
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProteolysis targeting chimeras (PROTACs) come of age: entering the third decade of targeted protein degradation
Michael J.
Bond
a and
Craig M.
Crews
 *abc
*abc
aDepartment of Pharmacology, Yale University, New Haven, CT 06511, USA. E-mail: craig.crews@yale.edu
bDepartment of Molecular, Cellular, and Developmental Biology, Yale University, New Haven, CT 06511, USA
cDepartment of Chemistry, Yale University, New Haven, CT 06511, USA
First published on 19th March 2021
Abstract
With the discovery of PROteolysis TArgeting Chimeras (PROTACs) twenty years ago, targeted protein degradation (TPD) has changed the landscape of drug development. PROTACs have evolved from cell-impermeable peptide-small molecule chimeras to orally bioavailable clinical candidate drugs that degrade oncogenic proteins in humans. As we move into the third decade of TPD, the pace of discovery will only accelerate. Improved technologies are enabling the development of ligands for “undruggable” proteins and the recruitment of new E3 ligases. Moreover, enhanced computing power will expedite identification of active degraders. Here we discuss the strides made in these areas and what advances we can look forward to as the next decade in this exciting field begins.
 Michael J. Bond | Michael Bond obtained his BS in Molecular & Cell Biology and Chemical Biology from the University of Connecticut (UConn) in 2016. During his time at UConn he worked in the labs of Dr. Charles Giardina and Dr. Dennis Wright uncovering the molecular target of a novel mitotic arresting agent. After completion of his undergraduate studies, Michael moved to Yale University to pursue a PhD. He is currently a 5th year doctoral student in the lab of Dr. Craig Crews working on PROTAC development. |
 Craig M. Crews | Craig Crews graduated from the University of Virginia with a BA in Chemistry and received his doctorate from Harvard University in 1993. On the faculty of Yale University since 1995, his research program focuses on developing novel chemical biology technologies to co-opt cellular machinery to answer basic biological questions and address pathological conditions. Kyprolis®/carfilzomib, a second generation proteasome inhibitor based on his lab's work was approved for the treatment of relapsed/refractory multiple myeloma in 2012. Two drug candidates based on his lab's PROTAC technology are currently being tested in clinical trials for breast and prostate cancers. |
Introduction
The initial 2001 study demonstrating the ability of PROteolysis TArgeting Chimeras (PROTACs) to hijack the ubiquitin proteasome system (UPS) offered a new chemical biology approach to control protein function, which continues to impact drug discovery today.1 From their humble beginnings as chimeric peptides, PROTACs have evolved into potent small molecules that can degrade a variety of intracellular and transmembrane proteins in cells, mice, and recently humans2–7 (clinical trials NCT04072952 and NCT03888612). The success of PROTACs helped launch the field of Targeted Protein Degradation (TPD), which has recently moved beyond the proteasome. For example, Lysosome Targeting Chimeras (LYTACs) harness the lysosomal degradation pathway to induce degradation of extracellular proteins8 and the Macroautophagy Degradation Targeting Chimera (MADTAC) platforms, AUTACs and ATTECs hijack the autophagy pathway, thus potentially enabling the targeted destruction of entire organelles and protein aggregates.9,10 These novel and exciting TPD strategies will not be discussed herein, however there are several reviews and perspectives highlighting these nascent heterobifunctional modalities.4,11,12 Instead, we will focus on how the molecules that started the TPD revolution, PROTACs, continue to evolve.The ubiquitin-proteasome system (UPS)
The ubiquitin-proteasome system (UPS) is the primary pathway in eukaryotic cells by which intracellular proteins are degraded.13 To ensure that this system is tightly controlled, ubiquitin, a highly evolutionarily conserved 76 amino acid (8.6 kDa) polypeptide, is used to mark proteins for destruction. Once labelled with ubiquitin, proteins can be recognized and degraded by the 26S proteasome, a large barrel-shaped multi-subunit protein complex consisting of six proteolytic sites.14 Although this system has been extensively reviewed elsewhere, a brief overview will be provided below for context.15Ubiquitination of proteins is carried out by a cascade of ubiquitinating enzymes named E1, E2, and E3. E1 enzymes activate ubiquitin by using ATP to adenylate the ubiquitin C-terminus. Once the activated ubiquitin thioester is formed, the ubiquitin is conjugated to an E2 enzyme via a trans-thioesterification. Next, the E2 enzyme binds an E3 ligase, which is typically a large protein complex consisting of a substrate adaptor and accessory proteins. Depending on the class of E3 ligase, the ubiquitin is next transferred directly from the E2 to a lysine on the surface of the substrate, or the ubiquitin is sequentially passed from the E2 to the E3 to the substrate. Ubiquitin itself contains seven lysine residues, which allows for iterative rounds of ubiquitination by the E3 ligase to form polyubiquitin chains. Proteins marked with K48-linked polyubiquitin can then be recognized and degraded by the 26S proteasome.
PROTAC mechanism
PROTACs are heterobifunctional molecules that can simultaneously engage an E3 ubiquitin ligase and a protein of interest (POI) to be degraded (Fig. 1).2,3,5,16–18 The E3 ligase recruiting element (E3RE) and the ligand for the POI are joined by a chemical linker, whose composition and length can have dramatic effects on PROTAC activity. Upon binding its targets, PROTACs induce the formation of a ternary complex, enabling the E3 ligase to ubiquitinate the POI on surface lysine residues.19,20 Once the POI is sufficiently ubiquitinated, it can be recognized and degraded by the 26S proteasome.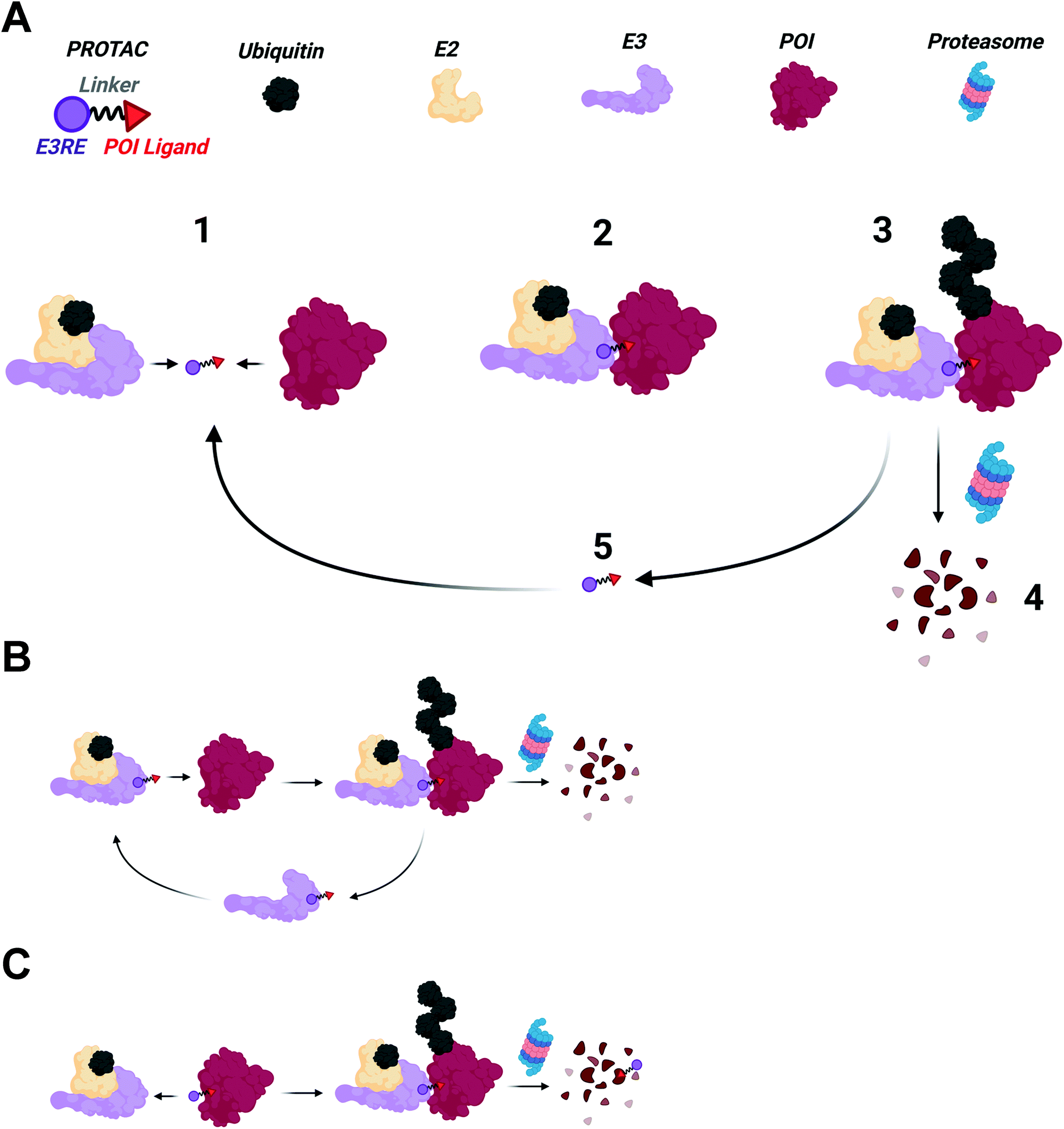 | ||
| Fig. 1 (A) PROTAC mechanism. (1) PROTACs engage their binding partners forming a ternary complex (2) stabilized by de novo protein–protein interactions. (3) The POI is then ubiquitinated by the E3 ligase, resulting in proteasomal degradation of the POI (4). (5) Noncovalent PROTACs can dissociate from the ternary complex and induce multiple cycles of degradation. (B) PROTACs that covalently bind the E3 ligase, but reversibly bind the POI can also undergo catalytic rounds of degradation. (C) PROTACs that covalently bind the POI cannot participate in catalytic rounds of degradation. PROTACs that bind in a covalent, reversible manner to either the E3 ligase or POI would have the catalytic cycle shown in A. | ||
Shifting paradigms
Over the past twenty years, PROTACs and the emerging TPD field have changed how academia and industry imagine drug discovery. Historically, drug development has relied on an “occupancy-driven” pharmacological model.17,21 In this paradigm, drug action is a direct result of its sustained occupancy of a binding site within the target protein. The fraction of bound drug is directly proportional to the compound's affinity for its target. Therefore, high affinity ligands (low nanomolar to picomolar) are necessary for specific biological activity at tolerable doses. While numerous academic and industrial research campaigns have been undertaken against a variety of pharmacological targets, development of specific, high affinity ligands for many disease targets remains challenging.PROTACs and other TPD technologies may circumvent the issues associated with “occupancy-driven” pharmacology since they act through an “event-driven” paradigm.3,18,21,22 Rather than compound activity correlating solely to affinity, activity instead correlates to the ability of PROTACs and similar molecules to elicit desired biological events, namely ubiquitination. A PROTAC's ability to induce ubiquitination is dependent on the transient formation of a E3 ligase–PROTAC–POI ternary complex. The ternary complex need not be sustained by the PROTAC any longer than is needed for target ubiquitination to occur, after which target destruction proceeds regardless of PROTAC binding. In fact, dissociation of the PROTAC is desirable since it permits the degrader to form additional ternary complexes with remaining POI molecules, inducing multiple rounds of degradation.19 Excitingly, PROTACs with weak binary interactions for a POI can still be potent degraders if a stable ternary complex is formed. This was exemplified by our work on foretinib-based PROTACs.20,23 Despite foretinib's ∼10 μM affinity for p38α kinase, foretinib-based PROTACs can potently degrade p38α with a DC50 of 210 nM. This case study suggests that transient binding of a modest affinity ligand can allow for sufficient ternary complex formation to induce ubiquitination and subsequent degradation of proteins with modest affinity POI ligands.
