
Arsenene monolayer as an outstanding anode material for (Li/Na/Mg)-ion batteries: density functional theory†
 *a
*a
Abstract
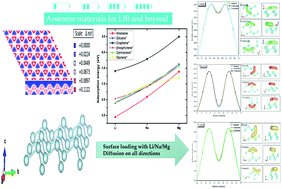
Arsenene, a single-layer arsenic nanosheet with a honeycomb structure, has recently attracted increasing attention due to its numerous exceptional properties. In this study, density functional theory (DFT) calculations were employed to investigate and compare the interactions of Li, Na and Mg ions with the Arsenene monolayer for the purpose of using it as an anode in lithium, sodium, and magnesium ion rechargeable batteries. The results indicated that the Li, Na and Mg adatoms preferentially adsorbed on the valley sites, with negative adsorption energies of −2.55, −1.91 and −1.10 eV, respectively. This strong binding between the alkali metals and the Arsenene monolayer is an important factor for battery applications. Also, it was found that Arsenene exhibited high theoretical specific capacities of up to 1430 mA h g−1 for Li and Mg and 1073.18 mA h g−1 for Na, which are extremely higher values than those of commercially used graphite anodes (372 mA h g−1) in Li-ion batteries. Furthermore, the diffusion barrier energies of the Li, Na and Mg ions were calculated using the nudged elastic band method. The activation energy barriers of these ions showed isotropic behavior for the different pathways (X, Y, and diagonal directions), where the obtained values were 0.16, 0.05 and 0.016 eV for Li, Na, and Mg ions, respectively. Our findings indicate that the high capacity, low open circuit voltage, and ultrahigh barrier diffusion make Arsenene a good candidate for application as an anode material for rechargeable batteries.
 Please wait while we load your content...
Please wait while we load your content...

