
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pyrene-based metal organic frameworks: from synthesis to applications
F. Pelin
Kinik†
 ,
Andres
Ortega-Guerrero†
,
Daniele
Ongari
,
Christopher P.
Ireland
and
Berend
Smit
*
,
Andres
Ortega-Guerrero†
,
Daniele
Ongari
,
Christopher P.
Ireland
and
Berend
Smit
*
Laboratory of Molecular Simulation (LSMO), Institut des Sciences et Ingénierie Chimiques (ISIC), Ecole Polytechnique Fédérale de Lausanne (EPFL), Rue de l'Industrie 17, CH-1951 Sion, Valais, Switzerland. E-mail: berend.smit@epfl.ch
First published on 21st January 2021
Abstract
Pyrene is one of the most widely investigated aromatic hydrocarbons given to its unique optical and electronic properties. Hence, pyrene-based ligands have been attractive for the synthesis of metal–organic frameworks (MOFs) in the last few years. In this review, we will focus on the most important characteristics of pyrene, in addition to the development and synthesis of pyrene-based molecules as bridging ligands to be used in MOF structures. We will summarize the synthesis attempts, as well as the post-synthetic modifications of pyrene-based MOFs by the incorporation of metals or ligands in the structure. The discussion of promising results of such MOFs in several applications; including luminescence, photocatalysis, adsorption and separation, heterogeneous catalysis, electrochemical applications and bio-medical applications will be highlighted. Finally, some insights and future prospects will be given based on the studies discussed in the review. This review will pave the way for the researchers in the field for the design and development of novel pyrene-based structures and their utilization for different applications.
 F. Pelin Kinik | Fatma Pelin Kinik received her BSc degree (2015) in Chemical Engineering from Istanbul Technical University (Turkey). She received her MSc degree (2017) in Chemical & Biological Engineering Department of Koç University (Turkey). Currently, she is a PhD student under the supervision of Prof. Berend Smit at EPFL, focusing on hydrogen generation using MOFs by heterogeneous catalysis and photocatalysis. |
 Andres Ortega-Guerrero | Andres Ortega-Guerrero received his undergraduate degree in electronics engineering from Universidad del Valle in Cali, Colombia. Later he received a MSc on Nanoscience and Nanotechnology from Katholieke Universiteit Leuven, Belgium and Technische Universität Dresden, Germany. Currently, he is a PhD student in Berend Smit's group, investigating the electronic and optical properties of MOFs for photocatalysis using computational methods. |
 Daniele Ongari | Daniele Ongari received his diploma in chemical engineering from Politecnico di Milano. In 2019, he completed his PhD under the supervision of Prof. Berend Smit at EPFL. His research focuses on the investigation of microporous materials, in particular MOFs and COFs, using computational methods to assess their performances for molecular adsorption and catalysis. |
 Christopher P. Ireland | Christopher P. Ireland obtained his PhD under the supervision of Prof. Matthew J. Rosseinsky at the University of Liverpool, where his work involved the synthesis of metal oxides for photocatalytic and optical applications. After time at UCL and the University of Cambridge, he was awarded a Marie Curie Fellowship in 2015 to move to the LSMO group at EPFL, led by Prof. Berend Smit. His research interests at EPFL involves the synthesis and characterization of metal–organic frameworks for environmental applications, such as carbon capture, and photocatalytic hydrogen generation. |
 Berend Smit | Berend Smit received a MSc in Chemical Engineering in 1987 and a MSc in Physics both from the Technical University in Delft. In 1990, he received a cum laude PhD in Chemistry from Utrecht University. He was a (senior) Research Physicists at Shell Research before he joined the University of Amsterdam. In 2004, he was elected Director of the European Center of Atomic and Molecular Computations (CECAM) Lyon France. In 2007 he became Professor of Chemical Engineering and Chemistry at U. C. Berkeley and Faculty Chemist at LBNL. Since July 2014 he is full professor at EPFL. |
1 Introduction
A chromophore is a molecule which is capable of absorbing particular wavelengths of visible light, giving rise to some changes in electron density after the excitation process. Chromophore organic molecules have unique characteristics; such as high charge carrier mobility, light absorption, solution processability, thermal stability, and availability for the coordination with metal ions.1 Among chromophoric materials, conjugated π-systems are perfect candidates for applications based on fluorescence, phosphorescence, or charge separation.2 Pyrene is a highly symmetrical four-benzene-ringed polycyclic aromatic hydrocarbon, which is one of the most studied chromophore molecules after its discovery in the residue of the distillation of coal tar in 1837.3 After it became commercially available, the first use of pyrene was for the synthetic dye industry thanks to its intense yellow colour. With the discovery of its favorable properties as being a “fluorophore” with excellent emission properties, having long excited-state lifetime and successful electron hole-pair dissociation, pyrene has been one of the most investigated aromatic hydrocarbons.4Pyrene molecule allows the addition of different functional groups by traditional synthetic techniques such as formylation/acetylation, bromination, de-tert-butylation, oxidation, and borylation reactions.1 In this way, pyrene-based molecules have been used as substrates in the synthesis of molecules such as nanographenes and metal cages.5 In addition, the optical properties of pyrene have been exploited in different materials such as organic light-emitter diodes (OLEDs) due to its emission characteristics, as well as organic photovoltaics (OPVs) and solar cells due to its absorption properties.1
Owing to their promising optical and structural characteristics, pyrene-based molecules have attracted the researchers in the field of metal–organic frameworks (MOFs) to be used as ligands. MOFs are crystalline materials consist of metal-based building blocks linked by organic bridging ligands. By the careful combining of these metal nodes with these ligands, MOFs with desired properties can be obtained for target applications. The optical characteristics of pyrene as a MOF-ligand give pyrene-based MOFs some promising optical properties. These include its excimer formation combined with long-lived excited states, high fluorescence quantum yield, exceptional distinction of the fluorescence bands for monomer and excimer, and the sensitivity of its excitation spectra to micro-environmental changes.4 The coordination of pyrene-based ligands with metal centres can give rise to new photophysical and photochemical properties, which cannot be observed if the pyrene molecule is isolated.6 These properties have been successfully incorporated in MOF structures to the design of an extensive variety of luminescence and photocatalytic applications. The functionalization of pyrene with different groups including carboxylates, phosphonates, and sulfonates facilitates the generation of different MOF structures with diverse metal-ion coordination chemistry. Thanks to the structural variability and aromaticity of pyrene-based ligands, MOFs with promising features (e.g., large surface area, suitable pore size and shape, π–π stacking interactions) can be obtained for the adsorption and separation of different molecules. Moreover, the introduction of pyrene-based ligands into the MOF structures can enhance the electron transfer efficiency due to the strong π–π interactions between the pyrene and the reactant molecule, resulting in promising activity in heterogeneous catalytic applications. Consequently, pyrene-based MOFs are a growing field with published structures in the literature displaying interesting topological, optical, and physicochemical properties covering a suprisingly large number of different applications.
This increasing number of studies of pyrene-based MOFs in different fields has motivated this review. One can find reviews relate other chromophoric MOF families, such as porphyrin-based MOFs.7–9 In this review article, the reader can find information about the design and the development of pyrene-based molecules as organic linkers for MOF synthesis. The synthesis of pyrene-based MOFs and their post-synthetic modification using different methods (e.g., atomic layer deposition in MOFs to install metal ions (AIM), solvothermal deposition in a MOF (SIM), or solvent-assisted ligand incorporation (SALI)) are covered in detail. The nature of pyrene proposes different optical characteristics by exploiting its ground and excited state properties, which makes this molecule appealing for many applications. In addition, pyrene-based MOFs can lead to unique topology-based properties as a result of combining pyrene molecule with different transition metal ions. MOFs offer a great platform to combine different electronic, physicochemical, and topological properties in one porous material. Therefore, a specific application can take advantage of either one or couple of these characteristics at the same time. In this review, we discuss the most promising results of such pyrene-based MOFs combining different properties in several applications; including luminescence sensing, photocatalysis, adsorption and separation of molecules, heterogeneous catalysis, electrochemistry applications and bio-medical applications. This review can pave a way for researchers working on the development of novel pyrene ligands and pyrene-based MOFs for different applications.
2 Design and synthesis of pyrene-based metal organic frameworks (MOFs)
2.1 Pyrene-based building blocks as organic linkers in MOFs
Thanks to its fascinating characteristics, pyrene has been considered as a starting material for creating building blocks for MOFs, which makes this molecule even more attractive. By using pre-existing pyrene as the core, different electrophilic aromatic substitutions at different positions can be applied by direct or indirect methods, resulting in (i) 1-substituted pyrenes, (ii) 1,3,6,8-tetrasubstituted pyrenes, (iii) 1,6- and 1,8-disubstituted pyrenes, (iv) 1,3-disubstituted pyrenes, (v) 2,7-disubstituted pyrenes, and (vi) 4,5,9,10-tetrasubstituted pyrenes (Fig. 1).4 Among the substituted pyrene molecules, 1,3,6,8-tetra substituted pyrenes are the most studied ones for designing building blocks to be used in MOF synthesis. Being the most electron-rich and active centers, 1-, 3-, 6- and 8-positions on the pyrene core are the most favourable connection points for the electrophilic substitution, based on both experimental results and the calculations of molecular orbitals.4 According to theoretical calculations, the energy of the 1-, 3-, 6-, and 8-positions are lower by 8.8 kcal mol−1 than the 2- and 7-positions.1 There are several in-depth articles on the design and synthesis of substituted pyrenes that can participate in creating different molecular architectures.4,10,11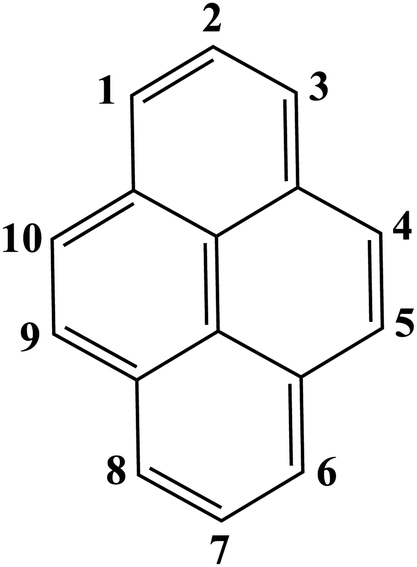 | ||
| Fig. 1 Illustration of the aromatic substitutions at different positions of pyrene. | ||
The bromine atom of a C–Br bond can easily be transformed to a C–C bond, C–N bond, or other group by Pd-catalyzed cross-coupling reactions.1 Given this, bromopyrenes have been thoroughly explored as key precursors for constructing building block materials. The synthesis of 1,3,6,8-tetrabromopyrene by Suzuki or Sonogashira coupling reactions gave rise to a increasing number of functionalized pyrene structures, which can be used in the synthesis of MOFs as organic ligands.4 The interest in the design of pyrene-based building blocks for MOF synthesis has tremendously increased after the synthesis of some novel pyrene-based tetratopic ligands. An important milestone was the synthesis of 1,3,6,8-tetrakis(p-benzoic acid)pyrene (TBAPy) ligand, which was obtained using a standard Suzuki–Miyaura reaction between 1,3,6,8-tetrabromopyrene and (4-(ethoxycarbonyl)phenyl)boronic acid, to design a fluorescent-MOF to respond to small guest molecules.12 TBAPy is a fluorescent ligand where the pyrene core is relatively rigid, and the benzoate groups substituted at the 1-, 3-, 6-, and 8-positions have a degree of rotational freedom. The carboxylate groups make this molecule attractive to link to the metal in a MOF. To date, different types of metals including alkali earth metals, transition metals, and post-transition metals have been coordinated with TBAPy ligand, resulting in 3D networks. These structures will be discussed in detail in the following sections. Following the interest on TBAPy, the researchers lately have focused on the synthesis of bulkier ligands by the incorporation of longer aromatic chains, such as biphenyl carboxylate and phenylethynyl carboxylate groups. Although tetra-substituted pyrenes are the most studied ligands for pyrene-based MOF synthesis, some other-mono and -di coordinated carboxylate-pyrene ligands and pyrene itself have also been studied for MOF synthesis. The functionalization of 2- and 7-positions on pyrene towards electrophilic aromatic substitution are less favorable than 1-, 3-, 6- and 8-positions, yet they can be activated selectively if a very bulky electrophile is employed.10 Pyrene-2,7-dicarboxylic acid (H2PDC) is as an example of the 2,7-disubstituted building blocks used in MOF synthesis. A Zn-MOF synthesized with H2PDC (IRMOF-14)13 has been particularly interesting thanks to its metal-substituted analogues M-IRMOF-14 (M = Zn, Cd, Be, Mg, Ca, Sr) with different chemical bonding, electronic structure, and optical properties.14Table 1 summarizes the structures and short names of pyrene-based ligands used in the synthesis of different MOFs to date.