Degrading “hard to drug” proteins
Advances in structural biology over the past 60 years have yielded thousands of disease-relevant protein structures. From these studies a clear pattern emerged: “druggable” proteins possess well-defined deep grooves and pockets that small molecules can occupy.24 However, other proteins with shallow or no binding pockets have classically been viewed as “undruggable.” Since small molecule ligands are components of PROTACs, one might have assumed that these proteins are also “undegradable.” However, several studies have showed that PROTACs are indeed capable of degrading these proteins, and as advances in ligand discovery continue, the repertoire of effective PROTACs will continue to shorten the list of “undruggable” disease targets (Fig. 2).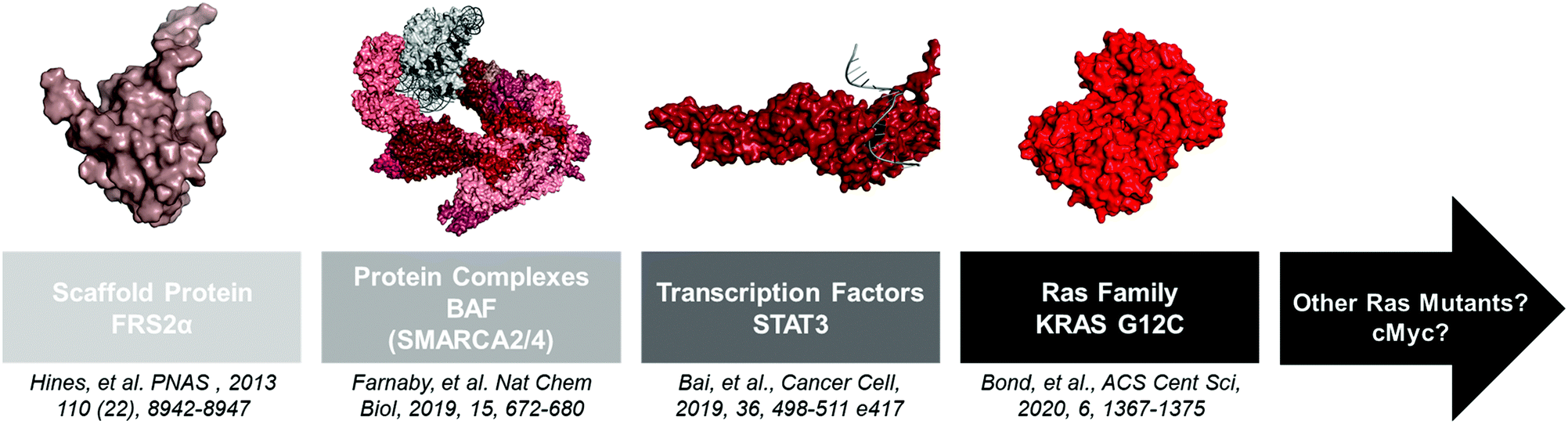 | ||
| Fig. 2 A non-extensive timeline of “hard to drug” proteins degraded with PROTACs. The insights gained from these studies will help the TPD community tackle other proteins like KRAS G12D and c-Myc. FRS2α PDB: 2MFQ; BAF PDB: 6LTJ; STAT3 PDB: 1BG1; KRAS G12C PDB: 6UTO. | ||
Scaffolding proteins
Scaffolding proteins that lack enzymatic activity are ideal PROTAC targets because they are generally impervious to small molecule inhibition. Our lab first demonstrated the ability of PROTACs to eliminate scaffolding proteins using phosphoPROTACs to degrade fibroblast growth factor receptor substrate 2α (FRS2α).25 FRS2α is a myristoylated, non-enzymatic scaffolding protein that participates in mitogen-activated protein kinase (MAPK) signaling during neuronal differentiation upon nerve growth factor (NGF) stimulation.26 Hines et al. developed a chimeric cell penetrable peptide consisting of a phospho-dependent FRS2α recognition sequence linked to a Von-Hippel Lindau (VHL) E3 ligase recognition sequence termed TrkAPPFRS2α.25 When PC12 cells were co-treated with TrkAPPFRS2α and NGF, the FRS2α TrkAPPFRS2α recognition sequence was phosphorylated, enabling TrkAPPFRS2α to simultaneously bind FRS2α and VHL. This resulted in proteasomal-dependent FRS2α degradation and a blockade of neuronal differentiation. Although high concentrations (>50 μM) of the peptide-based phosphoPROTACs were necessary to achieve robust degradation, this study demonstrated the feasibility of PROTAC-mediated elimination of a “hard to drug” protein.Multicomponent protein complexes
Large multiprotein complexes have traditionally been viewed as “undruggable” because inhibition of one subunit may not be entirely deleterious to complex function. Moreover, the targeting of large protein–protein interactions (PPIs) has proved challenging using small molecules.27 However, PROTACs and other TPD technologies may circumvent this issue. If the most ligandable subunit is targeted via a PROTAC, degradation of multiple complex members can be achieved through “bystander” ubiquitination.20 Alternatively, degradation of one subunit may destabilize the entire complex, which can also lead to degradation of the remaining complex subunits through the natural protein quality control machinery. The recent development of PROTACs targeting the BAF chromatin remodeling complex showcases the ability of TPD to modulate disease-relevant multicomponent protein complex activity.28The BAF/PBAF chromatin remodeling complex modulates the position of nucleosomes on genomic DNA, thereby regulating many cellular processes including gene expression, DNA replication, and DNA repair.29 Due to its role in DNA maintenance, the complex is highly mutated in human cancers (∼20%). Key components of this complex are the mutually exclusive ATPase subunits SMARCA2 and SMARCA4,30 which are promising targets for therapeutic intervention in cancers based on genetic knockdown studies.31 Interestingly, bromodomain (BD) inhibitors of SMARCA2/4 are not antiproliferative, suggesting that chromatin remodeling can occur irrespective of BD function.32–34 Therefore, elimination of the entire SMARCA2/4 protein by use of PROTACs and destabilization of the BAF complex may phenocopy genetic knockdown.
To test this, Farnaby et al. developed PROTACs using one of the aforementioned BD inhibitors.28 After solving the crystal structure of the BD inhibitor in complex with the SMARCA2BD to identify a point of linker attachment, the inhibitor was conjugated to a ligand for VHL using PEG linkers of various lengths. From this library, an active PROTAC was identified and biophysically characterized for its ability to induce a SMARCA2BD-PROTAC-VHL ternary complex. A crystal structure for this complex was solved and insights gained from this structure were used to rapidly develop a more potent SMARCA2/4 degrader, ACBl1. This selective degrader was then shown to be antiproliferative in AML cell lines, while the parent inhibitor did not attenuate cell viability. Excitingly, treatment with ACBl1 showed loss of other BAF components such as ACTLA6 and PHF10, suggesting bystander ubiquitination and/or destabilization of the entire complex upon SMARCA2/4 degradation. Although SMARCA4 degradation may not be therapeutically useful due to the tumor suppressor function it plays in many cancers, the development of a SMARCA2/4 degrader illustrates that PROTACs can be used to inhibit the function of large multicomponent protein complexes, as well as demonstrating the benefits of structure-guided PROTAC design (see Improving the efficiency of PROTAC identification). Concurrent with the publication of ACBl1, a dual ATPase inhibitor for SMARCA2/4 was developed that also induces cell death in AML cells. Future studies should be conducted to determine if PROTACs using these ligands will be more selective for SMARCA2 degradation than ACBI1. Other “undruggable” chromatin remodeling complexes have also been degraded with PROTACs and this has been reviewed elsewhere.4,5
Transcription factors
Transcription factors (TFs) are DNA binding proteins that promote changes in gene expression.35 Therefore, their dysregulation is associated with a myriad of diseases, especially cancers. TFs have been difficult to drug because their natural substrate, DNA, is negatively charged and compounds that mimic this interaction tend to have poor cell permeability.36 Additionally, some TFs, like STAT3, dimerize or form larger protein complexes that are difficult to drug due to the large surface area of the PPIs.35 PROTACs and other TPD technologies can overcome these limitations of traditional inhibitors since binding to any part of the transcription factor may be enough to elicit degradation.The recent development of a STAT3-targeting PROTAC is a great example of how PROTACs and similar technologies can be used to degrade “undruggable” TFs.37 Aberrant STAT3 activity has been linked to the incidence of several cancers including leukemias and lymphomas.38 STAT3 activation has long been held to be dimerization dependent, which is facilitated by phosphorylation of Tyr705.39 pTyr705 of one monomer is recognized by the SH2 domain of the other monomer. STAT3 ligands that bind to the SH2 domain have been discovered, but have low utility as inhibitors since monomeric STAT3 can still translocate into the nucleus and regulate gene expression.40,41 Additionally, these ligands have only modest selectivity for STAT3 over the SH2 domains in STAT1 and STAT4 (∼20 fold).
To address these issues, Bai et al. optimized a STAT3 ligand, SI-109, and subsequently linked it to the cereblon (CRBN) recruiter lenalidomide to create a specific STAT3-degrading PROTAC named SD-36.37 SD-36 is capable of degrading STAT3 in leukemia cell lines, with corresponding significant decreases in STAT3-regulated genes and induction of cell death. SD-36 degrades only STAT3, sparing STAT1 and STAT4, representing another example of how a promiscuous ligand can be developed into a more specific degrader.20 SD-36 was also able to eliminate tumors in mice, with complete remissions that lasted until the end of the 125-day experiment. This study, along with our lab's recent work on the TRAFTAC TPD system, showcase attractive new methodologies for degrading hard-to-drug TFs.42
RAS proteins
RAt Sarcoma (RAS) proteins are mutated in approximately 25% of human cancers and have long been attractive targets for cancer therapeutics.43 However, their lack of druggable pockets and high affinity for GTP have inhibited drug discovery efforts, thereby placing RAS proteins in the category of “undruggable.” Despite this, recent progress has been made targeting the KRAS G12C mutant prevalent in lung adenocarcinoma.43 The point mutation to cysteine enables the opportunity for covalent ligand development. Work by the Shokat group identified vinyl sulphone and acrylamide molecules that covalently bind KRAS G12C and form a drug stabilized pocket.44,45 This discovery re-invigorated the field of small molecule RAS inhibitor discovery, leading to the development of several covalent KRAS G12C inhibitors currently in clinical trials.46–49 Among them, AMGEN's sotorasib (AMG 510), the leading clinical candidate, and Mirati Therapeutics’ adagrasib (MRTX849) have shown potent in vitro and in vivo inhibition of KRAS G12C signaling and efficacy in early-stage clinical trials.46 However, recent studies have suggested that cancer cells can acquire resistance to KRAS G12C inhibitors by hyperactivation of other growth pathways.50,51 Therefore, alternative therapeutic strategies, like PROTACs, need to be explored.Several TPD systems using VHL fusion proteins have been developed to induce endogenous KRAS G12C degradation and tumor cell death in cells and in mice, suggesting KRAS degradation as a viable option for cancer therapy.52–54 The first attempt to induce KRAS G12C degradation with PROTACs recruited CRBN via pomalidomide and bound KRAS G12C using ARS1620, a tool KRAS G12C inhibitor developed by Wellspring Biosciences.47,55 Zeng et al. used a flow cytometry-based assay to identify a compound capable of degrading stably overexpressed GFP-KRAS G12C, named XY-4-88. Interestingly, XY-4-88 did not induce degradation of endogenous KRAS G12C, suggesting that degradation of the GFP-KRAS G12C fusion protein was due to ubiquitination of GFP and not KRAS G12C.
Our lab has also been interested in degrading KRAS G12C with PROTACs. Based on data from Zeng et al., we opted to test degraders that recruited VHL and used the synthetically tractable MRTX849 as the KRAS G12C warhead.49,56 We synthesized a small library of PROTACs and identified LC-2 as the first PROTAC capable of degrading endogenous KRAS G12C.57 LC-2 induces rapid, sustained degradation in both homozygous and heterozygous KRAS G12C-expressing cell lines with maximal degradation (DMax) values ranging from 40–90% and DC50 (concentration at which 1/2DMax was reached) values in the high nanomolar range (0.25–0.76 μM). However, KRAS G12C degradation with LC-2 does not inhibit downstream signaling as effectively as the parent MRTX849 inhibitor. Additionally, LC-2 is no more antiproliferative than MRTX849. This is due to the covalent nature of LC-2 that does not allow for catalytic rounds of KRAS G12C degradation, which has been observed in cases of target engagement by non-covalent PROTACs.19 Despite this mechanistic limitation on its potency, the identification of LC-2 confirms that “undruggable” RAS mutants can be degraded using the PROTAC technology. This is particularly exciting considering recent discoveries of cyclic peptides targeting KRAS G12D,58 a more prevalent oncogenic mutation. Macrocyclic PROTACs were recently described, so optimized, cell-permeable versions of KRAS G12D binding macrocycles could serve as warheads for future KRAS G12D PROTACs.59 Finally, our KRAS G12C story, and other recently published studies, highlight that not all E3 ligases can degrade all protein targets and that multiple E3 ligases should be tested during PROTAC development.60,61
Advances in ligand development
To expand the reach of TPD to degrade other “hard to drug” proteins, novel ligands for these targets need to be identified. Due to advancements in proteomics, chemical synthesis, and molecular biology, tools now exist to identify ligands for almost any protein. This is underscored by the recent Target 2035 campaign that aims to identify small molecule tool compounds for the entire human proteome within the next 15 years.62 The technologies that will lead the way in the discovery of novel ligands for PROTAC development will be chemoproteomics to identify covalent binding molecules and DNA encoded library (DEL) screens to identify reversible ligands63–65 (Fig. 3).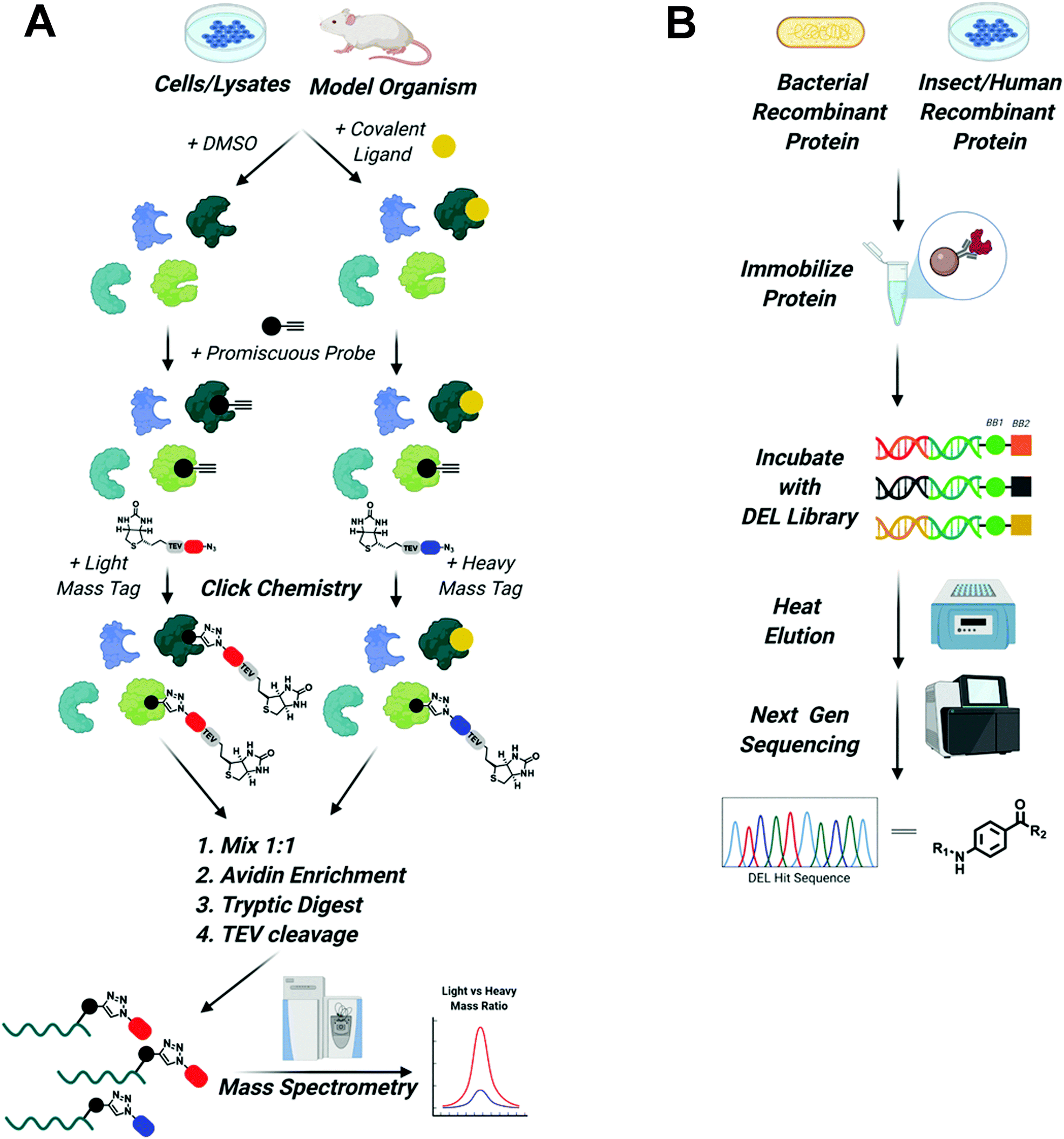 | ||
Fig. 3 Screening platforms that will enhance TPD in the next decade. (A) Chemo proteomic competition experiments using promiscuous probes will uncover covalent binders of difficult to drug POIs and E3 ligases. Briefly, whole cells, cell lysate, or model organisms are treated with DMSO or the covalent ligand of interest. Proteins are then harvested and treated with a promiscuous probe outfitted with a terminal alkyne. Next, click chemistry is used to install a biotinylated light mass tag in the DMSO sample or a biotinylated heavy mass tag in the covalent ligand treated sample. The samples are then mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, proteins are enriched by avidin pulldown, digested with trypsin, and finally biotin/avidin are removed via TEV cleavage. Purified proteins retaining the light and heavy tags are identified using mass spectrometry. Proteins with large light/heavy ratios are prospective targets of the covalent ligand (figure modified from Spradlin et al.93). (B) DEL screening will be used to identify reversible ligands for POIs and E3 ligases. Epitope tagged protein can be purified from bacteria, insect, or human cells and then immobilized on affinity resin. Immobilized protein is then incubated with DNA barcoded libraries of millions to billions of compounds. Samples are washed and then bound molecules are eluted with mild heating. Eluted compounds are identified using next generation sequencing and can then be synthesized off DNA and tested in biophysical assays to assess binding. :1 ratio, proteins are enriched by avidin pulldown, digested with trypsin, and finally biotin/avidin are removed via TEV cleavage. Purified proteins retaining the light and heavy tags are identified using mass spectrometry. Proteins with large light/heavy ratios are prospective targets of the covalent ligand (figure modified from Spradlin et al.93). (B) DEL screening will be used to identify reversible ligands for POIs and E3 ligases. Epitope tagged protein can be purified from bacteria, insect, or human cells and then immobilized on affinity resin. Immobilized protein is then incubated with DNA barcoded libraries of millions to billions of compounds. Samples are washed and then bound molecules are eluted with mild heating. Eluted compounds are identified using next generation sequencing and can then be synthesized off DNA and tested in biophysical assays to assess binding. | ||
It is now possible to identify covalent inhibitors in the context of purified proteins, cell lysates, whole cells, and even model organisms like mice,65 using competition between large electrophilic libraries and highly reactive probes that covalently modify a majority of the proteome.63 Briefly, ligandable hot spots are identified by first treating cells or protein lysates with libraries containing electrophilic compounds that specifically react with nucleophilic side chains like cysteine, lysine, or tyrosine. DMSO-treated samples are used as control. Next, samples are treated with a highly reactive pan-specific molecule (i.e. iodoacetamide for cysteine) functionalized with an alkyne. Click chemistry can then be used to install a biotinylated “light” or “heavy” mass tag, that can be used for subsequent affinity purification. Samples are mixed 1:1 and then purified using avidin agarose. Proteins are then digested with trypsin following TEV cleavage of the tag to remove biotin/avidin. Finally, relative amounts of tagged proteins, as determined by the ratio of light vs. heavy protein, can be analyzed using mass spectrometry. A decreased signal in ligand vs DMSO treated samples for a given protein suggests the library compound is binding to a ligandable hotspot within that protein. This molecule can then serve as the basis for the design and characterization of novel covalent ligands. Importantly, because the majority of the proteome can be evaluated in the same experiment, insights are gained into the specificity of the molecule for each identified ligandable site.
