
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic asymmetric synthesis of 3,2′-pyrrolinyl spirooxindoles via conjugate addition/Schmidt-type rearrangement of vinyl azides and (E)-alkenyloxindoles†
Ziwei
Zhong
 ,
Zhijie
Xiao
,
Xiaohua
Liu
,
Weidi
Cao
* and
Xiaoming
Feng
*
,
Zhijie
Xiao
,
Xiaohua
Liu
,
Weidi
Cao
* and
Xiaoming
Feng
*
Key Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, China. E-mail: wdcao@scu.edu.cn; xmfeng@scu.edu.cn
First published on 28th September 2020
Abstract
A catalytic asymmetric conjugate addition/Schmidt-type rearrangement of vinyl azides and (E)-alkenyloxindoles was realized. It afforded a variety of optically active 3,2′-pyrrolinyl spirooxindoles with high yields (up to 98%), and excellent diastereo- and enantioselectivities (up to 98% ee, >19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr), even at the gram-scale in the presence of a chiral N,N′-dioxide–nickel(II) complex. In addition, a possible catalytic cycle and transition state model were proposed to rationalize the stereoselectivity.
:1 dr), even at the gram-scale in the presence of a chiral N,N′-dioxide–nickel(II) complex. In addition, a possible catalytic cycle and transition state model were proposed to rationalize the stereoselectivity.
Introduction
Aza-spirocyclic fragments are ubiquitous in numerous natural products and pharmaceuticals.1 As a significant subset of such aza-spirocylic compounds, 3,2′-pyrrolidinyl spirooxindoles and their analogues (pyrrolines), which bear stereocenters at C3′ and C5′-positions of the pyrrolidine core, exhibit notable biological activities,2 such as the inhibitor of HIV-1,2d inhibitor of AGEs2e and inhibitor targeting Brr2 (Scheme 1a).2f Thus, the enantioselective construction of these useful skeletons has attracted great interest in the past decades. Catalytic asymmetric [3 + 2] cycloaddition of oxindole derivatives containing the nitrogen sources at the C3 position is demonstrated to be the most common and efficient approach for building the five-membered pyrrolidinyl ring.3 The related cycloaddition can undergo different construction pathways, for example, enantioselective 1,3-dipolar cycloaddition between azomethine ylides and electron deficient alkenes is well established (Scheme 1b, path a).3g,h In contrast to the rapid development with an azomethine ylide as the nitrogen-partner, electron-poor iminooxindole as the nitrogen-partner is less developed. The desired skeleton can be constructed by [3 + 2] annulation with cyclopropane or allylic silane as 1,3-dipoles, and a cascade reaction with nitroalkane-mesylate (Scheme 1b, path b).3i–k Regardless of these advances, more discoveries of novel asymmetric methodologies leading to 3,2′-pyrrolidinyl spirooxindole skeletons would be expected. | ||
| Scheme 1 Asymmetric synthesis of 3,2′-pyrrolidinyl spirooxindoles and their analogues. | ||
Vinyl azide,4 featuring both alkene and azide motifs conjugated together, has emerged as a versatile building block due to its unique properties, in the synthesis of nitrogen heterocyclic compounds, for instance 1-pyrroline via formal 1,3-dipolar cycloaddition with an alkene.4d,e Despite prominent advances, catalytic asymmetric reactions of vinyl azides are extremely rare.5 To the best of our knowledge, the only related work is a visible-light-induced catalytic asymmetric [3 + 2] photocycloaddition of vinyl azides with α,β-unsaturated N-acylpyrazoles reported by Meggers and coworkers.5b And enantiomerically pure 1-pyrrolines were obtained with a chiral rhodium complex as the catalyst (Scheme 2a). Recently, Chiba's group took advantage of this strategy to realize the synthesis of racemic 3,2′-pyrrolinyl spirooxindoles. High yields and dr values could be achieved in the presence of stoichiometric BF3·Et2O; by contrast, lower diastereoselectivities were obtained for some substrates with the use of 10 mol% of TiCl4 as the catalyst (Scheme 2b, left).6a It was worth mentioning that Wan's group used vinyl azides and diazooxindoles constructing a similar racemic skeleton via Rh(II)-catalyzed [1 + 1 + 3] annulation at a higher temperature.6b
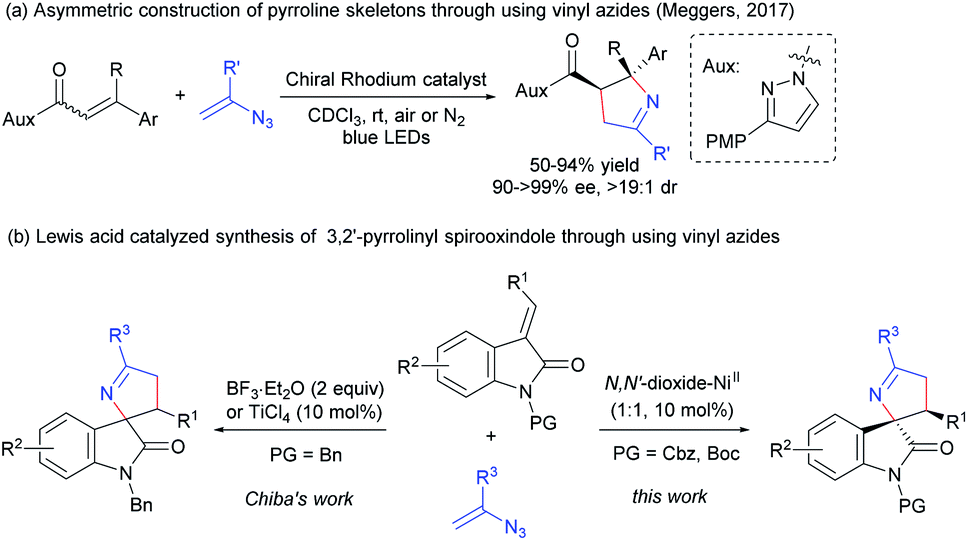 | ||
| Scheme 2 1,3-Dipolar cycloaddition with vinyl azide as the nitrogen source. | ||
Given the performance of chiral N,N′-dioxide–metal complexes7 in activation and stereocontrol of (E)-alkenyloxindoles, we envisage that with careful choice of these kinds of chiral ligands and metal salts, the catalytic asymmetric cycloaddition of (E)-alkenyloxindoles with vinyl azide as the nitrogen source would be suitable for enantioselective construction of chiral 3,2′-pyrrolinyl spirooxindole skeletons (Scheme 1b, path c). Herein, we wish to present a chiral N,N′-dioxide–NiII complex mediated catalytic asymmetric conjugate addition/Schmidt-type rearrangement of vinyl azides with (E)-alkenyloxindoles (Scheme 2b, right). Both α-aliphatic and α-aromatic substituted vinyl azides could be transformed into 3′-carbonyl-5′-substituted 3,2′-pyrrolinyl spirooxindoles in good yield with high to excellent diastereoselectivity and enantioselectivity under mild reaction conditions.
Results and discussion
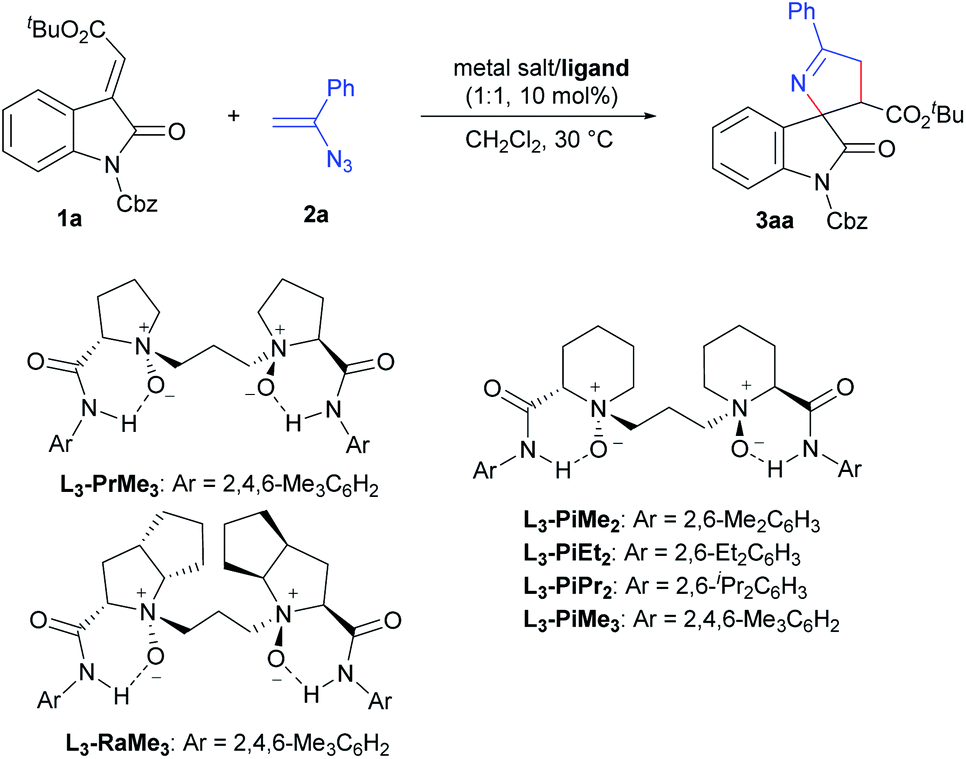
In our initial screening, we selected (E)-alkenyloxindole 1a and vinyl azide 2a as the model substrates to optimize the reaction conditions (Table 1) in view of that 3′-carbonyl substituents are common in the bioactive molecules shown in Scheme 1a. Firstly, various metal salts, such as Sc(OTf)3, Mg(OTf)2 and Ni(OTf)2, were evaluated by coordinating with (S)-pipecolic acid derived N,N′-dioxide ligand L3-PiMe3 (entries 1–3). It was found that the complex Ni(OTf)2 could give the desired product 3aa with better results (entry 3, 85% yield, 80% ee) in CH2Cl2 at 30 °C. The exploration of the counterions of the nickel salts (entries 3–5) suggested that the non-coordinated counterion (BF4−) led to a little increase in the enantioselectivity (entry 5, 86% yield, 83% ee). Then, we chose Ni(BF4)2·6H2O as the metal salt to investigate the effect of the chiral backbone of the N,N′-dioxide ligands. This revealed that L3-PiMe3 was superior to both L-proline derived L3-PrMe3 and L-ramipril derived L3-RaMe3 (entries 5–7). The use of L3-PiMe2 without a methyl group at the para-position of the phenyl group of the amide moiety improved the enantioselectivity to 86% (entry 8). Further exploration of the amide moiety of the N,N′-dioxide ligand showed that the complex L3-PiEt2/Ni(BF4)2·6H2O could give the best result (entry 9 vs. entry 10, 88% yield, 91% ee). Comparatively, when a chiral bisoxazoline ligand, BINAP or chiral phosphoric acid was used as the ligand, moderate yields and low ee values were achieved (see the ESI† for details). Other parameters such as solvents and additives were investigated as well; however, no better results were obtained (see the ESI† for more details). Enhancing the amount of vinyl azide 2a (2.0 equiv.) resulted in an improvement of the yield to 95% with 92% ee (entry 11). Upon lowering the catalyst loading to 5 mol%, the yield was reduced to 86% with the enantioselectivity being maintained (entry 12). Therefore, the optimal reaction conditions were established as 1a (0.1 mmol), 2a (0.2 mmol), and L3-PiEt2/Ni(BF4)2·6H2O (1:1, 10 mol%), in CH2Cl2 at 30 °C for 24 h. It was worth mentioning that the diastereoselectivity could be well controlled during the conditional screening process (>19:1 dr).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Metal salt | Ligand | Yieldb (%) | drc | eed (%) |
|
a Unless otherwise noted, all the reactions were carried out with 1a (0.10 mmol), 2a (0.10 mmol) and metal salt/ligand (1:1, 10 mol%) in CH2Cl2 (1.0 mL) at 30 °C for 24 h.
b Isolated yield of 3aa.
c Determined by 1H NMR.
d Determined by HPLC analysis on a chiral stationary phase.
e
2a (2.0 equiv.) was used.
f 5 mol% catalyst loading.
|
|||||
| 1 | Sc(OTf)3 | L3-PiMe3 | 48 | >19:1 |
25 |
| 2 | Mg(OTf)2 | L3-PiMe3 | 80 | >19:1 |
77 |
| 3 | Ni(OTf)2 | L3-PiMe3 | 85 | >19:1 |
80 |
| 4 | Ni(NTf2)2 | L3-PiMe3 | 87 | >19:1 |
80 |
| 5 | Ni(BF4)2·6H2O | L3-PiMe3 | 86 | >19:1 |
83 |
| 6 | Ni(BF4)2·6H2O | L3-PrMe3 | 80 | >19:1 |
82 |
| 7 | Ni(BF4)2·6H2O | L3-RaMe3 | 76 | >19:1 |
70 |
| 8 | Ni(BF4)2·6H2O | L3-PiMe2 | 84 | >19:1 |
86 |
| 9 | Ni(BF4)2·6H2O | L3-PiEt2 | 88 | >19:1 |
91 |
| 10 | Ni(BF4)2·6H2O | L3-PiPr2 | 95 | >19:1 |
71 |
| 11e | Ni(BF4)2·6H2O | L3-PiEt2 | 95 | >19:1 |
92 |
| 12e,f | Ni(BF4)2·6H2O | L3-PiEt2 | 86 | >19:1 |
92 |
With the optimized conditions in hand, the substrate scope was then evaluated (Table 2). Various (E)-alkenyloxindoles 1 bearing different ester groups (R1) could be transformed into the corresponding products 3aa–3fa in 90–95% yields, >19:1 dr and 85–92% ee (entries 1–6), and the enantioselectivities would slightly decrease if less steric hindrance of the ester groups was used (entries 2–4). Regardless of the electronic effect or steric hindrance positions of the substituents on the phenyl ring, this asymmetric reaction proceeded smoothly to afford 3ga–3na with good results (entries 7–14, 81–98% yields, >19:1 dr and 85–95% ee). It was worth mentioning that (E)-alkenyloxindoles bearing electron withdrawing halogen groups (F, Cl, and Br) at the C5-position delivered 3ga–3ia with higher ee values (entries 7–9) than those with electron donating groups (entries 10 and 11, 3ja and 3ka). N-Boc protected (E)-alkenyloxindoles were also tolerated well, giving 3oa and 3pa in 80% yield with 98% ee and 82% yield with 88% ee, respectively (entries 15 and 16), and the slightly lower yields were attributed to the decomposition of 1o and 1p through deprotection of the Boc group in the presence of the Lewis acid. Furthermore, the absolute configuration of 3ja was determined to be (3S,3′R) by the X-ray diffraction analysis.8
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Yieldb (%) | drc | eed (%) |
|
a Unless otherwise noted, all the reactions were carried out with 1 (0.10 mmol, PG = Cbz), 2a (0.20 mmol) and L3-PiEt2/Ni(BF4)2·6H2O (1:1, 10 mol%) in CH2Cl2 (1.0 mL) at 30 °C for 24 h.
b Isolated yield of 3.
c Determined by 1H NMR.
d Determined by HPLC analysis on a chiral stationary phase.
e PG = Boc. Boc = tert-butoxycarbonyl and Cbz = benzyloxycarbonyl.
|
|||||
| 1 | CO2tBu | H | 95 (3aa) | >19:1 |
92 |
| 2 | CO2Me | H | 91 (3ba) | >19:1 |
85 |
| 3 | CO2Et | H | 90 (3ca) | >19:1 |
89 |
| 4 | CO2iPr | H | 92 (3da) | >19:1 |
89 |
| 5 | CO2Ph | H | 92 (3ea) | >19:1 |
86 |
| 6 | CO2Bn | H | 91 (3fa) | >19:1 |
91 |
| 7 | CO2tBu | 5 F | 93 (3ga) | >19:1 |
91 |
| 8 | CO2tBu | 5-Cl | 87 (3ha) | >19:1 |
91 |
| 9 | CO2tBu | 5-Br | 85 (3ia) | >19:1 |
91 |
| 10 | CO2tBu | 5-Me | 95 (3ja) | >19:1 |
88 |
| 11 | CO2tBu | 5-OMe | 91 (3ka) | >19:1 |
87 |
| 12 | CO2tBu | 6-CF3 | 88 (3la) | >19:1 |
95 |
| 13 | CO2tBu | 7-F | 81 (3ma) | >19:1 |
94 |
| 14 | CO2tBu | 5,6-F2 | 98 (3na) | >19:1 |
90 |
| 15e | CO2tBu | H | 80 (3oa) | >19:1 |
98 |
| 16e | COPh | H | 82 (3pa) | >19:1 |
88 |

The scope of vinyl azides was next examined under the standard conditions (Table 3). A battery of 3,2′-pyrrolinyl spirooxindoles (3ab–3aj) were obtained in high to excellent yields and ee values regardless of the electron-rich or -deficient groups attached to the aryl group of vinyl azides. The reaction of electron-donating 2-methoxy-substituted or electron-withdrawing 2-chloro-substituted vinyl azides gave excellent yields and enantioselectivities (91–95% yields, 92–94% ee, 3ab and 3ac). In contrast, the same substituents at the meta-position of the phenyl rings delivered the products 3ad and 3ae with a lower ee value (81–82% ee). Compared with those vinyl azides involving electron-donating groups (Me and nBu) at the para-position of the aryl group (entries 5 and 6), vinyl azides bearing electron-withdrawing halogen groups (F and Cl) and ester groups exhibited higher reactivities (87–93%) with excellent enantioselectivities (entries 7–9, 94–96% ee, 3ah–3aj). The condensed-ring and heteroaromatic substrates were also tolerated in this reaction, readily affording spiropyrrolines 3ak and 3al in 85–95% yields and 90–94% ee, albeit with a lower dr value for 3al (83:17 dr). Furthermore, α-alkyl substituted vinyl azides, such as with cyclohexyl and benzyl, were applicable as well, and offered the corresponding products 3am and 3an in good yields (86–88%) with excellent stereocontrol (entries 12 and 13, 93–95% ee, >19:1 dr).
|
|
||||
|---|---|---|---|---|
| Entrya | R3 | Yieldb (%) | drc | eed (%) |
|
a Unless otherwise noted, all the reactions were carried out with 1a (0.10 mmol), 2 (0.20 mmol) and L3-PiEt2/Ni(BF4)2·6H2O (1:1, 10 mol%) in CH2Cl2 (1.0 mL) at 30 °C for 24 h.
b Isolated yield of 3.
c Determined by 1H NMR.
d Determined by HPLC analysis on a chiral stationary phase. Cbz = benzyloxycarbonyl.
|
||||
| 1 | 2-MeOC6H4 | 95 (3ab) | >19:1 |
92 |
| 2 | 2-ClC6H4 | 91 (3ac) | >19:1 |
94 |
| 3 | 3-MeOC6H4 | 90 (3ad) | >19:1 |
81 |
| 4 | 3-ClC6H4 | 92 (3ae) | >19:1 |
82 |
| 5 | 4-MeC6H4 | 92 (3af) | >19:1 |
84 |
| 6 | 4-nBuC6H4 | 91 (3ag) | >19:1 |
80 |
| 7 | 4-FC6H4 | 93 (3ah) | >19:1 |
95 |
| 8 | 4-ClC6H4 | 87 (3ai) | >19:1 |
96 |
| 9 | 4-CO2EtC6H4 | 88 (3aj) | >19:1 |
94 |
| 10 | 2-Naphthyl | 95 (3ak) | >19:1 |
94 |
| 11 | 3-Thienyl | 85 (3al) | 83:17 |
90 |
| 12 | Cyclohexyl | 88 (3am) | >19:1 |
95 |
| 13 | Benzyl | 86 (3an) | >19:1 |
93 |

Subsequently, we also tested various types of electron-deficient alkenes to broaden the synthetic scope. Alkylidene malonates 4 reacted with 2a smoothly upon switching the catalyst to Mg(NTf2)2/L3-PiMe3 in THF, and the corresponding chiral 1-pyrroline derivatives 5 could be achieved with good yields and high enantioselectivities (Scheme 3). Other electron-deficient alkenes were also examined, such as chromenes, chalcone and so on, but a low reactivity was found or a hetero-Diels–Alder reaction tended to occur (see the ESI† for details).
 | ||
| Scheme 3 The conjugate addition/Schmidt-type rearrangement of alkylidene malonates with vinyl azide. | ||
To evaluate the synthetic value of the catalytic system, a scale-up experiment was carried out (Scheme 4a). The (E)-alkenyloxindole 1a (3.0 mmol) reacted with vinyl azide 2a (6.0 mmol) smoothly and gave the desired product 3aa in 89% yield, 92% ee and >19:1 dr in the presence of the L-PiEt2/Ni(BF4)2·6H2O complex (10 mol%). Treatment of the product 3ai with Pd/C under a hydrogen atmosphere got rid of the protecting group (Cbz) to afford 6, which could be further transformed into the functionalized pyrrolidine product 7 in 95% yield with excellent stereoselectivity through hydrogenation (Scheme 4b).9
 | ||
| Scheme 4 (a) Scale-up synthesis of 3aa; (b) transformation of the product 3ai. | ||
To gain mechanistic insight into the reaction, control experiments were performed as shown in Scheme 5. Phenyl-2H-azirine 8 generated from vinyl azide 2a has been reported as a possible 1,3-dipole precursor in [3 + 2] cycloaddition.4 When azirine 8 was used to react with 1a under the standard conditions, no reaction occurred, ruling out the reaction pathway with the aryl-2H-azirine (8) intermediate (Scheme 5a). Moreover, when N-benzyl (E)-alkenyloxindole 1q was explored in this catalytic system, only 19% yield and 27% ee of the desired product were obtained (Scheme 5b), which indicated that the coordinating group unit of the nitrogen protecting group played an important role in achieving high reactivity and enantioselectivity in the current catalytic system. The activation mode might be different from that in a previous report using N-Bn or N-H based (E)-alkenyloxindole in the racemic version.6a
 | ||
| Scheme 5 Control experiments. | ||
Based on the results of the control experiment, X-ray structure of the product 3ja8 and Ni(BF4)2·6H2O/L3-PiEt2 complex,10 a possible reaction mechanism was proposed (Scheme 6). Firstly, (E)-alkenyloxindole 1j was activated by bidentate coordination with the Ni(BF4)2·6H2O/L3-PiEt2 complex.11 Vinyl azide prefers to undergo asymmetric 1,4-conjugate addition to 1j from its β-Re face because the β-Si face was shielded by the nearby 2,6-diethyl phenyl group of L3-PiEt2. The subsequent cyclization of Re–Si attack results in an (1′S, 2′R, 3′R)-azidocyclobutane (int-II), which was detected by high resolution mass spectrometry (see the ESI† for more details). In the following Schmidt-type rearrangement, the more electron-rich α-carbon group (C3) shifts to the nitrogen atom with the release of N2 to form int-III through antiperiplanar migration, and the stereochemistry of C3 is retained.12 Finally, the imidization of int-III affords the desired product (3S, 3′R)-3ja.
 | ||
| Scheme 6 The proposed catalytic cycle and working mode. | ||
Conclusions
We have successfully developed the Lewis acid catalyzed asymmetric synthesis of 3,2′-pyrrolinyl spirooxindoles via conjugate addition/Schmidt-type rearrangement of vinyl azides and (E)-alkenyloxindoles. The catalytic system of the chiral N,N′-dioxide/Ni(BF4)2·6H2O complex benefited this reaction efficiently to afford diverse chiral spiropyrroline derivatives in good yields with high stereoselectivities (up to 98% yield, >19:1 dr and 98% ee), which provided a new method for the synthesis of chiral nitrogen heterocyclic compounds with vinyl azides as the nitrogen source. Besides, a possible catalytic cycle along with the working mode was proposed to elucidate the reaction process and chiral induction. Further investigations on the catalytic asymmetric synthesis of aza-spirocylic compounds by using vinyl azides are ongoing in our laboratory.
Safety notice: vinyl azides are classified as organic azides, and act as versatile building blocks in organic synthesis. However, one should keep in mind the inherent toxicity, instability, shock sensitivity, and explosive power of azides. Great care must be taken when handling these compounds, particularly during concentration and the physical handling of isolated products, due to the explosive potential of the azide functionality.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate the National Natural Science Foundation of China (No. 21801174) for financial support.Notes and references
- (a) B. D. Morris and M. R. Prinsep, J. Nat. Prod., 1999, 62, 688 CrossRef CAS; (b) M. S. M. Pearson, N. Floquet, C. Bello, P. Voge, R. Plantier-Royon, J. Szymoniak, P. Bertus and J. B. Behr, Bioorg. Med. Chem., 2009, 17, 8020 CrossRef CAS; (c) A. Khan, P. Wood and J. Moskal, US Pat., 20110306586, December 15, 2011; (d) Y. Hayashi, K. Sankar, H. Ishikawa, Y. Nozawa, K. Mizoue and H. Kakeya, Bioorg. Med. Chem. Lett., 2009, 19, 3863 CrossRef CAS; (e) Y. Hayashi, M. Shoji, J. Yamaguchi, K. Sato, S. Yamaguchi, T. Mukaiyama, K. Sakai, Y. Asami, H. Kakeya and H. Osada, J. Am. Chem. Soc., 2002, 124, 12078 CrossRef CAS; (f) Y. Zheng, C. M. Tice and S. B. Singh, Bioorg. Med. Chem. Lett., 2014, 24, 3673 CrossRef CAS; (g) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748 CrossRef CAS.
- (a) Y. A. Ivanenkov, S. V. Vasilevski, E. K. Beloglazkina, M. E. Kukushkin, A. E. Machulkin, M. S. Veselov, N. V. Chufarova, E. S. Chernyaginab, A. S. Vanzcool, N. V. Zyk, D. A. Skvortsov, A. A. Khutornenko, A. L. Rusanov, A. G. Tonevitsky, O. A. Dontsova and A. G. Majouga, Bioorg. Med. Chem. Lett., 2015, 25, 404 CrossRef CAS; (b) W. Tan, X. T. Zhu, S. Zhang, G. J. Xing, R. Y. Zhu and F. Shi, RSC Adv., 2013, 3, 10875 RSC; (c) G. Bhaskar, Y. Arun, C. Balachandran, C. Saikumar and P. T. Perumal, Eur. J. Med. Chem., 2012, 51, 79 CrossRef CAS; (d) K. Hagiwara, T. Murakami, G. Xue, Y. Shimizu, E. Takeda, Y. Hashimoto, K. Honda, Y. Kondoh, H. Osada, Y. Tsunetsugu-Yokota and Y. Aida, Biochem. Biophys. Res. Commun., 2010, 394, 721 CrossRef CAS; (e) A. Kaur, S. Baldev, B. Vyas and O. Silakari, Eur. J. Med. Chem., 2014, 79, 282 CrossRef CAS; (f) M. Ito, M. Iwatani, T. Yamamoto, T. Tanaka, T. Kawamoto, D. Morishita, A. Nakanishi and H. Maezaki, Bioorg. Med. Chem. Lett., 2017, 25, 4753 CrossRef CAS.
- (a) Y. M. Cao, F. F. Shen, F. T. Zhang and R. Wang, Chem.–Eur. J., 2013, 19, 1184 CrossRef CAS; (b) F. Tan and H. G. Cheng, Targets Heterocycl. Syst., 2017, 21, 158 CAS; (c) D. I. Jiang, S. D. Dong, W. F. Tang, T. Lu and D. Du, J. Org. Chem., 2015, 80, 11593 CrossRef CAS; (d) L. Chen, Z. J. Wu, M. L. Zhang, D. F. Yue, X. M. Zhang, X. Y. Xu and W. C. Yuan, J. Org. Chem., 2015, 80, 12668 CrossRef CAS; (e) P. Yang, X. Wang, F. Chen, Z. B. Zhang, C. Chen, L. Peng and L. X. Wang, J. Org. Chem., 2017, 82, 3908 CrossRef CAS; (f) K. Q. Chen, Y. Li, C. L. Zhang, D. Q. Sun and S. Ye, Org. Biomol. Chem., 2016, 14, 2007 RSC; (g) X. Fang and C. J. Wang, Org. Biomol. Chem., 2018, 16, 2591 RSC; (h) H. Z. Gui, Y. Wei and M. Shi, Chem.–Asian J., 2020, 15, 1225 CrossRef CAS; (i) J. P. MacDonald, B. H. Shupe, J. D. Schreiber and A. K. Franz, Chem. Commun., 2014, 50, 5242 RSC; (j) S. Hajra and B. Jana, Org. Lett., 2017, 19, 4778 CrossRef CAS; (k) A. A. Akaev and E. M. Budynina, J. Org. Chem., 2019, 84, 3340 CrossRef CAS.
- (a) M. O. Forster and S. H. Newman, J. Chem. Soc., Trans., 1910, 97, 2570 RSC; (b) M. O. Forster and S. H. Newman, J. Chem. Soc., Trans., 1911, 99, 1277 RSC; (c) N. Jung and S. Bräse, Angew. Chem., Int. Ed., 2012, 51, 12169 CrossRef CAS; (d) E. A. Anderson and X. Bi, Chem. Soc. Rev., 2017, 46, 7208 RSC; (e) H. Hayashi, A. Kaga and S. Chiba, J. Org. Chem., 2017, 82, 11981 CrossRef CAS.
- (a) N. Thirupathi, F. Wei, C. H. Tung and Z. H. Xu, Nat. Commun., 2019, 10, 3158 CrossRef; (b) X. Q. Huang, X. Y. Li, X. L. Xie, K. Harms, R. Riedel and E. Meggers, Nat. Commun., 2017, 8, 2245 CrossRef; (c) T. Nakanishi, J. Kikuchi, A. Kaga, S. Chiba and M. Terada, Chem.–Eur. J., 2020, 26, 8230 CrossRef CAS; (d) M. Han, M. Yang, R. Wu, Y. Li, T. Jia, Y. J. Gao, H. L. Ni, P. Hu, B. Q. Wang and P. Cao, J. Am. Chem. Soc., 2020, 142, 13398 CrossRef CAS.
- (a) X. Zhu and S. Chiba, Chem. Commun., 2016, 52, 2473 RSC; (b) R. X. Yi, L. L. Qian and B. S. Wan, Chin. J. Catal., 2019, 40, 177 CrossRef CAS.
- For reviews of N,N′-dioxide ligands, see: (a) X. H. Liu, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2011, 44, 574 CrossRef CAS; (b) X. H. Liu, L. L. Lin and X. M. Feng, Org. Chem. Front., 2014, 1, 298 RSC; (c) X. H. Liu, H. F. Zheng, Y. Xia, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2017, 50, 2621 CrossRef CAS; (d) X. H. Liu, S. X. Dong, L. L. Lin and X. M. Feng, Chin. J. Chem., 2018, 36, 791 CrossRef CAS; (e) Z. Wang, X. H. Liu and X. M. Feng, Aldrichimica Acta, 2020, 53, 3 Search PubMed; (f) D. Zhang, Z. S. Su, Q. W. He, Z. K. Wu, Y. Q. Zhou, C. J. Pan, X. H. Liu and X. M. Feng, J. Am. Chem. Soc., 2020, 37(142), 15975 CrossRef; (g) X. B. Lin, Z. Tan, W. K. Yang, W. Yang, X. H. Liu and X. M. Feng, CCS Chem., 2020, 2, 1423 CrossRef; (h) J. Xu, Z. W. Zhong, M. Y. Jiang, Y. Q. Zhou, X. H. Liu and X. M. Feng, CCS Chem., 2020, 2, 1894 CrossRef.
- For the X-ray structure of the product: CCDC: 1977126 (3ja).
- The absolute configuration of 5 was determined to be (3S,3′R,5′R) from NOESY spectra.
- For the X-ray structure of the catalyst (Ni(BF4)2·6H2O/L3-PiEt2 J. Guo, X. H. Liu and X. M. Feng, Chem. Commun., 2018, 54, 12254 RSC ): , CCDC: 1831759.
- (a) J. Guo, Y. B. Liu, X. Q. Li, X. H. Liu, L. L. Lin and X. M. Feng, Chem. Sci., 2016, 7, 2717 RSC; (b) X. J. Lian, S. S. Guo, G. Wang, L. L. Lin, X. H. Liu and X. M. Feng, J. Org. Chem., 2014, 79, 7703 CrossRef CAS; (c) H. F. Zheng, P. He, Y. B. Liu, Y. L. Zhang, L. L. Lin, X. H. Liu and X. M. Feng, Chem. Commun., 2014, 50, 8794 RSC; (d) M. S. Xie, X. X. Wu, G. Wang, L. L. Lin and X. M. Feng, Acta Chim. Sin., 2014, 72, 856 CrossRef CAS; (e) Y. H. Zhou, Y. Lu, X. Y. Hu, H. J. Mei, L. L. Lin, X. H. Liu and X. M. Feng, Chem. Commun., 2017, 53, 2060 RSC; (f) Y. H. Zhou, L. L. Lin, X. H. Liu, X. Y. Hu, Y. Lu, X. Y. Zhang and X. M. Feng, Angew. Chem., Int. Ed., 2018, 57, 9113 CrossRef CAS.
- (a) X. M. Zhang, Y. Q. Tu, F. M. Zhang, Z. H. Chen and S. H. Wang, Chem. Soc. Rev., 2017, 46, 2272 RSC; (b) X. J. Wen, X. Y. Li, X. Luo, W. J. Wang, S. Song and N. Jiao, Chem. Sci., 2020, 11, 4482 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C{1H} and 19F{1H} NMR, HPLC spectra, and CD spectra (PDF). X-ray crystallographic data for 3ja (CIF). CCDC 1977126. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03776a |
| This journal is © The Royal Society of Chemistry 2020 |