| Pyrene-based ligands | Structures | MOFs | Metals used in MOFs | Ref. |
|---|---|---|---|---|
| 2,7-Pyrenedicarboxylic acid (PDC) |
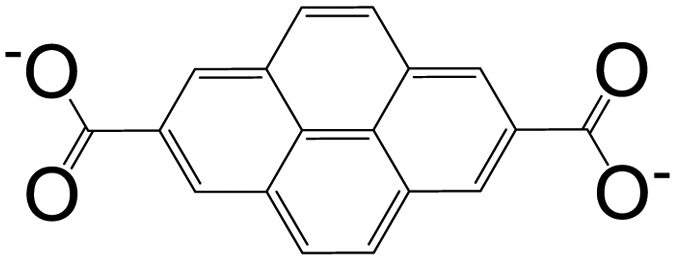
|
IRMOF-14 | Zn(II) | 13 |
| MOF-80 | Tb(III) | 43 | ||
| Sr(μ-DEF)(μ-PDC) | Sr(II) | 45 | ||
| NU-400 | Zr(IV) | 65 | ||
| 1,3,6,8-Pyrenetetrasulfonate (L3) |
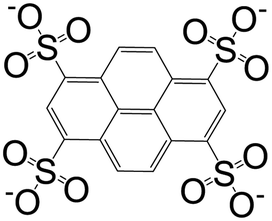
|
[Ag4(L3)(Pyr)4(H2O)2[Dy4(μ-pytet)(μ4-pytet)2(DMF)20(H2O)2]·4DMF·4H2O]n | Ag(I) | 44 |
| Dy(III) | 66 | |||
| 2-(Pyrene-1-carboxamido)terephthalic acid |
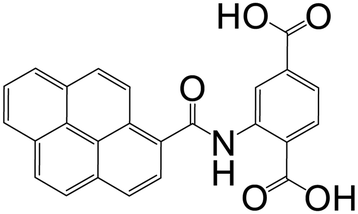
|
CPP-17-X | Gd(III), Eu(III), and Tb(III) | 67 |
| Octaethyl pyrene-1,3,6,8-tetraphosphonate |
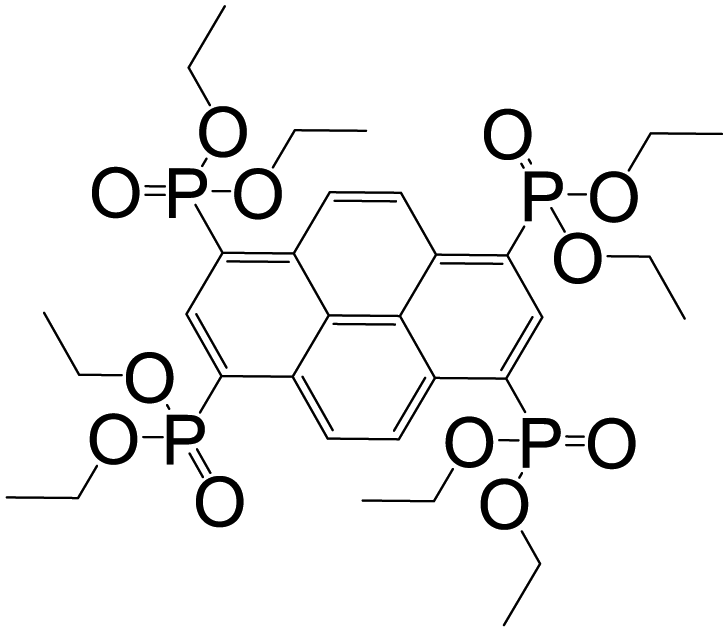
|
CALF-25 | Ba(II) | 53 |
| Pyrene-1,3,6,8-tetracarboxylic acid (PTCA) |
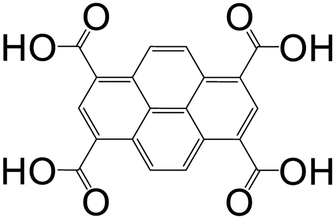
|
Mg-PTCA | Mg(II) | 68 |
| Ni-PTCA | Ni(II) | 69 | ||
| Zn-PTCA | Zn(II) | 70 | ||
| Cd-PTCA | Cd(II) | 71 | ||
| NU-1106 | Zr(IV) | 72 | ||
| 1,6-Pyrenedithiolate (PDT) |
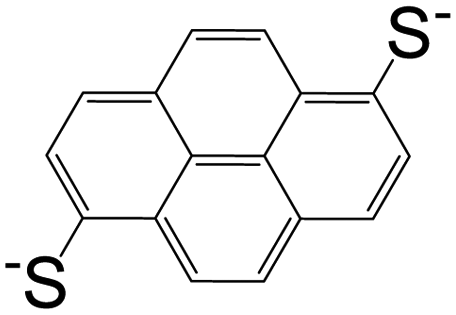
|
[Cd(PDT)2][Cd(en)3] | Cd(II) | 52 |
| 1,6-Bis(diphenyl phosphino)pyrene |
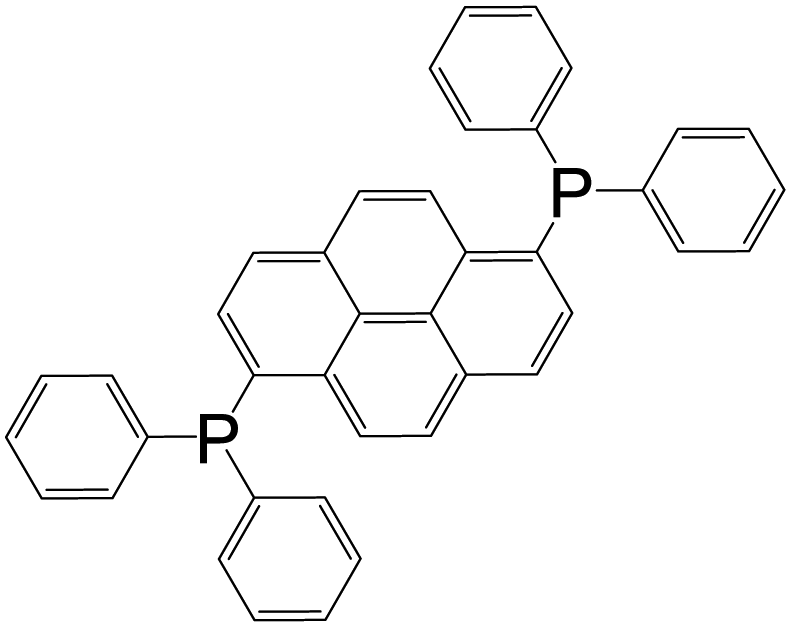
|
[Ag2L2(OTf)2] | Ag(I) | 48 |
| [Cu2L2(MeCN)4]2+ | Cu(I) | |||
| [Cu2L2I2] | Cu(I) | |||
| 3,3′,3′′,3′′′′-(Pyrene-1,3,6,8-tetrayl)tetrabenzoic acid (PTTB) |
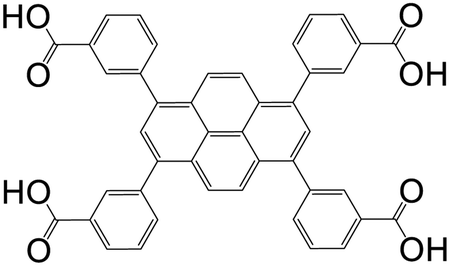
|
UTSA-72 | Zn(II) | 73 |
| 1,3,6,8-Tetrakis(4-methylthiophenyl)pyrene (L8) |
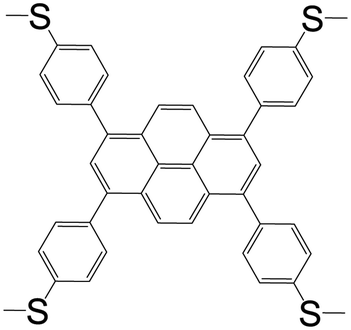
|
L82BiBr3 | Bi(III) | 46 |
| (4,4′-(2,8-Di-tert-butyl-4,10-dihydropyreno[4,5-d:9,10-d′]diimi-dazole-5,11-diyl)dibenzoic acid) (CIP) |
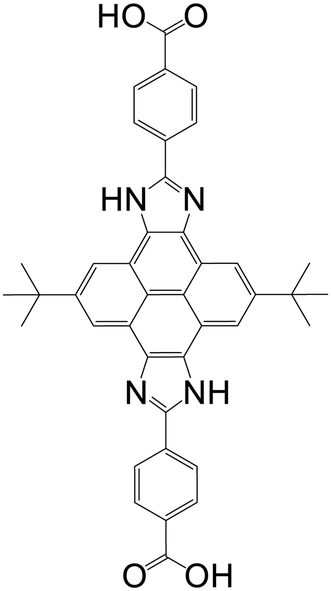
|
MAS-1 | Cu(II) | 47 |
| 1,3,6,8-Tetrakis(4-carboxylphenyl)pyrene (TBAPy) |
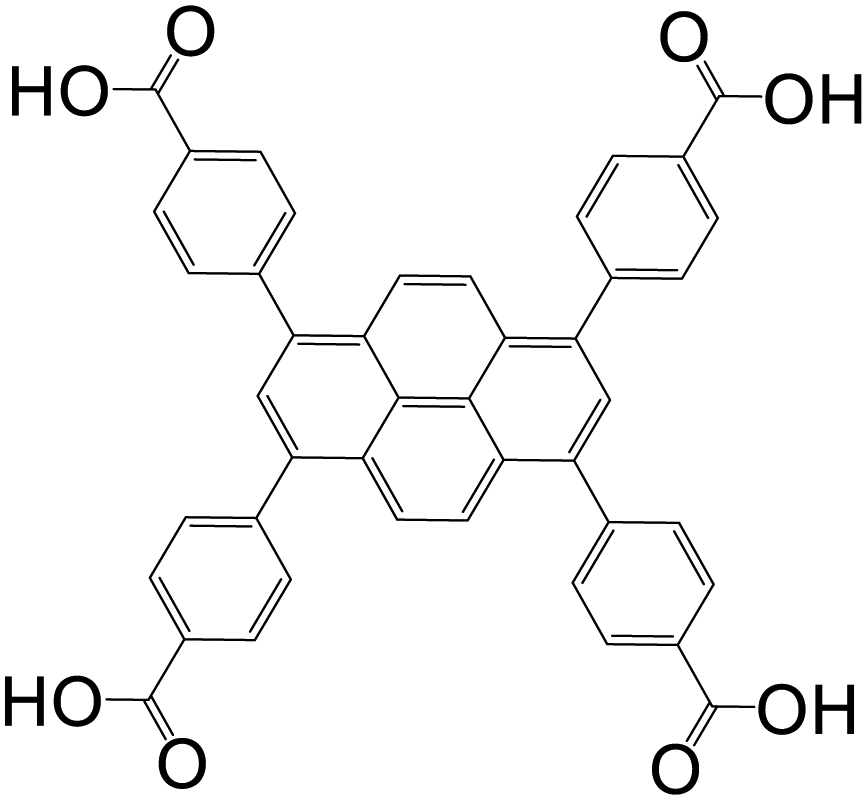
|
ROD-7 | In(III) | 12 |
| Zn-TBAPy | Zn(II) | 60 | ||
| NU-1000 | Zr(IV) | 74 | ||
| NU-901 | Zr(IV) | 75 | ||
| Ni-TBAPy | Ni(II) | 76 | ||
| ROD-6 | Mn(II) | 77 | ||
| ROD-8 | Cd(II) | 57 | ||
| NU-505(Zn, Ni) | Zn(II), Ni(II) | 78 | ||
| Zn-TBAPy | Zn(II) | 61 | ||
| NU-1300 | U(VI) | 79 | ||
| MLM 1–3 | Zr(IV) | 80 | ||
| MIT-26 | Mg(II) | 51 | ||
| UOF-1, UOF-2 | U(VI) | 81 | ||
| Cd-TBAPy | Cd(II) | 58 | ||
| Zr(TBAPy)5(TCPP) | Zr(IV) | 82 | ||
| SION-7 | Mg(II) | 83 | ||
| SION-19 | Zn(II) | 84 | ||
| AlPyr-MOF | Al(III) | 85 | ||
| Bi-TBAPy | Bi(III) | 86 | ||
| Bi-TBAPy | Bi(III) | 87 | ||
| Fe-TBAPy | Fe(II) | 88 | ||
| Cd-TBAPy | Cd(II) | 59 | ||
| Yb-TBAPy | Yb(III) | 89 | ||
| JXNU-5 | Eu(III) | 90 | ||
| SION-8 | Ca(II) | 91 | ||
| Zn-TBAPy | Zn(II) | 62 | ||
| Bi-NU-901 | Bi(III) | 92 | ||
| bioMOF 1 | Zn(II) | 93 | ||
| Zn-TBAPy | Zn(II) | 94 | ||
| M-TBAPy (M = Co, Zn) | Co(II), Zn(II) | 95 | ||
| TDL-Mg | Mg(II) | 96 | ||
| ACM-1 | Ti(IV) | 97 | ||
| SION-82 | Sr(II) | 98 | ||
| 1,3,6,8-Tetrakis(3,5-isophthalic acid)pyrene (TIAPy) or 1,3,6,8-tetrakis(3,5-dicarboxyphenyl)pyrene (tdcppy) |
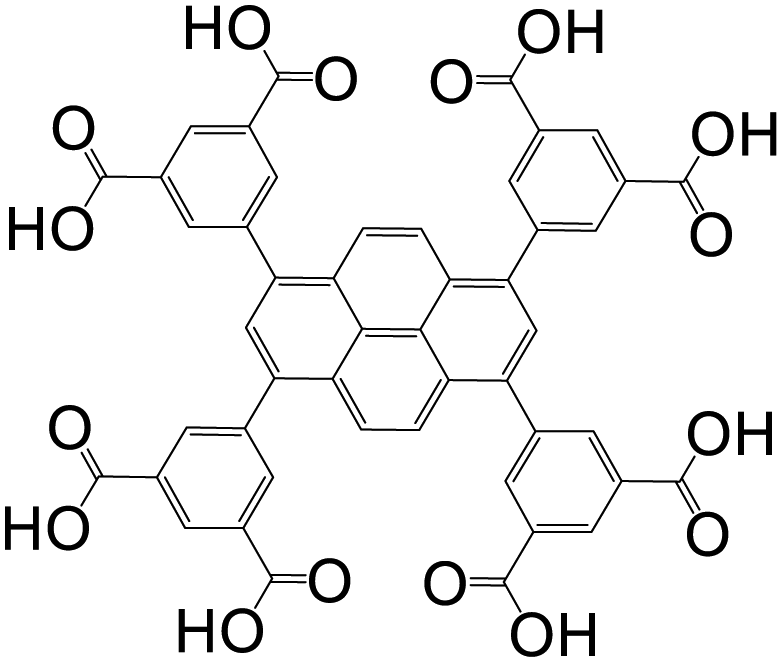
|
JUC-118 | Zn(II) | 49 |
| MgMOF | Mg(II) | 99 | ||
| Co-tdcppy | Co(II) | 100 | ||
| JUC-119 | Eu(III) | 101 | ||
| JUC-138 | In(III) | 102 | ||
| 1,3,6,8-Tetrakis(4-(α-carboxy-(R)-α-methyl)methoxy-2,6-dimethylphenyl)pyrene (PLA) |
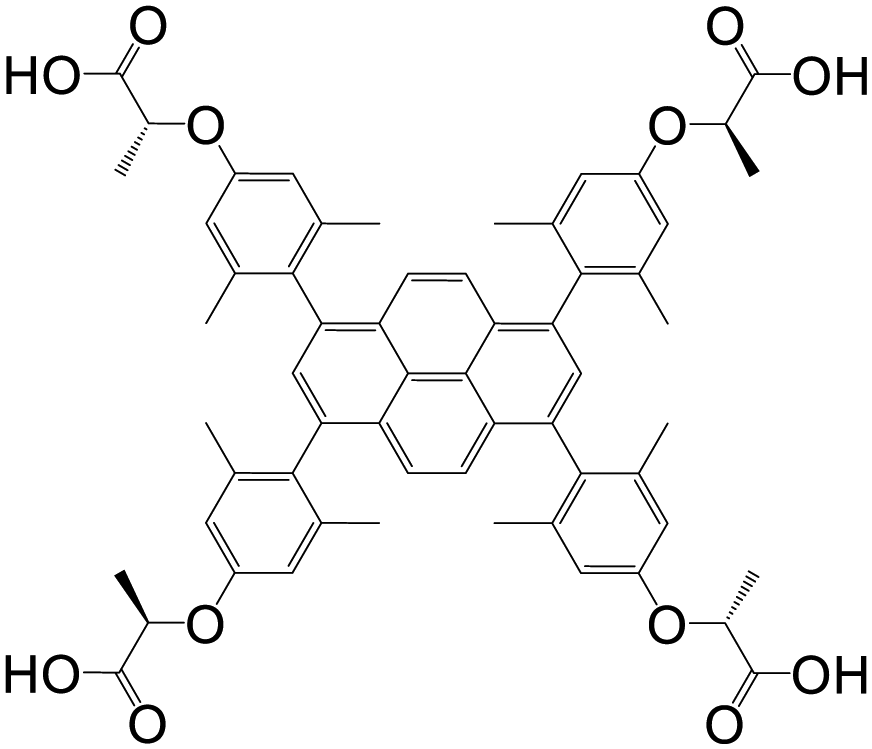
|
Zn-PLA | Zn(II) | 103 |
| 3,3′,3′′,3′′′-(Pyrene-1,3,6,8-tetrayltetrakis(benzene-4,1-diyl))tetraacrylate (PTSA) |
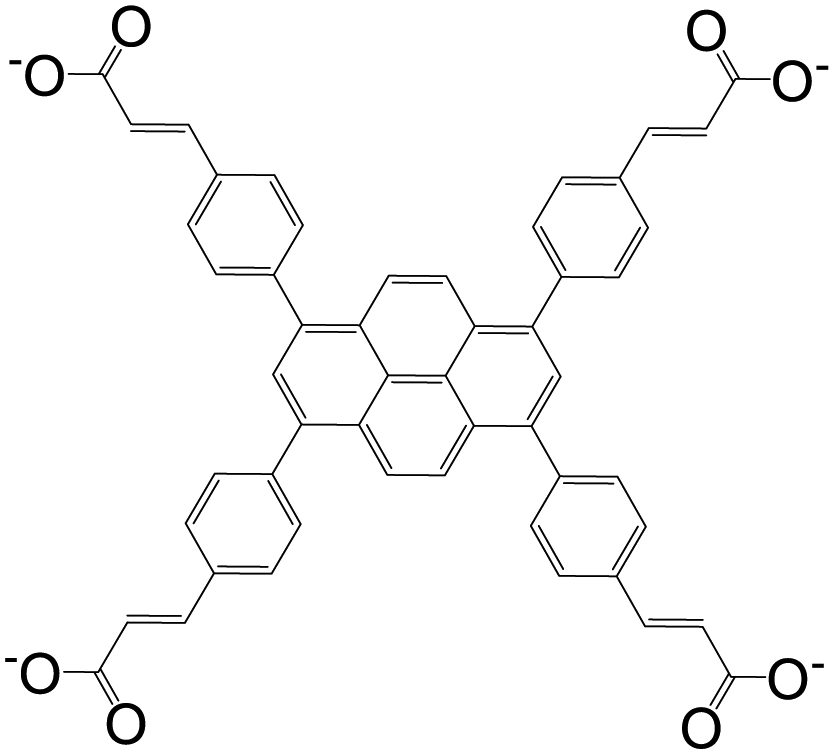
|
BUT-72 | Zr(IV) | 55 |
| 6,6′,6′′,6′′′-(Pyrene-1,3,6,8-tetrayl)tetrakis(2-naphthoate)(PTNA) |
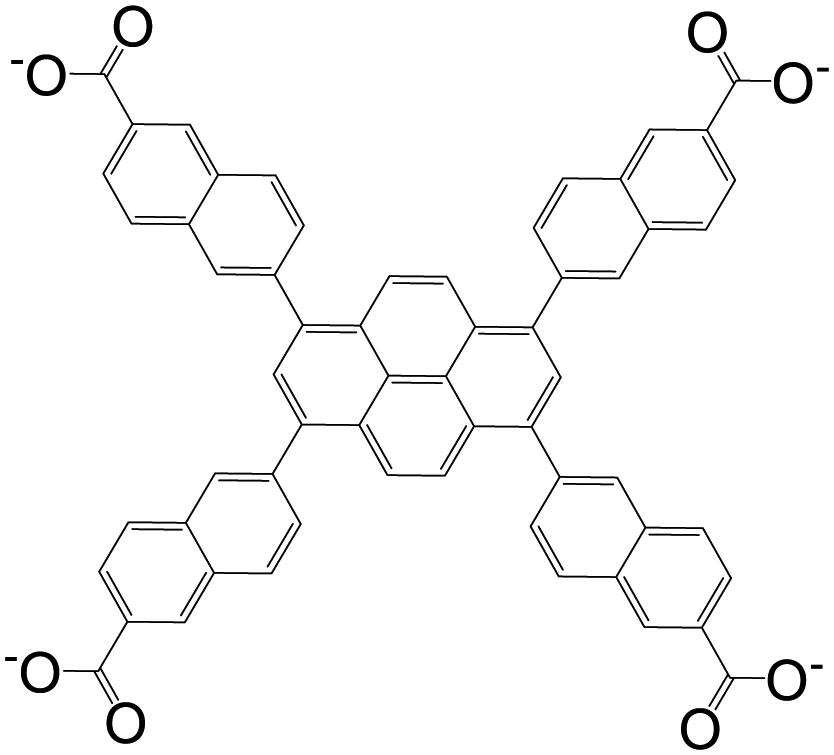
|
BUT-73 | Zr(IV) | 55 |
| NU-1003 | Zr(IV) | 104 | ||
| 4,4′,4′′,4′′′-(Pyrene-1,3,6,8-tetrayltetrakis(ethyne-2,1-diyl))tetrabenzoic acid (PTBA) |
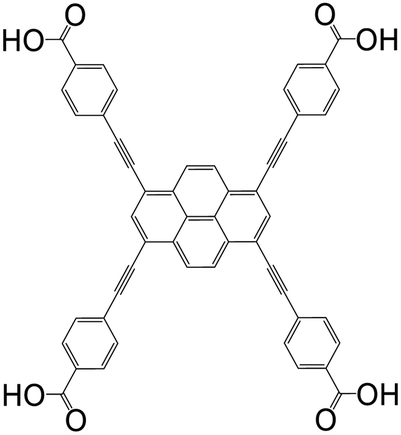
|
NU-1100 | Zr(IV) | 105 |
| Zrptba | Zr(IV) | 54 | ||
| 1,3,6,8-Tetrakis(2,6-dimethyl-4-(α-carboxy)methoxyphenyl)pyrene |
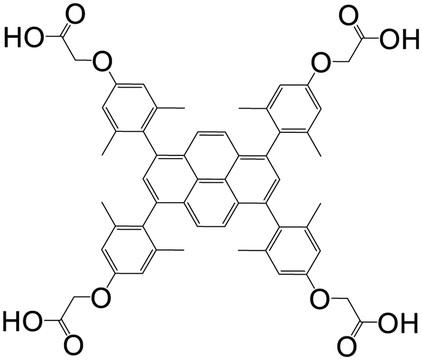
|
Mn-L | Mn(II) | 106 |
| 7,7′,7′′,7′′′-(Pyrene-1,3,6,8-tetrayl)tetrakis(9,9-dimethyl-9H-fluorene-2-carboxylic acid) |
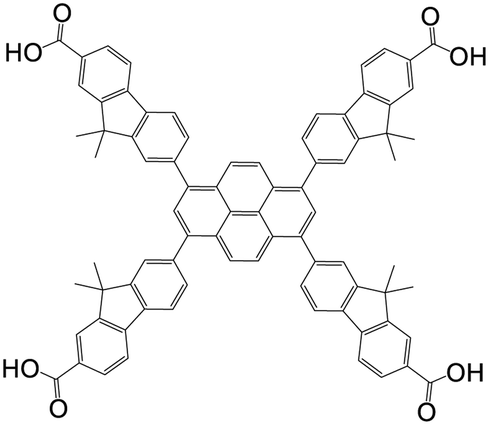
|
NU-1004 | Zr(IV) | 107 |
| 4,4′,4′′,4′′′-(pyrene-1,3,6,8-tetrayltetrakis(9,9-dimethyl-9H-fluorene-7,2-diyl))tetrabenzoic acid (Py-FP) |
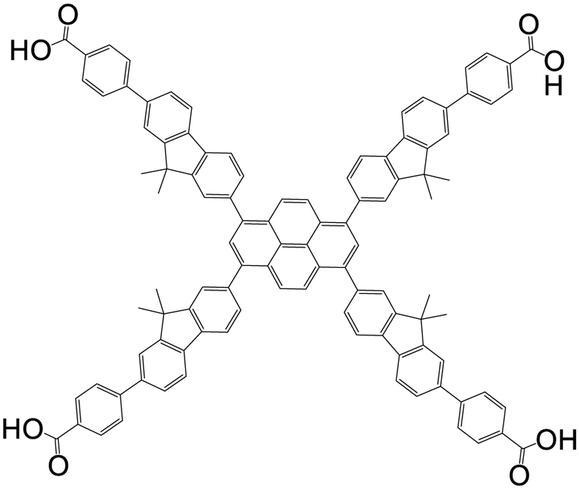
|
NU-1105 | Zr(IV) | 108 |
| 1,3,6,8-Tetrakis(4′-carboxy[1,1′-biphenyl]-4-yl-)pyrene (TCBPPy) |
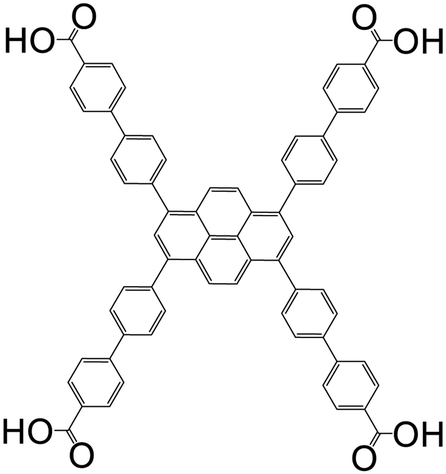
|
LMOF-1 | Zr(IV) | 109 |
| 4′,4′′′,4′′′′′,4′′′′′′′-(Pyrene-1,3,6,8-tetrayl)tetrakis(2′,5′-dimethyl-[1,1′-biphenyl]-4-carboxylic acid) (Py-XP) |
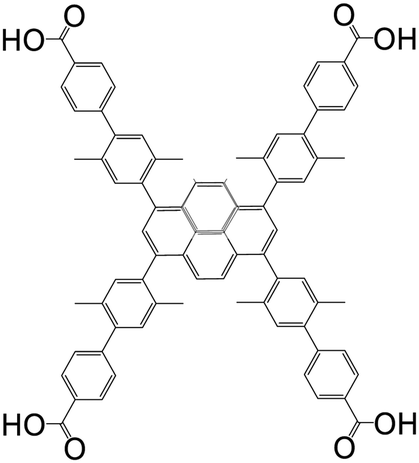
|
NU-1101 | Zr(IV) | 50 |
| 4,4′,4′′,4′′′-((2,7-Di-tert-butylpyrene-4,5,9,10-tetrayl)tetrakis(ethyne-2,1-diyl))-tetrabenzoate (BPETB) |
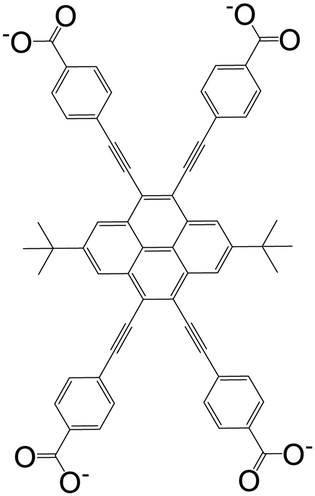
|
PCN-822(Zr, Hf) | Zr(IV) | 110 |
| Hf(IV) | ||||
| 4,4′,4′′,4′′′-((Pyrene-1,3,6,8-tetrayltetrakis(benzene-4,1-diyl))tetrakis(ethyne-2,1-diyl))tetrabenzoic acid (Py-PTP) |
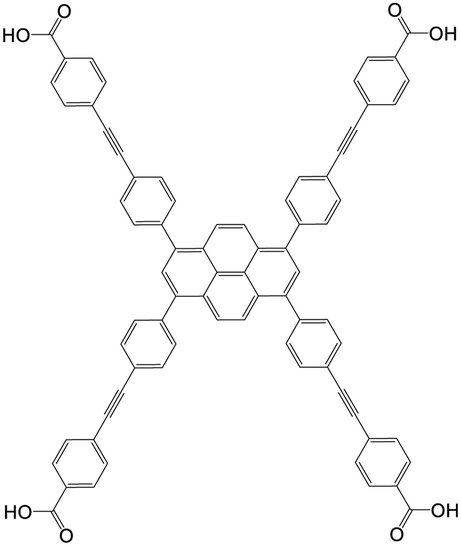
|
NU-1006 | Zr(IV) | 107 |
| NU-1103 | Zr(IV) | 50 | ||
| 4,4′,4′′,4′′′-((1E,1′E,1′′E,1′′′E)-(Pyrene-1,3,6,8-tetrayltetrakis(benzene-4,1-diyl))tetrakis(ethene-2,1-diyl))tetrabenzoic acid |
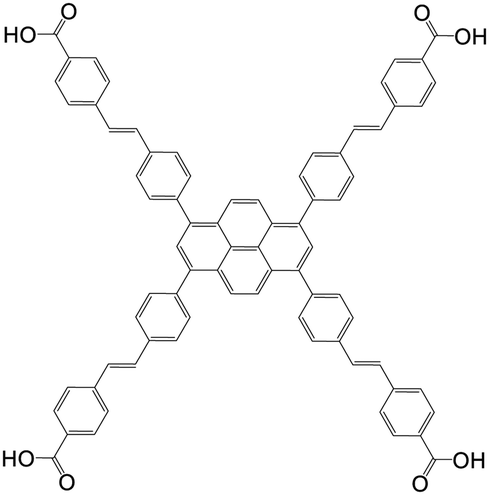
|
NU-1005 | Zr(IV) | 107 |
| 4′,4′′,4′′′,4′′′′-(Pyrene-1,3,6,8-tetrayl)tetrakis(2′,2′′,5′,5′′-tetramethyl-[1,1′,4′,1′′-terphenyl]-4-carboxylic acid |
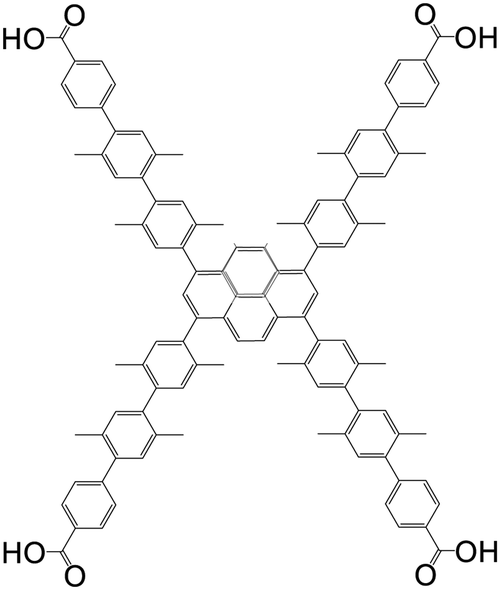
|
NU-1007 | Zr(IV) | 107 |
2.2 Synthesis of pyrene-based MOFs
The first use of pyrene molecule with MOFs was in 1995, where it was intercalated into a copper(I) coordination polymer as a guest molecule.15 The solvation of crystal data demonstrated that pyrene molecules could be placed in between the phazine stacks in a one-dimensional chain thanks to their suitable size and shape. The use of pyrene as a guest molecule in a MOF was performed in many other studies in the following years, to take advantage of the favorable characteristics of pyrene for different applications.16–39 The pyrene molecule can be included in MOFs also by (i) its attachment to an organic linker, and (ii) its use as a building block for the construction of the MOF after inserting desired functionalizations.2 In this review, we will mainly focus on the use of pyrene-based organic building blocks in the synthesis of MOFs. The use of pyrene chromophore for constructing MOF structures was first performed by the incorporation of 2,7-diazapyrene40,41 and naphtho(2,3-a)pyrene42 molecules as building blocks into 1D and 2D coordination polymers. An important study for the use of pyrene molecule in MOF synthesis was the study of Eddaoudi et al.,13 where the systematic design of pore size and functionality in isoreticular MOFs were investigated for methane storage. This study was the first example of a highly porous, 3D pyrene-based metal–organic network. The increasing number of attempts for designing new pyrene-based building blocks has resulted in an increasing number of novel MOF structures. Early studies mainly focused on the design and synthesis of novel pyrene-based MOFs.43–49 With the increasing attention, investigations have been concentrated on the structural properties,50–52 and stability of pyrene-based structures in different environments,53–56 in order to open a road for other researchers in the field. With the better understanding of the structures, the possible applications of these structures have started to be considered, which will be discussed in the following sections.An interesting feature of pyrene-based MOFs is that the use of the same metal and the ligand in the synthesis can result in different topologies and structural properties. A well-known example is cadmium-based pyrene-MOFs, in which the syntheses have been performed with a specific cadmium salt (Cd(NO3)2·4H2O) and TBAPy ligand, resulting in three different MOFs with different structures.57–59 ROD-8 is a 3D rod-type MOF with 1D porous channels and it crystallizes in the monoclinic space group P21/c.57 Although the positions of the oxygens in carboxylates in TBAPy are crystallographically fixed, the pyrene core with four branching phenyl groups can exhibit disorder with two different positions as shown in Fig. 2(a). Depending on the distribution of the two positions of the ligand, ROD-8 can be described by the two extreme ordered derived nets, lrk and lrl, which are both derived from the basic net lrj. ROD-8 was found to be promising for CH4 capture by virtue of its structural properties. Another example is Cd-TBAPy, which is a 2D layered framework with 1D channels (Fig. 2(b)) crystallized in triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) .58 Owing to its π-conjugated 2D layered structure and visible-light absorption edge of ≈600 nm, both water reduction and oxidation under visible-light irradiation can be performed with Cd-TBAPy. The last case is [Cd(TBAPy)(H2O)2]·4(H2O)n(S1), a 3D structure with 1D chains (Fig. 2(c)) crystallizes in a monoclinic space group P21/n.59S1 behaves as a thermometer for fluorescence sensing of temperature in a wide temperature range. Interestingly, the resulting MOFs perform well for such different applications based on their structural characteristics. It should be highlighted that not only the starting materials but also the synthesis conditions and solvents used in the synthesis of ROD-8 and Cd-TBAPy are very similar: they were both synthesized at 120 °C for 72 h using a mixture of DMF/dioxane/H2O mixed solvent. In the case of S1, the conditions are slightly different: a DMF/H2O solution was used and the synthesis was performed at 140 °C for 72 h. Nonetheless, the main difference between the three cases were the concentrations and the ratios of the metal salt and the ligand.
.58 Owing to its π-conjugated 2D layered structure and visible-light absorption edge of ≈600 nm, both water reduction and oxidation under visible-light irradiation can be performed with Cd-TBAPy. The last case is [Cd(TBAPy)(H2O)2]·4(H2O)n(S1), a 3D structure with 1D chains (Fig. 2(c)) crystallizes in a monoclinic space group P21/n.59S1 behaves as a thermometer for fluorescence sensing of temperature in a wide temperature range. Interestingly, the resulting MOFs perform well for such different applications based on their structural characteristics. It should be highlighted that not only the starting materials but also the synthesis conditions and solvents used in the synthesis of ROD-8 and Cd-TBAPy are very similar: they were both synthesized at 120 °C for 72 h using a mixture of DMF/dioxane/H2O mixed solvent. In the case of S1, the conditions are slightly different: a DMF/H2O solution was used and the synthesis was performed at 140 °C for 72 h. Nonetheless, the main difference between the three cases were the concentrations and the ratios of the metal salt and the ligand.
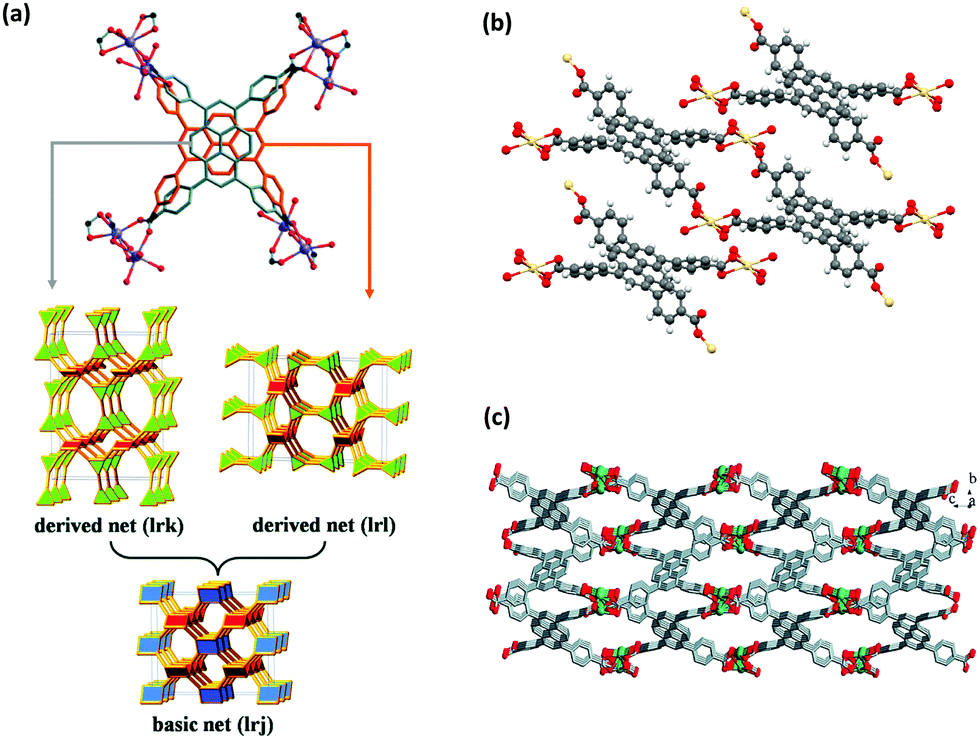 | ||
| Fig. 2 (a) The 3D structure of ROD-8.57 Although the positions of the carboxylate-O in TBAPy are crystallographically fixed, the voids allow the pyrene core with four branching phenyl groups to rattle between the two extreme positions of perpendicular orientation (demonstrated in silver and orange). Therefore, the overall 3D MOF experiences a type of framework disorder. The two positions can be regularly or randomly distributed in the framework, resulting in derived lrk and lrj nets. (b) The structural representation of 2D layers of Cd-TBAPy.58 (c) The 3D structure of S1.59 Reprinted with permission from ref. 57 and 59. | ||
Another example is Zn-based pyrene-MOFs, where the syntheses have been performed with Zn-salts and TBAPy ligand. Due to the different solution environments, different Zn-TBAPy-MOFs with different characteristics have been obtained.60–62 The structural comparisons between [Zn(TBAPy)1/2(H2O)2],62 [Zn2(TBAPy)(H2O)2]·(Guests)x),60 [Zn2(TBAPy)],60 and [Zn2(TBAPy)(H2O)2]·3.5DEF61 showed that the use of an ionic liquid in the synthesis of [Zn(TBAPy)1/2(H2O)2] resulted in a large separation between the parallel pyrene cores, allowing intercalation of the other layers to form the highest dense packing of chromophores. It is well known that the structure and morphology of MOFs depend on not only the reactants but also many synthetic condition parameters; including solvent type, pH value of the reaction mixture, time, the molar ratio of starting materials, the presence of counterions and pressure.63 These parameters are deterministic on the structural chemistry of ligands and the assembly process of ligands with metal centres, leading to products with diverse structures.63 This can explain the structural variety of Cd-TBAPy and Zn-TBAPy MOFs well.
When the pyrene-based MOFs in which the metal ions with the same coordination environment are considered, the situation is even more interesting. Similar to the cases above, the reaction solvents and conditions used in the synthesis of TBAPy MOFs based on In(III),12 Zn(II),60 Mg(II)68 and Zr(IV)74 are comparable (DMF and H2O and/or dioxane and/or HCl). Within these frameworks, all aforementioned metal ions are octahedrally coordinated; however, none of these frameworks is isostructural to each other. This is due to the formation of different forms of metal nodes (chains or clusters) as well as different orientation of TBAPy ligand around the metal atoms. This was proved by the investigation of ([Zn2(TBAPy)(H2O)2]·(Guests)x).60 When the axial coordinated H2O molecule to Zn(II) was removed upon activation, the MOF has a different topology in which Zn(II) was found to be tetrahedrally coordinated. This clearly explains that the role of the central metal atom (and thus the formation of metal clusters or chains) is also important for the final topology of the framework. In addition, the orientation of the TBAPy ligand around the metal atom is dependent on the available coordination sites of the metal atom. Based on these examples, it can be deduced that the structural differences unveil the need for more dedicated studies for the exploration of the synthesis conditions of pyrene-based MOFs.