Recently, the Nomura lab has showcased the power of this approach to identify a covalent ligand for Myc, an infamous, “undruggable” protein.66 Myc controls genes important in cell proliferation, metabolism, and cell survival. Its amplification is a hallmark of tumorigenesis, making Myc a very attractive target for therapeutics.67 However, like most transcription factors, it is predominantly disordered and lacks traditional druggable pockets. Using the approach summarized above, Boike et al. identified EN4, which covalently binds C171 of Myc with good specificity (8 off targets) in cells.66 EN4 is able to decrease Myc-associated gene expression and decrease tumor cell viability in cellular and in vivo models at high micromolar concentrations. Interestingly, C171 lies within a disordered region of Myc, suggesting that chemoproteomics can be used to identify ligandable hotspots in proteins that are intrinsically disordered.66 EN4 binding alone induces proteasomal dependent degradation of Myc at high micromolar concentrations. Myc targeting PROTACs, particularly reversible-covalent degraders, using EN4 could allow for more potent degradation of Myc. It will be interesting to see if other disordered proteins are also destabilized by covalent ligands and whether their ligands can be used as warheads for PROTAC development. Regardless of the outcome, covalent fragment screening will continue to be at the forefront of ligand discovery in the TPD field for the foreseeable future.
Although covalent ligands are becoming more prominent in drug discovery, reversible ligands remain the primary focus of drug discovery efforts in academia and industry. As highlighted by LC-2, reversible POI ligands are invaluable for TPD applications since they allow for catalytic degradation of proteins (although covalent E3 ligase ligands can permit catalytic degradation, as will be discussed in the next section). Therefore, robust high throughput screens for reversible ligands are necessary. A promising candidate is DNA encoded library (DNA) screens.68 DEL screens are a powerful combination of combinatorial synthetic chemistry and molecular biology.64 High fidelity reactions like reductive aminations, Pd/C cross couplings, amide formations, SNAR, etc. can be set up in parallel, allowing massive libraries to be constructed. Each reaction can be coupled to the addition of a nucleotide to a growing DNA strand, which enables each compound to be individually “barcoded”. Therefore, libraries of millions or billions of compounds can be screened simultaneously against a target. Typically, this is done using epitope-tagged purified protein that is immobilized onto the surface of a resin.69 However, to enable ligand identification in the presence of protein binding partners, epitope tagged protein could be expressed in cells either by CRISPR gene editing or plasmid transfection and then immobilized to the corresponding antibody resins. Once immobilized, compounds are incubated with the target protein. After several rounds of washing, bound compounds are then eluted using heat. Ideally, heating the sample will free the DEL compounds, but will not denature the protein, keeping it immobilized as to not interfere with downstream analysis. Eluted compounds are then identified by performing deep sequencing of their built-in DNA “barcodes”.64,68,69 Although there are limitations to DEL screening, namely the fact that compounds are not purified so the true identity of the ligand is not known (although this can easily be determined by testing predicted final compounds alongside reaction byproducts in a biophysical binding assay), it is an ideal system for PROTAC and TPD degrader development because the position for linker attachment on the molecule is already known once ligands are identified: the site at which the DNA barcode is attached to the molecule must be solvent exposed to have enabled binding of the target protein. Therefore, a linker emanating from the same chemical functionality as the DNA should be solvent exposed and likely capable of inducing ternary complex formation. Now that several companies have amassed large DEL libraries, the next decade should see an increase in compounds identified by this methodology. Due to the ability of DEL screening to inform sites of linker attachment, ligands identified by this system should make immediate impacts in the TPD field.
Expanding recruited E3 ligases
Despite evidence from our lab and others that multiple E3 ligases, encompassing all three E3 ligase families, can participate in TPD, the identification of ligands for the recruitment of E3 ligases has, until very recently, lagged behind identification of degradable POIs.70 The reasons for the disconnect are understandable. To generate an active molecule that induces POI degradation all one needs is a ligand that binds the protein at any binding site. Libraries of such compounds; those approved by the FDA; those that had failed clinical trials; and those developed solely as tool compounds already exist. To develop an E3RE one needs to identify a molecule that occupies the substrate recognition site of the E3 ligase (or E3 ligase adaptor protein), but does not abolish activity of the E3 ligase complex. While this rational design is possible, it has remained challenging due to a lack of structural knowledge of E3 ligase recognition elements or “degrons.” Additionally, high throughput screens are generally unable to distinguish between compounds that disrupt E3 ligase–substrate interactions and those that ablate protein activity. Therefore, the majority of E3 ligases used in TPD to date, even those that were recently identified, have been discovered through serendipity. However, advances in screening technologies and a better understanding of E3 ligase–substrate interactions may increase the number of E3REs available for TPD applications in the next decade (Fig. 4). Nonetheless, serendipity, especially in the form of elucidating the function of natural products, may continue to provide the field with more E3REs.71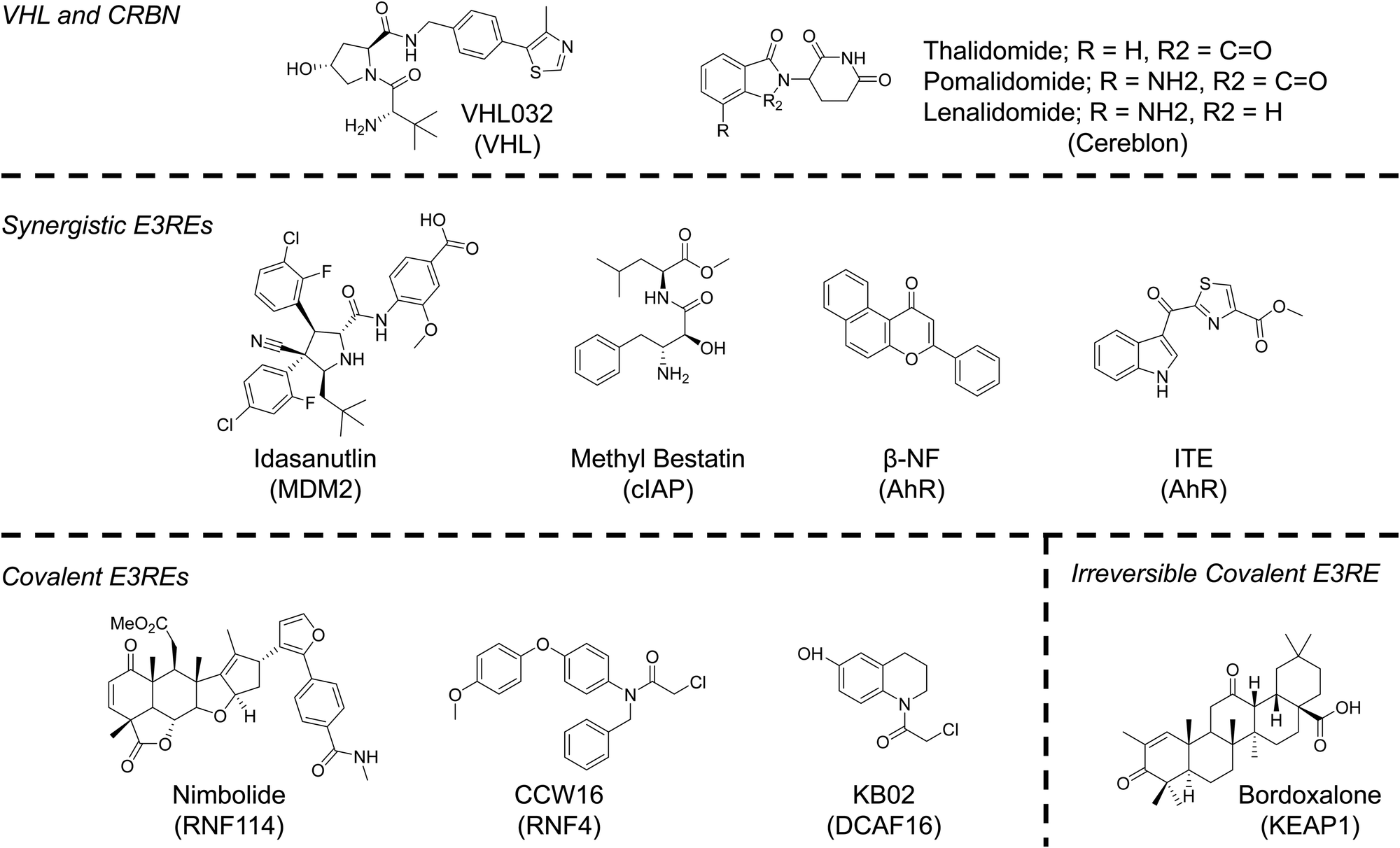 | ||
| Fig. 4 Chemical structures of E3 ligase recruiting elements (E3REs) used in TPD. | ||
“Old Reliable” E3REs – cereblon and VHL
The majority of reported PROTACs have recruited either cereblon (CRBN) or VHL. These are the preferred E3 ligases of the TPD community since they are ubiquitously expressed and potent small-molecule ligands of CRBN and VHL have been published, enabling PROTACs recruiting these E3 ligases to be active in many cell lines. Thalidomide, a drug that has had a checkered history as a teratogenic treatment for morning sickness turned cancer therapy, was serendipitously discovered to bind CRBN in 2010.72 In the decade since, thalidomide and its analogs (pomalidomide and lenalidomide) have become synonymous with TPD. Not only can these molecules be used to recruit CRBN, but they can also serve as molecular glues that change the surface of CRBN, facilitating the recognition of neo-substrates, such as SALL4, the likely source of its teratogenicity, and Ikaros zinc finger proteins, the reason for its cytotoxicity in lymphocytes.4,73,74 Molecular glues are another important UPS hijacking technology that have been extensively reviewed elsewhere.2,4,75 However, as will be discussed in subsequent sections, the more we learn about the mechanisms of action of PROTACs and of molecular glues, the more the line between these two modalities is blurring.Unlike CRBN, the recruitment of VHL for PROTACs resulted from a deliberate effort to identify E3REs. Early PROTACs recruited VHL using a peptide known to mimic the degron of VHL's natural substrate HIFα.1 This knowledge, combined with high-resolution determination of the peptides’ binding mode determined by X-ray crystallography, allowed our lab to develop a small molecule capable of hijacking VHL for TPD.76,77 The VHL ligand mimics the hydroxyproline of HIFα. Inversion of the stereochemistry at this hydroxyproline abolishes VHL binding. Incorporation of this diastereomeric VHL ligand into a PROTAC molecule ablates activity, unambiguously establishing a PROTAC's mechanism of action. This simple stereochemical change that preserves physiochemical properties (e.g. permeability) is a major advantage of the VHL ligand as abolishing binding of other E3REs requires more consequential chemical changes.
Since the initial discovery, VHL ligand analogs have been developed with improved potency and physiochemical properties.78,79 However, the most exciting new VHL ligands may actually be those that are weaker binders. We first demonstrated that weak VHL ligands (∼10 μM) can induce protein degradation of Halotag fusion proteins, although activity was modest (∼50% DMax).80 Building on this work, Han et al. recently synthesized a VHL ligand with ∼10 μM affinity and developed a potent, nanomolar degrader of the androgen receptor (AR).81 This study reaffirms that event driven molecules do not necessarily need high affinity ligands for activity. One caveat of this work is that a high affinity ligand for AR was needed to achieve degradation. However, these data are encouraging because it suggests that low micromolar E3REs identified from screens could be linked to already existing high affinity ligands to develop next generation PROTACs.
Synergistic E3REs – MDM2, cIAP, and AhR
Although less common than VHL and CRBN, the E3 ligases MDM2 and cIAP have been used for many years in TPD applications. In fact, the MDM2 E3RE nutlin was used to develop the first all small molecule PROTAC in 2008.82 The original nutlin compounds had poor physiochemical properties, which were exacerbated when developed into PROTACs. Recent efforts have identified more soluble and potent MDM2-p53 PPI inhibitors that may increase the use of MDM2 future TPD efforts.83Specific and Nongenetic Inhibitor of Apoptosis Protein (IAP)-dependent Protein Erasers (SNIPERs) are a TPD technology that recruits the cellular inhibitor of apoptosis protein (cIAP) using methyl bestatin analogs.84 These molecules are said to be distinct from PROTACs because SNIPERs induce the degradation of both the E3 ligase cIAP and the POI. Although this limits the catalytic ability of SNIPERs, simultaneous degradation of cIAP and an oncogene can have synergistic effects on apoptosis induction.85 A similar phenomenon has been observed by our lab and others with MDM2 recruiting PROTACs. Recruitment of MDM2 stabilizes its native substrate p53, the resultant upregulation of which in conjunction with a targeted oncogene's degradation can enhance antiproliferative effects compared to oncogene degradation alone.86,87
A lesser known E3 ligase whose recruitment and inhibition with PROTACs may result in synergistic activity is the aryl hydrocarbon receptor (AhR). The AhR is a ligand-activated transcription factor important in upregulating genes associated with metabolism of exogenous molecules and inflammatory signaling.88 However, when activated AhR can also bind CUL4B and act as an atypical E3 ligase substrate adaptor.89,90 The AhR is activated by a number of synthetic molecules and natural metabolites, most notably α-napthoflavone (α-NF), β-naphthoflavone (β-NF) and 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE).88 Taking advantage of the ligand induced E3 ligase activity of AhR, Ohoka et al., developed AhR-recruiting degraders that target cellular retinoic acid binding proteins (CRABPs) and BRD proteins by linking all-trans retinoic acid or JQ1 to one of the aforementioned AhR ligands.91 Modest, proteasome- and AhR-dependent degradation was observed for both proteins. Degradation of BRD proteins leads to more cell death in MCF-7 breast cancer cells compared to co-treatment of β-NF and JQ1. Activation of AhR with small molecule ligands can result in its autoubiquitination and degradation, which was observed at high concentrations of AhR-recruiting PROTAcs, suggesting they behave like SNIPERs. Since AhR overexpression is oncogenic in many tumors, co-degradation of AhR and another oncogene may enhance apoptosis induction.88 The potential for synergistic anti-tumorigenic activity is a major benefit to recruiting novel E3 ligases.
Covalent E3REs – DCAF16, RNF114, & RNF4
Employing the chemoproteomics approach outlined in a previous section, three new E3 ligases have recently been added to the TPD E3 ligase toolbox. Hijacking DCAF16 and RNF114 were serendipitous discoveries, much like CRBN, whereas RNF4 was a more targeted study, like that seen with VHL.In 2019, Zhang et al. set out to identify novel E3REs using a library of previously characterized electrophilic scout fragments.92 For their screen, scout fragments bearing chloroacetamide or acrylamide warheads were tethered to the SLF ligand, a high affinity binder of FKBP12. Proteasome- and neddylation-dependent degradation of FLAG-FKBP12_NLS was observed in stably expressing HEK293 cells for one compound, KBO2-SLF. Interestingly, only nuclear FLAG-FKBP12_NLS was depleted. This data suggested a nuclear cullin-RING E3 ligase was responsible for the observed degradation. Due to lack of signal in competitive chemoproteomics experiments, a FLAG pulldown approach was used to enrich proteins associated with KBO2-SLF compared to DMSO. These experiments identified DCAF16, an understudied nuclear E3 ligase. Control experiments with shRNA and CRISPR/Cas9 knockout confirmed a dependency on DCAF16 for KBO2-SLF activity. Although the exact cysteine modified by KBO2-SLF could not be identified, the utility of DCAF16 to degrade endogenous proteins was showcased with the design and testing of KBO2-JQ1. This PROTAC was capable of degrading BRD4 at modest mid-μM concentrations. The success of DCAF-recruiting PROTACs suggests the exciting possibility that recruitment of some E3 ligases may allow for cellular compartment-specific degradation.
Concurrent with the discovery of KBO2, nimbolide was reported to be useful for TPD.93 Nimobolide is a terpenoid natural product that has been reported to have antitumorigenic properties by a previously unknown mechanism. Due to the presence of an electrophilic cyclic enone, a chemoproteomics strategy was employed to identify possible targets to explain nimbolide's mechanism of action. From this screen, C8 of RNF114 was identified and verified as the primary target of nimbolide. RNF114 is a ring-finger containing protein that has E3 ligase function and is implicated in immune signaling and tumorigenesis.94 A notable substrate is the tumor suppressor p21WAF1/CIP1 94. Interestingly, excess nimbolide was found to block p21WAF1/CIP1 recruitment and ubiquitination, but did not ablate the catalytic activity of the E3 ligase. Therefore, nimbolide was tethered to the BRD inhibitor JQ1 to test whether RNF114 could be used for TPD applications. Excitingly, the resulting PROTAC, XH2, was capable of degrading BRD4 at low-nM concentrations in a proteasome-dependent manner. XH2, like nimbolide, was also able to stabilize p21 and other RNF114 substrates, therefore, RNF114-recruiting PROTACs may lead to synergistic effects like those seen with MDM2-recruiting PROTACs in specific cancer subtypes. Interestingly, BRD2 and BRD3 were spared by XH2 highlighting that recruitment of novel E3 ligases may enable addition degradation specificity. Nimbolide has also been used to recruit RNF114 to degrade BCR-Abl with greater specificity for that fusion oncoprotein than observed with phthalimide-based PROTACs.95 Nimbolide has not been widely utilized to degrade other proteins, most likely due to the fact that is difficult to synthesize and, instead is typically isolated from Neem tree leaves.93 To enhance the utility of RNF114 in TPD applications, a fully-synthetic RNF114 E3RE has been identified and used successfully to induce BRD4 and BCR-ABL degradation.96 The identification of this compound should increase the use of RNF114 in PROTAC design.
RNF4 is an E3 ligase that plays a key role in DNA repair by recognizing and ubiquitinating SUMOylated proteins, thereby targeting them for degradation.97 Due to the commercial availability of purified RNF4, Ward et al. set out to use a covalent ligand strategy to identify RNF4 ligands.98 From competitive assays with rhodamine-labelled electrophiles, several lead compounds were identified. The most promising ligand, TRH 1-23 was found to covalently bind RNF4 in a dose-dependent manner at Cys132/Cys135 without impairing RNF4 catalytic activity. To test whether this compound was a suitable RNF4 E3RE, TRH 1-23 was linked to JQ1 to form CCW 38-3 and the resulting PROTAC was capable of degrading BRD4 in a proteasome-dependent manner in 231MFP breast cancer cells. Again, CCW 38-3 showed enhanced specificity over the parent inhibitor JQ1, as BRD2 and BRD3 were spared by the PROTAC.
Not only are these studies valuable because they increase the number of E3 ligases that can be recruited using the PROTAC technology, but they also suggest that covalent engagement of E3 ligases can still allow for potent, catalytic degradation. This is exemplified by KBO2-based PROTACs, which only occupy 10–40% of cellular DCAF16.92 Covalent ligands may prime E3 ligases for inducing degradation as a pseudo-binary complex, rather than a ternary complex, would be needed for degradation. However in-depth mechanistic studies are needed to confirm this.
Reversible-covalent E3REs – KEAP1
Reversible-covalent (RC) molecules have received increased attention in recent years as they combine the specificity of covalent ligands with the pharmacokinetic properties of reversible inhibitors. PROTACs bearing RC POI ligands have been described for BTK targeting PROTACs.99,100 Interestingly, the RC BTK PROTACs are not as efficacious as covalent PROTACs, despite the fact that RC PROTACs should retain their catalytic nature since the PROTAC can dissociate. This suggests that ubiquitination and degradation may be on a faster time scale than dissociation, or may even impede dissociation.To determine if RC E3REs could be used for TPD, Tong et al. linked the natural product bardoxolone to JQ1.101 Bardoxolone contains a reversible α-cyanoenone hetero-Michael acceptor that modifies the KEAP1 E3 ligase. PROTACs bearing a peptide that recruits KEAP1 were previously developed, demonstrating KEAP1 utility in TPD. In agreement with these studies, the bardoxolone-JQ1 PROTAC, CDDO-JQ1, was capable of inducing proteasome- and neddylation-dependent degradation of BRD4. At higher concentrations CDDO-JQ1 has a SNIPER-like mechanism and induces the degradation of KEAP1, which could limit its utility in cancers since KEAP1 has tumor suppressor functions.102 Nonetheless, this study establishes RC E3REs as viable ligands for PROTAC development. Taken together, the studies described for covalent and RC E3REs also demonstrate that BRD4 proteins are susceptible to TPD by a variety of E3 ligases and should continue to serve as a great model system for the discovery of new E3REs over the next decade.
Tissue specific and disease specific degradation
There are >600 E3 ligases within the human proteome, some of which are projected to have tissue and disease specific expression.103 Over the next decade, identification of ligands for these E3 ligases will enable tissue/disease specific degradation that could improve potential toxicities associated with systemic target degradation. The power of sparing proteins based on E3 ligase expression has already been demonstrated using VHL-, CRBN-, and cIAP-recruiting PROTACs. At the end of 2019 and beginning of 2020, three degrader molecules targeting BCL-XL were reported.104–106 BCL-XL is an antiapoptotic BCL-2 family protein and a clinically relevant target in the treatment of cancers. Navitoclax (ABT263), is an experimental BCL-XL inhibitor that shows promising in vivo activity, but has yet to receive FDA approval because of dose-limiting thrombocytopenia induced by on-mechanism degradation of BCL-XL in platelets.107 Fortunately, the expression of VHL, CRBN, and cIAP are low in platelets. Therefore, researchers at the University of Florida and MD Anderson Cancer Center developed BCL-XL targeting PROTACs recruiting these E3 ligases. All three bifunctional molecules potently degrade BCL-XL in cancer cells, but do not have an effect on BCL-XL in platelets. In addition, the degraders are selective for BCL-XL over BCL-2, which also binds ABT263. Moreover, the VHL based PROTAC, DT2216, had improved anti-tumor effects in vivo compared to ABT263 with significantly less platelet toxicity. Finally, the CRBN based PROTAC, PZ15227, was shown to enhance the senolytic activity of ABT263 in mice while sparring platelets.106 These studies cleverly took advantage of the low expression of these commonly recruited E3 ligases in platelets to develop potent PROTACs with enhanced toxicity profiles compared to the parent warhead.The studies described above are exciting, however the expression of VHL, CRBN, and cIAP are rather broad, necessitating the discovery of ligands for E3 ligases that have more specific expression profiles. These E3 ligases could be systematically identified by searching all perspective E3 ligases (found here103) in the Human Protein Atlas database.108 The Human Protein Atlas combines RNA expression profiling with immunostaining to provide detailed tissue expression data for a majority of the human genome. It is a great platform for triaging perspective tissue specific E3 ligases.
Another exciting possibility that may be realized within the next decade is the recruitment of E3 ligases with disease-specific expression (Fig. 5). PROTACs engaging these E3 ligases should have decreased cytotoxicity since the target protein would be spared in healthy cells. The Melanoma Antigen (MAGE) family of proteins are exciting candidates to induce tumor-specific degradation. MAGE proteins have been reviewed in depth elsewhere,109–111 but briefly: type I MAGEs are cancer testis antigens whose expression is normally restricted to male reproductive tissues but become re-expressed during tumorigenesis. In 2010, MAGEs were found to bind and activate RING domain containing proteins to form MAGE-RING E3 Ligases (MRLs),112 thus MAGE proteins can also act as substrate recognition domains for MRLs.113 Development of ligands for MAGEs could therefore be used to induce tumor-specific degradation of a target protein.
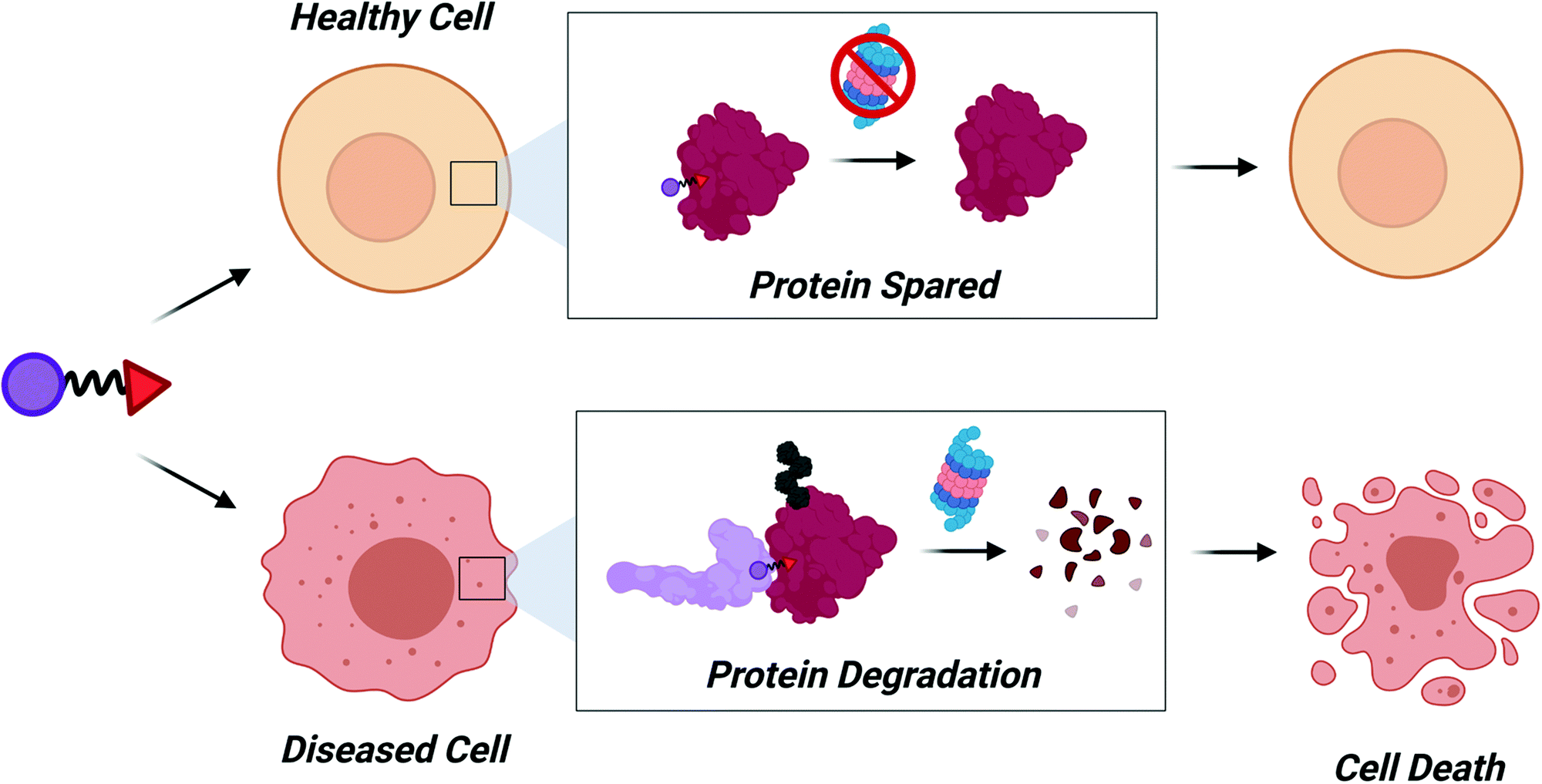 | ||
| Fig. 5 Cell specific degradation. Identification of E3REs for E3 ligases with disease- and tumor-specific expression will allow for increased control of protein degradation. Diseased cells will die as a result of degradation while healthy cells would be spared, decreasing risk of cytotoxicity. | ||
Recently, a ligand for MAGE-A11 was identified.114 MAGE-A11 binds the E3 ligase HUWE1 and PCF11, a member of an mRNA 3′ end processing complex.115 HUWE1 induces K63 polyubiqutination of PCF11 in a MAGE-A11 dependent manner, which inhibits the formation of the mRNA processing complex. This results in aberrant polyadenylation of complex substrates, like cyclin D2, leading to activation of oncogenes like Rb, which promotes tumorigenesis. Although the identified of a MAGE-A11 ligand may not be useful for TPD applications because MAGE-A11 promotes K63 ubiquitination rather than the K48 ubiquitination necessary for degradation, this study reveals a potential mechanism by which MAGE proteins interact with their substrates. MAGE-A11 recognizes a solvent exposed α-helix of PCF11 via its conserved MAGE homology domain (MHD). Although MHDs across the family can vary widely in sequence identity, the relative fold – a dual-winged helix – is conserved. Therefore, it is likely that the majority of MAGEs recognize solvent exposed α-helices of their substrates. With this knowledge, screening of peptides within solvent exposed α-helices of MAGE substrates could identify peptide “degrons” that could be used to screen libraries by proximity-based screening assays (e.g. TR-FRET) or fluorescence polarization. These studies would focus on identifying ligands for MAGEs that induce K48 polyubiquitination. Since such molecules will bind at the site of substrate recognition, they should be amenable to TPD applications and the development of tumor specific degraders.
Improving the efficiency of PROTAC identification
Despite the 20 years of advancement that have been outlined thus far, the identification of active PROTAC molecules continues to be accomplished mainly by an arduous, empirical fashion. Libraries of PROTAC molecules that vary (1) in attachment point to the E3RE or POI ligand, (2) linker length, and/or (3) linker composition are synthesized. Each compound is then screened in cells for activity, typically measured by the disappearance of protein via western blot. Based on these results, a new library of PROTACs is then made with minor changes to the structure (i.e. one or two atom differences in linker length) of the lead, parental PROTAC. This iterative process is repeated until an optimized PROTAC is developed that induces potent, nanomolar POI degradation. The time and reagent cost could be greatly reduced if we had a targeted approach to developing heterobifunctional degraders. Just as structure-guided design greatly accelerated small molecule discovery in the past three decades,116 a similar advance in the TPD field could greatly expedite identification of active degraders in the near future.Ternary complex structures
The importance of ternary complex formation in PROTAC function had been known for several years, as illustrated by our group's findings that even PROTACs that weakly bind (>1 μM) POIs can induce POI degradation provided the PROTAC forms strong ternary complexes with the POI and recruited E3 ligase.20 However, the full significance of this complex formation was not truly appreciated until the elucidation of the first PROTAC ternary complex crystal structure. In 2017, Gadd et al. solved the structure of MZ1 bound to VHL and BRD4BD2.117 Two striking features of this crystal structure were the many MZ1-induced de novo PPIs between VHL and BRD4BD2 as well as the collapse of MZ1's PEG linker. In parallel, Isothermal Titration Calorimetry (ITC) experiments demonstrated that MZ1-mediated BRD4BD2-MZ1-VHL ternary complex formation displays positive cooperativity.117 Biophysical and structural studies of BRD4BD2-MZ1-VHL complex suggest that such positive cooperativity is driven by the formation of the de novo PPIs. The formation of these interactions is facilitated by collapse of the PEG linker that enables VHL and BRD4BD2 to come into close proximity.In the past several years nearly 10 different ternary complex crystal structures have been solved.28,59,118,119 As witnessed in the BRD4BD2-MZ1-VHL ternary complex, unifying characteristics of these structures are de novo PPIs and the collapse of flexible linkers. Therefore, PROTACs have been optimized using these crystal structures by maximizing induced PPIs and developing rigid linkers that fix the PROTAC in a more active conformation. The first example of this was the transformation of MZ1 to AT1, which has improved BRD4 selectivity.117 Due to the collapse of the PEG linker, the VHL ligand and JQ1 of MZ1 were brought in close proximity upon binding BRD4. Therefore, the authors identified a vector for linker attachment that would enable the use of a shorter linker, which was hypothesized to better discriminate between BRD4, BRD2, and BRD3 due to decreased flexibility. The resulting molecule, AT1 retained reasonable binding to VHL and BRD4, but did not induce degradation of other BRD proteins like its parent PROTAC MZ1. Similarly, in the development ACBl1, the previously discussed SMARCA2/4 degrader, enhancing rigidity was used to improve potency.28 Thanks to the structural data for PROTAC1, the authors were able to arrive at a potent PROTAC with just two iterative design modifications, the most important being replacement of the flexible PEG linker with a more ridged 1,4 substituted phenyl ring. Finally, structure guided design enabled the synthesis of a PROTAC bearing a macrocyclic linker capable of potent, selective BRD4 degradation.59 This opens up the possibility of using macrocycles as recruiting elements as well as the use of macrocycle chemistry to improve PROTAC selectivity based on structural data. These studies, highlight that linker rigidity may be crucial to driving down potency in some systems and structural insights can help guide where that rigidity can be incorporated. As more rigid degraders are discovered the opportunity will also arise to biophysically compare binding kinetics of flexible and rigid PROTACs to discern how rigidity impacts association/dissociation of ternary complexes for a given E3 ligase/POI pair.
From these structure-guided design efforts it is tempting to believe that the more positively cooperative a PROTAC is, and the more rigid the linker, the better it will induce degradation. However, this does not appear to be the case. Numerous uncooperative or negatively cooperative PROTACs have been biophysically characterized and crystallized.118–120 Interestingly, even in the case where ternary complexes are not positively cooperative, novel PPIs are generated, as exemplified by the recent publication of cIAP-BTK PROTAC ternary complexes.119 Potent POI ligands and E3REs or inherent linker flexibility may enable these novel PPIs to form despite the negatively cooperative system. As with positively cooperative systems, negatively cooperative cIAP-BTK ternary complex PPIs drive specificity and flexible, adaptable linker conformations influence degradation profiles, which our lab has also observed for VHL-p38α PROTACs.23 From these studies, there is a clear path toward improving the efficiency of PROTAC identification: optimize novel PPIs between E3 ligases and POIs.
The ternary complex structures amassed thus far sew a unifying theme among different TPD technologies. Although the literature tries to paint PROTACs and molecular glues as distinct entities, these compounds function via a nearly identical mechanism: both modalities induce novel PPIs. Although molecular glues can be distinguished by the fact that they lack affinity for the POI in the absence of the E3 ligase and therefore require positivity cooperativity to induce degradation, these TPD approaches should be treated as variations on a theme rather than distinct classes of degrader molecules.
Mathematical modeling and computational PROTAC design
The insights gleaned from PROTAC ternary complexes do not only allow for structure-guided design of more potent compounds, but also reveal insight into the molecular bases of ternary complex formation. This has facilitated computational-based approaches to design programs that predict PROTAC-dependent ternary complex formation. Cooperativity, along with an intimate understanding of the equilibrium affinities that effect ternary complex formation, could provide a metric to characterize and predict PROTAC-mediated ternary complex assembly. Fortunately, mathematical expressions to characterize formation of ternary complexes have been solved, enabling prediction of both maximal ternary complex concentrations, and the kinetics of association and dissociation, on the basis of respective binary and ternary affinities.121 This suite will be a valuable tool to elucidate SAR trends during PROTAC development.The above model describes ternary complex formation at equilibrium. Although this will be useful for many systems, it may not accurately describe degradation kinetics for proteins with rapid turnover or in which deubiquitination occurs rapidly. Binding equilibria may not be reached as the pool of protein to be ubiquitinated is in constant flux. In an attempt to account for these more complex systems, a kinetic proofreading model for TPD molecules was recently developed.122 The system was developed using the commercially available MATLAB SimBiology software and used previously published BTK degradation data to develop the model. This program predicts how changes in permeability of a PROTAC will affect its DC50 and DMax. Selectivity can also be predicted, based on binding affinities of the PROTAC for the E3 ligase and POI, as well as the stability of the ternary complex (determined by the dissociation kinetics of ternary complex formation). The model can also be used to predict protein recovery after transient PROTAC treatment by comparing degradation kinetics to protein turnover. Along these same lines, this model can be used to predict PK and PD properties of PROTACs. Based on the kinetics of degradation, protein levels over different dosing regimens can be modeled to help identify which treatment schedule may be most beneficial. Therefore, this kinetic proofreading model will be invaluable as more degraders move into the clinic.
The computational models above will be beneficial to characterizing and ranking tested PROTAC molecules. However, to more fully expedite PROTAC discovery, programs are needed that will allow researchers to dock proposed degrader molecules and visualize the resultant ternary complex. These in silico modeling programs are already in development and have showed promising results. The two available models can be run using the Molecular Operating Environment (MOE) or Rosetta.123–125 Development of the MOE based software was a pioneering achievement of in silico docking of PROTAC ternary complexes. The program uses four different methods to predict ternary complex formation: (1) Sampling the entire ternary complex at once, (2) sampling PROTAC conformations independently, (3) one binary interaction is modeled, to which second protein's binding pose is modeled, and (4) PROTAC conformations are sampled independently of E3 ligase and POI, with E3-POI arrangements determined by independent protein–protein docking.123 This program was able to accurately predict active PROTACs based on previously published BRD degraders. Recent improvements to the MOE program allow for a fifth prediction method. Clustering of ternary complex ensembles has been found to increase the accuracy of identifying viable ternary complex structures.124 Additionally, two crystal structures of binary protein–ligand complexes can be used to predict optimal linker lengths to bridge the gap between the E3 and POI.
The new Rosetta program, PRosettaC, uses similar strategies to dock PROTACs into ternary complexes.125 Its protocol consists of sampling the distance between two anchor points on the E3RE or POI ligand. This can be based on already existing ternary complexes or one can use binary complex structures. Then, global protein–protein docking is used to predict the nascent E3 ligase–POI interactions. Local docking, followed by conformational sampling of PROTAC linkers, and finally clustering of complex poses, allows for the identification of plausible ternary complex structures. Excitingly, this program was able to accurately predict the formation of the BTK-CRBN E3 ligase complex, which had yet to be predicted by in silico docking.125 These data suggest that this program may be amenable to predicting ternary complexes of novel E3 ligase and POI ternary complexes. However, caution should be taken when interpreting data from either the MOE or PRosettaC programs, especially when considering new E3 ligase and POI ternary complexes. Crystallography artefacts, such as crystal packing interfaces, may unduly influence the PROTAC configurations within the top ensembles.
Discussion and future perspective
As we enter the third decade of TPD, PROTACs will continue to be at the cutting edge of targeted degradation research. Thanks to technological advances in proteomics, sequencing, and combinatorial chemistry, a bounty of reversible and covalent ligands will be identified. These compounds will comprise the next generation of PROTAC molecules. More “undruggable” proteins may become degradable and new E3 ligases with tissue specific and tumor specific expression patterns may be recruited. Our ability to rapidly identify active PROTAC molecules will continue to improve as we enhance our understanding of the structural underpinnings of their activity. These insights can then be incorporated into existing computational models to improve in silico prediction of PROTAC efficacy.Considering the strides in ternary complex structure determination and in silico PROTAC docking, one can imagine a robust workflow for expediting PROTAC identification (Fig. 6). First, binary protein–ligand co-crystal structures can be determined or can be accessed via the Protein Data Bank. If these co-crystal structures do not exist, programs like AlphaFold can be used to predict protein structure and compounds can be subsequently docked.126 Then, programs like PRosettaC, can be used to model possible linkers and predict novel PPIs. These predictions can also be combined with machine learning to investigate optimal linker lengths and compositions.127 Molecular Dynamics simulations will also play a role in ranking PROTAC structures computationally.23,117 Top hits from in silico modeling can then be synthesized and tested. Subsequently, the most active PROTACs can be selected for high-resolution structure determination in complex with POI and E3 by X-ray crystallography. As the resolution of cryo-electron microscopy continues to improve, it may too be an effective technique for determining the structure of ternary complexes.128 Finally, optimized PROTACs with improved potency and PK/PD can be synthesized and tested.
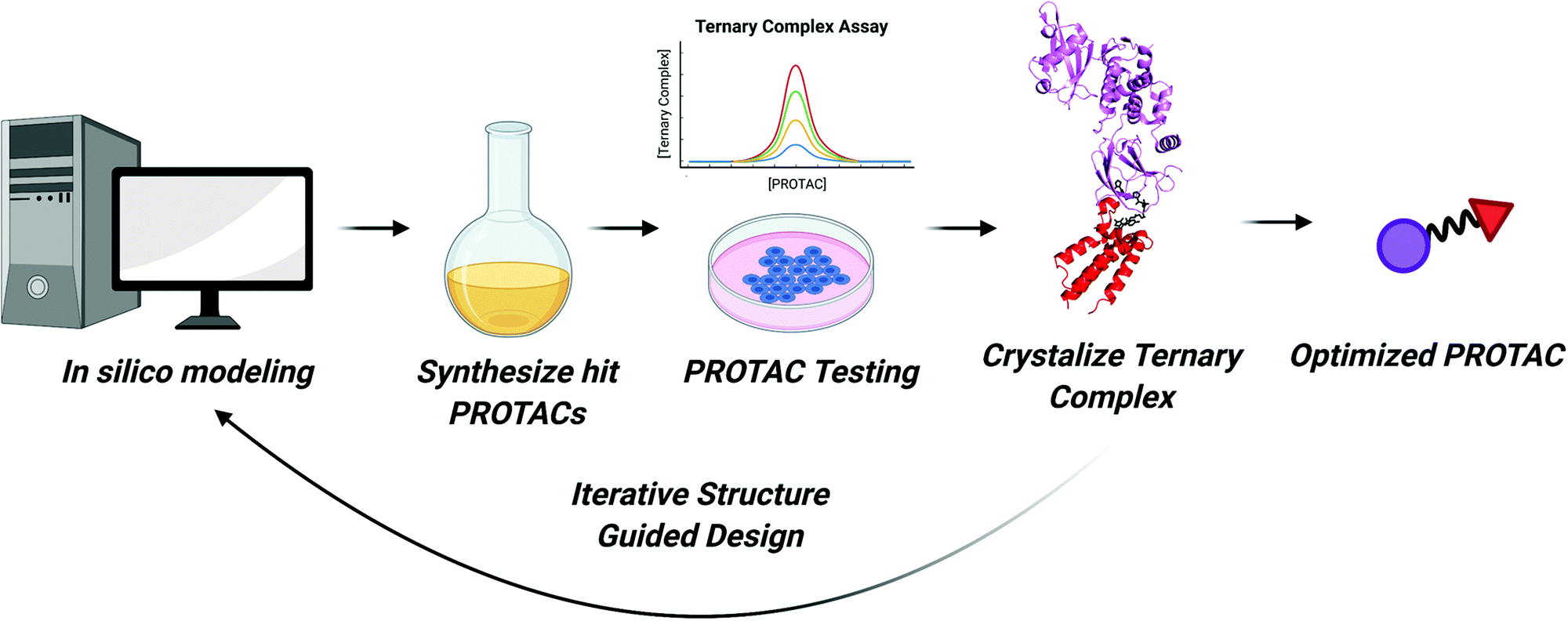 | ||
| Fig. 6 Flowchart for expedited PROTAC discovery. Improved in silico modeling of ternary complexes can predict which PROTACs will form suitable ternary complexes to induce ubiquitination. The top predicted PROTACs can then be synthesized. Next, molecules will be tested for engagement in in vitro ternary complex assays and for their ability to degrade the POI in cells. For cell active PROTACs, ternary complex structures can be solved enabling structure guided design of more potent PROTACs. This cycle can allow for iterative rounds of PROTAC design until an optimized PROTAC molecule is developed. | ||
There are even more exciting possibilities for PROTACs and the field of TPD beyond what has been covered in this review. The integration of PROTACs with other technologies has the potential to drastically change the landscape of TPD. For example, PROTACs could be used to enhance responses to immunotherapy. Antigens presented on major histocompatibility (MHC) receptors recognized by immune cells arise from the peptides of degraded proteins.129 Peptides from proteins degraded with PROTACs have been shown to be presented on MHC receptors.130 There is currently a major effort in the immunology field to identify neoantigens that are associated with cancers and other diseases.131 If the protein source of these neoantigens can be identified and ligands for them developed (or if they already exist), PROTACs could be used to induce their degradation and increase antigen presentation on MHC receptors. In combination with immune checkpoint inhibitors, these PROTACs could enhance the ability of the immune system to identify and eliminate cancers. Additionally, PROTACs against immune checkpoint proteins, like PD-L1, as well as PROTACs that modulate T cell receptor function have been developed.132 These compounds could themselves become useful small molecule immunotherapies that may have improved pharmacokinetic and toxicity profiles compared to antibody-based inhibitors.
Integration of TPD with other technologies may also have a dramatic impact on how PROTACs are administered to patients. Before tumor specific E3 ligases are identified and harnessed, PROTACs could be delivered to tumors using a recently published antibody drug conjugate approach.133 Alternatively, PROTACs can be embedded onto the surface of gold nanoparticles that can penetrate deep into tumors and other tissues.134 Finally, pro-drug PROTACs could be beneficial by improving clinical delivery and metabolic stability of PROTACs.135
The TPD field will continue to evolve at a rapid pace in the next decade. As a result, new, imaginative, recruiting elements will expand the number of cellular components that can be manipulated with TPD. This is highlighted by recent reports of RNA-PROTACs that use small molecule RNA mimics to bind and induce degradation of RNA binding proteins.136 RNA itself can also now be degraded using newly developed heterobifunctional molecules, Ribonuclease targeting chimeras (RIBOTACs), which have timely implications for degrading viral SARS-CoV-2 RNA.137,138 Additionally, DNA can now be used as a recruiting element to degrade “undruggable” transcription factors using our lab's newly developed TRAFTAC technology.42 We are also learning about novel modes of TPD, as seen with the recent publications of a covalent natural product molecular glue and a small molecule that induces BCL6 polymerization and degradation.139,140 It is possible that these mechanisms may also become prominent areas of TPD research. However, it is likely that PROTACs, and the TPD field they inspired, will continue pushing the boundaries of drug discovery for the foreseeable future.
Conflicts of interest
C. M. C. is founder, shareholder, and consultant to Arvinas, Inc., which supports research in his laboratory.Acknowledgements
We would like to thank Dr John Hines and Dr Brian Linhares for their careful reading and editing of this manuscript. C. M. C. is funded by the NIH (R35CA197589) and is supported by an American Cancer Research Professorship. M. J. B. acknowledges support from the NIH (F31CA232477 and 5T32GM067543).References
- K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews and R. J. Deshaies, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8554–8559 CrossRef CAS PubMed.
- G. M. Burslem and C. M. Crews, Cell, 2020, 181, 102–114 CrossRef CAS PubMed.
- M. Pettersson and C. M. Crews, Drug Discovery Today Technol., 2019, 31, 15–27 CrossRef PubMed.
- L. M. Luh, U. Scheib, K. Juenemann, L. Wortmann, M. Brands and P. M. Cromm, Angew. Chem., Int. Ed., 2020, 59(36), 15448–15466 CrossRef CAS PubMed.
- D. A. Nalawansha and C. M. Crews, Cell Chem. Biol., 2020, 27, 998–1014 CrossRef CAS PubMed.
- A. Mullard, Nat. Rev. Drug Discovery, 2019, 18, 895 Search PubMed.
- J. Cleveland, Cancer Discovery, 2020, 10(8), 1084 Search PubMed.
- S. M. Banik, K. Pedram, S. Wisnovsky, G. Ahn, N. M. Riley and C. R. Bertozzi, Nature, 2020, 584, 291–297 CrossRef CAS PubMed.
- D. Takahashi, J. Moriyama, T. Nakamura, E. Miki, E. Takahashi, A. Sato, T. Akaike, K. Itto-Nakama and H. Arimoto, Mol. Cell, 2019, 76, 797–810 CrossRef CAS PubMed.
- Z. Li, C. Wang, Z. Wang, C. Zhu, J. Li, T. Sha, L. Ma, C. Gao, Y. Yang, Y. Sun, J. Wang, X. Sun, C. Lu, M. Difiglia, Y. Mei, C. Ding, S. Luo, Y. Dang, Y. Ding, Y. Fei and B. Lu, Nature, 2019, 575, 203–209 CrossRef CAS PubMed.
- Y. Ding, Y. Fei and B. Lu, Trends Pharmacol. Sci., 2020, 41, 464–474 CrossRef CAS PubMed.
- D. Takahashi and H. Arimoto, Autophagy, 2020, 16, 765–766 CrossRef CAS PubMed.
- D. Nandi, P. Tahiliani, A. Kumar and D. Chandu, J. Biosci., 2006, 31, 137–155 CrossRef CAS PubMed.
- I. Livneh, V. Cohen-Kaplan, C. Cohen-Rosenzweig, N. Avni and A. Ciechanover, Cell Res., 2016, 26, 869–885 CrossRef CAS PubMed.
- G. Kleiger and T. Mayor, Trends Cell Biol., 2014, 24, 352–359 CrossRef CAS PubMed.
- S. L. Paiva and C. M. Crews, Curr. Opin. Chem. Biol., 2019, 50, 111–119 CrossRef CAS PubMed.
- J. Salami and C. M. Crews, Science, 2017, 355, 1163–1167 CrossRef CAS PubMed.
- A. C. Lai and C. M. Crews, Nat. Rev. Drug Discovery, 2017, 16, 101–114 CrossRef CAS PubMed.
- D. P. Bondeson, A. Mares, I. E. Smith, E. Ko, S. Campos, A. H. Miah, K. E. Mulholland, N. Routly, D. L. Buckley, J. L. Gustafson, N. Zinn, P. Grandi, S. Shimamura, G. Bergamini, M. Faelth-Savitski, M. Bantscheff, C. Cox, D. A. Gordon, R. R. Willard, J. J. Flanagan, L. N. Casillas, B. J. Votta, W. den Besten, K. Famm, L. Kruidenier, P. S. Carter, J. D. Harling, I. Churcher and C. M. Crews, Nat. Chem. Biol., 2015, 11, 611–617 CrossRef CAS PubMed.
- D. P. Bondeson, B. E. Smith, G. M. Burslem, A. D. Buhimschi, J. Hines, S. Jaime-Figueroa, J. Wang, B. D. Hamman, A. Ishchenko and C. M. Crews, Cell Chem. Biol., 2018, 25, 78–87 CrossRef CAS PubMed.
- M. Toure and C. M. Crews, Angew. Chem., Int. Ed., 2016, 55, 1966–1973 CrossRef CAS PubMed.
- G. M. Burslem, B. E. Smith, A. C. Lai, S. Jaime-Figueroa, D. C. McQuaid, D. P. Bondeson, M. Toure, H. Dong, Y. Qian, J. Wang, A. P. Crew, J. Hines and C. M. Crews, Cell Chem. Biol., 2018, 25, 67–77 CrossRef CAS PubMed.
- B. E. Smith, S. L. Wang, S. Jaime-Figueroa, A. Harbin, J. Wang, B. D. Hamman and C. M. Crews, Nat. Commun., 2019, 10, 131 CrossRef PubMed.
- S. Surade and T. L. Blundell, Chem. Biol., 2012, 19, 42–50 CrossRef CAS PubMed.
- J. Hines, J. D. Gough, T. W. Corson and C. M. Crews, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8942–8947 CrossRef CAS PubMed.
- S. H. Ong, G. R. Guy, Y. R. Hadari, S. Laks, N. Gotoh, J. Schlessinger and I. Lax, Mol. Cell. Biol., 2000, 20, 979–989 CrossRef CAS PubMed.
- M. R. Arkin, Y. Tang and J. A. Wells, Chem. Biol., 2014, 21, 1102–1114 CrossRef CAS PubMed.
- W. Farnaby, M. Koegl, M. J. Roy, C. Whitworth, E. Diers, N. Trainor, D. Zollman, S. Steurer, J. Karolyi-Oezguer, C. Riedmueller, T. Gmaschitz, J. Wachter, C. Dank, M. Galant, B. Sharps, K. Rumpel, E. Traxler, T. Gerstberger, R. Schnitzer, O. Petermann, P. Greb, H. Weinstabl, G. Bader, A. Zoephel, A. Weiss-Puxbaum, K. Ehrenhofer-Wolfer, S. Wohrle, G. Boehmelt, J. Rinnenthal, H. Arnhof, N. Wiechens, M. Y. Wu, T. Owen-Hughes, P. Ettmayer, M. Pearson, D. B. McConnell and A. Ciulli, Nat. Chem. Biol., 2019, 15, 672–680 CrossRef CAS PubMed.
- C. Hodges, J. G. Kirkland and G. R. Crabtree, Cold Spring Harbor Perspect. Med., 2016, 6(8), 1–24 Search PubMed.
- R. St Pierre and C. Kadoch, Curr. Opin. Genet. Dev., 2017, 42, 56–67 CrossRef PubMed.
- T. Oike, H. Ogiwara, Y. Tominaga, K. Ito, O. Ando, K. Tsuta, T. Mizukami, Y. Shimada, H. Isomura, M. Komachi, K. Furuta, S. Watanabe, T. Nakano, J. Yokota and T. Kohno, Cancer Res., 2013, 73, 5508–5518 CrossRef CAS PubMed.
- C. L. Sutherell, C. Tallant, O. P. Monteiro, C. Yapp, J. E. Fuchs, O. Fedorov, P. Siejka, S. Muller, S. Knapp, J. D. Brenton, P. E. Brennan and S. V. Ley, J. Med. Chem., 2016, 59, 5095–5101 CrossRef CAS PubMed.
- B. S. Gerstenberger, J. D. Trzupek, C. Tallant, O. Fedorov, P. Filippakopoulos, P. E. Brennan, V. Fedele, S. Martin, S. Picaud, C. Rogers, M. Parikh, A. Taylor, B. Samas, A. O'Mahony, E. Berg, G. Pallares, A. D. Torrey, D. K. Treiber, I. J. Samardjiev, B. T. Nasipak, T. Padilla-Benavides, Q. Wu, A. N. Imbalzano, J. A. Nickerson, M. E. Bunnage, S. Muller, S. Knapp and D. R. Owen, J. Med. Chem., 2016, 59, 4800–4811 CrossRef CAS PubMed.
- B. Vangamudi, T. A. Paul, P. K. Shah, M. Kost-Alimova, L. Nottebaum, X. Shi, Y. Zhan, E. Leo, H. S. Mahadeshwar, A. Protopopov, A. Futreal, T. N. Tieu, M. Peoples, T. P. Heffernan, J. R. Marszalek, C. Toniatti, A. Petrocchi, D. Verhelle, D. R. Owen, G. Draetta, P. Jones, W. S. Palmer, S. Sharma and J. N. Andersen, Cancer Res., 2015, 75, 3865–3878 CrossRef CAS PubMed.
- J. H. Bushweller, Nat. Rev. Cancer, 2019, 19, 611–624 CrossRef CAS PubMed.
- N. J. Yang and M. J. Hinner, Methods Mol. Biol., 2015, 1266, 29–53 CrossRef CAS PubMed.
- L. Bai, H. Zhou, R. Xu, Y. Zhao, K. Chinnaswamy, D. McEachern, J. Chen, C. Y. Yang, Z. Liu, M. Wang, L. Liu, H. Jiang, B. Wen, P. Kumar, J. L. Meagher, D. Sun, J. A. Stuckey and S. Wang, Cancer Cell, 2019, 36, 498–511 CrossRef CAS PubMed.
- F. Zhu, K. B. Wang and L. Rui, Cancers, 2019, 12 Search PubMed.
- H. Yu and R. Jove, Nat. Rev. Cancer, 2004, 4, 97–105 CrossRef CAS PubMed.
- J. Yang and G. R. Stark, Cell Res., 2008, 18, 443–451 CrossRef CAS PubMed.
- J. Chen, L. Bai, D. Bernard, Z. Nikolovska-Coleska, C. Gomez, J. Zhang, H. Yi and S. Wang, ACS Med. Chem. Lett., 2010, 1, 85–89 CrossRef CAS PubMed.
- K. T. G. Samarasinghe, S. Jaime-Figueroa, K. Dai, Z. Hu and C. M. Crews, bioRxiv, 2020 DOI:10.1101/2020.10.12.336529.
- I. A. Prior, P. D. Lewis and C. Mattos, Cancer Res., 2012, 72, 2457–2467 CrossRef CAS PubMed.
- J. M. Ostrem, U. Peters, M. L. Sos, J. A. Wells and K. M. Shokat, Nature, 2013, 503, 548–551 CrossRef CAS PubMed.
- J. Rudolph and D. Stokoe, Angew. Chem., Int. Ed., 2014, 53, 3777–3779 CrossRef CAS PubMed.
- Nat. Biotechnol., 2019, 37, 1247 Search PubMed.
- M. R. Janes, J. Zhang, L. S. Li, R. Hansen, U. Peters, X. Guo, Y. Chen, A. Babbar, S. J. Firdaus, L. Darjania, J. Feng, J. H. Chen, S. Li, S. Li, Y. O. Long, C. Thach, Y. Liu, A. Zarieh, T. Ely, J. M. Kucharski, L. V. Kessler, T. Wu, K. Yu, Y. Wang, Y. Yao, X. Deng, P. P. Zarrinkar, D. Brehmer, D. Dhanak, M. V. Lorenzi, D. Hu-Lowe, M. P. Patricelli, P. Ren and Y. Liu, Cell, 2018, 172, 578–589 CrossRef CAS PubMed.
- J. Canon, K. Rex, A. Y. Saiki, C. Mohr, K. Cooke, D. Bagal, K. Gaida, T. Holt, C. G. Knutson, N. Koppada, B. A. Lanman, J. Werner, A. S. Rapaport, T. San Miguel, R. Ortiz, T. Osgood, J. R. Sun, X. Zhu, J. D. McCarter, L. P. Volak, B. E. Houk, M. G. Fakih, B. H. O’Neil, T. J. Price, G. S. Falchook, J. Desai, J. Kuo, R. Govindan, D. S. Hong, W. Ouyang, H. Henary, T. Arvedson, V. J. Cee and J. R. Lipford, Nature, 2019, 575, 217–223 CrossRef CAS PubMed.
- J. B. Fell, J. P. Fischer, B. R. Baer, J. F. Blake, K. Bouhana, D. M. Briere, K. D. Brown, L. E. Burgess, A. C. Burns, M. R. Burkard, H. Chiang, M. J. Chicarelli, A. W. Cook, J. J. Gaudino, J. Hallin, L. Hanson, D. P. Hartley, E. J. Hicken, G. P. Hingorani, R. J. Hinklin, M. J. Mejia, P. Olson, J. N. Otten, S. P. Rhodes, M. E. Rodriguez, P. Savechenkov, D. J. Smith, N. Sudhakar, F. X. Sullivan, T. P. Tang, G. P. Vigers, L. Wollenberg, J. G. Christensen and M. A. Marx, J. Med. Chem., 2020, 63, 6679–6693 CrossRef CAS PubMed.
- M. B. Ryan, F. Fece de la Cruz, S. Phat, D. T. Myers, E. Wong, H. A. Shahzade, C. B. Hong and R. B. Corcoran, Clin. Cancer Res., 2020, 26, 1633–1643 CrossRef CAS PubMed.
- J. Y. Xue, Y. Zhao, J. Aronowitz, T. T. Mai, A. Vides, B. Qeriqi, D. Kim, C. Li, E. de Stanchina, L. Mazutis, D. Risso and P. Lito, Nature, 2020, 577, 421–425 CrossRef CAS PubMed.
- N. Bery, A. Miller and T. Rabbitts, Nat. Commun., 2020, 11, 3233 CrossRef CAS PubMed.
- L. M. Simpson, T. J. Macartney, A. Nardin, L. J. Fulcher, S. Roth, A. Testa, C. Maniaci, A. Ciulli, I. G. Ganley and G. P. Sapkota, Cell Chem. Biol., 2020, 27, 1164–1180 CrossRef PubMed.
- S. Roth, T. J. Macartney, A. Konopacka, K. H. Chan, H. Zhou, M. A. Queisser and G. P. Sapkota, Cell Chem. Biol., 2020, 27, 1151–1163 CrossRef PubMed.
- M. Zeng, Y. Xiong, N. Safaee, R. P. Nowak, K. A. Donovan, C. J. Yuan, B. Nabet, T. W. Gero, F. Feru, L. Li, S. Gondi, L. J. Ombelets, C. Quan, P. A. Janne, M. Kostic, D. A. Scott, K. D. Westover, E. S. Fischer and N. S. Gray, Cell Chem. Biol., 2020, 27, 19–31 CrossRef CAS PubMed.
- J. Hallin, L. D. Engstrom, L. Hargis, A. Calinisan, R. Aranda, D. M. Briere, N. Sudhakar, V. Bowcut, B. R. Baer, J. A. Ballard, M. R. Burkard, J. B. Fell, J. P. Fischer, G. P. Vigers, Y. Xue, S. Gatto, J. Fernandez-Banet, A. Pavlicek, K. Velastagui, R. C. Chao, J. Barton, M. Pierobon, E. Baldelli, E. F. Patricoin, 3rd, D. P. Cassidy, M. A. Marx, I. I. Rybkin, M. L. Johnson, S. I. Ou, P. Lito, K. P. Papadopoulos, P. A. Janne, P. Olson and J. G. Christensen, Cancer Discovery, 2020, 10, 54–71 CrossRef CAS PubMed.
- M. J. Bond, L. Chu, D. A. Nalawansha, K. Li and C. M. Crews, ACS Cent. Sci., 2020, 6, 1367–1375 CrossRef CAS PubMed.
- Z. Zhang, R. Gao, Q. Hu, H. Peacock, D. M. Peacock, S. Dai, K. M. Shokat and H. Suga, ACS Cent. Sci., 2020, 6, 1753–1761 CrossRef CAS PubMed.
- A. Testa, S. J. Hughes, X. Lucas, J. E. Wright and A. Ciulli, Angew. Chem., Int. Ed., 2020, 59, 1727–1734 CrossRef CAS PubMed.
- C. Steinebach, Y. L. D. Ng, I. Sosic, C. S. Lee, S. Chen, S. Lindner, L. P. Vu, A. Bricelj, R. Haschemi, M. Monschke, E. Steinwarz, K. G. Wagner, G. Bendas, J. Luo, M. Gutschow and J. Kronke, Chem. Sci., 2020, 11, 3474–3486 RSC.
- A. C. Lai, M. Toure, D. Hellerschmied, J. Salami, S. Jaime-Figueroa, E. Ko, J. Hines and C. M. Crews, Angew. Chem., Int. Ed., 2016, 55, 807–810 CrossRef CAS PubMed.
- A. J. Carter, O. Kraemer, M. Zwick, A. Mueller-Fahrnow, C. H. Arrowsmith and A. M. Edwards, Drug Discovery Today, 2019, 24, 2111–2115 CrossRef CAS PubMed.
- A. M. Roberts, C. C. Ward and D. K. Nomura, Curr. Opin. Biotechnol., 2017, 43, 25–33 CrossRef CAS PubMed.
- T. Kodadek, N. G. Paciaroni, M. Balzarini and P. Dickson, Chem. Commun., 2019, 55, 13330–13341 RSC.
- B. F. Cravatt, A. T. Wright and J. W. Kozarich, Annu. Rev. Biochem., 2008, 77, 383–414 CrossRef CAS PubMed.
- L. Boike, A. G. Cioffi, F. C. Majewski, J. Co, N. J. Henning, M. D. Jones, G. Liu, J. M. McKenna, J. A. Tallarico, M. Schirle and D. K. Nomura, Cell Chem. Biol., 2021, 28(1), 4–13 CrossRef CAS PubMed.
- M. Gabay, Y. Li and D. W. Felsher, Cold Spring Harbor Perspect. Med., 2014, 4(6), 1–13 Search PubMed.
- G. Veggiani, M. C. R. Gerpe, S. S. Sidhu and W. Zhang, Pharmacol. Ther., 2019, 199, 139–154 CrossRef CAS PubMed.
- D. Madsen, C. Azevedo, I. Micco, L. K. Petersen and N. J. V. Hansen, Prog. Med. Chem., 2020, 59, 181–249 Search PubMed.
- P. Ottis, M. Toure, P. M. Cromm, E. Ko, J. L. Gustafson and C. M. Crews, ACS Chem. Biol., 2017, 12, 2570–2578 CrossRef CAS PubMed.
- D. K. Nomura and T. J. Maimone, Curr. Top. Microbiol. Immunol., 2019, 420, 351–374 CAS.
- T. Ito, H. Ando, T. Suzuki, T. Ogura, K. Hotta, Y. Imamura, Y. Yamaguchi and H. Handa, Science, 2010, 327, 1345–1350 CrossRef CAS PubMed.
- M. E. Matyskiela, S. Couto, X. Zheng, G. Lu, J. Hui, K. Stamp, C. Drew, Y. Ren, M. Wang, A. Carpenter, C. W. Lee, T. Clayton, W. Fang, C. C. Lu, M. Riley, P. Abdubek, K. Blease, J. Hartke, G. Kumar, R. Vessey, M. Rolfe, L. G. Hamann and P. P. Chamberlain, Nat. Chem. Biol., 2018, 14, 981–987 CrossRef CAS PubMed.
- G. Lu, R. E. Middleton, H. Sun, M. Naniong, C. J. Ott, C. S. Mitsiades, K. K. Wong, J. E. Bradner and W. G. Kaelin, Jr., Science, 2014, 343, 305–309 CrossRef CAS PubMed.
- Y. Che, A. M. Gilbert, V. Shanmugasundaram and M. C. Noe, Bioorg. Med. Chem. Lett., 2018, 28, 2585–2592 CrossRef CAS PubMed.
- D. L. Buckley, J. L. Gustafson, I. Van Molle, A. G. Roth, H. S. Tae, P. C. Gareiss, W. L. Jorgensen, A. Ciulli and C. M. Crews, Angew. Chem., Int. Ed., 2012, 51, 11463–11467 CrossRef CAS PubMed.
- D. L. Buckley, I. Van Molle, P. C. Gareiss, H. S. Tae, J. Michel, D. J. Noblin, W. L. Jorgensen, A. Ciulli and C. M. Crews, J. Am. Chem. Soc., 2012, 134, 4465–4468 CrossRef CAS PubMed.
- J. Frost, C. Galdeano, P. Soares, M. S. Gadd, K. M. Grzes, L. Ellis, O. Epemolu, S. Shimamura, M. Bantscheff, P. Grandi, K. D. Read, D. A. Cantrell, S. Rocha and A. Ciulli, Nat. Commun., 2016, 7, 13312 CrossRef CAS PubMed.
- P. Soares, M. S. Gadd, J. Frost, C. Galdeano, L. Ellis, O. Epemolu, S. Rocha, K. D. Read and A. Ciulli, J. Med. Chem., 2018, 61, 599–618 CrossRef CAS PubMed.
- D. L. Buckley, K. Raina, N. Darricarrere, J. Hines, J. L. Gustafson, I. E. Smith, A. H. Miah, J. D. Harling and C. M. Crews, ACS Chem. Biol., 2015, 10, 1831–1837 CrossRef CAS PubMed.
- X. Han, L. Zhao, W. Xiang, C. Qin, B. Miao, T. Xu, M. Wang, C. Y. Yang, K. Chinnaswamy, J. Stuckey and S. Wang, J. Med. Chem., 2019, 62, 11218–11231 CrossRef CAS PubMed.
- A. R. Schneekloth, M. Pucheault, H. S. Tae and C. M. Crews, Bioorg. Med. Chem. Lett., 2008, 18, 5904–5908 CrossRef CAS PubMed.
- Y. Fang, G. Liao and B. Yu, Acta Pharm. Sin. B, 2020, 10, 1253–1278 CrossRef CAS PubMed.
- Y. Itoh, M. Ishikawa, M. Naito and Y. Hashimoto, J. Am. Chem. Soc., 2010, 132, 5820–5826 CrossRef CAS PubMed.
- M. Naito, N. Ohoka and N. Shibata, Drug Discovery Today Technol., 2019, 31, 35–42 CrossRef PubMed.
- J. Hines, S. Lartigue, H. Dong, Y. Qian and C. M. Crews, Cancer Res., 2019, 79, 251–262 CrossRef CAS PubMed.
- A. C. Qin, H. Jin, Y. Song, Y. Gao, Y. F. Chen, L. N. Zhou, S. S. Wang and X. S. Lu, Cell Death Dis., 2020, 11, 805 CrossRef CAS PubMed.
- I. A. Murray, A. D. Patterson and G. H. Perdew, Nat. Rev. Cancer, 2014, 14, 801–814 CrossRef CAS PubMed.
- F. Ohtake, A. Baba, I. Takada, M. Okada, K. Iwasaki, H. Miki, S. Takahashi, A. Kouzmenko, K. Nohara, T. Chiba, Y. Fujii-Kuriyama and S. Kato, Nature, 2007, 446, 562–566 CrossRef CAS PubMed.
- F. Ohtake, Y. Fujii-Kuriyama, K. Kawajiri and S. Kato, J. Steroid Biochem. Mol. Biol., 2011, 127, 102–107 CrossRef CAS PubMed.
- N. Ohoka, G. Tsuji, T. Shoda, T. Fujisato, M. Kurihara, Y. Demizu and M. Naito, ACS Chem. Biol., 2019, 14, 2822–2832 CrossRef CAS PubMed.
- X. Zhang, V. M. Crowley, T. G. Wucherpfennig, M. M. Dix and B. F. Cravatt, Nat. Chem. Biol., 2019, 15, 737–746 CrossRef CAS PubMed.
- J. N. Spradlin, X. Hu, C. C. Ward, S. M. Brittain, M. D. Jones, L. Ou, M. To, A. Proudfoot, E. Ornelas, M. Woldegiorgis, J. A. Olzmann, D. E. Bussiere, J. R. Thomas, J. A. Tallarico, J. M. McKenna, M. Schirle, T. J. Maimone and D. K. Nomura, Nat. Chem. Biol., 2019, 15, 747–755 CrossRef CAS PubMed.
- M. S. Rodriguez, I. Egana, F. Lopitz-Otsoa, F. Aillet, M. P. Lopez-Mato, A. Dorronsoro, S. Lobato-Gil, J. D. Sutherland, R. Barrio, C. Trigueros and V. Lang, Cell Death Dis., 2014, 5, e1399 CrossRef CAS PubMed.
- B. Tong, J. N. Spradlin, L. F. T. Novaes, E. Zhang, X. Hu, M. Moeller, S. M. Brittain, L. M. McGregor, J. M. McKenna, J. A. Tallarico, M. Schirle, T. J. Maimone and D. K. Nomura, ACS Chem. Biol., 2020, 15, 1788–1794 CrossRef CAS PubMed.
- M. Luo, J. N. Spradlin, S. M. Brittain, J. M. McKenna, J. A. Tallarico, M. Schirle, T. J. Maimone and D. K. Nomura, bioRxiv, 2020 DOI:10.1101/2020.07.12.198150.
- R. Vyas, R. Kumar, F. Clermont, A. Helfricht, P. Kalev, P. Sotiropoulou, I. A. Hendriks, E. Radaelli, T. Hochepied, C. Blanpain, A. Sablina, H. van Attikum, J. V. Olsen, A. G. Jochemsen, A. C. Vertegaal and J. C. Marine, Cell Death Differ., 2013, 20, 490–502 CrossRef CAS PubMed.
- C. C. Ward, J. I. Kleinman, S. M. Brittain, P. S. Lee, C. Y. S. Chung, K. Kim, Y. Petri, J. R. Thomas, J. A. Tallarico, J. M. McKenna, M. Schirle and D. K. Nomura, ACS Chem. Biol., 2019, 14, 2430–2440 CrossRef CAS PubMed.
- R. Gabizon, A. Shraga, P. Gehrtz, E. Livnah, Y. Shorer, N. Gurwicz, L. Avram, T. Unger, H. Aharoni, S. Albeck, A. Brandis, Z. Shulman, B. Z. Katz, Y. Herishanu and N. London, J. Am. Chem. Soc., 2020, 142, 11734–11742 CrossRef CAS PubMed.
- W. H. Guo, X. Qi, X. Yu, Y. Liu, C. I. Chung, F. Bai, X. Lin, D. Lu, L. Wang, J. Chen, L. H. Su, K. J. Nomie, F. Li, M. C. Wang, X. Shu, J. N. Onuchic, J. A. Woyach, M. L. Wang and J. Wang, Nat. Commun., 2020, 11, 4268 CrossRef PubMed.
- B. Tong, M. Luo, Y. Xie, J. N. Spradlin, J. A. Tallarico, J. M. McKenna, M. Schirle, T. J. Maimone and D. K. Nomura, Sci. Rep., 2020, 10, 15543 CrossRef CAS PubMed.
- K. Taguchi and M. Yamamoto, Front. Oncol., 2017, 7, 85 CrossRef PubMed.
- B. Medvar, V. Raghuram, T. Pisitkun, A. Sarkar and M. A. Knepper, Physiol. Genomics, 2016, 48, 502–512 CrossRef CAS PubMed.
- S. Khan, X. Zhang, D. Lv, Q. Zhang, Y. He, P. Zhang, X. Liu, D. Thummuri, Y. Yuan, J. S. Wiegand, J. Pei, W. Zhang, A. Sharma, C. R. McCurdy, V. M. Kuruvilla, N. Baran, A. A. Ferrando, Y. M. Kim, A. Rogojina, P. J. Houghton, G. Huang, R. Hromas, M. Konopleva, G. Zheng and D. Zhou, Nat. Med., 2019, 25, 1938–1947 CrossRef CAS PubMed.
- X. Zhang, Y. He, P. Zhang, V. Budamagunta, D. Lv, D. Thummuri, Y. Yang, J. Pei, Y. Yuan, D. Zhou and G. Zheng, Eur. J. Med. Chem., 2020, 199, 112397 CrossRef CAS PubMed.
- Y. He, X. Zhang, J. Chang, H. N. Kim, P. Zhang, Y. Wang, S. Khan, X. Liu, X. Zhang, D. Lv, L. Song, W. Li, D. Thummuri, Y. Yuan, J. S. Wiegand, Y. T. Ortiz, V. Budamagunta, J. H. Elisseeff, J. Campisi, M. Almeida, G. Zheng and D. Zhou, Nat. Commun., 2020, 11, 1996 CrossRef CAS PubMed.
- W. H. Wilson, O. A. O'Connor, M. S. Czuczman, A. S. LaCasce, J. F. Gerecitano, J. P. Leonard, A. Tulpule, K. Dunleavy, H. Xiong, Y. L. Chiu, Y. Cui, T. Busman, S. W. Elmore, S. H. Rosenberg, A. P. Krivoshik, S. H. Enschede and R. A. Humerickhouse, Lancet Oncol., 2010, 11, 1149–1159 CrossRef CAS PubMed.
- P. J. Thul and C. Lindskog, Protein Sci., 2018, 27, 233–244 CrossRef CAS PubMed.
- J. L. Weon and P. R. Potts, Curr. Opin. Cell Biol., 2015, 37, 1–8 CrossRef CAS PubMed.
- A. K. Lee and P. R. Potts, J. Mol. Biol., 2017, 429, 1114–1142 CrossRef CAS PubMed.
- R. R. Florke Gee, H. Chen, A. K. Lee, C. A. Daly, B. A. Wilander, K. Fon Tacer and P. R. Potts, J. Biol. Chem., 2020, 295, 16121–16155 CrossRef CAS PubMed.
- J. M. Doyle, J. Gao, J. Wang, M. Yang and P. R. Potts, Mol. Cell, 2010, 39, 963–974 CrossRef CAS PubMed.
- C. T. Pineda, S. Ramanathan, K. Fon Tacer, J. L. Weon, M. B. Potts, Y. H. Ou, M. A. White and P. R. Potts, Cell, 2015, 160, 715–728 CrossRef CAS PubMed.
- S. W. Yang, X. Huang, W. Lin, J. Min, D. J. Miller, A. Mayasundari, P. Rodrigues, E. C. Griffith, C. T. Gee, L. Li, W. Li, R. E. Lee, Z. Rankovic, T. Chen and P. R. Potts, Nat. Commun., 2020, 11, 4931 CrossRef CAS PubMed.
- S. W. Yang, L. Li, J. P. Connelly, S. N. Porter, K. Kodali, H. Gan, J. M. Park, K. F. Tacer, H. Tillman, J. Peng, S. M. Pruett-Miller, W. Li and P. R. Potts, Mol. Cell, 2020, 77, 1206–1221 CrossRef CAS PubMed.
- R. L. M. van Montfort and P. Workman, Essays Biochem., 2017, 61, 431–437 CrossRef PubMed.
- M. S. Gadd, A. Testa, X. Lucas, K. H. Chan, W. Chen, D. J. Lamont, M. Zengerle and A. Ciulli, Nat. Chem. Biol., 2017, 13, 514–521 CrossRef CAS PubMed.
- R. P. Nowak, S. L. DeAngelo, D. Buckley, Z. He, K. A. Donovan, J. An, N. Safaee, M. P. Jedrychowski, C. M. Ponthier, M. Ishoey, T. Zhang, J. D. Mancias, N. S. Gray, J. E. Bradner and E. S. Fischer, Nat. Chem. Biol., 2018, 14, 706–714 CrossRef CAS PubMed.
- J. Schiemer, R. Horst, Y. Meng, J. I. Montgomery, Y. Xu, X. Feng, K. Borzilleri, D. P. Uccello, C. Leverett, S. Brown, Y. Che, M. F. Brown, M. M. Hayward, A. M. Gilbert, M. C. Noe and M. F. Calabrese, Nat. Chem. Biol., 2021, 17, 152–160 CrossRef CAS PubMed.
- A. Zorba, C. Nguyen, Y. Xu, J. Starr, K. Borzilleri, J. Smith, H. Zhu, K. A. Farley, W. Ding, J. Schiemer, X. Feng, J. S. Chang, D. P. Uccello, J. A. Young, C. N. Garcia-Irrizary, L. Czabaniuk, B. Schuff, R. Oliver, J. Montgomery, M. M. Hayward, J. Coe, J. Chen, M. Niosi, S. Luthra, J. C. Shah, A. El-Kattan, X. Qiu, G. M. West, M. C. Noe, V. Shanmugasundaram, A. M. Gilbert, M. F. Brown and M. F. Calabrese, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E7285–E7292 CrossRef PubMed.
- B. Han, J. Biol. Chem., 2020, 295, 15280–15291 CrossRef PubMed.
- D. W. Bartlett and A. M. Gilbert, J. Pharmacokinet. Pharmacodyn., 2020 DOI:10.1007/s10928-020-09722-z.
- M. L. Drummond and C. I. Williams, J. Chem. Inf. Model., 2019, 59, 1634–1644 CrossRef CAS PubMed.
- M. L. Drummond, A. Henry, H. Li and C. I. Williams, J. Chem. Inf. Model., 2020, 60, 5234–5254 CrossRef CAS PubMed.
- D. Zaidman, J. Prilusky and N. London, J. Chem. Inf. Model., 2020, 60, 4894–4903 CrossRef CAS PubMed.
- A. W. Senior, R. Evans, J. Jumper, J. Kirkpatrick, L. Sifre, T. Green, C. Qin, A. Zidek, A. W. R. Nelson, A. Bridgland, H. Penedones, S. Petersen, K. Simonyan, S. Crossan, P. Kohli, D. T. Jones, D. Silver, K. Kavukcuoglu and D. Hassabis, Nature, 2020, 577, 706–710 CrossRef CAS PubMed.
- F. Imrie, A. R. Bradley, M. van der Schaar and C. M. Deane, J. Chem. Inf. Model., 2020, 60, 1983–1995 CrossRef CAS PubMed.
- M. Peplow, ACS Cent. Sci., 2020, 6, 1274–1277 CrossRef CAS PubMed.
- M. Wieczorek, E. T. Abualrous, J. Sticht, M. Alvaro-Benito, S. Stolzenberg, F. Noe and C. Freund, Front. Immunol., 2017, 8, 292 Search PubMed.
- S. M. Jensen, G. K. Potts, D. B. Ready and M. J. Patterson, Front. Immunol., 2018, 9, 2697 CrossRef PubMed.
- M. Arnaud, M. Duchamp, S. Bobisse, P. Renaud, G. Coukos and A. Harari, Curr. Opin. Biotechnol., 2020, 65, 52–59 CrossRef CAS PubMed.
- B. Cheng, Y. Ren, H. Cao and J. Chen, Eur. J. Med. Chem., 2020, 199, 112377 CrossRef CAS.
- M. A. Maneiro, N. Forte, M. M. Shchepinova, C. S. Kounde, V. Chudasama, J. R. Baker and E. W. Tate, ACS Chem. Biol., 2020, 15, 1306–1312 CrossRef CAS PubMed.
- Y. Wang, L. Han, F. Liu, F. Yang, X. Jiang, H. Sun, F. Feng, J. Xue and W. Liu, Colloids Surf., B, 2020, 188, 110795 CrossRef CAS PubMed.
- M. Wei, R. Zhao, Y. Cao, Y. Wei, M. Li, Z. Dong, Y. Liu, H. Ruan, Y. Li, S. Cao, Z. Tang, Y. Zhou, W. Song, Y. Wang, J. Wang, G. Yang and C. Yang, Eur. J. Med. Chem., 2020, 209 DOI:10.1016/j.ejmech.2020.112903.
- A. Ghidini, A. Clery, F. Halloy, F. H. T. Allain and J. Hall, Angew. Chem., Int. Ed., 2021, 60(6), 3163–3169 CrossRef CAS.
- M. G. Costales, B. Suresh, K. Vishnu and M. D. Disney, Cell Chem. Biol., 2019, 26, 1180–1186 CrossRef CAS PubMed.
- H. S. Haniff, Y. Tong, X. Liu, J. L. Chen, B. M. Suresh, R. J. Andrews, J. M. Peterson, C. A. O'Leary, R. I. Benhamou, W. N. Moss and M. D. Disney, ACS Cent. Sci., 2020, 6, 1713–1721 CrossRef CAS PubMed.
- M. Slabicki, H. Yoon, J. Koeppel, L. Nitsch, S. S. Roy Burman, C. Di Genua, K. A. Donovan, A. S. Sperling, M. Hunkeler, J. M. Tsai, R. Sharma, A. Guirguis, C. Zou, P. Chudasama, J. A. Gasser, P. G. Miller, C. Scholl, S. Frohling, R. P. Nowak, E. S. Fischer and B. L. Ebert, Nature, 2020, 588, 164–168 CrossRef CAS PubMed.
- Y. Isobe, M. Okumura, L. M. McGregor, S. M. Brittain, M. D. Jones, X. Liang, R. White, W. Forrester, J. M. McKenna, J. A. Tallarico, M. Schirle, T. J. Maimone and D. K. Nomura, Nat. Chem. Biol., 2020, 16, 1189–1198 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |