains (Fig. 12A). In the “wet” method, a double electric layer consisting of the Stern and diffused layer was formed on the calcite/water boundary. The micelles of SA were formed, and SA of free molecules or molecules from micelles dissociated in a basic aqueous solution (pH = 10.14) (Fig. 12B). The stearate anions can be chemisorbed on primary centers of Ca2+ ions or participate in ion exchange with OH− ions from secondary surface centers (Fig. 12B). With increasing adsorption density, vertical orientation of the adsorbed SA and stearate anions with respect to the surface is achieved, and the trans-conformation of the hydrocarbon chains is maintained due to the interactions between the hydrocarbon chains and the thickness of the double electrical layer. Such a double electric layer further influences the interaction of calcite with stearate anions and SA molecules (Fig. 12C).
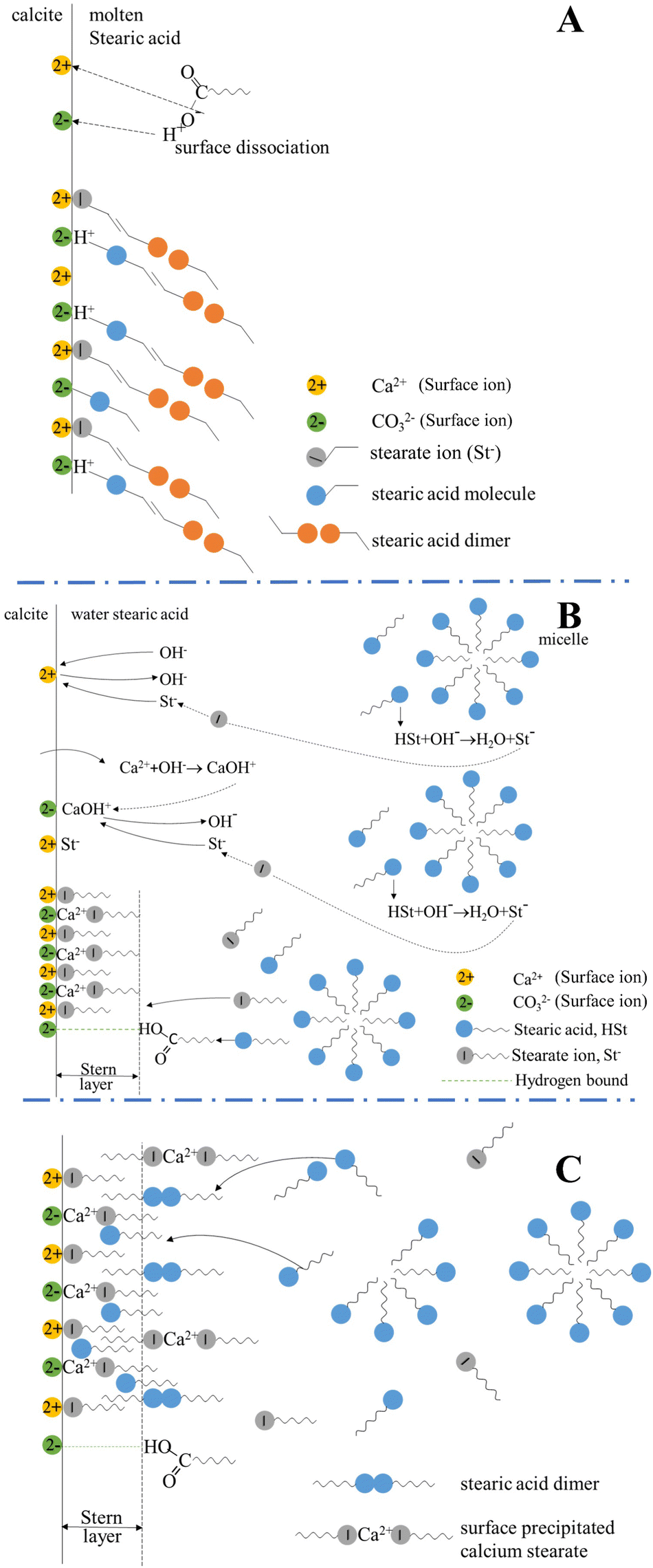 | ||
| Fig. 12 Schematic diagram illustrating the surface modification of calcite using stearic acid (SA) via the “dry process” and “wet process.” (A) Surface dissociation of molten SA on calcite and possible structure of the adsorbed layer on calcite in the “dry” method. (B) Interaction in the calcite/water and SA system, and the structure of the Stern layer in the “wet” method at a SA concentration of c < 1.5 phr (phr = mass of SA (g) per 100 g of limestone, 8.8 < pH < 10.14). (C) Structure of the adsorbed layer (the Stern layer and a part of the diffuse layer) in the calcite/water and SA system at a SA concentration of c > 1.5 phr (pH = 8.7–8.8). Slightly modified and reprinted from ref. 205. Copyright (2013), with permission from Elsevier. | ||
In addition to the above-mentioned “dry” or “wet” processes, a mechanochemical method involving the simple mixing of CaCO3 and organic acids via ball milling can be applied to modify the surface of CaCO3 NPs. The ball milling technique not only reduces the size of the CaCO3 NPs via mechanical force, but also enhances the chemical activity for the attachment of modifiers to their surface.206,207 Dry ball milling and wet ball milling have been used for the surface modification of CaCO3 NPs. Dry ball milling requires high energy consumption and a long milling time, while wet ball milling requires relatively low intensity. For example, Deepika et al.208 used a wet ball milling method to produce SA-modified CaCO3 NPs, wherein the hydrophobicity, reactivity and dispersion of the CaCO3 NPs were reported to be improved.209 The coating efficacy has been found to correlate with respect to the reactivity of CaCO3 and the modifiers in the ball milling approach.210 Yoğurtcuoğlu et al.207 also revealed that sodium oleate can be chemically adsorbed on the surface of CaCO3 NPs via wet ultra-fine grinding. In contrast, when the aqueous suspension of fatty acid salt (a mixture of RCOONa, sodium stearate or sodium oleate, RCOONa) was added into an aqueous suspension of CaCO3, the former dissociated sodium and carboxylate ions (RCOO−), which in turn then hydrolyzed and generated respective carboxylic acids (RCOOH). However, solid CaCO3 NPs partially dissolved and produced Ca2+ and CO32−, which hydrolyzed to HCO3− and OH−. It was suggested that the free Ca2+ at the surface of the CaCO3 NPs reacted electrostatically with RCOO−, while the OH− reacted with RCOOH due to the formation of hydrogen bonding (Fig. 13).193 Consequently, the CaCO3 NPs convert from hydrophilic to hydrophobic via the adsorption of a large amount of (RCOO)2Ca organic long chains. For example, the surface modification of CaCO3 NPs can be achieved using Ca(OH)2 and CO2via carbonation and modified in situ in the presence of calcium stearate ((CH3(CH2)16COO)2Ca, (RCOO)2Ca).33 (RCOO)2Ca absorbs on the surface of the CaCO3 NPs, or further adsorbs and forms a second layer over the RCOO–CaCO3 NPs. It may also transfer and be adsorbed on the surface of unmodified CaCO3 NPs.
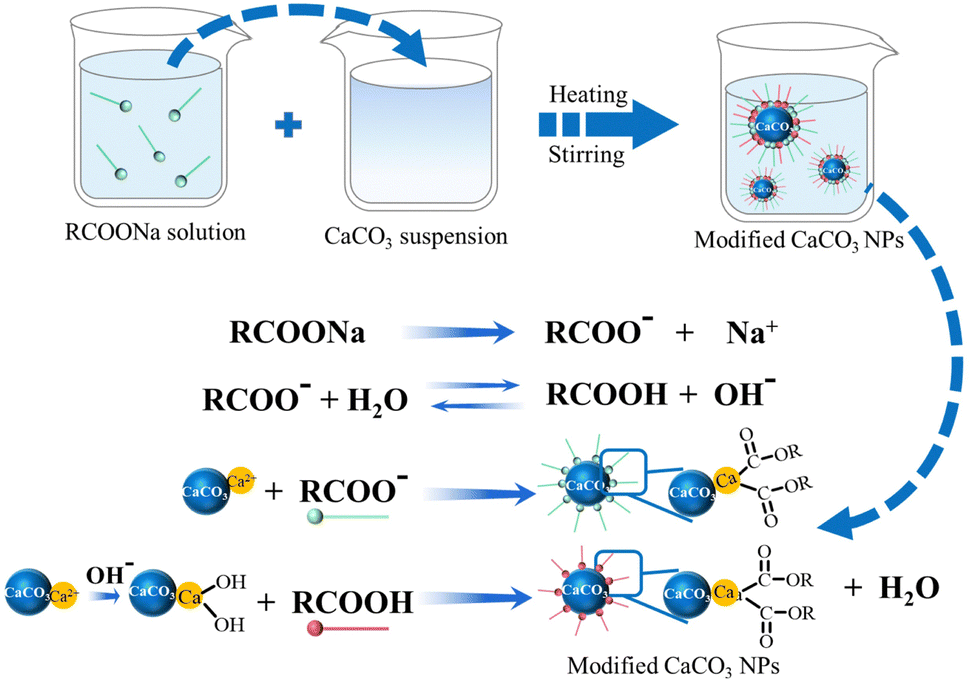 | ||
| Fig. 13 Schematic diagram showing the surface modification of CaCO3 NPs with fatty acid salt (RCOONa). (RCOO− adsorbed on Ca2+; RCOOH adsorbed on OH−via hydrogen bonding (shown in red)). Designed and illustrated by the authors of the present Review based on the study reported in ref. 193. | ||
Many types of anionic surfactants have been used to modify the surface of CaCO3 NPs via physical and chemical adsorption or chemical reactions. Anionic surfactants used to achieve this include SDS, 2-ethylhexylsulfosuccinate (AOT), fluorosurfactants (e.g., Zonyl® TBS),211 alkylbenzene sulfonic acid (ABSA),212 and laureth sulfonic acid (LSA).212 The electrostatic attraction between the positively-charged CaCO3 NPs and negatively-charged anionic surfactant headgroups results in monolayer adsorption of the surfactant at the NP–water interface.213 When CaCO3 NPs interact with amphiphilic compounds, for example linear alkylbenzene sulfonic acid (LABSA) and branched alkylbenzene sulfonic acid (BABSA), a monolayer of the surfactant is formed on the surface of the CaCO3 NPs due to electrostatic interactions and the hydrophobic effect.212 The surface of LABSA-modified CaCO3 NPs with a uniform arrangement of linear alkyl chains on it exhibits better hydrophobicity than that of BABSA-modified CaCO3 NPs. Nevertheless, LABSA and BABSA are both non-biodegradation in the environment. Two types of anionic surfactants, LSA surfactants, such as magnesium laureth sulfate (LSA-Mg) and calcium laureth sulfate (LSA-Ca), have also been used for the surface modification of CaCO3 NPs.214 However, the hydrophobicity of the modified CaCO3 NPs was found to be very compatible with hydrophobic polymers. A more recent study revealed that the use of an anionic fluorinated surfactant Zonyl TBS appeared to be more efficient in improving the hydrophobicity of CaCO3 NPs than the use of anionic hydrocarbon surfactants such as ABSA and LSA.211 Basically, anionic ABSA, LSA, and fluorinated surfactants exhibit similar adsorption on the surface of CaCO3 NPs (Fig. 14A). In a region of low anionic surfactant concentration, with an increase in anionic surfactant concentration, the surface of the CaCO3 NPs becomes increasingly more hydrophobic due to the gradual formation of a complete monolayer of an anionic surfactant on the positively-charged CaCO3 NPs via electrostatic interactions (Fig. 14A-b). However, after that, a further increase in surfactant concentration brings about a reverse change from hydrophobic to hydrophilic due to the formation of a bilayer of surfactant molecules on the surface of the CaCO3 NPs (Fig. 14A-b and c).
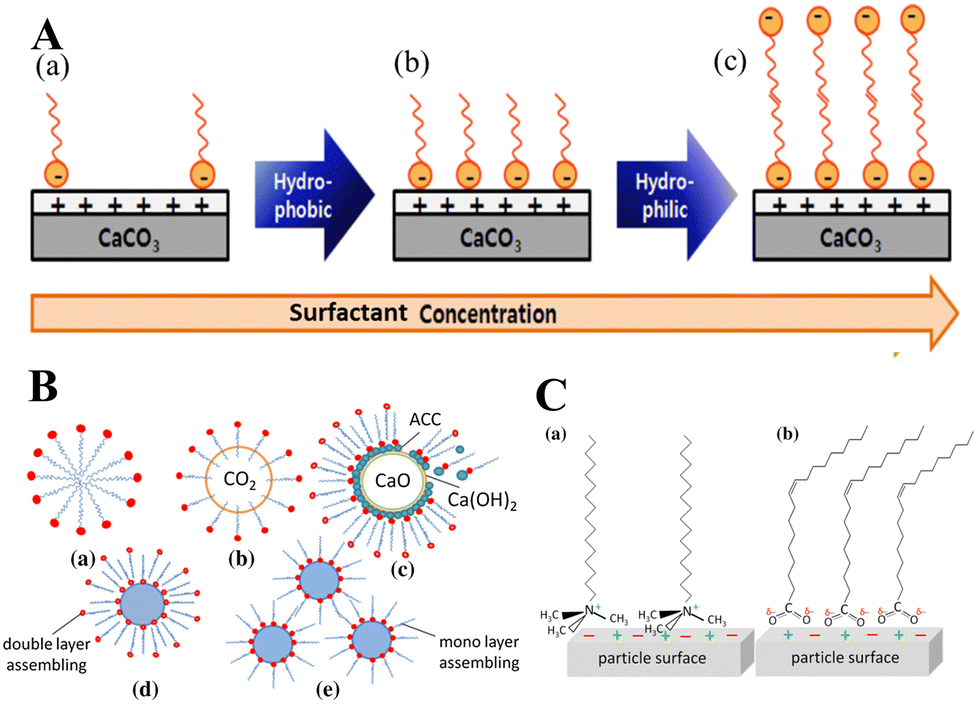 | ||
| Fig. 14 Schematic diagram illustrating the adsorption behavior of anionic and cationic surfactants on the surface of CaCO3 NPs. (A) The adsorption behavior of anionic ABSA, LSA, and fluorinated surfactants on the surface of CaCO3 NPs. (a) Adsorption of surfactant molecules on the surface of CaCO3 NPs; (b) formation of a monolayer saturated with surfactant molecules on the surface of CaCO3 NPs; (c) formation of a bilayer on the surface of CaCO3 NPs. Reprinted from ref. 212. Copyright (2014), with permission from Elsevier. (B) Assembly of the surfactant (CTAB and oleate) in the CaO–CO2–H2O system; a surfactant–water, b surfactant–CO2 bubbles, and c surfactant–CaO–Ca(OH)2–ACC; d surfactant–CaCO3 before filtration; and e surfactant–CaCO3 after filtration and washing. (C) Interaction of (a) CTAB and (b) oleate on the CaCO3 particle surface. (B and C) Reprinted by permission from [Springer Nature Customer Service Centre GmbH]: [Springer Nature] [ref. 195], [Copyright] (2015). | ||
The interaction of CaCO3 with a surfactant is remarkably affected by the size, ionic charge, and structure of the organic modifiers. A cationic surfactant reacts with negatively-charged CO32− on the surface of CaCO3 NPs. The modification can be in situ achieved in the process of bubbling carbonation (CaO–H2O–CO2) (Fig. 14B).195 When CaO particles and CO2 bubbles are introduced into an aqueous solution containing cationic surfactant (CTAB (R–N+–(CH3)3)), a fraction of the CTAB (R–N+–(CH3)3) assembles on the surface of the CO2 bubbles and the Ca(OH)2-covered CaO particles (Fig. 14B-a–c). An ACC shell is formed via reaction of the CO32− ions with the Ca(OH)2 layer on the CaO particle surface (Fig. 14B-c). The first layer of surfactant molecules adsorbs with the hydrophilic heads onto the surface of CaCO3. On this first layer, the second layer of surfactant molecules is assembled, forming a double layer structure (Fig. 14B-d). In the process of surface modification, the quaternary ammonium head (R–N+– (CH3)3) of CTAB interacts with the negatively-charged CO32− sites on the CaCO3 surface (Fig. 14C-a). In contrast, the polar carboxylate head (–COO–) with high charge density of the anionic surfactant (R-COO−) sodium oleate physically binds to or forms a complex with (coplanar) Ca2+ at the surface of CaCO3 (Fig. 14C-b). The R-COO− binds more strongly to the CaCO3 surface compared with R–N+–(CH3)3, with the largest quaternary ammonium head of the four alkyl groups bound to the central nitrogen atom. Small and linear surfactants with high charge density have been found to preferentially interact with the CaCO3 surface, while bulky surfactants need to structurally fit to the CaCO3 surface to ensure efficient interaction.195
3.2 Inorganic modification of CaCO3 NPs
Although the surface modification of CaCO3 NPs has thus far mainly focused on organic modification, the inorganic modification of CaCO3 NPs has been attempted so as to adjust their poor acid resistance and surface reaction activity.215 The surface of CaCO3 NPs modified by SiO2 can form SiO2@CaCO3 composite NPs with a core–shell structure.27 The SiO2 particles are tightly bound to CaCO3via Si–O–Ca bonds. The product exhibits the integrated properties of SiO2 and the CaCO3 NPs, showing improved surface reaction activity and acid- and heat-resistance of the CaCO3 NPs.215 In the surface modification of CaCO3 using aqueous hexafluorosilicic acid (H2SiF6) solution, amorphous SiO2 was generated from the hydrolysis of silicon hexafluoride (SiF62−), with release of F− anions. CaF2 subsequently precipitated from the reaction between F− anions and Ca2+ cations, which originated from partial dissolution of the CaCO3 surface at pH = 5–7. As a result, a layer of inorganic coating, consisting of amorphous SiO2 and crystalline CaF2 was formed on the CaCO3 surface. No partially fluorinated CaCO3 with extended structure has been detected.215SiO2@CaCO3 composite NPs have been successfully prepared via surface deposition,216 sol–gel,192 and mechanical methods.217 For example, in the preparation of SiO2@CaCO3 core–shell NPs via surface deposition, Na2SiO3 solution added to Ca(OH)2 slurry was carbonized at pH 9.0 by controlling the rate of CO2.216 The reaction was stopped when the pH value of the system dropped to 7.0, and was then aged for 2 h to obtain SiO2@CaCO3 core–shell NPs. SiO2 layers with a thickness of around 5 nm were observed to be coated continuously on the surface of the CaCO3 core. SiO2@CaCO3 core–shell NPs can also be produced using the sol–gel method via hydrolysis and condensation of tetraethyl orthosilicate (TEOS) in ethanol/water under alkaline conditions.218 A study has shown the preparation of SiO2@CaCO3 core–shell NPs via a mechanochemical method. In a study by Cui et al.,217 hydrated SiO2, 30 wt% sodium polyacrylate solution, and grinding media were mixed in water, and the mixture was ground for 90 min. Then, CaCO3 was mixed into the mixture, kept at pH 10 by the appropriate addition of ammonia, then the mixture was ground for 40 min. The mechanochemical process activates the SiO2 and CaCO3 NPs. Under alkaline conditions, the surface of CaCO3 yields Ca2+ and the surface of SiO2 yields silicon hydroxyl (Si–OH). Thus, SiO2 and CaCO3 then combine via the reaction of their hydroxy functional groups to form SiO2@CaCO3 core–shell NPs.
SiO2@CaCO3 core–shell NPs can be further functionalized to improve surface properties and endow the inorganic components with reactivity, thus improving the dispersion of CaCO3 particles and interface compatibility with organic matrices. Recently, a one-pot reaction to coat CaCO3 NPs with SiO2 and surface silanization using a bis-(γ-triethoxysilylpropyl)-tetrasulfide coupling agent has been reported.27 In this process, water was used as a solvent for the selected neutral silica sol solution, as the thermal motion of the SiO2 sol particles intensifies the Brownian motion at a relatively mild temperature (80 °C), increasing the probability of the occurrence of collisions between the SiO2 and CaCO3 NPs. This leads to instability of the sol system and the condensation of SiO2 particles on the surface of CaCO3 NPs to form a dense SiO2 coating. The further surface silanization of the SiO2@CaCO3 core–shell NPs endows the CaCO3 NPs with more reactive points, thus enhancing the crosslinking density of the composite.
4. CaCO3-Based nanostructured biomaterials
CaCO3 NPs and surface-modified derivatives prepared via controlled synthesis can be further engineered into a variety of CaCO3-based nanostructured biomaterials (Fig. 15), which can be used in bone tissue engineering, hemostatic agents, drug/gene/protein nanocarriers, diagnostic and therapeutic agents, and theranostic nanoplatforms. Firstly, CaCO3 NPs can be incorporated into a polymer matrix to generate a bioceramic that exhibits pronounced strength, excellent biocompatibility, bioactivity, and high osteoconductivity. Secondly, porous CaCO3 microspheres can be combined with biomacromolecules such as collagen and chitosan (CS) to form CaCO3-mineralized scaffolds and organic–inorganic hydrogels,75,219 which have uses in bone repair, osteoregeneration, and wound dressing. Thirdly, the combination of pH-sensitive gas-generating CaCO3 NPs with imaging contrast agents such as elements or organic fluorophores, bioimaging contrast agents for US imaging, MRI, fluorescence imaging, and multimodal imaging agents can be constructed for use in medical diagnosis (Table 3).56 Fourthly, CaCO3 functionalized using lipids or polymers shows enhanced physical stabilization and can act as a binding site for targeting agents to form porous or core–shell nanocarriers for use in oncosis, chemotherapy, PDT, and PTT.220,221 In addition, pH-sensitive CO2-generating CaCO3 NPs can be loaded with anticancer drugs and imaging agents, as a designed novel nanotheranostic platform for use in US imaging-guided chemotherapy and PDT,222,223 MRI-guided chemotherapy, and fluorescence imaging-guided chemotherapy and PDT.42,44,55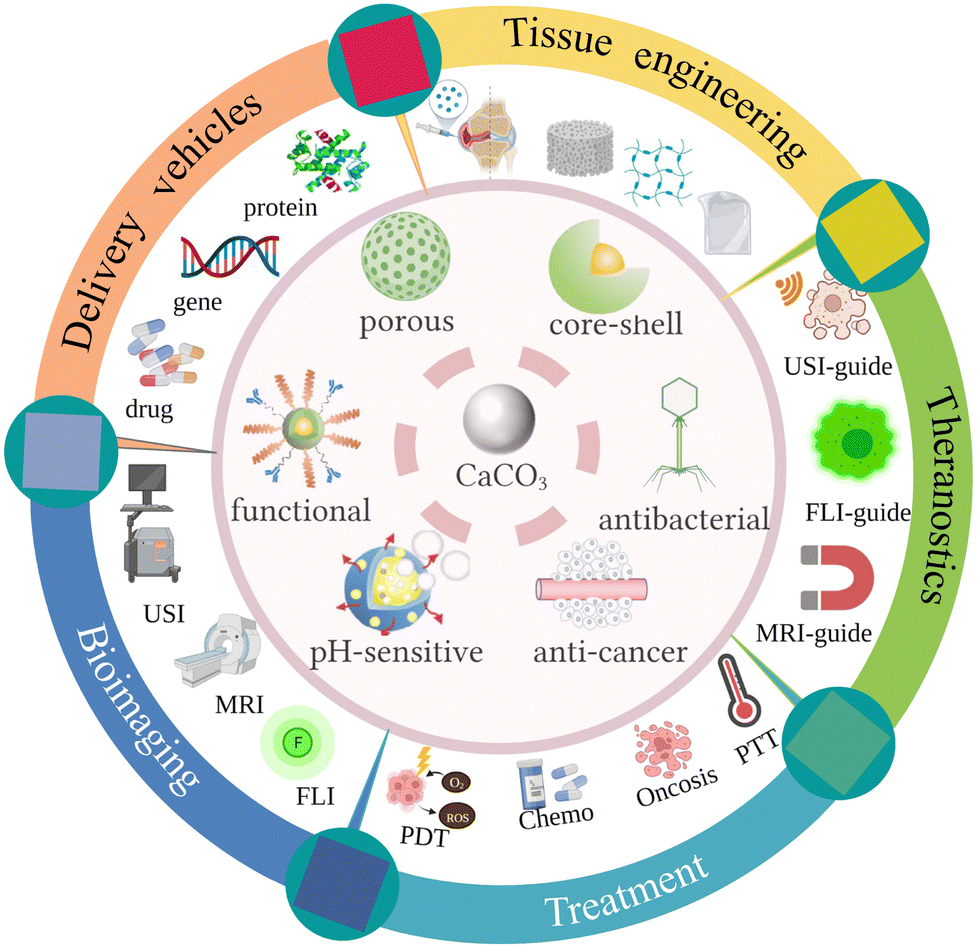 | ||
| Fig. 15 Schematic diagram showing emerging CaCO3-based nanostructured biomaterials (Created with https://BioRender.com). | ||
| CaCO3-Based nanocarriers | Synthesis methods | Therapeutic compound | Physicochemical characterization | Targeting ligand | Stimulus sensitivity | Therapy modalities | Ref. |
|---|---|---|---|---|---|---|---|
| Ce6: chlorin e6; Ce6-BMNs: Ce6-loaded bubble-generating mineralized NPs; DHA: dihydroartemisinin; HA: hyaluronic acid; HB: Hypocrellin B; GAG: glycosaminoglycan; GSH: glutathione; ICG: indocyanine green; NCOF: nanoscale covalent organic framework; NIR: near-infrared light; NMOF: nanoscale metal-organic framework; OVA: ovalbumin; PAA: polyacrylic acid; PDA: polydopamine; rGO-TEPA: tetraethylenepentamine-graphene; TCPP 4,4,4,4-(porphine-5,10,15,20-tetrayl) tetrakis(benzoic acid) | |||||||
| PAA/CaCO3 NPs | Template method | DOX | Average size:120 nm | N/A | pH | Chemotherapy | 49 |
| Heat treatment in a high-purity CO2 atmosphere | DOX loading capacity: 1.18 g of DOX per g of NPs | ||||||
| pH-sensitive sustained release | |||||||
| Vaterite CaCO3 nanoplate assemblies with exposed high-energy (001) facets | Solvothermal reaction of Ca (CH3COO)2 (c = 7 mM) in DMF/H2O mixed solvent (3:1 v/v) at 120 °C for 6 h |
DOX | Diameters ranging from 0.8 to 1.9 μm | N/A | pH | Chemotherapy | 265 |
| A maximum DOX loading capacity: 65% | |||||||
| Encapsulation efficiency: 80% | |||||||
| Sustainable release | |||||||
| CaCO3/rGO-TEPA NPs | Mineralized CaCO3 crystals in solution with rGO-TEPA | DOX | Spherical and a few cubic shaped crystals with approximately 2–4 μm in size | N/A | pH | Chemotherapy | 266 |
| DOX loading efficiency: 94.7% | |||||||
| pH-sensitive release and mild storage-release: pH 7.4:13.8%, pH 5.0:91.7% |
|||||||
| CaCO3-HA-HB NPs | CaCO3 NPs cross-linked HA through physical adsorption | HB | Hydrodynamic diameter: 410 nm | HA | pH | Photodynamic therapy | 267 |
| Enhancement of PDT cytotoxicity to cancer cells | NIR | ||||||
| HB loading capacity: 20 mg per g CaCO3 NPs as the original concentration of HB was 2 mg mL−1 | |||||||
| Cellular internalization of NPs by MCF-7 cells: 85% | |||||||
| OVA@CaCO3 NPs | Starch-templating method | OVA | Size: ≈500 nm | N/A | N/A | Therapeutic vaccines | 40 |
| The high loading rate of 19.6% | |||||||
| Increased antigen cross-presentation | |||||||
| Elicited CD8 T cell proliferation; Potent cytotoxic T lymphocytes responses, and excellent tumor rejection | |||||||
| Fe3O4@PDA@CaCO3/ICG NPs | Fe3O4 NPs: traditional alkaline precipitation method | IGG | Average hydrodynamic diameter: 134.4 nm | Magnetic field | Combinatorial photothermal/photodynamic therapy | 50 | |
| ICG: adsorbed onto PDA nanoshell through electrostatic adsorption under mildly acidic conditions established by the hydrolysis of CaCl2 | PDA | Decomposed in response to acidic TME and triggered drug release | NIR | ||||
| Increased the therapeutic bioavailability | pH | ||||||
| Low systemic toxicity | |||||||
| NMOF@DHA@CaCO3 NPs | Coprecipitation | DHA | Average sizes: 382 ± 23 nm in length and 182 ± 37 nm in width | N/A | GSH | Chemodynamic/oncosis/photodynamic therapy | 268 |
| Seed-growth strategy | TCPP | pH-triggered dissolution of outer CaCO3 mineralized layer; GSH-triggered degradation of NMOF core | pH | ||||
| Ablate the tumor completely | |||||||
| CaCO3@COF-BODIPY-2I@GAG NPs | Reversible imine bond formation at room temperature | BODIPY2I | Hydrodynamic diameter: 319.4 nm | GAG | NIR | Photodynamic and Ca2+-overload synergistic therapy | 269 |
| Bonding defect functionalization method | A significantly enhanced and selective antitumor effect on the colon tumor | pH | |||||
| Cu2O@CaCO3@HA NPs | In situ gas diffusion method | Cu2O | Average hydrodynamic diameter: 180 | HA | H2S | Photothermal/photodynamic/chemodynamic/Ca2+-overload mediated therapy | 270 |
| Turn “cold” tumors “hot” by reprogramming TAMs from pro-tumoral M2 phenotype to tumoricidal M1 phenotype | pH | ||||||
| Alg-CaCO3-PDA-PGED NPs | Biomineralization | Plasmid DNA | Hydrodynamic particle size: 429 nm | N/A | NIR | Dual-modal ultrasound and photoacoustic imaging-guided photothermal heating-enhanced gene therapy | 271 |
| PDA | Gene transfection efficiency with irradiation:46.10% | pH | |||||
| pH-responsive strong echogenic signals | |||||||
| DOX-CaCO3 hybrid NPs | Block copolymer templated in situ mineralization approach | DOX | Mean hydrodynamic diameter: ∼200 nm | N/A | pH | Ultrasound imaging-guided chemotherapy | 222 |
| pH-dependent generation of the echogenic signals | |||||||
| Ce6–BMNs | Anionic block copolymer-templated in situ mineralization method | Ce6 | Mean hydrodynamic diameter: 354 nm | N/A | pH | Ultrasound imaging-guided photodynamic therapy | 223 |
| pH-dependent generation of the echogenic signals | |||||||
| Ce6@CaCO3-PDA-PEG NPs | Gas diffusion procedure | Ce6; imaging ions (e.g., Mn2+) | Average size: ∼170 nm | N/A | NIR | Magnetic resonance/photoacoustic imaging-guided photodynamic therapy | 42 |
| Simultaneous loading of both imaging and therapeutic molecules | pH | ||||||
| pH-sensitive sustained release | |||||||
4.1 Bone regeneration and bone tissue engineering
To fabricate CaCO3-based bone substitutive cements for bone regeneration, the chosen material needs to mimic the unique structure and strength of bone and exhibit bone-forming bioactivity.233 The main bottleneck in fabricating CaCO3 ceramics is the difficulty in the sintering of CaCO3, as CaCO3 decomposes to CaO and CO2 between 600 °C and 700 °C. The feasibility of sintering cements entirely made of CaCO3 has already been demonstrated.225 Employing a highly reactive ACC phase and a metastable vaterite crystalline phase, a calcareous cement was prepared that during setting recrystallized in full agreement with Ostwald's rule of stages. The structural analysis of the phase composition during the setting and hardening periods provided evidence of the (re)crystallization of the initial metastable CaCO3 phases mostly into aragonite and/or calcite phases. The compressive strength of the CaCO3 cements (Rcomp ≤ 13 MPa) was poor, however, such mechanical properties do not hinder the use of these materials in in vivo applications such as bone filling, especially in low mechanical stress locations. Such CaCO3 cements release Ca2+ and CO32− ions or CO2 during the in vivo biodegradation with good cytocompatibility.
To achieve satisfactory clinical use, adequate tuning of the composition of CaCO3 cements is required, which requires a good understanding of their (re)crystallization to optimize CaCO3 bone cements. The adsorption of octanoic acid on the CaCO3 surface within the microstructure of CaCO3 cements has been shown to have no effect on the CaCO3 (re)crystallization progress.229 This observation opens up the possibility of functionalizing CaCO3 cements with other biomolecules such as proteins, growth factors, stem cells or antibiotics, which may enhance the protein adhesion, bactericidal effects, or drug release of cements without modifying the crystallization pathway of CaCO3. These CaCO3 cements functionalized with biomolecules exhibit high solubility, fast setting kinetics, and bioactive properties. Besides this, the creation of macropores in CaCO3 ceramics is highly recommended because they enable tissue ingrowth and accelerated osteointegration. Pore size and porosity play important roles in tissue ingrowth, defining the internal surface area available for cell adhesion, spreading and expansion, adsorption of biological metabolites, and bioresorbability at controlled rates that can match that of bone repair.234 Porous CaCO3 ceramics can be obtained via strategic methods, such as the carbonation of Ca(OH)2,234 introduction of sintering agents at low melting temperature,226 and sintering under a CO2 atmosphere.227 CaCO3 with macropores of over 100 μm can be prepared using NaCl as a pore generator by means of the carbonation of Ca(OH)2/NaCl composite solutions.234 CaCO3 composite ceramics were fabricated via the fast sintering of CaCO3 at 650 °C using the sintering agent phosphate-based glass (50P2O5·18CaO·12MgO·20Na2O), which has a low melting temperature.226 When implanted for in vivo study, the increase in bone formation and the associated degradation of the CaCO3 composite ceramics were pronouncedly greater than those of a biphasic CaP ceramic in the early implantation stage for more prolonged implantation times.226 Highly pure and monodisperse calcite can be assembled without any sintering aid under flowing CO2 at atmospheric pressure to fabricate porous CaCO3 ceramics with porosities in the range of 10–75%.227 The obtained CaCO3 ceramics exhibit faster resorption than β-tricalcium phosphate. Furthermore, both resorption and new bone formation are affected by the pore structure. However, such CaCO3 ceramics cannot promote new bone formation during long-term implantation.
The poor compressive strength of CaCO3 cements and ceramics has thus far hindered their utilization. However, in the biosphere, the precipitation of CaCO3 occurs in many living organisms in which CaCO3 acts as a structural support and solid endurable framework. In such organisms, the brittleness of pure CaCO3 is overcome by the integration of organic and inorganic materials. Biogenic CaCO3 has been considered and made into bioactive and biocompatible nacre-, and coral-based bone substitutes or porous ceramics.235,236 To enhance the compressive strength of CaCO3 cements into the range at which clinical applications (>30 MPa) are feasible and reasonable, ductile polymers have been used to reinforce brittle CaCO3 cements. CaCO3-based organic–inorganic cements exhibit strength and toughness. Myszka et al.231 reinforced brittle CaCO3 cement with a ductile polymeric 2-hydroxyethylmethacrylate. The compressive strength of CaCO3-based cements was observed to increase to 33 MPa upon the addition of 50 wt% of 2-hydroxyethylmethacrylate to a simulated body fluid solution after two weeks of hardening.
Several enrichment strategies have been reported in the literature. The first method is the direct formation of CaCO3 during hydrogel formation by delivery of CO32− and Ca2+.238 The second method is alternately soaking hydrogels in solutions of CO32− and carbonate Ca2+.239 Implantation of CaCO3-mineralized agarose hydrogels into cranial defects in rats was found to promote bone regeneration, in which CaCO3 was in the form of a mixture of calcite and vaterite. The third method involves incubation in solutions of Ca2+ and CO32− ions.240 The fourth is the diffusion of CO2 into hydrogels containing Ca2+,241 and double-diffusion systems where Ca2+ and CO32− ions diffuse into hydrogels from different reservoirs.242 Gong et al.243 developed injectable fibrin–glue composite hydrogels containing BMP-2 loaded on CaCO3 microspheres. In them, casein and heparin were dissolved in solutions of Ca2+ and CO32−, respectively. CaCO3 microspheres were co-functionalized with casein and heparin via strong affinity and thus showed osteoinductivity and specific binding sites for BMP-2 to achieve dual delivery of growth factors and Ca to promote bone regeneration. These CaCO3-based injectable nanostructured hydrogels can be injected into defects with minimum surgical invasiveness.
A novel strategy to generate CaCO3 formation inside hydrogels has been reported using enzymes such as urease, which catalyzes the conversion of urea and water to form HCO3− and NH4+. HCO3− can undergo deprotonation to form CO32−, which then subsequently reacts with Ca2+ to form CaCO3. The pH of the environment is raised by the formation of NH4+, which promotes the precipitation of CaCO3. Rauner et al.244 incorporated urease in polymer co-networks based on 2-hydroxyethyl acrylate and triethylene glycol dimethacrylate to form CaCO3 crystals within the hydrogel network. The urease-entrapped polymer co-networks were subsequently incubated in an aqueous solution containing urea and CaCl2. This resulted in the deposition of CaCO3 in the form of aragonite and calcite within the hydrogel network. Enzymatic urease-mediated mineralization of gellan gum hydrogels with CaCO3 and Mg-enriched CaCO3 has been reported for bone regeneration,245 wherein the prepared hydrogels stimulate the adhesion and growth of osteoblast-like cells.
Furthermore, CaCO3 as a crosslinking agent can be used to prepare injectable composite hydrogels. Sodium alginate is a polysaccharide extracted from natural brown algae that readily combines with Ca2+ to form Ca2+-crosslinked alginate hydrogels. However, these materials lack injectable properties due to their fast gelation time when the Ca2+ source is a CaCl2 solution. The use of a CaCO3 solution, a weak electrolyte, leads to a slower gelation rate due to the slow release of Ca2+. The formation of injectable CaCO3-crosslinked alginate composite hydrogels based on CaCO3 as a Ca2+ source has been reported.48 These CaCO3-crosslinked alginate hydrogels were prepared by blending poloxamers with alginate and crosslinked with CaCO3via chemical and physical double crosslinking. The CaCO3-crosslinked alginate hydrogels exhibit both higher mechanical strength and injectable properties compared with those of Ca2+–alginate hydrogels. However, their shear performance needs to be improved for clinical use of the in situ hydrogels, especially in the use for knee joints.
Natural coralline porous CaCO3 scaffolds have been used for bone substitution since 1970.249 Coral-derived scaffolds composed of aragonite have been reported to perform better than CaP materials when implanted in vivo250 and enhance osteoblast growth and osteogenic differentiation in vitro.251 CaCO3 can be entrapped on fibrous scaffolds by in situ synthesis via the mixing of Ca2+ and CO32− ions, providing a calcium source for bone repair.252 From this material, a porous structure can be obtained by mineral dissolution and can also be coated with functional molecules such as antibacterial and anti-inflammatory drugs or functional enzymes.
A scaffold based on electrospun polymeric polycaprolactone (PCL) fibers entrapping porous CaCO3 (vaterite) (PCL/CaCO3) has been designed for tissue regeneration with drug delivery and release functionalities.253 Due to the affinity of tannic acid (TA, C76H52O46) for Ca2+, it can be loaded on PCL/CaCO3 scaffolds to improve their antioxidant properties. The in vivo recrystallization of vaterite to calcite induces the release of TA. PCL/CaCO3 scaffolds with capabilities of carrying biologically-active molecules and release functionalities are promising candidates for implants in bone and tissue regeneration. The CaCO3 mineralized scaffolds can alter their surface hydrophobicity, enhancing cell adhesion on the surface. For example, piezoelectric poly(3-hydroxybutyrate) (PHB) and poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) fibrous scaffolds have been assembled via electrospinning and mineralized with CaCO3 in vaterite and calcite phases in situ using US.45 The CaCO3-mineralized PHB and PHBV scaffolds converted their surfaces from hydrophobic (for nonmineralized) to hydrophilic (for CaCO3-mineralized), thus enhancing osteoblast cell adhesion and proliferation, and facilitating apatite formation and osteoblast cell growth.
The scaffolds that are used to entrap CaCO3 are generally limited to polymer matrices obtained via electrospinning. However, recently, rapid prototyping processes have been used to create polymer/CaCO3 scaffolds with hierarchically porous structures. For example, Woldetsadik et al.254 prepared hierarchically porous CaCO3 scaffolds via a template-free, scalable supercritical CO2 process The supercritical-CO2 based process introduces nanoscale features to the hierarchically porous CaCO3 scaffolds. These hierarchically porous CaCO3 scaffolds exhibit a well-defined interconnected pore structure, as well as mechanical properties (e.g., hardness and Young's modulus) that are comparable to those of natural bone.
4.2 Hemostatic agents
Bleeding occurs in many clinical conditions, such as skin damage, perforation, and irregular bleeding.255 Hemostatic materials thus face the challenging requirements of rapid hemostasis, biosafety, and adaptability to different bleeding sites. In traditional Chinese medicine, powdered cuttlebone, a hemostatic material that mainly consists of CaCO3, can be orally administered to rapidly stop bleeding.256,257 It is important to note that only 0.15 mM of CaCO3 can be dissolved in water at room temperature, which is lower than the concentration of Ca2+ in human blood (2.15–2.65 mM).52 CaCO3, in the form of microspheres that exhibit high porosity and strong adsorption capacity, can be employed in irregular-shaped wounds. While the use of CaCO3 microspheres alone does not stop rapid bleeding, CaCO3–CS composites,256 CaCO3–CS acetate hydrogels,51 thrombin (Thr)-loaded CaCO3 microspheres,52 and self-propelled CaCO3-based hemostatic particles258 have been developed and successfully employed in wound healing. Ca2+ ions together with Thr, are involved in the activation of coagulation factor XIII in the coagulation system. Ca2+ also stops bleeding by increasing clot rigidity.259 CaCO3–CS composites have been prepared via a freeze-drying process.256 These composites can accelerate wound healing; healing wounds over 9 days compared to the 12 days taken for pure CaCO3 to achieve the same goal. However, the low water solubility of CS in neutral pH solution limits its further application in clinical wound healing. Recently, a chitosan acetate–CaCO3 hydrogel was developed to be used as a wound dressing, as illustrated in Fig. 16.51 In the composite hydrogel, the protons (H+) released from the charged amino groups (–NH3+) of chitosan acetate chains reacted with CaCO3, producing Ca2+ and amino groups (–NH2). CaCO3 reduces the acidity of the chitosan acetate, decreasing the potential for skin irritation. Moreover, upon reacting with CaCO3, the –NH3+ group of the chitosan acetate is no longer positively charged. As a consequence, Ca2+ ions from CaCO3 cross-link with the –NH2 groups of the chitosan acetate to form a strong and stiff colloidal hydrogel via electrostatic interactions (Fig. 16). In addition, when the hydrogel attaches to a wound site, Ca2+ ions participate in hemostasis, accelerating the formation of blood clots by favoring the conversion of prothrombin to Thr, as well as catalyzing many other coagulation-related reactions that promote the blood coagulation process.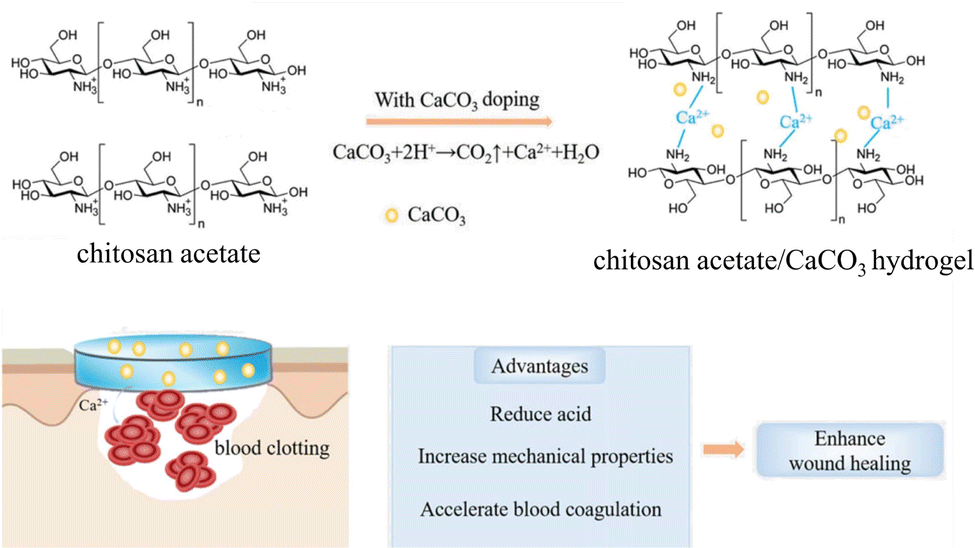 | ||
| Fig. 16 Schematic illustration of reaction occurring between chitosan acetate and CaCO3. The protons (H+) from the protonated amino groups of chitosan acetate react with CaCO3 producing Ca2+ and CO2. The Ca2+ contribute, together with the CaCO3 particles, to cross-link chitosan chains by coordination bonds involving amino and hydroxy (not shown) groups, increasing the mechanical properties. When the soft hydrogel adhered stably to a wound site, the released Ca2+ and remaining CaCO3 accelerated blood coagulation Adapted and reprinted from ref. 51. Copyright (2018), with permission from John Wiley and Sons under the terms of the Creative Commons Attribution Non-Commercial (CC BY-NC 3.0) License. | ||
Thr-loaded TA–CaCO3 microspheres (Thr@TA–CaCO3) have been prepared, wherein TA acts as a linker that connects the CaCO3 microspheres and Thr,52 and then when this material is used, Thr directly acts on the last step of the blood coagulation process together with coagulation factors to form insoluble fibrin. In particular, varying the amount of adsorbed Thr allowed control of the hemostatic time and the hemostatic ability of Thr@TA–CaCO3. For example, the Thr@TA–CaCO3 material when used reduced 94.9% of blood loss in an arterial injury in a rabbit.52
However, such CaCO3-based hemostatic materials require an external force to prevent blood flow to ultimately reach deep bleeding sites, resulting in coagulation in the superficial layers of the wound only. Attempts have thus been made to develop a mobile CaCO3-based powder hemostat with self-propelling properties so that the prepared material can overcome hemodynamic forces.255 CaCO3 rapidly produces CO2 in acidic solutions, forming porous MPs that adsorb protein, thus making it an ideal substance for making self-fueled particles. Thr-loaded CaCO3 particles with self-driving properties have been reported,258 prepared via the mixing of porous CaCO3 MPs with organic tranexamic acid (TXA–NH3+) in a 1:1 molar ratio and then injected into a buffered saline solution or whole blood. To create propelling particles that clot blood, Thr was adsorbed onto porous CaCO3 MPs. Once the Thr-loaded self-fueled CaCO3 MPs came into contact with blood, large amounts of microbubbles were generated and forced the MPs to move around in the bleeding wound. The Thr-loaded self-fueled CaCO3 MPs were pulled into the bleeding wound, allowing them to reach the deep bleeding site, thus increasing their efficiency. However, the microbubbles released from CaCO3 dissolution are mostly non-direction-selective. Thus, a significant number of MPs moved into the superficial layers of the wound, resulting in a reduction in the efficiency of the CaCO3-based hemostat. Additionally, acid–base reactions consumed the CaCO3 vector. A possible solution to this issue is to improve the movement of CaCO3-based hemostatic MPs so that they can be driven in a desired direction. Biphasic Janus self-propelled hemostatic particles (MSS@CaCO3) were prepared via the uniaxial growth of flower-like CaCO3 crystals on microporous starch (MSS) that was modified to be negatively charged (Fig. 17).255 The as-synthesized hemostatic Janus particles were then loaded with Thr (MSS@CaCO3T) and powered by the internal component CaCO3, in collaboration with protonated TXA (Fig. 17A–C). The prepared Janus MSS@CaCO3T self-propelled itself against flowing blood via a bubble detachment mechanism (Fig. 17D). CaCO3 loading on such unilateral Janus MSS@CaCO3T particles resulted in the preparation of a motorized hemostat with self-propelling properties that were shown to be able to reach deep bleeding sites (Fig. 17E and F) of the liver and femoral artery hemorrhage models, wherein the hemorrhages were rapidly controlled in ≈50 s and ≈3 min, respectively.
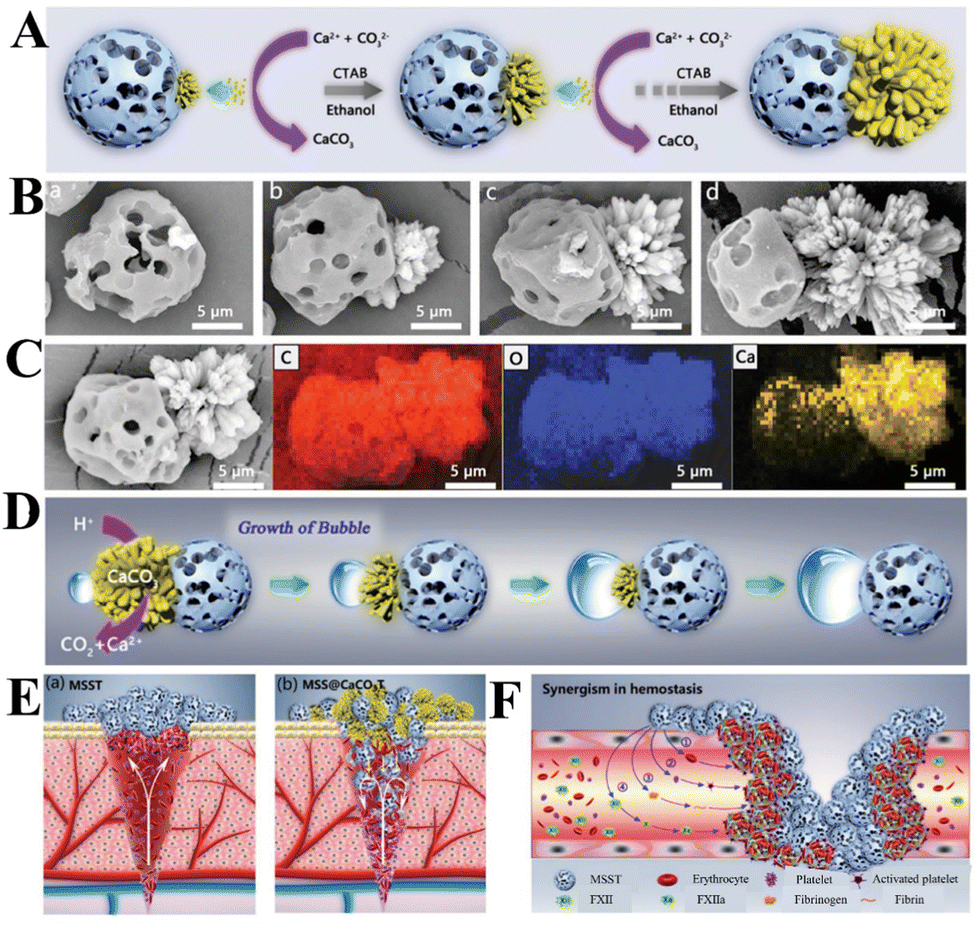 | ||
| Fig. 17 Schematic diagram showing synthetic procedure for preparing Janus MSS@CaCO3T particles for the control of perforating and irregular hemorrhages. (A) Illustration of the growth of flower-like CaCO3 crystals on negative-potential-charged microporous starch (MSS) during the synthesis of the Janus MSS@CaCO3 particles. (B) SEM images of MSS@CaCO3 Janus particles with different ratios of [MSS]/[CaCO3]: (a) 1:0.1, (b) 1:0.5, (c) 1:1, and (d) 1:2. (C) EDS mapping of the C, O, and Ca elements of the MSS@CaCO3 Janus particles with an aspect ratio of 1:1. (D) Illustration of bubble growth over a Janus particle during its self-propelling motion. (E) Illustration of the proposed hemostatic process of (a) MSST and (b) Janus MSS@CaCO3T in hemorrhage models. (F) Illustration of the proposed blood coagulation mechanism. Reprinted from ref. 255. Copyright (2020), with permission from John Wiley and Sons under the terms of the Creative Commons Attribution Non-Commercial (CC BY-NC 3.0) License. | ||
4.3 pH-Sensitive CaCO3 NPs as bioimaging contrast agents
US is one of the most powerful non-invasive, diagnostic imaging tools that is currently available. Tumor and normal body tissues have similar ultrasonic properties, thus making it difficult to distinguish between them.260 To this aim, CaCO3 has been used to construct enhanced contrast agents to improve the resolution of ultrasound imaging. The developed CaCO3-based enhanced contrast agents for US imaging have been shown to exhibit strong echogenic signals at the site of tumor tissue and excellent echo persistence.222 The prepared pH-sensitive CaCO3 NPs are stable at neutral pH and decompose to generate hyperechogenic CO2 bubbles under acidic conditions. The CaCO3 NPs can be activated under stereotypical tumoral acidic conditions.261 In addition, by combining CaCO3 NPs with paramagnetic ions such as Gd3+, pH-responsive CaCO3-based composites can be used for MR/US dual-modal imaging.4.4 Drug and gene nanocarriers for therapy
Inorganic NPs, such as SiO2, CaP, tricalcium phosphate, hydroxyapatite, and layered double hydroxides are promising drug and gene nanocarriers that exhibit high drug and gene loading.272 However, the poor biodegradability of these inorganic NPs and the lack of a controllable pH-responsive mechanism often hinder their clinical application.267 Organic NPs, such as dendrimers, liposomes, micelles, and polymer NPs, display excellent pH sensitivity, yet their use under physiological conditions is limited because of their organic composition. These limitations are not applicable to CaCO3 NPs due to their nontoxicity, biocompatibility, biodegradability, and acid sensitivity, as well as their finely controlled synthesis and ease of functionalization, thus meaning that they can be used for pH-responsive drug/gene/protein delivery (Fig. 18).95,273 The structure of the CaCO3 NPs is well maintained in normal tissues, while drug release can be triggered in the acidic environments of tumor tissues (pH 6.5–6.8) and endosomes/lysosomes (pH 4.5–6.5) (Fig. 18A and B).273 The positively-charged Ca2+ that are exposed on the surface of the CaCO3 NPs have a high affinity toward negatively-charged groups, thus making it possible to functionalize them with targeting agents to advance precision-targeted therapy and controlled drug release (Fig. 18C).274 Thus, the pH-sensitive properties of CaCO3 NPs provide a new possibility for targeting and controlling the release of drugs and genes in the human body (Table 3). | ||
| Fig. 18 Schematic diagram showing CaCO3-based drug and gene nanocarriers. (A) The pH-dependent solubility of CaCO3: equilibrium relationship between CO2 and CaCO3. Data were taken from ref. 264. (Ksp, the solubility product; Kasn, the association constant of calcium and bicarbonate; Ka, the equilibrium constant of carbonate and bicarbonate). (B) Drug-loaded CaCO3 NPs: slow dissolution at normal physiological pH (7.4) and faster dissolution and release of drugs at acidic pH (<6.5) in the tumor environment. (C) Function and characteristics of CaCO3 NPs for their use as a nanocarrier. | ||
(1) Porous CaCO3 nanocarriers. Porous CaCO3 nanocarriers exhibit a high specific surface area and high drug loading capacity, which make them suitable for use in drug delivery. Zhang et al.275 studied the biocompatibility of porous CaCO3 toward HeLa cells, with the results implying that porous CaCO3 shows good biocompatibility. For maximizing therapeutic activity by increasing the concentration of CaCO3, the effect of high dosages of reactive oxygen species (ROS) and glutathione (GSH) should be judiciously considered. Haruta et al.276 performed an insulin-loading study, which took advantage of the non-toxicity and biocompatibility of porous CaCO3. In this context, animal and clinical studies have shown that the nasal delivery of insulin loaded with porous CaCO3 can act quickly to control blood sugar levels, especially in diabetics, but longer-term toxicity tests are needed to ensure safety in humans. Ibuprofen, nifedipine, losartan potassium, metronidazole benzoate, and other poorly water-soluble drugs have been effectively loaded onto porous CaCO3via solvent evaporation, with the loading capacity equivalent to the pore capacity of the carrier and no drug loss experienced during the process.277
(2) Hybrid CaCO3/active molecular single crystals. CaCO3 single crystals are an ideal nanoplatform to construct smart vehicles for drug-targeted delivery due to their capability to adsorb and entrap drugs. The pH-sensitive solubility of CaCO3 can be exploited to release entrapped molecules only when the dissolution of the crystals occurs to achieve zero leakage of drugs at physiological pH. In pioneering research, complete structural and biological characterization of doxorubicin (DOX)/CaCO3 single crystals has led to the following points being highlighted: (i) calcite can load DOX molecules efficiently; (ii) DOX molecules are entrapped along specific crystallographic directions of calcite; (iii) the release of DOX molecules is pH-responsive and occurs preferentially close to the surface of cancer cells; and (iv) the released drug molecules are uptaken by the cancer cells, which leads to their death.278 Hybrid calcite single crystals can also be used as micro-carriers for the controlled local release of retinoic acid, which is entrapped within single crystal lattice of calcite. The release of retinoic acid occurred only in the proximity of stem cells, upon dissolution of the hybrid calcite crystals that were dispersed in a fibrin scaffold. The environment provided by this composite scaffold enabled differentiation towards neuronal cells that form a 3D neuronal network.279
(3) Organic–inorganic hybrid nanocarriers. The functionalization of CaCO3-based nanocarriers by polyelectrolytes to form organic–inorganic hybrid nanocarriers via a layer-by-layer assembly method not only leads to the formation of a material with increased drug loading capacity and controlled drug release properties, but improved thermodynamic stability and tumor targeting capability.280 In a research, CaCO3 and lentinan have been employed in a biomineralization inspired process to produce CaCO3–lentinan composites with a hierarchical composite pore structure via the hierarchical assembly of NPs.281 The hierarchical CaCO3–lentinan composites were observed to obviously reduce the release rate and prolong the release time of the anticancer drug DOX. Reduced graphene oxide–tetraethylenepentamine (rGO–TEPA) sheet matrices have been mineralized with CaCO3 to produce a CaCO3/rGO–TEPA drug carrier with a hollow structure and rough surface. CaCO3/rGO–TEPA exhibited a DOX loading efficiency of 94.7% and release efficiencies at pH 7.4 and 5.0 of 13.8% and 91.7%, respectively.266 Mesoporous PAA/CaCO3 NPs have also been synthesized that exhibit a high drug loading capacity for DOX, achieved due to the electrostatic interactions between the negatively-charged carboxyl groups (–COO−) of PAA and positively-charged DOX NPs.282
CaCO3-based nanocarriers can be functionalized with targeting agents, such as peptides, antibodies, polymers, and aptamers, to achieve targeted delivery and controlled drug release simultaneously. Surface modification of the NPs with PEG leads to their higher stability in the bloodstream as well as preventing them from being captured by macrophages, and/or adjustment of their active targeting capability so that these carriers can accumulate at a specific site.283 For example, mesoporous CaCO3 NPs were functionalized via a layer-by-layer assembly technique using SA and CS as alternating assembly materials and FA as a target molecule and then surface modified with PEG before being loaded with DOX.273 Due to the acid-mediated targeting of cells by mesoporous CaCO3 NPs, DOX-loaded FA-targeted polyelectrolyte hybrid mesoporous CaCO3 NPs exhibited significant cell-inhibitory effects. Analogously, porous CaCO3 NPs of 250 nm in size acted as a carrier to encapsulate and deliver the photosensitizer Hypocrellin B in the body for photo-dynamic therapy.267
In addition, anticancer drugs can be encapsulated via in situ CaCO3 mineralization, preventing a loss of drugs during circulation and the need for post treatment. DOX@CaCO3–hesperidin nanospheres were prepared using hesperidin as a crystallization pathway modifier at room temperature.284 The crystallization of CaCO3 was initiated by the gradual diffusion of CO2 and NH3 into a mixed solution of CaCl2, hesperidin, and DOX. DOX@CaCO3–hesperidin nanospheres, with a narrow size range of ∼200 nm, showed a drug loading efficiency of 83%, a drug loading content of 14 wt%, and pH-responsive drug release performance. A small number of cells were killed at pH = 7.4 (cell viability = 99 ± 9%), indicating very little DOX release. However, when the pH value was decreased to pH = 5.0, the cell viability dropped to 38%.
(4) CaCO3-Based synergistic therapy nanoplatform. Construction of a multifunctional stimuli-responsive CaCO3-based nanoplatform enabling improved intratumoral penetration of therapeutics and reversal of multiple-drug resistance is an important goal in achieving effective cancer treatment. For example, a nanocomposite consisting of CaCO3-modified magnetic polydopamine (PDA) NPs loaded with indocyanine green (ICG), namely Fe3O4@PDA@CaCO3/ICG (FPCI) NPs, were developed to integrate the photothermal capability of PDA with the photodynamic capability of ICG.50 In particular, CaCO3 not only entraps ICG in the form of a stable aggregate to evade blood clearance, but also facilitates the controlled release of ICG in response to the acidic TME via a self-decomposition process. The release of ICG was subsequently activated through the acidolysis of CaCO3 in the presence of protons (H+). Therefore, the acidic TME serves as a key to switch on the dissolution of the CaCO3 layer and release the entrapped ICG. Subsequently, the PDA nanoshell and released ICG function as NIR light-energy photoabsorbers for achieving photothermal therapy, while ICG also serves as a photosensitizer for inducing PDT.
A CaCO3-based multifunctional nanoplatform can be used to realize synergistic therapies (Table 3). A multifunctional therapeutic agent makes it possible to achieve a combination of PTT, PDT, Ca2+ interference, and chemodynamic therapy. To this aim, a nanoscale covalent organic framework (NCOF)-based nanoagent, CaCO3@COF–BODIPY-2I@GAG, was embedded with CaCO3 NPs and surface modified with a photosensitizer (PS) (BODIPY-2I) and a glycosaminoglycan (GAG) targeting agent for CD44 receptors on digestive tract tumor cells (Fig. 19A). Under light irradiation, the surface covalently attached BODIPY-2I generates 1O2, while the NCOF matrix-protected CaCO3 NPs are safely delivered into tumor cells via endocytosis without any premature leakage. Moreover, the pH-sensitive CaCO3 NPs dissolve in the endo/lysosomes (pH = 5.0) and release Ca2+ (Fig. 19A–C). The CaCO3@COF–BODIPY-2I@GAG coated with GAG, a specific targeting agent for CD44 receptors on digestive tract tumor cells, significantly promoted the uptake in HCT-116 cells. The CaCO3@COF–BODIPY-2I@GAG therapeutic agent thus displays significantly enhanced antitumor efficiency via synergistic PDT and 1O2-triggered Ca2+ overload both in vitro and in vivo.
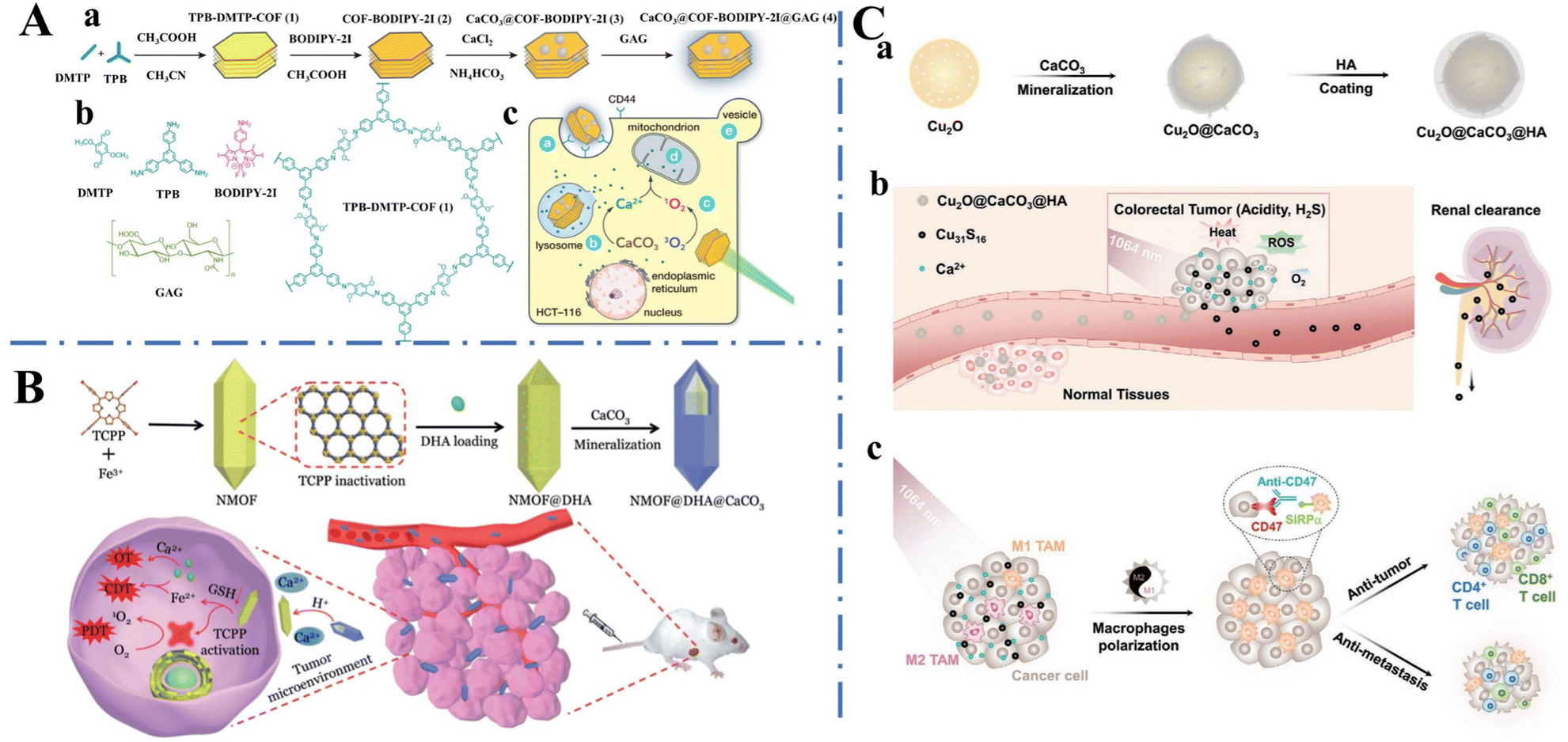 | ||
| Fig. 19 Schematic diagram of a CaCO3-based multifunctional nanoplatform. (A) A glycosylated covalent organic framework equipped with BODIPY and CaCO3 for synergistic tumor therapy. (a) Synthetic procedure of CaCO3@COF–BODIPY-2I@GAG. (b) Molecular structure of DMTP, TPB, BODIPY-2I, GAG, and TPB–DMTP–COF. (c) Synergistic intracellular Ca2+ overload and PDT. Reprinted from ref. 269. Copyright (2020), with permission from John Wiley and Sons under the terms of the Creative Commons Attribution Non-Commercial (CC BY-NC 3.0) License. (B) Preparation of the nanoplatforms and the programmed drug release for cancer therapy. Reproduced from ref. 268. Copyright (2019), with permission from John Wiley and Sons under the terms of the Creative Commons Attribution Non-Commercial (CC BY-NC 3.0) License. (C) A colorectal tumor microenvironment-activated bio-decomposable and metabolizable Cu2O@CaCO3 nanocomposite for synergistic oncotherapy a) Synthetic route of Cu2O@CaCO3@HA. (b) The tumor microenvironment-triggered bio-decomposition, anti-tumor responses, and renal clearance of Cu2O@CaCO3@HA. (c) The antitumor immune responses activated by the tumor microenvironment in combination with CD47 blockade. Reprinted from ref. 270. Copyright (2020), with permission from John Wiley and Sons under the terms of the Creative Commons Attribution Non-Commercial (CC BY-NC 3.0) License. | ||
Triple synergistic therapy in the form of chemodynamic, oncosis, and PDT, can be achieved using CaCO3 mineralized nanoplatforms. A NMOF@DHA@CaCO3 nanoplatform was constructed from a Fe-TCPP [(4,4,4,4-(porphine-5,10,15,20-tetrayl) tetrakis (benzoic acid)] NMOF (nanoscale MOF) coated with a CaCO3 mineralized layer (Fig. 19B).268 Dihydroartemisinin (DHA) was encapsulated within the pores of the NMOF coated with the CaCO3 mineralized layer, preventing leakage during its transport in the bloodstream. When the nanoplatform reaches tumor sites, the outer CaCO3 layer dissolved in the weakly acidic TME to generate NMOF@DHA and Ca2+. Then NMOF@DHA enters cancer cells and the Fe3+ of NMOF is reduced to Fe2+ due to the high concentration of intracellular glutathione (GSH), resulting in structural collapse of the NMOF structure. With this structural collapse, the release of DHA and the activation of TCPP contributed to the synergistic treatment via Fe2+–DHA-mediated chemodynamic, Ca2+–DHA-mediated oncosis, and TCPP-mediated PDT. This triple synergistic therapeutic action allows the highest cancer therapeutic efficiency to be achieved by the system, promoting the complete ablation of the tumor. Cu2O can be used as a precursor for preparing Cu2−xS therapeutic agents to perform promising H2S-activated PTT/PDT. Classic research evidence has demonstrated that the upregulation of the cystathionine-β-synthase enzyme increases the production of endogenous H2S in the acidic microenvironment of colorectal tumors. In this pursuit, acidic dissolvable CaCO3 was selected as a protective shell to prevent Cu2O from reacting with sulfide to produce active Cu2−xS before reaching the colorectal cancer sites. A core–shell Cu2O@CaCO3 nanocomposite was first prepared (Fig. 19C-a),270 before functionalizing it with HA (Cu2O@CaCO3@HA) to endow the Cu2O@CaCO3 nanocomposite with desirable targeting ability and biocompatibility. Once the Cu2O@CaCO3@HA reaches colorectal cancer sites, the CaCO3 mineralized layer dissolves and releases Ca2+, resulting in intracellular Ca2+ overload, thus resulting in Ca2+ interference therapy (Fig. 19C–b). Subsequently, the nanocomposite decomposes into Cu31S16 NPs when the exposed Cu2O comes into contact with H2S, exhibiting prominent photothermal performance, photocatalytic properties, and Fenton-like activity toward PTT/PDT/chemodynamic therapy. The hyperthermia and oxidative stress generated from the Cu2O@CaCO3 nanocomposite can efficiently reprogram macrophages from the M2 phenotype to the M1 phenotype and initiate a vaccine-like immune effect after primary tumor removal (Fig. 19C–c), further inducing an immune-favorable TME and intense immune responses toward anti-CD47 antibodies to simultaneously inhibit colorectal cancer distant metastasis and recurrence via immunotherapy.
More interestingly, mild photothermal heating (∼43 °C) increased the permeability of the cell membrane, thereby enhancing cellar uptake and gene transfection efficiency. Thus, the intergration of mild photothermal heating and biomineralized CaCO3-based gene carrier enhanced gene therapy could lead to improved therapeutic effects without causing inflammation in the body. The assembled structures of polysaccharides are supposed to induce the crystal nucleation and growth of CaCO3via electrostatic interactions or hydrogen bonds. One-dimensional (1D) Alg–CaCO3 NPs were produced by assembling micelles of polysaccharide SA as a template for their mineralization (Fig. 20).271 Then, a PDA coating was applied to conjugate the cationic polymer PGED (ethylenediamine functionalized poly(glycidyl methacrylate)) to the surface of the 1D Alg–CaCO3 NPs via a Schiff base/Michael addition reaction to endow the NPs with mild hyperthermia and p53 gene delivery functions. Under NIR-light irradiation, mild hyperthermia was triggered to enhance cellular uptake of the NPs. The pH-responsive degradation of the Alg–CaCO3–PDA–PGED gene carrier further promoted gene release, thereby enhancing gene transfection efficiency. Meanwhile, tumor cell apoptosis was induced effectively without causing any pro-inflammatory responses in the body. In addition, taking advantage of the photothermal effect of PDA and the generation of CO2 bubbles from CaCO3 dissolution in the acidic TME, dual-modal US imaging and photoacoustic imaging (PAI) was realized to monitor and guide the therapy. The CaCO3-based multifunctional platform thus represents a desirable system for the construction of photothermal gene carriers for enhanced therapeutic effects in an inflammation-free manner.
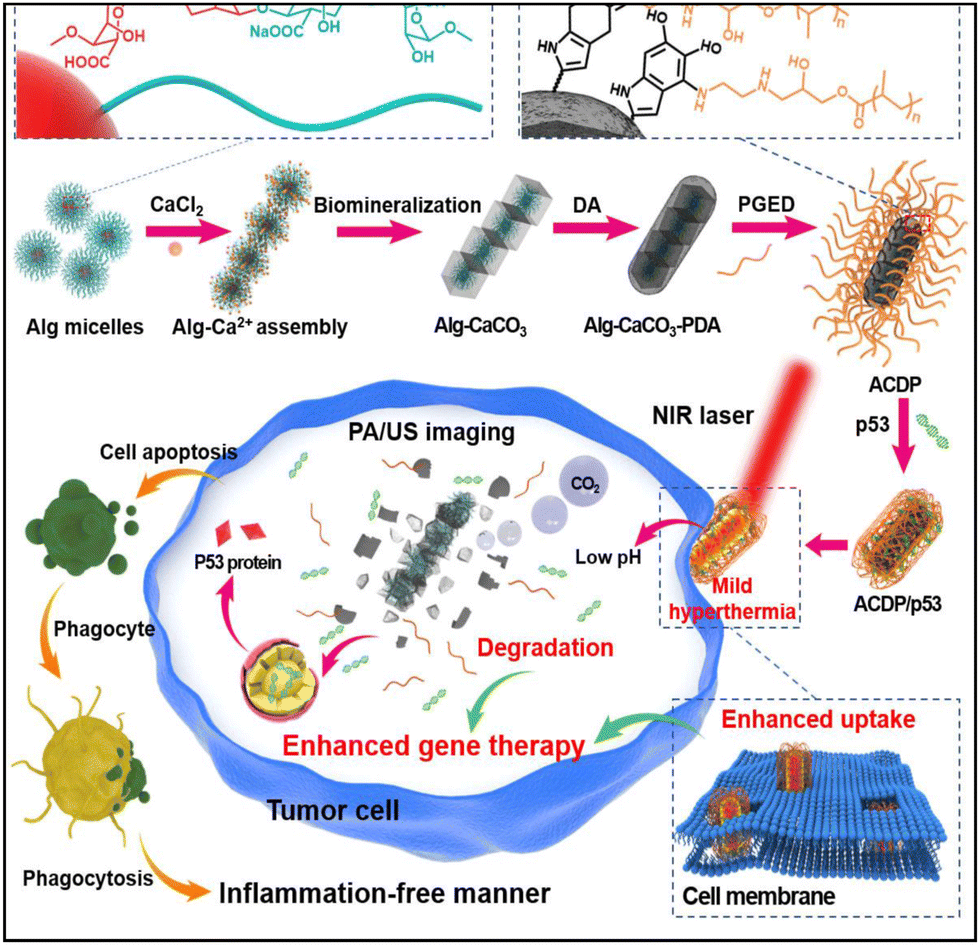 | ||
| Fig. 20 Schematic diagram of the preparation of an Alg–CaCO3–PDA–PGED gene carrier and its application in mild hyperthermia-enhanced gene therapy. Alg: alginate; PDA: polydopamine; PGED: ethylenediamine functionalized poly (glycidyl methacrylate). Reprinted from ref. 271. Copyright (2021), with permission from Elsevier. | ||
CaCO3-based nanocarriers can be further designed for efficient co-delivery of gene and drugs. Alg-modified CaCO3 NPs have been used for the co-delivery of a therapeutic gene (p53 plasmid) and DOX. The Alg/CaCO3/DNA/DOX NPs exhibited a high cell inhibition rate of around 80%, indicating that the Alg/CaCO3/DNA/DOX NPs effectively mediated gene transfection and delivered the drug to the cells. To further impart active targeting properties to CaCO3-based delivery systems, Liang et al.288 introduced a biotin moiety to a CaCO3-based nanocarrier and prepared heparin–biotin/heparin/CaCO3/CaP hybrid NPs via co-precipitation. p53 and DOX were co-loaded simultaneously on the hybrid NPs during their co-precipitation. Due to the presence of the CaCO3/CaP inorganic component and biotin moiety in the polymer chain, the heparin–biotin/heparin/CaCO3/CaP/DNA/DOX NPs exhibited both pH sensitivity and tumor-targeting properties, respectively.
CaCO3 MPs are intrinsically hydrophilic with a net neutral charge, meaning that they can enhance ovalbumin–CaCO3 nanocomposite (OVA@CaCO3) translocation across a mucus barrier. Furthermore, they can be surface modified via simple adsorption with ligands that specifically target receptors on antigen-presenting cells for targeted vaccine delivery.292 Peptide nanofiber–CaCO3 composite MPs have been synthesized via a precipitation reaction between CaCl2 and Na2CO3 and the growing CaCO3 cores captured antigenic peptide OVA nanofibers within them.292 The peptide nanofiber–CaCO3 composite MPs can be used as self-adjuvant oral vaccine delivery vehicles. They were found to efficiently penetrate the mucus barrier and localize to immune inductive sites where they elicited the production of the protective IgA isotype. Encapsulation of the nanofibers into CaCO3 microparticles not only protected the nanofibers from the harsh gastric environment but also ensured efficient delivery and antigen dose sparing for oral delivery. Traditional CaCO3 particles with bulk structures remain unsuitable for efficient antigen loading. Hierarchical CaCO3 NPs (≈500 nm) were fabricated using the soft template antigen OVA (Fig. 21A and B). The crystallization of CaCO3 was affected by the presence of the OVA soft template at the start of particle formation, leading to the formation of primary grains (≈30 nm) of vaterite. The vaterite grains were further stacked into hierarchical NPs (Fig. 21C), inside which OVA was embedded NPs (Fig. 21D). The hierarchical OVA@CaCO3 NPs were shown to express high-performance antigen delivery and cross-presentation (Fig. 21E).40 Taking advantage of the unique physicochemical properties of crystalline vaterite, cluster structure, and its high loading with OVA, the OVA@CaCO3 NPs can efficiently ferry the cargo antigen to dendritic cells and blast lysosomes for antigen escape to the cytoplasm (Fig. 21F). After navigation to acidic lysosomes, the OVA@CaCO3 NPs dissolved, generating drastic CO2 release that blasts the lysosomal membranes mechanically (Fig. 21G). The CO2 produced from the dissolution of CaCO3 induces physical stress and autophagy via the LC3/Beclin 1 pathways.
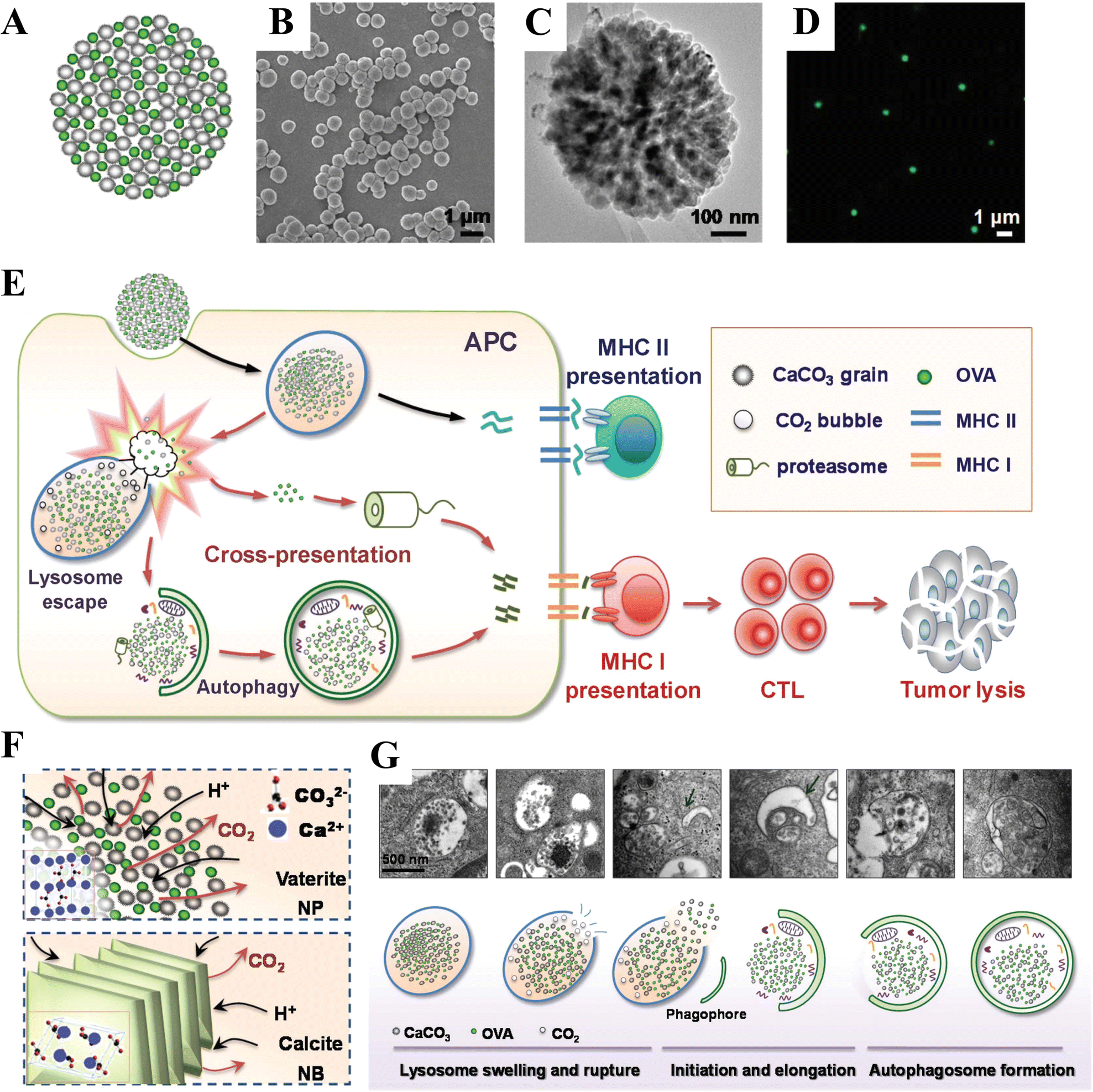 | ||
| Fig. 21 Schematic diagram illustrating characterization of an ovalbumin@CaCO3 nanoparticles (OVA@CaCO3 NP) and its application in potential antigen cross-presentation. (A) Schematic illustration, (B) SEM image, and (C) TEM image showing the hierarchical structure of an OVA@CaCO3 NP; (D) CLSM image demonstrating the successful doping of OVA (green) inside NPs. (E) Antigen cross-presentation induced by OVA@CaCO3 NP-related lysosome escape and autophagy. (F) The OVA@CaCO3 NP/NB (traditional CaCO3 nanobulk (NB, 500 nm, lamellae stacking, calcite crystal form)) dissolution mechanism. (F) TEM images and a corresponding scheme illustrating lysosome escape and autophagy. Reprinted from ref. 40. Copyright (2018), with permission from John Wiley and Sons under the terms of the Creative Commons Attribution Non-Commercial (CC BY-NC 3.0) License. | ||
4.5 Microenvironment-activated CaCO3-containing nanocomposites as diagnostic and therapeutic agents
To further enrich the properties of CaCO3, surface modification is an effective strategy by which to prepare a multifunctional CaCO3 diagnostic and therapeutic nanoplatform. The hollow CaCO3–PDA nanocomposite was prepared via a one-pot, dopamine-mediated biomineralization inspired method using a gas diffusion procedure (Fig. 22A).42 The as-synthesized CaCO3–PDA was then modified with PEG and loaded with the effective photosensitizer chlorin e6 (Ce6) to form Ce6@CaCO3–PDA–PEG hollow NPs (Fig. 22B). The photoactivity of the loaded Ce6 was quenched by the strong absorption of PDA at neutral pH, which was activated with recovered fluorescence and improved singlet oxygen generation ability at reduced pH within the TME (Fig. 22C–E). These Ce6@CaCO3–PDA–PEG hollow NPs are thus promising for use in fluorescence imaging-guided cancer PDT with reduced skin phototoxicity. In addition, the M-loaded (M = Fe3+, Zn2+, Mn2+, or Co2+) CaCO3–PDA–PEG NPs were formed NPs via metal ion chelation and then modified with PEG (Fig. 22F), with the resultant material showing excellent T1-contrasting ability for PAI (Fig. 22H). In particular, the CaCO3–PDA–PEG hollow NPs allow the simultaneous loading of both imaging (e.g., Mn2+) and therapeutic molecules (e.g., Ce6) for fluorescence/MRI/PAI-guided cancer therapy (Fig. 22E, G and H).
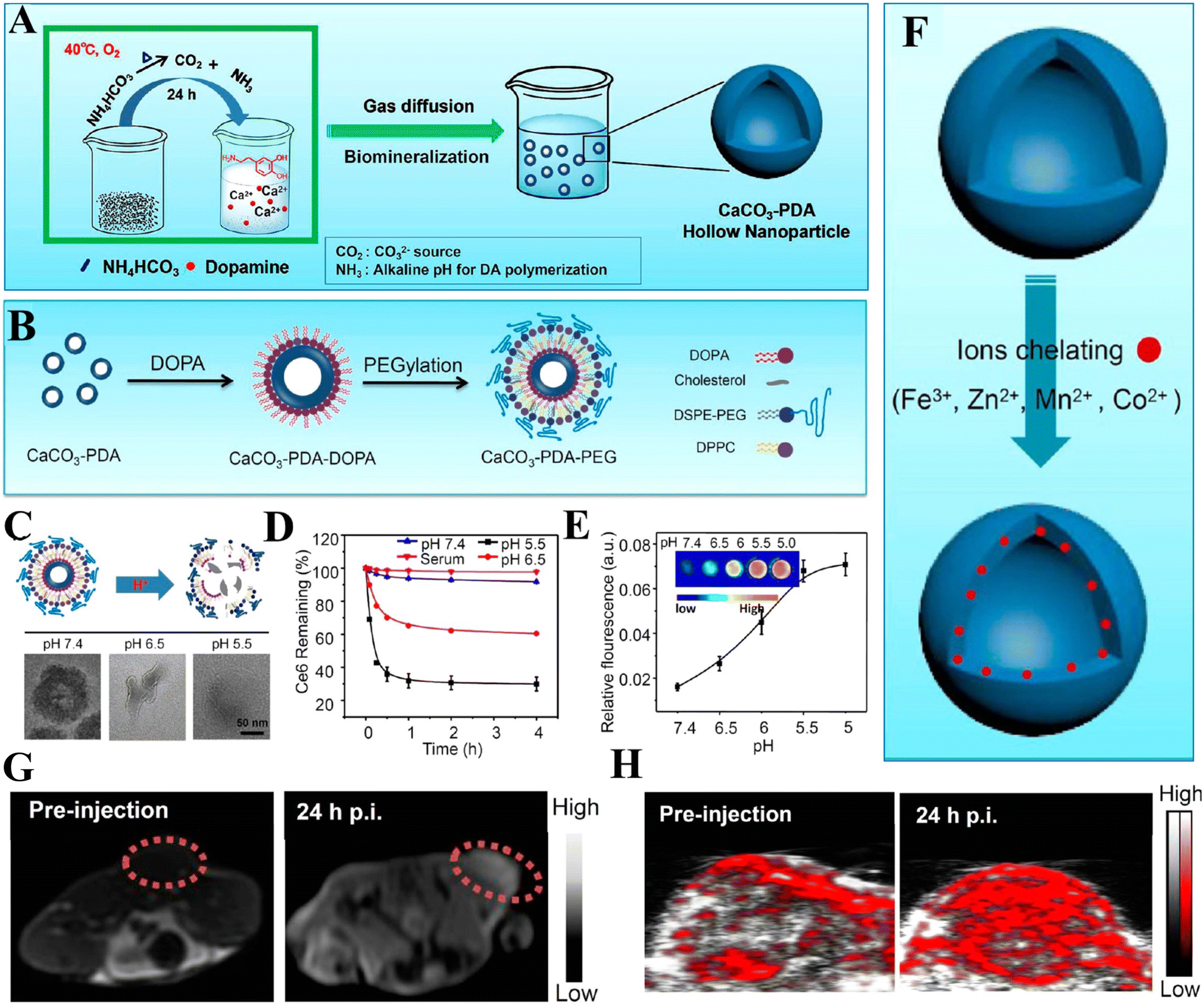 | ||
| Fig. 22 Schematic diagram showing preparation and application of CaCO3–PDA–PEG hollow NPs as a tumor acidic pH-activatable nanoplatform for multimodal imaging-guided cancer therapy. (A) Synthesis of CaCO3–PDA. (B) The PEGylation process of CaCO3–PDA. (C) The acid-responsive decomposition of CaCO3−PDA−PEG NPs and their corresponding TEM images after incubation in PBS at three different pH levels for 2 h. (D) Time-dependent release profiles of Ce6 from Ce6@CaCO3–PDA–PEG incubated in PBS or serum at different pH levels. (E) pH-dependent fluorescence intensity of Ce6@CaCO3−PDA−PEG after being incubated in PBS at various pH levels for 2 h. The inset shows the corresponding fluorescence image. (F) The metal ion-doping process of CaCO3–PDA–PEG. (G) T1-weighted MR images of 4T1 tumors taken before injection (left) and 24 h after i.v. injection (right) of CaCO3–PDA(Mn)–PEG. (H) Photoacoustic images of 4T1 tumors taken before injection (left) and 24 h after i.v. injection (right) of CaCO3–PDA–PEG. Ce6: chlorin e6; PDA: polydopamine; PEG: polyethylene glycol. Reprinted with permission from ref. 42. Copyright (2018) American Chemical Society. | ||
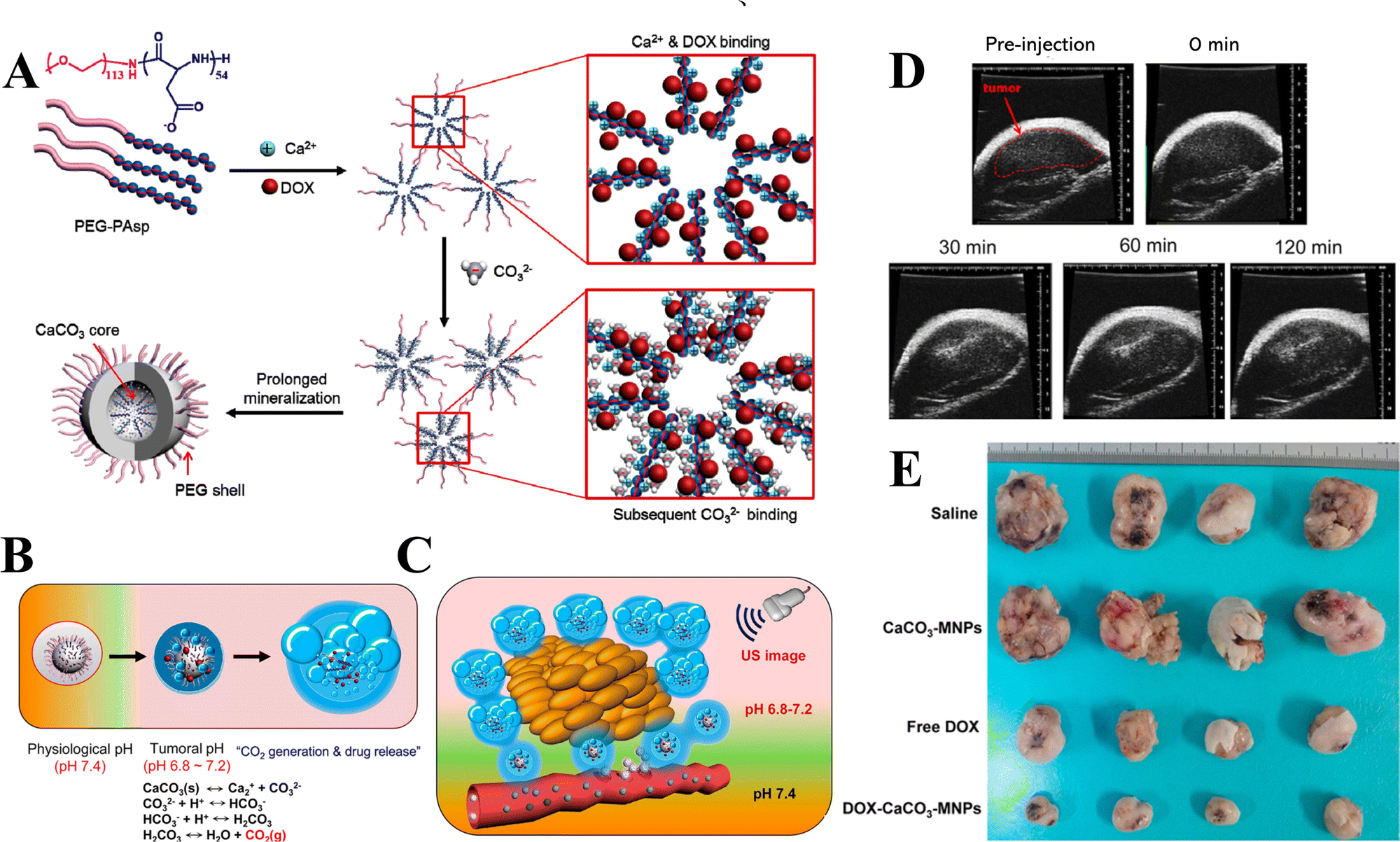 | ||
| Fig. 23 Schematic diagram of DOX–CaCO3–MNPs for simultaneous ultrasound imaging and therapy. (A) Fabrication process of the DOX–CaCO3–MNPs. (B) Mechanism of the CO2 generation and drug release of the DOX–CaCO3–MNPs. (C) Bubble generation and drug release after the accumulation of DOX–CaCO3–MNPs in tumor tissue. (D) In vivo US imaging of a squamous cell carcinoma tumor via the intratumoral injection of the DOX–CaCO3–MNPs. (E) Images of excised tumors of each group with controls (saline and CaCO3–MNPs), free DOX, and the DOX–CaCO3–MNPs 16 days post-treatment. Reprinted with permission from ref. 222. Copyright (2015) American Chemical Society. | ||
The combination of CaCO3-based US imaging and PDT functions into a rationally designed nanoplatform can allow the realization of the real-time monitoring of PDT. Ce6-Loaded bubble-generating mineralized NPs (Ce6-BMNs) comprising a Ce6-loaded CaCO3 core and hydrated PEG shell were fabricated for US imaging and PDT.223 The Ce6-BMNs effectively inhibited Ce6 release at physiological pH (7.4) and generated CO2 gas bubbles, simultaneously triggering the release of Ce6 at tumoral acidic pH for contrast-enhanced diagnostic US imaging and NIR-absorbing photosensitizers for remote PDT. Moreover, Ce6 release can be accelerated with CO2 bubble generation due to the dissolution of the CaCO3 mineral core in the tumoral environment, enhancing the delivery efficacy of Ce6 and the generation of singlet oxygen into the TME.
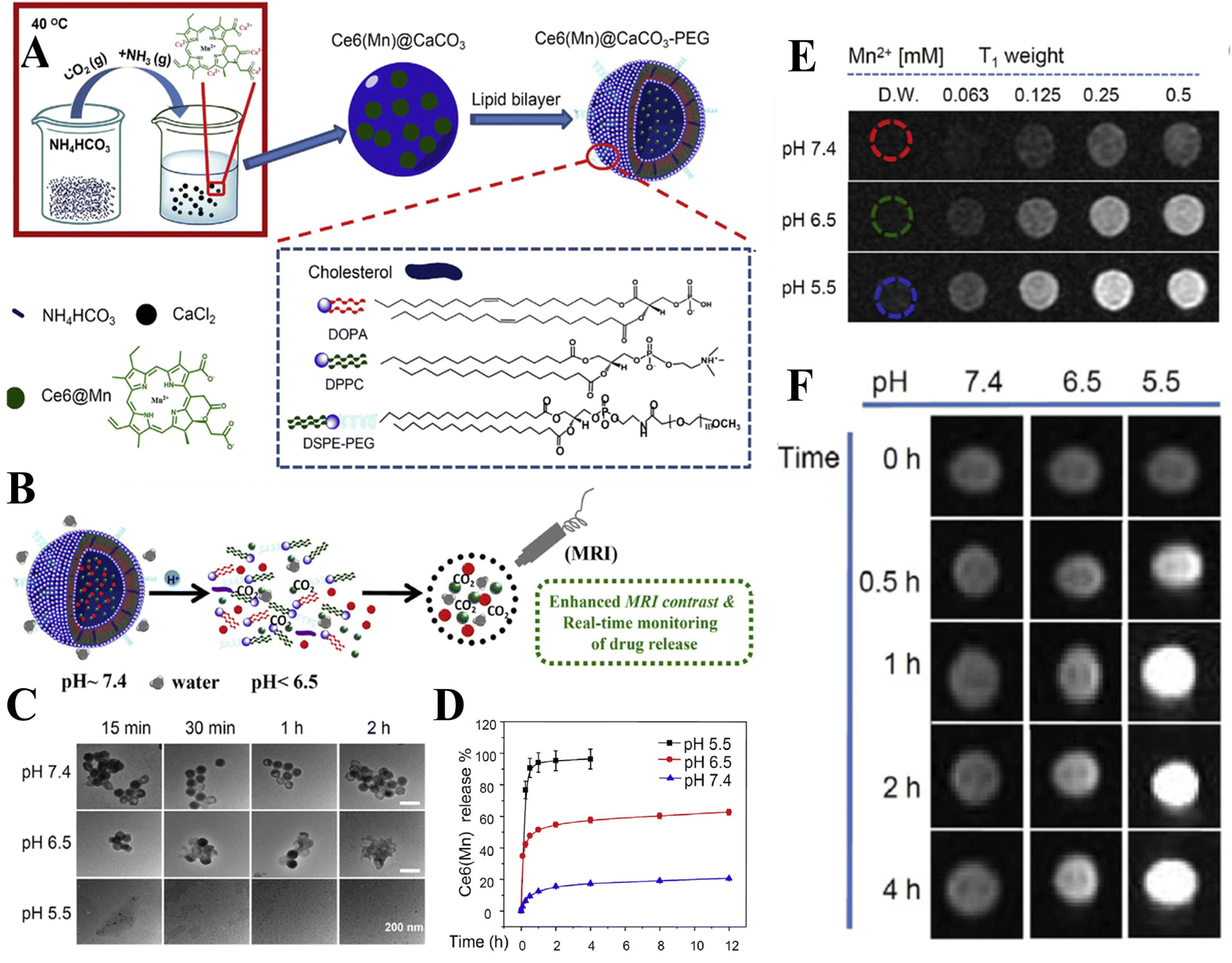 | ||
| Fig. 24 Schematic diagram of Ce6(Mn)@CaCO3–PEG NPs for MRI-monitored drug release. (A) Synthesis and structure of the Ce6(Mn)@CaCO3–PEG NPs. (B) pH-responsive decomposition of the Ce6(Mn)@CaCO3–PEG NPs. (C) TEM images of the Ce6(Mn)@CaCO3–PEG NPs after being immersed in PBS at three different pH levels. (D) Time-dependent Ce6(Mn) release from the NPs Ce6(Mn)@CaCO3–PEG NPs in PBS at different pH levels. (E) T1 relaxivities of Ce6(Mn)@CaCO3–PEG at different pH levels. (F) Time-dependent T1 MR images of Ce6(Mn)@CaCO3–PEG(DOX) after incubation in PBS at different pH levels. Reprinted from ref. 55. Copyright (2016), with permission from Elsevier. | ||
5. CaCO3 for mimetic nacre composites
In nature, living organisms can produce hierarchically structured lightweight CaCO3-based materials with outstanding structures, good stiffness, remarkable strength, and toughness.7 Such organisms include mollusks, echinoderms, calcisponges, corals, and certain types of algae.6,20,297,298 Recent studies have revealed that CaCO3 plays a role in the integrated visual system of chitons,299 in the bioceramic hard buoyancy tanks of cuttlefish,8 and in both the light-focusing eye lenses and mechanical support of brittlestars.9 More recently, Li et al.300 discovered that high-magnesium calcite [CaMg(CO3)2] armor overlays the exoskeletons of major workers of the leaf-cutter ant Acromyrmex echinatior, affording them mechanical protection. Nacre (mother-of-pearl) is a lightweight hierarchically CaCO3-based material with high strength and toughness, which has been the subject of much research attention toward the preparation of macroscopic artificial materials that mimic its micro/nanostructure.301,302It is well-known that nacre, the pearly internal layer of many mollusk shells, exhibits a bricks-and-mortar structure comprising 95% aragonite and 5% organic components (proteins and polysaccharides) by volume (Fig. 25).294 Nacre is formed by the deposition of crystal precursors (Ca2+ and CO32−) as well as ACC to form aragonite tablets on the surface of an insoluble matrix under the regulation of soluble biomacromolecules such as polysaccharides and proteins (Fig. 25B).296 The aragonite tablets and thin sheets of organic matrix are alternately arranged to form a highly organized, ‘bricks-and-mortar’ or ‘brick-wall’ microstructure,294 wherein each aragonite-rich plate is uniformly aligned vertically in the c-axis [001] direction (Fig. 25A). The organization of aragonite tablets and organic composites in the nacre and the formed hierarchical structure have been shown to significantly increase the toughness of nacre composites.303 For example, the shell of Strombus gigas comprises 99% aragonite and 1% organic phase (proteins and chitin) by volume. The nacre in this shell with a “crossed lamellar microarchitecture” is 3000 times tougher than single crystals of aragonite.303,304
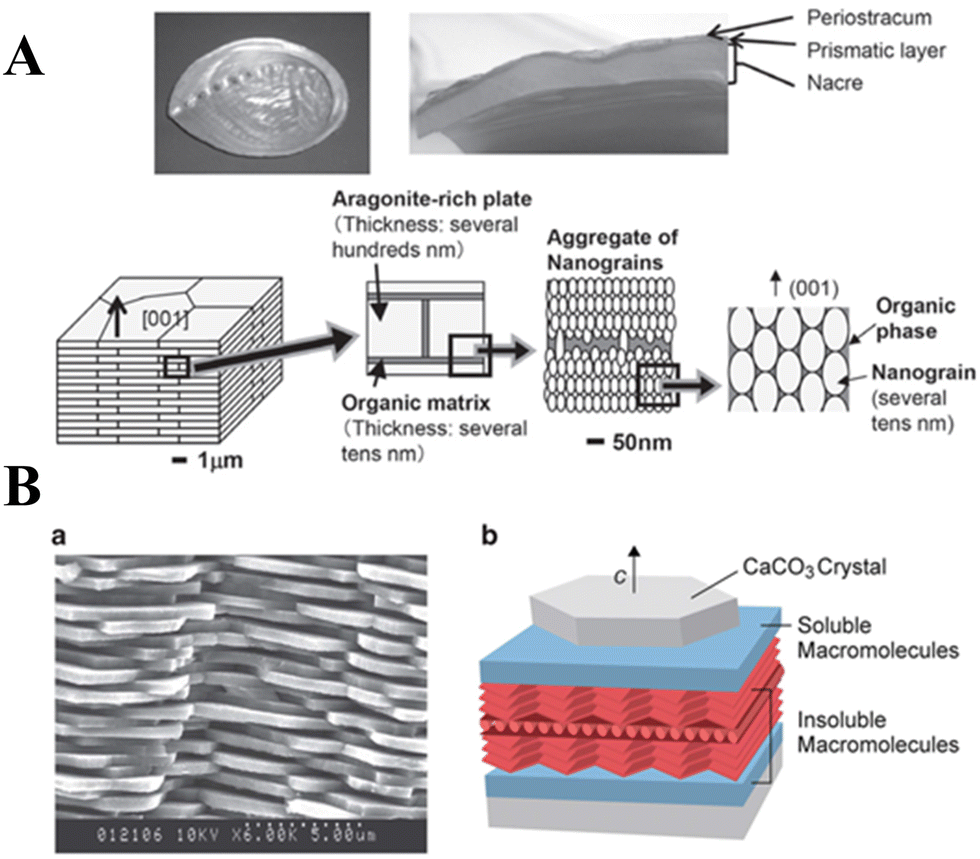 | ||
| Fig. 25 Schematic diagram showing the bricks-and-mortar structure of nacre. (A) Photographs of the abalone H. gigantea: bottom view (top left) and cross-section (top right) showing nacre at the inner surface. The platelet consists of ‘nanograins,’ aragonite particles on the order of several tens of nanometers, and the organic phase surrounding the nanograins. Reprinted from ref. 294. Copyright (2011), with permission from National Institute for Materials Science. (B) (a) Scanning electron microscopy (SEM) image of the fractured nacre. (b) Schematic illustration of the composite structure of nacre. Reprinted from ref. 295 and 296. Copyright (2000), with permission from John Wiley and Sons under the terms of the Creative Commons Attribution Non-Commercial (CC BY-NC 3.0) License. | ||
The outstanding mechanical properties of CaCO3-based nacre materials can be primarily attributed to two key factors. First, the fractions of the stiff minerals such as aragonite within the nacres are relatively high. Second, the aragonite is typically assembled into ordered microstructures to slow crack propagation.305 Stimulated by the remarkable mechanical performance of such a nacre, significant efforts have made to biomimetically engineer CaCO3 materials into hierarchical structured composites that exhibit exceptional stiffness, strength, and toughness.306 Over the past few decades, many nacre-inspired structural materials have indeed been synthesized.307–309 The strategies used to prepare these materials can be categorized as self-assembly,310,311 layer-by-layer,312 and freezing-induced assembly-and-mineralization techniques.313,314 The focus here is on studies and advances made over the last decade or so in using CaCO3 as an inorganic building block to produce nacre mimetic materials. Self-assembly seems to be a promising and economical strategy for the large-scale production of CaCO3-based nacre.
The challenging issue in this arena lies in the lack of methods available for preparing CaCO3 in a thin tablet form with a thickness of <500 nm.315 Li et al.315 developed a vacuum-evaporation-induced self-assembly method by which to synthesize single-crystalline CaCO3 nanotablets in large quantities as primary building blocks for constructing nacreous inorganic–organic hybrid films. The CaCO3 NPs were first obtained in the presence of CTAB (R–N+–(CH3)3) and polyoxyethylene sorbitan monolaurate surfactants in a water–ethylene glycol co-solvent. The CaCO3 NPs were in the size range of 1–3 nm and appeared to be stable for months in a mixed water/ethylene glycol solution. When CaCO3 NPs were prepared in the absence of surfactants, CaCO3 nanotablets were formed instantaneously. It was suggested that the formation of CaCO3 nanotablets could be formed during the washing treatment of CaCO3 NPs. Nacre-like CaCO3–gelatine inorganic–organic hybrid films have also been prepared using CaCO3 nanotablets as inorganic building blocks and gelatine (a protein derived from bovine skin) as a gluing matrix via a casting-then-assembling process. Typically, the ultimate tensile strength of the prepared CaCO3 (33 wt%)–gelatine films is around 97 MPa, comparable to that of nacre.
A layer-by-layer deposition method has been used to combine hard inorganic CaCO3 NPs with a soft organic phase in the design of a multilayered material featuring precisely ordered nanoscale building blocks.306 By mimicking the natural layer-by-layer approach employed to fabricate nacre, Finnemore and coworkers used the interplay between polymer-mediated CaCO3 growth and the layer-by-layer deposition of porous organic films to successfully replicate nacre for the first time using CaCO3.312 Use of an (NH4)2CO3 diffusion technique employing a PAA-containing aqueous solution of CaCl2 and MgCl2 yielded stable polymer-induced liquid precursor droplets, which were used to wet a carboxyl-terminated substrate and coalesce into an ACC film rich in Mg and PAA (Fig. 26A). The ACC was then crystallized by exposure to high humidity, resulting in a calcite structure featuring stacked crystalline layers interconnected with porous organic films. An advantage of this approach to prepare artificial nacre is the fabrication of the porous organic inter-crystalline layers (Fig. 26A). These pores allow the propagation of the crystallization of CaCO3 across the organic layers, providing vertical crystalline CaCO3 continuity, which enhances the mechanical stability of the resultant nacre. In terms of its morphology, growth route, and iridescence, the artificial nacre prepared was found to be comparable to natural nacre (compare Fig. 26B–a–c with d–g). In particular, a comparison of the SEM image of the fractured surfaces of natural (Fig. 26B-b) and artificial (Fig. 26B–e) nacre reveal very similar multilayers of 400 nm-thick calcite tablets with a nanogranular texture, characteristic of nacre.
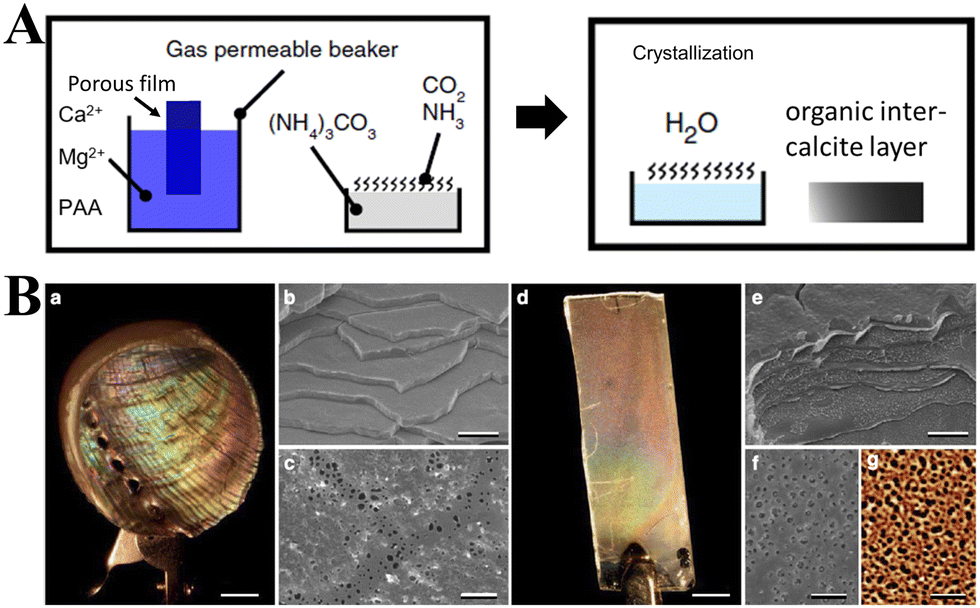 | ||
| Fig. 26 Biomimetic layer-by-layer assembly of artificial nacre. (A) Artificial nacre synthesis. (B) Comparison between biogenic and artificial nacre. (a) Photograph showing the bright iridescence of nacre. (b) Fractured surface SEM image of a stack of mineral tablets. (c) Organic inter-crystalline film that allows for vertical crystal continuity between tablets. (d) Artificial nacre, exhibiting a similar coloration to that in (a). (e) SEM image of a fractured surface showing seven aligned CaCO3 tablets separated by organic films. f) SEM image of a polymer (PVP) film on calcite showing a similar pore distribution to that in (c). (g) Atomic force microscopy (AFM) height image of the porous film. Adapted and reprinted by permission from [Springer Nature Customer Service Centre GmbH]: [Springer Nature]: [ref. 312], [Copyright] (2012). | ||
Considering that mollusks construct their nacre via mineralization in a preformed laminated matrix, Mao and colleagues developed a freezing-induced assembly-and-mineralization process (Fig. 27) to fabricate CaCO3-based nacre-like materials using mesoscale tactics in which the nanostructure and the microstructure are controlled simultaneously.314 Using a freezing-induced assembly process (Fig. 27A and B), a chitosan matrix with a predesigned laminated structure was fabricated, which was subsequently acetylated (Fig. 27C) and transformed to β-chitin to avoid unnecessary swelling or dissolution. The acetylated matrix was then mineralized in the presence of PAA and Mg2+ by decomposing Ca(HCO3)2 in a peristaltic pump-driven circulatory system (Fig. 27D). Synthetic nacre was finally obtained via silk fibroin infiltration and hot-pressing of the mineralized chitin matrix (Fig. 27E). The resultant millimeter-thick synthetic nacre comprised alternating organic layers and aragonite platelet layers (91 wt%), exhibiting remarkable ultimate strength and fracture toughness (Fig. 27F and G).
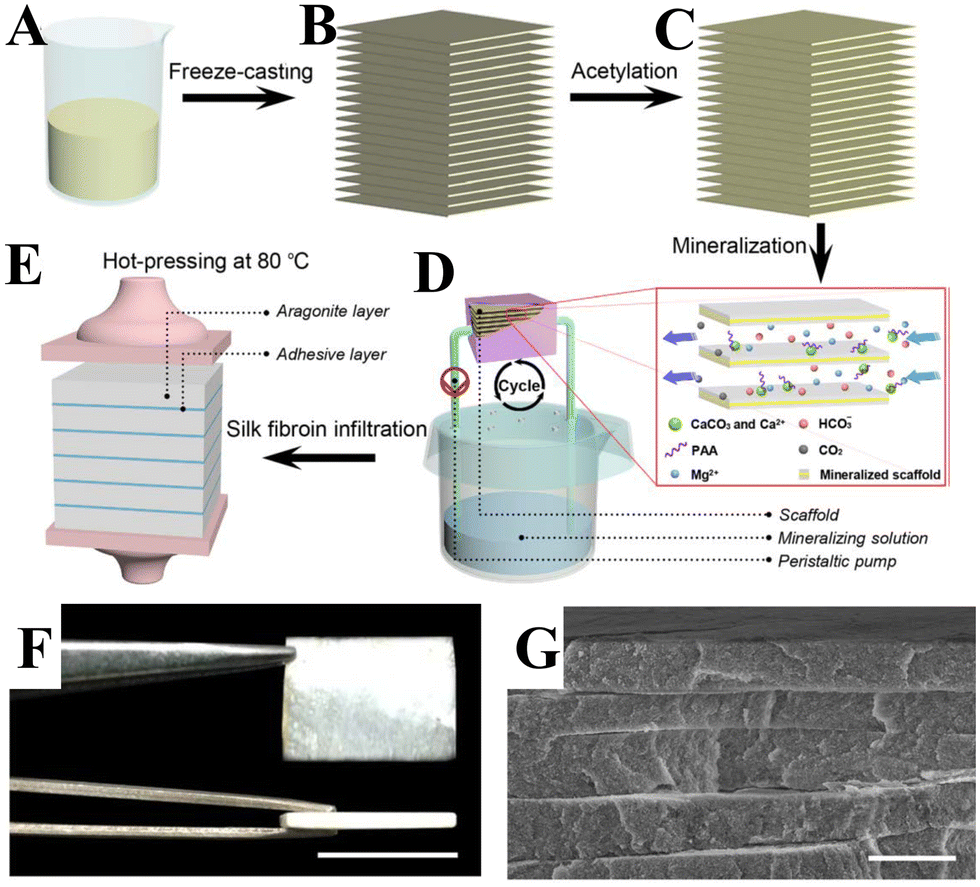 | ||
| Fig. 27 Schematic diagram showing fabrication scheme of the synthetic nacre. (A) Starting solution, chitosan/acetic acid solution. (B) Freeze-casted laminated chitosan matrix. (C) Matrix after acetylation, where chitosan is converted to β-chitin. (D) Mineralization of the matrix. (E) Laminated synthetic nacre is obtained after silk fibroin-infiltration and hot-pressing processes. (F) The as-prepared synthetic nacre. (G) SEM image of a cross section of the synthetic nacre. Scale bar: 1 cm for F; 3 mm for G. From ref. 314. Copyright (2016), Adapted and reprinted with permission from AAAS. | ||
6. Engineering CaCO3 into environmental and energy nanostructured materials
6.1 Environmental remediation
CaCO3 removes heavy metals from wastewater primarily via adsorption and precipitation processes. At a low metal concentration, the adsorption process mainly occurs via the replacement of Ca2+ by heavy metal ions,326 whereas at high metal concentrations the precipitation process is dominant.327 Traditional wastewater treatment with CaCO3 from geological sources (geo-CaCO3, e.g., calcite and aragonite) can reduce the residual Pb2+ content of the water to 1 mg L−1. Yet, there are still issues in using this material to meet the new standards stipulated by the Centers for Disease Control & Prevention, which are that discharge must contain no more than 0.05 mg L−1 of Pb2+.328 Geo-CaCO3 has an intrinsically dense crystal structure, which results in it exhibiting low pollutant removal efficiency and the potential to generate a significant amount of sludge. Recent studies have shown that biogenic CaCO3 (bio-CaCO3) derived from marine shells, eggshells, and oyster shells, exhibits better performance than geo-CaCO3 in terms of wastewater treatment, especially in improved Pb2+ removal efficiency and producing less sludge.323,326,329 The hierarchical porous organic–inorganic hybrid structure of bio-CaCO3 allows deeper penetration of Pb2+ into the CaCO3 material via pores/slits in the particles, with it eventually becoming immobilized. Meanwhile, the organic functional groups of bio-CaCO3 (e.g., –COOH) assist in the adsorption of Pb2+ (Fig. 28A). For example, bio-CaCO3 derived from oyster shells shows remarkable performance in removing Pb2+ from wastewater.326 Since to some extent Ca2+ and CO32− are dissolved on the surface of CaCO3, the generation of CO32− leads to the supersaturation of Pb2+, thus favoring the nucleation and growth of PbCO3 and Pb3(CO3)2(OH)2. Pb2+ forms a complex with the functional groups of bio-CaCO3, followed by the interaction of Pb2+ with the bio-CaCO3. As a result, the maximum adsorption capacity of bio-CaCO3 toward Pb2+ is three times that of geo-CaCO3, reaching 1667 mg g−1. The adsorption of Cd2+ on bio-CaCO3 (CaCO3 formed by Bacillus subtilis) is a spontaneous endothermic process that can be described in terms of its kinetics as a pseudo-second order process.322 Similarly, during the removal of heavy metal ions from water, CaCO3 in the form of vaterite continuously releases Ca2+ due to participating in an ion exchange reaction with metal ions. In this process, Pb2+ and Ca2+ ions exchange and recrystallize. For this reason, vaterite prepared from oyster shells exhibits the following removal efficiencies for ions: Pb2+ (99.9%), Cr3+ (99.5%), Fe3+ (99.3%), and Cu2+ (57.1%).323 The organic–inorganic hierarchical microstructure and organic matter of bio-CaCO3 are conducive to the adsorption of rare earth elements, resulting in rare earth elements being recycled from wastewater. Recently, taking the advantages of mussel-inspired PDA chemistry, Zhou et al.330 modified the surface of bio-CaCO3 from waste oyster shells with PDA via a facile oxidative polymerization route to increase its organic matter content, and eventually promote its Eu3+ adsorption capacity. The antioxidation properties of the organic matter of amino- or thiol-containing molecules of the groups used to modify bio-CaCO3, as well as the high surface area provided by bio-CaCO3 hierarchical structure, significantly improve the affinity of PDA to bio-CaCO3 (Fig. 28B), which is anchored on bio-CaCO3via bidentate metal coordination. The abundant catechol and amine groups of the uniform PDA coating on bio-CaCO3 are then able to capture a large amount of Eu3+ ions from solution. In contrast, the surface of geo-CaCO3 is smooth, with only a few dopamine molecules anchored on its tight surface.
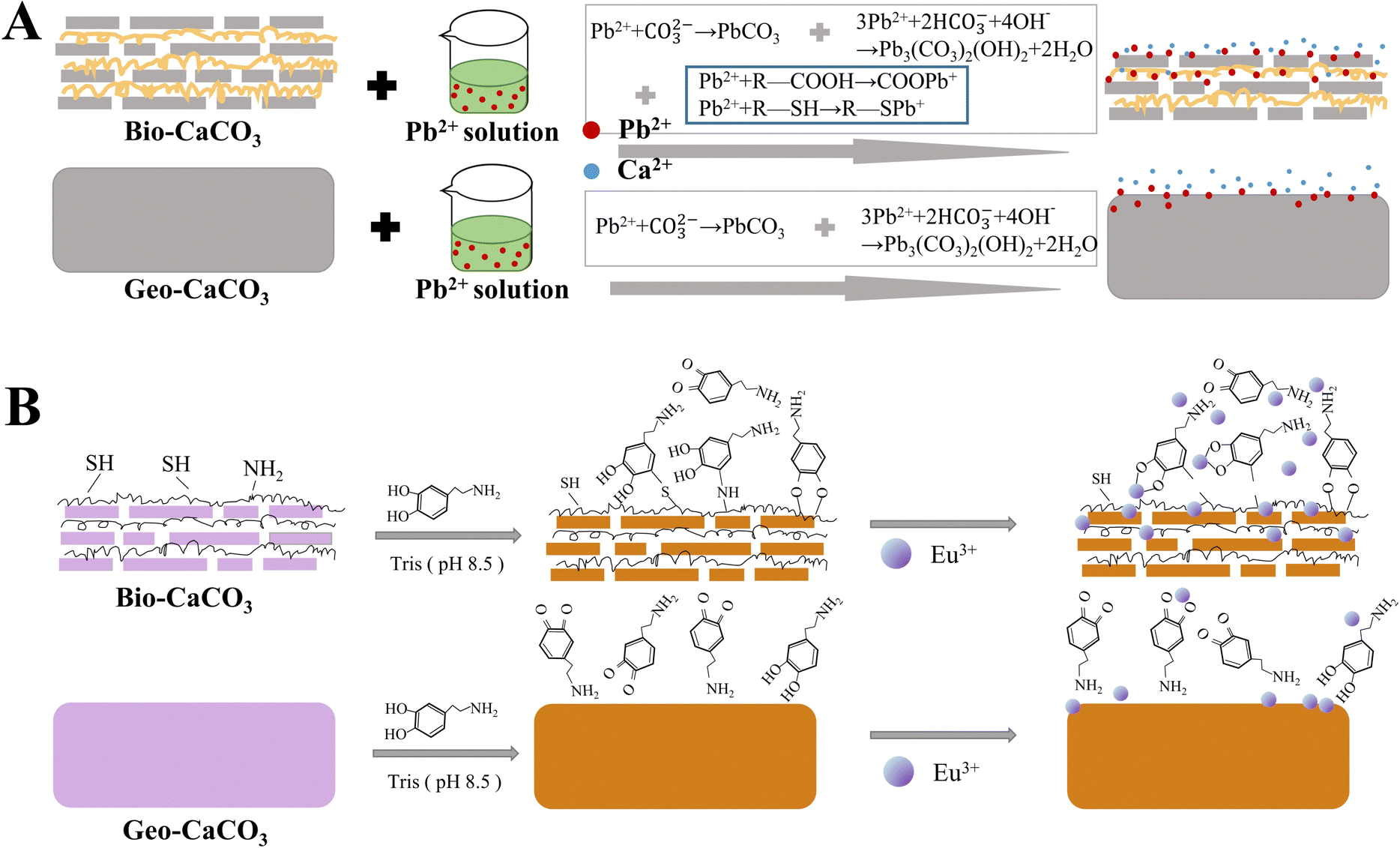 | ||
| Fig. 28 Schematic diagram illustrating bio-CaCO3 with a hierarchical organic–inorganic composite structure that enhances the removal of metal ions from wastewater. (A) Schematic illustration of Pb2+ sequestration on bio-CaCO3 and geo-CaCO3. Reprinted and adapted with permission from ref. 326. Copyright (2017) American Chemical Society. (B) Schematic illustration of PDA coating on bio-CaCO3 and geo-CaCO3, and the corresponding Eu3+ adsorption mechanism. Reprinted and adapted from ref. 330. Copyright (2018), with permission from Elsevier. | ||
As for the adsorption and precipitation of heavy metals on CaCO3 surfaces, a puzzling issue is that generally CaCO3 is hard to dissolve (Ksp = 3.36 × 10−9), and thus only slowly reacts with metal ions.325,331 Such low level of potential interaction means that a large amount of CaCO3 has to be used in an application to achieve a high removal efficiency. A recent study demonstrated that ball milling can be used to improve the reactivity of CaCO3 with Pb2+.209 During the ball milling process, the surface of CaCO3 is constantly activated due to continuous mechanical collisions, extrusion, and shearing. The originally stable CaCO3 is thus broken up into smaller particles with more active fresh surfaces exposed, thus improving its solubility and chemical reactivity in solution. In the ball milling process, CO32− generated from the hydrolysis of CaCO3 (Ksp = 3.36 × 10−9) was reacted with Pb2+ from acidic mine drainage to form the more stable material PbCO3 (Ksp = 7.40 × 10−14). This process allowed less CaCO3 to be used to achieve effective Pb2+ removal from the drainage and also led to less sludge being produced. However, the reactivity of the activated CaCO3 was not sufficient to precipitate Zn2+ (ZnCO3, Ksp = 1.46 × 10−10) at the same time. Such a difference might lead to the development of a new way of efficiently separating Pb2+ from Zn2+.
In addition, different crystal structures of CaCO3 exhibit different electron densities around the Ca atoms. As such, this inevitably affects the replacement of Ca atoms by heavy metal ions. This has been evidenced in a few new studies that have shown that crystal phase is related to both reactivity with and selectivity toward heavy metal ions.323,332,333 Magnetic mesoporous CaCO3 nanocomposites (MCCRs) with vaterite and aragonite phases exhibit stronger adsorption ability toward Cd2+ than for Pb2+, while MCCRs with a calcite phase show better adsorption of Pb2+ than Cd2+.316 These adsorption differences may be mainly due to the different degrees of matching of the crystal lattice between Pb2+- and Cd2+-bearing precipitates and the different crystal phases of CaCO3. These findings provide valuable information for the design and enhancement in the reactivity and selectivity of CaCO3-based adsorbents toward heavy metal ions for application in environmental remediation.
CaCO3/porous carbon composites are harmless to humans and hold the potential to be used in the removal of heavy metals from water and soil. A hierarchical porous carbon sorbent has recently been successfully fabricated via the pyrolysis of rice straw in the presence of CaCO3 NPs for the effective removal of metal ions from water.334 The calcite–biochar composites prepared at 700 °C exhibit the efficient removal of Pb (99.9%). In the process, calcite acted as a catalyst for the catalytic carbonization of biochar and contributed toward changes in yield, pH, texture, and the surface functional groups of the CaCO3–biochar composites. For the calcite–biochar composites, calcite formed a stronger bond with Pb under alkaline conditions. Upon an increase in pH in aqueous solution, more Pb2+ was likely to be precipitated by the CO32– of the calcite.
A common challenge when using CaCO3-based adsorbents is that they are difficult to separate from wastewater after use and often require further treatment of the sludge to be retrieved. To tackle this issue, a carbonate-based mesoporous magnetic adsorbent (IO@CaCO3) consisting of needle-like iron oxide (IO) and calcite was synthesized via a hydrothermal synthetic strategy by Islam et al.317 Due to synergistic effects between needle-like IO and CaCO3, the IO@CaCO3 material almost completely removed (99.99%) heavy metal ions from wastewater in only 9 min. The adsorption of the metal ions on IO@CaCO3 was attributed to a specific adsorption mechanism (instead of non-specific Coulombic attraction), such as ion-exchange reactions that take place between the anionic metal ions (Cr(VI) and As(V)) and anionic surface groups (OH− and CO32−) or between the cationic metal ions (Pb2+) and cations (Ca2+) on the surface of the IO@CaCO3 adsorbent. As it is magnetic, IO@CaCO3 can be easily separated from solution using an external magnetic field, enabling it to be effectively recycled.
Many ions can potentially interfere with the removal of phosphate from water. The hydrolysis of CaCO3 results in cationic species, such as Ca2+, CaHCO3+, and CaOH+, being produced at a pH of <8 (CaCO3 = Ca2+ + CO32−, pK = 3.25; CO32− + H2O = HCO3− + OH−, pK = 3.67; Ca2+ + HCO3− = CaHCO3+, pK = −0.82; Ca2+ + OH− = CaOH+, pK = −1.40). CaCO3-based adsorbents have been developed for the removal of phosphate from water. An abundance of Ca2+ ions has been shown to facilitate the removal of phosphate ions from water in the presence of CaCl2. In addition, biochar–calcite composites appeared to outperform calcite alone in the removal of phosphate ions from water. The reduced phosphate adsorption in the presence of HCO3− may have been due to the increased competition for adsorption sites from HCO3−. More recently, with the aim of removing and recovering phosphate from water, Zhou et al.337 used calcite and rice husk biochar at a pyrolysis temperature of 700 °C, pyrolysis time of 2.3 h, and a rice husk: calcite ratio of 4.2:1 (w/w) to prepare a rice husk biochar–calcite composite. CaCO3 was converted into CaO at a high pyrolysis temperature, which then reacted with SiO2 to form a calcium silicate, wollastonite (CaSiO3), and increased the distribution of active Ca ions. The CaSiO3 may contribute to remove phosphate from aqueous media via the precipitation of phosphate to CaHPO4 under acidic conditions or Ca3(PO4)2 under neutral conditions. The removal of phosphate by the rice husk biochar–calcite composite was shown to occur synergistically via pore filling, electrostatic interactions, and precipitation. The design of ecofriendly and cost-effective CaCO3-based composites is thus a promising approach by which to remove phosphate and other organic and inorganic pollutants from wastewater.
Highly flexible free-standing CaCO3 films with only 5% sodium alginate (NaAlg) have been developed for oil–water separation (Fig. 29A).338 The free-standing CaCO3 films were prepared by strongly mixing CaCl2 and alginate together in water to form a Ca–Alg network, then Na2CO3 was added to the mixture to combine with Ca2+ to produce CaCO3, followed by filtering to obtain CaCO3/CaAlg hybrid films.341 These CaCO3/CaAlg hybrid films exhibit a coarse surface with micropores and hydrophilicity. The superhydrophilicity and the highly rough surfaces of the films result in them having underwater superoleophobic properties with ultralow oil adhesion. Hence, a self-cleaning function was realized when black crude oil that was adhered to a prewetted CaCO3 film was easily removed after its immersion in water.
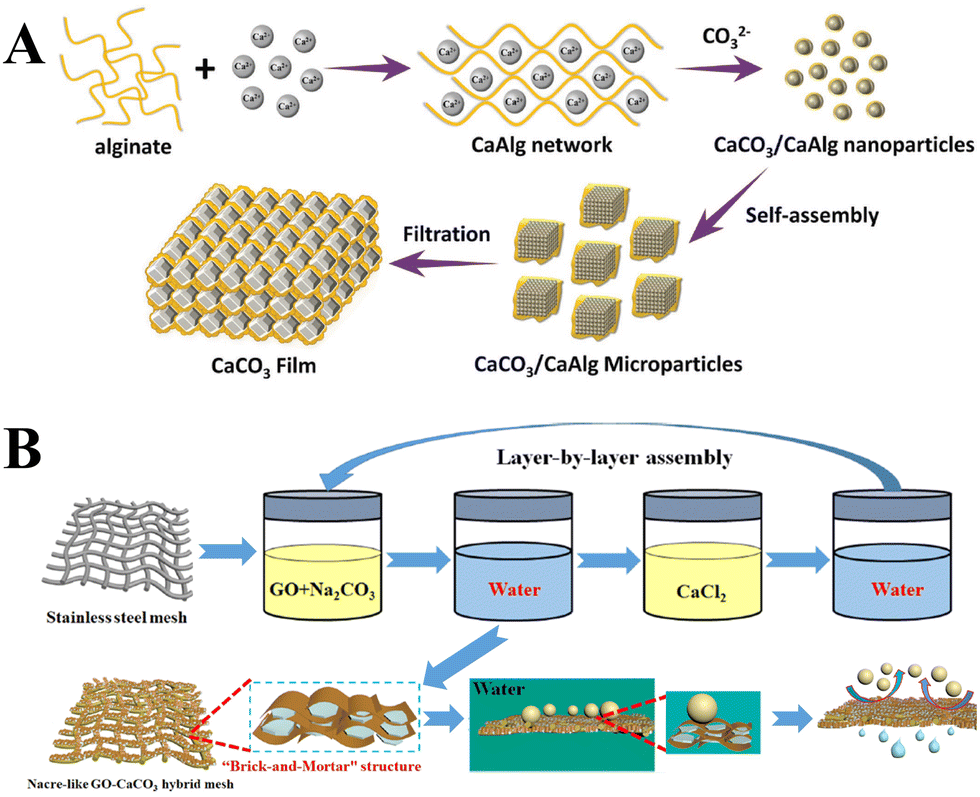 | ||
| Fig. 29 Schematic diagram showing preparation of CaCO3 hybrid materials for oil–water separation. (A) Flexible free-standing CaCO3 film. Reprinted from ref. 338. Copyright (2017), with permission from John Wiley and Sons under the terms of the Creative Commons Attribution Non-Commercial (CC BY-NC 3.0) License. (B) A nacre-like graphene oxide (GO)–CaCO3 hybrid mesh for oil–water separation. Reproduced with permission from ref. 339. Copyright (2020) American Chemical Society. | ||
More recently, superhydrophilic/underwater superoleophobic cellulose films (FP@SA/CaCO3) with good oil–water separation performance have also been developed.342 Ca2+ was used to crosslink an SA film grafted on the surface of filter paper (FP) to improve the interface stability of the membrane. NaAlg–Ca was obtained via an ion-exchange mechanism between –COONa functional group and Ca2+ and the crosslinking reaction between the NaAlg molecular chains improved the stability of the material. During alternating soaking processes, the NaAlg–Ca coated composite membrane captured CO32− to form CaCO3 NPs, and the generated CaCO3 NPs adhered stably to the membrane surface via NaAlg–Ca ionic bonds. CaCO3 particles increase the surface roughness of FP@NaAlg/CaCO3 films and are beneficial for promoting the superhydrophilicity of a material. For a petroleum ether–hexane–toluene–soybean oil–dichloroethane–water mixture, the separation efficiency of the FP@NaAlg/CaCO3 films remained above 99% even after 20 cycles, indicating that they exhibit excellent stability and antifouling performance.
Furthermore, CaCO3 can be formed on the surfaces of various meshes for the fabrication of superhydrophilic/superoleophobic CaCO3-based hybrid films. The CaCO3 coating of the surfaces of such hybrid films increases their dimensional stability, stiffness, and heat resistance.342 On stainless steel mesh, a superhydrophilic and underwater superoleophobic GO–CaCO3 hybrid mesh was fabricated via a layer-by-layer self-assembly method (Fig. 29B). Interestingly, the GO nanosheets in the material regulate the growth of the CaCO3 nanocrystals between the GO oxide layers to construct “bricks-and-mortar” structures. The GO–CaCO3 hybrid mesh exhibits strong mechanical properties, with a Young's modulus of 25.4 ± 2.6 GPa and a high separation efficiency of >99% toward a series of oil–water mixtures and a flux of 179 640 L m−2 h−1 for cyclohexane–water mixtures. A hierarchical CaCO3 thin coating with structural continuity and uniformity has been deposited on a nylon mesh via seeded mineralization by the slow diffusion of CO2 into CaCl2 solution in the presence of PAA.343 These hierarchical CaCO3 thin coatings on the nylon mesh exhibit underwater superoleophobicity, very low adhesion of oil in water, and stiffness and strength similiar to the prismatic-type biominerals found in mollusc shells and can thus be employed in oil–water separation. This conventional mineralization process is complex and requires the pretreatment of the polymer surface and the addition of organic additives. Tang et al.344 deposited CaCO3 on stainless steel mesh via bacterially (Bacillus subtilis) induced biomineralization and subsequent surface modification with NaAlg. This bacterially-induced CaCO3 biomineralization occurs at room temperature and in air without human intervention, thus representing an eco-friendly biomineralization strategy that is easy to conduct. The formed NaAlg/CaCO3–stainless steel mesh with micro/nanostructured CaCO3 coating and low surface energy exhibits oil fluxes in the range of 0.2–9.12 × 104 L m−2 h−1 and separation efficiencies of >94.8% toward various oil–water mixtures, showing great potential for use in oil–water applications under harsh conditions, such as slurry and dust environments, as well as at low temperature.
Eggshell, which is composed of around 96% CaCO3, can be used as a support and template upon which to immobilize metal NPs. For example, Ag NP-loaded eggshell catalysts have been prepared for the oxidation of volatile organic compounds.345 The strong interaction between the Ag NPs and eggshell resulted in the decomposition temperature of the eggshell (i.e., CaCO3) being lower than that of pure eggshell. The layered eggshell membrane (proteins and polysaccharides) played a crucial role in the dispersion of Ag NPs on eggshell supports via strong metal–protein bonding interactions. The hierarchical porous structure of the eggshell increases the contact between volatile organic compounds and Ag NPs and enhances mass and energy transmission.
The CaCO3 in the eggshell provides active carbonate radicals (˙CO3−), which favor the degradation of organic compounds or microorganisms.346 CaCO3 provides the key chemical species (such as CO32−, HCO3−, OH−) in aqueous solution, which can react with ˙OH− to give rise to carbonate radicals, ˙CO3−. By employing eggshell as a CaCO3 source and template, eggshell-derived CaCO3/CuS and CaCO3/PbS nanocomposites as photocatalytic systems have been reported (Fig. 30).346,347 Using eggshell as a support allows the immobilization of CuS or PbS NPs onto a material with which to achieve the purification of wastewater (organic degradation and bacteria inactivation), thus preventing the agglomeration of the NPs. Moreover, CaCO3 provides highly reactive radical species (˙CO3−), which are essential in catalytic reactions and antibacterial applications. CaCO3/CuS nanocomposites can be excited using NIR light. Their outstanding photocatalytic degradation of 4-nitrophenol (4-NP) and antibacterial activity against E. coli and S. aureus can be attributed to synergistic photocatalytic/PTT/PDT effects (Fig. 30A).347 CaCO3/PbS nanocomposites have been used for solar light-assisted photodegradation of tetracycline hydrochloride (TC-HCl) (Fig. 30B),346 a process in which ˙CO3− species make a remarkable contribution. CaCO3-based photocatalysts thus hold promise for use in further studies on the design of cost-effective environmentally-benign systems for more sustainable development.
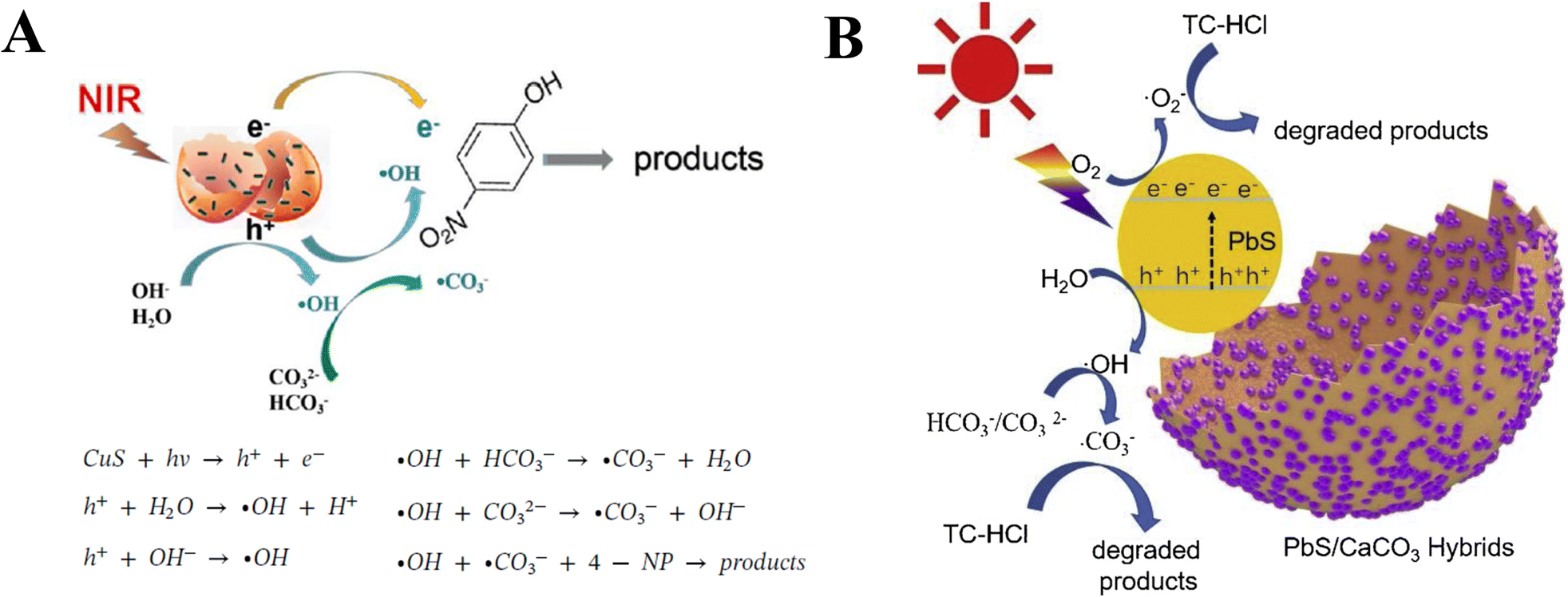 | ||
| Fig. 30 Schematic diagram of CaCO3-modified photocatalytically active semiconductor nanocomposites. (A) Schematic representation of 4-nitrophenol (4-NP) degradation using CaCO3/CuS. Reprinted from ref. 347. Copyright (2020), with permission from Elsevier; (B) illustrative degradation of TC-HCl over a CaCO3/PbS catalyst. Reprinted from ref. 346. Copyright (2020), with permission from Elsevier. | ||
6.2 Energy materials
Phase change materials (PCMs) allow controllable and conditional energy storage and release.348 Microencapsulated phase change materials (MEPCMs) with a rigid shell not only avoid the leakage associated with solid–liquid PCMs, but also enhance their heat transfer and thermal response.349 CaCO3 is rigid and dense, which gives it desirable thermal conductivity (2.167 W m−1 K) properties. The synthesis of CaCO3 as an inorganic shell is cost-effective and can be achieved under mild synthetic conditions. Accordingly, CaCO3-based MEPCMs do not exhibit the flammability, low thermal conductivity and mechanical strength, and poor thermochemical stability that are usually associated with organic shell materials, such as polystyrene,350 phenolic resin,351 and acrylic polymer.352 Various paraffin PCMs, such as those based on n-alkanes, have been encapsulated with CaCO3 or modified CaCO3, including CaCO3@n-eicosane,348 CaCO3@n-nonadecane,353 CaCO3@n-octadecane,354 CaCO3@paraffin,355,356 CaCO3@n-tetradecane,357 GO-modified CaCO3@paraffin,358 and Ce3+-doped CaCO3@paraffin.359 The thermal conductivity of CaCO3-based MEPCMs increases with an increase in the content of the CaCO3 shell.On the surface of the PCM core, the CaCO3 shell is usually formed via interfacial precipitation between water-soluble Ca2+ and CO32−. Such a precipitation reaction is usually fast, and the self-assembly of CaCO3 precursors at an oil–water interface is difficult to control, leading to the resultant CaCO3-based MEPCMs exhibiting poor microstructures. A CaCO3 shell resulting from the precipitation of Ca2+ and CO32− onto the surface of oil droplets of PCMs via a self-assembly process has been investigated in various emulsion systems containing nonionic, cationic, anionic or complex surfactants.348,349,354 For example, for CaCO3@n-octadecane, CaCO3 precipitated via self-assembly on the surfaces of n-octadecane micelles in an oil-in-water emulsion in the presence of Tween 80 and Span 80 mixed nonionic surfactants.348 Such a CaCO3 material assembled via complexation between Ca2+ and the hydroxyl groups of the mixed nonionic surfactants, followed by the precipitation of CO32− at the interface of the micelles. Furthermore, a compact and thick highly thermally conductive CaCO3 shell that was formed in a crystalline phase of vaterite can induce α-form crystallization via heterogeneous nucleation, thus enhancing the crystallinity of n-octadecane and improving its thermal conductivity. Moreover, it also enhanced the reliability, durability, and anti-osmosis properties of the CaCO3@n-octadecane microcapsules. The stereochemical and geometric relationship between molecules of the anionic surfactant SDBS and CaCO3 nuclei has been shown to favor the expression of specific crystalline facets, leading to different types of crystals being produced. Different concentrations of sodium dodecyl benzenesulfonic acid (SDBS) led to the synthesis of CaCO3 shell with different morphologies.348 At low concentrations of SDBS (<2.0 mmol L−1), a calcite shell is formed on an n-eicosane core, whereas at high concentrations of SDBS (>5.0 mmol L−1), a vaterite shell is formed. The CaCO3@n-eicosane MEPCMs present rhombohedral and spherical morphologies, with the spherical CaCO3@n-eicosane MEPCMs exhibited a higher encapsulation efficiency and energy-storage efficiency than their rhombohedral counterparts.
CaCO3@n-Tetradecane MEPCMs have been prepared via a self-assembly technique using sodium dodecyl sulfate (SDS) and alkylphenol polyoxyethylene ether (OP-10) as mixed templates in an oil-in-water emulsion system.357 In this process, OP-10 acts as an nonionic emulsifier to increase the stability of the emulsion system, thus decreasing the particle size of the CaCO3@n-tetradecane MEPCMs. As a result of complexation between Ca2+ and SO42− of the SDS, a large amount of concentrated Ca2+ can be self-assembled on the surface of the n-tetradecane micelles, with the formed CaCO3 shell adopting a mixture of vaterite and calcite crystalline phases. By changing the core/shell mass ratio and the mass ratio of the SDS and OP-10 surfactants, a series of CaCO3@n-tetradecane MEPCMs were obtained. The CaCO3@n-tetradecane MEPCMs exhibit high thermal conductivity, thermal storage performance, good thermal stability, and show great potential for use in energy storage.
A CaCO3 shell has also been used in the microencapsulation of a paraffin core to enhance thermal conductivity and to make it adjustable over a wide phase change temperature. For example, for CaCO3@paraffin MEPCMs produced via a self-assembly pathway from a mixture of Tween 80 and Span 80 as a templating agent by changing the weight ratio of the paraffin core, the phase change temperature of the CaCO3@paraffin MEPCMs can be adjusted over a temperature range of 25–50 °C.355 The MEPCMs also exhibited significant enhancement in thermal conductivity, primarily due to the thermally conductive CaCO3 shell. Jiang and co-workers fabricated a series of CaCO3@paraffin materials with styrene–maleic anhydride (SMA) and different pH values via a self-assembly process.356 The negatively charged carboxyl groups of the SMA molecules on the surface of paraffin droplets attracted positively charged Ca2+ ions to form calcite. The side chains of the SMA molecules were not conducive to forming vaterite crystals with a cationic coordination number of 12. In the paraffin emulsion, at high pH CaCO3 crystals nucleated and grew rapidly, resulting in their irregular shape and the uneven particle size distribution of the MEPCMs. Low pH was not conducive to CaCO3 deposition, thus the number of CaCO3 particles deposited on the surface of paraffin emulsion was reduced. Moreover, CaCO3@paraffin MEPCMs in a paraffin emulsion of pH 7 showed a high encapsulation efficiency of around 56.6%.
However, using CaCO3 as a shell for MEPCMs, the encapsulation ratio was found to be low and did not prevent leaking of the core. Emir et al.360 for the first time coated their thermal conductivity. The thermal storage capacities of CaCO3@n-heptadecane and MEPCMs with Ag shells to produce multilayered Ag@CaCO3@n-heptadecane MEPCMs to improve Ag@CaCO3@n-heptadecane were found to be comparable to those of other paraffin-based materials microencapsulated with CaCO3. Jiang and coworkers reported GO-modified paraffin MEPCMs with a CaCO3 shell.358 The paraffin was first emulsified with SMA to obtain negatively-charged paraffin droplets. The paraffin particles adsorbed Ca2+ on their surface, and the Ca2+ and the negatively-charged GO were then attracted to each other via an electrostatic interaction. The CaCO3 shell formed after the addition of Na2CO3 was wrapped by and interconnected to the network of the GO. The GO-modified CaCO3@paraffin MEPCMs with perfect spherical core–shell structures were found to exhibit a high encapsulation ratio (73.19%) to improve the prevention of the core from leakage. In addition, the CaCO3 shell was found to be compatible with GO, with GO sharing some of the external force and transfer, as well as consuming some of the external energy. Thus, the GO-modified CaCO3@paraffin MEPCMs exhibited good thermal stability, thermal conductivity (0.857 W m−1 K), and mechanical properties.
CaCO3-based MEPCMs can also be used as light-harvesting systems. However, the poor solar light-harvesting capability of the CaCO3 shell limits the solar photothermal energy conversion and storage of these materials. A general route to achieving high photothermal energy conversion efficiency was developed in which energy-efficient photothermal materials were incorporated into CaCO3-based MEPCMs. For example, CaCO3/Fe3O4@n-docosane MEPCMs were successfully fabricated via a nonaqueous (O/W) emulsion-templated self-assembly technology using three types of surfactants: cationic cetrimonium bromide, anionic SDS, and nonionic PEO–PPO–PEO triblock copolymer (Fig. 31). The precipitation reaction rate of CaCO3 can be adjusted and controlled in a nonaqueous (O/W) emulsion system prepared by formamide as the dispersion medium, thereby promoting the formation of a core-shell structure. Indeed, the use of a CaCO3/Fe3O4 composite shell in CaCO3/Fe3O4@n-docosane MEPCMs has been shown to enhance the efficiency of solar photothermal energy conversion and storage,349 as the unique optical properties of the Fe3O4 NPs lead to an acceleration in the photothermal energy conversion and storage of the material. In this way, the photothermal conversion efficiency of the material was improved by 47.9%, which is much higher than that of CaCO3@n-docosane MEPCMs.
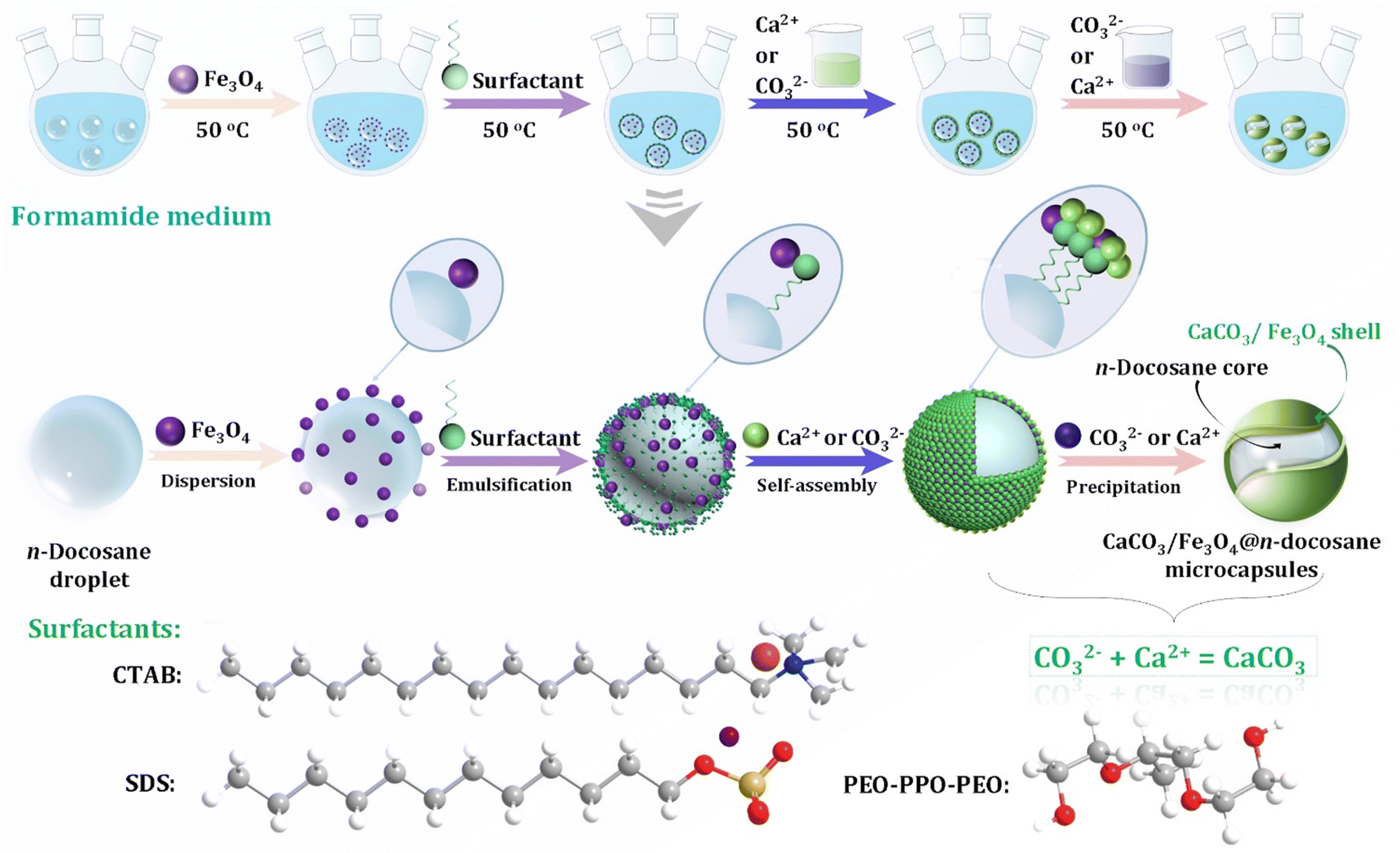 | ||
| Fig. 31 Schematic diagram illustrating novel synthetic strategy for constructing CaCO3/Fe3O4@n-docosane microencapsulated phase change materials. Reprinted from ref. 349. Copyright (2021), with permission from Elsevier. | ||
In addition, CaCO3 has been shown to act as a promising substrate for producing low-cost fluorescent materials that exhibit good luminescence properties.361 In this arena, CaCO3 has been used as a matrix to synthesize CaCO3:Eu3+ phosphors362 and Tb3+/Eu3+ co-doped CaCO3 phosphors.363 CaCO3 integrated with rare earth elements can be introduced into MEPCMs to enhance their thermal energy storage and photoluminescence. Ce3+-Doped CaCO3@paraffin MEPCMs have been designed via a self-assembly precipitation method to synthesize materials that exhibit thermal energy storage and photoluminescence properties.359 Such dual-function Ce3+-doped CaCO3@paraffin MEPCMs might have potential applications in high-tech smart fiber or textiles systems, as well as in thermal and photosensitive sensors.
7. Amorphous calcium carbonate
ACC was first reported in the twentieth century by Addadi et al.364,365 Since then, ACC has been discovered in many organisms, such as earthworms,21 in the teeth of chitons,366 nacre,367 and in the exoskeletons of crustaceans.368 ACC is a precursor of crystalline CaCO3 in mollusks and sea urchins188 and has been classified into proto-calcite, proto-aragonite and proto-vaterite types.369,370 ACC can be further divided into transient precursor and hydrated forms. The stable hydrated form that contains at least one mole of water for every mole of CaCO3 is usually the form adopted by synthetic ACC,365 whereas the transient phase is essentially anhydrous and has thus far only been found in biominerals.58,61Preparation conditions such as pH, temperature, solvent, and additives influence the composition, structure, hydration, and stability of ACC.94 However, how organisms accurately control ACC formation under ambient conditions still remains unclear.94,371 Recently, various inorganic ions such as Mg2+ and PO43−,64,372 polymers,67 and macromolecules372 have been used to stabilize ACC in a short time frame. The preparation conditions and use of additives in the formation and stabilization of ACC have been shown to result in good control over the structure and morphology of the resultant ACC-based materials. Taking advantage of its flexible properties as an amorphous precursor,373 or its mechanical properties in biomineral hybrid structures,374,375 ACC can be used to design strong organic–inorganic hybrid materials. ACC NPs are active and high in energy, thus making them prone to hydrolysis in an intracellular environment.25,30 Monodispersed ACC NPs have been reportedly used as templates to prepare a preloaded drug-delivery carrier and theranostic system.39
7.1 Preparation and stabilization of ACC
Several methods have thus far been reported to prepare ACC, such as the direct mixing of a Ca2+–CO32− aqueous reaction system with or without additives,60,376 gas diffusion techniques,369,377 the hydrolysis of carbonate,378,379 and using a miniemulsion.62 The preparation protocols, presence of co-precipitated ions such as CO32− and Ca2+, temperature, pH, solvent, and drying conditions are factors that have been shown to affect the preparation of ACC and impact upon its further transformation into crystalline CaCO3.380 Synthetic ACC contains water and its lifetime is related to its hydrate content as it transforms into a stable polymorph via a dissolution–(re)precipitation process accompanied by a loss of all of its water content.61As the preparation of ACC usually involves the mixing of two aqueous solutions containing Ca2+ and CO32−, traces of both salt from the counter ions and water are inevitably present in the final ACC material.380 A fast-freezing technique can be employed to isolate ACC from mixed CaCl2–Na2CO3. Dried pure ACC has been prepared by quenching aqueous saturated colloidal CaCO3 suspension in liquid nitrogen (N2) and then sublimating the solvent under vacuum.381 Such treatments removed water from the reaction mixture quickly and prevented transformation of the ACC, resulting in an ACC with extended stability of up to 6 weeks when exposed to atmospheric conditions. Recently, it has been found that the instantaneous carbonation of Ca(OH)2 aerosols with CO2 followed by rapid drying of the ACC aerosols prevented the crystallization and resulted in the formation of a pure, stable, and dry ACC.380 The ACC exhibited good stability against further crystallization for up to 3 weeks in air (∼33% relative humidity). It has been demonstrated that mechanochemical treatment of ACC crystals leads to the structural collapse of ACC to smaller particles and the formation of defects in its structure.382 The addition of Na2CO3 during milling was shown to lead to the incorporation of Na+ cations into the ACC structure. As they were not easily incorporated into the lattice of the material, the Na+ cations kinetically hindered the recrystallization of the resulting ACC. Moreover, the Ca in the ACC structure exhibited distorted octahedral coordination of six oxygen atoms, with an average coordination number of 5 reported for ball-milled ACC.
The drying process can affect the stability of ACC.69 An ACC suspension obtained from two aqueous solutions containing Ca2+ and CO32− was quenched in an organic solvent such as ethanol, isopropanol or acetone as part of its drying process.69 The organic solvent was shown to isolate ACC from the water and slow its crystallization,383 with monodisperse ACC NPs in the range of 100–200 nm produced from a water–ethanol system.384 Furthermore, the formation and transformation of ACC in a mixed solvent of ethanol and water has been found to be affected by the pH of the solution. The solubility of ACC has been reported to decrease with an increase in the pH value of an aqueous solution from 5 to 12.5, and affect the formation and stability of ACC. A porous ACC, with a pore volume of ∼0.86 cm3 g−1 and a pore-size distribution centered at around 8–9 nm has been developed from CaO and CO2 using methanol as a solvent.383 This ACC with a low water content (0.56 mol of H2O per mol of CaCO3) remains amorphous and retains its highly porous structure for over 3 weeks under semi-air-tight storage conditions.
The inclusion of additives, such as Mg2+,64 PO43−,67,372,385 SiO2,69 carboxyl species,74,356 and polymers,386 into ACC bulk have been well documented to improve the stability of ACC significantly. Mg2+ has been found in almost all biogenic ACC.387 The incorporation of Mg2+ in ACC significantly slows its transformation into crystalline phases.388 Such an effect of Mg2+ can be attributed to strong and well-hydrated Mg2+ cations that can delay the nucleation and growth of calcite and can either be incorporated within the calcite lattice to create a barrier to prevent the further growth of calcite nuclei.389,390 Anions such as SiO44−, PO43−, and OH− also influence the stability of ACC. The role of SiO44− in stabilizing ACC has been evidenced by the presence of SiO44− in cystoliths.391 In these cystoliths, a high SiO44− content in the ACC correlated with high thermal stability. Indeed, the negatively-charged tetrahedral SiO44− in the ACC may conceivably destabilize calcite formation both by preventing regular structural packing and by perturbing the charge equilibrium.391 Thus, the stabilization of ACC appears to be strictly related to the destabilization of calcite by “geometric frustration” of its crystal lattice. The PO43− ion also has a tetrahedral structure and has been suggested to stabilize ACC in lobster carapace.392 It has also been reported that PO43− ions form a coating around ACC domains or enter into the ACC framework, thus preventing its transformation into calcite.372 Moreover, PO43− ions reduce particle nucleation and growth rate by binding to a crystal nucleus, thus effectively inhibiting the further crystallization of ACC.65 The role of OH− ions in the ACC stabilizer is negligible when they are added after the precipitation of ACC as OH− ions are not able to alter the surface of the ACC NPs due to the ion association between Ca2+ and CO32− being much stronger than that between Ca2+ and OH−. However, when OH− ions are incorporated in the bulk of ACC NPs rather than adsorbing on their surface, they have a remarkable influence on particle size.371
By inducing various carboxyl-based additives, such as citric acid (CA), adipic acid (AA), and hexanoic acid (HAA), into a suspension of ACC in methanol, followed by a fast drying process, the adsorption and incorporation of these additives can improve the stability of ACC.68 In this process, the CA, AA and HA molecules adsorb onto the surface of the ACC NPs via electrostatic interactions (Fig. 32A, B and D), with the adsorbed CA and AA molecules forming hydrogen bonds via their carboxyl groups (Fig. 32A and B), while HA cannot form further interactions due to it only containing one carboxyl group (Fig. 32D). The concentrations of citrate in ACC extracted from exoskeletons and gastroliths has been found to be higher than those of high-molecular weight phosphoenolpyruvate and 3-phosphoglycerate.393 Citrate strongly chelates Ca2+, thus reducing the activity of free Ca2+ in solution. ACC synthesized in the presence of citrate is thus more stable than pure ACC. Besides this, the adsorption and incorporation of citrate in ACC increases both the lifetime and thermal stability ACC. An increase in the lifetime of the ACC corresponds to an increase in the adsorption and incorporation of citrate in ACC NPs that has the effect of slowing the dehydration and decomposition of ACC.68
 | ||
| Fig. 32 Schematic diagram showing possible interactions in carboxyl-based additive-stabilized ACC NPs. (A) Citric acid (CA); (B) adipic acid (AA); (C) poly(acrylic acid) (PAA) and (D) hexanoic acid (HAA) (the red color indicates additives attached to the surface of ACC NPs via electrostatic interactions; the light green color indicates hydrogen bonds). Reprinted from ref. 69. Copyright (2020), with permission from Elsevier. | ||
According to the “non-classical” nucleation pathway, small CaCO3 pre-nucleation clusters of 0.6–2 nm in size exist in a stearic acid monolayer as a template deposited on a supersaturated 9 mM Ca(HCO3)2 solution.80,394 It is possible to stabilize such small CaCO3 entities using specific ligands to produce monolayer-protected CaCO3 clusters. For example, Sun and coworkers reported a solvothermal method by which to prepare monolayer-protected ACC clusters using 10,12-pentacosadiynoic acid (PCDA) as the ligand, ethanol as the solvent, and NaHCO3 decomposition as a CO2 source.395 The ethanol both solubilizes the ligand and assists in the formation of kinetically stable ACC. Two main reactions successively occur, according to eqn (10) and (11). The PCDA chains are bound to the ACC core via chelation between their terminal –COOH groups and Ca2+ ions. The chemical formula of a monolayer protected ACC cluster with an average size of ∼4.9 nm is suggested to be: (CaCO3)7(H2O)4(PCDA)3.
 | (10) |
| CaCl2 + CO2 + NH4OH → CaCO3·xH2O(ACC) + NH4Cl | (11) |
The formation of ACC can also be influenced by many types of polymers and macromolecules, such as PDA,396 PAA,69 proteins94 and dendrimers.397 The inhibition in the transformation of ACC to a crystalline phase might involve the confining of the hydrated ACC within a hydrophobic polymer coating so that the water cannot escape. The small ACC particles are therefore isolated from the aqueous environment, making ACC stable.365 Wang et al.396 demonstrated that ACC NPs can be stabilized via a dopamine polymerization process at room temperature using an aqueous solution of CaCl2, dimethyl carbonate, and dopamine. The Ca2+ ions were found to strongly interact with the flexible chains of the PDA, which were not only incorporated into but also coated the surface of the primary ACC NPs. The PDA network stabilizes the ACC, preventing the ACC NPs from growing bigger. Moreover, the PDA coating inhibits the dissolution of the ACC dissolution, thus slowing the subsequent Ostwald ripening. Moreover, such a coating creates isolated confinement spaces for ACC that prevent the contact and merger of ACC NPs, which further restricts the possible solid-phase transformation of ACC to a crystalline phase.
Similarly, PAA, a long-chain polymer additive, can stabilize ACC NPs by surrounding them to form a shell-like structure around the central ACC NP core (Fig. 32C).68 Although the carboxyl groups on PAA could form hydrogen bondings with another ACC–PAA NP, the bulky PAA molecules possibly have relatively weak bonds (Fig. 32C). The long carbon chain of PAA prevents the further aggregation of the ACC–PAA NPs. When very small amounts of double hydrophilic block copolymers (DHBCs) comprising poly(ethylene oxide) (PEO) and PAA blocks are used as controlling agents, the adsorption layer of the block copolymer protects the liquid precursor of ACC from coalescence and/or coagulation.386 Subsequently, spherical NPs of ACC with a narrow size distribution were obtained. The PAA not only interacts with Ca2+ but also induces the formation of CaCO3-rich liquid phases, referred to as PILP phases.398,399 Instead of being incorporated into the ACC bulk, PAA adsorbs to the ACC surface, which results in the ACC nanospheres being highly stable.400 Biomacromolecules, polysaccharides, and proteins can also bind to the surface of ACC, isolating it from water and inhibiting its dissolution and subsequent crystallization to crystalline phases.401,402 Such bio-macromolecules can also bind strongly to free Ca2+, thus inhibiting or slowing its interaction with HCO3− and CO32− during the formation and transformation of ACC.21 Dendrimers are monodisperse macromolecules with a regular and highly branched 3D structure that can prolong the incubation time of ACC due to dendritic effects. By alternating the concentration of a carboxylic acid-terminated G0.5 poly(amidoamine) dendrimer that acts as a nucleation site at a fixed initial pH of 12 ± 0.2 at 15 °C with a CaCl2–Na2CO3 liquid–liquid phase, spherical ACC NPs have been produced.397 The surface of the dendrimer was proposed to act as a nucleation site, whereas its surface branch cells were found to compete with CO32−. In the reaction, G0.5 acts as a stabilizer and inhibitor for ACC, preventing its crystallization to form calcite and vaterite, possibly resulting from the effects of the G0.5 dendrimer and its coordination of Ca2+ ions.
In the absence of biologically relevant additives such as divalent cations, negatively-charged polymers, and PO43−, the binding between positively-charged Ca2+ ions and negatively-charged CO32− ions resulted in the formation of CaCO3 complex ions. The ACC phase separated from solution via a spinodal decomposition process wherein the overall concentration of the CaCO3 complexes determined the average particle size of the ACC (Fig. 33A).403 To tune the particle size of ACC and promote the phase separation process, the additives must be able to interact with Ca2+ or CO32− ions simultaneously to induce the occurrence of long-range interactions between the CaCO3 complex ions (Fig. 33B). The first way to this involves a cooperative ion-association process wherein the additives are attracted to either Ca2+ or CO32− ions and provide additional effective interactions between the CaCO3 complex ions. Such a cooperative ion-association process can be achieved by charged polymers with a long chain length, which attract either Ca2+ or CO32− ions and their associated CaCO3 complex ions along the chain (Fig. 33B). A similar result can be achieved using ions such as PO43− that are able to form a dense structure with Ca2+ ions.403
 | ||
| Fig. 33 Schematic diagram showing inherent mechanisms of ACC stabilization. (A) In the absence or presence of additives that are not able to influence the formation of ACC. (B) In the presence of charged polymers and PO43− ions that are able to influence the formation of ACC. Adapted and reprinted from ref. 403. Copyright (2018), with permission from the Royal Society of Chemistry. | ||
The lifespan of ACC is controlled by the type, structural arrangement, and quantities of the hydrous components (i.e., mobile vs. rigid water, hydroxide ions) present in its structure.404 The mobile water present within the structure of ACC is readily lost at temperatures of <150 °C, whereas rigid and less accessible water and hydroxide ions can only be removed at temperatures of >250 °C. Concomitant with the dehydration process, structural reorganization of the ACC has been observed from NMR analyses405 and molecular modeling,406 indicating that with increasing temperature a more ordered and stable atomic network forms. Besides the role that the structural arrangement of ACC plays in its lifespan, ACC that precipitates slowly in the range of pH 9–10 has been suggested to be more stable.370
7.2 Functionality and application of amorphous CaCO3-based materials
Generally, ACC plays three major roles in biominerals. First, it acts as a transient, shapeable, amorphous precursor phase for crystalline CaCO3.407 Second, ACC is a rapidly accessible store of Ca for skeletal growth, as well as for the on demand production and repair of load-bearing structures.408 For example, in the case of arthropods,409 freshwater crayfish usually store protein-stabilized ACC in their gastroliths and use it to build their exoskeleton during molting.94,393 Third, owing to its isotropic nature and ability to incorporate high concentrations of trace elements, ACC functions as a structural material. For example, crustaceans use ACC to stiffen exoskeletal cuticles. Owing to its adjustable properties as an amorphous precursor,373 and the mechanical properties that it provides in biomineral hybrid structures,374,375 ACC has been used as a precursor or final component for the design of new materials. In addition, because of their excellent transparent properties, strength, stiffness, and hardness, hydrogel films57 with optical transparency and increased hardness can be created by incorporating ACC as an isotropic filler material in polymer phases.386 Furthermore, ACC-based nanomaterials have shown great potential for use in biomedical applications as they are small in size and flexible toward further engineering.By incorporation of 2–3 nm-sized ACC NPs into PAA, transparent, stable, and crack-free ACC/PAA thin films have been prepared.60 Under ambient conditions, PAA and ACC are hybridized together (Fig. 34A), which results in the formation of ACC/PAA thin films consisting of ACC, PAA, and water. The complexation with a large amount of PAA and water molecules strongly inhibits the further crystallization of ACC in these ACC/PAA thin films. The ACC/PAA thin films possessed nano-segregated structures to form a new class of glassy functional materials.
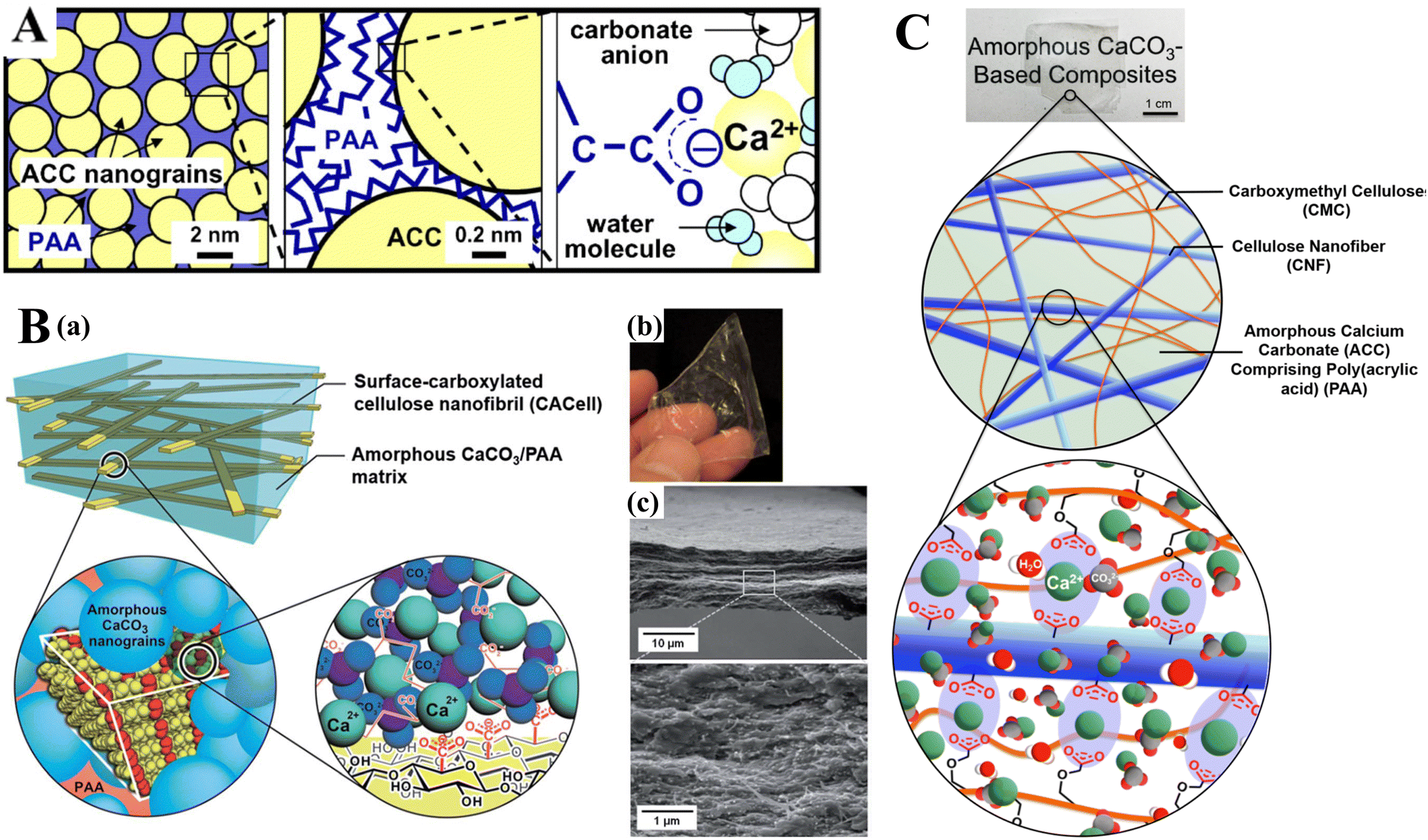 | ||
| Fig. 34 Schematic diagram of novel ACC-based organic/inorganic hybrid films. (A) Nanosegregated amorphous composite consisting of ACC and PAA. Reprinted from ref. 60. Copyright (2008), with permission from John Wiley and Sons under the terms of the Creative Commons Attribution Non-Commercial (CC BY-NC 3.0) License. (B) Materials design and structures of CACell/ACC/PAA composites. (a) Schematic illustration of the design of CACell/ACC/PAA composites, showing the ACC/PAA matrix reinforced with the CACell network. Carboxyl groups on the nanofibril surfaces bound to calcium ions in the nanosegregated ACC/PAA matrix. (b) The prepared CACell/ACC/PAA composite film. (c) SEM images of a cross-section of the CACell/ACC/PAA composite. Reprinted from ref. 410. Copyright (2014), with permission from the Royal Society of Chemistry. (C) ACC-based (CMC/CNF/ACC) transparent films with biomimetic structures. Reprinted with permission from ref. 411. Copyright (2018) American Chemical Society. (CACell/ACC/PAA: carboxylated cellulose/amorphous calcium carbonate/poly (acrylic acid); CMC/CNF/ACC: carboxymethyl cellulose/cellulose nanofiber/amorphous calcium carbonate.) | ||
Moreover, nanofibrillar cellulose-reinforced ACC/PAA free-standing composite films have been obtained by soaking a hydrogel sheet of carboxylated cellulose (CACell) in a colloidal ACC dispersion and drying it (Fig. 34B).410 The CACell/ACC/PAA free-standing composite films were shown to mimic the organic/organic/inorganic nanocomposite structures of crustacean exoskeletons, in which the ACC component binds to chitin nanofibrils at the interface via acidic chitin-binding peptides (Fig. 34B-a). In such films, the reinforcing fibrils of the carboxylated cellulose capable of interacting with Ca2+ directly bind to ACC, which results in a strengthening of the interface of the fibrils and ACC (Fig. 34B-c). The stiff yet flexible and strong transparent CACell/ACC/PAA free-standing composite films exhibit elastic flexibility similar to those of natural crustacean exoskeletons (Fig. 34B-b).
Another example concerns the design and preparation of transparent and mechanically-tough CMC/CNF/ACC composite films via incorporating water-dispersible cellulose derivatives, namely carboxymethyl cellulose (CMC) and surface-decorated crystalline cellulose nanofibers (CNFs) into ACC/PAA composites (Fig. 34C).411 The incorporation of ACC into cellulose-based materials was found to enhance the mechanical properties of the resultant CMC/CNF/ACC composite films while maintaining their high transparency and environmental friendliness. Molecular-scale interactions between CMC and ACC via intermolecular forces between the carboxylate groups and Ca2+ were thought to enhance the stiffness and hardness of the flexible CMC/CNF/ACC composite films. Indeed, a CMC/CNF/ACC composite film with a ratio of 40, 40, and 20 wt% of CMC, CNFs, and ACC, respectively, showed a Young's modulus of 15.8 ± 0.93 GPa and a tensile strength of 268 ± 20 MPa. An appropriate amount of ACC interacting with the polymer fulfills the space between the organic matrices.
000 g mol−1) in water, an ACC/PAA supramolecular hydrogel can be obtained.70 The resultant ACC/PAA hydrogel is a complex of ACC NPs physically cross-linked by PAA chains via electrostatic interactions between COO− and Ca2+. The hydrogel is a dough-like material, soft but tough, and can be shaped into films, cylinders, and stars. It can also be stretched into very long fibers with plastic deformation without any elastic recovery. Moreover, when the hydrogel is broken in half and ressembled, it can rapidly self-heal within 5 s. A SEM image of the freeze-dried ACC/PAA hydrogel shows that it has a porous internal structure. In addition, very small ACC NPs of 1.5–3 nm can be identified from the TEM image of the dry gel. Considering the increasing environmental concerns arising from the petroleum-based production of conventional plastics, the ACC/PAA hydrogel may be used as a new environmentally-friendly and sustainable plastic material or “mineral plastic.”
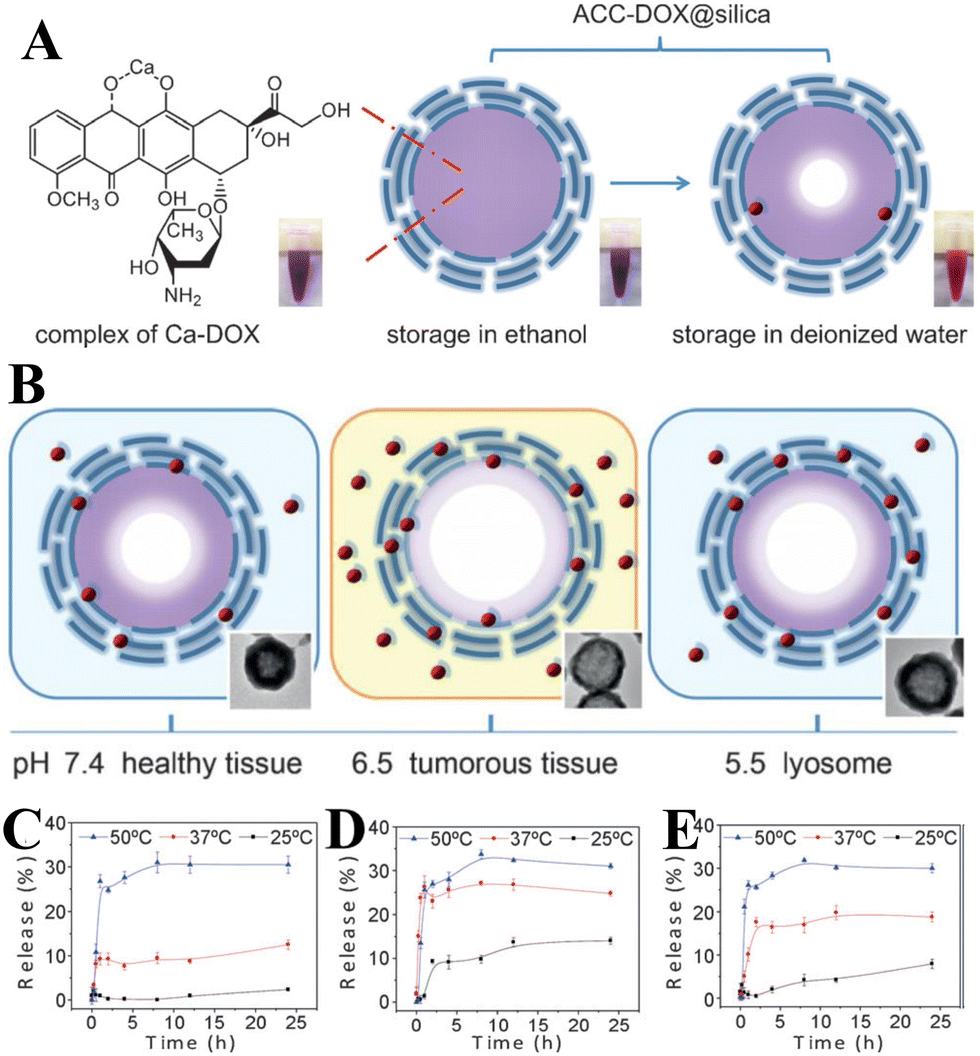 | ||
| Fig. 35 Schematic diagram of ACC–DOX@silica nanoreactor for pH-responsive delivery in cancer therapy. (A) The ACC–DOX@silica nanoreactor and its stability in ethanol and water. The inset images (from left to right) show the suspensions of ACC–DOX in ethanol, ACC–DOX@silica in ethanol, and ACC–DOX@silica in water. (B) The release behavior of ACC–DOX NPs under various pH conditions. Cumulative release of DOX from ACC–DOX@silica suspensions in various aqueous buffers at (C) pH 7.4, (D) pH 6.5, and (E) pH 5.5 at different temperatures of 25, 37, and 50 °C. (A), (C), (D), (E) Reprinted from ref. 415. Copyright (2015), with permission from John Wiley and Sons under the terms of the Creative Commons Attribution Non-Commercial (CC BY-NC 3.0) License. (B) Reprinted from ref. 95. Copyright (2018), with permission from MDPI under the terms of the Creative Commons Attribution License. | ||
However, the thick silica shell may induce adverse effects such as an increase in nanocarrier size, inadequate drug release, and hard degradation in vivo.416 Therefore, a more appropriate biocompatible and biodegradable stabilizer is desirable for stabilizing the ACC NPs and achieving sustained drug release. When ACC–DOX was surface modified with phospholipid (PL) (containing an appropriate amount of DSPE–PEG and/or DSPE–PEG–FA), the PL substance acted as a biocompatible and water-resistant coating to improve the half-life period of the modified PL/ACC–DOX NPs and maintain the water responsiveness of the ACC (Fig. 36A).417 The PL also acts as a diffusion barrier to prevent potential drug leakage. Using a facile solvent-diffusion method, mitoxantrone (MIT)-preloaded PL–ACC (PL/ACC–MIT) hybrid NPs containing the shielding polymer PEG and targeting moiety FA were prepared.72 The PL/ACC–MIT NPs were shown to have satisfactory stability toward various aqueous environments with minimal drug leakage and good targeting capability. Xu et al.272 developed tunable pH-responsive DOX@ACC/PAA NPs (pH 7.4–5.6) by encapsulating DOX in PAA-stabilized ACC NPs. The chelation between DOX and Ca2+and the hydrogen bonding between DOX and PAA, enabled efficient DOX encapsulation into the ACC/PAA NPs. The resultant monodisperse DOX@ACC/PAA NPs were found to be small in size (62 ± 10 nm), have good serum stability, and high drug encapsulation efficiency (>80%) and loading capacity (>9%). Importantly, the release of DOX from the NPs could be further modulated to higher pH response ranges (pH 7.7–6.0) via the doping of specific amounts of Sr3+ or Mg2+ into the DOX@ACC/PAA NPs. The resultant Sr3+- or Mg2+-doped DOX@ACC/PAA NPs could be potentially utilized for specific domains of cells or tissues with a less acidic microenvironment.
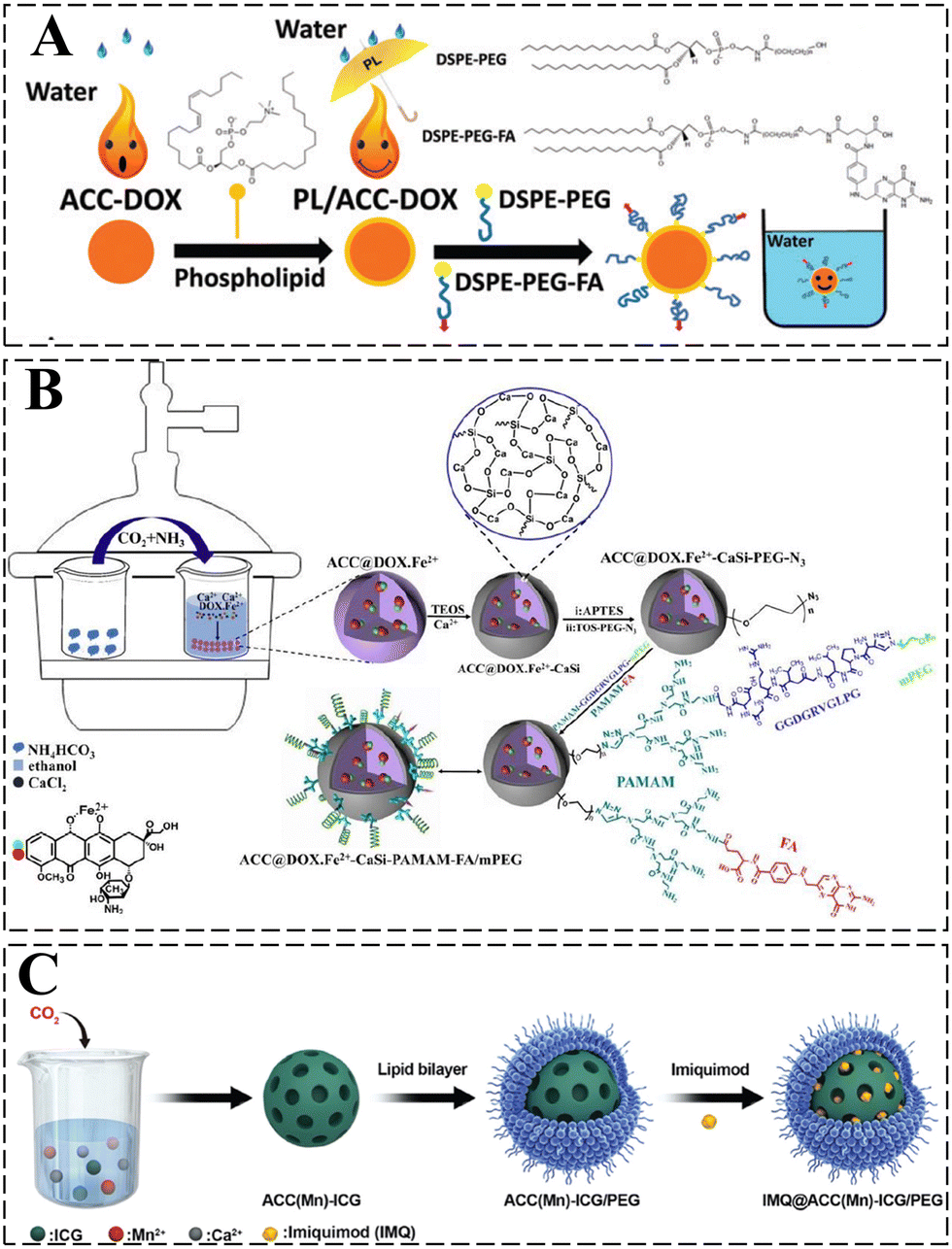 | ||
| Fig. 36 Schematic diagram of ACC NPs for pH-responsive drug delivery. (A) PL/ACC–DOX hybrid NPs for controllable burst drug release and enhanced tumor penetration. Reprinted from ref. 417. Copyright (2018), with permission from the Royal Society of Chemistry. (B) TME-activatable Fe–DOX preloaded ACC nanoformulation triggers ferroptosis in target tumor cells. Reprinted with permission from ref. 73. Copyright (2020), with permission from AAAS under the terms of the Creative Commons Attribution NonCommercial License 4.0 (CC-BY-NC). (C) IMQ@ACC(Mn)–ICG/PEG NPs as immunoadjuvants for multimodal imaging and enhanced photoimmunotherapy. Reprinted from ref. 420. Copyright (2020), with permission from the Royal Society of Chemistry. ACC: amorphous CaCO3; ICG: indocyanine green; IMQ: imiquimod; PEG: polyethylene glycol; PL: phospholipid. | ||
To address inadequate drug release and poor drug penetration in cancer therapy, if the high instability of ACC could be harnessed while realizing drug burst release within specific targeted sites, it would be a beneficial material to use to improve drug release within cells in a water-responsive manner. To this aim, Wang et al.418 locked ACC within a superhydrophobic oleic acid (OA) shell and then modified the shell with a PEG corona to form PEG/OA–ACC. The PEG/OA–ACC material exhibits greatly enhanced stability of ACC under neutral conditions and the high aqueous instability of ACC was demonstrated to realize drug burst release in an acidic environment within cancer cells, which resulted in a comparable anticancer effect being achieved to that of free drugs. Similarly, lipase-triggered water-responsive “Pandora's box” (MS/ACC–DOX) NPs have been constructed by coating DOX-preloaded ACC–DOX NPs with monostearin (MS).419 During its application, the high aqueous instability of ACC led to a burst release of the encapsulated DOX, inducing apoptosis and cytotoxicity to kill tumor cells. The released MS/ACC–DOX NPs from the dead or dying cells continued to water-responsively release DOX after being unpacked in a “Pandora’s box” manner, inducing apoptosis on untreated neighboring cells (neighboring effect). The severe neighboring effect of MS/ACC–DOX NPs with increased cytotoxicity and continuously released free DOX molecules from the exterior to the interior of the solid tumor contributed to enhance DOX penetration into deep tumor matrix, resulting in elevating antitumor efficacy.
ACC NPs can be simultaneously loaded with multiple molecules to achieve multimodal cancer treatment. The ACC substrate can synergize with the therapeutic interaction between DOX and Fe2+ to form a TME-activatable Fe–DOX preloaded ACC nanoformulation with complementary ferroptosis/apoptosis-inducing capability. For example, a tumor-targeted ACC-based nanoplatform (ACC@DOX.Fe2+–CaSi–PAMAM–FA/mPEG) was developed for ferroptosis therapy (Fig. 36B).73 ACC@DOX–Fe2+ cores were first established via a gas diffusion method, followed by coating with a thin layer of CaSi (Fig. 36B). The ACC@DOX·Fe2+–CaSi material was then simultaneously conjugated with folate-modified and PEGylated polyamidoamine (PAMAM) dendrimers, which offered a functional balance between circulation longevity and tumor-specific uptake. The DOX and Fe2+ were released intracellularly in a self-regulated manner via the acidity-triggered degradation of ACC, where DOX amplifies the ferroptosis effects of Fe2+ by producing H2O2. In addition, the ACC NPs have the ability to clear H+ from the tumor tissue, inhibiting the polarization of tumor-associated macrophages to M2-like phenotypes. Mn2+-Doped ACC NPs have been designed as a multicarrier to co-deliver photosensitive ICG and the toll-like-receptor-7 agonist imiquimod (IMQ) to achieve pH-triggered controllable drug delivery and tumor-specific enhanced photoimmunotherapy guided by multimodal imaging (Fig. 36C).420 In this study, ICG was chelated with Ca2+ ions and was finally embedded in ACC in the form of a Ca–ICG complex. ACC(Mn)–ICG NPs were modified with a self-assembled lipid bilayer, followed by loading of IMQ to form IMQ@ACC(Mn)–ICG/PEG NPs (Fig. 36C). ACC NPs scavenged the H+ of the tumor to reverse the immunosuppression of the tumor environment, effectively inhibiting tumor metastases. The IMQ@ACC(Mn)–ICG/PEG NPs heavily accumulated in tumor tissues and significantly ablated the tumors after fluorescence/MRI-guided laser irradiation. These bioresponsive theranostic NPs based-on biodegradable ACC could thus become an effective tool for use in cancer theranostics.
8. Concluding remarks
The past decade has witnessed tremendous advances in experimental strategies for controlling the preparation and modification of CaCO3 MNPs, as well as the stabilization of synthetic ACC. Focus has been on the investigation of the nucleation and growth mechanisms of specific CaCO3 polymorphs and the development of new methods by which to engineer CaCO3 into multifunctional nanostructured materials. Thus, various CaCO3-based materials have been developed and used for bone repair, wound healing, bioimaging, theranostics, environmental maintenance, and clean energy production and storage. Despite impressive achievements, there are still open challenges in terms of developing new methodologies for controlled synthesis, surface modification, and engineering of CaCO3 into nanostructured materials and their applications.Previous studies have indicated that although CaCO3 precipitation is not a simple process, the insights discussed above might play a role in predicting a particular form or shape of CaCO3 precipitation to some extent. The control of the crystal size and morphology of CaCO3 MNPs particles can be achieved even by slight variations in the temperature, pH, mixing of reactants, or by the addition of different organic or inorganic additives to reactions. Although it remains of major interest, special attention has been paid to polymorphism, which can be regulated via subtle interplay between thermodynamics and kinetic factors.104 For example, high supersaturation causes the initial formation of small particles and metastable phases, such as ACC or vaterite, which then transforms via solution-mediated process into very stable calcite. However, if organic additives, including surfactants, macromolecules, and polyelectrolytes, or certain inorganic additives are present, the unstable phases can be stabilized more or less. In such systems, the shape of CaCO3 particles could be significantly affected by the concentration and nature of additives, and particular crystal planes can be expressed as a consequence of specific interactions between additive and growing crystal surfaces. Formation of spherical or rounded particles could be a result of inhibited or spherulitic growth in the presence of additives and the aggregation of smaller primary particles. Under the conditions of high initial supersaturation, the interplay of kinetics of nucleation, crystal growth, dissolution, and ageing appear to be critical for the physical and chemical characteristics of the final CaCO3 precipitate. In contrast, a lower initial supersaturation typically causes the direct formation of thermodynamically stable polymorph calcite, either by heterogeneous or homogeneous nucleation. The deviation from stable rhombohedral morphology in the absence of additives might be a consequence of a nonstoichiometric distribution of constituent ions (Ca2+ and CO32−) or H+ and OH− and the dominant adsorption of ions that are in excess on specific crystal planes. The remarkable influence of additional inorganic ions on the CaCO3 precipitation and its products has captured particular attention recently, and yet the understanding of it is still in infancy. It is now known that low temperatures and the presence of orthophosphate promote the formation of hydrated forms, such as ikaite (or monohydrocalcite). Higher temperatures and the presence of Mg2+, promote the formation of aragonite and other metastable phases. Unfortunately, although the CO2 carbonation process conducted using an economically and environmentally-friendly process provides CaCO3 MNPs with a wide particle size distribution range, shapes and polymorphs, it cannot be well controlled. In contrast, CaCO3 particles of various sizes (nanometer to micrometer), polymorph composition, and morphologies (needle-like, rod-like, whisker-like, spheres, porous spheres, hollow spheres, etc.) can be synthesized using different reactants, additives, and even different experimental set ups, for example Ca2+–H2O/organic solvents/additives/CO32− precipitation systems, W/O reverse emulsion, or biomimetic CaCO3 formation. However, the production of CaCO3 in such systems is still hard to achieve on a large scale.
An in-depth understanding of the basic principles of CaCO3 nucleation and growth is essential for achieving tunable synthesis. Currently, the predominant nucleation and growth mechanisms are still questionable. The classical nucleation mechanism, which assumes the formation of critically sized nuclei of a new solid phase, is challenged by the non-classical concept. On the contrary, the non-classical mechanism assumes the initial formation of thermodynamically-stable prenucleation clusters or the formation of a dense liquid precursor phase via liquid–liquid phase separation. Further studies are thus required to address the following issues. How can the pathway of a certain specific crystallization process be controlled? Which crystallization process is dominant under a specific set of conditions? In addition, the current research on non-classical CaCO3 crystallization is mainly based on the assumption and the conditions of a homogeneous nucleation, so further questions are: what is the exact crystallization mechanism and growth process under heterogeneous nucleation? What is the exact mode of the molecular interactions between the substrates and precursors during the nucleation?
So far, the surface modification of CaCO3 MNPs has been predominantly accomplished either via surface adsorption and coating with coupling agents, organic acids, or anionic and cationic surfactants, or using solid inorganic SiO2. Such modification tunes the surface polarity, hydro- and oleophilicity, stability, and reactivity of the CaCO3 particles. The surface-modified CaCO3 can then be further engineered into functional hierarchical nanostructured materials, which show great potential in numerous fields, ranging from biomaterials and biomedicine to environmental remediation and energy storage. A characteristic of CaCO3 NPs is their sensitivity to pH, which allows them to release the cargo conditionally at specific sites. Nevertheless, there is still much need for future work to enable better tune the pH-sensitivity of the CaCO3 NPs. In this context, as the subtle changes in a TME are not only down to pH, other physiological factors, such as low-oxygen levels, abnormal cysteine metabolism or concentrations of relevant ions, are worth considering so as to match these factors to the features of CaCO3 materials. At present, most of the biomedical applications of CaCO3 have only reached the pre-clinical stage. Biomimetically ordered arrangements of CaCO3 nanocrystals, as a prototype of advanced organic–inorganic composites, have led to nacre-like structures being produced, but their density and uniformity are far from those of the nacre in seashells or mollusks. It is thus expected that the aforementioned technologies of synthetic CaCO3 will be extended to utilize biogenic CaCO3 resources (e.g., eggshell or mollusk shells), which are available in large quantities in the agriculture and food industries, with the aim of achieving good recyclability, sustainability, and a low-carbon footprint.
ACC also has great potential for use as a smart and pH-responsive hydrogel or drug carrier. Over the last decade, much effort has been made to develop an effective way of stabilizing ACC, however, the long-term stability of ACC has yet to be accomplished. Both the intrinsic properties of ACC and the adjacent environmental conditions (temperature, pressure, humidity, etc.) have been found to be critical in determining its stability. The correlation of the synthesis conditions with the ACC stability and the interactions between the additives and the resultant ACC still remain unclear. An unanswered question that remains is related to the inhibition of dehydration and transformation of ACC into crystalline CaCO3. To tackle the complexity of ACC systems, it is necessary to further investigate the mechanisms of the biomineralization processes of specific organisms that are responsible for accurately controlling the formation and stabilization of ACC. Such detailed studies will be helpful toward the design and subsequent production of functional biomimetic CaCO3 nanostructures and/or composites in an environmentally-friendly and even sustainable manner.
Author contributions
Yu-Qin Niu: investigation, writing original draft, writing-review and editing; Jia-Hui Liu: reviewing, revision and editing; Cyril Aymonier: revision and editing; Simona Fermani: revision and editing; Damir Kralj: reviewing, revision and editing; Giuseppe Falini: reviewing, revision and editing; Chun-Hui Zhou: conceiving the project, conceptualization, discussion, supervision, writing and reviewing, revision and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to acknowledge the financial support from the National Natural Science Foundation of China (22072136). G. F. and S. F. thank the Bluebio ERANET for funding (project CASEAWA; grant number 161B0949).Notes and references
- J. W. Morse, R. S. Arvidson and A. Luttge, Chem. Rev., 2007, 107, 342–381 CrossRef CAS PubMed.
- W. Shi, S. Zhang, Y. Wang, J. Wang, B. Li, X. Liu, X. Liu, Q. Wang and J. Cheng, ChemistrySelect, 2018, 3, 6050–6055 CrossRef CAS.
- H. Ehrlich, M. Motylenko, P. V. Sundareshwar, A. Ereskovsky, I. Zgłobicka, T. Noga, T. Płociński, M. V. Tsurkan, E. Wyroba, S. Suski, H. Bilski, M. Wysokowski, H. Stöcker, A. Makarova, D. Vyalikh, J. Walter, S. L. Molodtsov, V. V. Bazhenov, I. Petrenko, E. Langer, A. Richter, E. Niederschlag, M. Pisarek, A. Springer, M. Gelinsky, D. Rafaja, A. Witkowski, D. C. Meyer, T. Jesionowski and K. J. Kurzydłowski, Adv. Funct. Mater., 2016, 26, 2503–2510 CrossRef CAS.
- H. Ehrlich, E. Brunner, P. Simon, V. V. Bazhenov, J. P. Botting, K. R. Tabachnick, A. Springer, K. Kummer, D. V. Vyalikh, S. L. Molodtsov, D. Kurek, M. Kammer, R. Born, A. Kovalev, S. N. Gorb, P. G. Koutsoukos and A. Summers, Adv. Funct. Mater., 2011, 21, 3473–3481 CrossRef CAS.
- A. Kertmen, I. Petrenko, C. Schimpf, D. Rafaja, O. Petrova, V. Sivkov, S. Nekipelov, A. Fursov, A. L. Stelling, K. Heimler, A. Rogoll, C. Vogt and H. Ehrlich, Int. J. Mol. Sci., 2021, 22, 12588 CrossRef CAS PubMed.
- Y. Politi, T. Arad, E. Klein, S. Weiner and L. Addadi, Science, 2004, 306, 1161–1164 CrossRef CAS PubMed.
- J. N. Murphy, C. M. Schneider, K. Hawboldt and F. M. Kerton, Matter, 2020, 3, 2029–2041 CrossRef.
- T. Yang, Z. Jia, H. Chen, Z. Deng, W. Liu, L. Chen and L. Li, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 23450–23459 CrossRef CAS PubMed.
- J. Aizenberg, A. Tkachenko, S. Weiner, L. Addadi and G. Hendler, Nature, 2001, 412, 819–822 CrossRef CAS PubMed.
- P. Simon, W. Pompe, D. Gruner, E. Sturm, K. Ostermann, S. Matys, M. Vogel and G. Rodel, ACS Biomater. Sci. Eng., 2022, 8, 526–539 CrossRef CAS PubMed.
- H. Ehrlich, E. Bailey, M. Wysokowski and T. Jesionowski, Biomimetics, 2021, 6, 46 CrossRef CAS PubMed.
- M. Wysokowski, P. Zaslansky and H. Ehrlich, ACS Biomater. Sci. Eng., 2020, 6, 5357–5367 CrossRef CAS PubMed.
- M. Wysokowski, I. Petrenko, R. Galli, C. Schimpf, D. Rafaja, J. Hubalkova, C. G. Aneziris, S. Dyshlovoy, G. von Amsberg, H. Meissner, Y. M. Yakovlev, K. R. Tabachnick, A. L. Stelling and H. Ehrlich, Appl. Phys. A, 2020, 126, 727 CrossRef CAS.
- J. Aufort and R. Demichelis, Cryst. Growth Des., 2020, 20, 8028–8038 CrossRef CAS.
- Y.-B. Hu, D. A. Wolf-Gladrow, G. S. Dieckmann, C. Völker and G. Nehrke, Mar. Chem., 2014, 162, 10–18 CrossRef CAS.
- Z. Y. Zou, W. J. E. M. Habraken, G. Matveeva, A. C. S. Jensen, L. Bertinetti, M. A. Hood, C. Y. Sun, P. U. P. A. Gilbert, I. Polishchuk, B. Pokroy, J. Mahanid, Y. Politi, S. Weiner, P. Werner, S. Bette, R. Dinnebier, U. Kolb, E. Zolotoyabko and P. Fratzl, Science, 2019, 363, 396–400 CrossRef CAS PubMed.
- T. Mass, A. J. Giuffre, C. Y. Sun, C. A. Stifler, M. J. Frazier, M. Neder, N. Tamura, C. V. Stan, M. A. Marcus and P. Gilbert, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E7670–E7678 CrossRef CAS PubMed.
- A. Akiva-Tal, S. Kababya, Y. S. Balazs, L. Glazer, A. Berman, A. Sagi and A. Schmidt, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14763–14768 CrossRef CAS PubMed.
- Y. U. Gong, C. E. Killian, I. C. Olson, N. P. Appathurai, A. L. Amasino, M. C. Martin, L. J. Holt, F. H. Wilt and P. U. Gilbert, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6088–6093 CrossRef CAS PubMed.
- R. T. DeVol, C. Y. Sun, M. A. Marcus, S. N. Coppersmith, S. C. Myneni and P. U. Gilbert, J. Am. Chem. Soc., 2015, 137, 13325–13333 CrossRef CAS PubMed.
- M. E. Hodson, L. G. Benning, B. Demarchi, K. E. Penkman, J. D. Rodriguez-Blanco, P. F. Schofield and E. A. Versteegh, Geochem. Trans., 2015, 16, 4 CrossRef PubMed.
- F. C. Meldrum, Int. Mater. Rev., 2003, 48, 187–224 CrossRef CAS.
- Y. Boyjoo, V. K. Pareek and J. Liu, J. Mater. Chem. A, 2014, 2, 14270–14288 RSC.
- T. Yang, J. Fu, L. Ma, H. Du, X. Yue, B. Zhao and C. Wang, Mater. Sci. Eng., C, 2020, 114, 111019 CrossRef CAS PubMed.
- H. Bahrom, A. A. Goncharenko, L. I. Fatkhutdinova, O. O. Peltek, A. R. Muslimov, O. Y. Koval, I. E. Eliseev, A. Manchev, D. Gorin, I. I. Shishkin, R. E. Noskov, A. S. Timin, P. Ginzburg and M. V. Zyuzin, ACS Sustainable Chem. Eng., 2019, 7, 19142–19156 CrossRef CAS.
- T. M. Stawski, T. Roncal-Herrero, A. Fernandez-Martinez, A. Matamoros-Veloza, R. Kroger and L. G. Benning, Phys. Chem. Chem. Phys., 2018, 20, 13825–13835 RSC.
- Y. Yu, J. Zhang, H. Wang and Z. Xin, Polymers, 2020, 12, 2668 CrossRef CAS PubMed.
- N. A. J. M. Sommerdijk and G. de With, Chem. Rev., 2008, 108, 4499–4550 CrossRef CAS PubMed.
- F. C. Meldrum and H. Colfen, Chem. Rev., 2008, 108, 4332–4432 CrossRef CAS PubMed.
- L. Štajner, J. Kontrec, B. Njegić Džakula, N. Maltar-Strmečki, M. Plodinec, D. M. Lyons and D. Kralj, J. Cryst. Growth, 2018, 486, 71–81 CrossRef.
- J. A. Lopez-Berganza and R. M. Espinosa-Marzal, Cryst. Growth Des., 2016, 16, 6186–6198 CrossRef CAS.
- Y.-Y. Kim, J. D. Carloni, B. Demarchi, D. Sparks, D. G. Reid, M. E. Kunitake, C. C. Tang, M. J. Duer, C. L. Freeman, B. Pokroy, K. Penkman, J. H. Harding, L. A. Estroff, S. P. Baker and F. C. Meldrum, Nat. Mater., 2016, 15, 903–910 CrossRef CAS PubMed.
- C. Han, Y. Hu, K. Wang and G. Luo, Powder Technol., 2019, 356, 414–422 CrossRef CAS.
- Z. Liu, H. Onay, F. Guo, J. Chen, L. Poltorak, P. Hedayati and E. J. R. Sudhölter, Energy Fuels, 2021, 35, 1358–1370 CrossRef CAS.
- D. Volodkin, Adv. Colloid Interface Sci., 2014, 207, 306–324 CrossRef CAS PubMed.
- N. T. Enyedi, J. Makk, L. Kotai, B. Berenyi, S. Klebert, Z. Sebestyen, Z. Molnar, A. K. Borsodi, S. Leel-Ossy, A. Demeny and P. Nemeth, Sci. Rep., 2020, 10, 8696 CrossRef CAS PubMed.
- A. Y. Cai, Y. J. Zhu and C. Qi, Adv. Mater. Interfaces, 2020, 7, 2000819 CrossRef CAS.
- V. Vergaro, I. Pisano, R. Grisorio, F. Baldassarre, R. Mallamaci, A. Santoro, G. P. Suranna, P. Papadia, F. P. Fanizzi and G. Ciccarella, Materials, 2019, 12, 1481 CrossRef CAS PubMed.
- Y. Zhao, Z. Luo, M. H. Li, Q. Y. Qu, X. Ma, S. H. Yu and Y. L. Zhao, Angew. Chem., Int. Ed., 2015, 54, 919–922 CrossRef CAS PubMed.
- S. Wang, D. Ni, H. Yue, N. Luo, X. Xi, Y. Wang, M. Shi, W. Wei and G. Ma, Small, 2018, 14, e1704272 CrossRef PubMed.
- Y. Zhao, L. N. Lin, Y. Lu, S. F. Chen, L. Dong and S. H. Yu, Adv. Mater., 2010, 22, 5255–5259 CrossRef CAS PubMed.
- Z. Dong, L. Feng, Y. Hao, M. Chen, M. Gao, Y. Chao, H. Zhao, W. Zhu, J. Liu, C. Liang, Q. Zhang and Z. Liu, J. Am. Chem. Soc., 2018, 140, 2165–2178 CrossRef CAS PubMed.
- L. Kang, M. Cui, F. Jiang, Y. Gao, H. Luo, J. Liu, W. Liang and C. Zhi, Adv. Energy Mater., 2018, 8, 1801090 CrossRef.
- G. Begum, T. N. Reddy, K. P. Kumar, K. Dhevendar, S. Singh, M. Amarnath, S. Misra, V. K. Rangari and R. K. Rana, ACS Appl. Mater. Interfaces, 2016, 8, 22056–22063 CrossRef CAS PubMed.
- R. V. Chernozem, M. A. Surmeneva, S. N. Shkarina, K. Loza, M. Epple, M. Ulbricht, A. Cecilia, B. Krause, T. Baumbach, A. A. Abalymov, B. V. Parakhonskiy, A. G. Skirtach and R. A. Surmenev, ACS Appl. Mater. Interfaces, 2019, 11, 19522–19533 CrossRef CAS PubMed.
- M. Fujioka-Kobayashi, K. Tsuru, H. Nagai, K. Fujisawa, T. Kudoh, G. Ohe, K. Ishikawa and Y. Miyamoto, J. Tissue Eng. Regen. Med., 2018, 12, 2077–2087 CAS.
- X. Li, X. Yang, X. Liu, W. He, Q. Huang, S. Li and Q. Feng, Prog. Nat. Sci.: Mater. Int., 2018, 28, 598–608 CrossRef CAS.
- N. Xu, J. Xu, X. Zheng and J. Hui, ChemistryOpen, 2020, 9, 451–458 CrossRef CAS PubMed.
- H. Shi, L. Li, L. Zhang, T. Wang, C. Wang, D. Zhu and Z. Su, CrystEngComm, 2015, 17, 4768–4773 RSC.
- P. Xue, M. Hou, L. Sun, Q. Li, L. Zhang, Z. Xu and Y. Kang, Acta Biomater., 2018, 81, 242–255 CrossRef CAS PubMed.
- Y. Zhou, H. Li, J. Liu, Y. Xu, Y. Wang, H. Ren and X. Li, Polym. Adv. Technol., 2019, 30, 143–152 CrossRef CAS.
- P. Shi, D. Zhou, Y. Zhu, B. Peng, N. Shao and X. Zan, ACS Appl. Bio Mater., 2021, 4, 1030–1037 CrossRef CAS.
- M. Kim, J. H. Lee, S. E. Kim, S. S. Kang and G. Tae, ACS Appl. Mater. Interfaces, 2016, 8, 8409–8418 CrossRef CAS PubMed.
- J. Lee, H. S. Min, D. G. You, K. Kim, I. C. Kwon, T. Rhim and K. Y. Lee, J. Controlled Release, 2016, 223, 197–206 CrossRef CAS PubMed.
- Z. Dong, L. Feng, W. Zhu, X. Sun, M. Gao, H. Zhao, Y. Chao and Z. Liu, Biomaterials, 2016, 110, 60–70 CrossRef CAS PubMed.
- H. Huang, W. Zhang, Z. Liu, H. Guo and P. Zhang, Ultrasonics, 2020, 108, 106198 CrossRef CAS PubMed.
- B. Cantaert, D. Kuo, S. Matsumura, T. Nishimura, T. Sakamoto and T. Kato, ChemPlusChem, 2017, 82, 107–120 CrossRef CAS PubMed.
- J. H. E. Cartwright, A. G. Checa, J. D. Gale, D. Gebauer and C. I. Sainz-Diaz, Angew. Chem., Int. Ed., 2012, 51, 11960–11970 CrossRef CAS PubMed.
- L. B. Gower, Chem. Rev., 2008, 108, 4551–4627 CrossRef CAS PubMed.
- Y. Oaki, S. Kajiyama, T. Nishimura, H. Imai and T. Kato, Adv. Mater., 2008, 20, 3633–3637 CrossRef CAS.
- I. Buljan Meić, J. Kontrec, D. Domazet Jurašin, A. Selmani, B. Njegić Džakula, N. Maltar-Strmečki, D. M. Lyons, M. Plodinec, M. Čeh, A. Gajović, M. D. Sikirić and D. Kralj, CrystEngComm, 2018, 20, 35–50 RSC.
- R. K. Pai and S. Pillai, CrystEngComm, 2008, 10, 865–872 RSC.
- H. Du, C. Courrégelongue, J. Xto, A. Böhlen, M. Steinacher, C. N. Borca, T. Huthwelker and E. Amstad, Chem. Mater., 2020, 32, 4282–4291 CrossRef CAS.
- S. Y. Yang, H. H. Chang, C. J. Lin, S. J. Huang and J. C. Chan, Chem. Commun., 2016, 52, 11527–11530 RSC.
- C. Rao, M. Li, X. Sun, M. Li, X. Lian, H. Wang, L. Jia, B. Niu and W. Li, Mater. Chem. Phys., 2020, 255, 123552 CrossRef CAS.
- M. Milovanovic, M. T. Unruh, V. Brandt and J. C. Tiller, J. Colloid Interface Sci., 2020, 579, 357–368 CrossRef CAS PubMed.
- G. B. Cai, G. X. Zhao, X. K. Wang and S. H. Yu, J. Phys. Chem. C, 2010, 114, 12948–12954 CrossRef CAS.
- D. J. Tobler, J. D. Rodriguez-Blanco, K. Dideriksen, N. Bovet, K. K. Sand and S. L. S. Stipp, Adv. Funct. Mater., 2015, 25, 3081–3090 CrossRef CAS.
- R. Sun, C.-W. Tai, M. Strømme and O. Cheung, Microporous Mesoporous Mater., 2020, 292, 109736 CrossRef CAS.
- S. T. Sun, L. B. Mao, Z. Y. Lei, S. H. Yu and H. Colfen, Angew. Chem., Int. Ed., 2016, 55, 11765–11769 CrossRef CAS PubMed.
- S.-C. Huang, T. Minami, K. Naka and Y. Chujo, Polym. Compos., 2015, 36, 330–335 CrossRef CAS.
- C. Wang, M. Han, X. Liu, S. Chen, F. Hu, J. Sun and H. Yuan, Int. J. Nanomed., 2019, 14, 1503–1517 CrossRef CAS PubMed.
- C. C. Xue, M. H. Li, Y. Zhao, J. Zhou, Y. Hu, K. Y. Cai, Y. L. Zhao, S. H. Yu and Z. Luo, Sci. Adv., 2020, 6, 2008732 Search PubMed.
- L. Shi, M. Tang, Y. Muhammad, Y. Tang, L. He, W. Wang, Z. Tong and L. Li, CrystEngComm, 2021, 23, 3033–3042 RSC.
- C. Qi, J. Lin, L. H. Fu and P. Huang, Chem. Soc. Rev., 2018, 47, 357–403 RSC.
- Y. I. Svenskaya, H. Fattah, O. A. Inozemtseva, A. G. Ivanova, S. N. Shtykov, D. A. Gorin and B. V. Parakhonskiy, Cryst. Growth Des., 2018, 18, 331–337 CrossRef CAS.
- O. A. Jimoh, K. S. Ariffin, H. B. Hussin and A. E. Temitope, Carbonates Evaporites, 2017, 33, 331–346 CrossRef.
- B. Rugabirwa, D. Murindababisha, Y. Li, Y. Hong, Y. Su, H. Wang and J. Li, ACS Sustainable Chem. Eng., 2019, 7, 6251–6258 CrossRef CAS.
- R. Salomão, L. M. M. Costa and G. M. Olyveira, Adv. Tissue Eng. Regen. Med., 2017, 3, 336–340 Search PubMed.
- D. Gebauer, A. Volkel and H. Colfen, Science, 2008, 322, 1819–1822 CrossRef CAS PubMed.
- D. Gebauer and H. Cölfen, Nano Today, 2011, 6, 564–584 CrossRef CAS.
- N. Wada, N. Horiuchi, M. Nakamura, K. Nozaki, A. Nagai and K. Yamashita, ACS Omega, 2018, 3, 16681–16692 CrossRef CAS.
- J. Jiang, C. Chen, B. Xiao, Z. Bai, C. Jiang, C. Yang, Y. Wu and X. Wang, CrystEngComm, 2017, 19, 7332–7338 RSC.
- V. Vergaro, E. Carata, F. Baldassarre, E. Panzarini, L. Dini, C. Carlucci, S. Leporatti, B. F. Scremin, D. Altamura, C. Giannini, F. P. Fanizzi and G. Ciccarella, Adv. Powder Technol., 2017, 28, 2445–2455 CrossRef CAS.
- J. Q. Qi, R. Guo, Y. Wang, X. W. Liu and H. L. Chan, Nanoscale Res. Lett., 2016, 11, 120 CrossRef PubMed.
- J. A. Juhasz-Bortuzzo, B. Myszka, R. Silva and A. R. Boccaccini, Cryst. Growth Des., 2017, 17, 2351–2356 CrossRef CAS.
- B. Njegić Džakula, J. Kontrec, M. Ukrainczyk, S. Sviben and D. Kralj, Cryst. Res. Technol., 2014, 49, 244–256 CrossRef.
- N. M. Mlinarić, J. Kontrec, B. N. Džakula, G. Falini and D. Kralj, Crystals, 2021, 11, 250 CrossRef.
- Z. M. Liu, C. Y. Shao, B. Jin, Z. S. Zhang, Y. Q. Zhao, X. R. Xu and R. K. Tang, Nature, 2019, 574, 394–398 CrossRef CAS PubMed.
- Z. Mu, K. Kong, K. Jiang, H. Dong, X. Xu, Z. Liu and R. Tang, Science, 2021, 372, 1466–1470 CrossRef CAS.
- P. J. M. Smeets, A. R. Finney, W. Habraken, F. Nudelman, H. Friedrich, J. Laven, J. J. De Yoreo, P. M. Rodger and N. Sommerdijk, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E7882–E7890 CrossRef CAS PubMed.
- E. M. Pouget, P. H. H. Bomans, J. A. C. M. Goos, P. M. Frederik, G. de With and N. A. J. M. Sommerdijk, Science, 2009, 323, 1455–1458 CrossRef CAS PubMed.
- Y. Y. Kim, E. P. Douglas and L. B. Gower, Langmuir, 2007, 23, 4862–4870 CrossRef CAS PubMed.
- H. Du and E. Amstad, Angew. Chem., Int. Ed., 2020, 59, 1798–1816 CrossRef CAS PubMed.
- A. D. Trofimov, A. A. Ivanova, M. V. Zyuzin and A. S. Timin, Pharmaceutics, 2018, 10, 167 CrossRef CAS PubMed.
- A. Sarkar and S. Mahapatra, Cryst. Growth Des., 2010, 10, 2129–2135 CrossRef CAS.
- I. Buljan Meić, J. Kontrec, D. Domazet Jurašin, B. Njegić Džakula, L. Štajner, D. M. Lyons, M. Dutour Sikirić and D. Kralj, Cryst. Growth Des., 2017, 17, 1103–1117 CrossRef.
- W. Ostwald, Z. Phys. Chem., 1897, 22, 289–330 CrossRef CAS.
- G. Montes-Hernandez, F. Renard, N. Findling and A. L. Auzende, CrystEngComm, 2015, 17, 5725–5733 RSC.
- T. Zheng, H. Yi, S. Zhang and C. Wang, J. Cryst. Growth, 2020, 549, 125870 CrossRef CAS.
- B. V. Parakhonskiy, A. M. Yashchenok, S. Donatan, D. V. Volodkin, F. Tessarolo, R. Antolini, H. Mohwald and A. G. Skirtach, ChemPhysChem, 2014, 15, 2817–2822 CrossRef CAS PubMed.
- L. Brečević and D. Kralj, Kinetics and mechanisms of crystal growth in aqueous systems, Interfacial Dynamics, 2000 Search PubMed.
- D. Konopacka-Łyskawa, Crystals, 2019, 9, 223 CrossRef.
- Y. Mori, T. Enomae and A. Isogai, Mater. Sci. Eng., C, 2009, 29, 1409–1414 CrossRef CAS.
- R. Evík, M. Pérez-Estébanez, A. Viani, P. Aek and P. Mácová, Powder Technol., 2015, 284, 265–271 CrossRef.
- S. Yamanaka, Y. Sugawara, T. Oiso, T. Fujimoto, Y. Ohira and Y. Kuga, CrystEngComm, 2015, 17, 1773–1777 RSC.
- A. T. Nagaraja, S. Pradhan and M. J. McShane, J. Colloid Interface Sci., 2014, 418, 366–372 CrossRef CAS PubMed.
- G. J. Price, M. F. Mahon, J. Shannon and C. Cooper, Cryst. Growth Des., 2011, 11, 39–44 CrossRef CAS.
- Ç. M. Oral and B. Ercan, Powder Technol., 2018, 339, 781–788 CrossRef.
- B. Rugabirwa, D. Murindababisha, H. Wang and J. Li, Cryst. Growth Des., 2018, 19, 242–248 CrossRef.
- C. Y. Tai and C.-k Chen, Chem. Eng. Sci., 2008, 63, 3632–3642 CrossRef CAS.
- J. Jiang, Y. Ma, T. Zhang, Z. Liang and Z. Cui, RSC Adv., 2015, 5, 80216–80219 RSC.
- A. U. Badnore and A. B. Pandit, Chem. Eng. Process., 2015, 98, 13–21 CrossRef CAS.
- K. Murai, T. Kinoshita, K. Nagata and M. Higuchi, Langmuir, 2016, 32, 9351–9359 CrossRef CAS PubMed.
- B. Parakhonskiy, M. V. Zyuzin, A. Yashchenok, S. Carregal-Romero, J. Rejman, H. Mohwald, W. J. Parak and A. G. Skirtach, J. Nanobiotechnol., 2015, 13, 53 CrossRef PubMed.
- D. Kralj, J. Kontrec, L. Brecevic, G. Falini and V. Nothig-Laslo, Chemistry, 2004, 10, 1647–1656 CrossRef CAS PubMed.
- L. N. Plummer and E. Busenberg, Geochim. Cosmochim. Acta, 1982, 46, 1011–1040 CrossRef CAS.
- D. Kralj, L. Brečević and A. E. Nielsen, J. Cryst. Growth, 1990, 104, 793–800 CrossRef CAS.
- B. Njegić Džakula, S. Fermani, Z. Dubinsky, S. Goffredo, G. Falini and D. Kralj, Chem. – Eur. J., 2019, 25, 10616–10624 CrossRef PubMed.
- Y. Zhao, W. Du, L. Sun, L. Yu, J. Jiao and R. Wang, Colloid Polym. Sci., 2013, 291, 2191–2202 CrossRef CAS.
- H. Tang, J. Yu, X. Zhao and D. H. L. Ng, J. Alloys Compd., 2008, 463, 343–349 CrossRef CAS.
- J. Kontrec, N. Tomašić, N. Matijaković Mlinarić, D. Kralj and B. Njegić Džakula, Crystals, 2021, 11, 1075 CrossRef CAS.
- T. E. Reilly, L. N. Plummer, P. J. Phillips and E. Busenberg, Water Resour. Res., 1994, 30, 421–433 CrossRef.
- J. L. Bischoff, J. A. Fitzpatrick and R. J. Rosenbauer, J. Geol., 1993, 101, 21–33 CrossRef CAS.
- D. Kralj and L. Brecevic, Colloids Surf., A, 1995, 96, 287 CrossRef CAS.
- L. Brečević and A. E. Nielsen, J. Cryst. Growth, 1989, 98, 504–510 CrossRef.
- Q. Hu, J. Zhang, H. Teng and U. Becker, Am. Mineral., 2012, 97, 1437–1445 CrossRef CAS.
- J. Kawano, N. Shimobayashi, A. Miyake and M. Kitamura, J. Phys.: Condens. Matter, 2009, 21, 425102 CrossRef PubMed.
- M. Ma, Y. Wang, X. Cao, W. Lu and Y. Guo, Cryst. Growth Des., 2019, 19, 6972–6988 CrossRef CAS.
- Y.-B. Hu, M. Wolthers, D. A. Wolf-Gladrow and G. Nehrke, Cryst. Growth Des., 2015, 15, 1596–1601 CrossRef CAS.
- J. Chen and L. Xiang, Powder Technol., 2009, 189, 64–69 CrossRef CAS.
- R. Ševčík, M. Pérez-Estébanez, A. Viani, P. Šašek and P. Mácová, Powder Technol., 2015, 284, 265–271 CrossRef.
- S. Sovova, A. Abalymov, M. Pekar, A. G. Skirtach and B. Parakhonskiy, J. Mater. Chem. B, 2021, 9, 8308–8320 RSC.
- J.-P. Andreassen, J. Cryst. Growth, 2005, 274, 256–264 CrossRef CAS.
- J.-P. Andreassen and M. J. Hounslow, AIChE J., 2004, 50, 2772–2782 CrossRef CAS.
- E. Altay, T. Shahwan and M. Tanoğlu, Powder Technol., 2007, 178, 194–202 CrossRef CAS.
- S. Gopi, V. K. Subramanian and K. Palanisamy, Mater. Res. Bull., 2013, 48, 1906–1912 CrossRef CAS.
- S. Fermani, B. Njegić Džakula, M. Reggi, G. Falini and D. Kralj, CrystEngComm, 2017, 19, 2451–2455 RSC.
- D. Gebauer, Minerals, 2018, 8, 179 CrossRef.
- R. Demichelis, A. Schuitemaker, N. A. Garcia, K. B. Koziara, M. De La Pierre, P. Raiteri and J. D. Gale, Annu. Rev. Mater. Sci., 2018, 48, 327–352 CrossRef CAS.
- J. J. De Yoreo and P. G. Vekilov, Rev. Mineral. Geochem., 2003, 54, 57–93 CrossRef CAS.
- T. N. Ramesh, S. A. Inchara and K. Pallavi, J. Chem. Sci., 2015, 127, 843–848 CrossRef CAS.
- X. Ji, G. Li and X. Huang, Mater. Lett., 2008, 62, 751–754 CrossRef CAS.
- J. P. Zou, H. Z. Yang, P. Xiao and Y. F. Pan, J. Inorg. Mater., 2016, 31, 711–718 CrossRef CAS.
- N. Hanafy, M. Giorgi, C. Nobile, M. Cascione and S. Leporatti, Beni-Suef Univ. J. Basic Appl. Sci., 2015, 4, 60–70 Search PubMed.
- J. Bolze, B. Peng, N. Dingenouts, P. Panine, T. Narayanan and M. Ballauff, Langmuir, 2002, 18, 8364–8369 CrossRef CAS.
- D. Pontoni, J. Bolze, N. Dingenouts, T. Narayanan and M. Ballauff, J. Phys. Chem. B, 2003, 107, 5123–5125 CrossRef CAS.
- A.-R. Ibrahim, J. B. Vuningoma, X. Hu, Y. Gong, D. Hua, Y. Hong, H. Wang and J. Li, J. Supercrit. Fluids, 2012, 72, 78–83 CrossRef CAS.
- C. Y. Wang, P. Xiao, J. Z. Zhao, X. Zhao, Y. H. Liu and Z. C. Wang, Powder Technol., 2006, 170, 31–35 CrossRef CAS.
- M. Ukrainczyk, J. Kontrec, V. Babić-Ivančić, L. Brečević and D. Kralj, J. Powder Technol., 2007, 171, 192–199 CrossRef CAS.
- L. Du, Y. Wang and G. Luo, Particuology, 2013, 11, 421–427 CrossRef CAS.
- F. C. Donnelly, F. Purcell-Milton, V. Framont, O. Cleary, P. W. Dunne and Y. K. Gun'ko, Chem. Commun., 2017, 53, 6657–6660 RSC.
- C. W. Turner and D. W. Smith, Ind. Eng. Chem. Res., 1998, 37, 439–448 CrossRef CAS.
- G. Zhu, H. Li, S. Li, X. Hou, D. Xu, R. Lin and Q. Tang, J. Cryst. Growth, 2015, 428, 16–23 CrossRef CAS.
- W. Chuajiw, K. Takatori, T. Igarashi, H. Hara and Y. Fukushima, J. Cryst. Growth, 2014, 386, 119–127 CrossRef CAS.
- X. Chen, Y. Zhu, Y. Guo, B. Zhou, X. Zhao, Y. Du, H. Lei, M. Li and Z. Wang, Colloid Surf., A, 2010, 353, 97–103 CrossRef CAS.
- N. Matijaković, G. Magnabosco, F. Scarpino, S. Fermani, G. Falini and D. Kralj, Crystals, 2019, 9, 16 CrossRef.
- Y. Lai, L. Chen, W. Bao, Y. Ren, Y. Gao, Y. Yin and Y. Zhao, Cryst. Growth Des., 2015, 15, 1194–1200 CrossRef CAS.
- D. Walsh, B. Lebeau and S. Mann, Adv. Mater., 1999, 11, 324–328 CrossRef CAS.
- Y. Fukui and K. Fujimoto, J. Mater. Chem., 2012, 22, 3493–3499 RSC.
- T. Beuvier, B. Calvignac, G. J.-R. Delcroix, M. K. Tran, S. Kodjikian, N. Delorme, J.-F. Bardeau, A. Gibaud and F. Boury, J. Mater. Chem., 2011, 21, 9757–9761 RSC.
- N. McCann, D. Phan, M. Attalla, G. Puxty, D. Fernandes, W. Conway, X. Wang, R. Burns, I. van Altena, G. Lawrance and M. Maeder, Energy Procedia, 2009, 1, 995–1002 CrossRef CAS.
- J. Kontrec, M. Ukrainczyk, B. N. Džakula and D. Kralj, Cryst. Res. Technol., 2013, 48, 622–626 CAS.
- Y. Shen, A. Xie, Z. Chen, W. Xu, H. Yao, S. Li, L. Huang, Z. Wu and X. Kong, Mater. Sci. Eng., A, 2007, 443, 95–100 CrossRef.
- C. Kosanović, S. Fermani, G. Falini and D. Kralj, Crystals, 2017, 7, 355 CrossRef.
- Y. Y. Kim, R. Darkins, A. Broad, A. N. Kulak, M. A. Holden, O. Nahi, S. P. Armes, C. C. Tang, R. F. Thompson, F. Marin, D. M. Duffy and F. C. Meldrum, Nat. Commun., 2019, 10, 5682 CrossRef CAS PubMed.
- R. Stepić, L. Jurković, K. Klementyeva, M. Ukrainczyk, M. Gredičak, D. M. Smith, D. Kralj and A. S. Smith, Cryst. Growth Des., 2020, 20, 2853–2859 CrossRef.
- S. Weiner and L. Addadi, J. Mater. Chem., 1997, 7, 689–702 RSC.
- L. Addadi, J. Moradian, E. Shay, N. G. Maroudas and S. Weiner, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 2732–2736 CrossRef CAS PubMed.
- L. A. Gower and D. A. Tirrell, J. Cryst. Growth, 1998, 191, 153–160 CrossRef CAS.
- S. Borukhin, L. Bloch, T. Radlauer, A. H. Hill, A. N. Fitch and B. Pokroy, Adv. Funct. Mater., 2012, 22, 4216–4224 CrossRef CAS.
- C. A. Orme, A. Noy, A. Wierzbicki, M. T. McBride, M. Grantham, H. H. Teng, P. M. Dove and J. J. DeYoreo, Nature, 2001, 411, 775–779 CrossRef CAS PubMed.
- Y. M. Guo, F. F. Wang, J. Zhang, L. Yang, X. M. Shi, Q. L. Fang and X. M. Ma, Res. Chem. Intermed., 2013, 39, 2407–2415 CrossRef CAS.
- N. Wada, N. Horiuchi, M. Nakamura, K. Nozaki, A. Nagai and K. Yamashita, Cryst. Growth Des., 2018, 18, 872–878 CrossRef CAS.
- G. Montanari, L. Z. Lakshtanov, D. J. Tobler, K. Dideriksen, K. N. Dalby, N. Bovet and S. L. S. Stipp, Cryst. Growth Des., 2016, 16, 4813–4821 CrossRef CAS.
- F. C. Meldrum and H. Cölfen, Chem. Rev., 2008, 108, 4332–4432 CrossRef CAS PubMed.
- B. Njegić-Džakula, L. Brečević, G. Falini and D. Kralj, Cryst. Growth Des., 2009, 9, 2425–2434 CrossRef.
- P. J. Smeets, K. R. Cho, R. G. Kempen, N. A. Sommerdijk and J. J. De Yoreo, Nat. Mater., 2015, 14, 394–399 CrossRef CAS PubMed.
- H. Deng, S. Wang, X. Wang, C. Du, X. Shen, Y. Wang and F. Cui, Regener. Biomater., 2015, 2, 187–195 CrossRef CAS PubMed.
- S. Xu, H. Zhang, B. Qiao and Y. Wang, Cryst. Growth Des., 2021, 21, 7306–7325 CrossRef CAS.
- H. Colfen and S. Mann, Angew. Chem., Int. Ed., 2003, 42, 2350–2365 CrossRef PubMed.
- M. Jehannin, A. Rao and H. Colfen, J. Am. Chem. Soc., 2019, 141, 10120–10136 CrossRef CAS PubMed.
- D. Gebauer, M. Kellermeier, J. D. Gale, L. Bergstrom and H. Colfen, Chem. Soc. Rev., 2014, 43, 2348–2371 RSC.
- R. Demichelis, P. Raiteri, J. D. Gale, D. Quigley and D. Gebauer, Nat. Commun., 2011, 2, 590 CrossRef PubMed.
- J. Rieger, M. Kellermeier and L. Nicoleau, Angew. Chem., Int. Ed., 2014, 53, 12380–12396 CAS.
- P. Bots, L. G. Benning, J.-D. Rodriguez-Blanco, T. Roncal-Herrero and S. Shaw, Cryst. Growth Des., 2012, 12, 3806–3814 CrossRef CAS.
- Y. Politi, R. A. Metzler, M. Abrecht, B. Gilbert, F. H. Wilt, I. Sagi, L. Addadi, S. Weiner and P. U. P. A. Gilbert, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17362–17366 CrossRef CAS PubMed.
- J. Seto, Y. Ma, S. A. Davis, F. Meldrum, A. Gourrier, Y. Y. Kim, U. Schilde, M. Sztucki, M. Burghammer, S. Maltsev, C. Jager and H. Colfen, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 3699–3704 CrossRef CAS PubMed.
- L. Granasy, T. Pusztai, G. Tegze, J. A. Warren and J. F. Douglas, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 72, 011605 CrossRef PubMed.
- J. D. Rodriguez-Blanco, S. Shaw and L. G. Benning, Nanoscale, 2011, 3, 265–271 RSC.
- W.-L. Tan, A. L. Ahmad, C. P. Leo and S. S. Lam, J. CO2 Util., 2020, 42, 101333 CrossRef CAS.
- J. Pedrosa, J. A. F. Gamelas, A. F. Lourenço and P. J. Ferreira, Colloid Surf., A, 2016, 497, 1–7 CrossRef CAS.
- Y. U. Liang, S. Sun, H. A. O. Ding and X. Hou, Surf. Rev. Lett., 2020, 27, 1950224 CrossRef CAS.
- A. Sarkar, A. K. Ghosh and S. Mahapatra, J. Mater. Chem., 2012, 22, 11113 RSC.
- A. Barhoum, L. Van Lokeren, H. Rahier, A. Dufresne and G. Van Assche, J. Mater. Sci., 2015, 50, 7908–7918 CrossRef CAS.
- G. Cheng, X. Yu, G. Ding and C. Xu, Asian J. Chem., 2013, 25, 5558–5560 CrossRef CAS.
- L. C. Li and Y. Zhang, Adv. Mater. Res., 2009, 79-82, 1967–1970 CAS.
- Z. Tang, G. Cheng, Y. Chen, X. Yu and H. Wang, Adv. Powder Technol., 2014, 25, 1618–1623 CrossRef CAS.
- J. Lee, S. H. Jo and J. Lim, J. Ind. Eng. Chem., 2019, 74, 63–70 CrossRef CAS.
- N. Shimpi, A. Mali, D. P. Hansora and S. Mishra, Nanosci. Nanoeng., 2015, 3, 8–12 CAS.
- Y. Zhou, S. Wang, Y. Zhang and Y. Zhang, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 1226–1236 CrossRef CAS.
- L. M. Racca, L. C. Bertolino, C. R. Nascimento, A. M. F. de Sousa, L. Y. Reznik, L. Yokoyama and A. L. N. da Silva, J. Nanopart. Res., 2019, 21, 232 CrossRef CAS.
- J. Kim, S. K. Bea, Y. H. Kim, D.-W. Kim, K.-Y. Lee and C.-M. Lee, Biotechnol. Bioprocess Eng., 2015, 20, 794–799 CrossRef CAS.
- Z. Cao, M. Daly, L. Clémence, L. M. Geever, I. Major, C. L. Higginbotham and D. M. Devine, Appl. Surf. Sci., 2016, 378, 320–329 CrossRef CAS.
- S. R. Mihajlović, D. R. Vučinić, Ž. T. Sekulić, S. Z. Milićević and B. M. Kolonja, Powder Technol., 2013, 245, 208–216 CrossRef.
- C. Jeon, S. Park, J.-H. Bang, S. Chae, K. Song and S.-W. Lee, Coatings, 2018, 8, 43 CrossRef.
- E. Yoğurtcuoğlu and M. Uçurum, Powder Technol., 2011, 214, 47–53 CrossRef.
- Deepika, S. K. Hait and Y. Chen, J. Coat. Technol. Res., 2014, 11, 273–282 CrossRef CAS.
- C. Zeng, H. Hu, X. Feng, K. Wang and Q. Zhang, Chemosphere, 2020, 249, 126227 CrossRef CAS PubMed.
- Deepika, S. K. Hait, J. Christopher, Y. Chen, P. Hodgson and D. K. Tuli, Powder Technol., 2013, 235, 581–589 CrossRef CAS.
- D. Kim, J. Lee, S. Lee and J. Lim, Colloid Surf., A, 2018, 536, 213–223 CrossRef CAS.
- E. Song, D. Kim, B. J. Kim and J. Lim, Colloid Surf., A, 2014, 461, 1–10 CrossRef CAS.
- Z. G. Cui, Y. Z. Cui, C. F. Cui, Z. Chen and B. P. Binks, Langmuir, 2010, 26, 12567–12574 CrossRef CAS PubMed.
- E. M. Song, D. W. Kim and J. C. Lim, J. Ind. Eng. Chem., 2015, 28, 351–358 CrossRef CAS.
- D. S. Kim and C. K. Lee, Appl. Surf. Sci., 2002, 202, 15–23 CrossRef CAS.
- S. Zhang and X. Li, Powder Technol., 2004, 141, 75–79 CrossRef CAS.
- C. Cui, H. Ding, L. Cao and D. Chen, Pol. J. Chem. Technol., 2015, 17, 128–133 CrossRef CAS.
- L. Jiang, K. Pan and Y. Dan, Colloid Polym. Sci., 2006, 285, 65–74 CrossRef CAS.
- A. M. Ferreira, A. S. Vikulina and D. Volodkin, J. Controlled Release, 2020, 328, 470–489 CrossRef CAS PubMed.
- S. Sharma, A. Verma, B. V. Teja, G. Pandey, N. Mittapelly, R. Trivedi and P. R. Mishra, Colloid Surf., B, 2015, 133, 120–139 CrossRef CAS PubMed.
- G. Choukrani, B. Maharjan, C. H. Park, C. S. Kim and A. R. Kurup Sasikala, Mater. Sci. Eng., C, 2020, 106, 110226 CrossRef CAS PubMed.
- K. H. Min, H. S. Min, H. J. Lee, D. J. Park, J. Y. Yhee, K. Kim, I. C. Kwon, S. Y. Jeong, O. F. Silvestre, X. Chen, Y.-S. Hwang, E.-C. Kim and S. C. Lee, ACS Nano, 2015, 9, 134–145 CrossRef CAS PubMed.
- D. J. Park, K. H. Min, H. J. Lee, K. Kim, I. C. Kwon, S. Y. Jeong and S. C. Lee, J. Mat. Chem. B, 2016, 4, 1219–1227 RSC.
- Y. Fujita, T. Yamamuro, T. Nakamura, S. Kotani, C. Ohtsuki and T. Kokubo, J. Biomed. Mater. Res., 1991, 25, 991–1003 CrossRef CAS PubMed.
- C. Combes, B. Miao, R. Bareille and C. Rey, Biomaterials, 2006, 27, 1945–1954 CrossRef CAS PubMed.
- F. P. He, J. Zhang, F. W. Yang, J. X. Zhu, X. M. Tian and X. M. Chen, Mater. Sci. Eng., C, 2015, 50, 257–265 CrossRef CAS PubMed.
- S. Umemoto, T. Furusawa, H. Unuma, M. Tajika and T. Sekino, Dent. Mater. J., 2021, 40, 1202–1207 CrossRef PubMed.
- Q. W. Zhong, W. H. Li, X. P. Su, G. Li, Y. Zhou, S. C. Kundu, J. M. Yao and Y. R. Cai, Colloid Surf., B, 2016, 143, 56–63 CrossRef CAS PubMed.
- J. Rodríguez-Sánchez, B. Myszka, A. R. Boccaccini and D. K. Dysthe, J. Am. Ceram. Soc., 2019, 102, 6980–6990 CrossRef.
- A. Diez-Escudero, M. Espanol and M.-P. Ginebra, Synthetic bone graft substitutes: Calcium-based biomaterials, Woodhead Publishing, 2020 Search PubMed.
- B. Myszka, K. Hurle, K. Zheng, S. E. Wolf and A. R. Boccaccini, J. Mater. Chem. B, 2019, 7, 3403–3411 RSC.
- P. Opitz, L. Besch, M. Panthöfer, A. Kabelitz, R. E. Unger, F. Emmerling, M. Mondeshki and W. Tremel, Adv. Funct. Mater., 2021, 31, 2007830 CrossRef CAS.
- S. Kim and C. B. Park, Adv. Funct. Mater., 2013, 23, 10–25 CrossRef CAS.
- Y. Lee, Y. M. Hahm, S. Matsuya, M. Nakagawa and K. Ishikawa, J. Mater. Sci., 2007, 42, 5728–5735 CrossRef CAS.
- C. T. Begley, M. J. Doherty, R. A. B. Mollan and D. J. Wilson, Biomaterials, 1995, 16, 1181–1185 CrossRef CAS PubMed.
- Y. Barbotteau, J. L. Irigaray and J. F. Mathiot, Phys. Med. Biol., 2003, 48, 3611–3623 CrossRef CAS PubMed.
- S. Rossler, R. Unbehau, T. Gemming, B. Kruppke, H. P. Wiesmann and T. Hanke, Sci. Rep., 2020, 10, 118 CrossRef CAS PubMed.
- M. Xie, M. O. Olderoy, J. P. Andreassen, S. M. Selbach, B. L. Strand and P. Sikorski, Acta Biomater., 2010, 6, 3665–3675 CrossRef CAS PubMed.
- Y. Suzawa, T. Funaki, J. Watanabe, S. Iwai, Y. Yura, T. Nakano, Y. Umakoshi and M. Akashi, J. Biomed. Mater. Res. Part A, 2010, 93A, 965–975 CAS.
- N. H. Munro and K. M. McGrath, Chem. Commun., 2012, 48, 4716–4718 RSC.
- H. Li, H. L. Xin, D. A. Muller and L. A. Estroff, Science, 2009, 326, 1244–1247 CrossRef CAS PubMed.
- O. Grassmann and P. Lobmann, Chem. – Eur. J., 2003, 9, 1310–1316 CrossRef CAS PubMed.
- Y. H. Gong, Y. L. Zhang, Z. N. Cao, F. Ye, Z. F. Lin and Y. Li, Biomater. Sci., 2019, 7, 3614–3626 RSC.
- N. Rauner, M. Meuris, S. Dech, J. Godde and J. C. Tiller, Acta Biomater., 2014, 10, 3942–3951 CrossRef CAS PubMed.
- T. E. L. Douglas, A. Lapa, S. K. Samal, H. A. Declercq, D. Schaubroeck, A. C. Mendes, P. V. der Voort, A. Dokupil, A. Plis, K. De Schamphelaere, I. S. Chronakis, E. Pamula and A. G. Skirtach, J. Tissue Eng. Regen. Med., 2017, 11, 3556–3566 CrossRef CAS PubMed.
- M. Li, H. Ma, F. Han, D. Zhai, B. Zhang, Y. Sun, T. Li, L. Chen and C. Wu, Adv. Mater., 2021, 9, e2104829 CrossRef PubMed.
- Q. Huang, Y. Liu, Z. Ouyang and Q. Feng, Bioact. Mater., 2020, 5, 980–989 CrossRef PubMed.
- C. Dhand, S. T. Ong, N. Dwivedi, S. M. Diaz, J. R. Venugopal, B. Navaneethan, M. H. Fazil, S. Liu, V. Seitz, E. Wintermantel, R. W. Beuerman, S. Ramakrishna, N. K. Verma and R. Lakshminarayanan, Biomaterials, 2016, 104, 323–338 CrossRef CAS PubMed.
- Y. C. Wu, T. M. Lee, K. H. Chiu, S. Y. Shaw and C. Y. Yang, J. Mater. Sci.: Mater. Med., 2009, 20, 1273–1280 CrossRef CAS PubMed.
- V. Viateau, M. Manassero, L. Sensebe, A. Langonne, D. Marchat, D. Logeart-Avramoglou, H. Petite and M. Bensidhoum, J. Tissue Eng. Regener. Med., 2016, 10, E177–187 CrossRef CAS PubMed.
- C. T. Tran, C. Gargiulo, H. D. Thao, H. M. Tuan, L. Filgueira and D. Michael Strong, Cell Tissue Bank., 2010, 12, 247–261 CrossRef PubMed.
- B. Kruppke, J. Farack, S. Weil, E. D. Aflalo, D. Polakova, A. Sagi and T. Hanke, J. Biomed. Mater. Res., Part A, 2020, 108, 694–708 CrossRef CAS PubMed.
- M. S. Saveleva, A. N. Ivanov, M. O. Kurtukova, V. S. Atkin, A. G. Ivanova, G. P. Lyubun, A. V. Martyukova, E. I. Cherevko, A. K. Sargsyan, A. S. Fedonnikov, I. A. Norkin, A. G. Skirtach, D. A. Gorin and B. V. Parakhonskiy, Mater. Sci. Eng., C, 2018, 85, 57–67 CrossRef CAS PubMed.
- A. D. Woldetsadik, S. K. Sharma, S. Khapli, R. Jagannathan and M. Magzoub, ACS Biomater. Sci. Eng., 2017, 3, 2457–2469 CrossRef CAS PubMed.
- Q. Li, E. Hu, K. Yu, R. Xie, F. Lu, B. Lu, R. Bao, T. Zhao, F. Dai and G. Lan, Adv. Funct. Mater., 2020, 30, 2004153 CrossRef CAS.
- W. He, X. Huang, J. Zhang, Y. Zhu, Y. Liu, B. Liu, Q. Wang, X. Huang and D. He, Materials, 2021, 14, 3350 CrossRef CAS PubMed.
- J. K. Jang, O. S. Lee, T. J. Kang and S. C. Lim, Food Sci. Biotechnol., 2013, 22, 99–105 CrossRef.
- J. R. Baylis, J. H. Yeon, M. H. Thomson, A. Kazerooni, X. Wang, A. E. St John, E. B. Lim, D. Chien, A. Lee, J. Q. Zhang, J. M. Piret, L. S. Machan, T. F. Burke, N. J. White and C. J. Kastrup, Sci. Adv., 2015, 1, e1500379 CrossRef PubMed.
- B. T. Turner, Jr. and M. C. Maurer, Biochemistry, 2002, 41, 7947–7954 CrossRef PubMed.
- M. L. P. Vidallon, A. M. Douek, A. Quek, H. McLiesh, J. Kaslin, R. F. Tabor, A. I. Bishop and B. M. Teo, Part. Part. Syst. Charact., 2020, 37, 1900471 CrossRef CAS.
- C. Qi, J. Lin, L. H. Fu and P. Huang, Chem. Soc. Rev., 2018, 47, 357–403 RSC.
- M. Kim, J. H. Lee, S. E. Kim, S. S. Kang and G. Tae, ACS Appl. Mater. Interfaces, 2016, 8, 8409–8418 CrossRef CAS PubMed.
- Z. Wei, X. Lin, M. Wu, B. Zhao, R. Lin, D. Zhang, Y. Zhang, G. Liu, X. Liu and J. Liu, Sci. Rep., 2017, 7, 5370 CrossRef PubMed.
- S. L. Goss, K. A. Lemons, J. E. Kerstetter and R. H. Bogner, J. Pharm. Pharmacol., 2007, 59, 1485–1492 CrossRef PubMed.
- J. Zhang, Y. Li, H. Xie, B. L. Su, B. Yao, Y. Yin, S. Li, F. Chen and Z. Fu, ACS Appl. Mater. Interfaces, 2015, 7, 15686–15691 CrossRef CAS PubMed.
- J. Li, H. Jiang, X. Ouyang, S. Han, J. Wang, R. Xie, W. Zhu, N. Ma, H. Wei and Z. Jiang, ACS Appl. Mater. Interfaces, 2016, 8, 30027–30036 CrossRef CAS PubMed.
- Q. Q. Dong, J. L. Li, L. Y. Cui, H. L. Jian, A. H. Wang and S. Bai, Colloid Surf., A, 2017, 516, 190–198 CrossRef CAS.
- X. Y. Wan, H. Zhong, W. Pan, Y. H. Li, Y. Y. Chen, N. Li and B. Tang, Angew. Chem., Int. Ed., 2019, 58, 14134–14139 CrossRef CAS PubMed.
- Q. Guan, L. L. Zhou, F. H. Lv, W. Y. Li, Y. A. Li and Y. B. Dong, Angew. Chem., Int. Ed., 2020, 59, 18042–18047 CrossRef CAS PubMed.
- M. Chang, Z. Hou, D. Jin, J. Zhou, M. Wang, M. Wang, M. Shu, B. Ding, C. Li and J. Lin, Adv. Mater., 2020, 32, 2004647 CrossRef CAS PubMed.
- Y. Liu, B. Yu, X. Dai, N. Zhao and F. J. Xu, Biomaterials, 2021, 274, 120885 CrossRef CAS PubMed.
- C. Y. Xu, Y. F. Yan, J. C. Tan, D. H. Yang, X. J. Jia, L. Wang, Y. S. Xu, S. Cao and S. T. Sun, Adv. Funct. Mater., 2019, 29, 1808146 CrossRef.
- J. J. Xing, Y. Q. Cai, Y. K. Wang, H. F. Zheng and Y. J. Liu, Adv. Polym. Technol., 2020, 8749238 CAS.
- S. Maleki Dizaj, M. Barzegar-Jalali, M. H. Zarrintan, K. Adibkia and F. Lotfipour, Expert Opin. Drug Delivery, 2015, 12, 1649–1660 CrossRef PubMed.
- Y. Zhang, P. Ma, Y. Wang, J. Du, Q. Zhou, Z. Zhu, X. Yang and J. Yuan, World J. Nano Sci. Eng., 2012, 02, 25–31 CrossRef CAS.
- S. Haruta, T. Hanafusa, H. Fukase, H. Miyajima and T. Oki, Diabetes Technol. Ther., 2003, 5, 1–9 CrossRef CAS PubMed.
- D. Preisig, D. Haid, F. J. O. Varum, R. Bravo, R. Alles, J. Huwyler and M. Puchkov, Eur. J. Pharm. Biopharm., 2014, 87, 548–558 CrossRef CAS PubMed.
- G. Magnabosco, M. Di Giosia, I. Polishchuk, E. Weber, S. Fermani, A. Bottoni, F. Zerbetto, P. G. Pelicci, B. Pokroy, S. Rapino, G. Falini and M. Calvaresi, Adv. Healthcare Mater., 2015, 4, 1510–1516 CrossRef CAS PubMed.
- M. Barbalinardo, M. Di Giosia, I. Polishchuk, G. Magnabosco, S. Fermani, F. Biscarini, M. Calvaresi, F. Zerbetto, G. Pellegrini, G. Falini, B. Pokroy and F. Valle, J. Mat. Chem. B, 2019, 7, 5808–5813 RSC.
- R. Kurapati and A. M. Raichur, J. Mat. Chem. B, 2013, 1, 3175–3184 RSC.
- X. Ma, S. Yuan, L. Yang, L. Li, X. Zhang, C. Su and K. Wang, CrystEngComm, 2013, 15, 8288–8299 RSC.
- H. Z. Shi, L. Li, L. Y. Zhang, T. T. Wang, C. G. Wang, D. X. Zhu and Z. M. Su, CrystEngComm, 2015, 17, 4768–4773 RSC.
- S. Maleki Dizaj, S. Sharifi, E. Ahmadian, A. Eftekhari, K. Adibkia and F. Lotfipour, Expert Opin. Drug Delivery, 2019, 16, 331–345 CrossRef CAS PubMed.
- T. Yang, Z. Wan, Z. Liu, H. Li, H. Wang, N. Lu, Z. Chen, X. Mei and X. Ren, Mater. Sci. Eng., C, 2016, 63, 384–392 CrossRef CAS PubMed.
- D. Zhao, C. Q. Wang, R. X. Zhuo and S. X. Cheng, Colloids Surf., B, 2014, 118, 111–116 CrossRef CAS PubMed.
- Y. Wu, W. Gu, J. Tang and Z. P. Xu, J. Mat. Chem. B, 2017, 5, 7194–7203 RSC.
- C. Q. Wang, J. L. Wu, R. X. Zhuo and S. X. Cheng, Mol. Biosyst., 2014, 10, 672–678 RSC.
- P. Liang, C.-Q. Wang, H. Chen, R.-X. Zhuo and S.-X. Cheng, Polym. Int., 2015, 64, 647–653 CrossRef CAS.
- P. Shi, S. Luo, B. Voit, D. Appelhans and X. Zan, J. Mat. Chem. B, 2018, 6, 4205–4215 RSC.
- Q. Chen, C. Wang, X. Zhang, G. Chen, Q. Hu, H. Li, J. Wang, D. Wen, Y. Zhang, Y. Lu, G. Yang, C. Jiang, J. Wang, G. Dotti and Z. Gu, Nat. Nanotechnol., 2019, 14, 89–97 CrossRef CAS PubMed.
- H. Ruan, Q. Hu, D. Wen, Q. Chen, G. Chen, Y. Lu, J. Wang, H. Cheng, W. Lu and Z. Gu, Adv. Mater., 2019, 31, e1806957 CrossRef PubMed.
- J. D. Snook, C. B. Chesson, A. G. Peniche, S. M. Dann, A. Paulucci, I. V. Pinchuk and J. S. Rudra, J. Mater. Chem. B, 2016, 4, 1640–1649 RSC.
- V. Lauth, B. Loretz, C.-M. Lehr, M. Maas and K. Rezwan, Chem. Mater., 2016, 28, 3796–3803 CrossRef CAS.
- H. Kakisawa and T. Sumitomo, Sci. Technol. Adv. Mater., 2011, 12, 064710 CrossRef PubMed.
- T. Kato, T. Suzuki and T. Irie, Chem. Lett., 2000, 186–187 CrossRef CAS.
- A. Sugawara-Narutaki, Polym. J., 2012, 45, 269–276 CrossRef.
- G. Falini, S. Fermani and S. Goffredo, Semin. Cell Dev. Biol., 2015, 46, 17–26 CrossRef PubMed.
- A. Junaidi, J. Anim. Vet. Adv., 2007, 6, 591–594 Search PubMed.
- L. Li, M. J. Connors, M. Kolle, G. T. England, D. I. Speiser, X. H. Xiao, J. Aizenberg and C. Ortiz, Science, 2015, 350, 952–956 CrossRef CAS PubMed.
- H. Li, C. Y. Sun, Y. Fang, C. M. Carlson, H. Xu, A. Jesovnik, J. Sosa-Calvo, R. Zarnowski, H. A. Bechtel, J. H. Fournelle, D. R. Andes, T. R. Schultz, P. Gilbert and C. R. Currie, Nat. Commun., 2020, 11, 5792 CrossRef CAS PubMed.
- J. Wang, Q. Cheng and Z. Tang, Chem. Soc. Rev., 2012, 41, 1111–1129 RSC.
- Y. Y. Zhang, S. S. Gong, Q. Zhang, P. Ming, S. J. Wan, J. S. Peng, L. Jiang and Q. F. Cheng, Chem. Soc. Rev., 2016, 45, 2378–2395 RSC.
- S. Kamat, X. Su, R. Ballarini and A. H. Heuer, Nature, 2000, 405, 1036–1040 CrossRef CAS PubMed.
- K. Iwase and K. Mori, Cryst. Growth Des., 2020, 20, 2091–2098 CrossRef CAS.
- A. Xin, Y. Su, S. Feng, M. Yan, K. Yu, Z. Feng, K. Hoon Lee, L. Sun and Q. Wang, Adv. Mater., 2021, 33, e2006946 CrossRef PubMed.
- N. A. Yaraghi and D. Kisailus, Annu. Rev. Phys. Chem., 2018, 69, 23–57 CrossRef CAS PubMed.
- G. M. Luz and J. F. Mano, Philos. Trans. R. Soc., A, 2009, 367, 1587–1605 CrossRef CAS PubMed.
- A. R. Studart, Chem. Soc. Rev., 2016, 45, 359–376 RSC.
- H. Zhao, Z. Yang and L. Guo, NPG Asia Mater., 2018, 10, 1–22 CrossRef CAS.
- P. Das, J. M. Malho, K. Rahimi, F. H. Schacher, B. Wang, D. E. Demco and A. Walther, Nat. Commun., 2015, 6, 5967 CrossRef PubMed.
- B. Zhu, N. Jasinski, A. Benitez, M. Noack, D. Park, A. S. Goldmann, C. Barner-Kowollik and A. Walther, Angew. Chem., Int. Ed., 2015, 54, 8653–8657 CrossRef CAS PubMed.
- A. Finnemore, P. Cunha, T. Shean, S. Vignolini, S. Guldin, M. Oyen and U. Steiner, Nat. Commun., 2012, 3, 966 CrossRef PubMed.
- H. Bai, Y. Chen, B. Delattre, A. P. Tomsia and R. O. Ritchie, Sci. Adv., 2015, 1, e1500849 CrossRef PubMed.
- L. B. Mao, H. L. Gao, H. B. Yao, L. Liu, H. Colfen, G. Liu, S. M. Chen, S. K. Li, Y. X. Yan, Y. Y. Liu and S. H. Yu, Science, 2016, 354, 107–110 CrossRef CAS PubMed.
- X. Q. Li and H. C. Zeng, Adv. Mater., 2012, 24, 6277–6282 CrossRef CAS PubMed.
- P. L. Wang, T. T. Shen, X. Y. Li, Y. Y. Tang and Y. J. Li, ACS Appl. Nano Mater., 2020, 3, 1272–1281 CrossRef CAS.
- M. S. Islam, W. S. Choi, B. Nam, C. Yoon and H.-J. Lee, Chem. Eng. J., 2017, 307, 208–219 CrossRef CAS.
- Q. Q. Shi, Y. T. Wang, X. Zhang, B. X. Shen, F. M. Wang and Y. F. Zhang, Fuel Process. Technol., 2020, 199, 106247 CrossRef CAS.
- S. Mallakpour, A. Abdolmaleki and F. Tabesh, Ultrason. Sonochem., 2018, 41, 572–581 CrossRef CAS PubMed.
- X. Zhang, D. Shi, X. Li, Y. Zhang, J. Wang and J. Fan, Chemosphere, 2019, 224, 390–397 CrossRef CAS PubMed.
- J. J. Jacob, R. Varalakshmi, S. Gargi, M. A. Jayasri and K. Suthindhiran, npj Clean Water, 2018, 1, 1 CrossRef CAS.
- R. Liu and B. Lian, J. Hazard. Mater., 2019, 378, 120707 CrossRef CAS PubMed.
- P. Lin, H. Wu, S. Hsieh, J. Li, C. Dong, C. Chen and S. Hsieh, Chemosphere, 2020, 254, 126903 CrossRef CAS PubMed.
- X. Ma, L. Li, L. Yang, C. Su, K. Wang and K. Jiang, J. Cryst. Growth, 2012, 338, 272–279 CrossRef CAS.
- X. Ma, L. Li, L. Yang, C. Su, K. Wang, S. Yuan and J. Zhou, J. Hazard. Mater., 2012, 209–210, 467–477 CrossRef CAS PubMed.
- X. Zhou, W. Liu, J. Zhang, C. Wu, X. Ou, C. Tian, Z. Lin and Z. Dang, ACS Appl. Mater. Interfaces, 2017, 9, 35785–35793 CrossRef CAS PubMed.
- T. Wen, Y. Zhao, T. Zhang, B. Xiong, H. Hu, Q. Zhang and S. Song, Chemosphere, 2020, 246, 125842 CrossRef CAS PubMed.
- H. Henry, M. F. Naujokas, C. Attanayake, N. T. Basta, Z. Q. Cheng, G. M. Hettiarachchi, M. Maddaloni, C. Schadt and K. G. Scheckel, Environ. Sci. Technol., 2015, 49, 8948–8958 CrossRef CAS PubMed.
- Y. Du, F. Lian and L. Zhu, Environ. Pollut., 2011, 159, 1763–1768 CrossRef CAS PubMed.
- X. Zhou, W. Liu, C. Tian, S. Mo, X. Liu, H. Deng and Z. Lin, Chem. Eng. J, 2018, 351, 816–824 CrossRef CAS.
- H. Merrikhpour and M. Jalali, Clean Technol. Environ. Policy, 2012, 14, 845–855 CrossRef CAS.
- B. Xiong, T. Zhang, Y. Zhao, T. Wen, Q. Zhang, S. Bao and S. Song, Sci. Total Environ., 2020, 698, 134270 CrossRef CAS PubMed.
- M. Zaharia, F. Bucatariu, F. Doroftei, D. Loghin, A. Vasiliu and M. Mihai, Colloid Surf., A, 2021, 613, 126084 CrossRef CAS.
- S. Ramola, T. Belwal, C. J. Li, Y. Y. Wang, H. H. Lu, S. M. Yang and C. H. Zhou, Sci. Total Environ., 2020, 709, 136171 CrossRef CAS PubMed.
- Y. Lei, J. Remmers, M. Saakes, R. van der Weijden and C. Buisman, ACS Sustainable Chem. Eng., 2019, 7, 7362–7368 CrossRef CAS PubMed.
- Y. Lei, S. Narsing, M. Saakes, R. D. van der Weijden and C. J. N. Buisman, Environ. Sci. Technol., 2019, 53, 10774–10780 CrossRef CAS PubMed.
- S. Ramola, T. Belwal, C. J. Li, Y. X. Liu, Y. Y. Wang, S. M. Yang and C. H. Zhou, J. Cleaner Prod., 2021, 299, 126802 CrossRef CAS.
- X. Yuan, W.-C. Nie, C. Xu, X.-H. Wang, Q. Xiao, F. Song, X.-L. Wang and Y.-Z. Wang, Adv. Funct. Mater., 2018, 28, 1704956 CrossRef.
- J. Dai, L. Wang, Y. Wang, S. Tian, X. Tian, A. Xie, R. Zhang, Y. Yan and J. Pan, ACS Appl. Mater. Interfaces, 2020, 12, 4482–4493 CrossRef CAS PubMed.
- R. K. Gupta, G. J. Dunderdale, M. W. England and A. Hozumi, J. Mater. Chem. A, 2017, 5, 16025–16058 RSC.
- J. D. Dai, L. L. Wang, Y. Wang, S. J. Tian, X. H. Tian, A. H. Xie, R. L. Zhang, Y. S. Yan and J. M. Pan, ACS Appl. Mater. Interfaces, 2020, 12, 4482–4493 CrossRef CAS PubMed.
- J. Yang, J. Y. Cui, A. T. Xie, J. D. Dai, C. X. Li and Y. S. Yan, Colloids Surf., A, 2021, 608, 125583 CrossRef CAS.
- M. Li, Y. Chen, L. B. Mao, Y. Jiang, M. F. Liu, Q. Huang, Z. Yu, S. Wang, S. H. Yu, C. Lin, X. Y. Liu and H. Colfen, Langmuir, 2018, 34, 2942–2951 CrossRef CAS PubMed.
- S. K. Tang, X. T. Chang, M. Y. Li, T. Ge, S. C. Niu, D. S. Wang, Y. C. Jiang and S. B. Sun, Chem. Eng. J., 2021, 405, 126597 CrossRef CAS.
- Y. Guo, D.-P. Yang, M. Liu, X. Zhang, Y. Chen, J. Huang, Q. Li and R. Luque, J. Mater. Chem. A, 2019, 7, 8832–8844 RSC.
- X. Zhang, J. Huang, Z. Kang, D.-P. Yang and R. Luque, Mol. Catal., 2020, 484, 110786 CrossRef CAS.
- X. H. Zhang, M. H. Liu, Z. W. Kang, B. Q. Wang, B. Wang, F. Y. Jiang, X. S. Wang, D.-P. Yang and R. Luque, Chem. Eng. J., 2020, 388, 124304 CrossRef CAS.
- S. Yu, X. Wang and D. Wu, Energy Fuels, 2014, 28, 3519–3529 CrossRef CAS.
- H. Liu, X. Tian, M. Ouyang, X. Wang, D. Wu and X. Wang, Renewable Energy, 2021, 179, 47–64 CrossRef CAS.
- Y. Jiang, D. Wang and T. Zhao, J. Appl. Polym. Sci., 2007, 104, 2799–2806 CrossRef CAS.
- L. Sánchez, P. Sánchez, A. de Lucas, M. Carmona and J. F. Rodríguez, Colloid Polym. Sci., 2007, 285, 1377–1385 CrossRef.
- Y. Ma, J. Zong, W. Li, L. Chen, X. Tang, N. Han, J. Wang and X. Zhang, Energy, 2015, 87, 86–94 CrossRef CAS.
- Q. Zhang, C. Liu and Z. Rao, ChemistrySelect, 2019, 4, 8482–8492 CrossRef CAS.
- S. Yu, X. Wang and D. Wu, Appl. Energy, 2014, 114, 632–643 CrossRef CAS.
- T. Wang, S. Wang, R. Luo, C. Zhu, T. Akiyama and Z. Zhang, Appl. Energy, 2016, 171, 113–119 CrossRef CAS.
- Z. Jiang, W. Yang, F. He, C. Xie, J. Fan, J. Wu and K. Zhang, Langmuir, 2018, 34, 14254–14264 CrossRef CAS PubMed.
- Y. Fang, T. Zou, X. Liang, S. Wang, X. Liu, X. Gao and Z. Zhang, ACS Sustainable Chem. Eng., 2017, 5, 3074–3080 CrossRef CAS.
- Z. Jiang, W. Yang, F. He, C. Xie, J. Fan, J. Wu and K. Zhang, ACS Sustainable Chem. Eng., 2018, 6, 5182–5191 CrossRef CAS.
- H. Wei, F. He, Y. Li, Q. Zhang, Y. Zhou, H. Yan, R. He, J. Fan and W. Yang, ACS Sustainable Chem. Eng., 2019, 7, 18854–18862 CrossRef CAS.
- S. Emir and H. Paksoy, Energy Storage, 2020, 3, e214 Search PubMed.
- Y. Pan, M. Wu and Q. Su, Mater. Res. Bull., 2003, 38, 1537–1544 CrossRef CAS.
- H. He, J. Li, L. Liu, S. Bao, X. Chen, P. Zhang, W. Zhang and X. Lai, Ceram. Int., 2018, 44, 5070–5075 CrossRef CAS.
- H. Zhu, B. Qian, X. Zhou, Y. Song, K. Zheng, Y. Sheng and H. Zou, J. Lumin., 2018, 203, 441–446 CrossRef CAS.
- E. A. Minchin, Fortschr. Zool., 2022, 2, 171–274 Search PubMed.
- L. Addadi, S. Raz and S. Weiner, Adv. Mater., 2003, 15, 959–970 CrossRef CAS.
- M. J. Root, Calcif. Tissue Int., 1990, 47, 112–116 CrossRef CAS PubMed.
- N. Nassif, N. Pinna, N. Gehrke, M. Antonietti, C. Jager and H. Colfen, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12653–12655 CrossRef CAS PubMed.
- Y. Levi-Kalisman, S. Raz, S. Weiner, L. Addadi and I. Sagi, Adv. Funct. Mater., 2002, 12, 43–48 CrossRef CAS.
- R. S. K. Lam, J. M. Charnock, A. Lennie and F. C. Meldrum, CrystEngComm, 2007, 9, 1226–1236 RSC.
- D. Gebauer, P. N. Gunawidjaja, J. Y. P. Ko, Z. Bacsik, B. Aziz, L. Liu, Y. Hu, L. Bergström, C.-W. Tai, T.-K. Sham, M. Edén and N. Hedin, Angew. Chem., Int. Ed., 2010, 49, 8889–8891 CrossRef CAS PubMed.
- Z. Zou, X. Yang, M. Albéric, T. Heil, Q. Wang, B. Pokroy, Y. Politi and L. Bertinetti, Adv. Funct. Mater., 2020, 30, 2000003 CrossRef CAS.
- Z. Zou, J. Xie, E. Macías-Sánchez and Z. Fu, Cryst. Growth Des., 2020, 21, 414–423 CrossRef.
- S. Kajiyama, T. Nishimura, T. Sakamoto and T. Kato, Small, 2014, 10, 1634–1641 CrossRef CAS PubMed.
- S. Matsumura, S. Kajiyama, T. Nishimura and T. Kato, Small, 2015, 11, 5127–5133 CrossRef CAS PubMed.
- L. K. Grunenfelder, S. Herrera and D. Kisailus, Small, 2014, 10, 3207–3232 CrossRef CAS PubMed.
- J. Jiang, M. R. Gao, Y. H. Qiu and S. H. Yu, Nanoscale, 2010, 2, 2358–2361 RSC.
- C. Zhong and C. C. Chu, Langmuir, 2009, 25, 3045–3049 CrossRef CAS PubMed.
- I. Sondi, S. D. Skapin and B. Salopek-Sondi, Cryst. Growth Des., 2008, 8, 435–441 CrossRef CAS.
- M. Faatz, F. Grohn and G. Wegner, Mater. Sci. Eng., C, 2005, 25, 153–159 CrossRef.
- J. M. Xto, C. N. Borca, J. A. van Bokhoven and T. Huthwelker, Chem. Commun., 2019, 55, 10725–10728 RSC.
- J. Ihli, A. N. Kulak and F. C. Meldrum, Chem. Commun., 2013, 49, 3134–3136 RSC.
- S. Leukel, M. Panthöfer, M. Mondeshki, G. Kieslich, Y. Wu, N. Krautwurst and W. Tremel, Chem. Mater., 2018, 30, 6040–6052 CrossRef CAS.
- R. Sun, P. Zhang, É. G. Bajnóczi, A. Neagu, C.-W. Tai, I. Persson, M. Strømme and O. Cheung, ACS Appl. Mater. Interfaces, 2018, 10, 21556–21564 CrossRef CAS PubMed.
- S. F. Chen, H. Colfen, M. Antonietti and S. H. Yu, Chem. Commun., 2013, 49, 9564–9566 RSC.
- J. Ihli, Y. Y. Kim, E. H. Noel and F. C. Meldrum, Adv. Funct. Mater., 2013, 23, 1575–1585 CrossRef CAS.
- B. Guillemet, M. Faatz, F. Grohn, G. Wegner and Y. Gnanou, Langmuir, 2006, 22, 1875–1879 CrossRef CAS PubMed.
- F. J. Zhu, T. Nishimura, T. Sakamoto, H. Tomono, H. Nada, Y. Okumura, H. Kikuchi and T. Kato, Chem. – Asian J., 2013, 8, 3002–3009 CrossRef CAS PubMed.
- E. Loste, R. M. Wilson, R. Seshadri and F. C. Meldrum, J. Cryst. Growth, 2003, 254, 206–218 CrossRef CAS.
- J. D. Rodriguez-Blanco, S. Shaw, P. Bots, T. Roncal-Herrero and L. G. Benning, J. Alloys Compd., 2012, 536, S477–S479 CrossRef CAS.
- S. Zhang, O. Nahi, L. Chen, Z. Aslam, N. Kapur, Y. Y. Kim and F. C. Meldrum, Adv. Funct. Mater., 2022, 32, 2201394 CrossRef CAS.
- A. Gal, S. Weiner and L. Addadi, J. Am. Ceram. Soc., 2010, 132, 13208–13211 CAS.
- A. Al-Sawalmih, C. Li, S. Siegel, P. Fratzl and O. Paris, Adv. Mater., 2009, 21, 4011–4015 CrossRef CAS.
- A. Sato, S. Nagasaka, K. Furihata, S. Nagata, I. Arai, K. Saruwatari, T. Kogure, S. Sakuda and H. Nagasawa, Nat. Chem. Biol., 2011, 7, 197–199 CrossRef CAS PubMed.
- E. M. Pouget, P. H. H. Bomans, J. A. C. M. Goos, P. M. Frederik, G. D. With and N. A. J. M. Sommerdijk, Science, 2009, 323, 1455–1458 CrossRef CAS PubMed.
- S. Sun, D. Gebauer and H. Colfen, Chem. Commun., 2016, 52, 7036–7038 RSC.
- S.-S. Wang and A.-W. Xu, Cryst. Growth Des., 2013, 13, 1937–1942 CrossRef CAS.
- W. Shi, Z. Ma, Y. Mu, J. Wang, B. Li, X. Wang, Z. Teng and X. Liu, RSC Adv., 2017, 7, 45113–45120 RSC.
- L. B. Gower and D. J. Odom, J. Cryst. Growth, 2000, 210, 719–734 CrossRef CAS.
- Y. Jiang, M. Kellermeier, D. Gebaue, Z. Lu, R. Rosenberg, A. Moise, M. Przybylski and H. Colfen, Nat. Commun., 2017, 8, 15933 CrossRef CAS PubMed.
- Z. Zou, L. Bertinetti, Y. Politi, P. Fratzl and W. Habraken, Small, 2017, 13, 1603100 CrossRef PubMed.
- J. Su, X. Liang, Q. Zhou, G. Zhang, H. Wang, L. Xie and R. Zhang, Biochem. J., 2013, 453, 179–186 CrossRef CAS PubMed.
- D. J. Tobler, J. D. R. Blanco, K. Dideriksen, K. K. Sand, N. Bovet, L. G. Benning and S. L. S. Stipp, Procedia Earth Planet. Sci., 2014, 10, 143–148 CrossRef CAS.
- Z. Zou, I. Polishchuk, L. Bertinetti, B. Pokroy, Y. Politi, P. Fratzl and W. Habraken, J. Mat. Chem. B, 2018, 6, 449–457 RSC.
- D. J. Tobler, J. D. Rodriguez Blanco, H. O. Sørensen, S. L. S. Stipp and K. Dideriksen, Cryst. Growth Des., 2016, 16, 4500–4508 CrossRef CAS.
- J. Ihli, W. C. Wong, E. H. Noel, Y. Y. Kim, A. N. Kulak, H. K. Christenson, M. J. Duer and F. C. Meldrum, Nat. Commun., 2014, 5, 3169 CrossRef PubMed.
- M. Saharay, A. O. Yazaydin and R. J. Kirkpatrick, J. Phys. Chem. B, 2013, 117, 3328–3336 CrossRef CAS PubMed.
- E. Beniash, J. Aizenberg, L. Addadi and S. Weiner, Proc. R. Soc. B, 1997, 264, 461–465 CrossRef CAS.
- T. Y. J. Han and J. Aizenberg, Chem. Mater., 2008, 20, 1064–1068 CrossRef CAS.
- S. Raz, O. Testeniere, A. Hecker, S. Weiner and G. Luquet, Biol. Bull., 2002, 203, 269–274 CrossRef CAS PubMed.
- T. Saito, Y. Oaki, T. Nishimura, A. Isogai and T. Kato, Mater. Horiz., 2014, 1, 321–325 RSC.
- D. Kuo, T. Nishimura, S. Kajiyama and T. Kato, ACS Omega, 2018, 3, 12722–12729 CrossRef CAS PubMed.
- H. Ping, H. Xie, Y. M. Wan, Z. X. Zhang, J. Zhang, M. Y. Xiang, J. J. Xie, H. Wang, W. M. Wang and Z. Y. Fu, J. Mater. Chem. B, 2016, 4, 880–886 RSC.
- M. Milovanovic, L. Mihailowitsch, M. Santhirasegaran, V. Brandt and J. C. Tiller, J. Mater. Sci., 2021, 56, 15299–15312 CrossRef CAS.
- C. Xu, Y. Yan, J. Tan, D. Yang, X. Jia, L. Wang, Y. Xu, S. Cao and S. Sun, Adv. Funct. Mater., 2019, 29, 1808146 CrossRef.
- Y. Zhao, Z. Luo, M. Li, Q. Qu, X. Ma, S. H. Yu and Y. Zhao, Angew. Chem., Int. Ed., 2015, 54, 919–922 CrossRef CAS PubMed.
- T. L. Liu, L. L. Li, X. Teng, X. L. Huang, H. Y. Liu, D. Chen, J. Ren, J. Q. He and F. Q. Tang, Biomaterials, 2011, 32, 1657–1668 CrossRef CAS PubMed.
- C. Wang, X. Liu, S. Chen, F. Hu, J. Sun and H. Yuan, Chem. Commun., 2018, 54, 13080–13083 RSC.
- C. Wang, S. Chen, Q. Yu, F. Hu and H. Yuan, J. Mater. Chem. B, 2017, 5, 2068–2073 RSC.
- C. Wang, S. Chen, Y. Wang, X. Liu, F. Hu, J. Sun and H. Yuan, Adv. Mater., 2018, 30, e1706407 CrossRef PubMed.
- M. Wang, B. Zhou, L. Wang, F. Zhou, N. Smith, D. Saunders, R. A. Towner, J. Song, J. Qu and W. R. Chen, J. Mat. Chem. B, 2020, 8, 8261–8270 RSC.
| This journal is © The Royal Society of Chemistry 2022 |