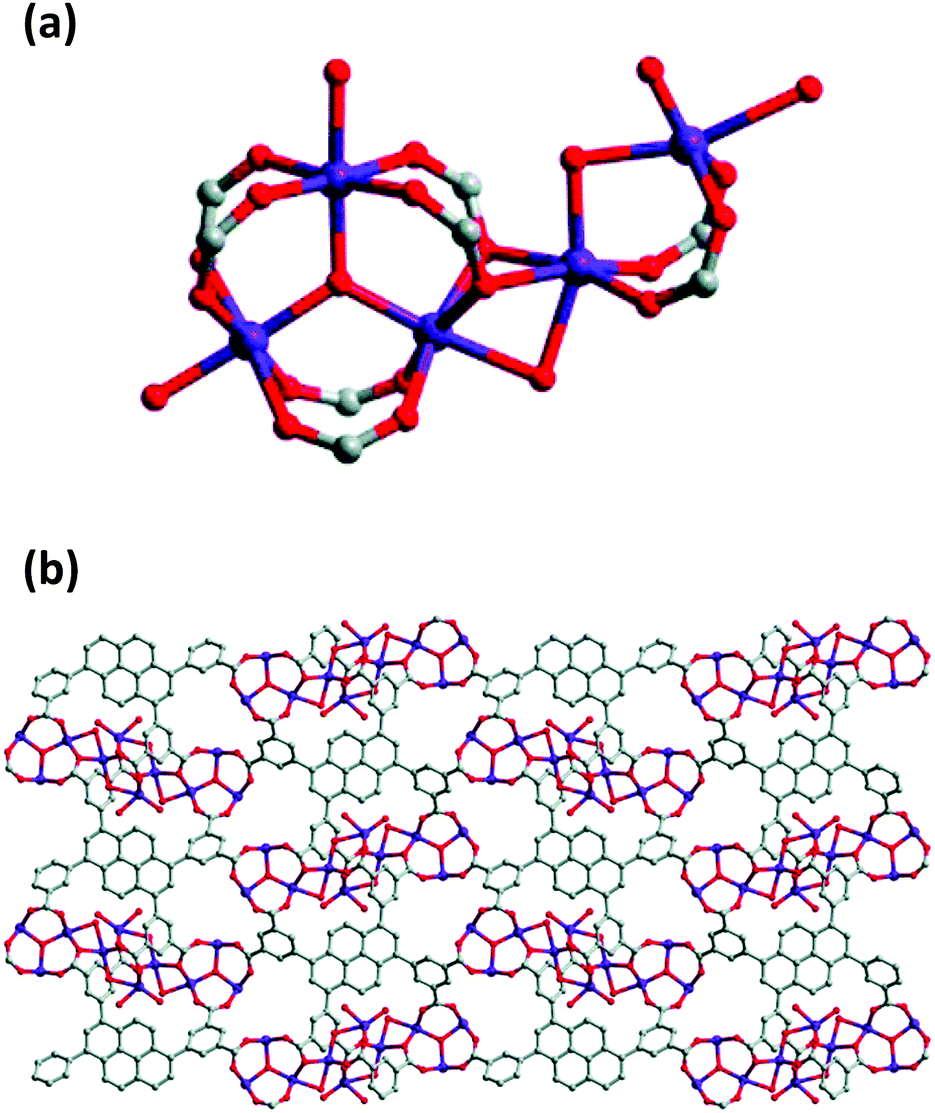
The interest in pyrene-based MOFs has tremendously increased with the synthesis of NU-1000, a zirconium-based MOF consisting of eight-connected octahedral [Zr6(μ3-O)4(μ3-OH)4(OH)4(H2O)4]8+ nodes bridged by the carboxylate groups of TBAPy ligands (Fig. 3(a) and (b)).74,111 The extended 3D structure of NU-1000 has uniform hexagonal channels with the diameter of 31 Å and triangular channels with the diameter of 10 Å (Fig. 3(c)), in addition to the smaller pores with the diameter of 8 Å separating the ab layers along the c direction (Fig. 3(d)).111 Due to its attractive properties such as having a high Brauner–Emmett–Teller (BET) surface area (2320 m2 g−1) and pore volume (1.4 cm3 g−1), good thermal stability, fluorescence properties and the suitability of the structure for metalation or other types of functionalization, NU-1000 has drawn attention since its first synthesis. As a result, the chemical and structural properties of NU-1000 have been deeply studied experimentally and computationally; by exploring its synthesis conditions,112–117 structural stability,118,119 proton topology and surface chemistry,120–123 and designing organic linkers for its better crystallinity.124
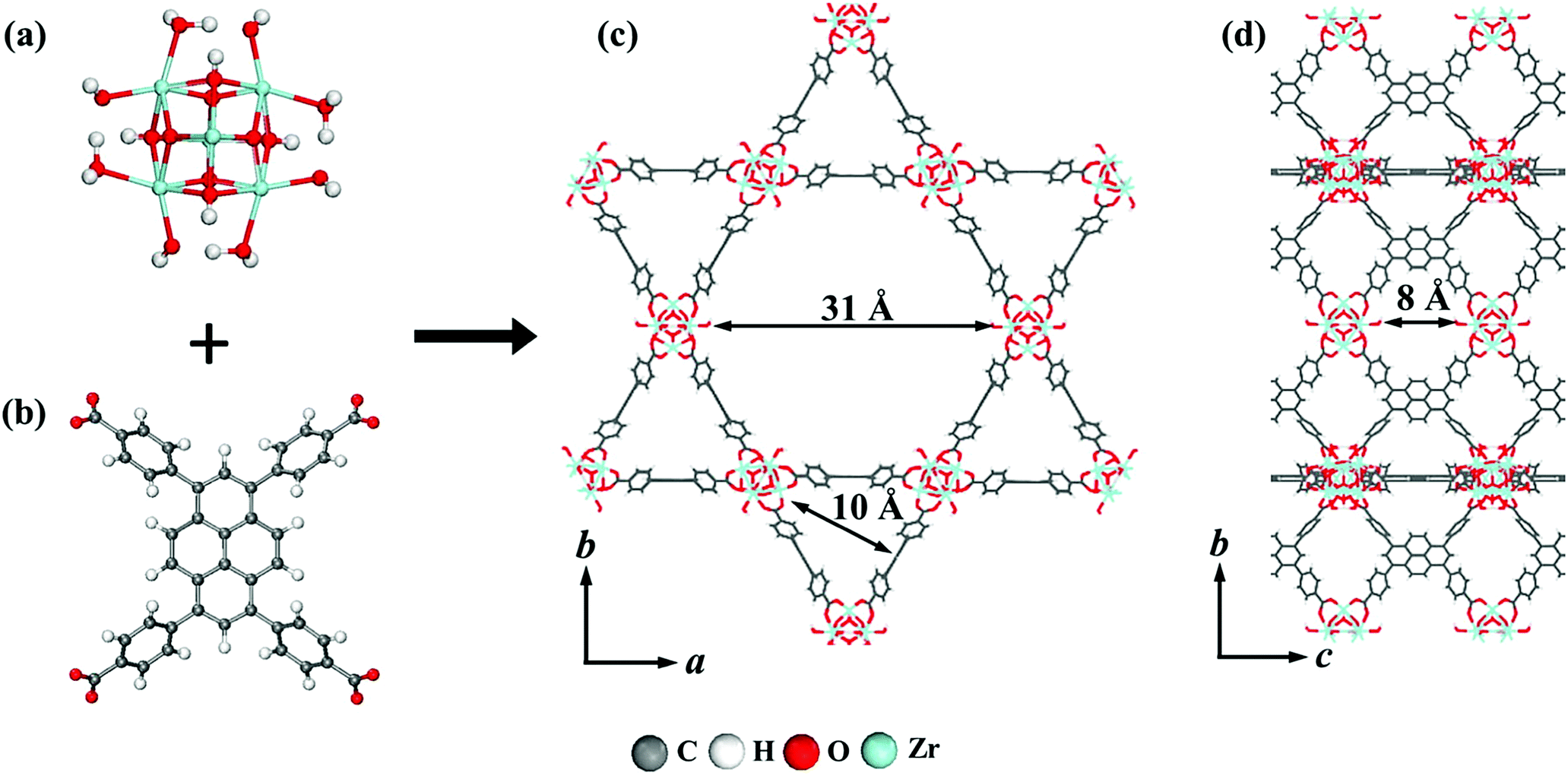 | ||
| Fig. 3 The structure of NU-1000 (a) the Zr node, (b) TBAPy ligand, (c) the hexagonal and triangular channels along the c direction, and (d) the small pore connecting the ab layers along the c direction. Reprinted with permission from ref. 111. | ||
2.3 Post-synthetic modification of pyrene-based MOFs
Post-synthetic modification has been a favourable approach for tuning the physical and chemical properties of MOFs by introducing different functionalities into the structure after the framework is synthesized. Post-synthetic modifications can be performed by the modification of the linkers through covalent bond formation or cleavage, or the modification of the metal clusters by introducing the formation and/or cleavage of coordination bonds.125 In the case of pyrene-based MOFs, the most common post-synthetic modifications are (i) the incorporation of metal ions via atomic layer deposition in a MOF (AIM),126–128 (ii) solvothermal deposition in a MOF (SIM),129 and (iii) solvent-assisted ligand incorporation (SALI).130–132 These methods are especially attractive for the post-synthetic modification of TBAPy-based MOFs due to the presence of eight terminal (i.e. non-bridging) –OH groups on the ligand, which makes the structure suitable for channel modification.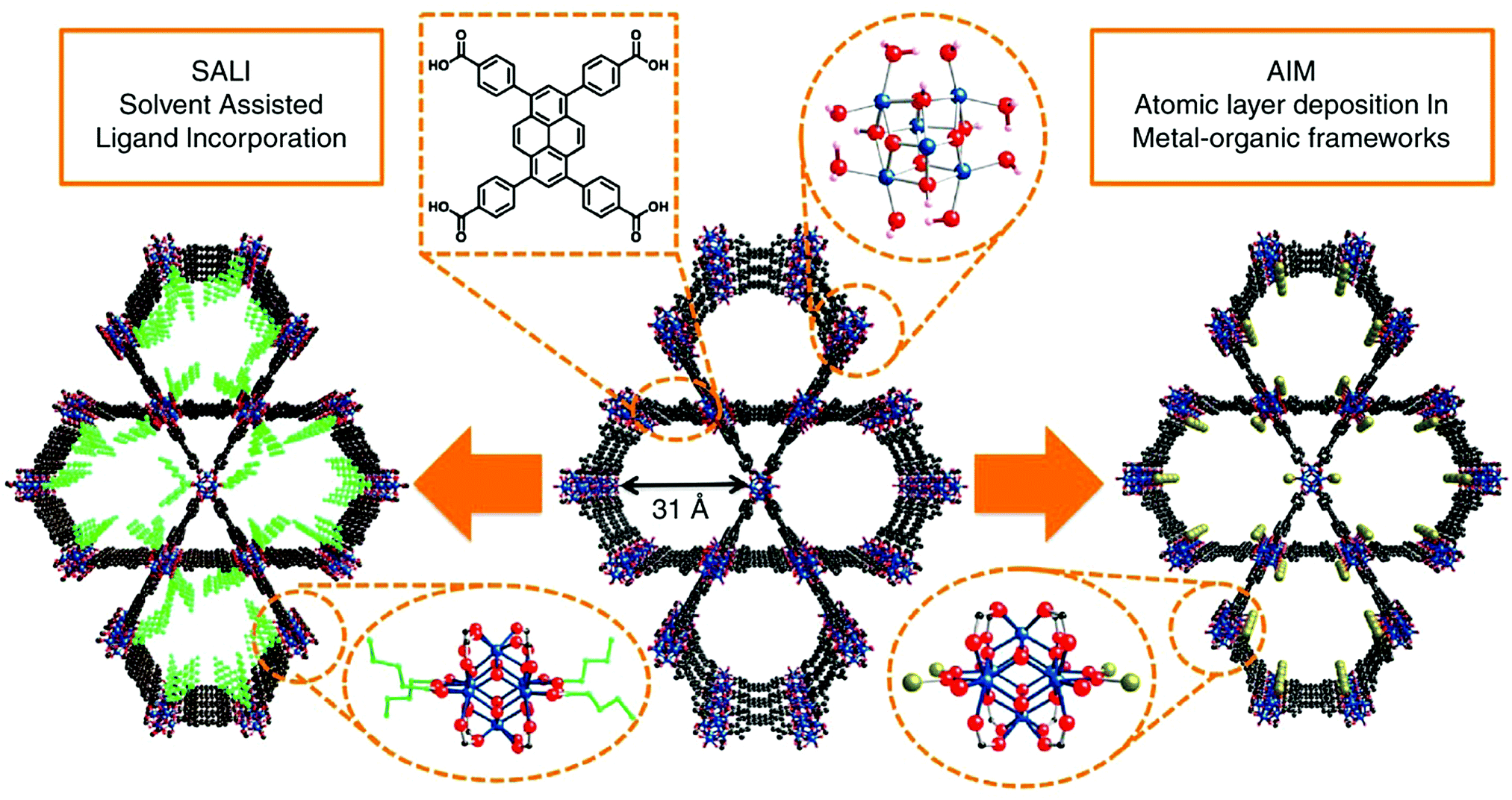 | ||
| Fig. 4 Post-synthetic functionalization of NU-1000 by AIM and SALI. The ligand incorporation into NU-1000 by SALI is represented in green, and the gold sphere shows the metal cluster introduced into NU-1000 by AIM. Color code: Zr: blue; O: red; and C: black. Reprinted with permission from ref. 112. | ||
In a typical AIM experiment, the metal precursor is used as the first reactant while the co-reactant can differentiate depending on the desired resulting product (e.g. H2O vapor can be used for the single-metal site or metal-oxide formation, whereas H2S can be pulsed for the metal-sulfide formation). A custom-made stainless steel sample holder containing MOF powder is placed in the ALD chamber. Then, the sample is heated up to 125 °C before exposure to reagents to remove physisorbed water. After heating, the metal precursor is introduced and maintained at a temperature around 100–140 °C, where the pulses of metal precursor follows the time sequence of t1–t2–t3: t1 is the precursor pulse time, t2 is the substrate exposure time, and t3 is the inert gas purge time. To ensure full metalation of the metal sites of the MOF throughout the microcrystals, the metal pulsing cycle is repeated couple of times. Then, the sample is subsequently exposed to co-reactant pulses with a specific t1–t2–t3 time sequence. Following the AIM, the sample is thermally activated. Although AIM is a useful method for the metalation of nodes, the MOF should have some characteristics to be considered for AIM, including relatively good stability and labile sites for replacement. Hence, AIM was first performed on NU-1000, due to its advantageous properties: (i) having mesoporous pores to ease the diffusion of ALD reactants, (ii) having thermal and hydrolytical stability, and (iii) the presence of functional groups (–OH groups) on Zr6 nodes to ensure the metalation. As a proof of concept, the first attempt of AIM with NU-1000 was performed with Zn and Al in an ALD reactor at 110 and 140 °C, resulting in Zn-AIM and Al-AIM successfully.74 PXRD measurements showed the retained crystal structure after the metalation while BET measurements indicated a decrease in surface area of NU-1000 from 2320 m2 g−1 to 1580 and 1160 m2 g−1 for Zn-AIM and Al-AIM, respectively. The metalation of NU-1000 has been performed with different single metals and metal clusters up to date thanks to AIM.126,136–138 Some of them were found to be very promising for different applications, mainly for catalysis, which will be discussed in Section 3. Furthermore, the bonding energetics of metal vapor adsorption onto NU-1000 for the metalation of the nodes has been investigated using different experimental techniques and density functional theory (DFT).139
The other common method for cluster metalation in a pyrene-based MOF is solvothermal deposition in MOF (SIM), which is complementary to gas-phase metalation of the node (AIM). In this method, the modifying metal species are dissolved in a non-reactive solvent (e.g. dimethylformamide (DMF), methanol or heptane) followed by soaking of the MOF into the solution.140 In some cases, the metal precursors are only stable in air- and water-free environments, therefore the deposition is performed under inert conditions (i.e. in an Argon filled glovebox) by using anhydrous solvents.141–144 The solution can be left at room temperature,141–145 or heated at a certain temperature129,146,147 for a specific amount of time, to provide the formation of metal species on the structure. The resulting powder is washed and then activated in a drying oven. In order to perform SIM efficiently, it is important to have a the metal source which is reactive toward –OH and H2O functionalities on the metal node, and the structural integrity should be unaffected from the metalation to preserve the original characteristics. The metal should be soluble in the solvent; however, it should be highlighted that the formation of coordination complexes between the metal source and the solvent should be inhibited.148 A color change can be observed in the samples, indicating the successful incorporation of metal species. As in the case of AIM, Zr-nodes of NU-1000 have been modified with different species; such as ions,146,149 oxides,141,142,145 sulfides,143 and single-metal atoms144,147 to date for different target applications.
Although both AIM and SIM are successful methods for incorporating metal species into the structure, there are some structural differences in the resulting samples, causing different performances on a specific application. In order to investigate this, Li et al.149 made a study where the incorporation of Co(II) ions to the Zr6 nodes of NU-1000 was performed via SIM and AIM, denoted as Co-SIM + NU-1000 and Co-AIM + NU-1000, respectively. Both catalysts were first subjected to an activation process, by heating at 230 °C with a flow of 48 mL min−1 O2 (10% diluted with He) and 96 min−1 of C3H8 (3%, diluted with Ar) until the propane conversion is constant. After activation, both materials were found to be active toward the oxidative dehydrogenation of propane to propene, at reaction temperatures as low as ≈200 °C. However, catalytic activity as well as propene selectivity of these two catalysts were different under the same experimental conditions due to the differences in the Co species generated in Co-SIM + NU-1000 and Co-AIM + NU-1000 materials upon activation, which was analyzed by in situ X-ray absorption spectroscopy. Upon activation with O2, a new Co⋯Co interaction appeared for Co-AIM + NU-1000, which is absent in Co-SIM + NU-1000. This observation indicates that the selection of the post-modification method can be of importance depending on the target application.
Owing to its high thermal and chemical stability as well as the presence of the terminal –OH groups on the Zr metal nodes, NU-1000 has been found promising for the functionalization with different moieties by SALI. Deria et al.130 used the approach SALI with NU-1000 for the first time in the literature, by attaching perfluoroalkyl carboxylic acids of varying chain length (1, 3, 7, and 9) within the mesoporous channels of the MOF. SALI was performed by soaking NU-1000 in a concentrated solution of fluoroalkane carboxylic acid ligand in DMF at 60 °C for 18–24 h. The resultant materials were named as SALI-n (e.g., SALI-1–SALI-9), where n is defined based on the length of the carboxylic acid ligand used in the synthesis. The N2 adsorption isotherms and BET surface area analyses of SALI-n samples indicated a systematic decrease in surface area from 2320 m2 g−1 for NU-1000 to 1710 and 870 m2 g−1 for SALI-1 and SALI-9, respectively. In addition, the pore sizes and pore volumes of the samples decreased with increasing chain length of the modifying ligand. Nevertheless, CO2 adsorption studies showed that perfluoroalkane-functionalized nodes in the SALI-n system increased the affinity toward CO2, resulting in systematically higher values for Qst with increasing chain length. Followed by this study, different ligands have been introduced to NU-1000 by SALI and the resulting materials were found to be promising for separation, catalysis and storage.131 Furthermore, SALI modification can be followed by AIM or SIM to deposit single metal sites on the incorporated ligand. SALI method can expand the pyrene-based MOFs in several directions by the incorporation of various types of charged anchoring moieties, multiple light harvesters or redox active moieties for the design of functional arrays or photoredox antenna systems.151
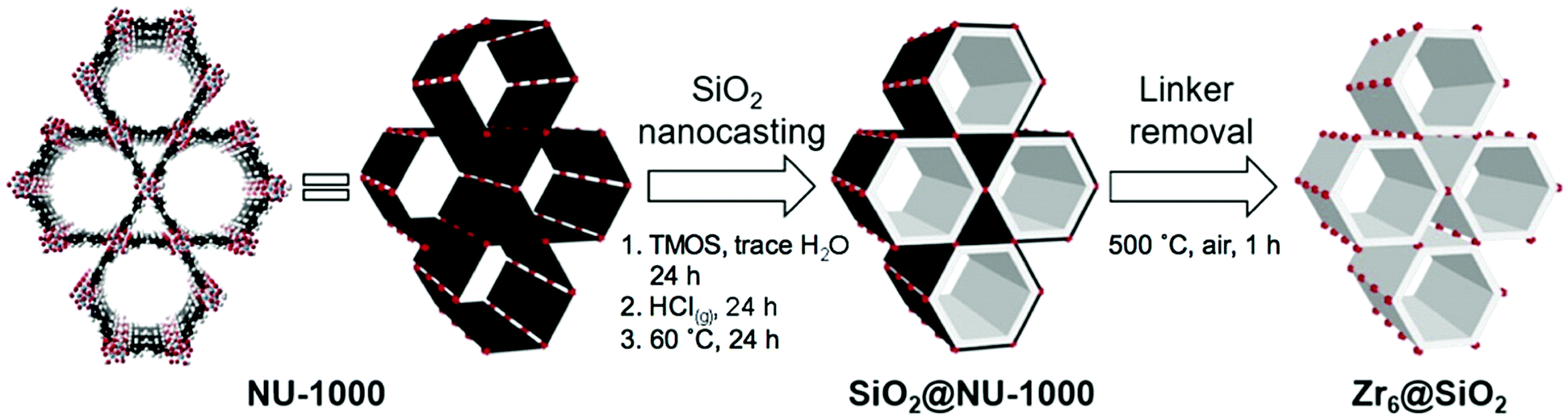 | ||
| Fig. 5 The schematic representation for the nanocasting process of NU-1000 with silica. The image on the left represents the NU-1000 structure with Zr-clusters (red) and TBAPy linkers (black). The white layer in the middle image represents silica. Reprinted with permission from ref. 150. | ||
3 Applications of pyrene-based MOFs
3.1 Luminescence sensing
A celebrated feature of MOFs is the control of their physical properties by the selection of the organic and inorganic units. These properties include photoemission, which arises following the absorption of radiative excitation energy. The emission can take place by fluorescence (emission from a singlet excited state to the ground state) or by phosphorescence (emission from a triplet excited state to the ground state) (Fig. 6). MOFs including these emitting properties are known as “luminescent MOFs”.154 The origin of the luminescence in MOFs can be associated with a variety of mechanisms due to the structural diversity of MOFs.155 This can include ligand-based emission,66 metal-based emission,67,89 emission due to excimer formation,156 or guest induced emission.155,157 Pyrene-based MOFs have been attractive for sensing applications due to the fluorescence properties of pyrene. Pyrene has a very characteristic violet emission colour when illuminated under UV light in dilute solutions, due to its monomer emission occurring between 370 and 420 nm, arising from the π → π* transitions. Pyrene molecules can present an additional band called “excimer” emission occurring at longer wavelengths (ranging from 425 to 550 nm, centered around 460 nm) as a result of one electronically excited pyrene molecule interacting with a second one in its ground electronic state.158 The pyrene excimer emission is an excited-state process that depends on the orientation and proximity between the two pyrene molecules, which usually tends to form strong π–π stacking.159,160 The two main factors affecting the luminescence properties of pyrene excimers include the π–π orbital overlapping and interplanar distance.160,161 This property can be used in porous materials containing pyrene to develop sensors for the detection of guest molecules such as gases, organic molecules, and metals, when pyrenes are localised in the structure (Table 2). Since pyrene is a florescent probe whose spectrum is responsive to the polarity of its environment, different environmental parameters can be also detected by utilizing the excimer fluorescence of pyrene. In addition, pyrene molecules can also exhibit excimer emission and emission from higher excited states.162 Stylianou et al.12 reported the increase of the lifetime emission of a coordinated pyrene indium MOF (In-TBAPy), compared to the isolated pyrene molecules. Likewise, the emission wavelength, lifetime, and intensity of In-TBAPy can be tuned with the selection of guest species and loadings. The structure and chemical nature of guest molecules have a direct influence on the luminescence properties of pyrene-based MOFs.99,101,106,163 Moon et al.164 used SALI to incorporate 5(6)-carboxynaphthofluorescein (CNF) within NU-1000 to modify the optical properties of the material. The new MOF, NU-1000-CNF, displayed visually colorimetric pH sensing ability, where it has reversible colour change when exposed to solutions or vapors of varying pH.164 Such optical response diversity and sensitivity in pyrene-MOFs with luminescence have placed them as potential luminescent probe materials, due to the fact that they can serve as sensory systems of analytes that are bound to the MOFs by virtue of their porosity.109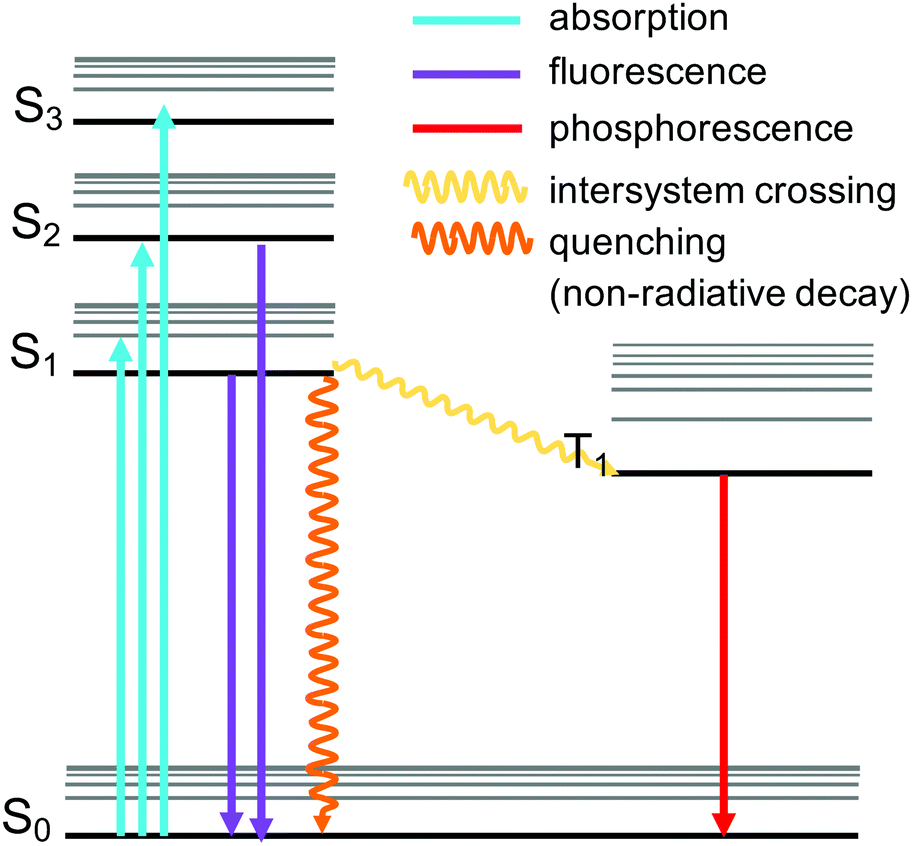 | ||
| Fig. 6 Jablonski diagram of the pyrene MOFs involved in luminescence sensing. | ||
| Application | Analyte | MOF name | Mechanism | Excitation (nm) | Ref. |
|---|---|---|---|---|---|
| Explosives | 2,4,6-Trinitrotoluene (TNT) | Mn-L | Quenching fluorescence | 330 | 106 |
| Nitrobenzene (NB) | LMOF-1 | Quenching fluorescence | 370 | 109 | |
| Nitrobenzene (NB) | MgMOF | Quenching fluorescence | 300 | 99 | |
| 2,4,6-Trinitrophenol (TNP) | Pyrene-tagged UiO-66-NH2 | Quenching fluorescence | 360 | 163 | |
| 2,4,6-Trinitrotoluene (TNT) | UiO-66-Py | Quenching fluorescence | 365 | 165 | |
| Gas and toxic molecules | H2S | Zr(TBAPy)5(TCPP) | Turn-on fluorescence | 365 | 168 |
| O2 | YbIII-TBAPy | Quenching fluorescence | 355 | 89 | |
| Acetone | Bio-MOF Zn-MOF (1) | Quenching fluorescence | 280 | 94 | |
| Cr2O72− | NU-1000 | Quenching fluorescence | 400 | 170 | |
| Ions | Cu2+ | CPP-16 | Quenching fluorescence | 335 | 156 |
| OH− | JUC-119 | Stokes shift luminescence | 300 | 101 | |
| Cu2+ | MIL-53-L | Quenching fluorescence | 337 | 171 | |
| Hg2+ | SALI-MAA-3eq MOF | Quenching fluorescence | 365 | 172 | |
| Biomolecules | Histidine | Zn-PLA | Quenching fluorescence | 355 | 103 |
| 1-Hydroxypyrene | NU-1000 | Quenching fluorescence | 360 | 173 | |
| Uric acid | Hf-UiO-66-Py | Quenching fluorescence | 340 | 166 | |
| Cholesterol | Rh6G@NU-1000-CMCD | Turn-on fluorescence | 470 | 167 | |
| Biothiols | Bi-TBAPy | Turn-on fluorescence | 396 | 87 | |
| Temperature | 100–300 K | {[Cd(TBAPy)(H2O)2]·4(H2O)}n(1) | Temperature-dependent emission | 355 | 59 |
| 80–450 K | SION-7 | Temperature-dependent emission | 405 | 83 | |
| 293–460 K | Zn(TBAPy)1/2(H2O)2 | Temperature-dependent emission | 315 | 62 | |
| 20–440 K | M2(TBAPy)(H2O)2 [M = Co, Zn] | Temperature-dependent emission | 420 | 95 | |
| Polycyclic | Acenaphthylene | NU-1000 | Quenching fluorescence | 395 | 174 |
| Aromatic | Pyrene | NU-1000 | Excimer fluorescence | 395 | 174 |
| Hydrocarbons | Fluoranthene | NU-1000 | Exciplex fluorescence | 395 | 174 |
| pH | pH 1–10 | NU-1000-CNF | Colorimetric | 164 | |
Table 2 shows different applications of pyrene-based MOFs in luminescence sensing applications. One of the applications is the sensing of nitro-aromatic explosive molecules. This application exploits the π-donor nature of the pyrene linkers with the electron-deficient nature of nitroaromatics, in order to sense the analytes via fluorescence quenching mechanism. Bajpai et al.106 synthesized the Mn-L MOF for nitroaromatic explosives sensing. Mn-L displayed a gradual decrease in fluorescence intensity upon increasing the concentration of nitroaromatic molecules. Thus, the photoexcited MOF electrons located in the conduction band of Mn-L interact with the LUMO of the analytes via charge transfer, leading to the diminution of the fluorescence. Different nitroaromatic explosives displaced a linear relationship of the Stern–Volmer curve, yet 2,4,6-trinitrotoluene (TNT) presented higher value of Stern–Volmer quenching constant along with higher quenching efficiency (74%). Similarly, other MOFs have been exploiting this charge-transfer interaction displaying selective sensitivity for explosives like nitrobenzene,99,109 2,4,6-trinitrophenol,163 and 2,4,6-trinitrotoluene.165
The quenching fluorescence mechanism can also be translated to applications involving analytes with different chemistry. The design of interacting sites, the chemistry of the analyte, and the cavities of MOFs play a role in the selective sensing. Huang et al.94 successfully synthesized an anionic MOF featuring a combination of mononuclear and tetranuclear zinc clusters and a mix-and-match strategy of two different types of organic ligands (TBAPy and 6-benzylaminopurine). This MOF exhibited exclusive luminescence quenching effect by acetone as a result of the formation of small cavities, leading to the binding of acetone by multiple hydrogen bonds with the MOF. Another example of the rational design for sensing application exploiting fluorescence quenching is sensitivity to chiral analytes. Chandrasekhar et al.103 developed a Zn-MOF (Zn-PLA) based on a pyrene-tetralactic acid linker, which can selectively sense histidine via fluorescing quenching. The nature of the chiral organic linkers in Zn-PLA imparted by the lactic acid moieties allowed the specific discrimination of histidine among all other amino acids, and the enantiodiscrimination of the D and L forms of histidine. Like the above, different rational design of pyrene-based MOFs has been applied for sensing applications exploiting quenching fluorescence of different molecules, like bio-molecules, ions, and gas and toxic molecules (Table 2).
Although in most cases the selectivity comes from the combination of the metal cluster and linker properties of the MOF itself, some cases require the use of post-synthetic modifications. This is the case of applications where require the inclusion of pyrene moieties in Zr-based MOFs,156,166 or the inclusion of polymers. Gong et al.167 designed a MOF where carboxymethyl β-cyclodextrin (β-CMCD) was grafted onto the NU-1000 MOF (NU-1000-CMCD) as an energy transfer bridge and to capture cholesterol recognition. Unlike the previous cases, the sensing mechanism in NU-1000-CMCD is a turn-on fluorescence. Fig. 7 shows a schematic of the structural and dimensional parameters of NU-1000-CMCD. The β-CMCD acts as a host–guest recognition for rhodamine 6G (Rh6G) or cholesterol; if Rh6G is attached to β-CMCD the luminescence is quenched due to the fluorescent resonance energy transfer (FRET) while the cholesterol induces the fluorescence restoration of the pyrene ligand. NU-1000 can also be used as a fluorescence turn-on probe for selectively and sensitively detecting H2S and its derivatives S2−, if tetrakis(4-carboxyphenyl)porphyrin (TCPP) is installed as linker via the solvothermal method.168 The installation of TCPP in NU-1000 causes the natural quenching fluorescence of the pyrene ligands, as a result of TCPP–TBAPy interaction. The fluorescence is restored in Zr(TBAPy)5(TCPP) due to N–S bond formation of S2− with N atoms in the porphyrin moiety. This suggests the role of N atoms in the porphyrin for the detection of H2S derivatives S2− in aqueous solutions. The tunability of MOFs allows a plethora of applications for smart design involving fluorescence and photophysical mechanisms.
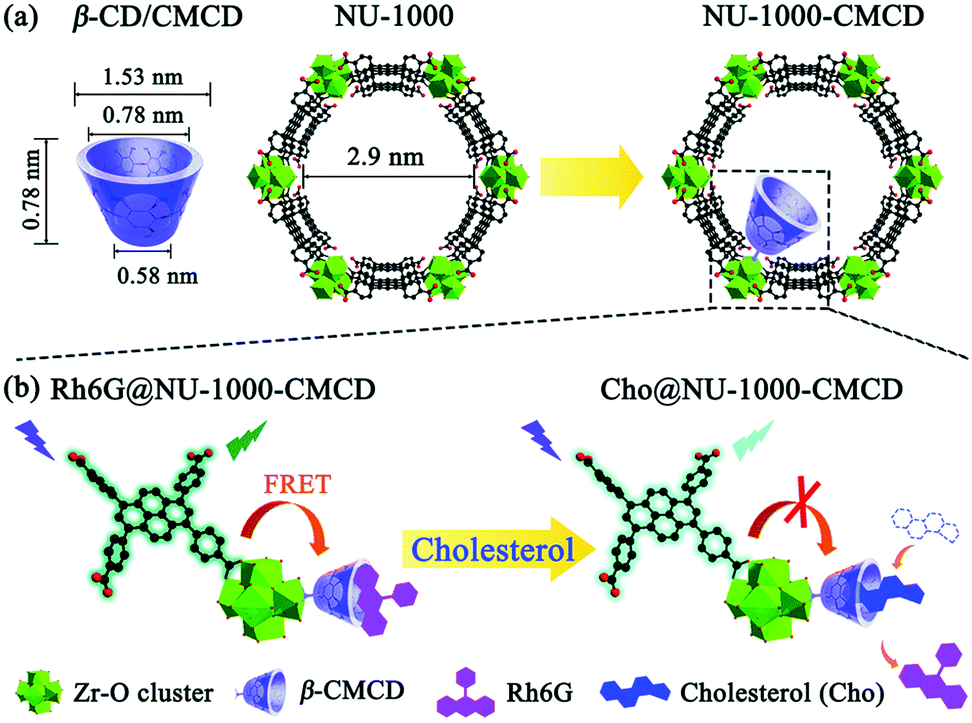 | ||
| Fig. 7 (a) Structural and dimensional parameters of β-CD/CMCD and NU-1000 as well as NU-1000-CMCD. (b) Schematic illustration of the proposed sensing mechanism for cholesterol detection with β-CMCD as the recognition sites using a Rh6G@NU-1000-CMCD probe. Reprinted with permission from ref. 167. | ||
The luminescence in pyrene-based MOFs can also be exploited for other applications such as temperature sensing. Materials exhibiting tunable fluorescence emission upon heating or cooling are considered smart materials as their optical properties can be exquisitely controlled by adjusting the external temperature.83 It is known that the pyrene-based MOF topology plays an important role in the interchromophoric interactions behavior of the excited stated,169 yet the temperature is also an important factor. Gładysiak et al.83 reported a pyrene-based MOF, SION-7, which can display either monomer or excimer fluorescence emission due to the temperature-dependent extent of interchromophoric interactions between the TBAPy3-ligands within the framework. The same phenomena has been also reported in Cd, Co, and Zn TBAPy-based MOFs where the temperature is a key parameter in the interchromophoric interactions.59,95 These materials display a linearly decreasing emission intensity dependence to the increase of temperature in the ranges of 100 to 300 K ({[Cd(TBAPy)(H2O)2]·4(H2O)}n) and 100 to 400 K (M2(TBAPy)(H2O)2 [M = Co, Zn]). Zeng et al.59 reported a relative thermal sensitivity of 0.319 K−1 with a maximum value of 0.32 K−1 at 300 K for {[Cd(TBAPy)(H2O)2]·4(H2O)}n, these values were comparable to those of the lanthanide coordination polymers. The emission intensities in these pyrene-based MOFs increase at lower temperatures since intramolecular rotations and non-radiative relaxations processes are restricted.59 On the contrary, higher temperatures will promote more electron transitions by thermal energy activation within the excited states with varying vibrational levels hence more non-radiative decays process can occur.83 Likewise, the MOF topology and porosity can directly lead to changes in the photophysical properties. The formation of pyrene excimers as a result of the MOF topology can be regulated by temperature-dependent spacing change or be hindered by solvent molecules interactions.83
3.2 Photocatalysis
Photocatalysis is a type of catalysis involving the acceleration of a photoreaction by adding substances (catalysts) that participate in the chemical reaction without being consumed. Photocatalysis, while varying in details in terms of reactions and mechanisms, can be described by four steps: (i) light absorption to generate electron-hole pairs carriers; (ii) separation of excited charge carriers; (iii) transfer of electrons and holes to the photocatalyst; and (iv) utilization of charge carriers for redox reactions.175 The organic chromophore energy transfer mediator and light harvesting properties of pyrene have been exploited in the design of heterogeneous photocatalyst. Table 3 shows the main applications in the field of photocatalysis using pyrene-based MOFs. MOFs have shown to be excellent candidates for the sorption and degradation of chemical warfare agents thanks to their porous and tunable nature.176 A recurrent use of pyrene-based MOFs is the sulfur mustard degradation. Omar K. Farha and Joseph T. Hupp have pioneered the field with the application of NU-1000 for the selective oxidation of 2-chloroethyl ethyl sulfide (CEES) via singlet oxygen (1O2) generation without producing the toxic sulfone.176 This approach exploits the use of pyrene-based linkers as UV photosensitizers in order to absorb light and generate an excited singlet state. This singlet can later be transformed into a triplet state via singlet to triplet inter-system crossing.177,178 Energy transfer processes can take place from the pyrene triplet state to ground state triplet oxygen, thereby yielding singlet oxygen. The 3D crystal structure of MOFs provides a platform to heterogenized and isolated pyrene moieties in an ordered array, which can enhance the efficiency of singlet oxygen generation by preventing aggregation and photo-degradation.176 NU-1000 inherits the photophysical properties of pyrene and under UV LED irradiation, 0.2 mmol CEES can be completely and selectively oxidized to CEESO within 15 min using 1 mol% of NU-1000 (2.16 mg, 2 μmol by pyrene unit).176 When compared with 1.35 mg (2 μmol, 1 mol%) of the isolated linker, TBAPy, the complete conversion occurs after 30 min confirmed by gas chromatography and the selectivity of the photo oxidation reaction was corroborated by NMR experiments. The half-time of the NU-1000 MOF is 6.2 min while the half-time of the linker (TBAPy) is 10.5 min for complete conversion of CEES. The improved rate of reaction was associated with the rigid framework of pyrene linkers in NU-1000 preventing deactivation by aggregation.| MOF Name | Photocatalysis | Co-Catalyst | Ref. |
|---|---|---|---|
| NU-1000 | Sulfur mustard degradation | — | 176 and 192 |
| NiS-AIM | HER | — | 184 |
| JUC-138 | Azure B photodegradation | — | 102 |
| Br-BDP@NU-1000 | Sulfur mustard degradation | — | 179 |
| NU-1000-PCBA | Sulfur mustard degradation | — | 180 |
| CdS@NU-1000 | HER | CdS | 189 |
| NU-1000 | Photocatalytic atom-transfer radical addition | 181 | |
| Cd-TBAPy | Water reduction and oxidation | Pt and CoPi | 190 |
| MoSx-SIM | HER | — | 193 |
| Bi-TBAPy | HER | Pt | 191 |
| PPIX@NU-1000 | Sulfur mustard degradation | — | 194 |
| FeSC12-AIM | Nitrate reduction | — | 182 |
| ACM-1 | HER | Pt | 97 |
| NU-400 | Sulfur mustard degradation | — | 65 |
| PCN-822(Hf) | Amine oxidation | — | 110 |
| NU-1000 | 4-Methoxybenzyl alcohol oxidation | — | 146 |
| NU-1000-U | 4-Methoxybenzyl alcohol oxidation | — | 146 |
| FDH@Rh-NU-1006 | CO2 reduction | NAD enzyme | 183 |
Zr6 node topology of NU-1000 can offer a platform to tune the photophysical and photochemical properties of the MOF via post-synthetic modifications.121 Atilgan et al.179 incorporated the BODIPY chromophore onto the Zr6 metal node of NU-1000 via SALI. The resulting MOF named Br-BDP@NU-1000 (0.2 mol%, 0.4 μmol) presented approximately a half-time of 2 min for 0.2 mmol CEES complete photo-oxidation into CEESO, which is much faster than that of NU-1000. Importantly, the incorporation of BODIPY molecules allowed the material to display CEES photocatalytic activity under green LED irradiation, in contrast the UV LED sourced is needed for NU-1000. The post-synthetic modification of Zr6 node has been a recurrent strategy to tune the singlet oxygen production including photosensitizers like fullerene180 and porphyrin.80 Other pyrene-based MOFs have also been used for photodegradation of pollutants as in the case of Azure B photodegradation in JUC-138.102 A different mechanism of organic transformation was reported by Zhang et al.,181 where the photocatalytic atom-transfer radical addition (ATRA) of perfluoroalkyl iodides onto olefins occurs in NU-1000. They showed that NU-1000 photochemically generates the reactive radical species by sensitisation of the perfluoroalkyl iodides through a photoinduced energy-transfer pathway. Importantly, the authors reported the importance of the visible-light source where the best performance (93% isolated yield) was obtained with a LED with a center wavelength of 405 nm, using 2.5 mol% of NU-1000 which is rationalized by the strong absorption of NU-1000 at 405 nm. NU-1000 has also been used for other organic transformations, such as a nitrate reduction photocatalyst by iron thiolate cluster incorporation into Zr6 node of the NU-1000 MOF.182 Choi et al.182 showed how this new MOF (named as FeSC12-AIM) in an aqueous solution containing nitrate (30 ppm of N, calculated by nitrogen mass) and ascorbic acid as a sacrificial electron donor, can photochemically transform NO3− to NH4+ as a result of the photoexcitation of the pyrene linker to the iron thiolated cluster. In general, pyrene-based MOFs can be used for photocatalytic organic transformation reactions involving singlet oxygen or photoinduced energy transfer reactions.
The photo-sensitizing properties of pyrene-based MOFs have also been exploited in promising energy applications like photocatalytic CO2 reduction183 and H2 production.184 Theoretical and experimental studies have shown key features of photocatalytic systems like absorption,185 charge transfer,186,187 and charge-separation187,188 present in pyrene-based MOFs. Inspired by natural photosynthesis, Chen et al.183 encapsulated formate dehydrogenase (FDH) in a Zr pyrene-based MOF, NU-1006, anchored with Rh-based electron mediator in order to couple co-enzyme regeneration with enzymatic CO2 reduction for efficient CO2 fixation system. The material FHD@Rh-NU-1006 facilitates ultrafast photoinduced electron transfer, leading to CO2 reduction to formic acid when combined with the co-enzyme, nicotinamide adenine dinucleotide (NAD). Other energy-related photo-catalytic applications with pyrene-based MOFs have focused on the synthesis of materials for hydrogen evolution reaction (HER) (Table 3). Peters et al.184 reported the anchoring of NiSx in NU-1000 via AIM, where the new material NiS-AIM can catalytically produce H2 upon light illumination. The pyrene-based linkers act as a UV photosensitizer and the nickel sulfide-functionalized node catalyzes the reduction of water to generate hydrogen gas with a rate of 3.1 mmol g−1 h−1 under UV light (LED, maximum 390 nm). The visible light absorption profile of NiS-AIM was improved by the use of rose bengal dye as a photosensitizer. The porous nature of the MOF allows the diffusion of the dye, leading to an improvement in the light-absorption (at 550 nm) and enhancement in the H2 production (4.8 mmol g−1 h−1) upon visible light irradiation.184 Another approach to improve NU-1000 HER photocatalysis was the dual modification by the incorporation of CdS nanaparticles (as a co-catalyst) along with reduced graphene oxide (RGO).189 The addition of RGO led to more efficient charge separation and further accelerated the transfer of photogenerated electrons, than in the case where RGO is not considered. The material, CdS@NU-1000/1%RGO, presented a 12.1 times higher H2 generation rate than that of commercial CdS under visible light. Remarkably, CdS@NU-1000/1%RGO displayed a significantly enhanced photostability compared to CdS.
Although the tunability of the optical properties of NU-1000 has been explored, there are other TBAPy-based MOFs with HER photocatalytic activity. TBAPy linkers have been combined with metal clusters such as Cd, Bi, and Ti which have led to materials with compelling HER properties. The cadmium MOF (Cd-TBAPy) is a 2D layer material exhibiting visible-light absorption around 600 nm and dual functions of water reduction and oxidation when combined with Pt and CoPi co-catalyst, respectively.190 After loading suitable co-catalysts, the hydrogen and oxygen evolution rate can reach up to 4.3 and 81.7 μmol h−1, respectively. In this case, the proper combination of TBAPy and the transition metal can lead to materials exhibiting better visible light absorption than pyrene itself. The bismuth (Bi-TBAPy)191 and titanium (ACM-1)97 MOFs are other examples of pyrene-based MOFs with good HER when combined with co-catalyst. Both MOFs exhibit a typical ligand-to-metal charge transfer (LMCT) leading to intrinsic charge separation confirmed by XPS spectra and DFT calculations. Bi-TBAPy and ACM-1 optimal H2 production rates are 140 μmol g−1 h−1 and 67 μmol h−1, respectively, when combined with Pt nanoparticles. It is noteworthy to mention that ACM-1, unlike Bi-TBAPy, can present a reduced HER (5.9 μmol h−1) without the presence of co-catalyst. The 1D titanium rod metal cluster in ACM-1 leads to position the Ti–O orbitals at the conduction band of the material, resulting in an efficient electron transfer from the organic ligand.97 Despite the number of pyrene-based MOFs for photocatalytic applications is still small, the unmatched control over MOF photophysical properties have led to materials suitable for different application including the synergy of pyrene linkers with different metal clusters.
3.3 Adsorption and separation of molecules
MOFs have been widely studied for adsorption and separation applications due to their geometrically and crystallographically well-defined framework structures with high permanent porosity, large surface area, and remarkable thermal and mechanical stability. MOFs can be designed for desired separation and capture processes by tuning the microporous channels and introducing functional groups. There are several pyrene-based MOFs in the literature that have been investigated for adsorption and separation owing to different reasons, such as (i) preferential binding sites on the carboxylates and aromatic rings on the linker,90,195 (ii) permanent porosity and framework stability in the presence of water,78 (iii) suitable pyrene-pyrene distance for the adsorption and separation of gas molecules,57,77,85 (iv) the presence of rotatable units connected to pyrene ligand, resulting in enhanced loading of the guest molecules in the structure,60 (v) donor–acceptor interaction between the adsorbate molecule and pyrene,91 and (vi) the possibility of using functional groups within the MOF as structure-directing agents.84 In this section, we will discuss pyrene-based MOFs that have been investigated for adsorption and separation of different molecules and Table 4 will be summarizing all the performances.| MOF name | Molecules | Adsorption performance | Ref |
|---|---|---|---|
| Energy-related gases | |||
| Ni-TBAPy | CO2 and CH4 (@298 K, 1 bar) | 15 and 10 cm3 g−1 | 76 |
| ROD-6 | CO2 and N2 (@298 K, 1 bar) | 39.36 and 3 cm3 g−1 | 196 |
| ROD-7 | CO2 and N2 (@298 K, 1 bar) | 34.09 and 2 cm3 g−1 | 12 and 196 |
| ROD-8 | CO2, CH4 and N2 (@298 K, 1 bar) | 40.21, 17.16 and 2.5 cm3 g−1 | 57 |
| NU-1100 | CO2 (@298 K, 30 bar), CH4 and H2 (@298 K, 65 bar) | 26.2, 16.8 and 6 mmol g−1 | 105 |
| UTSA-72 | CO2, CH4 and N2 (@296 K, 1 bar) | 21.7, 4.4 and 1.1 cm3 g−1 | 73 |
| Mg-PTCA | CO2 (@195 K, 1 bar), H2 (@77 K, 1 bar), and O2 (@77 K, 0.19 bar) | 160.5, 92.2 and 200.1 cm3 g−1 | 68 |
| Co-tdcppy | CO2 (@298 K, 1 bar) | 61.6 cm3 g−1 | 100 |
| ZnLi-PTCA | CO2, CH4 and N2 (@298 K, 1 bar) | 60.9, 15.9 and 3.6 cm3 g−1 | 70 |
| Cd-PTC | CO2 and CH4 (@298 K, 1 bar) | 58 and 7 cm3 g−1 | 71 |
| Ni-PTCA | CO2, CH4 and N2 (@298 K, 1 bar) | 65, 20 and 5 cm3 g−1 | 69 |
| NU-1000-SALI-9 | CO2 (@273 K, 0.15 bar) | 6.2 cm3 cm−3 | 130 |
| NU-901-SALI-BA-3,5-NH2 | CO2 (@273 K, 1 bar) | 123 cm3 g−1 | 199 |
| PEI(50)@NU-1000 | CO2 and N2 (@298 K, 0.15 bar) | 1.06 and 0.0073 mmol g−1 | 200 |
| NU-505-Zn | N2 (@77 K, 1 bar) | 400 cm3 g−1 | 78 |
| Al-PyrMOF | CO2 and N2 (@313 K, 1 bar) | 2.29 and 0.2 mmol g−1 | 85 |
| Organic and inorganic pollutants | |||
| NU-1000 | (SeO32−) and (SeO42−) | 102 and 62 mg g−1 in <1 min | 204 |
| NU-1000 | Sb(III) and Sb(V) | 136.97 and 287.88 mg g−1 in 10 h | 203 and 205 |
| NU-1000 | (Cr2O27−) | 76.8 mg g−1 in 3 min | 206 |
| NU-1000 | Atrazine | 36 mg g−1 in <1 min | 207 |
| NU-1000 | Glyphosate | 8.97 mg g−1 in 3 min | 208 |
| NU-1000 | (ReO4−) | 210 mg g−1 in 5 min | 209 |
| NU-1000 | 2-CEES and DMMP | 4.197 and 1.70 mmol g−1 | 210 |
| NU-1300 | Proteins: Cyt-c and a-La | 90 and 6% of Cyt-c and a-La in 20 h | 79 |
| UOF-1 and UOF-2 | Cs+ | 108 mg and 96 mg g−1 in 60 min | 81 |
| SION-82 | VOCs | 107 mg g−1 in equilibrium | 98 |
| Hydrocarbons | |||
| Zn-TBAPy | mX and pX (@303 K, 15 bar) | 3.36 and 2.11 mmol g−1 | 60 |
| NU-1105 | Propane (@273 K, 40 bar) | 700 cm3 g−1 | 108 |
| JXNU-5 | Acetylene (C2H2) (@298 K, 1 bar) | 55.9 cm3 g−1 | 90 |
| bioMOF 1-NH2 | C2H2, C2H4 and C2H6 (@298 K, 1 bar) | 55.6, 52.4 and 51 cm3 g−1 | 93 |
| NU-1000 | R134-a | 17 mmol g−1 | 214 |
| NU-1000 | Propane (@273 K) and isobutane (@298 K) | 0.49 and 0.76 g g−1 | 216 |
| Carbohydrates and carbohydrate-derived aromatics | |||
| IL/NU-1000 | Coumaric and ferulic acids | 66 and 72 mg g−1 in [Ch][Lys], and 70 and 95 mg g−1 in [EOA][OAc] | 220 |
| NU-1000 | Phenolics (4-hydroxybenzaldehyde, 4-hydroxybenzoic acid, vanillin, vanillic acid, syringaldehyde, and syringic acid) | 354, 325, 371, 391, 340, and 373 mg g−1, respectively | 218 |
| NU-1000 | HMF and furfural | 240 and 467 mg g−1 | 219 |
| NU-1000 | Cellobiose and maltose | 1260 and 1383 mg g−1 | 221 |
| Radioactive gases | |||
| NU-1106 | Xe and Kr (@298 K, 1 bar) | 38 and 16 cm3 g−1 | 72 |
| SION-8 | I2 | 340 mg g−1 in 200 h | 91 |
| NU-1000 | I2 | 1.45 g g−1 in 60 h | 224 |
Pyrene-based MOFs can have different structural characteristics based on the size of pyrene ligand, or its stacking in the framework, resulting in different gas adsorption performances.68,73 UTSA-72 is a 2D microporous paddle-wheel MOF which was constructed from Zn2(COO)4 clusters connected by PTTB linkers.73 Due to the presence of two kinds of cavities of 2.9 × 2.9 Å2 and 2.4 × 4.6 Å2, UTSA-72 was investigated toward the separation of CO2 from CH4 and N2. The higher adsorption capacity of CO2 than those of CH4 and N2 resulted in moderately high CO2/N2 (33.4) and CO2/CH4 (7.2) selectivity. Another pyrene-based MOF, NU-1100 was synthesized based on Zr6O4(OH)412+ clusters and a pyrene-based tetratopic ligand PTBA,105 which is a much bigger ligand in size compared to PTTB. This is due to the presence of ethynyl groups in between the pyrene core and benzoic acids. Consequently, the CH4 gravimetric deliverable capacity of NU-1100 has been found significantly higher (0.24 g g−1) than most promising CH4-storage materials such as HKUST-1 (0.154 g g−1), PCN-14 (0.136 g g−1), and UTSA-20 (0.134 g g−1). These studies were followed by other TBAPy-MOFs with rod SBUs including Mn(II), In(III) and Cd(II) metals (denoted as ROD-6,196 ROD-7,12 and ROD-8,57 respectively). Although its lower surface area, the CO2 uptake capacity of ROD-6 (7.73 wt%) surpasses that of ROD-7 (6.70 wt%) and is comparable to that of ROD-8 (7.90 wt%) at ambient conditions. This can be explained by the stronger interactions between the CO2 molecules and the framework, resulting in a slightly increasing isosteric heat of adsorption (20–21 kJ mol−1)77 ROD-8 was further investigated toward CH4 adsorption, due to two main reasons: (i) the nearest pyrene-pyrene distance (4.35 Å) fits well to the CH4 kinetic diameter of ca. 3.8 Å, and (ii) the isosteric heat of adsorption is close to the calculated optimal value of 18.8 kJ mol−1 for CH4 adsorbents. As a result, CH4 uptake in ROD-8 was obtained as 0.77 wt% at 298 K and 1 bar, which outperforms some of the reported MOFs to date.
Besides structural characteristics, it is well known that the presence of open-metal sites favors the adsorption of specific gases, such as CO2.76 Pyrene-based MOFs can be synthesized as to have open metal sites in the structure, based on the design of the ligand. A Co-based pyrene MOF constructed from 1,3,6,8-tetra(3,5-dicarboxyphenyl)pyrene (tdcppy) ligand was found to have additional cobalt sites in the structure, which are acting as open metal sites (Fig. 8).100 Single-crystal X-ray diffraction analysis showed that while six carboxylate groups on the pyrene participate in composing the cobalt trigonal prismatic SBUs, the other two carboxylate groups bridge cobalt sites dangled on cobalt trigonal prismatic clusters, providing open metal sites for CO2 uptake. In addition, anionic MOFs can be constructed using pyrene-based ligands, such as PTCA.69–71 It was found out that the cations arising from the decomposition of the solvent molecule used in the synthesis act as the counter ions in the pores and favor the separation of CO2 from CH4 and N2.
 | ||
| Fig. 8 (a) Open metal sites dangled on the cobalt trigonal prismatic SBU of the Co-based pyrene MOF, Co-tdcppy. Atom colour code: purple, Co; grey, C; red, O. (b) The structure of Co-based pyrene MOF viewed along the b direction. Modified and reprinted with permission from ref. 100. | ||
The presence of water in flue gas stream is an important problem for industrial settings for the capture of CO2. Therefore, the selection of metal and ligand is highly important for the design of robust structures upon water exposure.78,105 It is important to expand the understanding of the interaction of water with the pyrene-based MOF structures to rationally design feasible structures. Very recently, a detailed computational and experimental study was performed by Boyd et al.85 to understand the effect of having an “adsorbaphore” in the MOF structure for CO2 separation in industrial conditions. Adsorbaphore term was introduced to specify the common pore shape and chemistry of a binding site in a MOF that provides optimal interactions to preferentially bind to a particular guest molecule. A library of 325![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 hypothetical MOFs was generated and each MOF was screened to investigate its CO2/N2 selectivity and CO2 working capacity. It was identified that the presence of two parallel pyrene and porphyrin rings (“adsorbaphores”) with interatomic spacings of approximately 7 Å favors the binding of CO2, while excluding the presence of water molecules due to their low Henry coefficient. Based on these findings, a porphyrin-based MOF (Al-PMOF) and a pyrene-based MOF (Al-PyrMOF) were synthesized with tetrakis(4-carboxyphenyl)porphyrin (TCPP) and TBAPy ligands, which are coordinated to one-dimensional Al(III)-oxygen rods. (Fig. 9(a)). Al is an attractive choice as the metal due to being less costly and earth-abundant compared to other well-investigated metals, and it ensures a strong bond with the carboxylate O-atoms of the ligands resulting in enhancing the thermal and hydrolytic stability of a MOF. Both experiments and calculations proved that while the stacked rings of porphyrin and pyrene are preferential adsorption sites for CO2 molecules, water molecules do not prefer to bind in between them. Moreover, the affinity of the MOF toward CO2 is higher than that of N2, resulting in good separation of CO2/N2 mixtures (Fig. 9(b)). Thanks to its highly stable framework in the presence of water, Al-PyrMOF showed a remarkable CO2 working capacity in a humid environment compared to those of other commercially available materials (zeolite 13× and activated carbon) and well-studied water-stable MOF (UiO-66-NH2) (Fig. 9(c)).
000 hypothetical MOFs was generated and each MOF was screened to investigate its CO2/N2 selectivity and CO2 working capacity. It was identified that the presence of two parallel pyrene and porphyrin rings (“adsorbaphores”) with interatomic spacings of approximately 7 Å favors the binding of CO2, while excluding the presence of water molecules due to their low Henry coefficient. Based on these findings, a porphyrin-based MOF (Al-PMOF) and a pyrene-based MOF (Al-PyrMOF) were synthesized with tetrakis(4-carboxyphenyl)porphyrin (TCPP) and TBAPy ligands, which are coordinated to one-dimensional Al(III)-oxygen rods. (Fig. 9(a)). Al is an attractive choice as the metal due to being less costly and earth-abundant compared to other well-investigated metals, and it ensures a strong bond with the carboxylate O-atoms of the ligands resulting in enhancing the thermal and hydrolytic stability of a MOF. Both experiments and calculations proved that while the stacked rings of porphyrin and pyrene are preferential adsorption sites for CO2 molecules, water molecules do not prefer to bind in between them. Moreover, the affinity of the MOF toward CO2 is higher than that of N2, resulting in good separation of CO2/N2 mixtures (Fig. 9(b)). Thanks to its highly stable framework in the presence of water, Al-PyrMOF showed a remarkable CO2 working capacity in a humid environment compared to those of other commercially available materials (zeolite 13× and activated carbon) and well-studied water-stable MOF (UiO-66-NH2) (Fig. 9(c)).
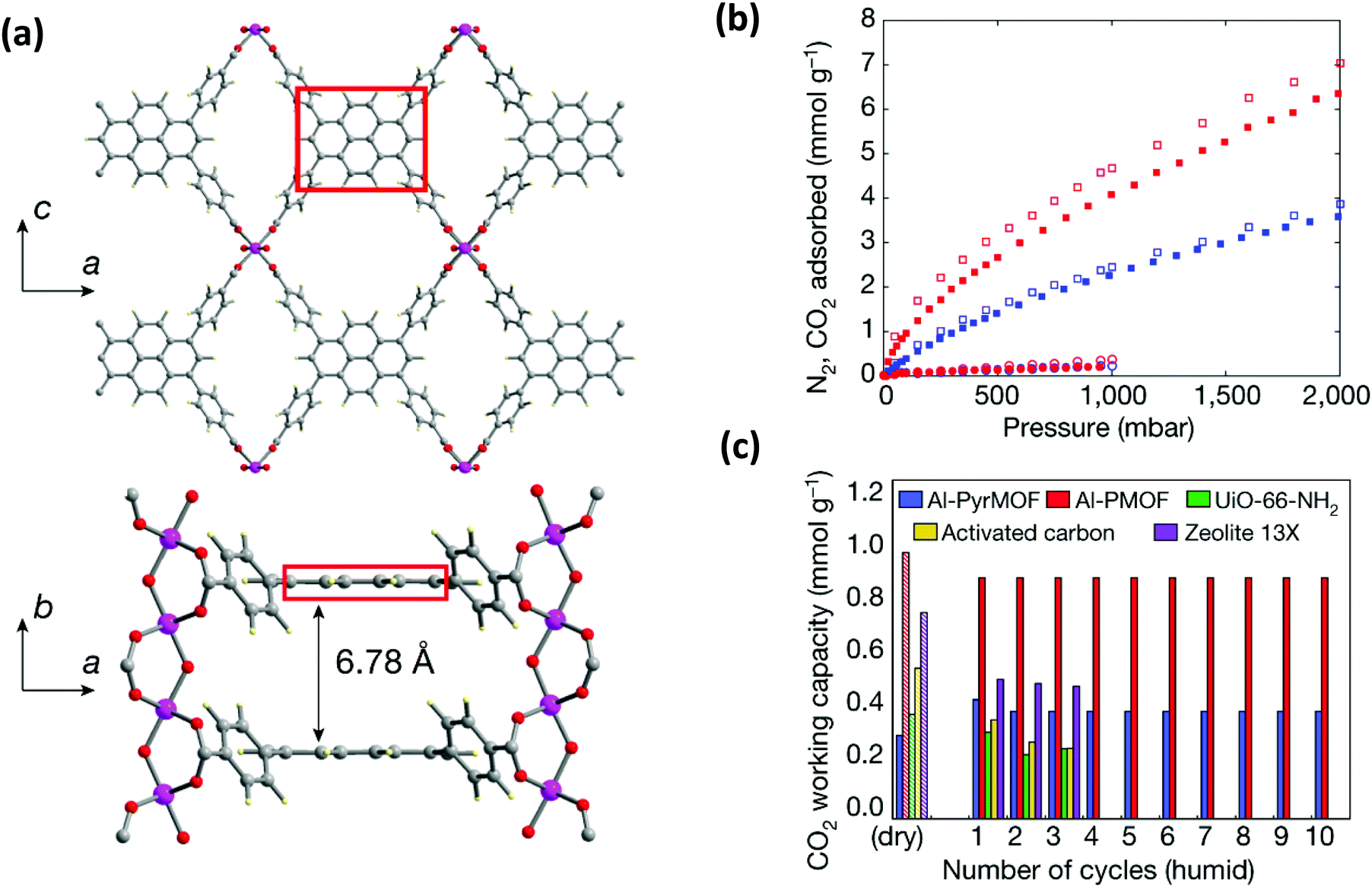 | ||
| Fig. 9 (a) The representation of the three-dimensional non-interpenetrated structure of Al-PyrMOF that is constructed from Al(III) rods connected by TBAPy ligands. The distance between two adsorbaphores (shown in red box) is calculated as 6.78 Å, which is very close to the computational value. Atom colour code: pink, Al; grey, C; blue, N; red, O; pale yellow, H. (b) Experimental CO2 (filled squares), experimental N2 (filled circles), computational CO2 (open squares) and computational N2 (open circles) single-component adsorption isotherms collected on activated Al-PMOF (red) and Al-PyrMOF (blue) at 313 K. (c) Recycling ability of Al-PyrMOF in comparison with other materials under dry and humid (85% relative humidity) conditions, with 85/15 v/v of N2/CO2 (313 K and 1 bar). Modified and reprinted with permission from ref. 85. | ||
NU-1000 has been a well-investigated member of pyrene-based MOFs because of its high chemical and mechanical stability, which arises from the strong ionic bonding between Zr and carboxylate oxygen atoms. It has large surface area which can lead to high gas adsorption capacities, and selective porosity which is a favorable characteristic for gas separation applications. Therefore, NU-1000 has been investigated both computationally and experimentally for the capture of different gases.197 The adsorption isotherms and the determination of the isosteric heats of adsorption of several gases (H2, D2, Ne, N2, CO, CH4, C2H6, Ar, Kr, and Xe) on NU-1000 have been demonstrated, where the adsorption sites were predicted and understood via DFT calculations.195,198 Results showed that the adsorption of the gas molecules starts with strong adsorption sites near Zr atoms, followed by the filling of other adsorption sites on the nodes and organic framework. NU-1000 is also the perfect candidate for post-synthetic modifications thanks to the presence of functional groups (–OH groups) on Zr6 nodes. Deria et al.130 investigated the functionalization of NU-1000 with perfluoroalkane groups (SALI-n, where n = 1, 3, 7, 9 depending on the varying chain length of the perfluoroalkyl carboxylic acids) for improving the interactions with CO2. The selection of perfluoroalkene groups was based on two main reasons: (i) fluorinated MOFs have been attractive candidates due to their hydrophobicity, which is an important feature for post combustion CO2 capture, and (ii) the presence of C–F dipoles is expected to improve the interactions between the framework and the quadrupole of CO2. As a result, SALI-n samples showed higher isosteric heat of adsorption values with increasing chain length, where the value of NU-1000 (17 kJ mol−1) was doubled in SALI-9 (34 kJ mol−1). SALI was applied to another pyrene-based Zr-MOF, NU-901, by the incorporation of amine moieties which are well-known functional groups for enhancing the interactions of CO2 molecules with the framework.199 CO2 isotherms showed that NU-901-SALI-BA-3,5-NH2 presented better CO2 adsorption than those of activated NU-1000 and NU-901. It also showed better CO2 uptake compared to that of NU-901-BA-NH2, where the amine functionalization was introduced during the synthesis by the use of 4-aminobenzoic acid as the ligand. The inferior performance of NU-901-BA-NH2 can be attributed to the position of the 4-amino benzoic acid, which significantly blocks the microporous channels and inhibits the access to the amino groups. In addition to SALI, post-synthetic modifications on NU-1000 have been also performed by the impregnation of amine molecules (polyethyleneimine) in the pores, resulting in approximately 37 times higher selectivity toward CO2 than that of the pristine NU-1000.200
Inorganic anions released from mining and industrial production activities are one of the major toxic pollutants in water, creating serious environmental and health problems. Since they are usually large molecules in size, the selection of the MOF based on the size of the analytes is the key criteria. NU-1000 has been studied for the removal of different toxic anions from water, not only because of its water stability but also thanks to its pore sizes. NU-1000 has uniform hexagonal channels with diameter 31 Å and triangular channels with diameter 10 Å aligned parallel to the c-axis, which are connected via small pores of 8 Å. Therefore, it has large enough pores for the diffusion of the anions. The anions in different sizes, including (i) selenite (SeO32−, 4.82 Å) and selenate (SeO42−, 5.20 Å),204 (ii) antimonite (Sb(OH)3, 3.05 Å) and antimonate (Sb(OH)6−, 7.36 Å),203,205 and (iii) dichromate (Cr2O72−, 6.00 Å)206 could be successfully removed from the aqueous solutions by NU-1000. The removal performance toward the anions SeO32−, SeO42−, Sb(III) and Sb(V) was remarkably better compared to some other Zr-MOFs, such as UiO-66 functionalized with different groups (–NH2, –OH, and –SO3H) and UiO-67. In the case of Cr2O72−, the uptake capacity of NU-1000 could surpass not only other Zr-MOFs but also some well studied MOFs, such as HKUST-1, ZIF-8, and MIL-100(Fe). These results highlight the importance of both large pore size and substantial bridging sites in the nodes of NU-1000 for the fast and efficient adsorption of different inorganic anions. In all cases, the presence of terminal hydroxyl groups on the Zr6 nodes can be replaced by the anions and work as efficient adsorptive sites as demonstrated in Fig. 10. ICP-OES analysis established that no zirconium is lost to solution during the adsorption of the anions. Moreover, NU-1000 framework remains intact after the adsorption, proved by different characterization techniques; including PXRD, FTIR and surface area measurements.
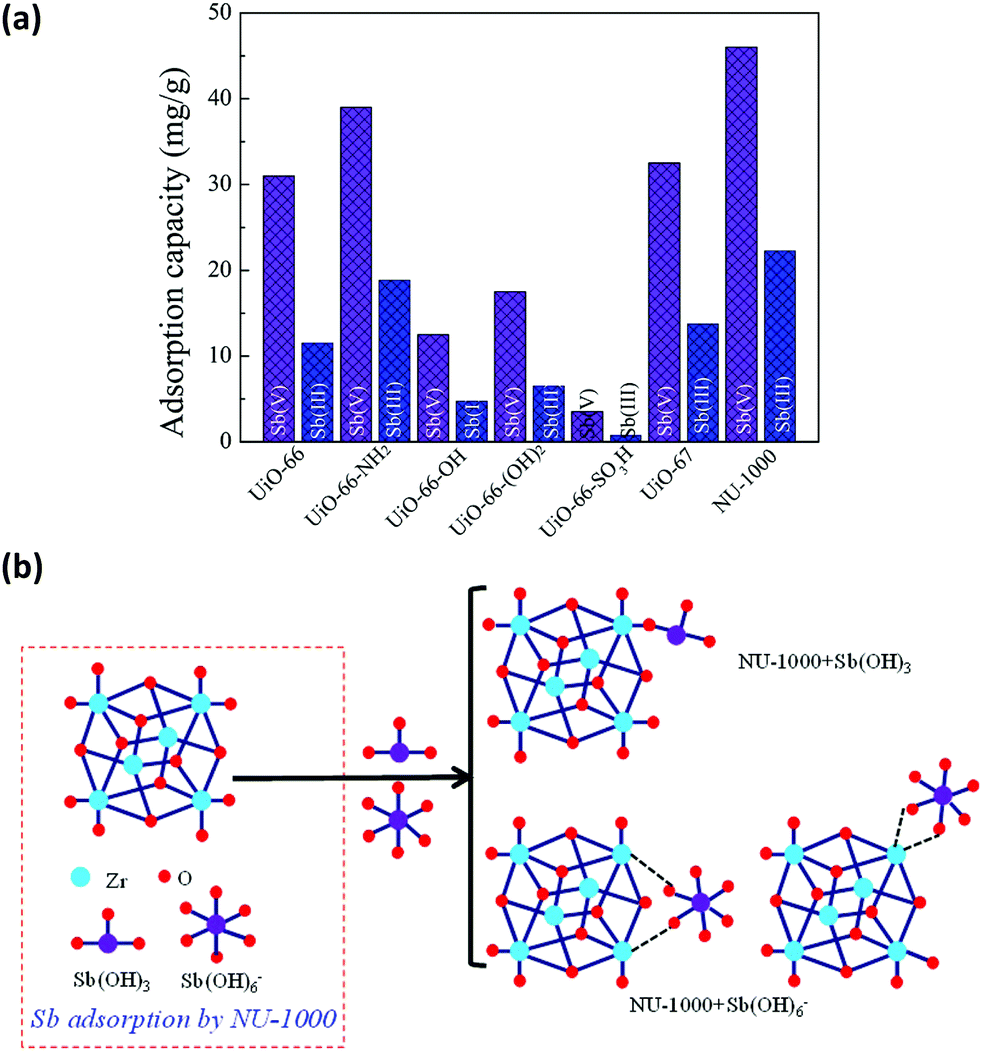 | ||
| Fig. 10 (a) Adsorption rates of Sb(OH)3 and Sb(OH)6− in different Zr-MOFs, (b) different binding possibilities of Sb(OH)3 and Sb(OH)6− to the node of NU-1000. Reprinted with permission from ref. 205. | ||
NU-1000 is also a good candidate for the removal of organic pollutant molecules, including volatile organic compounds (VOCs) and herbicides, which are harmful to both environment and human health. Thanks to π–π stacking interactions between the VOCs and pyrene-based ligand, SION-82 synthesized by Sudan et al.98 was found to capture some common VOCs (benzene, pyridine, and thiophene) effectively. SION-82 captured benzene efficiently (107 mg g−1) in dry conditions, and no uptake decrease was observed in the presence of high relative humidity for at least six cycles. Atrazine is a widely used herbicide in the agricultural industry, which contaminates the surface and groundwater supplies. Akpinar et al.207 studied the effect of the linkers in Zr-MOFs on atrazine uptake capacity and uptake kinetics. Results showed that although UiO-67, DUT-52, and NU-1000 have similar pore sizes and surface areas, NU-1000 showed an exceptional atrazine uptake because of π–π interactions between the pyrene and the atrazine. Another organic pollutant resulting from the use of herbicides is glyphosate, which causes damage to human cells and increases the risk of erosion and contamination of surface and groundwater sources. Pankajakshan et al.208 studied the adsorption of glyphosate using NU-1000 and UiO-67. The selection of these MOFs were because of the higher affinity of Lewis acid Zr metal nodes for the phosphate functional group of glyphosate, which is a Lewis base. The presence of wide pores (31 Å) in NU-1000 allows the glyphosate solution to interact better with the metal nodes compared to UiO-67, resulting in better adsorption performance.
The nuclear weapon industry and the use of toxic chemical warfare agents (CWAs) result in extremely toxic pollutants. Therefore, efficient and safe removal is highly important. Technetium-99 (Tc-99) is a fission product resulting from nuclear weapon activities and it is in the form of a highly soluble, environmentally mobile, and volatile pertechnetate ion (TcO4−) in the oxidizing environment. Drout et al.209 evaluated NU-1000 as a sorbent for aqueous perrhenate (ReO4−), which is the non-radioactive simulant for TcO4−. Single-crystal X-ray diffraction showed that ReO4− ions bind in a unique chelating mode in both small pore and mesopore of NU-1000 in addition to two non-chelating modes in each pore, resulting in a promising adsorption efficiency. Asha et al.210 investigated NU-1000 toward the capture of two CWA simulants, 2-chloroethyl ethyl sulfide (2-CEES, the simulant of sulfur mustard gas), and dimethyl methyl phosphonate (DMMP, the simulant of sarin). NU-1000 performed efficient and reusable adsorption of the simulants from the aqueous medium. The remarkable performance of NU-1000 is due to the chemical interactions between the thioether and chloro groups of 2-CEES, and phosphate groups of DMMP, with the Zr–OH groups of the MOF. Further studies showed that DMMP can be effectively adsorbed even from the air at ambient conditions and decomposed in the pores of the MOF.211
Very recently, water-stable anionic pyrene-based uranium MOFs have drawn attention with the aim of efficient separation of organic dyes and biomolecules,79 as well as the separation of fission products produced in the nuclear fuel cycle.81 The combination of a uranium salt with a highly symmetrical carboxylic pyrene-based ligand eventuated in different porous uranyl frameworks. Li et al.79 designed a 3D water-stable pyrene-based uranium MOF, NU-1300. Thanks to its anionic nature, NU-1300 shows a remarkable performance for the selective adsorption of positively charged molecules compared to negatively charged ones. This was proved by the study of selective adsorption of the cationic dyes (methylene blue (MEB), JanusGreen B (JB), ethyl violet (EV)) over an anionic dye (resorufin sodium salt (RS)). While 85% of RS remained in the solution, 10% of MEB, and 20% of JB and EV remained in the solution at the end of 42 h. The capture of anionic dyes was also evidenced by the changes in the absorbance of the supernatant solutions. The cation capture ability of NU-1300 was further confirmed by performing the separation of two proteins, a cationic enzyme (cytochrome c, Cyt-c) selectively over the anionic protein (a-lactabumin, a-La) analyzed by UV-Vis spectra. Similarly, Ai et al.81 synthesized two porous pyrene-based uranium MOFs, UOF-1 and UOF-2, for the adsorption of radioactive Cs+ cations from the aqueous solutions. Both MOFs were able to capture Cs+ cations from the aqueous solution efficiently due to their anionic frameworks. Single-crystal XRD analysis of as-synthesized samples indicates that [(CH3)2NH2]+ cations are present in the channels of both MOFs as the counter ions. The crystallinity of structures remained the same after the adsorption of Cs+ cations thanks to their exchange with [(CH3)2NH2]+ cations in the channels. Upon adsorption, two adjacent [UO2(COO)3]− SBUs share Cs atoms to form a 1D chain, and then these 1D chains are further linked by a TBAPy ligand to construct a 3D extended architecture.
Pyrene-based MOFs are also favorable for the separation of hydrocarbon mixtures thanks to the favorable interaction of some hydrocarbon molecules with the pyrene.217 The separation of acetylene (C2H2) from CO2 is an important industrial hydrocarbon separation process with a major difficulty originating in the similar molecular shapes of C2H2 and CO2. Moreover, the presence of water or moisture is usually a significant drawback for this industrial separation process. Based on these necessities, a pyrene-based MOF, JXNU-5 was constructed from TBAPy ligand and one-dimensional europium carboxylate rods by Liu et al.90 The presence of strong π–π stacking interactions between conjugated pyrenes resulted in water-resistant JXNU-5. Structural investigations demonstrated that the carboxylate oxygen atoms of the europium carboxylate rods are exposed on the pore wall, which are desirable sites for the acidic H atoms of C2H2 molecules but are unfavorable sites for CO2 molecules with two electronegative O atoms. Consequently, JXNU-5 could adsorb more C2H2 (55.9 cm3 g−1) then CO2 (34.8 cm3 g−1) at 298 K and 1 atm. The separation of C2 hydrocarbons including C2H2, C2H4 (ethylene) and C2H6 (ethane) from CH4 is as important to ensure more effective use of natural gas and utilization of C2 hydrocarbons for other useful processes. Huang et al.93 synthesized (Me2NH2)2[Zn6(TBAPy)2(Ade-NH2)4(μ4-O)](DMF)10 (H2O)8(bioMOF 1-NH2) by the coordination of Zn(II) with TBAPy and 2-aminoadenine (Ade-NH2). The structure exhibits one-dimensional tubular channels with exposed Watson–Crick faces and nitrogen-rich purines, providing strong adsorption sites for C2 hydrocarbons. The additional amine groups positioned toward the pores enhanced the adsorption capacity of more polarizable C2 molecules by contrast with CH4.
Different studies on NU-1000 have been performed by Yabushita et al. for the separation of aromatic compounds from hydrocarbons in aqueous solution, including (i) furanics (e.g. 5-hydroxymethylfurfural (HMF) and furfural)219 (ii) phenolics (e.g. 4-hydroxybenzaldehyde, 4-hydroxybenzoic acid, vanillin, vanillic acid, syringaldehyde, and syringic acid),218 and (iii) coumaric and ferulic acids.220 In all studies, aromatic compounds have successfully been separated from the sugars that they were derived from. The adsorption of sugars such as glucose, fructose, and xylose was excluded in all cases when they are in competition with the aromatics. However, results showed that the use of ionic liquid (IL) can enhance the uptake of NU-1000 toward monomeric sugars as a result of the generation of new adsorption sites by the incorporation of IL into the MOF space.220 NU-1000 can be further used for the molecular recognition of different types of sugars, which may be crucial for the selective synthesis of glucose from biomass-based feedstocks via cellulose depolymerization.221 Previous studies on enzyme-catalyzed depolymerization of cellulose showed that the presence of cellobiose can result in product inhibition and glucose product degradation. Therefore, it is promising that NU-1000 can adsorb cellobiose efficiently while the adsorption of glucose was excluded. Electronic structure calculations demonstrated that the reason of the selective molecular recognition is the number of favorable CH–π interactions made by the axial CH groups on a single face of the molecule with pyrene units of the MOF, which is higher in the case of cellobiose compared to that of glucose (Fig. 11). Because of the same reason, the interaction of sugars with pyrene of NU-1000 are weaker than that of aromatics.221
 | ||
| Fig. 11 (a) Adsorption isotherms of carbohydrates on NU-1000 at 297 K, where c/csat in x-axis defines the equilibrium concentration relative to the saturation concentration. The inset shows isotherms in a low concentration range. (b) Illustration of the structures of different carbohydrates and their adsorption on NU-1000. The red protons are on an axial plane of each carbohydrate. The blue plates represent the adsorptive areas of pyrene units in which the hydrocarbons can interact with. Modified and reprinted with permission from ref. 221. | ||
Since the use of pyrene-based MOFs can be advantageous for the capture of radioactive gases, their stability under radiation conditions at nuclear energy reprocessing sites is highly important. Hanna et al.225 investigated the radiolytic stability of NU-1000 by exposing gamma irradiation. The PXRD pattern of NU-1000 showed no change after both high and low irradiation dose, confirming the structural stability of the MOF. Moreover, the surface area and morphology remained the same as demonstrated by N2 physisorption isotherms and SEM images. This can be attributed to (i) higher linker connectivity of NU-1000 making it more stable toward cleavage of the carboxylate oxygen-zirconium bonds, (ii) low density of Zr6O8 nodes in NU-1000, causing the absorption of a limited amount of radiation, and (iii) the absorption of radiation mainly by Zr-nodes then its transfer to the dangling –OH and H2O groups, rather than damaging TBAPy linkers.
3.4 Heterogeneous catalysis
Heterogeneous catalysis is one of the most extensively studied fields with MOFs due to their high chemical versatility, porosity allowing molecules diffusion, and the presence of catalytically active centers in the structure. The catalytic function can be implemented at the organic ligand or metal node, which can be done by the direct synthesis or post-synthetic modification such as AIM and SALI. In addition, some catalytically active species can be incorporated in the pores of the MOF by encapsulation. In this way, MOFs can either participate in the catalytic reaction by stabilizing transition states, orienting molecules, or presenting additional active sites. They can also act as a medium for dispersing the catalytic species.226 The inclusion of pyrene ligands in the structure of MOFs has drawn attention to heterogeneous catalysis due to its advantageous electronic properties. Studies on graphene and carbon nanotubes (CNT) showed that the introduction of pyrene into the catalytic systems can enhance the electron transfer efficiency due to the strong π−π interactions, resulting in better catalytic activity.227,228 Based on this, Tu et al.88 synthesized Fe-TBAPy for benzene hydrolxylation, considering that the 3.4 Å pores sandwiched between the pyrene cores of TBAPy could be the strong adsorption-sites because of the strong π−π interaction between the benzene and the pyrene. Therefore, as an emerging subfamily, pyrene-based MOFs have aroused interest in the last years and been investigated for several heterogeneous catalytic reactions computationally and experimentally. As yet, computational studies have focused on the study of energetic barriers, formation mechanisms and the post-synthetic modification of catalytic active sites on the Zr6 nodes in NU-1000 for different catalytic reactions; including ethylene dimerization,111,229,230 ethylene hydrogenation,231 alkyne semihydrogenation,232 alcohol dehydrogenation,233 hydrolysis of CWAs,234,235 and C–H bond activation.236–238 In addition, computational studies have been informative about the importance of the selection of transition metals for the post-synthetic functionalization on the node, and supports for the design of heterogeneous catalyst.239The interest in NU-1000 has increased to a very great extent thanks to the experimental post-synthetic modification methods, such as AIM, SIM, and SALI, to introduce different secondary metal components in the framework. The applicability of post-synthetic modifications is owing to the large mesopores of NU-1000, which allows efficient and homogeneous metalation on the zirconium nodes and promotes diffusive transport of substrates within the MOF during heterogeneous catalysis.141 Moreover, the stability of the structure in different environments as well as the high porosity and large enough channels of NU-1000 also enable the impregnation of guest molecules. In the following sections, the experimental heterogeneous catalytic reactions performed by NU-1000 will be focused mainly due to its rising popularity among all pyrene-based MOFs.
Due to their tendency to aggregate and instability in solutions, polyoxometalates (POMs) are one of the most studied species supported by NU-1000.244 POMs are anionic metal oxide clusters composed of group V or VI transition metals having acid–base and redox properties. NU-1000 has been found as an appropriate support for POMs thanks to its small triangular channels (12 Å) enabling the incorporation of large POMs while the larger (31 Å) hexagonal channels promote the substrate diffusion.245 For example, Ahn et al.246 showed that the incorporation of phosphotungstic acid (PTA, H3PW12O40) in NU-1000 can be successfully performed and can provide the tungsten oxide active sites for the acid-catalyzed reaction of o-xylene isomerization/disproportionation. The structure remained stable after the catalysis, showing the applicability of POM-incorporated MOFs (POM@MOFs) even for aggressive reactions such as hydrocarbon isomerization.
POMs are also successful catalysts for acid and oxidation reactions; therefore, POM@MOFs have been studied for the oxidative detoxification of a mustard gas simulant, CEES. The oxidation products can be either a singly oxidized sulfoxide (CEESO), or a doubly oxidized sulfone (CEESO2). While CEESO is considered nontoxic, CEESO2 has detrimental effects similar to the mustard gas. Therefore, the selectivity of the reaction is highly important. Two different POMs, H3PW12O40 and H5PV2Mo10O40, have been encapsulated in NU-1000 (named as PW12@NU-1000245,247,248 and PV2Mo10@NU1000,244 respectively) for the oxidation of CEES using hydrogen peroxide (H2O2) as the oxidant. Results showed that while 100% conversion of the parent agent is successfully done, the problem is the selectivity toward the desired non-toxic oxidation product.244 For example, Buru et al.245 showed that PW12@NU-1000 could successfully convert CEES in 20 min; however, the selectivity toward CEESO remained at 57%. They also investigated the performance of H3PW12O40 and NU-1000 individually, finding that while H3PW12O40 promoted the selective oxidation of CEES, NU-1000 is overoxidizing CEES by the formation of CEESO2. Based on this, the effect of the POM location and accessibility of substrates on the catalytic rate and selectivity have been investigated using the same system. The samples were activated in different ways, under vacuum at 120 °C for PW12@NU-1000-120, and by supercritical CO2 drying for PW12@NU-1000-scCO2.247 While 100% conversion in 20 min with 59% selectivity was observed for PW12@NU-1000-120, 100% conversion in <10 min with 90% selectivity was obtained for PW12@NU-1000-scCO2. Volumetric N2 sorption isotherms showed that PW12@NU-1000-scCO2 has a reduced mesopore volume, yet PW12@NU-1000-120 has reduced micropore volume. This can indicate that in PW12@NU-1000-120, the POM is situated in micropores and the mesopores of MOF are accessible, resulting in the formation of both CEESO and CEESO2 products. When the POM is situated in the mesopores as in the case of PW12@NU-1000-scCO2, diffusion of the substrate to POM in the pores is no longer hindered and singly-oxidized product is formed almost exclusively.
| Reaction | Reactant | Material name | Ref. |
|---|---|---|---|
| Hydrogenation | m-Nitrophenol | CoS-AIM | 128 |
| Ethylene | Ir(C2H4)2-supported NU-1000 | 249 and 250 | |
| Ethylene | Ni-AIM | 135 | |
| Ethene | ReOx-functionalized NU-1000 | 251 | |
| Ethylene | Pt-AIM | 253 | |
| Diphenylacetylene | Rh-Ga-NU-1000 | 254 | |
| Oxidation | Benzyl alcohol | Co-Al-NU-1000 | 255 |
| Methane | Cu-NU-1000 | 256 | |
| Propane | Co-SIM + NU-1000 | 129 | |
| Co-AIM + NU-1000 | |||
| Propane | CoAIM-M-SIM-NU-1000 (M = Ni, Zn, Al, Ti, Mo) | 257 | |
| 4-Methoxybenzyl alcohol | V-NU-1000 | 141 | |
| 4-Methoxybenzyl alcohol | Zr-NU-1000-V | 145 | |
| Dehydration | Ethanol | Al-NU-1000 | 258 |
| 2-Propanol | Si-NU-1000 | 259 | |
| Glucose | PO4/NU | 260 | |
| Oligomerization | Ethylene | Ni-Facac-AIM-NU-1000 | |
| Ni-Acac-AIM-NU-1000 | 261 | ||
| Ethylene | Cr-SIM-NU-1000 | 147 | |
| Epoxidation | Cyclohexene | Mo-SIM | 142 |
| Cyclohexene | Nb-AIM | ||
| Nb-SIM | 262 | ||
| Polymerization | Ethylene and 1-hexene | Hf-NU-1000-ZrBn | 263 |
| Hydrolysis | Nerve agent simulant | Ce-n-SIM-NU-1000 | |
| Ce-l-SIM-NU-1000 | 264 |
SALI-based functionalization of MOF nodes enables of the fine-tuning the electronic properties and/or the Brønsted acid character of the MOF catalyst. In the case of NU-1000, SALI approach takes advantage of the ability of the Zr6 nodes to be altered through the reaction between the free Zr–OH moieties (labile –OH and H2O ligands) and introduced nonstructural organic ligands.90 The functional ligands may be carboxylic acid containing molecules,265–269 phosphonic acid containing molecules,270 or perfluoroalkane chains,271 in which their addition in NU-1000 results in enhanced activity in different catalytic reactions. Moreover, the further modification of the ligands with varying electron-donating or electron-withdrawing groups can be performed to tune the electronic environment provided by the Zr6 nodes.267 SALI-modification of NU-1000 can be performed with an organic ligand,268 or a metal complex featuring an organic ligand.269 NU-1000 functionalized by 5,5-di-thio-bis(2-nitrobenzoic acid) ligand (DTNB@NU-1000) was found to be catalytically active for the degradation and subsequent detection of a nerve agent, which was observed by the release of the chromophore degradation product by visual inspection of filtered samples.268 The immobilization of an Ir(III) complex with a bis-phosphinite pincer ligand in NU-1000 was performed to enhance the catalytic activity toward the hydrogenation of liquid alkenes (1-decene and styrene) compared to that of homogeneous Ir(III)-complex, by preventing leaching of Ir from the structure.269 SALI-based ligand incorporation can be followed by anchoring catalytically active metal sites on the ligand attached to the Zr nodes. Madrahimov et al.270 synthesized NU-1000-bpy-NiCl2 by SALI-modification of NU-1000 with 5-methylphosphonate-2,2′-bipyridine (NU-1000-bpy), followed by the exposure of NU-1000-bpy material to a solution of anhydrous NiCl2. While NU-1000 and NU-1000-bpy showed no activity for liquid-phase ethylene dimerization, NU-1000-bpy-NiCl2 showed 95% conversion with 93% selectivity toward the desired product, 1-butene. Similarly, Berijani et al.266 immobilized L-tartaric acid on the Zr6 nodes of NU-1000 and then anchored Mo(IV)-complex on the ligand as the Lewis acid site, forming a chiral heterogeneous catalyst C-NU-1000-Mo. The catalyst could successfully perform the epoxidation of various prochiral alkenes to form enantiomers of epoxides, where the conversion of styrene to styrene epoxide was completed in 5 h with 86% selectivity. The deposition of metal ions on a specific location can be also provided by SALI-modification. Peters et al.265 modified NU-1000 with naphthalene dicarboxylate (NDC) linkers to block the small cavities where few-atom clusters of cobalt oxide preferentially grow. This approach can be highly useful where catalyst location matters for the enhanced catalytic reactivity. In addition to the deposition of metal ions, metal nanoparticles can be incorporated in SALI-modified NU-1000 as studied by Huang et al.271 NU-1000 has been modified with perfluoroalkane groups to provide a hydrophobic platform to encapsulate Pd nanoparticles in the pores. The resulting material Pd@F15-NU-1000 was found to be catalytically active (87% conversion with 21% selectivity) for the direct C–H arylation of indoles in water.
3.5 Electrochemistry applications
MOFs have recently attracted the attention of electrochemical applications like electrochemical catalysis,272 supercapacitors,273 batteries,274 and electrochromic devices.75 The presence of well defined porous channels is one feature of interest for ion transport. However, one major limitation of MOFs for being fully considered in electrical and electrochemical applications is their intrinsic low electrical and direction-dependent conductivity. Charge transport in MOFs presents a small electronic coupling and frontier orbitals overlap, given the separation between linkers.275 In this regard, pyrene-based MOFs like NU-901 and NU-1000 have been modified via post-synthetic modifications to improve the conductivity properties suitable for electrochemical applications. Wang et al.276 incorporated thiophene oligomers on the Zr6 node of NU-1000 via SALI to improve the electronic conductivity of the material. The length of the oligomers allowed the electrochemical conversion to a channel-anchored strands of polythiophene, resulting in a material with an improved conductivity without compromising the MOF porosity. Another strategy is to exploit the donor nature of the pyrene moiety combined with electron acceptor molecules. The porous topology of NU-1000 and NU-901 have been exploited to the encapsulation of acceptor moieties like metallacarboranes277 and buckminsterfullerene (C60)278 as guest molecules, tuning the conductivity via donor–acceptor interactions.Although different strategies can lead to conductive MOFs, it is important to understand the mechanisms of charge transport in MOFs. Charge propagation in MOFs can be as a result of ligand-node orbital energy overlap, π-stacking interactions and redox hopping. The last one is of great importance for electrocatalytic applications. Several studies have anchored redox active building blocks like metallocenes in NU-1000 for redox hopping transfer and as a redox shuttle in photoelectrochemical or electrocatalytic systems.279–281 Celis-Salazar et al.281 studied the electron diffusion and ion diffusion in the redox-hopping process in three metallocene-doped (M = Fe, Ru, Os) NU-1000 MOFs via chronoamperometric response measurements. Their results highlighted the importance of metal for electron diffusion, likewise ion mobility can lower due to ion-pairing associations. The electron redox hopping rates inside M-NU-1000 frameworks can be controlled through the modifications of the self-exchange rates of redox centers and proper selection of counteranions.281 Charge transport properties can be also affected by the topology of the MOF. In NU-1000 the redox-hopping-based charge transport is highly anisotropic, presenting a higher diffusion coefficient along the one-dimensional mesopores.282 These fundamental electronic properties and findings have direct implication for the design and further optimization of pyrene-based MOFs in electrochemical applications, where the understanding of the charge transport mechanism behavior is essential.
Based on redox-active and catalytic functionalities, electroactive pyrene-based MOFs can be synthesized as thin films via atomic layer deposition (ALD)283 or electrophoretic deposition (EPD).284 EPD thin-film technique distinguishes from the previously discussed ALD (Section 2.3.1) by the direct use of an electric field. EPF is based on the movement and deposition of charged particles under the electric field onto a conductive electrode, to develop thin or thick films and coatings. The thickness and morphology of the films can be controlled by simply adjusting the depositing time and applied potential.285 These thin films can be used as molecular switches,286,287 bias-switchable permselectivity anions membranes,279 and electrocatalysts for OER,283,288 HER,289,290 and CO2 reduction.291 Among these applications, electrocatalytic OER and HER are of interest since electrocatalytic water splitting can provide a much more sustainable and carbon-neutral route for energy generation. The intermittent nature of renewable power sources is a common problem for the technology design of energy-storage devices. Electrochemical water splitting is a promising pathway to overcome this because it produces the energy-dense and clean-burning fuel hydrogen. In such a scenario, intermittent energies such as wind and solar can be used to generate electricity, and when available drive the splitting of water at electrocatalyst electrodes in electrolyzers.292 Noh et al.289 anchored molybdenum sulfide (MoSx) in NU-1000 via SIM (MoSx-SIM) to support multiple catalyst active sites for electrocatalytic HER. MoSx-SIM presented a low electrocatalytic HER activity in electrode-supported thin-film form, due to the insulating nature of the thin film MOF which leaves the catalytic sites electrochemically isolated from the underlying electrode. To overcome such limitation, molecular viologen-type redox mediators were included to effectively diffuse in the MOF and to deliver the electrons from the underlying electrode to the MoSx catalytic units. Mechanistic studies showed that overall control of the rate of catalysis can be defined by mediator-to-catalyst electron transfer, solution-to-catalyst proton transfer, or both.289,290 Kung et al.283 performed the chemical growth of uniform thin films of NU-1000 on transparent fluorine-doped tin oxide (FTO) conducting glass, followed by the deposition of Co(II) ions, resulting in the oxidation catalyst Co-AIM NU-1000. Cyclic voltammetric (CV) experiments showed that Co-AIM-NU-1000 is electrochemically active. The charge transfer occurred by hole hopping through the repeating pyrene units, meanwhile the counterions can diffuse through the regular channels for the neutralization of the charges formed on the linkers. The pH was found to be important for the oxidation efficiency as the material electrocatalyzes the four-electron conversion of hydroxide ion to dioxygen at pH = 11 while it shows a poor activity at pH = 8.2, 9, and 10.
3.6 Bio-medical applications
MOFs have emerged in the field of bio-medicine applications. They are of particular interest because of their crystal porosity, tunable functionalities and properties, and availability to encapsulate molecules.293 Pyrene-based MOFs have also been envisioned and conceived as smart materials for biological applications. The luminescence properties of pyrene-based MOFs is one of the attractive properties for the detection of biomarkers related to diseases.87,166,173 The advantage of these MOFs based sensors is that they can be implemented as MOF-coated paper test strips.166 The optical properties of pyrene-based MOFs have also been considered as an X-ray computed tomography (CT) contrast agent. Robison et al.92 reported in vitro studies of a bismuth pyrene MOF (Bi-NU-901), showing ∼7 times better contrast intensity compared to that of a zirconium MOF (NU-901) with the same topology and ∼14 times better contrast than a commercially available CT contrast agent. Their results are of interest to bio-imaging applications for clinical tests given the need to have agents with the lowest dose possible, maximum contrast between tissue and background, and minimal toxicity. In this case, Bi-NU-901 could require much less material compared to contrast agents in current use.92 Another optical property of pyrene-based MOFs has been used in the field of photodynamic therapy, where the capability of singlet generation can be use to kill cancerous and precancerous cells after light activation.294 Zhao et al.294 designed pyrene-based MOF nanoparticles as a photodynamic therapy nanoplatform. The material is made from NU-1000 mixed with TCPP via ligand substitution, subsequently, its surface is functionalized with poly-(ethylene glycol) (PEG) and folic acid for biocompatibility and tumor targeting effect, respectively. The MOF nanoparticles could generate reactive oxygen species under 650 nm laser irradiation efficiently. The nanoparticles displayed low toxicity and good photodynamic effect with antitumor efficacy in vitro and in vivo experiments.294The natural crystalline porosity of MOFs has attracted the attention for the development of new and efficient drug delivery systems.296 Drug delivery systems are devices that enable a therapeutic substance to selectively reach its site of action without reaching the non-target cell, organs, or tissues. Such devices should accomplish a controlled and sustained release of the medicine, to minimize side effects and ensure the efficacy of the pharmaceutical upon reaching the desired target.297 MOFs are promising for this biomedical application since they can be modified to acquired functionalities to give biostability, target selectivity, and encapsulation of the cargo.61 There has been recent progress in using pyrene-MOFs as promising drug delivery systems. This includes applications like insulin delivery,298,299 and chemotherapy treatments.295,297,300,301 MOFs bring different advantages over traditional pharmaceuticals. In the case of oral insulin delivery nanocarriers, MOFs can exhibit higher insulin loading capacities as a result of their porosity. Chen et al.298 immobilised insulin in NU-1000 obtaining a high loading of ∼40 wt% in only 30 min. The pore topology and dimensionality allow the effective diffusion of insulin through the framework, and also exclude digestive insulin enzymes like pepsin under simulated stomach environment conditions. Most importantly, the nanocarriers exhibit the controlled release under conditions mimicking the delivery target point. These last features of selective target and controlled release are of great interest to cancer treatment applications. Zhao et al.295 synthesized PEG NU-1000 MOF for the encapsulation of an anticancer drug Doxorubicin (DOX) (Fig. 12). The material DOX@NU-1000@PEG displayed good biocompatibility and could inhibit tumor growth efficiently with a long-time release behavior for over 2 weeks. On the other hand, in some cases, it is necessary to control the release kinetics as in the case of 5-fluorouracil (5-FU) for breast cancer treatment. Hu et al.301 synthesized a Mg pyrene-based MOF (TDL-Mg) with suitable host–guest interactions with 5-FU, leading to a medically reasonable sustained release rate and curative effect on HeLa cancer cells. Likewise, the advantage of using a pyrene-based MOF like TDL-Mg is thanks to the natural fluorescent properties which were also exploited to monitored the 5-FU release using the “turn-on” fluorescence mechanism.
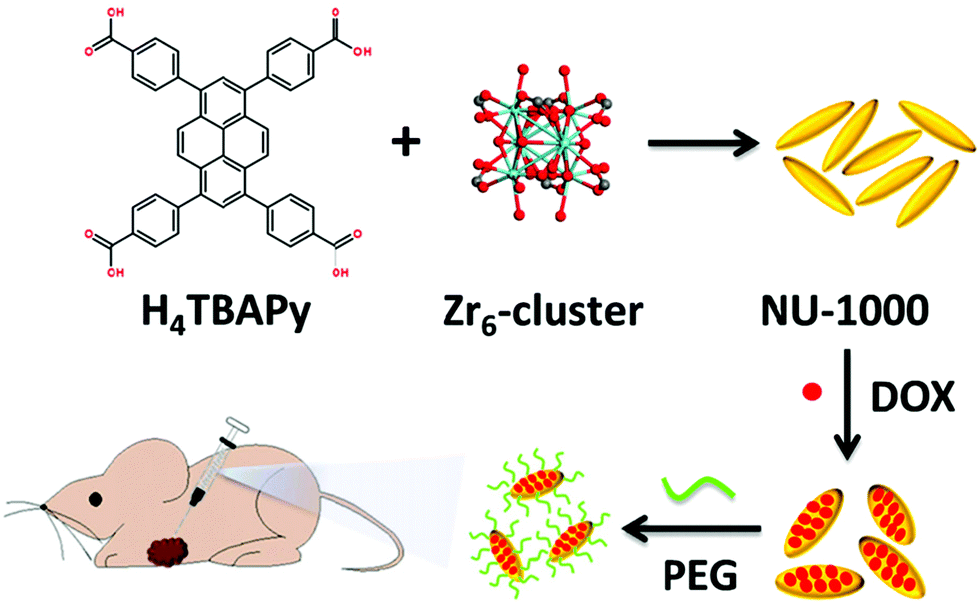 | ||
| Fig. 12 Preparation and application of NU-1000 nanoparticles for chemotherapy. Doxorubicin (DOX) molecules are encapsulated in the NU-1000 nanoparticles and functionalized with polyethylene glycol (PEG). Reprinted with permission from ref. 295. | ||
4 Concluding remarks and future prospects
Pyrene-based MOFs have attracted attention in different research fields as mentioned previously, given the promising structural, optical, and electronic properties of the pyrene moiety. Up to date, more than 20 different pyrene-based ligands have been combined with different metals (from transition metals to lanthanides), resulting in well-defined structures with varying structural characteristics such as porosity, pore size, and topology, which are the key components in many applications mentioned in this review. Pore surface engineering using post-modification methods (such as AIM, SIM, and SALI) has encouraged researchers to decorate metal nodes with various single metal sites, metal clusters, and linkers, resulting in the desired functionalization of the structures for target applications.There have been numerous attempts for synthesizing novel pyrene-based molecules, in addition to the studies exploring their combination with metals in coordination complexes. Nevertheless, pyrene-based MOFs are one of the relatively new subsets of the extensive MOF-related research studies. The curiosity on pyrene-based MOFs has increased tremendously with the continued development of novel pyrene-based ligands over the past few years. However, the challenges faced throughout the synthesis of pyrene derivatives (mainly during the coupling reactions), as well as in the prediction of the coordination environment of the pyrene-based ligands are still the main difficulties. Therefore, the research is still at an early stage in terms of the variety of pyrene linker structures and the number of reported pyrene-based MOFs. Most of the pyrene-based organic ligands rely on similar chemistry, where pyrene core is decorated with benzene-derivatives with different functionalities (mostly carboxylates) at -1, -3, -6 and -8 positions. Hence, the effect of different electrophilic aromatic substitutions on the coordination with metals should be unveiled by concentrating more on the ligand design. For example, electron-donating groups such as pyrazole or thiol can be considered as the next-step functionalization on the pyrene core, resulting in a novel MOF chemistry based on the interactions with metal nodes.
It is not easy to predict the resulting MOF structure as the MOF synthesis is complicated with too many variables and this is valid for all different MOF families. Therefore, the relationship between the synthesis conditions and the resulting topologies of MOFs are still not well understood. Different experimental protocols can lead to well distinctive topological MOF structures, despite the starting precursors are the same ligand and transition metal element. Therefore, there is a need for the fundamental understanding of the effect of different synthetic routes for the development of new MOFs with the same metal chemistry, as in the case of Zn-TBAPy MOFs. Pyrene-based MOFs is a good platform for the systematic investigation of the effect of reaction conditions on the resulting structures thanks to the possibility of synthesizing a significant number of structures with promising physicochemical features. Machine learning tools can be exploited to capture the importance of the synthesis parameters to gain a better understanding of pyrene-based MOF synthesis conditions and pay a better experimental design and optimal synthesis conditions. Consequently, computational hypothetical MOF generation and screening strategies can expand the library of pyrene-based MOFs that can lead the experimental attempts in the right direction. Such studies can lead to the prediction of crucial characteristics, such as the optical properties, surface chemistry, and pore shape required for target specific applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the National Centre of Competence in Research (NCCR) Materials' Revolution: Computational Design and Discovery of Novel Materials (MARVEL) of the Swiss National Science Foundation (SNSF) and the Swiss National Supercomputing Centre (CSCS) under project ID s1005. A. O.-G. acknowledges the financial support of the Swiss National Science Foundation (SNSF) under Grant 200021_172759. D. O. was supported by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No. 666983, MaGic).References
- X. Feng, J. Hu, C. Redshaw and T. Yamato, Chem. – Eur. J., 2016, 22, 11898–11916 CrossRef CAS PubMed.
- R. Haldar, L. Heinke and C. Wöll, Adv. Mater., 2020, 32, 1905227 CrossRef CAS.
- A. Laurent, Ann. Chim. Phys., 1837, 66, 136–137 Search PubMed.
- T. M. Figueira-Duarte and K. Mullen, Chem. Rev., 2011, 111, 7260–7314 CrossRef CAS.
- D. Zych, Molecules, 2019, 24, 2551 CrossRef PubMed.
- A. J. Howarth, M. B. Majewski and M. O. Wolf, Coord. Chem. Rev., 2015, 282, 139–149 CrossRef.
- B. J. Burnett, P. M. Barron and W. Choe, CrystEngComm, 2012, 14, 3839–3846 RSC.
- N. U. Day, C. C. Wamser and M. G. Walter, Polym. Int., 2015, 64, 833–857 CrossRef CAS.
- C. F. Pereira, M. M. Simões, J. P. Tomé and F. A. Almeida, Paz, Molecules, 2016, 21, 1348 CrossRef PubMed.
- J. M. Casas-Solvas, J. D. Howgego and A. P. Davis, Org. Biomol. Chem., 2014, 12, 212–232 RSC.
- E. Manandhar and K. J. Wallace, Inorg. Chim. Acta, 2012, 381, 15–43 CrossRef CAS.
- K. C. Stylianou, R. Heck, S. Y. Chong, J. Bacsa, J. T. A. Jones, Y. Z. Khimyak, D. Bradshaw and M. J. Rosseinsky, J. Am. Chem. Soc., 2010, 132, 4119–4130 CrossRef CAS.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- L.-M. Yang, P. Ravindran, P. Vajeeston and M. Tilset, Phys. Chem. Chem. Phys., 2012, 14, 4713–4723 RSC.
- T. Kuroda-Sowa, M. Munakata and H. Matsuda, S.-i. Akiyama and M. Maekawa, J. Chem. Soc., Dalton Trans., 1995, 2201–2208 RSC.
- L. Tchertanov and C. Pascard, Acta Crystallogr., Sect. B: Struct. Sci., 1997, 53, 904–915 CrossRef.
- F. H. Herbstein, Acta Crystallogr., Sect. B: Struct. Sci., 2001, 57, 517–519 CrossRef CAS.
- R. Sekiya and S.-i. Nishikiori, Chem. Commun., 2001, 2612–2613 RSC.
- B. D. Wagner, G. J. McManus, B. Moulton and M. J. Zaworotko, Chem. Commun., 2002, 2176–2177 RSC.
- T. Kuroda-Sowa, S. Q. Liu, Y. Yamazaki, M. Munakata, M. Maekawa, Y. Suenaga, H. Konaka and H. Nakagawa, Inorg. Chem., 2005, 44, 1686–1692 CrossRef CAS.
- R. Sekiya, S.-i. Nishikiori and K. Ogura, Inorg. Chem., 2006, 45, 9233–9244 CrossRef CAS.
- G. J. McManus, J. J. Perry, M. Perry, B. D. Wagner and M. J. Zaworotko, J. Am. Chem. Soc., 2007, 129, 9094–9101 CrossRef CAS.
- Q. Wang, J. Zhang, C.-F. Zhuang, Y. Tang and C.-Y. Su, Inorg. Chem., 2009, 48, 287–295 CrossRef CAS PubMed.
- L. Rajput and K. Biradha, CrystEngComm, 2009, 11, 1220 RSC.
- R. Sekiya, S.-i. Nishikiori and R. Kuroda, CrystEngComm, 2009, 11, 2251 RSC.
- L. Rajput and K. Biradha, New J. Chem., 2010, 34, 2415 RSC.
- R. Sekiya and S.-i. Nishikiori, Chem. Commun., 2012, 48, 5022 RSC.
- G. Mukherjee and K. Biradha, Chem. Commun., 2012, 48, 4293 RSC.
- M. Nishio, N. Motokawa, M. Takemura and H. Miyasaka, Dalton Trans., 2013, 42, 15898 RSC.
- G. Mukherjee and K. Biradha, Cryst. Growth Des., 2014, 14, 419–422 CrossRef CAS.
- J.-W. Ye, H.-L. Zhou, S.-Y. Liu, X.-N. Cheng, R.-B. Lin, X.-L. Qi, J.-P. Zhang and X.-M. Chen, Chem. Mater., 2015, 27, 8255–8260 CrossRef CAS.
- A. C. Tella, O. A. Ameen, P. A. Ajibade and L. O. Alimi, J. Porous Mater., 2015, 22, 1599–1605 CrossRef CAS.
- T. Delgado, M. Meneses-Sánchez, L. Piñeiro-López, C. Bartual-Murgui, M. C. Muñoz and J. A. Real, Chem. Sci., 2018, 9, 8446–8452 RSC.
- H.-R. Fu, X.-X. Wu, L.-F. Ma, F. Wang and J. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 18012–18020 CrossRef CAS.
- D. Shen, G. Wang, Z. Liu, P. Li, K. Cai, C. Cheng, Y. Shi, J.-M. Han, C.-W. Kung, X. Gong, Q.-H. Guo, H. Chen, A. C.-H. Sue, Y. Y. Botros, A. Facchetti, O. K. Farha, T. J. Marks and J. F. Stoddart, J. Am. Chem. Soc., 2018, 140, 11402–11407 CrossRef CAS.
- L. Zhao, J. Yang, M. Gong, Y. Zhang and J. Gu, J. Mater. Chem. C, 2020, 8, 3904–3913 RSC.
- M. Meneses-Sánchez, L. Piñeiro-López, T. Delgado, C. Bartual-Murgui, M. C. Muñoz, P. Chakraborty and J. A. Real, J. Mater. Chem. C, 2020, 8, 1623–1633 RSC.
- J. X. Li, Q. L. Guan, Y. Wang, Z. X. You, Y. H. Xing, F. Y. Bai and L. X. Sun, New J. Chem., 2020, 44, 1446–1454 RSC.
- R. Dalapati, S. Nandi and S. Biswas, Dalton Trans., 2020, 49, 8684–8692 RSC.
- A. J. Blake, N. R. Champness, A. N. Khlobystov, D. A. Lemenovskii, W.-S. Li and M. Schröder, Chem. Commun., 1997, 1339–1340 RSC.
- A. J. Blake, G. Baum, N. R. Champness, S. S. M. Chung, P. A. Cooke, D. Fenske, A. N. Khlobystov, D. A. Lemenovskii, W.-S. Li and M. Schröder, J. Chem. Soc., Dalton Trans., 2000, 4285–4291 RSC.
- G. L. Ning, L. P. Wu, K. Sugimoto, M. Munakata, T. Kuroda-Sowa and M. Maekawa, J. Chem. Soc., Dalton Trans., 1999, 2529–2536 RSC.
- N. L. Rosi, J. Kim, M. Eddaoudi, B. Chen, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 1504–1518 CrossRef CAS PubMed.
- F.-F. Li, J.-F. Ma, S.-Y. Song, J. Yang, Y.-Y. Liu and Z.-M. Su, Inorg. Chem., 2005, 44, 9374–9383 CrossRef CAS.
- C. A. Williams, A. J. Blake, C. Wilson, P. Hubberstey and M. Schröder, Cryst. Growth Des., 2008, 8, 911–922 CrossRef CAS.
- G. Huang, Y.-Q. Sun, Z. Xu, M. Zeller and A. D. Hunter, Dalton Trans., 2009, 5083–5093 RSC.
- C.-C. Tsai, T.-T. Luo, J.-F. Yin, H.-C. Lin and K.-L. Lu, Inorg. Chem., 2009, 48, 8650–8652 CrossRef CAS PubMed.
- J. Hu, M.-H. Nguyen and J. H. Yip, Inorg. Chem., 2011, 50, 7429–7439 CrossRef CAS.
- N. Zhao, F. Sun, H. He, J. Jia and G. Zhu, Cryst. Growth Des., 2014, 14, 1738–1743 CrossRef CAS.
- T. C. Wang, W. Bury, D. A. Gómez-Gualdrón, N. A. Vermeulen, J. E. Mondloch, P. Deria, K. Zhang, P. Z. Moghadam, A. A. Sarjeant, R. Q. Snurr, J. F. Stoddart, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2015, 137, 3585–3591 CrossRef CAS PubMed.
- S. S. Park, C. H. Hendon, A. J. Fielding, A. Walsh, M. OKeeffe and M. Dincă, J. Am. Chem. Soc., 2017, 139, 3619–3622 CrossRef CAS.
- M. Roy, A. A. Lonardo, D. N. Pham, A. Kreider-Mueller, J. A. Golen and D. R. Manke, CrystEngComm, 2019, 21, 4255–4257 RSC.
- J. M. Taylor, R. Vaidhyanathan, S. S. Iremonger and G. K. Shimizu, J. Am. Chem. Soc., 2012, 134, 14338–14340 CrossRef CAS.
- S. B. Kalidindi, S. Nayak, M. E. Briggs, S. Jansat, A. P. Katsoulidis, G. J. Miller, J. E. Warren, D. Antypov, F. Corà, B. Slater, M. R. Prestly, C. Martí-Gastaldo and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2015, 54, 221–226 CrossRef CAS.
- C. Dong, J. Bai, X.-L. Lv, W. Wu, J. Lv and J.-R. Li, Inorg. Chem., 2019, 58, 15909–15916 CrossRef CAS PubMed.
- L. Robison, R. J. Drout, L. R. Redfern, F. A. Son, M. C. Wasson, S. Goswami, Z. Chen, A. Olszewski, K. B. Idrees, T. Islamoglu and O. K. Farha, Chem. Mater., 2020, 32, 3545–3552 CrossRef CAS.
- R.-J. Li, M. Li, X.-P. Zhou, S. W. Ng, M. O'Keeffe and D. Li, CrystEngComm, 2014, 16, 6291–6295 RSC.
- Y. Xiao, Y. Qi, X. Wang, X. Wang, F. Zhang and C. Li, Adv. Mater., 2018, 30, 1803401 CrossRef PubMed.
- X. Zeng, R. Liu, D. Liu, Q. Liu and Y. Wang, Z. Anorg. Allg. Chem., 2019, 645, 1379–1383 CrossRef CAS.
- K. C. Stylianou, J. Rabone, S. Y. Chong, R. Heck, J. Armstrong, P. V. Wiper, K. E. Jelfs, S. Zlatogorsky, J. Bacsa, A. G. McLennan, C. P. Ireland, Y. Z. Khimyak, K. M. Thomas, D. Bradshaw and M. J. Rosseinsky, J. Am. Chem. Soc., 2012, 134, 20466–20478 CrossRef CAS.
- A. V. Vinogradov, H. Zaake-Hertling, A. S. Drozdov, P. Lönnecke, G. A. Seisenbaeva, V. G. Kessler, V. V. Vinogradov and E. Hey-Hawkins, Chem. Commun., 2015, 51, 17764–17767 RSC.
- J.-H. Qin, Y.-D. Huang, Y. Zhao, X.-G. Yang, F.-F. Li, C. Wang and L.-F. Ma, Inorg. Chem., 2019, 58, 15013–15016 CrossRef CAS.
- R. Seetharaj, P. Vandana, P. Arya and S. Mathew, Arab. J. Chem., 2019, 12, 295–315 CrossRef CAS.
- F. P. Kinik, A. Ortega-Guerrero, D. Ongari, C. P. Ireland and B. Smit, Mater. Cloud Archive, 2020, 156, 2020, DOI:10.24435/materialscloud:z5-ct.
- G. Ayoub, M. Arhangelskis, X. Zhang, F. Son, T. Islamoglu, T. Friščić and O. K. Farha, Beilstein J. Nanotechnol., 2019, 10, 2422–2427 CrossRef CAS.
- A. García-García, I. Oyarzabal, J. Cepeda, J. M. Seco, A. A. García-Valdivia, S. Gómez-Ruiz, A. Salinas-Castillo, D. Choquesillo-Lazarte and A. Rodríguez-Diéquez, New J. Chem., 2018, 42, 832–837 RSC.
- J. Park and M. Oh, CrystEngComm, 2016, 18, 8372–8376 RSC.
- Y.-L. Huang, Y.-N. Gong, L. Jiang and T.-B. Lu, Chem. Commun., 2013, 49, 1753–1755 RSC.
- Y.-L. Huang, L. Jiang and T.-B. Lu, ChemPlusChem, 2016, 81, 780 CrossRef CAS.
- Y.-L. Huang, D.-C. Zhong, L. Jiang, Y.-N. Gong and T.-B. Lu, Inorg. Chem., 2017, 56, 705–708 CrossRef CAS PubMed.
- L. Hu, X.-J. Hong, X.-M. Lin, J. Lin, Q.-X. Cheng, B. Lokesh and Y.-P. Cai, Cryst. Growth Des., 2018, 18, 7088–7093 CrossRef CAS.
- R. R. Maldonado, X. Zhang, S. Hanna, X. Gong, N. C. Gianneschi, J. T. Hupp and O. K. Farha, Dalton Trans., 2020, 49, 6553–6556 RSC.
- H. Alawisi, B. Li, Y. He, H. D. Arman, A. M. Asiri, H. Wang and B. Chen, Cryst. Growth Des., 2014, 14, 2522–2526 CrossRef CAS.
- J. E. Mondloch, W. Bury, D. Fairen-Jimenez, S. Kwon, E. J. DeMarco, M. H. Weston, A. A. Sarjeant, S. T. Nguyen, P. C. Stair, R. Q. Snurr, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 10294–10297 CrossRef CAS PubMed.
- C.-W. Kung, T. C. Wang, J. E. Mondloch, D. Fairen-Jimenez, D. M. Gardner, W. Bury, J. M. Klingsporn, J. C. Barnes, R. Van Duyne, J. F. Stoddart, M. R. Wasielewski, O. K. Farha and J. T. Hupp, Chem. Mater., 2013, 25, 5012–5017 CrossRef CAS.
- K. C. Stylianou, J. Bacsa, D. Bradshaw and M. J. Rosseinsky, Z. Anorg. Allg. Chem., 2014, 640, 2123–2131 CrossRef CAS.
- R.-J. Li, M. Li, X.-P. Zhou, D. Li and M. O'Keeffe, Chem. Commun., 2014, 50, 4047–4049 RSC.
- O. Karagiaridi, W. Bury, D. Fairen-Jimenez, C. E. Wilmer, A. A. Sarjeant, J. T. Hupp and O. K. Farha, Inorg. Chem., 2014, 53, 10432–10436 CrossRef CAS PubMed.
- P. Li, N. A. Vermeulen, X. Gong, C. D. Malliakas, J. F. Stoddart, J. T. Hupp and O. K. Farha, Angew. Chem., Int. Ed., 2016, 55, 10358–10362 CrossRef CAS PubMed.
- K. C. Park, C. Seo, G. Gupta, J. Kim and C. Y. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 38670–38677 CrossRef CAS.
- J. Ai, F.-Y. Chen, C.-Y. Gao, H.-R. Tian, Q.-J. Pan and Z.-M. Sun, Inorg. Chem., 2018, 57, 4419–4426 CrossRef CAS PubMed.
- L. Guo, M. Wang and D. Cao, Small, 2018, 14, 1703822 CrossRef PubMed.
- A. Gładysiak, T. N. Nguyen, R. Bounds, A. Zacharia, G. Itskos, J. A. Reimer and K. C. Stylianou, Chem. Sci., 2019, 10, 6140–6148 RSC.
- S. L. Anderson, P. G. Boyd, A. Gładysiak, T. N. Nguyen, R. G. Palgrave, D. Kubicki, L. Emsley, D. Bradshaw, M. J. Rosseinsky, B. Smit and K. C. Stylianou, Nat. Commun., 2019, 10, 1–8 CAS.
- P. G. Boyd, A. Chidambaram, E. García-Díez, C. P. Ireland, T. D. Daff, R. Bounds, A. Gładysiak, P. Schouwink, S. M. Moosavi, M. M. Maroto-Valer, J. A. Reimer, A. R. Navarro, T. K. Woo, S. Garcia, K. C. Stylianou and B. Smit, Nature, 2019, 576, 253–256 CrossRef CAS PubMed.
- Y. Xiao, X. Guo, J. Liu, L. Liu, F. Zhang and C. Li, Chin. J. Catal., 2019, 40, 1339–1344 CrossRef CAS.
- Q. L. Guan, Y. H. Xing, J. Liu, C. Han, C. Y. Hou and F. Y. Bai, J. Phys. Chem. C, 2019, 123, 23287–23296 CrossRef CAS.
- T. N. Tu, H. T. T. Nguyen, H. T. D. Nguyen, M. V. Nguyen, T. D. Nguyen, N. T. Tran and K. T. Lim, RSC Adv., 2019, 9, 16784–16789 RSC.
- Y. Hasegawa, T. Matsui, Y. Kitagawa, T. Nakanishi, T. Seki, H. Ito, Y. Nakasaka, T. Masuda and K. Fushimi, Chem. – Eur. J., 2019, 25, 12308–12315 CrossRef CAS.
- R. Liu, Q.-Y. Liu, R. Krishna, W. Wang, C.-T. He and Y.-L. Wang, Inorg. Chem., 2019, 58, 5089–5095 CrossRef CAS PubMed.
- A. Gładysiak, T. N. Nguyen, M. Spodaryk, J. Lee, J. B. Neaton, A. Züttel and K. C. Stylianou, Chem. – Eur. J., 2019, 25, 501–506 Search PubMed.
- L. Robison, L. Zhang, R. J. Drout, P. Li, C. R. Haney, A. Brikha, H. Noh, B. L. Mehdi, N. D. Browning, V. P. Dravid, Q. Cui, T. Islamoglu and O. K. Farha, ACS Appl. Bio Mater., 2019, 2, 1197–1203 CrossRef CAS PubMed.
- Y.-L. Huang, P.-L. Qiu, H. Zeng, H. Liu, D. Luo, Y. Y. Li, W. Lu and D. Li, Eur, J. Inorg. Chem., 2019, 4205–4210 CAS.
- Y.-L. Huang, P.-L. Qiu, J.-P. Bai, D. Luo, W. Lu and D. Li, Inorg. Chem., 2019, 58, 7667–7671 CrossRef.
- Y.-D. Huang, J.-H. Qin, X.-G. Yang, H.-R. Wang, F.-F. Li and L.-F. Ma, J. Solid State Chem., 2020, 285, 121252 CrossRef CAS.
- Z. Hu, C. Qiao, Z. Xia, F. Li, J. Han, Q. Wei, Q. Yang, G. Xie, S. Chen and S. Gao, ACS Appl. Mater. Interfaces, 2020, 12, 14914–14923 CrossRef CAS.
- A. Cadiau, N. Kolobov, S. Srinivasan, M. G. Goesten, H. Haspel, A. V. Bavykina, M. R. Tchalala, P. Maity, A. Goryachev, A. S. Poryvaev, M. Eddaoudi, M. V. Fedin, O. F. Mohammed and J. Gascon, Angew. Chem., Int. Ed., 2020, 32, 97–104 CAS.
- S. Sudan, A. Gładysiak, B. Valizadeh, J.-H. Lee and K. C. Stylianou, Inorg. Chem., 2020, 59, 9029–9036 CrossRef CAS PubMed.
- S. Xie, H. Wang, Z. Liu, R. Dai and L. Huang, RSC Adv., 2015, 5, 7121–7124 RSC.
- W.-Y. Gao, S. Palakurty, L. Wojtas, Y.-S. Chen and S. Ma, Inorg. Chem. Front., 2015, 2, 369–372 RSC.
- N. Zhao, F. Sun, S. Zhang, H. He, J. Liu, Q. Li and G. Zhu, Inorg. Chem., 2015, 54, 65–68 CrossRef CAS.
- N. Zhao, F. Sun, N. Zhang and G. Zhu, Cryst. Growth Des., 2017, 17, 2453–2457 CrossRef CAS.
- P. Chandrasekhar, A. Mukhopadhyay, G. Savitha and J. N. Moorthy, Chem. Sci., 2016, 7, 3085–3091 RSC.
- P. Li, S.-Y. Moon, M. A. Guelta, L. Lin, D. A. Gómez-Gualdrón, R. Q. Snurr, S. P. Harvey, J. T. Hupp and O. K. Farha, ACS Nano, 2016, 10, 9174–9182 CrossRef CAS.
- O. V. Gutov, W. Bury, D. A. Gomez-Gualdron, V. Krungleviciute, D. Fairen-Jimenez, J. E. Mondloch, A. A. Sarjeant, S. S. Al-Juaid, R. Q. Snurr, J. T. Hupp, T. Yildirim and O. K. Farha, Chem. – Eur. J., 2014, 20, 12389–12393 CrossRef CAS PubMed.
- A. Bajpai, A. Mukhopadhyay, M. S. Krishna, S. Govardhan and J. N. Moorthy, IUCrJ, 2015, 2, 552–562 CrossRef CAS PubMed.
- P. Li, Q. Chen, T. C. Wang, N. A. Vermeulen, B. L. Mehdi, A. Dohnalkova, N. D. Browning, D. Shen, R. Anderson, D. A. Gómez-Gualdrón, F. M. Cetin, J. Jagiello, A. M. Asiri, J. F. Stoddart and O. K. Farha, Chemistry, 2018, 4, 1022–1034 CrossRef CAS.
- P. Deria, D. A. Gómez-Gualdrón, W. Bury, H. T. Schaef, T. C. Wang, P. K. Thallapally, A. A. Sarjeant, R. Q. Snurr, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2015, 137, 13183–13190 CrossRef CAS PubMed.
- R. Zou, X. Ren, F. Huang, Y. Zhao, J. Liu, X. Jing, F. Liao, Y. Wang, J. Lin, R. Zou and J. Sun, J. Mater. Chem. A, 2015, 3, 23493–23500 RSC.
- Y. Zhang, J. Pang, J. Li, X. Yang, M. Feng, P. Cai and H.-C. Zhou, Chem. Sci., 2019, 10, 8455–8460 RSC.
- J. Ye, L. Gagliardi, C. J. Cramer and D. G. Truhlar, J. Catal., 2017, 354, 278–286 CrossRef CAS.
- T. C. Wang, N. A. Vermeulen, I. S. Kim, A. B. F. Martinson, J. F. Stoddart, J. T. Hupp and O. K. Farha, Nat. Protoc., 2016, 11, 149 CrossRef CAS.
- L. C. Gallington, I. S. Kim, W.-G. Liu, A. A. Yakovenko, A. E. Platero-Prats, Z. Li, T. C. Wang, J. T. Hupp, O. K. Farha, D. G. Truhlar, A. B. F. Martinson and K. W. Chapman, J. Am. Chem. Soc., 2016, 138, 13513–13516 CrossRef CAS PubMed.
- T. E. Webber, W.-G. Liu, S. P. Desai, C. C. Lu, D. G. Truhlar and R. L. Penn, ACS Appl. Mater. Interfaces, 2017, 9, 39342–39346 CrossRef CAS.
- F. Cheng, E. S. Marshall, A. J. Young, P. J. Robinson, J.-S. G. Bouillard, A. M. Adawi, N. A. Vermeulen, O. K. Farha, M. R. Reithofer and J. M. Chin, Chem. – Eur. J., 2017, 23, 15578–15582 CrossRef CAS PubMed.
- T. Islamoglu, K.-i. Otake, P. Li, C. T. Buru, A. W. Peters, I. Akpinar, S. J. Garibay and O. K. Farha, CrystEngComm, 2018, 20, 5913–5918 RSC.
- T. E. Webber, S. P. Desai, R. L. Combs, S. Bingham, C. C. Lu and R. L. Penn, Cryst. Growth Des., 2020, 20, 2965–2972 CrossRef CAS.
- J. E. Mondloch, M. J. Katz, N. Planas, D. Semrouni, L. Gagliardi, J. T. Hupp and O. K. Farha, Chem. Commun., 2014, 50, 8944 RSC.
- S. Yuan, J. Peng, Y. Zhang and Y. Shao-Horn, J. Phys. Chem. C, 2019, 123, 28266–28274 CrossRef CAS.
- X.-P. Wu, L. Gagliardi and D. G. Truhlar, Phys. Chem. Chem. Phys., 2018, 20, 1778–1786 RSC.
- N. Planas, J. E. Mondloch, S. Tussupbayev, J. Borycz, L. Gagliardi, J. T. Hupp, O. K. Farha and C. J. Cramer, J. Phys. Chem. Lett., 2014, 5, 3716–3723 CrossRef CAS PubMed.
- R. C. Klet, Y. Liu, T. C. Wang, J. T. Hupp and O. K. Farha, J. Mater. Chem. A, 2016, 4, 1479–1485 RSC.
- D. Yang, V. Bernales, T. Islamoglu, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and B. C. Gates, J. Am. Chem. Soc., 2016, 138, 15189–15196 CrossRef CAS PubMed.
- W.-G. Liu and D. G. Truhlar, Chem. Mater., 2017, 29, 8073–8081 CrossRef CAS.
- H. García and S. Navalón, Metal–Organic Frameworks: Applications in Separations and Catalysis, John Wiley & Sons, Ltd, Weinheim, Germany, 2018 Search PubMed.
- R. C. Klet, T. C. Wang, L. E. Fernandez, D. G. Truhlar, J. T. Hupp and O. K. Farha, Chem. Mater., 2016, 28, 1213–1219 CrossRef CAS.
- C.-W. Kung, J. E. Mondloch, T. C. Wang, W. Bury, W. Hoffeditz, B. M. Klahr, R. C. Klet, M. J. Pellin, O. K. Farha and J. T. Hupp, ACS Appl. Mater. Interfaces, 2015, 7, 28223–28230 CrossRef CAS PubMed.
- A. W. Peters, Z. Li, O. K. Farha and J. T. Hupp, ACS Nano, 2015, 9, 8484–8490 CrossRef CAS.
- Z. Li, A. W. Peters, V. Bernales, M. A. Ortuño, N. M. Schweitzer, M. R. DeStefano, L. C. Gallington, A. E. Platero-Prats, K. W. Chapman, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, ACS Cent. Sci., 2017, 3, 31–38 CrossRef CAS.
- P. Deria, J. E. Mondloch, E. Tylianakis, P. Ghosh, W. Bury, R. Q. Snurr, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2013, 135, 16801–16804 CrossRef CAS.
- P. Deria, W. Bury, J. T. Hupp and O. K. Farha, Chem. Commun., 2014, 50, 1965–1968 RSC.
- P. Deria, W. Bury, I. Hod, C.-W. Kung, O. Karagiaridi, J. T. Hupp and O. K. Farha, Inorg. Chem., 2015, 54, 2185–2192 CrossRef CAS.
- R. W. Johnson, A. Hultqvist and S. F. Bent, Mater. Today, 2014, 17, 236–246 CrossRef CAS.
- T. Islamoglu, S. Goswami, Z. Li, A. J. Howarth, O. K. Farha and J. T. Hupp, Acc. Chem. Res., 2017, 50, 805–813 CrossRef CAS PubMed.
- Z. Li, N. M. Schweitzer, A. B. League, V. Bernales, A. W. Peters, A. B. Getsoian, T. C. Wang, J. T. Miller, A. Vjunov, J. L. Fulton, J. A. Lercher, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2016, 138, 1977–1982 CrossRef CAS PubMed.
- I. S. Kim, J. Borycz, A. E. Platero-Prats, S. Tussupbayev, T. C. Wang, O. K. Farha, J. T. Hupp, L. Gagliardi, K. W. Chapman, C. J. Cramer and A. B. F. Martinson, Chem. Mater., 2015, 27, 4772–4778 CrossRef CAS.
- A. E. Platero-Prats, A. B. League, V. Bernales, J. Ye, L. C. Gallington, A. Vjunov, N. M. Schweitzer, Z. Li, J. Zheng, B. L. Mehdi, A. J. Stevens, A. Dohnalkova, M. Balasubramanian, O. K. Farha, J. T. Hupp, N. D. Browning, J. L. Fulton, D. M. Camaioni, J. A. Lercher, D. G. Truhlar, L. Gagliardi, C. J. Cramer and K. W. Chapman, J. Am. Chem. Soc., 2017, 139, 10410–10418 CrossRef CAS PubMed.
- A. Halder, S. Lee, B. Yang, M. J. Pellin, S. Vajda, Z. Li, Y. Yang, O. K. Farha and J. T. Hupp, J. Chem. Phys., 2020, 152, 084703 CrossRef CAS PubMed.
- J. M. Lownsbury, I. A. Santos-López, W. Zhang, C. T. Campbell, H. S. Yu, W.-G. Liu, C. J. Cramer, D. G. Truhlar, T. Wang, J. T. Hupp and O. K. Farha, J. Phys. Chem. C, 2016, 120, 16850–16862 CrossRef CAS.
- C. T. Lollar, J.-S. Qin, J. Pang, S. Yuan, B. Becker and H.-C. Zhou, Langmuir, 2018, 34, 13795–13807 CrossRef CAS PubMed.
- Y. Cui, M. Rimoldi, A. E. Platero-Prats, K. W. Chapman, J. T. Hupp and O. K. Farha, ChemCatChem, 2018, 10, 1772–1777 CrossRef CAS.
- H. Noh, Y. Cui, A. W. Peters, D. R. Pahls, M. A. Ortuño, N. A. Vermeulen, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2016, 138, 14720–14726 CrossRef CAS.
- H. Noh, C.-W. Kung, K.-I. Otake, A. W. Peters, Z. Li, Y. Liao, X. Gong, O. K. Farha and J. T. Hupp, ACS Catal., 2018, 8, 9848–9858 CrossRef CAS.
- C.-W. Kung, C. O. Audu, A. W. Peters, H. Noh, O. K. Farha and J. T. Hupp, ACS Energy Lett., 2017, 2, 2394–2401 CrossRef CAS.
- K.-i. Otake, Y. Cui, C. T. Buru, Z. Li, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2018, 140, 8652–8656 CrossRef CAS PubMed.
- J. G. Knapp, X. Zhang, T. Elkin, L. E. Wolfsberg, S. L. Hanna, F. A. Son, B. L. Scott and O. K. Farha, CrystEngComm, 2020, 22, 2097–2102 RSC.
- T. A. Goetjen, X. Zhang, J. Liu, J. T. Hupp and O. K. Farha, ACS Sustainable Chem. Eng., 2019, 7, 2553–2557 CrossRef CAS.
- T.-F. Liu, N. A. Vermeulen, A. J. Howarth, P. Li, A. A. Sarjeant, J. T. Hupp and O. K. Farha, Eur. J. Inorg. Chem., 2016, 4349–4352 CrossRef CAS.
- Z. Li, A. W. Peters, V. Bernales, M. A. Ortuño, N. M. Schweitzer, M. R. DeStefano, L. C. Gallington, A. E. Platero-Prats, K. W. Chapman, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, ACS Cent. Sci., 2017, 3, 31–38 CrossRef CAS PubMed.
- C. D. Malonzo, S. M. Shaker, L. Ren, S. D. Prinslow, A. E. Platero-Prats, L. C. Gallington, J. Borycz, A. B. Thompson, T. C. Wang, O. K. Farha, J. T. Hupp, C. C. Lu, K. W. Chapman, J. C. Myers, R. L. Penn, L. Gagliardi, M. Tsapatsis and A. Stein, J. Am. Chem. Soc., 2016, 138, 2739–2748 CrossRef CAS.
- P. Deria, J. E. Mondloch, O. Karagiaridi, W. Bury, J. T. Hupp and O. K. Farha, Chem. Soc. Rev., 2014, 43, 5896–5912 RSC.
- W. Zhao, Z. Wang, C. D. Malonzo, T. E. Webber, A. E. Platero-Prats, F. Sotomayor, N. A. Vermeulen, T. C. Wang, J. T. Hupp, O. K. Farha, R. L. Penn, K. W. Chapman, M. Thommes and A. Stein, Chem. Mater., 2018, 30, 1301–1315 CrossRef CAS.
- C. D. Malonzo, Z. Wang, J. Duan, W. Zhao, T. E. Webber, Z. Li, I. S. Kim, A. Kumar, A. Bhan, A. E. Platero-Prats, K. W. Chapman, O. K. Farha, J. T. Hupp, A. B. F. Martinson, R. L. Penn and A. Stein, Inorg. Chem., 2018, 57, 2782–2790 CrossRef CAS PubMed.
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef.
- W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Chem. Soc. Rev., 2017, 46, 3242–3285 RSC.
- W. Cho, H. J. Lee, G. Choi, S. Choi and M. Oh, J. Am. Chem. Soc., 2014, 136, 12201–12204 CrossRef CAS PubMed.
- A. Karmakar, P. Samanta, S. Dutta and S. K. Ghosh, Chem. – Asian J., 2019, 14, 4506–4519 CrossRef CAS PubMed.
- R. A. Bermúdez, Y. Colón, G. A. Tejada and J. L. Colón, Langmuir, 2005, 21, 890–895 CrossRef.
- F. M. Winnik, Chem. Rev., 1993, 93, 587–614 CrossRef CAS.
- Y. Ge, Y. Wen, H. Liu, T. Lu, Y. Yu, X. Zhang, B. Li, S.-T. Zhang, W. Li and B. Yang, J. Mater. Chem. C, 2020, 8, 11830–11838 RSC.
- G. K. Bains, S. H. Kim, E. J. Sorin and V. Narayanaswami, Biochem, 2012, 51, 6207–6219 CrossRef CAS.
- T. Itoh, Chem. Rev., 2012, 112, 4541–4568 CrossRef CAS PubMed.
- R. Dalapati and S. Biswas, Sens. Actuators, B, 2017, 239, 759–767 CrossRef CAS.
- S.-Y. Moon, A. J. Howarth, T. Wang, N. A. Vermeulen, J. T. Hupp and O. K. Farha, Chem. Commun., 2016, 52, 3438–3441 RSC.
- Y. Tang, H. Wu, J. Chen, J. Jia, J. Yu, W. Xu, Y. Fu, Q. He, H. Cao and J. Cheng, Dyes Pigm., 2019, 167, 10–15 CrossRef CAS.
- R. Dalapati and S. Biswas, Inorg. Chem., 2019, 58, 5654–5663 CrossRef CAS PubMed.
- M. Gong, J. Yang, Y. Li, Q. Zhuang and J. Gu, J. Mater. Chem. C, 2019, 7, 12674–12681 RSC.
- L. Guo, M. Wang and D. Cao, Small, 2018, 14, 1703822 CrossRef PubMed.
- P. Deria, J. Yu, T. Smith and R. P. Balaraman, J. Am. Chem. Soc., 2017, 139, 5973–5983 CrossRef CAS PubMed.
- Z.-J. Lin, H.-Q. Zheng, H.-Y. Zheng, L.-P. Lin, Q. Xin and R. Cao, Inorg. Chem., 2017, 56, 14178–14188 CrossRef CAS PubMed.
- C. Liu and B. Yan, Sens. Actuators, B, 2016, 235, 541–546 CrossRef CAS.
- J. Kim, J. S. Oh, K. C. Park, G. Gupta and C. Yeon Lee, Inorg. Chim. Acta, 2019, 486, 69–73 CrossRef CAS.
- Y. Zhou, Q. Yang, J. Cuan, Y. Wang, N. Gan, Y. Cao and T. Li, Analyst, 2018, 143, 3628–3634 RSC.
- D. Ning, Q. Liu, Q. Wang, X.-M. Du, Y. Li and W.-J. Ruan, Dalton Trans., 2019, 48, 5705–5712 RSC.
- S. Zhu and D. Wang, Adv. Energy Mater., 2017, 7, 1700841 CrossRef.
- Y. Liu, C. T. Buru, A. J. Howarth, J. J. Mahle, J. H. Buchanan, J. B. DeCoste, J. T. Hupp and O. K. Farha, J. Mater. Chem. A, 2016, 4, 13809–13813 RSC.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- K. K. Ng and G. Zheng, Chem. Rev., 2015, 115, 11012–11042 CrossRef CAS.
- A. Atilgan, T. Islamoglu, A. J. Howarth, J. T. Hupp and O. K. Farha, ACS Appl. Mater. Interfaces, 2017, 9, 24555–24560 CrossRef CAS PubMed.
- A. J. Howarth, C. T. Buru, Y. Liu, A. M. Ploskonka, K. J. Hartlieb, M. McEntee, J. J. Mahle, J. H. Buchanan, E. M. Durke, S. S. Al-Juaid, J. F. Stoddart, J. B. DeCoste, J. T. Hupp and O. K. Farha, Chem. – Eur. J., 2017, 23, 214–218 CrossRef CAS PubMed.
- T. Zhang, P. Wang, Z. Gao, Y. An, C. He and C. Duan, RSC Adv., 2018, 8, 32610–32620 RSC.
- H. Choi, A. W. Peters, H. Noh, L. C. Gallington, A. E. Platero-Prats, M. R. DeStefano, M. Rimoldi, S. Goswami, K. W. Chapman, O. K. Farha and J. T. Hupp, ACS Appl. Energy Mater, 2019, 2, 8695–8700 CrossRef CAS.
- Y. Chen, P. Li, J. Zhou, C. T. Buru, L. Đorđević, P. Li, X. Zhang, M. M. Cetin, J. F. Stoddart, S. I. Stupp, M. R. Wasielewski and O. K. Farha, J. Am. Chem. Soc., 2020, 142, 1768–1773 CrossRef CAS.
- A. W. Peters, Z. Li, O. K. Farha and J. T. Hupp, ACS Appl. Mater. Interfaces, 2016, 8, 20675–20681 CrossRef CAS.
- X. Li, J. Yu, D. J. Gosztola, H. C. Fry and P. Deria, J. Am. Chem. Soc., 2019, 141, 16849–16857 CrossRef CAS PubMed.
- J. Yu, J. Park, A. Van Wyk, G. Rumbles and P. Deria, J. Am. Chem. Soc., 2018, 140, 10488–10496 CrossRef CAS PubMed.
- E. Caballero-Mancebo, B. Cohen, S. Smolders, D. E. De Vos and A. Douhal, Adv. Sci., 2019, 6, 1901020 CrossRef CAS.
- I. Choudhuri and D. G. Truhlar, J. Phys. Chem. C, 2020, 124, 8504–8513 CrossRef CAS.
- P. P. Bag, X.-S. Wang, P. Sahoo, J. Xiong and R. Cao, Catal. Sci. Technol., 2017, 7, 5113–5119 RSC.
- Y. Xiao, Y. Qi, X. Wang, X. Wang, F. Zhang and C. Li, Adv. Mater., 2018, 30, 1803401 CrossRef PubMed.
- Y. Xiao, X. Guo, J. Liu, L. Liu, F. Zhang and C. Li, Chin. J. Catal., 2019, 40, 1339–1344 CrossRef CAS.
- C. T. Buru, M. B. Majewski, A. J. Howarth, R. H. Lavroff, C.-W. Kung, A. W. Peters, S. Goswami and O. K. Farha, ACS Appl. Mater. Interfaces, 2018, 10, 23802–23806 CrossRef CAS.
- H. Noh, Y. Yang, S. Ahn, A. W. Peters, O. K. Farha and J. T. Hupp, J. Electrochem. Soc., 2019, 166, H3154–H3158 CrossRef CAS.
- M. S. Lee, S. J. Garibay, A. M. Ploskonka and J. B. DeCoste, MRS Commun., 2019, 9, 464–473 CrossRef CAS.
- W. Zhang, Y. Ma, I. A. Santos-Loópez, J. M. Lownsbury, H. Yu, W.-G. Liu, D. G. Truhlar, C. T. Campbell and O. E. Vilches, J. Am. Chem. Soc., 2018, 140, 328–338 CrossRef CAS PubMed.
- R.-J. Li, M. Li, X.-P. Zhou, D. Li and M. O'Keeffe, Chem. Commun., 2014, 50, 4047–4049 RSC.
- S. Vandenbrande, T. Verstraelen, J. J. Gutiérrez-Sevillano, M. Waroquier and V. Van Speybroeck, J. Phys. Chem. C, 2017, 121, 25309–25322 CrossRef CAS PubMed.
- G. O. Vissers, W. Zhang, O. E. Vilches, W.-G. Liu, H. S. Yu, D. G. Truhlar and C. T. Campbell, J. Phys. Chem. C, 2019, 123, 6586–6591 CrossRef CAS.
- S. J. Garibay, I. Iordanov, T. Islamoglu, J. B. DeCoste and O. K. Farha, CrystEngComm, 2018, 20, 7066–7070 RSC.
- J. H. Kang, T.-U. Yoon, S.-Y. Kim, M.-B. Kim, H.-J. Kim, H.-C. Yang and Y.-S. Bae, Microporous Mesoporous Mater., 2019, 281, 84–91 CrossRef CAS.
- E. M. Dias and C. Petit, J. Mater. Chem. A, 2015, 3, 22484–22506 RSC.
- S. Kato, R. J. Drout and O. K. Farha, Cell Rep., 2020, 1, 100006 Search PubMed.
- S. Rangwani, A. J. Howarth, M. R. DeStefano, C. D. Malliakas, A. E. Platero-Prats, K. W. Chapman and O. K. Farha, Polyhedron, 2018, 151, 338–343 CrossRef CAS.
- A. J. Howarth, M. J. Katz, T. C. Wang, A. E. Platero-Prats, K. W. Chapman, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2015, 137, 7488–7494 CrossRef CAS PubMed.
- J. Li, X. Li, T. Hayat, A. Alsaedi and C. Chen, ACS Sustainable Chem. Eng., 2017, 5, 11496–11503 CrossRef CAS.
- Z.-J. Lin, H.-Q. Zheng, H.-Y. Zheng, L.-P. Lin, Q. Xin and R. Cao, Inorg. Chem., 2017, 56, 14178–14188 CrossRef CAS.
- I. Akpinar, R. J. Drout, T. Islamoglu, S. Kato, J. Lyu and O. K. Farha, ACS Appl. Mater. Interfaces, 2019, 11, 6097–6103 CrossRef CAS.
- A. Pankajakshan, M. Sinha, A. A. Ojha and S. Mandal, ACS Omega, 2018, 3, 7832–7839 CrossRef CAS PubMed.
- R. J. Drout, K. Otake, A. J. Howarth, T. Islamoglu, L. Zhu, C. Xiao, S. Wang and O. K. Farha, Chem. Mater., 2018, 30, 1277–1284 CrossRef CAS.
- P. Asha, M. Sinha and S. Mandal, RSC Adv., 2017, 7, 6691–6696 RSC.
- A. M. Plonka, Q. Wang, W. O. Gordon, A. Balboa, D. Troya, W. Guo, C. H. Sharp, S. D. Senanayake, J. R. Morris, C. L. Hill and A. I. Frenkel, J. Am. Chem. Soc., 2017, 139, 599–602 CrossRef CAS PubMed.
- E. Vargas L and R. Q. Snurr, Langmuir, 2015, 31, 10056–10065 CrossRef CAS PubMed.
- H. Chen and R. Q. Snurr, Langmuir, 2020, 36, 1372–1378 CrossRef CAS.
- J. Zheng, D. Barpaga, O. Y. Gutiérrez, N. D. Browning, B. L. Mehdi, O. K. Farha, J. A. Lercher, B. P. McGrail and R. K. Motkuri, ACS Appl. Energy Mater., 2018, 1, 5853–5858 CrossRef.
- X. Xia, M. Cao, Z. Liu, W. Li and S. Li, Chem. Eng. Sci., 2019, 204, 48–58 CrossRef CAS.
- H. Chen, Z. Chen, O. K. Farha and R. Q. Snurr, ACS Sustainable Chem. Eng., 2019, 7, 18242–18246 CrossRef CAS.
- Z. Bao, G. Chang, H. Xing, R. Krishna, Q. Ren and B. Chen, Energy Environ. Sci., 2016, 9, 3612–3641 RSC.
- M. Yabushita, P. Li, K. A. Durkin, H. Kobayashi, A. Fukuoka, O. K. Farha and A. Katz, Langmuir, 2017, 33, 4129–4137 CrossRef CAS.
- M. Yabushita, P. Li, H. Kobayashi, A. Fukuoka, O. K. Farha and A. Katz, Chem. Commun., 2016, 52, 11791–11794 RSC.
- M. Yabushita, G. Papa, P. Li, A. Fukuoka, O. K. Farha, B. A. Simmons and A. Katz, Fuel Process. Technol., 2020, 197, 106189 CrossRef CAS.
- M. Yabushita, P. Li, V. Bernales, H. Kobayashi, A. Fukuoka, L. Gagliardi, O. K. Farha and A. Katz, Chem. Commun., 2016, 52, 7094–7097 RSC.
- D. Banerjee, C. M. Simon, A. M. Plonka, R. K. Motkuri, J. Liu, X. Chen, B. Smit, J. B. Parise, M. Haranczyk and P. K. Thallapally, Nat. Commun., 2016, 7, 1–7 Search PubMed.
- M. Kawabe, K. Masuda and J. Yamaguchi, J. Phys. Soc. Jpn., 1968, 24, 1281–1285 CrossRef CAS.
- P. Chen, X. He, M. Pang, X. Dong, S. Zhao and W. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 20429–20439 CrossRef CAS.
- S. L. Hanna, D. X. Rademacher, D. J. Hanson, T. Islamoglu, A. K. Olszewski, T. M. Nenoff and O. K. Farha, Ind. Eng. Chem. Res., 2020, 59, 7520–7526 CrossRef CAS.
- J. Gascon, A. Corma, F. Kapteijn and F. X. Llabres i Xamena, ACS Catal., 2014, 4, 361–378 CrossRef CAS.
- P. D. Tran, A. Le Goff, J. Heidkamp, B. Jousselme, N. Guillet, S. Palacin, H. Dau, M. Fontecave and V. Artero, Angew. Chem., Int. Ed., 2011, 50, 1371–1374 CrossRef CAS.
- H. Lei, C. Liu, Z. Wang, Z. Zhang, M. Zhang, X. Chang, W. Zhang and R. Cao, ACS Catal., 2016, 6, 6429–6437 CrossRef CAS.
- J. Ye, L. Gagliardi, C. J. Cramer and D. G. Truhlar, J. Catal., 2018, 360, 160–167 CrossRef CAS.
- S. Pellizzeri, M. Barona, V. Bernales, P. Miró, P. Liao, L. Gagliardi, R. Q. Snurr and R. B. Getman, Catal. Today, 2018, 312, 149–157 CrossRef CAS.
- A. Benali, Y. Luo, H. Shin, D. Pahls and O. Heinonen, J. Phys. Chem. C, 2018, 122, 16683–16691 CrossRef CAS.
- S. P. Desai, J. Ye, T. Islamoglu, O. K. Farha and C. C. Lu, Organometallics, 2019, 38, 3466–3473 CrossRef CAS.
- M. A. Ortuño, V. Bernales, L. Gagliardi and C. J. Cramer, J. Phys. Chem. C, 2016, 120, 24697–24705 CrossRef.
- H. Chen, P. Liao, M. L. Mendonca and R. Q. Snurr, J. Phys. Chem. C, 2018, 122, 12362–12368 CrossRef CAS.
- M. L. Mendonca and R. Q. Snurr, ACS Catal., 2019, 10, 1310–1323 CrossRef.
- D. R. Pahls, M. A. Ortuño, P. H. Winegar, C. J. Cramer and L. Gagliardi, Inorg. Chem., 2017, 56, 8739–8743 CrossRef CAS PubMed.
- M. C. Simons, M. A. Ortuño, V. Bernales, C. A. Gaggioli, C. J. Cramer, A. Bhan and L. Gagliardi, ACS Catal., 2018, 8, 2864–2869 CrossRef CAS.
- C. A. Gaggioli, J. Sauer and L. Gagliardi, J. Am. Chem. Soc., 2019, 141, 14603–14611 CrossRef CAS PubMed.
- B. Yang, K. Sharkas, L. Gagliardi and D. G. Truhlar, Catal. Sci. Technol., 2019, 9, 7003–7015 RSC.
- J. Ye, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. C, 2018, 122, 26987–26997 CrossRef CAS.
- L. R. Redfern, Z. Li, X. Zhang and O. K. Farha, ACS Appl. Nano Mater., 2018, 1, 4413–4417 CrossRef CAS.
- P. Li, J. A. Modica, A. J. Howarth, E. Vargas, P. Z. Moghadam, R. Q. Snurr, M. Mrksich, J. T. Hupp and O. K. Farha, Chem, 2016, 1, 154–169 CAS.
- R. Gil-San-Millan, E. López-Maya, A. E. Platero-Prats, V. Torres-Pérez, P. Delgado, A. W. Augustyniak, M. K. Kim, H. W. Lee, S. G. Ryu and J. A. Navarro, J. Am. Chem. Soc., 2019, 141, 11801–11805 CrossRef CAS PubMed.
- C. T. Buru, M. C. Wasson and O. K. Farha, ACS Appl. Nano Mater., 2019, 3, 658–664 CrossRef.
- C. T. Buru, P. Li, B. L. Mehdi, A. Dohnalkova, A. E. Platero-Prats, N. D. Browning, K. W. Chapman, J. T. Hupp and O. K. Farha, Chem. Mater., 2017, 29, 5174–5181 CrossRef CAS.
- S. Ahn, S. L. Nauert, C. T. Buru, M. Rimoldi, H. Choi, N. M. Schweitzer, J. T. Hupp, O. K. Farha and J. M. Notestein, J. Am. Chem. Soc., 2018, 140, 8535–8543 CrossRef CAS PubMed.
- C. T. Buru, A. E. Platero-Prats, D. G. Chica, M. G. Kanatzidis, K. W. Chapman and O. K. Farha, J. Mater. Chem. A, 2018, 6, 7389–7394 RSC.
- C. T. Buru, J. Lyu, J. Liu and O. K. Farha, Front. Mater., 2019, 6, 152 CrossRef.
- D. Yang, S. O. Odoh, T. C. Wang, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and B. C. Gates, J. Am. Chem. Soc., 2015, 137, 7391–7396 CrossRef CAS.
- D. Yang, S. O. Odoh, J. Borycz, T. C. Wang, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and B. C. Gates, ACS Catal., 2016, 6, 235–247 CrossRef CAS.
- M. Rimoldi, J. T. Hupp and O. K. Farha, ACS Appl. Mater. Interfaces, 2017, 9, 35067–35074 CrossRef CAS PubMed.
- V. Bernales, D. Yang, J. Yu, G. Gümüslü, C. J. Cramer, B. C. Gates and L. Gagliardi, ACS Appl. Mater. Interfaces, 2017, 9, 33511–33520 CrossRef CAS.
- I. S. Kim, Z. Li, J. Zheng, A. E. Platero-Prats, A. Mavrandonakis, S. Pellizzeri, M. Ferrandon, A. Vjunov, L. C. Gallington, T. E. Webber, N. A. Vermeulen, R. L. Penn, R. B. Getman, C. J. Cramer, K. W. Chapman, D. M. Camaioni, J. L. Fulton, J. A. Lercher, O. K. Farha, J. T. Hupp and A. B. F. Martinson, Angew. Chem., Int. Ed., 2018, 57, 909–913 CrossRef CAS PubMed.
- S. P. Desai, J. Ye, J. Zheng, M. S. Ferrandon, T. E. Webber, A. E. Platero-Prats, J. Duan, P. Garcia-Holley, D. M. Camaioni, K. W. Chapman, M. Delferro, O. K. Farha, J. L. Fulton, L. Gagliardi, J. A. Lercher, R. L. Penn, A. Stein and C. C. Lu, J. Am. Chem. Soc., 2018, 140, 15309–15318 CrossRef CAS PubMed.
- A. B. Thompson, D. R. Pahls, V. Bernales, L. C. Gallington, C. D. Malonzo, T. Webber, S. J. Tereniak, T. C. Wang, S. P. Desai, Z. Li, I. S. Kim, L. Gagliardi, R. L. Penn, K. W. Chapman, A. Stein, O. K. Farha, J. T. Hupp, A. B. F. Martinson and C. C. Lu, Chem. Mater., 2016, 28, 6753–6762 CrossRef CAS.
- T. Ikuno, J. Zheng, A. Vjunov, M. Sanchez-Sanchez, M. A. Ortuño, D. R. Pahls, J. L. Fulton, D. M. Camaioni, Z. Li, D. Ray, B. L. Mehdi, N. D. Browning, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and J. A. Lercher, J. Am. Chem. Soc., 2017, 139, 10294–10301 CrossRef CAS PubMed.
- Z. Li, A. W. Peters, A. E. Platero-Prats, J. Liu, C.-W. Kung, H. Noh, M. R. DeStefano, N. M. Schweitzer, K. W. Chapman, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2017, 139, 15251–15258 CrossRef CAS PubMed.
- M. Rimoldi, V. Bernales, J. Borycz, A. Vjunov, L. C. Gallington, A. E. Platero-Prats, I. S. Kim, J. L. Fulton, A. B. F. Martinson, J. A. Lercher, K. W. Chapman, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, Chem. Mater., 2017, 29, 1058–1068 CrossRef CAS.
- M. Rimoldi, L. C. Gallington, K. W. Chapman, K. MacRenaris, J. T. Hupp and O. K. Farha, Chem. – Eur. J., 2017, 23, 8532–8536 CrossRef CAS PubMed.
- M. Yabushita, P. Li, T. Islamoglu, H. Kobayashi, A. Fukuoka, O. K. Farha and A. Katz, Ind. Eng. Chem. Res., 2017, 56, 7141–7148 CrossRef CAS.
- J. Liu, J. Ye, Z. Li, K.-I. Otake, Y. Liao, A. W. Peters, H. Noh, D. G. Truhlar, L. Gagliardi, C. J. Cramer, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2018, 140, 11174–11178 CrossRef CAS PubMed.
- S. Ahn, N. E. Thornburg, Z. Li, T. C. Wang, L. C. Gallington, K. W. Chapman, J. M. Notestein, J. T. Hupp and O. K. Farha, Inorg. Chem., 2016, 55, 11954–11961 CrossRef CAS PubMed.
- R. C. Klet, S. Tussupbayev, J. Borycz, J. R. Gallagher, M. M. Stalzer, J. T. Miller, L. Gagliardi, J. T. Hupp, T. J. Marks, C. J. Cramer, M. Delferro and O. K. Farha, J. Am. Chem. Soc., 2015, 137, 15680–15683 CrossRef CAS.
- J. Liu, L. R. Redfern, Y. Liao, T. Islamoglu, A. Atilgan, O. K. Farha and J. T. Hupp, ACS Appl. Mater. Interfaces, 2019, 11, 47822–47829 CrossRef CAS PubMed.
- A. W. Peters, K. Otake, A. E. Platero-Prats, Z. Li, M. R. DeStefano, K. W. Chapman, O. K. Farha and J. T. Hupp, ACS Appl. Mater. Interfaces, 2018, 10, 15073–15078 CrossRef CAS PubMed.
- K. Berijani, A. Morsali and J. T. Hupp, Catal. Sci. Technol., 2019, 9, 3388–3397 RSC.
- J. Liu, Z. Li, X. Zhang, K.-I. Otake, L. Zhang, A. W. Peters, M. J. Young, N. M. Bedford, S. P. Letourneau, D. J. Mandia, J. W. Elam, O. K. Farha and J. T. Hupp, ACS Catal., 2019, 9, 3198–3207 CrossRef CAS.
- M. C. de Koning, G. W. Peterson, M. van Grol, I. Iordanov and M. McEntee, Chem. Mater., 2019, 31, 7417–7424 CrossRef CAS.
- M. Rimoldi, A. Nakamura, N. A. Vermeulen, J. J. Henkelis, A. K. Blackburn, J. T. Hupp, J. F. Stoddart and O. K. Farha, Chem. Sci., 2016, 7, 4980–4984 RSC.
- S. T. Madrahimov, J. R. Gallagher, G. Zhang, Z. Meinhart, S. J. Garibay, M. Delferro, J. T. Miller, O. K. Farha, J. T. Hupp and S. T. Nguyen, ACS Catal., 2015, 5, 6713–6718 CrossRef CAS.
- Y.-B. Huang, M. Shen, X. Wang, P. Huang, R. Chen, Z.-J. Lin and R. Cao, J. Catal., 2016, 333, 1–7 CrossRef CAS.
- Q. Shi, S. Fu, C. Zhu, J. Song, D. Du and Y. Lin, Mater. Horiz., 2019, 6, 684–702 RSC.
- R. R. Salunkhe, Y. V. Kaneti, J. Kim, J. H. Kim and Y. Yamauchi, Acc. Chem. Res., 2016, 49, 2796–2806 CrossRef CAS.
- R. Zhao, Z. Liang, R. Zou and Q. Xu, Joule, 2018, 2, 2235–2259 CrossRef CAS.
- S. Patwardhan and G. C. Schatz, J. Phys. Chem. C, 2015, 119, 24238–24247 CrossRef CAS.
- T. C. Wang, I. Hod, C. O. Audu, N. A. Vermeulen, S. T. Nguyen, O. K. Farha and J. T. Hupp, ACS Appl. Mater. Interfaces, 2017, 9, 12584–12591 CrossRef CAS PubMed.
- C.-W. Kung, K. Otake, C. T. Buru, S. Goswami, Y. Cui, J. T. Hupp, A. M. Spokoyny and O. K. Farha, J. Am. Chem. Soc., 2018, 140, 3871–3875 CrossRef CAS PubMed.
- S. Goswami, D. Ray, K.-I. Otake, C.-W. Kung, S. J. Garibay, T. Islamoglu, A. Atilgan, Y. Cui, C. J. Cramer, O. K. Farha and J. T. Hupp, Chem. Sci., 2018, 9, 4477–4482 RSC.
- I. Hod, W. Bury, D. M. Gardner, P. Deria, V. Roznyatovskiy, M. R. Wasielewski, O. K. Farha and J. T. Hupp, J. Phys. Chem. Lett., 2015, 6, 586–591 CrossRef CAS PubMed.
- A. Van Wyk, T. Smith, J. Park and P. Deria, J. Am. Chem. Soc., 2018, 140, 2756–2760 CrossRef CAS PubMed.
- P. J. Celis-Salazar, M. Cai, C. A. Cucinell, S. R. Ahrenholtz, C. C. Epley, P. M. Usov and A. J. Morris, J. Am. Chem. Soc., 2019, 141, 11947–11953 CrossRef CAS PubMed.
- S. Goswami, I. Hod, J. D. Duan, C.-W. Kung, M. Rimoldi, C. D. Malliakas, R. H. Palmer, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2019, 141, 17696–17702 CrossRef CAS PubMed.
- C.-W. Kung, J. E. Mondloch, T. C. Wang, W. Bury, W. Hoffeditz, B. M. Klahr, R. C. Klet, M. J. Pellin, O. K. Farha and J. T. Hupp, ACS Appl. Mater. Interfaces, 2015, 7, 28223–28230 CrossRef CAS PubMed.
- I. Hod, W. Bury, D. M. Karlin, P. Deria, C.-W. Kung, M. J. Katz, M. So, B. Klahr, D. Jin, Y.-W. Chung, T. W. Odom, O. K. Farha and J. T. Hupp, Adv. Mater., 2014, 26, 6295–6300 CrossRef CAS PubMed.
- L. Besra and M. Liu, Prog. Mater. Sci., 2007, 52, 1–61 CrossRef CAS.
- P. R. McGonigal, P. Deria, I. Hod, P. Z. Moghadam, A.-J. Avestro, N. E. Horwitz, I. C. Gibbs-Hall, A. K. Blackburn, D. Chen, Y. Y. Botros, M. R. Wasielewski, R. Q. Snurr, J. T. Hupp, O. K. Farha and J. F. Stoddart, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11161–11168 CrossRef CAS PubMed.
- Q. Chen, J. Sun, P. Li, I. Hod, P. Z. Moghadam, Z. S. Kean, R. Q. Snurr, J. T. Hupp, O. K. Farha and J. F. Stoddart, J. Am. Chem. Soc., 2016, 138, 14242–14245 CrossRef CAS PubMed.
- Y. Diao, J. Hu, S. Cheng, F. Ma, M.-Q. Li, X. Hu, Y. Y. Li, J. He and Z. Xu, Inorg. Chem., 2020, 59, 5626–5631 CrossRef CAS PubMed.
- H. Noh, C.-W. Kung, K.-I. Otake, A. W. Peters, Z. Li, Y. Liao, X. Gong, O. K. Farha and J. T. Hupp, ACS Catal., 2018, 8, 9848–9858 CrossRef CAS.
- H. Noh, Y. Yang, X. Zhang, T. A. Goetjen, Z. H. Syed, Z. Lu, S. Ahn, O. K. Farha and J. T. Hupp, ChemElectroChem, 2020, 7, 509–516 CrossRef CAS.
- C.-W. Kung, C. O. Audu, A. W. Peters, H. Noh, O. K. Farha and J. T. Hupp, ACS Energy Lett., 2017, 2, 2394–2401 CrossRef CAS.
- A. G. Wallace and M. D. Symes, Trends Chem., 2019, 1, 247–258 CrossRef CAS.
- S. Kato, K.-i. Otake, H. Chen, I. Akpinar, C. T. Buru, T. Islamoglu, R. Q. Snurr and O. K. Farha, J. Am. Chem. Soc., 2019, 141, 2568–2576 CrossRef CAS.
- X. Zhao, Z. Zhang, X. Cai, B. Ding, C. Sun, G. Liu, C. Hu, S. Shao and M. Pang, ACS Appl. Mater. Interfaces, 2019, 11, 7884–7892 CrossRef CAS PubMed.
- X. Zhao, S. Liu, C. Hu, Y. Liu, M. Pang and J. Lin, ACS Appl. Bio Mater., 2019, 2, 4436–4441 CrossRef CAS PubMed.
- M. Pander, A. Żelichowska and W. Bury, Polyhedron, 2018, 156, 131–137 CrossRef CAS.
- M. H. Teplensky, M. Fantham, P. Li, T. C. Wang, J. P. Mehta, L. J. Young, P. Z. Moghadam, J. T. Hupp, O. K. Farha, C. F. Kaminski and D. Fairen-Jimenez, J. Am. Chem. Soc., 2017, 139, 7522–7532 CrossRef CAS PubMed.
- Y. Chen, P. Li, J. A. Modica, R. J. Drout and O. K. Farha, J. Am. Chem. Soc., 2018, 140, 5678–5681 CrossRef CAS.
- S. Wang, Y. Chen, S. Wang, P. Li, C. A. Mirkin and O. K. Farha, J. Am. Chem. Soc., 2019, 141, 2215–2219 CrossRef CAS PubMed.
- D. Chen, D. Yang, C. A. Dougherty, W. Lu, H. Wu, X. He, T. Cai, M. E. Van Dort, B. D. Ross and H. Hong, ACS Nano, 2017, 11, 4315–4327 CrossRef CAS PubMed.
- Z. Hu, C. Qiao, Z. Xia, F. Li, J. Han, Q. Wei, Q. Yang, G. Xie, S. Chen and S. Gao, ACS Appl. Mater. Interfaces, 2020, 12, 14914–14923 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |