
Graphene-based fabrics and their applications: a review
J. Molina
Departamento de Ingeniería Textil y Papelera, EPS de Alcoy, Universitat Politècnica de València, Plaza Ferrándiz y Carbonell s/n, 03801 Alcoy, Spain. E-mail: jamopue@doctor.upv.es; Fax: +34 966528438; Tel: +34 966528583
First published on 30th June 2016
Abstract
Graphene has emerged as a revolutionary material in different fields of science and engineering due to its extraordinary properties such as: high electron mobility, high thermal conductivity, mechanical properties, easy functionalization, etc. The field of textiles is continuously integrating new materials to provide fabrics with new functionalities, hence its incorporation on fabrics was a logical step. Its application to the field of textiles has been recently reported, which has allowed the development of textiles with different functionalities such as: antistatic, UV-protecting, electroconductive, photocatalytic, antibacterial, thermal conductivity, energy storage in flexible supercapacitors, electrodes for batteries, sensors, etc. Up to date no review has been written regarding graphene-based fabrics and their applications. The present review aims to fill the existing gap and provide perspectives into the preparation and applications of graphene-based fabrics and yarns.
 J. Molina | Javier Molina received his Bachelor Degree in Materials Science from Universitat Politècnica de València (UPV) in 2006. In 2011 obtained the PhD from UPV, and in 2012 he was awarded a Prize for his PhD. In 2013 he worked at Instituto Tecnológico de la Energía for six months. In 2013–2015 period, he spent one year as a Postdoctoral Researcher at Universidade do Minho (Portugal) and 9 months at UPV. His research focuses on electrochemistry, nanomaterials synthesis (such as conducting polymers, graphene materials, nanoparticles) and their applications for different purposes. He has published more than 30 papers in international peer-reviewed journals. |
1. Introduction
The field of textiles has experienced great development and a high degree of innovation during the last few years. Modern textiles can no longer be considered as mere garments since nowadays they incorporate new functionalities. These functionalities are normally provided by the development and application of new materials. The different applications developed include: flame resistance,1 thermal regulation,2 electrochromic,3 antimicrobial,4 UV protection,5 self-cleaning,6 solar energy harvesting,7 photonic,8 electrical conduction,9 or even catalysis,10 have been reported on textiles. Electrical conduction is of particular interest since its development allows the integration of computing, digital components and electronics in the fabrics. Different methods have been developed for conferring conductivity on fabrics, such as: the employment of metallic fibers,11 chemical metallization of fibers,12 extrusion of fibers with conducting particles,13 or the synthesis of conducting polymer film on the fabrics.14–16Recently, graphene has emerged as a revolutionary material due to its amazing properties such as: high electron mobility at room temperature (2.5 × 105 cm2 V−1 s−1), high thermal conductivity (above 3000 W m K−1), Young's modulus (1 TPa), intrinsic strength (130 GPa), impermeability to any gases, ability to sustain high electric currents densities, easy chemical functionalization, etc.17 Its isolation in 2004 by A. K. Geim and K. S. Novoselov,18 and the groundbreaking experiments they performed allowed them to be awarded the Nobel Prize in Physics in 2010.19 Possible applications of graphene materials pointed out in bibliography include: flexible electronics, photonics and optoelectronics, spintronics, composite materials, energy generation and storage, biomedical applications, sensors, etc.17,20 The number of papers published every year related to graphene has suffered an exponential evolution, as does the number of patents. European Union has devoted a great economic effort (1000 million €) to graphene research under the Graphene Flagship Horizon 2020 programme. The aim of this programme is “to take graphene and related layered materials from the realm of fundamental science to industrial and societal applications in the space of ten years”.
Having mentioned all the properties and possible applications of graphene materials family, the integration of graphene into textiles was a logical step in order to achieve not only conductive textiles but also multifunctional fabrics. Several reviews have been written about the production and application of graphene fibers that is another area of development in the field.21–24 The methods for the production of G fibers are normally based on the coagulation of a precursor solution. However, up to date, no review has been written about graphene-based fabrics and their applications. The aim of this review is to fill the existing gap with an evaluation of all the work performed in this area and their applications. The review is focused on the most widely employed approaches to obtain G-based fabrics: the chemical deposition of G, GO or RGO on fabrics and the production of GWFs by means of CVD technique.
2. Graphene-based fabrics
Mainly, three methods have been developed in bibliography for the production of G-based fabrics/yarns. The first one is the chemical coating of fabrics/yarns with G materials such as G, GO, RGO, etc. The second one consists in the CVD deposition process of G on a metallic mesh (Cu normally) that is later removed by an acid treatment, remaining after this process only the G-fabric structure, these type of fabrics are named graphene woven fabrics (GWF). And the third one, includes the production of G fibers and its application on fabrics that is not considered in the review since it has been widely covered in other reviews.21–24The most widely employed method is the first one due to its easy application, as well as scalability. As well, usually, the material employed to coat the fabrics is GO that is cheaper than G and can be obtained in larger quantities due to the process of production that consists in chemical oxidation methods.25,26 GO is an insulating material due to the disrupted sp2 structure produced by the oxidation of graphite during its synthesis. However, its conductivity can be partially restored after reduction of GO to produce RGO.25,26
2.1. Graphene-coated fabrics
This section provides a compilation of the work performed in bibliography employing simple coating of G, GO, RGO, etc. on fabrics or textile fibers mainly by aqueous solution impregnation (dip coating). As mentioned previously, GO is the most employed material due to its lower prize. In addition, GO has a negatively charged structure which makes it ideal for its dispersion in aqueous solutions.27 What is more, the negative charges of GO allow their interaction with the functional groups of the fabrics, thus increasing the fixation of GO. The posterior conversion to RGO by chemical, electrochemical, thermal or UV methods allows the partial restoration of conductivity as explained previously. However, the reduction is not complete and recalcitrant oxidized groups cannot be reduced, thus conferring a negative zeta potential to the surface of RGO.27 On the other hand, G has no charged surface and tends to precipitate in aqueous solutions. When employing G in solution to coat fabrics, the deposition has to be aided by dispersants to help in the dispersion and stability of the G solutions. A lot of work has been performed employing the coating of fabrics with G materials.28–85Fig. 1 shows the adsorption process of GO sheets on the surface of different fibers to form a GO layer. Thereafter, the reduction converts insulating GO to conducting RGO due to the partial restoration of the sp2 structure. GO on fabrics has a characteristic brown color, which is converted into black color after reduction. The process for fixing G is similar to this one but it does not include the reduction process. The interaction of GO, G or RGO with the fibers is essential to allow their fixation on the fibers surface and obtain a proper coating with good conductivity. Functional groups present on the fibers help in their fixation. Their interaction with the fibers can be through electrostatic interaction, van der Waals forces, hydrogen bonding, π–π interactions, hydrophobic interactions, etc. In order to boost their fixation, different chemicals and processes have been also employed for such purpose.
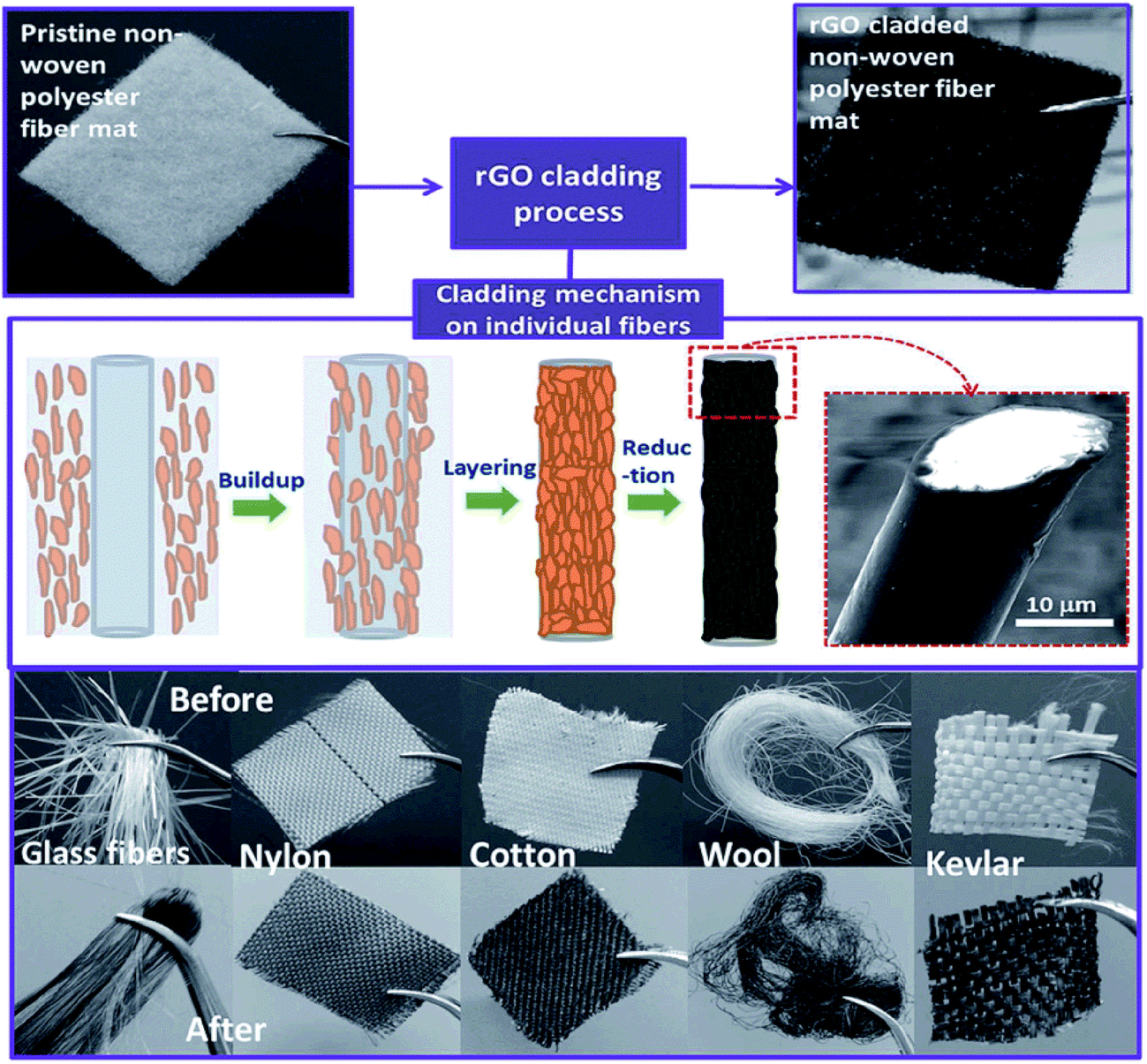 | ||
| Fig. 1 Schematic of the cladding process, mechanism and photographs of cladded materials. Reproduced from ref. 46 with permission of The Royal Society of Chemistry. | ||
BSA, which is a pH sensitive protein, has been widely employed for this objective.37,40,45,49,57,58,62 Below pH 4.9 it has positive charge and above pH 4.9 negative charge. When adsorbed on the fabrics, the positive charges of BSA helped in the adsorption process of GO (which has negative charges) and created electrostatic forces that allowed the self-assembly of GO on the fabrics. Molina et al. applied plasma treatment to generate negative charges on the surface of PES,45 these negative charges increased the BSA fixation that charged positively the surface of the fabrics, and allowed the self assembly of GO sheets. In addition, the high roughness produced by the plasma treatment also helped in the fixation process of GO sheets. Zhao et al. also applied plasma treatment in order to increase the wettability of nylon/lycra fabrics and facilitate GO adhesion.80
NCPCl has been employed to treat PES fibers and increase GO adsorption.34 The interaction between PES and NCPCl was due to hydrogen bond between carboxyl groups of PES and pyridinium group of the surfactant. A complete study taking into account the different interactions (and their energy) that can take place between the fiber surface and GO was performed. The most effective adsorption took place at pH 3 (where an adsorption capacity of 0.38 mg GO/g fiber was obtained).
Chitosan has been employed as dispersant and binding additive of GNSs.28 WPU has been also employed as dispersant and binding additive of GNPs. Hydrogen bonds were formed between –NH groups of WPU and –OH groups of GNPs.29,31 The employment of PU has been also reported,50 it was fixed on PET due to the interaction between the carbonyl group of PET and the –NH group of PU. Dipolar interactions or hydrogen bonding helped in the fixation of RGO on the fabrics. The use of resins has been also reported for increasing the fixation of G derivatives.52
PEI, which reacted with hydroxyl groups of cellulose, was employed to form a cationic layer on cotton fabrics. Thereafter layer-by-layer self-assembly approach was employed to coat samples with multiple alternate layers (up to 6) of PEDOT-PSS-GNS (polyanion) and chitosan (polycation) based on electrostatic interaction.30
SDS surfactant helped in the dispersion of GNRs in solution but also increased their affinity to cotton fibers and produced a more evenly distribution of GNRs on the surface of the fibers (Fig. 2).60 Sodium cholate surfactant has also been employed for the dispersion of GNSs.72,73
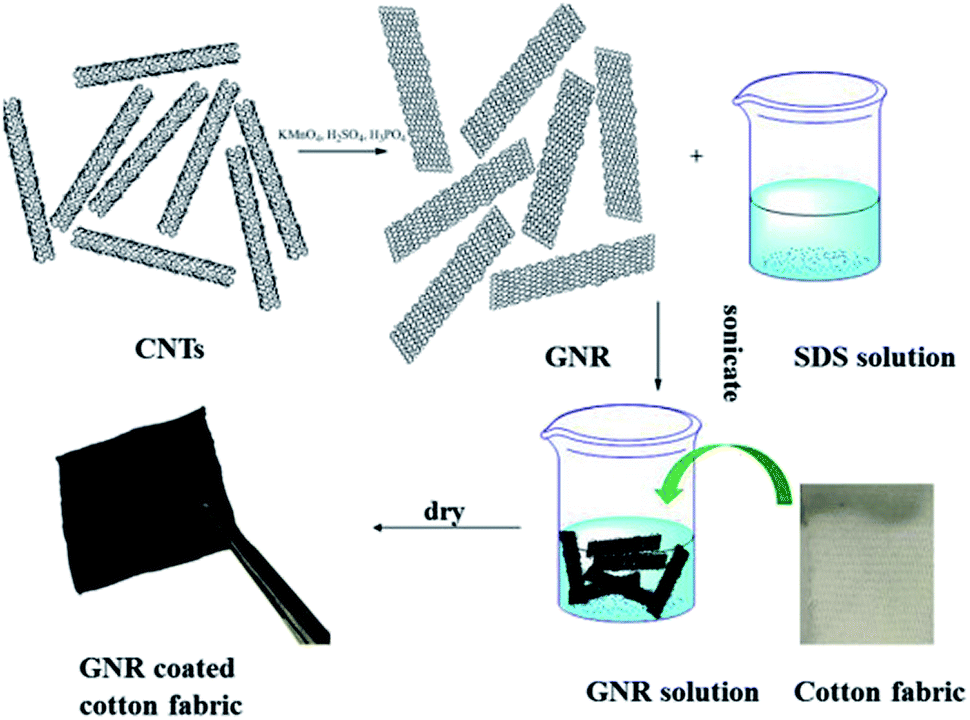 | ||
| Fig. 2 Schematic illustration of the fabrication process of GNR coated cotton fabric.60 Reprinted from Composites Science and Technology, 117, L. Gan, S. Shang, C. W. M. Yuen and S.-x. Jiang, Graphene nanoribbon coated flexible and conductive cotton fabric, page 210, Copyright (2015), with permission from Elsevier. | ||
Acid treatment has been also reported on carbon fibers to increase their hydrophilicity and increase ERGO adhesion.82
The dip coating method has been widely employed, and the majority of papers have employed this approach. The fabric is placed in contact with the solution containing the G material for a certain amount of time and after this, the fabric is dried. If necessary, reduction is performed afterwards. However, other methods for G and GO deposition have been employed, such as: vacuum filtration,32,61 brush coating,33,78 direct electrochemical deposition,82,84 electrophoresis,64,67,68 kinetic trapping method,47 wet transfer of monolayer films,42,75 or screen printing.63 The use of ultrasonication during the synthesis has also been reported during the dip coating process and allowed a homogeneous distribution of the coating on the fabrics.48,50,61,71,76
When employing GO, a reduction method is necessary to restore the conductivity of the G structure. The majority of works that employ GO as precursor, employ chemical reductants such as ascorbic acid,36,38,61,80 Na2S2O4,38,39,43–45,57 hydroiodic acid,37,46,49,51,59,62,65,85 hydrazine,38,40,46,48,50,58,59,67,68,74,76,84 NaBH4,38,69 NaOH,38,83 TiCl3,54,56 ammonium hydroxide,67 or hydroxylamine hydrochloride.81 However, other methods such as thermal reduction,71,77–79 electrochemical reduction,64,82,84 or UV reduction33 have been applied due to the advantage that no chemicals are employed and no by-products are generated by the reduction process. In the case of thermal reduction, the process has to be carried out in inert atmosphere and damage to the fibers can occur due to the elevated temperature. The electrochemical reduction needs a conductive substrate to carry the current flow and for this reason it is mainly applied on carbon cloth substrates. In a study, the effect of the reductant (NaBH4, N2H4, C6H8O6, Na2S2O4 and NaOH) on the electrical properties of RGO/cotton fabrics was performed by Shateri-Khalilabad et al.38 To reach proper levels of conductivity, different number of G/RGO layers are normally applied to the fabrics.
Different textile substrates have been employed in bibliography, Table 1 shows a summary of the work performed.28–85 As well, composition, different parameters employed during the synthesis of the coating, method of reduction, application of the fabric and properties of interest have been included in Table 1.
| Material composition | Properties | Method of synthesis | Reductant | Application | Ref. |
|---|---|---|---|---|---|
| GNS–chitosan/cotton fabrics | UPF: 465.8 (chitosan–GNS/cotton), 7.31 (cotton)UPF after laundering 10 times: 432.7 (chitosan–GNS/cotton), 7.28 (cotton) | GNS coating: dip coating (0.2% wt chitosan + 2% v/v acetic acid + 0.1–1% wt GNS, 2 h). Padding 2 times, drying 70 °C (10 min), curing 110 °C (10 min) | — | UV-blocking | 28 |
| GNPs-WPU/cotton fabrics | UPF: 356.74 (GNPs-WPU/cotton), 32.71 (cotton)UPF after laundering 10 times: 5% decrease | GNP coating: dip coating (WPU + 0.05–0.4% wt GNP, 1 h). Padding 2 times, drying 65 °C (5 min), curing 110 °C (5 min) | — | UV-blocking | 29 |
| PEDOT-G-PSS/chitosan–PEI/cotton fabric | UPF: 312 (PEDOT-G-PSS/chitosan/PEI/cotton), 92.4 (PEDOT-PSS-chitosan-PEI/cotton), 9.37 (cotton)UPF after laundering 10 times: 301.39 (PEDOT-G-PSS-chitosan/cotton)Electrical resistivity: 2.29 Ω m (PEDOT-GNS-PSS-chitosan/cotton), 208.4 Ω m (PEDOT-PSS-chitosan-PEI/cotton), 7.79 × 108 Ω m (cotton) | PEI coating: dip coating (0.01 M PEI, 2 h, 75 °C)Chitosan coating: dip coating (3% wt chitosan in 2% acetic acid)PEDOT/PSS/GNS coating: dip coating (1% wt PEDOT + 0.5% wt PSS + 10 mg L−1 (∼1% wt) GNS, 20 min), (1 to 6 coatings applied alternatively by layer-by-layer self-assembly: 1–6 coatings of PEDOT/PSS/GNS and 1–6 coatings of chitosan) | — | UV blocking, electrical conductivity | 30 |
| GNPs-WPU/cotton fabrics | UPF: 500 (GNPs-WPU/cotton), 8.19 (cotton)UPF after laundering 10 times: 2% increaseElectrical resistivity: 2.94 × 10−1 Ω m (GNPs-WPU/cotton) 1.15 × 107 Ω m (cotton)Far-infrared emissivity: 0.911 (wavelength 4–18 µm) (GNPs-WPU/cotton)Thermal conductivity: 50.633 W m−1 K−1 × 10−3 (0.48 g m−2 GNPs), 38.5 W m−1 K−1 × 10−3 (0 g m−2 GNPs) | GNP coating: dip coating (WPU + 0.8% wt GNP, 100 min). Padding 2 times, drying 70 °C (10 min), curing 120 °C (5 min), GNP content on fabrics: 240, 320, 480 mg m−2 (1, 2, 3 coating cycles) | — | UV-blocking, far-infrared emission, electrical conductivity, thermal conductivity | 31 |
| Pani–GO/cotton fabric | UPF: 445.21 (Pani–GO/cotton), 424.88 (GO/cotton), 29.43 (Pani/cotton), 6.86 (cotton)UPF after laundering 10 times: 412.63 (Pani–GO/cotton), 380.95 (GO/cotton), 29.21 (Pani/cotton), 6.82 (cotton)Electrical resistivity: 48.35 Ω cm (Pani–GO/cotton), 2084.91 Ω cm (Pani/cotton)Electrical resistivity after laundering 10 times: 52.37 Ω cm (Pani–GO/cotton), 2108.78 Ω cm (Pani/cotton) | GO coating: vacuum filtration deposition (5 g L−1 GO)Pani coating: adsorption (20 mL aniline + 80 mL ethanol, 90 min), oxidation by APS, 120 min (APS ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, HCl ratio 1:0.5, ratio respect aniline) :1, HCl ratio 1:0.5, ratio respect aniline) |
— | UV-blocking, electrical conductivity | 32 |
| RGO/wool fabric RGO/cotton fabric | Surface resistivity: 45 kΩ per square (RGO/wool fabric), 100.8 kΩ per square (RGO/cotton fabric)UPF: 5 (cotton), >50 (RGO/cotton fabric) | GO coating: brush coating (2 g L−1 GO, pH 4.5). Drying: 90 °C, 10 min. (Process repeated 5 times to increase GO adsorption) | UV light (8 passes UV irradiation line) | UV blocking, electrical conductivity | 33 |
| GO–NCPCl/PES fibers | GO weight uptake: 0.38 mg GO/g PES (pH 3), 0.14 mg GO/g PES (pH 4.5), negligible deposition (pH 9) | NCPCl coating: dip coating (10−4 M NCPCl, 293 K, 24 h)GO coating: dip coating (0.4 g L−1 GO, 293 K, 12000 s) |
— | — | 34 |
| GO/cotton fabric | Water contact angle: 143° (0.4 g L−1 GO), 135° (0.2 g L−1 GO), 121° (0.1 g L−1 GO), 60° (0.05 g L−1 GO), 0° (cotton) | GO coating: dip coating (0.05–0.4 g L−1 GO, 45 min) | — | Hydrophobicity | 35 |
| PMS–RGO/cotton RGO/cotton | Surface resistance: 91.8 kΩ per square (RGO/cotton), 112.5 kΩ per square (PMS–RGO/cotton)Surface resistance of RGO/cotton with number of RGO coatings: 1 (400.2 kΩ per square), 5 (12.6 kΩ per square), 10 (3.4 kΩ per square), 20 (0.84 kΩ per square)Water contact angle: 143.2° (RGO/cotton), 163° (PMS–RGO/cotton) | GO coating: dip coating (0.2 g L−1 GO, 30 min, repeated 3 times (multiple coatings, 1 to 20)PMS coating: dip coating (methyltrichlorosilane in hexane, 15 min). Curing 110 °C, 60 min | 0.05 M C6H8O6, 95 °C, 60 min | Hydrophobicity, electrical conductivity | 36 |
| RGO–BSA/nylon-6 yarns and fabrics (or PES or cotton yarns) | Conductivity: 1040 S m−1 (RGO–BSA/nylon-6 fabric) | BSA coating: dip coating (0.5% wt BSA, 10 min), drying 1 hour, washingGO coating: dip coating (1 g L−1 GO) | HI (2 mL HI + 5 mL acetic acid, 15 min) | Electrical conductivity | 37 |
| RGO/cotton fabrics | Surface resistance of RGO/cotton as function of reductant: NaBH4 (34600 kΩ cm−1), NaOH (23300 kΩ cm−1), N2H4 (62.7 kΩ cm−1), C6H8O6 (31.2 kΩ cm−1), Na2S2O4 (19.4 kΩ cm−1)Surface resistance with number of RGO coatings (Na2S2O4 reductant): 1 (201.1 kΩ cm−1), 10 (1.27 kΩ cm−1), 20 (0.374 kΩ cm−1) |
GO coating: dip coating (0.05% wt GO, 30 min) (multiple coatings, 1 to 20) | NaBH4, N2H4, C6H8O6, Na2S2O4 and NaOH (25 mM, 95 °C, 60 min). Best reductor: Na2S2O4 (30 min, 95 °C) | Electrical conductivity | 38 |
| RGO/polyarylate yarns | Electrical resistivity: 92.52 Ω cm−1 (it could be tuned from 102 Ω cm−1 to 109 Ω cm−1 depending on the RGO content)GO weight uptake: 8 mg GO/g fiber | GO coating: dip coating (0.3% wt GO, pH 2.13, 15 min) | 0.5% wt Na2S2O4, 363 K, 30 min | Electrical conductivity | 39 |
| RGO–BSA/silk fabrics RGO–BSA/silk yarns | Surface resistance: 386.6 kΩ per square (1 coating), 1.5 kΩ per square (5 coatings) (RGO–BSA/silk fabrics)Electrical conductivity: 3595 S m−1 (RGO–BSA/silk yarns) | BSA coating: dip coating (0.5% wt BSA, 10 min). Drying 30 °C, 15 minGO coating: dip coating (2 g L−1 GO, 30 min) (multiple coatings, 1 to 7) | Hydrazine, overnight | Electrical conductivity | 40 |
| PPy–GO/PES fabrics | Surface resistivity: 177 Ω per square (10% wt GO in solution), 385 Ω per square (20% wt GO in solution), 472 Ω per square (30% wt GO in solution) | PPy/GO coating: dip coating (0.02 M pyrrole + GO (10%, 20%, 30% wt respect to pyrrole mass), 30 min adsorption. Pyrrole oxidation: 0.05 M FeCl3, 150 min | — | Electrical conductivity | 41 |
| G/PP fibers G/PLA fibers | Sheet resistance: 1 kΩ per square (G/PP)Optical transparency: 89% (G/PP), 92% (PP) | G coating: monolayer G grown by CVD and transferred to the fibers by wet methods | — | Electrical conductivity | 42 |
| RGO/PES fabrics | Resistance: 26 kΩ cm2 (1 RGO coating), 700 Ω cm2 (2 RGO coatings), 23.15 Ω cm2 (3 RGO coatings), >1011 Ω cm2 (PES) | GO coating: dip coating (3 g L−1 GO, 30 min) (multiple coatings, 1 to 4) | 0.5% wt Na2S2O4, 90 °C, 30 min | Electrical conductivity, antistatic material | 43 |
| RGO/PES fabrics | Impedance modulus: 105 Ω (1 RGO coating), 19450 Ω (2 RGO coatings), 2157 Ω (3 RGO coatings), 667 Ω (4 RGO coatings), >1010 Ω (PES) |
GO coating: dip coating (3 g L−1 GO, 30 min) (multiple coatings, 1 to 4) | 0.5% wt Na2S2O4, 90 °C, 30 min | Electrical conductivity, antistatic material | 44 |
| RGO–BSA/plasma treated PES fabrics | Impedance modulus: 73 Ω (RGO–BSA/PES plasma treated), 4.6 × 105 Ω (RGO/PES–plasma + chemical reduction) 2.1 × 107 Ω (RGO/PES), >1011 Ω (RGO/PES–plasma) >1011 Ω (PES) | BSA coating: dip coating (0.5% wt BSA, pH 7, 15 min). Washing to remove BSA excessGO coating: dip coating (3 g L−1 GO, 30 min) (multiple coatings, 1 to 10) | 50 mM Na2S2O4, 90 °C, 30 min | Electrical conductivity, antistatic material | 45 |
| RGO/(PES, nylon, cotton, Kevlar mats; wool and glass fibers) | Electrical conductivity: 13 S cm−1 (RGO/Kevlar), 4.5 S cm−1 (RGO/nylon), 0.6 S cm−1 (RGO/glass), 0.1 S cm−1 (RGO/PES), 40 mS cm−1 (RGO/cotton), 10 mS cm−1 (RGO/wool) | GO coating: dip coating (0.37 g L−1 GO, 80 °C) | HI or hydrazine solution, 30 min | Electrical conductivity | 46 |
| FLG–graphite/PET fabric | Sheet resistance: 77.9 MΩ per square (2.5% wt FLG), 3.6 kΩ per square (7.4% wt FLG), 2.5 kΩ per square (10.7% wt FLG) | FLG coating: dip coating (graphite + heptane + water, sonication, 1 h) (kinetic trapping method) | — | Electrical conductivity | 47 |
| RGO/cotton fabric | Resistance: 4.1 × 1015 Ω (cotton), 8.45 × 1010 Ω (cotton/GO), 2.01 × 107 (cotton/RGO) | GO coating: dip coating (4 g L−1 GO, ultrasonication, 30 min) | 27% wt hydrazine solution, 100 °C, 24 h, reflux | Electrical conductivity | 48 |
| RGO–BSA/PES fibers RGO–BSA/cotton fibers | Conductivity: 10−1 S m−1 | BSA coating: dip coatingGO coating: dip coating | HI solution (2 mL HI + 5 mL acetic acid), 40 °C, 1–20 min | Electrical conductivity | 49 |
| RGO–PU/PET fabric (nonwoven) | Surface conductivity: 2.0 × 10−5 S per square (RGO–PU/PET fabric, 0.08% wt RGO in solution), 2.2 × 10−14 S per square (PET fabrics) | PU coating: dip coating (0.1% wt PU in DMF, 60 s)RGO coating: dip coating (0.001–15% wt RGO, 10 min, 0 °C, ultrasonication) | 1 mL L−1 hydrazine hydrate | Electrical conductivity, heat generation | 50 |
| Thiophenol/AgNPs–RGO/PU (electrospun PU fiber mats) | Surface resistivity: 3.5 × 102 Ω per square (RGO/PU), 10 Ω per square (thiophenol/AgNPs–RGO/PU) | GO coating: dip coating (500 mg L−1 GO, 2 h)AgNPs coating: dip coating (0.5–3% wt AgNPs, 30 min) (Diameter AgNPs ∼20 nm) | HI, 100 °C, 10 s | Electrical conductivity, thermal stability | 51 |
| G-resin/cotton fabrics MWCNTs-resin/cotton fabrics BN-resin/cotton fabrics | Thermal conductivity: 0.047 W m−1 K−1 (cotton), 0.078 W m−1 K−1 (11.1% G), 0.10 W m−1 K−1 (20.0% G), 0.14. W m−1 K−1 (33.3% G), 0.29 W m−1 K−1 (50.0% G) | GO coating: dip coating (0.5, 1, 2, 4% G in 4% wt Hercosett XC resin and 1.2% wt 3-(N,N-dimethylmyristylammonio)propanesulfonate) (11.1, 20.0, 33.3, 50% final content of G filler in the coatings, respectively)Drying and curing: 120 °C, 10 min | — | Thermal conductivity | 52 |
| Intumescent flame retardant-polyacrylamide GO/cotton fabrics | Temperature of decomposition: 324 °C (cotton), 351 °C (intumescent flame-retardant polyacrylamide–GO/cotton, 20 coatings)Time to ignition: 41 s (cotton), 64 s (intumescent flame-retardant polyacrylamide–GO/cotton, 20 coatings)Peak heat release rate: 77 kW m−2 (cotton), 153 kW m−2 (intumescent flame-retardant polyacrylamide–GO/cotton, 20 coatings) | Polyacrylamide coating: dip coating (0.1 g L−1 intumescent flame retardant-polyacrylamide, 10 s)GO coating: dip coating (0.02 g L−1 GO, 30 s) (multiple coatings) | — | Flame retardant | 53 |
| TiO2–PVP–GO/cotton fabric | Photocatalytic activity (degradation MB 10 mg L−1, UV irradiation): 87.14%Antibacterial activity: 99% (E. coli and S. aureus)Antifungal activity: 99% (C. albicans) | GO coating: dip coating (0.42% wt GO, 45 min, 70 °C)PVP coating: dip coating (2 g L−1 PVP, 10 min)TiO2 coating: dip coating (1.53 mL TiCl3, 100 mL H2O, 95 °C, 60 min)Curing: 130 °C, 3 min | 1.53 mL TiCl3/100 mL H2O | Photocatalytic activity, antibacterial, antifungal | 54 |
| GO/cotton fabrics | Photocatalytic activity: measured in 1.5 mg L−1 resazurin dye under UV light irradiationAntibacterial activity: 46%, 62%, 74% (Gram-negative bacteria for 6 h, 12 h and 24 h, respectively). 68%, 86%, 100% (Gram-positive bacteria for 6 h, 12 h and 24 h, respectively) | GO coating: dip coating (0.25 g in water, stirring 300 rpm, 24 h) | — | Antibacterial, photocatalytic fabrics | 55 |
| TiO2–PVP–GO/cotton fabric | Electrical resistance: 4 × 106 Ω per square (0.02% wt GO, 0.8 mL TiCl3), 3.6 × 10−3 Ω per square (0.5% wt GO, 0.8 mL TiCl3)Photocatalytic activity (degradation MB 10 mg L−1, UV irradiation): 87.14% (0.5% wt GO, 1.2 mL TiCl3)Antibacterial activity: 99.4% (E. coli), 99.4% (S. aureus) (0.5% wt GO, 1.2 mL TiCl3)Antifungal activity: 99.2% (C. albicans) (0.5% wt GO, 1.2 mL TiCl3) | GO coating: dip coating (0.5% wt GO, 45 min, 70 °C)PVP coating: dip coating (2 g L−1 PVP, 10 min)TiO2 coating: dip coating (0.8/1.2 mL TiCl3 in 100 mL H2O, 95 °C, 60 min)Curing: 130 °C, 3 min | 0.8 mL/1.2 mL TiCl3 (15% TiCl3 in 10% HCl) | Electrical conductivity, photocatalytic activity, antibacterial, antifungal | 56 |
| TiO2–RGO–BSA/plasma treated PES fabrics | Impedance modulus: 2102 Ω (1 RGO–BSA/PES plasma treated), 112 Ω (4 RGO–BSA/PES–plasma treated), >1011 Ω (PES)Charge transfer resistance: 727.3 Ω (1 RGO–BSA/PES plasma treated), 197.3 Ω (4 RGO–BSA/PES–plasma treated)Photocatalytic activity (degradation rhodamine B 10 mg L−1, UV irradiation): 70.4% (1 RGO coating), 77.9% (4 RGO coatings) | BSA coating: dip coating (0.5% wt BSA, pH 7, 10 min. Washing to remove BSA excess)GO coating: dip coating (3 g L−1 GO, 60 min) (multiple coatings, 1 to 4)TiO2 coating: dip coating (5 g L−1 TiO2, 2 min, padding 2 bar. Drying 100 °C) | 50 mM Na2S2O4, 90 °C, 30 min | Electrical conductivity, photocatalytic activity | 57 |
| RGO–BSA/cotton fabric | Surface resistance: 40 Ω per square (RGO–BSA/cotton), 510 Ω per square (RGO/cotton)Photocatalytic activity (degradation 10 mg L−1 MB, 100 min irradiation): 27% (RGO/cotton), 45% (RGO–BSA/cotton) | BSA coating: dip coating (0.15 g L−1 BSA, 5 min), 30 min drying 60 °C, 3 washingsGO coating: dip coating (0.1% wt GO, 30 min, 80 °C) (multiple coatings, 1 to 10) | 40 mM hydrazine, 100 °C, 30 min | Electrical conductivity, photocatalytic activity | 58 |
| RGO/nylon fabric | Electrical conductivity: 4.5 S cm−1 (RGO/nylon) 6 × 10−12 S cm−1 (nylon) | GO coating: dip coating (0.37 g L−1) | HI solution, hydrazine solution | Electrical conductivity, electrocardiogram monitoring | 59 |
| GNR/cotton fabric | Resistance: 80 Ω | GNR coating: dip coating (0.25 g L−1 GNR + 2.5 g L−1 SDS) | — | Electrical conductivity, strain sensor | 60 |
| RGO/silk fibers (capacitor) PtNPs–RGO/silk fibers (H2O2 sensor) enzyme–PtNPs–RGO/silk fiber mat (glucose sensor) | Conductivity: 57.9 S m−1 (RGO/silk)Sheet resistivity: 90 Ω per square (RGO/silk)Three-electrode configuration (1 M H2SO4): Capacitance: 17.75 mF cm−2 (RGO/silk)H2O2 sensor: 0–2.5 mM (linear range), 0.2 µM (detection limit), 0.56 mA mM−1 cm−2 (sensitivity)Glucose sensor: 10 µM–10 mM (linear range), 1 µM (detection limit), 150.8 µA mM−1 cm−2 (sensitivity) | GO coating: dip coating (50 mg silk fibers, 10 mg GO, ultrasonication 15 min, filtering)PtNPs synthesis: electrochemical synthesis (CV, 0 → −0.6 V, 50 mV s−1, 10 mM H2PtCl6 + 0.1 M HCl solution)Glucose oxidase fixation: dip coating (10 g L−1 glucose oxidase + 30 g L−1 BSA + 0.01 M PBS (pH 7.4). Exposure to glutaraldehyde (50 µL, 25%, 35 °C, 3 h) for cross-linking glucose oxidase to BSA | 10 g L−1 ascorbic acid, 100 °C, 2 h | Sensor (H2O2, glucose). Capacitor | 61 |
| RGO–BSA/cotton yarns RGO–BSA/PES yarns | Current sensitivity to 0.25 ppm NO2: −7% (RGO/cotton), −6% (RGO/PES) Current sensitivity to 1.25 ppm NO2: −12% (RGO/cotton), −12% (RGO/PES) | BSA coating: dip coating (0.5% wt BSA, 30 min. Drying 1 h. Washing) GO coating: dip coating (2 g L−1 GO) | HI (2 mL HI + 5 mL acetic acid) 40 °C, 10 min | NO2 gas sensor | 62 |
| G pellets/MWCNTs–cotton fabric | Surface electrical resistivity: 4.7 kΩ (3% wt G), 9.1 kΩ (1% wt G), 12.8 kΩ (0.5% wt G), 13 kΩ (0% wt G)Relative change in resistance: 50% (methanol exposure) 15% (acetone exposure) | GO coating: screen printing (G pellets 0.5, 1, 3% wt + MWCNTs 3% wt + aliphatic urethane acrylate + Esacure DP250 photoinitiator. Mechanical stirring 30 min. Application of the paste to the fabric. Curing 30 min with IR light) | — | Vapor sensor (acetone and methanol) | 63 |
| ERGO/carbon cloth | Charge transfer resistance: 2.5 Ω (carbon cloth), 0.6 Ω (ERGO/carbon cloth)Plateau discharge rates: 15.8 µA cm−2 (carbon cloth), 24.5 µA cm−2 (ERGO/carbon cloth)Discharge life: 230 h (carbon cloth), 330 h (ERGO/carbon cloth)Maximum power density: 19.5 mW m−2 (carbon cloth), 52.5 mW m−2 (ERGO/carbon cloth)Electrical energy converted: 1.75 J (carbon cloth), 5.34 J (ERGO/carbon cloth) | GO coating: electrophoresis on carbon cloth (0.2 g L−1 GO, 0.3 mA cm−2, 30 min). Reduction. Drying and UV sterilization, 3 h | Electrochemical reduction, −0.6 mA cm−2, 90 s | Anode for microbial fuel cell | 64 |
| RGO/cotton fabric | Surface resistance: 114 Ω per square (20 GO coatings), 55 Ω per square (20 GO coatings after acid treatment)Charge transfer resistance: 9.68 Ω cm2 (RGO/cotton), 6.74 Ω cm2 (Pt-coated electrode)Conversion efficiency I3−: 2.52% (RGO/cotton), 7.20% (Pt-coated electrode)Short circuit current: 9.08 mA cm−2 (RGO/cotton), 14.88 mA cm−2 (Pt-coated electrode)Open circuit voltage: 0.64 V (RGO/cotton), 0.66 V (Pt-coated electrode)Fill factor: 42.97 (RGO/cotton), 71.18 (Pt-coated electrode) | GO coating: dip coating (1% wt GO, 80 °C, 30 min) (multiple coatings, 1 to 20) | 0.1 M HI, 90 °C, vapor reduction | Cathode in dye sensitized solar cells | 65 |
| PPy/AQSA–RGO/PES filter cloth | Electrical conductivity: 0.7 kΩ cm−1 (PPy/RGO), 2 kΩ cm−1 (PPy)Degradation rate of MB: 59.2% (PPy), 64.0% (PPy/RGO), 74.6% (PPy/RGO with Fe2+ in solution) (conditions: −1 V (cathode), 0.2 mM Fe2+, 5 mg L−1 MB, 0.05 M Na2S2O4 supporting electrolyte, after 120 min of reaction) | RGO coating: dip coating (0.02 g L−1 RGO, 10 min)RGO/AQSA coating: dip coating (0.02 g L−1 RGO, 0.25 mM AQSA, 10 min)Spraying: APS solution (20 mg L−1, 50 mL)PPy coating: 0.5 mL, 90 °C, 15 min (vapor phase polymerization) | — | Electrocatalytic membrane (acting as cathode) | 66 |
| RGO–CNTs/carbon cloth-PET | Turn-on field at 10 µA cm−2: 0.26 V µm−1 (RGO–CNTs), 0.49 V µm−1 (CNTs), 0.43 V µm−1 (RGO)Current density at threshold field (0.55 V µm−1): 1 mA cm−2 (RGO–CNTs), 60 µA cm−2 (CNTs), 250 µA cm−2 (RGO) | RGO/CNTs coating: electrophoretic deposition (CNTs, 1 mA cm−2, 10 min); 1 g L−1 RGO (2.5 mA cm−2, 10 min) | Reduction with hydrazine hydrate (35% wt), ammonium hydroxide (25% wt), 90 °C, 1 h | Field emission device | 67 |
| RGO/carbon cloth + Ar plasma treatment | Turn-on field at 10 µA cm−2: 0.78 V µm−1 (0 s plasma), 0.59 V µm−1 (40 s plasma), 0.41 V µm−1 (3 min plasma)Threshold field: 0.96 V µm−1 (1 mA cm−2) (40 s plasma), 0.81 V µm−1 (1 mA cm−2) (3 min plasma). For 0 s plasma treatment, emission does not achieve the threshold value | RGO coating: electrophoretic deposition (2.4 mA cm−2, 5 s- 10 min)Plasma etching: Ar gas, 0.2 mbar, 150 W, 50 s- 5 min | Hydrazine hydrate (80%) (1:7 weight ratio with GO), 90 °C, 6 hours |
Field emission device | 68 |
| RGO/cotton fabrics | Sheet resistance: 560 Ω per squareThree-electrode configuration (1 M Na2SO4): Specific capacitance: 40 F g−1 (5 mV s−1, 0 V → 1 V)Capacitance retention: 90% (1000 cycles at 0.85 A g−1) | GO coating: dip coating (2 g L−1 GO, 30 min) (20 coatings) | 0.5 M NaBH4, 12 h | Capacitor | 69 |
| PPy/GO/cotton fabrics | Electrical conductivity: 0.9 S cm−1 (PPy/cotton), 1.12 S cm−1 (PPy/GO/cotton)Three-electrode configuration (1 M Na2SO4): Capacitance (50 mV s−1): 24.3 F g−1 (PPy/cotton), 35.7 F g−1 (PPy/GO/cotton) | GO coating: dip coating (5 g L−1 GO, 30 min)PPy coating: dip coating (2 mM pyrrole, 2 h (pyrrole adsorption), 0.08 M FeCl3 addition (oxidation, 4 h, 0–5 °C, N2 gas flow) | — | Capacitor, electrical conductivity | 70 |
| PPy/RGO/cotton fabrics | Electrical conductivity: 1.2 S cm−1 (PPy/RGO/cotton)Two-electrode configuration (fabric/2 M NaCl/fabric):Capacitance: 336 F g−1 (PPy/RGO/cotton), 234 F g−1 (PPy/cotton)Energy density: 21.1 W h kg−1 (at 0.6 mA cm−2)Capacitance retention: 64% (PPy/RGO/cotton), 35% (PPy/cotton) (500 cycles at 0.6 mA cm−2) | GO coating: dip coating (2 g L−1 GO, 30 min, ultrasonication). ReductionPPy coating: dip coating (1 M pyrrole, 30 min (adsorption), 0.5 M FeCl3 adding (oxidation, 2 h, ice bath)) | Thermal reduction, 250 °C, 2 h, N2 atmosphere | Supercapacitor, electrical conductivity | 71 |
| MnO2–GNS/PES fabrics | Sheet resistance: 700 Ω per square (35 cycles of deposition)Three-electrode configuration (0.5 M Na2SO4):Specific capacitance: 315 F g−1 (2 mV s−1, 0 V → 0.85 V)Two-electrode configuration (fabric/0.5 M Na2SO4/SWCNTs fabric):Maximum power density: 110 kW kg−1Energy density: 12.5 W h kg−1 Capacitance retention: 95% (5000 cycles at 2.2 A g−1) | GNS coating: dip coating (∼3 g L−1 GNS + 0.5 g L−1 sodium cholate). MnO2 coating: electrodeposition (20 mM Mn(NO3)2 + 100 mM NaNO3, 60 min, 0.1 mA cm−2) (35 coatings) | — | Supercapacitor | 72 |
| PEDOT/PSS–MnO2–GNS/PES fabrics SWCNTs–MnO2–GNS/PES fabrics | Sheet resistance: 700 Ω per squareThree-electrode configuration (0.5 M Na2SO4):Specific capacitance: 380 F g−1 (0.1 mA cm−2) (PEDOT/PSS–MnO2–GNS/PES fabrics)Capacitance retention after: 93% (MnO2–GNS/PES fabrics), 95% (PEDOT/PSS–MnO2–GNS/PES fabrics), 96% (SWCNTs–MnO2–GNS/PES fabrics) (3000 cycles at 1 mA cm−2) | GNS coating: dip coating (0.15 g L−1 + sodium cholate surfactant) (multiple coatings)MnO2 coating: electrochemical deposition (20 mM Mn(NO3)2 + 100 mM NaNO3, deposition under constant current of 100 µA cm−2, 90 min)SWCNTs coating: dip coating (0.2 g L−1 SWCNTs)PEDOT/PSS coating: dip and dry (2–3 times) | — | Supercapacitor | 73 |
| Pani–GO/CNT fibers TiO2–RGO/CNT fibers | Three-electrode configuration (1 M H2SO4):Capacitance: 229.5 F cm−3 (Pani–GO/CNT fibers), 186 F cm−3 (Pani–RGO/CNT fibers) (20 mV s−1)Photocurrent (at 100 mW cm−2 irradiation): 132 µA cm−2 (TiO2–RGO/CNT fibers), 25 µA cm−2 (TiO2/CNT fibers) | GO coating: dip coating (0.05 g L−1 GO + 1–2% DMF, 2 h, 2 times)Pani coating: electrosynthesis (0.05 M aniline + 1 M H2SO4, 0.75 V, 10 min)TiO2 coating: metal–organic CVD (10 min) | Hydrazine (95 °C, 1 h, hydrazine:GO wt ratio (7:10) |
Capacitor, photoelectrode | 74 |
| (G-V2O5/MWCNTs)10/PES fabrics coated with Ni/Cu/Ni/Au | Three-electrode configuration (2 M KCl):Specific capacitance (1 mV s−1): 2590 F g−1 (G-V2O5/MWCNTs), 1600 F g−1 (V2O5/MWCNTs) (3 nm V2O5), 510 F g−1 (V2O5/MWCNTs) (20 nm V2O5)Power density: 800 W kg−1Energy density: 96 W h kg−1Capacitance retention: >97% (5000 cycles, 20 mV s−1) | G coating: G transfer methodV2O5/MWCNTs coating: spray coating (multiple coatings, 1 to 10) | — | Supercapacitor | 75 |
| RGO/polyamide 66 nanofibers (electrospun) | Volume resistance: 330 Ω cm (d GO < 200 nm), 1.1 Ω cm (d GO 200–450 nm), 14.3 Ω cm (d GO > 450 nm), 1670 Ω cm (d GO 200–450 nm, microfiber fabric, 1 coating), 0.5 Ω cm (d GO 200–450 nm, microfiber fabric, 5 coatings)Two-electrode configuration (fabric/polyamide 66 + 1 M H2SO4/fabric):Specific capacitance: 280 F g−1 (d GO 200–450 nm), 65.4 F g−1 (d GO < 200 nm), 95.3 F g−1 (d GO > 450 nm), 130 F g−1 (d GO 200–450 nm, micrometer fabric, 5 GO coatings) (10 mV s−1)Specific energy: 10 W h kg−1 (at 0.5 A g−1) (d GO 200–450 nm)Specific power: 1500 W kg−1 (3 A g−1) (d GO 200–450 nm) | GO coating: dip coating (0.5 g L−1 GO (diameter GO < 200 nm, 200–450 nm or >450 nm), ultrasonication 2 h) | Hydrazine vapor, 120 °C. Oven 200 °C, 2 h (to remove C–N bonding) | Supercapacitor | 76 |
| RGO/cotton fabrics | Sheet resistance: 910 Ω per square (RGO/cotton), 1.09 × 108 Ω per square (GO/cotton)Two-electrode configuration (fabric/separator + 6 M KOH/fabric):Capacitance: 87.53 mF cm−2 (2 mV s−1), 81.4 F g−1 (2 mV s−1)Volumetric capacitance: 5.53 F cm−3 (62.5 mA cm−3)Capacitance retention: 89.82% after 1000 cycles (2 mV s−1), 90.5% after 100 bending cycles (2 mV s−1)Energy density: 767.36 µW h cm−2 (31.26 mW cm−3) | GO coating: dip coating (4 g L−1 GO) (5 coatings) | Thermal reduction and carbonization of cotton fabrics, 300 °C, 2 h, Ar atmosphere | Supercapacitor | 77 |
| RGO/cotton fabrics | Electrical resistance: 225 Ω cm−1Mass loading: 1.08 mg cm−2Two-electrode configuration (fabric/cotton + 6 M KOH aqueous electrolyte or 2.0 M EMIMBF4 in acetonitrile/fabric):Specific capacitance: 326.8 F g−1 (6 M KOH, 10 mV s−1), 15.6 F g−1 (2 M EMIMBF4, 10 mV s−1)Power density: 1.5 kW kg−1 (6 M KOH, 3 A g−1)Energy density: 7.13 W h kg−1 (6 M KOH, 3 A g−1), 12.3 W h kg−1 (2 M EMIMBF4/acetonitrile)Capacitance retention: 93.8% (after 1500 cycles, 100 mV s−1, 6 M KOH), 93% (after 1500 cycles, 100 mV s−1, 2 M EMIMBF4) | GO coating: brush coating (2 g L−1 GO) (50 coatings) | Thermal reduction and carbonization of cotton fabrics, 300 °C, 2 h, Ar atmosphere | Supercapacitor | 78 |
| RGO/carbon fabrics | Mass loading: 0.6–0.8 mg cm−2Three electrode configuration (1 M H3PO4):Specific capacitance: 414 F g−1 (5 mV s−1)Capacitance retention: 93% (after 1000 cycles)Two-electrode configuration (fabric/PVA–H3PO4/fabric): Device capacitance: 70.4 F g−1 (5 mV s−1)Energy density: 5.8 W h kg−1 (27.7 kW kg−1) | GO coating: dip coating (5 coatings) | Thermal reduction, 160 °C, 2 h, Ar atmosphere | Supercapacitor | 79 |
| RGO/nylon lycra fabrics PPy–RGO/nylon lycra fabrics | Surface resistivity: 240 Ω per square (25 cycles)RGO loading (25 cycles): 2.3 mg cm−2PPy loading: 2.8 mg cm−2Two electrode configuration (fabric/separator + 1 M Li2SO4/fabric):Capacitance: 12.3 F g−1 (RGO), 15.5 F g−1 (RGO, 50% strain), 114 F g−1 (PPy–RGO), 125 F g−1 (PPy–RGO, 50% strain) (5 mV s−1)Capacitance retention: 76% (RGO), 89% (RGO, 50% strain), 74% (PPy/RGO), 79% (PPy/RGO, 50% strain) (after 2000 cycles, 0.1 A g−1)Energy density: 2.53 W h kg−1 (5 mV s−1) | Plasma treatment of the fabric: previous to GO coatingGO coating: dip coating (3 g L−1 GO, 30 min) (25 coatings)PPy coating: dip coating (pyrrole + Na2NDS (30 min, ice bath). Addition of APS (oxidation). Reaction time: 2 h) | 0.1 M L-ascorbic acid, 95 °C, 60 min | Supercapacitor | 80 |
| MWCNTs–PPy–RGO/PES non-woven fabrics | Conductivity: 0.4 S cm−1 (RGO/PES)Two electrode configuration (fabric/separator + 1 M KCl/fabric):Capacitance: 305 F g−1 (MWCNTs–PPy–RGO/PES) 290 F g−1 (PPy–RGO/PES), 118 F g−1 (RGO/PES), 72.6 F g−1 (PPy/PES), 222.9 F g−1 (MWCNTs/PES) (5 mV s−1)Capacitance retention: 83.4% (PPy–RGO/PES), 94.5% (MWCNTs–PPy–RGO/PES) (after 1000 cycles, 80 mV s−1) | GO coating: dip coating (GO 5 g L−1 + hydroxylamine, hydrochloride 0.125 M, 5 min)PPy coating: dip coating (FeCl3·6H2O solution, 5 min). Exposure to pyrrole vapor (polymerization during 5 h)MWCNTs: dip coating (2 g L−1 MWCNTs, 5 min) | Hydroxylamine, hydrochloride, 130 °C, 30 min | Supercapacitor | 81 |
| ERGO/carbon fibers | Three-electrode configuration (1 M Na2SO4):Specific capacitance: 3.4 µF cm−1 (10 mV s−1, single fiber), 10.3 µF cm−2, 22.6 µF cm−1 (10 mV s−1, single fiber, acid treatment)Two-electrode configuration (yarn/PVA–H3PO4/yarn):Specific capacitance: 307 mF cm−2, 13.5 mF cm−1 (250 fibers) (0.05 mA cm−1)Capacitance retention: 85% after 5000 cycles (250 fibers, 0.05 mA cm−1)Energy density: 1.09 µW h cm−1 (27.2 µW cm−1)Power density: 748.6 µW cm−1 | Carbon fiber treatment: H2SO4/HNO3 (3:1 in volume, 10 h)GO coating: electrochemical deposition (3 g L−1 GO, 0.1 M LiClO4, −1.2 V, 10 min) |
Electrochemical reduction | Supercapacitor | 82 |
| PPy–MnO2–RGO/stainless steel yarns | Three-electrode configuration (1 M Na2SO4):Specific capacitance: 36.6 mF cm−1, 486 mF cm−2Two-electrode configuration (yarn/PVA–H3PO4/yarn):Specific capacitance: 31 mF cm−1, 411 mF cm−2Energy density: 0.0092 mW h cm−2, 1.1 mW h cm−3Capacitance retention: 92% (after 4950 cycles, 80 mA cm−3) | RGO coating: deep coating (6 g L−1 GO (6 mL) + 1 M NaOH (20 µL) + yarns. Autoclave, 180 °C, 12 h)MnO2 coating: electrochemical deposition (0.02 M Mn(NO3)2 + 0.01 M NaNO3. 0.92 V, 45 min)PPy coating: electrochemical deposition (0.1 M p-toluenesulfonic acid + 0.3 M sodium toluene sulfonate + 0.5% pyrrole monomer (v/v) at 0 °C. 0.8 V, 1.5 min) | NaOH | Supercapacitor | 83 |
| ERGO–Ni/cotton yarns | Electrical resistance: 1.6 Ω cm−1Three electrode configuration (1 M Na2SO4):Capacitance: 292.3 F cm−3, 311 F g−1 (87.9 mA cm−3)Two electrode configuration (yarn/PVA–LiCl/yarn):Capacitance: 0.11 F cm−1Maximum energy density: 6.1 mW h cm−3Maximum power density: 1400 mW cm−3Capacitance retention: 82% (after 10000 cycles, 439.6 mA cm−3) |
Ni coating: electroless deposition (60 min)GO coating: electrochemical deposition (3 g L−1 GO + 0.1 M LiClO4, −1.2 V, 10 min) | Electrochemical reduction + hydrazine vapor reduction, 60 °C, 3 h | Supercapacitor | 84 |
| RGO–Pani/nitrogen-doped carbon fiber cloth | Two-electrode configuration (fabric/separator–1 M H2SO4/fabric):Specific capacitance: 1145 F g−1 (RGO–Pani/N-doped carbon cloth), 1050 F g−1 (Pani/N-doped carbon cloth), 940 F g−1 (GO–Pani/N-doped carbon cloth), 520 F g−1 (Pani)Maximum energy density: 25.4 W h kg−1 (at 52.5 kW kg−1)Maximum power density: 92.2 kW kg−1 (at 20.3 W h kg−1)Capacitance retention: 94% (after 5000 cycles) | Nitrogen doping carbon fiber cloth: cold plasma treatment N2/O2, 2 minPani coating: dip coating (aniline + 1 M H2SO4, 10 min) APS oxidation (24 h, ratio aniline:APS (4:1))GO coating: dip coating (0.5 g L−1, 15 min) |
HI reduction | Supercapacitor | 85 |
2.2. Applications of graphene coated fabrics
Tian et al.28 coated cotton fabrics with GNSs with the aid of chitosan as dispersant and binding additive. With a low G content in the coating solution (<1% wt) the UV-blocking properties of the substrate increased a 60-fold when compared with bare cotton. Qu et al.29 coated cotton fabrics with GNPs. WPU was employed to aid the dispersion of GNPs at different content (0.05–0.4% wt). Hydrogen bonds were formed between –NH groups of WPU and –OH groups of GNPs. With a small GNPs content a high UPF was achieved (356.74, 10-fold increase when compared to bare cotton fabric). Tian et al.30 deposited by electrostatic layer-by-layer self-assembly PEDOT-PSS (polyanion) and chitosan (polycation). GNS were also employed to dope PEDOT and enhance its electronic and UV-shielding properties. First of all, a cationic layer was formed on cotton fabrics with PEI coating, which reacted with hydroxyl groups of cellulose fibers. Thereafter the anionic layer (GNS/PEDOT/PSS) was deposited by electrostatic self-assembly. After washing and drying, the cationic layer (chitosan) was applied. This process was repeated from 1 to 6 times. The presence of GNS produced a decrease of the electrical resistivity of 2 orders of magnitude. The UPF also increased notably due to the presence of GNS (92.4 vs. 312, without and with GNSs, respectively).
Hu et al.31 coated cotton fabrics with GNPs. Fig. 3a shows the cotton fabric prior to the GNPs deposition and Fig. 3b shows the fabric after the coating was applied. The UPF value achieved in this case was 500 (0.8% wt GNPs), a 60-fold increase when compared to bare cotton fabrics. In addition, the fabrics also showed increasing far-infrared emissivity as the GNPs content increased. The higher content of GNPs allows the heat to be converted into infrared light that could be detected by a thermograph (this can be observed from Fig. 3c to Fig. 3f). Far-infrared radiation boosts blood circulation and metabolism as well as it helps in recovering muscles form fatigue. For this reason, materials that emit far-infrared radiation have been employed for therapeutic and health purposes. Conductivity and thermal conductivity increased with the increasing G content, as well.
 | ||
| Fig. 3 Images of (a) uncoated cotton fabric, (b) graphene-coated fabric, (c–f) thermographs of graphene-coated fabrics with different coated weights (control cotton fabric, 240, 320, and 480 mg m−2 graphene-coated fabrics, respectively).31 Reprinted from Carbon, 95, X. Hu, M. Tian, L. Qu, S. Zhu and G. Han, Multifunctional cotton fabrics with graphene/polyurethane coatings with far-infrared emission, electrical conductivity, and ultraviolet blocking properties, page 631, Copyright (2015), with permission from Elsevier. | ||
Tang et al.32 coated cotton with GO by vacuum filtration deposition, thereafter Pani was chemically deposited on the surface of the GO/cotton fabric. Fabrics acted as UV-blocking and conductive materials. Since GO is an insulating material, Pani provided the conductivity. When GO was deposited on the surface of cotton, it provided a substrate where Pani could be homogeneously coated and agglomerations of Pani were avoided, which boosted the conductivity of the fabrics. Javed et al.33 coated cotton and wool fabrics by brush coating with GO. Later GO was reduced to RGO by UV light irradiation, avoiding the employment of additional chemicals to achieve the GO reduction. Strong GO adsorption was obtained on both fabrics due to strong van der Waals forces, hydrogen bonds and covalent bonds. Excellent UV blocking properties were also observed by the authors.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds which confere them a hydrophobic behavior in this part. This has been employed to produce hydrophobic fabrics. Tissera et al.35 coated cotton fabrics with GO by dipping method in solutions containing different GO concentrations. With the more concentrated solutions, thicker GO coatings were deposited on the fabrics, mid regions of GO were dominant (hydrophobic regions) and consequently more hydrophobicity was obtained. Shateri-Khalilabad and Yazdanshenas36 produced conductive superhydrophobic textiles by coating cotton with RGO and later with nanostructured PMS (a hydrophobic material in the form of filaments of 30–90 nm). GO was fixed on cotton through van der Waals and hydrogen bonds and was reduced by ascorbic acid. The reduction of GO to RGO produced the removal of functional groups and RGO gained hydrophobicity as quantified by contact angle measurements. The additional PMS coating provided nanoscale roughness and further increased the contact angle on the conductive fabrics. Thus, self-cleaning fabrics could be obtained. Fig. 4 shows images of water contact angle measurements performed on original cotton fabric (Fig. 4a), RGO–cotton (Fig. 4b) and PMS–RGO–cotton (Fig. 4c).
C bonds which confere them a hydrophobic behavior in this part. This has been employed to produce hydrophobic fabrics. Tissera et al.35 coated cotton fabrics with GO by dipping method in solutions containing different GO concentrations. With the more concentrated solutions, thicker GO coatings were deposited on the fabrics, mid regions of GO were dominant (hydrophobic regions) and consequently more hydrophobicity was obtained. Shateri-Khalilabad and Yazdanshenas36 produced conductive superhydrophobic textiles by coating cotton with RGO and later with nanostructured PMS (a hydrophobic material in the form of filaments of 30–90 nm). GO was fixed on cotton through van der Waals and hydrogen bonds and was reduced by ascorbic acid. The reduction of GO to RGO produced the removal of functional groups and RGO gained hydrophobicity as quantified by contact angle measurements. The additional PMS coating provided nanoscale roughness and further increased the contact angle on the conductive fabrics. Thus, self-cleaning fabrics could be obtained. Fig. 4 shows images of water contact angle measurements performed on original cotton fabric (Fig. 4a), RGO–cotton (Fig. 4b) and PMS–RGO–cotton (Fig. 4c).
 | ||
| Fig. 4 Red-dyed water droplets sitting on (a) the original cotton; (b) the graphene–cotton; (c) the PMS–graphene–cotton. The images on the right show corresponding goniometer images for 5 µL droplets.36 Cellulose, Preparation of superhydrophobic electroconductive graphene-coated cotton cellulose, 20, 2013, 969, M. Shateri-Khalilabad, M. E. Yazdanshenas, with permission of Springer. | ||
 | ||
| Fig. 5 Change in the surface conductivity of the composite fabrics with the increase in the weight fraction of RGO in the initial aqueous dispersion. The insets display SEM images of the composite fabrics prepared at RGO fractions of 0.001 (I), 0.030 (II) and 0.080 wt% (III). Reproduced from ref. 50 with permission of The Royal Society of Chemistry. | ||
The conductivity obtained also depends on the number of G/RGO layers applied and it is normal to apply a high number of G/RGO coatings on the fabrics to achieve appropriate levels of conductivity. Fig. 6 shows an evolution of the resistance of the cotton fabrics coated with RGO–BSA (CCF–RGO) and RGO (SCF–RGO). As it can be seen, BSA coating improves the fixation and conductivity of the fabric whichever the number of coatings applied. The number of G/RGO layers applied on the fabrics has been also indicated in Table 1 when available.
 | ||
| Fig. 6 Comparison of the decrease in resistance with dipping number of SCF–RGO (RGO/cotton) and CCF–RGO (RGO–BSA/cotton).58 Reprinted from Carbohydrate Polymers, 130, I. A. Sahito, K. C. Sun, A. A. Arbab, M. B. Qadir, S. H. Jeong, page 305, Copyright (2015), with permission from Elsevier. | ||
As explained previously, the reductant has also a marked influence on the conductivity obtained. Shateri-Khalilabad and Yazdanshenas38 coated cotton fabrics with GO and studied the effect of the reductant employed on the conductivity of RGO/cotton fabrics obtained. The reduction time is also important and it is a parameter that should be optimized when obtaining RGO coatings from GO since it depends on the type of reductant employed as well as the synthesis conditions. In this sense, Ha et al.49 studied the reduction process of RGO deposited on PES and cotton fibers aided by BSA coating. Authors employed Raman spectroscopy to study the reduction of GO by HI and showed that the conducting pathways on the fibers are formed within the first minute of reduction. Thereafter, conductivity remains stable till 20 min of reduction, and beyond this time, a decrease of conductivity was observed, maybe due to the deposition of I− that could act as scattering centers for the current flow.
The most important parameters of conducting fabrics have been summarized in Table 1 (conductivity, sheet resistance, electrical resistance, etc.). However, some aspects of the papers dealing principally on conductivity will be summarized following (special synthesis methods, special properties or applications, etc.).
Yun et al.37 obtained conductive and colored nylon-6 yarns through dying RGO with rhodamine 6G. The conductivity of the fabrics only varied a 9% in the 220–325 K, which demonstrated its applicability for real-life applications. Woltornist et al.47 coated PET simulated leather fabrics with a mixture of FLG and graphite obtained by ultrasonication. The approach employed was a kinetic trapping method, which consisted in a mixture of n-heptane and water. Both are poor solvents for FLG and graphite and both accumulate in the interface between the two solvents, the exfoliation of graphite was also facilitated in this way by means of ultrasonication. When a hydrophilic surface was present, FLG/graphite deposited on its surface. Fabrics were placed in contact with the mixture and were ultrasonicated to allow the deposit of the coating, weight uptakes as high as 15% of graphitic material on the fabrics were achieved. Graphite tended to accumulate in the interstices between the fibers and FLG were deposited on the surface of the fibers.
Other methods employed for obtaining conductive yarns include wet transfer of monolayer G. Neves et al.42 synthesized monolayer G by CVD on Cu foil and transferred it to PP and PLA fibers by wet method to produce conductive and transparent coatings on the fibers (fibers were immobilized on a rigid PET support). The surface of the fibers was previously treated with ultraviolet-ozone treatment to remove impurities from the surface of the fibers and produced a more uniform surface that promoted G adhesion.
Due to the resistive response of G conducting fabrics, they have been tested as electrothermal materials. The temperature of the fabrics can be tuned by varying the potential and shows applicability for heating garments (for instance for maintaining the body temperature of patients, etc.).50 Fig. 7 shows the evolution of the temperature of RGO–PU/PET fabrics depending on the applied potential.
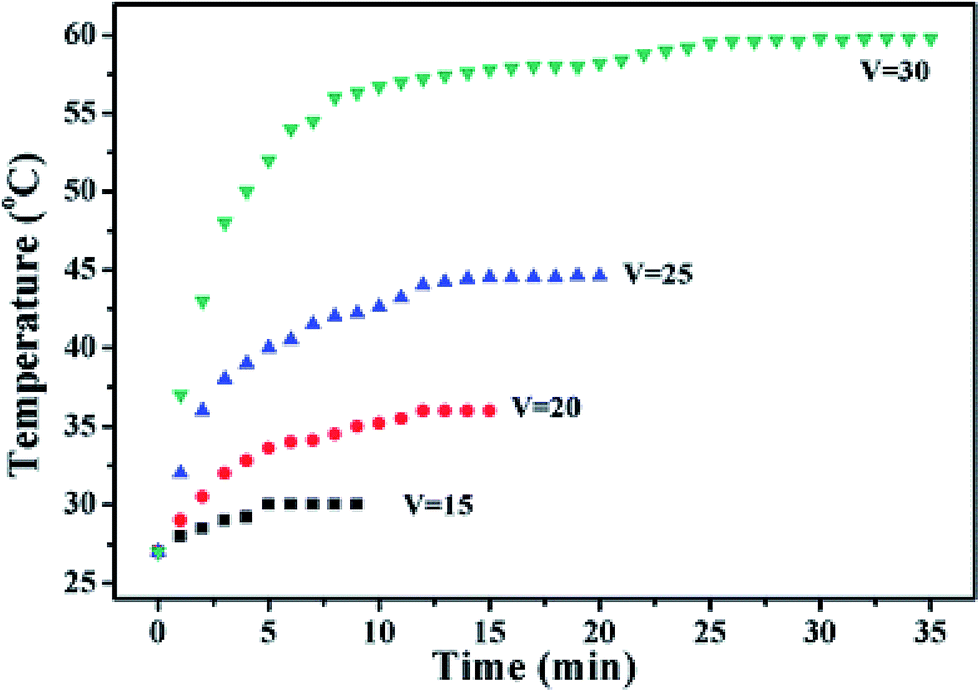 | ||
| Fig. 7 Time dependence of temperature for the composite fabrics prepared in an aqueous dispersion with an RGO content of 0.080 wt%. Reproduced from ref. 50 with permission of The Royal Society of Chemistry. | ||
Molina et al. published different works43–45 in which PES fabrics were coated with RGO and they performed a complete electrical and electrochemical characterization of these materials with non-traditionally employed techniques for the characterization of these materials such as: cyclic voltammetry, electrochemical impedance spectroscopy or scanning electrochemical microscopy. The RGO coatings demonstrated to be electroactive and were homogeneously distributed on the surface of the fabrics. In their electrochemical characterization using scanning electrochemical microscopy, its amphoteric behavior was observed. The RGO coatings could act either as a reductant or as an oxidant.44 In cyclic voltammetry characterization, only low scan rates allowed the observation of RGO redox processes due to the resistive nature of the coatings, composed of lots of RGO sheets that allowed the electrical flow through the fabric.
Hsiao et al.51 coated PU fiber mats, obtained by electrospinning, with GO, later GO was converted to RGO with hydriodic acid as the reducing agent. Electrical conduction of the fiber mats was further improved with the deposition of thiophenol-modified AgNPs. Fig. 8 shows a schematic representation of the production process, in which the functional groups of GO bonded with the functional groups of PU nanofibers by means of hydrogen bonding. AgNPs were fixed due to π–π interaction between RGO and thiophenol-modified AgNPs. Fig. 9a shows the PU nanofibers, and after GO deposition it can be seen that all the voids between the nanofibers were coated with GO (Fig. 9b). Fig. 9c show the cross section of the GO/PU nanocomposite. Fig. 9d and Fig. 9e show the surface and cross section of the RGO/PU composite, respectively.
 | ||
| Fig. 8 Schematic representation of the procedure for preparing the AgNP@RGO/WPU composites.51 Reprinted from Composites Science and Technology, 118, S.-T. Hsiao, C.-C. M. Ma, H.-W. Tien, W.-H. Liao, Y.-S. Wang, S.-M. Li and W.-P. Chuang, Preparation and characterization of silver nanoparticle-reduced graphene oxide decorated electrospun polyurethane fiber composites with an improved electrical property, page 173, Copyright (2015), with permission from Elsevier. | ||
 | ||
| Fig. 9 SEM images of the surface of (a) PU, (b) GO/PU, (c) cross section of GO/PU, (d) RGO/PU and (e) cross section of GO/PU composite.51 Reprinted from Composites Science and Technology, 118, S.-T. Hsiao, C.-C. M. Ma, H.-W. Tien, W.-H. Liao, Y.-S. Wang, S.-M. Li and W.-P. Chuang, Preparation and characterization of silver nanoparticle-reduced graphene oxide decorated electrospun polyurethane fiber composites with an improved electrical property, page 175, Copyright (2015), with permission from Elsevier. | ||
Hybrid conducting materials consisting of GO and a conducting polymer were reported by Molina et al.,41 who deposited PPy/GO hybrid material on PES fabrics by means of chemical oxidation of pyrrole/GO solution by FeCl3. Different GO contents were employed (10, 20 and 30% wt respect to pyrrole mass) to study the effect on the conductivity. During the polymerization, GO acted as a counter ion (negative charge) to neutralize the positive charges created in the structure of PPy (polarons and bipolarons). When PPy/GO was deposited, the fabric became conductive (177 Ω per square, for 10% wt GO content). When the GO content increased, the surface resistivity also increased slightly. An excessive amount of GO, increased electrical resistance due to its insulating nature. In addition, X-ray photoelectron spectroscopy measurements showed a decrease of the doping level of the polymer (N+/N) as the GO content increased, in accordance with the surface resistivity results. GO as counter ion has the advantage of its high size (between hundreds of nm and several µm) when compared with traditional organic counter ions, and cannot be expulsed from the PPy structure due to dedoping. Due to the immobilization of GO inside the polymer structure, cations would migrate to produce the compensation of charges inside PPy.
Graphene derivatives have been also employed as flame-retardant fillers to produce fabrics with more thermal stability and flame retardant properties. Huang et al.53 coated cotton fabrics with intumescent flame retardant-polyacrylamide/GO by layer-by-layer assembly. An increase in the temperature of decomposition and the time of ignition was observed. Conversely, the heat release rate was diminished.
Sahito et al.58 cationized a cotton fabric with BSA. With the BSA coating an increase of 67.74% of GO deposited was obtained when compared to bare cotton. Finally, GO was converted to RGO by hydrazine hydrate vapors. Electrical conductivity and photocatalytic activity in the degradation of a MB solution were improved with the BSA coating, due to the higher amount of RGO deposited. In addition the authors pointed out the possibility of employing the fabric in the future for textile-structured solar cells.
RGO-coated fabrics have been also employed as substrate materials onto which TiO2 NPs have been deposited to obtain photocatalytic fabrics.54,56,57 Under irradiation, TiO2 NPs produce electrons and holes. Electrons combine with O2 and produce O2−, and the combination of holes and water generates hydroxyl radicals. These highly active oxygen generated species can oxidize organic pollutants. The efficiency of photocatalytic processes is determined by the electron/hole pair life, which is around 10−9 s for TiO2 alone.54 The time needed for chemical interaction with organic matter is in the 10−8 to 10−3 s order; therefore a decrease of the photocatalytic efficiency is observed. In this sense, G materials act as conducting materials and enable the effective electron/hole separation due to their conducting pathways. In this way, the lifetime of the electron/hole pair is increased, as does the photocatalytic efficiency of these materials. Other advantage is that G materials can also extend light absorption, acting as an electron donor to produce more O2− radical species. In addition, G materials can also act as adsorbents of organic matter, thus facilitating the contact between the photocatalytic material and the pollutants, organic matter, bacteria, etc.54
Karimi et al.54 coated cotton fabrics with GO and PU. Thereafter TiCl3 was added, and acted as a reductant of GO and at the same time, TiO2 NPs (8–13 nm) were formed due to the oxidation of Ti3+. Authors performed a series of experiments varying the GO and TiCl3 concentrations. The effect of these parameters on the photocatalytic efficiency (in the degradation of MB dye solution) was measured and optimized with a mathematical model. The modified fabrics were also employed as antimicrobial (Escherichia coli and Staphylococcus aureus) and antifungal materials (Candida albicans). Cytotoxicity tests were performed and showed no hazard for health. In another article,56 the same authors published a similar study.
Molina et al.57 developed RGO-coated PES fabrics and coated them with commercial TiO2 NPs. An increase in the number of RGO coatings applied, produced an increase of the conductivity and a decrease of the charge transfer resistance of the fabrics in solution. Consequently, an increase in the photocatalytic efficiency (in rhodamine B degradation) was also observed.
Yapici et al.59 coated nylon fabrics with RGO to produce electrodes for obtaining electrocardiograms. The performance of these electrodes was very similar to the conventional electrodes employed for such purpose and presented several advantages over traditional electrodes (Ag/AgCl) such as: they alleviate the need for gel, provide comfort, wearability, reusability and easy integration in personal clothing.
Gan et al.60 coated cotton fabrics with GNRs obtained from MWCNTs unzipping by strong oxidants. Bending, stress–strain and washing experiments showed no significant degradation of the conductivity of the fabrics. A linear dependence of the resistance on the strain was observed when elongation was lower than 20%, indicating that the fabric could be employed as a strain sensor in smart textiles. GWFs have been more widely applied for this purpose due to their higher sensitivity as will be seen in Section 2.4.1.
Liang et al.61 coated silk fiber mats with GO via vacuum filtration. Later GO was reduced to RGO chemically. The morphology of the composite showed a loose structure on the top of the film and a compact structure at the bottom of the film, due to the accumulation of RGO sheets. The porous structure helped to the diffusion of analytes to the electrode. The composite material also showed capacitive properties due to the high surface area of the silk/RGO composite. Thereafter, PtNPs were deposited on the RGO/silk fiber mats via CV electrodeposition, a spiky structure in the form of flowers was obtained (Fig. 10a and b). Fig. 10c shows the EDX analysis which corroborates the presence of PtNPs on the surface of the fibers. The modified fibers were employed as electrode materials for H2O2 sensing (Fig. 10d). H2O2 is a by-product of many analyte-specific enzymes, as well as a known brain neuromodulator. After modification with glucose oxidase enzyme, the conducting fibers were also employed as glucose sensing material that could be employed for diabetes diagnosis.
 | ||
| Fig. 10 (a) An SEM image of a G/silk film decorated with Pt nanospheres. (b) A high-magnification SEM image of the G/silk film decorated with Pt nanosphere. The right inset right shows a Pt spiky nanosphere and the left inset shows a photograph of a spiky flower head that is similar to the Pt nanosphere. (c) EDX spectrum of the Pt nanosphere decorated G/silk film. (d) Calibration curve of H2O2 detection by the Pt nanosphere decorated G/silk film at 0.65 V vs. Ag/AgCl. The inset shows a magnified calibration curve at low H2O2 concentration (1 mM to 10 mM). Reproduced from ref. 61 with permission of The Royal Society of Chemistry. | ||
Yun et al.62 coated cotton and PES fibers with BSA to facilitate GO adsorption. After chemical reduction, the conductive fibers were embroidered in commercial fabrics and employed as gas sensors. The obtained sensors were 3-fold more sensitive to NO2 than when obtained in the form of flat RGO films, which can be attributed to the high surface area of the yarns. When exposed to NO2 as an oxidizing gas, the resistance of the RGO sensor decreased owing to the increased hole concentration resulting in the negative sign of the response. Washing and bending (1000 tests) did not affect the performance of the fibers and provided a constant response for 7 days monitoring (0.13% variation). Ethanol, ethylene, acetone, and CO2 gases in 10-fold concentrations did not cause interference in the NO2 determination.
Skrzetuska et al.63 coated cotton fabrics with G pellets (23 nm of thickness, 68 G layers on average) and MWCNTs by screen printing technique and the developed fabrics were employed as gas sensors. The change in the electrical resistance of the fabrics when exposed to different vapor gases was employed as an indication of the gas concentration. Methanol and acetone gases were employed to test the sensing properties of the screen printed electrodes, being more sensitive to methanol than towards acetone. The differences could be attributed to differences in the dipole moment, which is lower in the case of methanol (1.61 D vs. 2.91 D for acetone).
Liu et al.64 deposited electrochemically ERGO on carbon cloth. The fabrics were employed as anode materials for a Pseudomonas aeruginosa mediatorless microbial fuel cell. ERGO promoted the growth of bacteria growth due to its biocompatibility and enhanced electron transfer rate due to two mechanisms: direct electron transfer and through cell-excreted mediator-enabled electron transfer pathways. Pseudomonas aeruginosa produces pyocyanin, phenazine-1-carboxamide or phenazine-1-carboxylic acid that can function as redox mediators for the transfer of electrons between the bacteria and the anode electrode. The ERGO modification improved power density and energy conversion by 2.7 and 3 times, respectively.
Sahito et al.65 obtained RGO coated fabrics and employed them as counter electrode in dye sensitized solar cells. The counter electrode in this type of cells is usually coated with Pt to increase its electroactivity, the counter electrode with RGO is Pt free and hence it is cheaper. The energy conversion efficiency obtained with the RGO/cotton fabric was lower than that obtained with Pt counter electrode. However, there are several advantages with this type of electrodes, such as: the low cost, the simple production method, flexibility and biodegradability of the materials, which make this type of electrodes promising candidates for future technology of textile structured solar cells.
Zhao et al.66 developed PPy/RGO coated PES filter cloth membranes and employed them at the same time as membrane and cathode material. AQSA was employed as a dopant to improve the conductivity of the membrane and the generation of radicals. The filtration effect enhanced the contact between the membrane and the pollutants. The membrane was employed in an electro-Fenton system to degrade an organic pollutant (MB). H2O2 was produced on the cathode through O2 reduction, which reacted with Fe2+ to produce hydroxyl and hydroperoxyl radicals that were responsible for oxidizing organic matter. Hydroxyl radicals could also be directly produced on the cathode. RGO acted as an effective bridge between PPy and the catalyst.
Tian et al.74 wrapped CNT fibers with GO by means of π–π interaction, and later reduced to RGO. TiO2 was also deposited by CVD to produce photoelectrode materials. When depositing TiO2/RGO on CNTs, the photocurrents obtained were multiplied by a 5 factor when compared with TiO2/CNTs (132 vs. 25 µA cm−2). This was due to the deposition of more TiO2 NPs and the creation of donor–acceptor interfaces due to tight binding between RGO and TiO2 NPs.
 | ||
| Fig. 11 Schematic diagram of the mechanism that, by stably attaching ERGO additives, increased specific surface area featuring a unique wrinkled structure for adsorption of electrolyte ions contributing to the excellent electrochemical double-layers. (A) Pure carbon fiber electrode. (B) ERGO–carbon fiber electrode. Reproduced from ref. 82 with permission of The Royal Society of Chemistry. | ||
Yaghoubidoust et al.70 coated cotton fabrics with GO and later with PPy obtained by chemical oxidation, obtaining a double layer coating. The previous GO deposit enhanced conductivity of the fabric, it should be taken into account that GO could be partially reduced to RGO during the oxidation process of pyrrole to PPy. Specific capacitance was also improved by the GO coating as measured by CV. Xu et al.71 employed a similar approach, however in this case authors reduced GO to RGO to obtain more conductive coatings. Although the presence of RGO produced a thinner PPy coating (1.5 mg cm−2 vs. 6 mg cm−2), the capacitance was higher in the case of PPy/RGO coating. This can be attributed to a higher surface area, yielding increased electrode/electrolyte interface areas. In addition, RGO formed a conducting layer below the PPy layer, which facilitated the electron transfer between RGO and PPy. The presence of RGO coating stabilized PPy and increased its cycling stability due to the interaction between RGO and PPy (π–π interaction) which limited the swelling and shrinking of the PPy coating. A similar effect of stabilization of the conducting polymers has been observed by other authors.85 Zhao et al.80 coated nylon-lycra fabrics with RGO and later a PPy film was also deposited on the surface of the fabrics. In this case, the capacitive performance was even better under 50% stretching, since strain allowed a better contact between RGO sheets.
Liu et al.81 coated PES non-woven fabrics with RGO, PPy and MWCNTs and studied its performance as supercapacitor material. A synergistic effect was observed among all the components of the flexible fabric. The presence of RGO increased the surface area of PPy, and MWCNTs increased the conductivity, high rate performance and the stability.
Yu et al.85 deposited Pani nanowires array coating on nitrogen-doped carbon fiber cloth. Thereafter RGO was deposited on top of the Pani coating to buffer the volume change of Pani that suffers in the oxidation/reduction process, which contributed to enhance the long term stability of the supercapacitor. Strong π–π interaction allowed good electron transfer between Pani and the basal plane of RGO. Nanostructured Pani allowed a high electrode/electrolyte interface area and short diffusion lengths, and RGO decreased the internal resistance of the electrode.
Huang et al.83 deposited RGO, MnO2 and PPy successively on the surface of stainless steel fibers. The obtained fibers could be knitted into fabrics for wearable energy storage textiles. RGO improved charge transfer with MnO2 and increased capacitance. MnO2 contributed to the pseudocapacitance and PPy improved electron transfer and also participated in the pseudocapacitive charge storage. The yarns could be assembled in series and in parallel to meet the voltage and current requirements in real applications. In addition, the yarns could be knitted into patterns without loosing their properties.
Yu et al.72,73 published two works in which they coated fabrics with GNS/MnO2 coatings. Fabrics were previously coated with exfoliated GNS and after this, electrochemical deposition of MnO2 was performed. Materials with high specific capacitance were obtained (315 F g−1). MnO2 was employed as a typical active pseudocapacitive material which uses fast and reversible redox reactions at the surface of electroactive materials. GNS acted as a conductor that facilitated the electrochemical synthesis of MnO2NPs and facilitated fast electron transport between GNS and MnO2NPs. The morphology of the MnO2NPs (flower-like) enhanced the surface area and reduced ion diffusion length during charging/discharging. In addition, GNS acted as a double layer capacitor, accumulating electrical charges arising from ion absorption (double layer capacitance). In another work, authors employed a conductive wrapping of PEDOT/PSS or SWCNTs on the GNS–MnO2/PES fabrics.73 This wrapping further increased the specific capacitance (till 380 F g−1) and the rate capability, which can be attributed to shorter ion diffusion path and increased electronic conductivity. The additional conductive wrapping created a new electron transport path and also participated in increasing the capacitance through double layer capacitance (SWCNTs) or pseudocapacitance (PEDOT).
Shakier et al.75 obtained supercapacitor electrodes based on layer-by-layer assembly of G layers acting as conductive spacers between layers of V2O5 coated-MWCNTs. The G coatings provided an extra conductive pathway, avoided the agglomeration of MWCNTs and enhanced the specific capacitance by a 67%, till values as high as 2590 F g−1. Electrodes stored charge through electric double layer capacitance (MWCNTs) and through pseudocapacitive mechanism (V2O5). The optimal thickness of the V2O5 coating on MWCNTs was 3 nm, a higher thickness decreased the ratio of utilization, which decreased the specific capacitance.
Wang et al.76 employed nanofabrics of electrospun polyamide coated with RGO to produce supercapacitive materials. The nanofabrics increased the surface area of the fabric and avoided the aggregation of RGO, thus being advantageous when compared to microfabrics (capacitance when employing nanofabrics was 4.4 times higher than with microfabrics). GO with different sizes were coated on the fabrics to study the influence of this parameter on the capacitive behavior (diameters: <200 nm (S), 200–450 nm (M), >450 nm (L)). GO was fixed on polyamide through hydrogen bonding between GO functional groups and the amide groups of polyamide 66. The optimal size of GO nanosheets was 200–450 nm, because they had a size that could wrap the fibers and maintained the pores of the fabric. The lowest size (<200 nm) did not produce proper coatings due to an incomplete coverage of the fibers, and the highest size (>450 nm) blocked the pores of the fabric due to the larger size of GO nanosheets. Medium size GO nanosheets improved GO loading and fixation, surface area, conductivity and electrolyte could easily access, thus obtaining the highest capacitance values. Fig. 12 shows micrographs of the coatings obtained on the nanofabrics depending on the size of the GO sheets. A schematic representation is also included to clarify the effect of the GO sheets size.
 | ||
| Fig. 12 The SEM images of (a, e) PA66 nanofibers, (b, f) S-RGO/PA66-nano, (c, g) M-RGO/PA66-nano, and (d, h) L-RGO/PA66-nano.76 Reprinted from Carbon, 73, Y.-S. Wang, S.-M. Li, S.-T. Hsiao, W.-H. Liao, P.-H. Chen, S.-Y. Yang, H.-W. Tien, C.-C. M. Ma and C.-C. Hu, Integration of tailored reduced graphene oxide nanosheets and electrospun polyamide-66 nanofabrics for a flexible supercapacitor with high-volume- and high-area-specific capacitance, page 93, Copyright (2014), with permission from Elsevier. | ||
Ramadoss et al.79 coated carbon fabrics with GO and later reduced it to RGO by thermal treatment at low temperature. High values of specific capacitance were achieved (414 F g−1) and a good rate capability was also observed, which was attributed to a shorter diffusion path for electrolyte and electrons, a highly activated surface and improved electrical conductivity. Two pieces of the conducting fabric, were separated by filter paper separator and a flexible solid state supercapacitor was obtained employing a H3PO4/PVA gel as electrolyte. Bending did not cause significant change in its properties. 5 supercapacitors were mounted and charged, and they powered a LED for more than 15 minutes. A nanogenerator could also be integrated to harvest the energy from vibrational and mechanical deformation to charge the capacitor and this one supplied power to a photosensor. This demonstrates their application for wearable and portable devices.
Liu et al.84 coated cotton yarns with Ni by a polymer-assisted metal deposition method. Later ERGO was deposited by means of electrochemical methods and was further reduced with hydrazine. The fibers obtained showed high capacitance values and could be embroidered or weft due to the enhanced mechanical resistance provided by Ni coating. The fibers could be connected in series, in parallel or in a combination of both to meet operational voltage or power requirements.
2.3. Graphene woven fabrics
Another method that has been applied for the synthesis of G-based fabrics is the production of graphene woven fabrics.86–101 This method consists in growing G coating on a metallic mesh (normally Cu) by CVD (Fig. 13a).86 During the growth, Cu wires interconnect due to the high temperature employed in CVD growth (∼1000 °C). Thereafter, the Cu mesh is removed by FeCl3/HCl treatment and the G structure collapses (upper and lower part of the wires) to produce graphene micron-ribbons. The resulting structure has the form of the Cu mesh employed (Fig. 13b). Fig. 13c shows a photograph of the resulting GFWs structure. Fig. 13d shows a TEM micrograph and the diffraction pattern which shows a hexagonal symmetry. Two sets of the hexagonal spots can be observed with a rotation of 26° between them. This is due that the two sides of the GWF that collapsed are analyzed at the same time. The GWF structure obtained is polycrystalline with several G layers and surface wrinkles are present. The sheet resistance of GWFs can be tuned by varying the coverage of G by simply varying the parameters of the supporting Cu mesh (width of wires and distance between wires). Table 2 shows a compilation of the works that have dealt with GWFs and their applications. | ||
| Fig. 13 Fabrication of GWFs by CVD using copper wire meshes as substrates. (a) Schematic of steps for GWF preparation. (b) Macroscopic optical images (left), top-view SEM images (right) of copper meshes before (top) and after (bottom) graphene growth. Scale bars, 200 mm. (c) Optical images of GWF films floating on water and deposited on glass and PET. Scale bars, 5 mm. (d) TEM image of a GMR and selected area electron diffraction pattern from the region marked with a yellow box. Scale bars, 50 nm (left), 5 (1/nm) (right). Reprinted by permission from Macmillan Publishers Ltd: [Scientific Reports] (ref. 86), copyright (2012). | ||
| Material composition | Properties | Method of synthesis | Application | Reference |
|---|---|---|---|---|
| GWF/PDMS (sensor) GWF (solar cells) | Surface resistivity: 500–2500 Ω per square (GWF), 200–1200 Ω per square (GWF + HNO3 treatment)Optical transparency: 50–90%Linearity as strain sensor: strain < 5% (reversible) Change of electrical resistance under strain: 25-fold (2% strain), 230-fold (5% strain)Efficiency, solar energy conversion: 2.5% (GWF), 3% (GWF/Si), 3.6% (GWF/PEDOT), 3.8% (GWF/Si–HBr–Br2), 6.1% (GWF/Si–HNO3) | CVD synthesis on Cu mesh followed by Cu etching | GWF/polymer composites as strain sensors GWF/semiconductor in solar cells | 86 |
| GWF/PDMS | Change of electrical resistance under strain: 1-fold (0.5% strain), 5–10-fold (2% strain), 103 to 104-fold (8% strain)Gauge factor: 103 (2–6% strain), 106 (>7% strain)Linearity as strain sensor: strain < 10% (reversible)Stability: >100 cycles | CVD synthesis on Cu mesh followed by Cu etching | GWF/polymer composites as strain sensors | 87 |
| GWF/Ti–Au-PDMS | Sheet resistance: 1840 Ω per squareTransmittance: 92.4% (550 nm)Linearity as strain sensor: 3.025% (warp), 0.727% (weft) (round electrodes)Change of electrical resistance under strain: 60% on average | CVD synthesis on Cu mesh followed by Cu etching | GWF/polymer composites as strain sensors | 89 |
| G fibers/PVA G fibers/PVA–PDMS | Conductivity: 9.6 × 103 S m−1 (for 10% wt PVA)Tensile strength: 590 MPa (16% elongation)Limit of strain: 7.1% (to maintain conductivity)Gauge factor: 5.02 (1–6.3% strain)Stability: 200 cycles, bending radius 5.5 mm. 200 cycles, elongation 6% | CVD synthesis on Cu wires followed by Cu etchingPVA coating: 1, 3, 5, 10% PVA solutions. Drying: 3 h, room temperature | Strain sensor | 90 |
| GWF/PDMS | Change of electrical resistance under strain: 1–2-fold (1% strain), 5–10-fold (2% strain), 103 to 104-fold (8% strain)Gauge factor: 500 (2% strain), 10000 (>8% strain)Stability: 24% decrease of initial response after applying 2% strain for 1000 cycles. 20% strain can be achieved by adjusting growth parameters or 40% by changing growth and adopting oblique direction stretching |
CVD synthesis on Cu mesh followed by Cu etching | Sensor for human motion detection, sound signal acquisition, spatially resolved monitoring of external stress distribution | 91 |
| GWF/PDMS-medical tape | Sheet resistance: 400–500 Ω per squareChange of electrical resistance under strain: 10-fold (2% strain), 104-fold (8% strain) Reversibility: <30% strain | CVD synthesis on Cu mesh followed by Cu etching | Sensor for human motion detection: clenching, phonation, expression change, blink, breath, and pulse | 92 |
| GWF/PDMS | Detection limit: 0.3 rad m−1 (0% pre-stretching of PDMS film)Ratio of tolerance limit: 1000 (100 rad m−1)Limit: 800 rad m−1 (20% pre-stretching of PDMS film)Stability: 1000 cycles (0–100 rad m−1) (10% pre-strain, 45° winding angle) | CVD synthesis on Cu mesh followed by Cu etching | Torsion sensor | 93 |
| GWF/PDMS-tape | Change of electrical resistance for letters registration: >4% | CVD synthesis on Cu mesh followed by Cu etching | Acoustic sensor | 94 |
| PtNPs/GWF/n-Si | Efficiency, solar energy conversion: 3–5% (GWF/n-Si), 7.51% (GWFs/n-Si + solid electrolyte), 7.94% (PtNPs/GWF/n-Si), 10.02% (PtNPs/GWF/n-Si + solid electrolyte) | CVD synthesis on Cu mesh followed by Cu etchingPtNPs synthesis: H2PtCl6 10 mM, irradiation 82 mW cm−2, 3–15 minSolid electrolyte: PVA/HNO3 | Solar cells | 95 |
| GWF | Electrical conductivity: 66.7–127 S cm−1Electromagnetic shielding effectiveness: 12.86 dB (10 GHz)Lowest IR transmittance: 70.85% (4500 nm) (120 mesh)Highest IR transmittance: 87.85% (2500–6500 nm) (40 mesh) | CVD synthesis on Cu mesh followed by Cu etching | Electromagnetic shielding | 96 |
| GWF/polishing cloth GWF/polyethylene GWF/PET GWF/filter paper | Two-electrode configuration (fabric/PVA–H3PO4 gel/fabric):Areal capacitance (at 60 mV s−1): 9 mF cm−2 (GWF/polishing cloth), 3 mF cm−2 (GWF/filter paper), 2 mF cm−2 (GWF/polyethylene), 1 mF cm−2 (GWF/PET)Specific capacitance: 267 F g−1 (GWF/polishing cloth)Stability: 100% after 1000 cycles of charge/discharge (GWF/polishing cloth, GWF/filter paper, GWF/PET) | CVD synthesis on Cu mesh followed by Cu etching | Supercapacitor | 97 |
| GWF/porous carbon core GWF/porous carbon core N-doped MnO2–GWF/porous carbon core N-doped | Three-electrode configuration (Pt wrapped with the fabric in 1 M Na2S2O4):Sheet resistance: 10 Ω per squareSpecific surface area: 688 m2 g−1Total pore volume: 435 m3 g−1Areal capacitance: 44 mF cm−2Specific capacitance: 173 F g−1 (N-doped), 225 F g−1 (N-doped/MnO2)Stability: 95% after 1000 cycles of charge/discharge at 20 A g−1 | CVD synthesis on Ni mesh followed by Ni etching | Supercapacitor | 98 |
| Pani/GWF | Two-electrode configuration (fabric/PVA–H3PO4 gel/fabric):Areal capacitance (0.1 mA cm−2): 23 mF cm−2 (Pani–GWF), 2 mF cm−2 (GWF)Specific capacitance (0.1 mA cm−2): 771 F g−1 (Pani–GWF), 66.7 F g−1 (GWF)Highest energy density: 15 mW h m−2 (0.33 mW cm−2, Pani–GWF), 1.4 mW h m−2 (0.03 mW cm−2, GWF)Highest power density: 1 mW cm−2 (2.6 mW h m−2, Pani–GWF), 0.3 mW cm−2 (0.8 mW h m−2, GWF)Stability: 100% after 2000 cycles of charge/discharge | CVD synthesis on Cu mesh followed by Cu etchingPani coating: 0.5 M aniline + 0.1 M HCl, potentiostatic synthesis at +0.8 V, 15 min | Supercapacitor | 99 |
| Pani–GWF/PDMS | Two-electrode configuration (fabric/PVA–H3PO4 gel/fabric):Maximum static strain: 30%Maximum dynamic stretching rate: 60%/sMaximum specific capacitance (60 mV s−1): 8 mF cm−2 (20% pre-stretching, Pani–GWF/PDMS), 17 µF cm−2 (no pre-stretching, GWF/PDMS), 26 µF cm−2 (20% pre-stretching, GWF/PDMS)Stability: 100% after 1000 cycles of charge/discharge | CVD synthesis on Cu mesh followed by Cu etchingPani coating: 0.5 M aniline + 0.1 M HCl, potentiostatic synthesis at +0.8 V, 15 min | Supercapacitor | 100 |
| GWF/epoxy composite | Electrical conductivity: 2.9 S cm−1 (GWFs), 0.15 S cm−1 (GWF/epoxy) (0.19% wt G), 10−12 S cm−1 (epoxy)Fracture toughness: 1.67 MPa m1/2 (application force at 0° in the direction of the fibers) and 1.78 MPa m1/2 (45° direction fibers) (0.62% wt G) | CVD synthesis on Ni mesh followed by Ni etchingEpoxy application: 80 °C, 0.5 h. 120 °C, 2 h | Composite materials with conductivity and enhanced mechanical properties | 101 |
2.4. Applications of graphene woven fabrics
 | ||
| Fig. 14 GWF/polymer hybrid films. (a) Resistance–strain curves for GWF/PDMS hybrids along different directions. Inset shows the schematics and corresponding optical images. Scale bars, 300 mm. (b) Electromechanical properties of the GWF/PDMS films. Resistance change relative to the original value (ΔR/R0) recorded for a number of cycles at tensile strains of 2% and 5%. (c) Stretchable sensor fixed to a finger, and relative changes in resistance for finger motion. Insets show corresponding photographs. Reprinted by permission from Macmillan Publishers Ltd: [Scientific Reports] (ref. 86), Copyright (2012). | ||
In another work,87 the same authors performed a similar study and explained the mechanism underlying for such good performance of GWFs as strain sensors. Under strain, cracks in the structure begin to form at weak points. The crack length and crack density propagate with the increasing strain (Fig. 15). After removing the external force, the cracks disappear and the fractured GWF recovers to its initial position since it is stuck to PDMS substrate. These cracks are responsible for the change in resistance of the sensor. In this paper, very high gauge factors for GWF/PDMS were reported (106 for strain > 7%). The sensors were also employed for compression, torsion and shearing strain sensing, although the response was much lower than when employed for strain monitoring.
 | ||
| Fig. 15 A series of optical images showing the formation of crack and their evolution in GWF under different strain, and corresponding schematics. Adapted by permission from Macmillan Publishers Ltd: [Scientific Reports] (ref. 86), Copyright (2012). | ||
Wang et al.88 applied bio-inspired Voronoi polycrystalline micromechanics model and experimental validations to explain such high gauge factors. It was demonstrated that the successive cracking, the “fish-scale” like network structure of GWFs (overlapping of G crystals on the grain boundary), and the superlubricity between overlapped G flakes play a crucial paper.
Lee et al.89 produced piezoresistors by assembling GWF on PDMS where Ti–Au electrodes had been previously deposited (round and band type) to produce touch sensors.
The method employed for producing GWFs has also been employed for producting G fibers by Wang et al.90 The G fibers were coated with PVA to produce protected conducting fibers that were mounted on PDMS film to construct a strain sensor.
Yang et al.91 employed GWFs as strain sensors for human monitoring (human skin movement, pulse, jugular venous pressure, body movement), acoustic signal acquisition, and spatially resolved monitoring of external stress distribution. Thicker G coatings produced GWFs with a larger stretching range (>20%), however sensitivity was sacrificed (lower gauge factors). The higher stretching limit would allow to employ these sensors for monitoring activities such as walking, running and grasping. Wang et al.92 applied GWFs for similar body motion monitoring (clenching, phonation, expression change, blink, breath, and pulse). Authors pointed out applications such as lie detectors based on muscle deformation amplitude, or evaluation of the degree of fatigue by measuring the frequency and amplitude of the blinks, etc.
Torsion sensors have also been reported by Yang et al.,93 obtained by wrapping GWF/PDMS around a PDMS rod and applying torsion to the polymeric rod. Differences were observed when pre-stretching to the PDMS film was applied or not. When applying a 20% pre-stretching, the sensor could withstand torsions of up to 800 rad m−1 because waving of the GWFs was avoided and their tolerance to high strains was improved. Lower pre-tensions favoured the sensitivity due to a large gauge factor. The sensor could discriminate among forward and backward torsion by the change in resistance and the response was not dependent on the frequency of the applied load.
Wang et al.94 employed GWFs for sound signal acquisition and recognition. The sensor, which was very sensitive to tiny strains and vibrations, was located on a human throat and it was employed to register the muscular movement/vibration. It was used for collecting and recognizing the 26 english letters, some typical Chinese characters, phrases and animal sounds. The waveforms were specific of each letter and showed similar key features when performed by different people. The same signal wave forms were obtained without vocalization, which could help patients with problems in vocalization. In addition, authors pointed out the employment of these sensors in earthquake monitoring, animal communication and robotic voice development. The sensor was also employed to perform measurements with a loudspeaker, where the sensor was located on the loudspeaker membrane. The audio frequency and the collected signal had a matching synchronization.
 | ||
| Fig. 16 Schematic diagrams of (a) pristine GWF/n-Si solar cell, (b) in situ galvanic synthesis of Pt–GWF, and (c) Pt–GWF/n-Si solar cell. Reprinted from ref. 95 with permission of The Royal Society of Chemistry. | ||
Zang et al.97 obtained GWFs and deposited them on different substrates (polishing cloth, polyethylene, PET and filter paper) and studied their capacitive behavior. Supercapacitors with very low thickness (<1 mm) were mounted with PVA–H3PO4 as polymer gel electrolyte and the best results were obtained on polishing cloth. Deformation of the devices (fold, twist, knead, and curl) increased capacitance due to a better contact between the electrode and the gel electrolyte. Therefore, the supercapacitors could be shaped into different forms without loss in their capacitance.
Li et al.98 obtained GWFs composed of G layers and porous carbon on the core, hence integrating the conductivity of G and the porous characteristics of porous carbon. Depending on the synthesis conditions and cooling rate, the composition of the material could be adjusted. Thinner multilayer G films provided a larger specific surface area and voids for high-performance supercapacitor electrodes. Thicker deposits provided a better skeleton structure to be filled with other functional materials. The mean pore size of the material was in the 2–3 nm range, favouring the formation of an electrical double layer. N-doping enhanced the pseudocapacitive effect and improved the specific capacitance by a 150%. Capacitance could be further increased depositing other materials such as MnO2 on the surface of the GWFs.
Zang et al.99 coated GWFs with Pani obtained by potentiostatic deposition and employed the Pani/GWFs composite as supercapacitor material. GWF acted as support material onto which Pani could be deposited and also as a current collector for capacitance measurements. In addition, GWFs also acted as a double layer capacitor and Pani acted as a pseudo-capacitive material. The presence of Pani improved greatly the capacitance of GWFs (from 2 mF cm−2 to 23 mF cm−2) (Fig. 17). The supercapacitor showed great stability after 2000 cycles with no appreciable loss of capacitance (Fig. 17e); what is more, bending and curling even improved the areal capacitance since a better contact between the polymer gel electrolyte and the electrode was favored.
 | ||
| Fig. 17 Electrochemical performance of the GWF + PANI (15 min) film supercapacitor. (a) CV curves (60 mV s−1), (b) galvanostatic charge–discharge curves (0.1 mA cm−2), (c) areal capacitances (0.1 to 2 mA cm−2) of GWF and GWF + PANI. (d) Areal capacitance versus electropolymerization time. (e) Cycling stability of the flexible GWF + PANI supercapacitor. (f) Schematic diagram of the GWF electrodes. Reprinted from ref. 99 with permission of The Royal Society of Chemistry. | ||
In another study,100 the same authors employed the same approach to produce supercapacitors and tested the static and dynamic performance under strain. The GWFs were assembled on pre-stretched polymer substrates in order to improve flexibility and enlarge the strain range of the supercapacitors. Till a 30% strain, no change in the capacitance was observed. Pani was also electropolymerized on GWFs as explained previously in order to increase capacitance.99 No change in the capacitance was neither observed till a 30% strain. According to this, the maximum strain for the dynamic tests was restricted to 30%. The supercapacitors could withstand high rates of dynamic stretching (60%/s, strain frequency 1 Hz). The GWFs increased the surface area and the robustness, being more suitable for dynamic stretching deformation.
3. Conclusions and perspectives
Textile fabrics present different advantages when compared with sheet materials, such as their high surface area, flexibility, mechanical properties, etc., which make them attractive substrates onto which other functional materials can be deposited. On the other hand, graphene (G) and related materials have emerged as a revolutionary material in the field of materials science and physics due to their extraordinary properties. G materials provide a conductive platform that can be integrated into textiles by means of chemical deposition, by producing graphene woven fabrics (GWFs) or by integrating conductive fibers of G in the fabrics. A lot of work has been performed with G-based fibers and there are several reviews that deal with this topic.21–24 For this reason, this subject has not been included in the present review. The present review is focused on the chemical deposition of G materials on fabrics/yarns and the synthesis of GWFs and their applications.The chemical method is simple and can be scaled up by means of traditional techniques employed in textile industry such as soaking, drying, reducing with chemical compounds, etc. Graphene oxide (GO) is employed as the main G source since it is cheap and it can be produced in great quantities and for the moment is the only G derivative that could meet the large requirements of G for textile industry if this technology were to be established. GO is simply adsorbed on the surface of the fabrics by means of chemical interactions (electrostatic, hydrogen bonds, π–π interactions, etc.). GO is an insulating material, however, its conductivity is partially restored by means of chemical, thermal, electrochemical or UV reduction to produce reduced graphene oxide (RGO). However, the conductivity obtained with this procedure is lower than in the case of GWFs since RGO retains some degree of oxidation (functional groups) after reduction. The applications of these type of fabrics (composed only of G or with other functional materials) can be: UV protection, conducting fabrics, antistatic fabrics, IR emission, hydrophobicity, sensors for electrocardiogram acquisition, heat generation, thermal conduction, photocatalytic activity, electrocatalytic activity, antibacterial, antifungal, gas and liquid sensors, anode for microbial fuel cells, cathode for solar cells, field emission devices, capacitive materials for energy storage, etc.
With the GWFs, higher conductivity is obtained, however the synthesis route consists in chemical vapor deposition on a Cu mesh (most widely metal employed as substrate), the subsequent removal of the copper mesh by chemical dissolution and the transfer of the remaining G mesh to a substrate to support it. The GWFs are composed of several G layers with polycrystalline nature. Due to the high conductivity obtained, this method could be employed for applications where high conductivity is needed, being sensors one of the main uses of this type of textile structures. The high sensitivity of these fabrics as strain sensors, as demonstrated by their very high gauge factors, opens the door to new applications such as voice recognition, movement sensors, breath and pulse sensors, etc.
Regarding future developments, more effort should be devoted to increasing the adhesion of G/GO/RGO coatings. In the case of GO, given the negative charges of GO due to the presence of oxygen containing functional groups, cationization of the surface of the fabrics should be accomplished. This has been mainly attained by means of chemical compounds (already under investigation, being bovine serum albumin coatings the most employed).
The development in the area of energy applications will certainly be explored in more depth in the future and more applications will be envisaged. These applications include flexible supercapacitors composed of G alone or combined with other capacitive materials, their development will allow the implementation of electronic textiles with different functionalities. Other type of materials or devices that harvest energy from body temperature (thermoelectric materials) or body movement could also be integrated taking advantage of G thermal and electrical conductivity. The applications of G as electrode material for energy applications also include Li-ion batteries, Li–sulphur batteries, Li air/oxygen batteries, fuel cells, microbial fuel cells, enzymatic fuel cells or solar cells for example.103,104 Some of them have already been reported for G-based fabrics and other will be explored shortly.
The application of semiconducting nanoparticles on G fabrics should be studied in more depth, since mainly TiO2 nanoparticles have been applied for the moment.54,56,57,74 With the functionalization by other semiconductors, G fabrics could be employed as photocatalytic materials or could be integrated in solar cells to increase the energy conversion efficiency.61,87,95 Given its organic nature, the modification with organic dyes can be easily achieved through π–π interactions or hydrophobic interactions,105 and can be used as an approach to increase the bandwidth energy absorbance of G materials, which mainly absorb radiation in the UV-region, to visible regions where dyes absorb.106 This approach would also led to an increase in the energy conversion efficiency in solar cells.
Another area of research not covered by bibliography up to date is the employment of modified G materials such as nitrogen-doped G,107–109 metallic doped G110 that have been applied for the fields of sensors, catalysis, etc. The application of other 2-D materials such as transition metal dichalcogenides should also be subject of future study due to their interesting properties.111–114
Another possibility pointed out in bibliography is the production of bionic materials integrating nanomaterials (such as G or carbon nanotubes) on natural fibers (for example on silk produced by spiders).115
Abbreviations
| APS | Ammonium persulphate |
| AQSA | Anthraquinone-2-sulfonic acid sodium salt monohydrate |
| BN | Boron nitride |
| BSA | Bovine serum albumin |
| CNT | Carbon nanotube |
| CV | Cyclic voltammetry |
| CVD | Chemical vapor deposition |
| DMF | Dimethylformamide |
| EMIMBF4 | 1-Ethyl-3-methylimidazolium tetrafluoroborate |
| ERGO | Electrochemically reduced graphene oxide |
| FLG | Few layer graphene |
| G | Graphene |
| GNP | Graphene nanoplate |
| GNR | Graphene nanoribbon |
| GNS | Graphene nanosheet |
| GO | Graphene oxide |
| GWF | Graphene woven fabric |
| MB | Methylene blue |
| MWCNT | Multiwalled carbon nanotube |
| Na2NDS | Naphthalene-2,6-disulfonic acid disodium salt |
| NCPCl | N-Cetylpyridinium chloride |
| NP | Nanoparticle |
| Pani | Polyaniline |
| PBS | Phosphate buffer solution |
| PDMS | Polydimethylsiloxane |
| PEDOT | Poly(3,4-ethylenedioxythiophene) |
| PEI | Polyethyleneimine |
| PES | Polyester |
| PET | Poly(ethylene terephthalate) |
| PLA | Polylactic acid |
| PMS | Polymethylsiloxane |
| PP | Polypropylene |
| PPy | Polypyrrole |
| PSS | Poly(styrenesulfonate) |
| PU | Polyurethane |
| PVA | Polyvinyl alcohol |
| PVP | Poly(vinylpyrrolidone) |
| QD | Quantum dot |
| RGO | Reduced graphene oxide |
| SDS | Sodium dodecyl sulfonate |
| SWCNT | Single walled carbon nanotubes |
| UPF | Ultraviolet protection factor |
| WPU | Waterborne anionic aliphatic polyurethane |
| Wt | Weight |
Acknowledgements
J. Molina wishes to thank to the Spanish Ministerio de Ciencia e Innovación (contract CTM2011-23583) for the financial support. J. Molina is grateful to the Conselleria d’Educació, Formació i Ocupació (Generalitat Valenciana) for the Programa VALi + D Postdoctoral Fellowship (APOSTD/2013/056).References
- A. R. Horrocks, B. K. Kandola, P. J. Davies, S. Zhang and S. A. Padbury, Polym. Degrad. Stab., 2005, 88, 3–12 CrossRef CAS.
- Y. Shin, D. Yoo and K. Son, J. Appl. Polym. Sci., 2005, 96, 2005–2010 CrossRef CAS.
- A. Laforgue, J. Mater. Chem., 2010, 20, 8233–8235 RSC.
- N. K. Vu, A. Zille, F. R. Oliveira, N. Carneiro and A. P. Souto, Plasma Processes Polym., 2013, 10, 285–296 CrossRef.
- J. O. Carneiro, V. Teixeira, J. H. O. Nascimento, J. Neves and P. B. Tavares, J. Nanosci. Nanotechnol., 2011, 11, 1–8 CrossRef.
- M. Yu, Z. Wang, H. Liu, S. Xie, J. Wu, H. Jiang, J. Zhang, L. Li and J. Li, ACS Appl. Mater. Interfaces, 2013, 5, 3697–3703 CAS.
- R. He, T. D. Day, M. Krishnamurthi, J. R. Sparks, P. J. A. Sazio, V. Gopalan and J. V. Badding, Adv. Mater., 2013, 25, 1461–1467 CrossRef CAS PubMed.
- D. Graham-Rowe, Nat. Photonics, 2007, 1, 6–7 CrossRef CAS.
- R. F. Service, Science, 2003, 301, 909–911 CrossRef CAS PubMed.
- J. W. Lee, T. Mayer-Gall, K. Opwis, C. E. Song, J. S. Gutmann and B. List, Science, 2013, 341, 1225–1229 CrossRef CAS PubMed.
- O. K. E. Y. Bulgun, Int. J. Cloth. Sci. Tech., 2009, 21, 127–136 CrossRef.
- D. Akbarov, B. Baymuratov, P. Westbroek, R. Akbarov, K. Declerck and P. Kiekens, J. Appl. Electrochem., 2006, 36, 411–418 CrossRef CAS.
- A. Bhattacharyya and M. Joshi, Fibers Polym., 2011, 12, 734–740 CrossRef CAS.
- A. Kaynak, S. S. Najar and R. C. Foitzik, Synth. Met., 2008, 158, 1–5 CrossRef CAS.
- J. Molina, A. I. del Río, J. Bonastre and F. Cases, Eur. Polym. J., 2008, 44, 2087–2098 CrossRef CAS.
- J. Molina, M. F. Esteves, J. Fernández, J. Bonastre and F. Cases, Eur. Polym. J., 2011, 47, 2003–2015 CrossRef CAS.
- K. S. Novoselov, V. I. Fal’ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- A. K. Geim, Angew. Chem., Int. Ed., 2011, 50, 6967–6985 CrossRef PubMed.
- A. C. Ferrari, et al., Nanoscale, 2015, 7, 4598–4810 RSC.
- Z. Xu and C. Gao, Mater. Today, 2015, 18, 480–492 CrossRef CAS.
- F. Meng, W. Lu, Q. Li, J.-H. Byun, Y. Oh and T.-W. Chou, Adv. Mater., 2015, 27, 5113–5131 CrossRef CAS PubMed.
- H. Cheng, C. Hu, Y. Zhao and L. Qu, NPG Asia Mater., 2014, 6, e113 CrossRef CAS.
- G. Sun, X. Wang and P. Chen, Mater. Today, 2015, 18, 215–226 CrossRef CAS.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 2, 101–105 CrossRef PubMed.
- M. Tian, X. Tang, L. Qu, S. Zhu, X. Guo and G. Han, Mater. Lett., 2015, 145, 340–343 CrossRef CAS.
- L. Qu, M. Tian, X. Hu, Y. Wang, S. Zhu, X. Guo, G. Han, X. Zhang, K. Sun and X. Tang, Carbon, 2014, 80, 565–574 CrossRef CAS.
- M. Tian, X. Hu, L. Qu, S. Zhu, Y. Sun and G. Han, Carbon, 2016, 96, 1166–1174 CrossRef CAS.
- X. Hu, M. Tian, L. Qu, S. Zhu and G. Han, Carbon, 2015, 95, 625–633 CrossRef CAS.
- X. Tang, M. Tian, L. Qu, S. Zhu, X. Guo, G. Han, K. Sun, X. Hu, Y. Wang and X. Xu, Synth. Met., 2015, 202, 82–88 CrossRef CAS.
- K. Javed, C. M. A. Galib, F. Yang, C.-M. Chen and C. Wang, Synth. Met., 2014, 193, 41–47 CrossRef CAS.
- J. A. Moleon, A. Ontiveros-Ortega, E. Gimenez-Martin and I. Plaza, Dyes Pigm., 2015, 122, 310–316 CrossRef CAS.
- N. D. Tissera, R. N. Wijesena, J. R. Perera, K. M. N. de Silva and G. A. J. Amaratunge, Appl. Surf. Sci., 2015, 324, 455–463 CrossRef CAS.
- M. Shateri-Khalilabad and M. E. Yazdanshenas, Cellulose, 2013, 20, 963–972 CrossRef CAS.
- Y. J. Yun, W. G. Hong, W.-J. Kim, Y. Jun and B. H. Kim, Adv. Mater., 2013, 25, 5701–5705 CrossRef CAS PubMed.
- M. Shateri-Khalilabad and M. E. Yazdanshenas, Carbohydr. Polym., 2013, 96, 190–195 CrossRef CAS PubMed.
- B. Fugetsu, E. Sano, H. Yu, K. Mori and T. Tanaka, Carbon, 2010, 48, 3340–3345 CrossRef CAS.
- Z. Lu, C. Mao and H. Zhang, J. Mater. Chem. C, 2015, 3, 4265–4268 RSC.
- J. Molina, A. Zille, J. Fernández, A. P. Souto, J. Bonastre and F. Cases, Synth. Met., 2015, 204, 110–121 CrossRef CAS.
- A. I. S. Neves, T. H. Bointon, L. V. Melo, S. Russo, I. de Schrijver, M. F. Craciun and H. Alves, Sci. Rep., 2015, 5, 09866 CrossRef CAS PubMed.
- J. Molina, J. Fernández, J. C. Inés, A. I. del Río, J. Bonastre and F. Cases, Electrochim. Acta, 2013, 93, 44–52 CrossRef CAS.
- J. Molina, J. Fernández, A. I. del Río, J. Bonastre and F. Cases, Appl. Surf. Sci., 2013, 279, 46–54 CrossRef CAS.
- J. Molina, J. Fernández, M. Fernandes, A. P. Souto, M. F. Esteves, J. Bonastre and F. Cases, Synth. Met., 2015, 202, 110–122 CrossRef CAS.
- Y. A. Samad, Y. Li, S. M. Alhassan and K. Liao, RSC Adv., 2014, 4, 16935–16938 RSC.
- S. J. Woltornist, F. A. Alamer, A. McDannald, M. Jain, G. A. Sotzing and D. H. Adamson, Carbon, 2015, 81, 38–42 CrossRef CAS.
- W. Gu and Y. Zhao, Adv. Mater. Res., 2011, 331, 93–96 CrossRef CAS.
- D. H. Ha, S. Jung, H.-J. Kim, D. Kim, W.-J. Kim, S. N. Yi, Y. Jun and Y. J. Yun, Synth. Met., 2015, 204, 90–94 CrossRef CAS.
- X. Liu, Z. Qin, Z. Dou, N. Liu, L. Chen and M. Zhu, RSC Adv., 2014, 4, 23869–23875 RSC.
- S.-T. Hsiao, C.-C. M. Ma, H.-W. Tien, W.-H. Liao, Y.-S. Wang, S.-M. Li and W.-P. Chuang, Compos. Sci. Technol., 2015, 118, 171–177 CrossRef CAS.
- A. Abbas, Y. Zhao, J. Zhou, X. Wang and T. Lin, Fibers Polym., 2013, 14, 1641–1649 CrossRef CAS.
- G. Huang, J. Yang, J. Gao and X. Wang, Ind. Eng. Chem. Res., 2012, 51, 12355–12366 CAS.
- L. Karimi, M. E. Yazdanshenas, R. Khajavi, A. Rashidi and M. Mirjalili, Appl. Surf. Sci., 2015, 332, 665–673 CrossRef CAS.
- K. Krishnamoorthy, U. Navaneethaiyer, R. Mohan, J. Lee and S.-J. Kim, Appl. Nanosci., 2012, 2, 119–126 CrossRef CAS.
- L. Karimi, M. E. Yazdanshenas, R. Khajavi, A. Rashidi and M. Mirjalili, Cellulose, 2014, 21, 3813–3827 CrossRef CAS.
- J. Molina, F. Fernandes, J. Fernández, M. Pastor, A. Correia, A. P. Souto, J. O. Carneiro, V. Teixeira and F. Cases, Mater. Sci. Eng., B, 2015, 199, 62–76 CrossRef CAS.
- I. A. Sahito, K. C. Sun, A. A. Arbab, M. B. Qadir and S. H. Jeong, Carbohydr. Polym., 2015, 130, 299–306 CrossRef CAS PubMed.
- M. K. Yapici, T. Alkhidir, Y. A. Samad and K. Liao, Sens. Actuators, B, 2015, 221, 1469–1474 CrossRef CAS.
- L. Gan, S. Shang, C. W. M. Yuen and S.-x. Jiang, Compos. Sci. Technol., 2015, 117, 208–214 CrossRef CAS.
- B. Liang, L. Fang, Y. Hu, G. Yang, Q. Zhu and X. Ye, Nanoscale, 2014, 6, 4264–4274 RSC.
- Y. J. Yun, W. G. Hong, N.-J. Choi, B. H. Kim, Y. Jun and H.-K. Lee, Sci. Rep., 2015, 5, 10904 CrossRef CAS PubMed.
- E. Skrzetuska, M. Puchalski and I. Krucińska, Sensors, 2014, 14, 16816–16828 CrossRef PubMed.
- J. Liu, Y. Qiao, C. X. Guo, S. Lim, H. Song and C. M. Li, Bioresour. Technol., 2012, 114, 275–280 CrossRef CAS PubMed.
- I. A. Sahito, K. C. Sun, A. A. Arbab, M. B. Qadir and S. H. Jeong, Electrochim. Acta, 2015, 173, 164–171 CrossRef CAS.
- F. Zhao, L. Liu, F. Yang and N. Ren, Chem. Eng. J., 2013, 230, 491–498 CrossRef CAS.
- R. Roy, A. Jha, D. Banerjee, N. S. Das and K. K. Chattopadhyay, AIP Adv., 2013, 3, 012115 CrossRef.
- U. N. Maiti, S. Maiti, N. S. Das and K. K. Chattopadhyay, Nanoscale, 2011, 3, 4135–4141 RSC.
- L.-L. Xu, M.-X. Guo, S. Liu and S.-W. Bian, RSC Adv., 2015, 5, 25244–25249 RSC.
- F. Yaghoubidoust, D. H. B. Wicaksono, S. Chandren and H. Nur, J. Mol. Struct., 2014, 1075, 486–493 CrossRef CAS.
- J. Xu, D. Wang, Y. Yuan, W. Wei, L. Duan, L. Wang, H. Bao and W. Xu, Org. Electron., 2015, 24, 153–159 CrossRef CAS.
- G. Yu, L. Hu, M. Vosgueritchian, H. Wang, X. Xie, J. R. McDonough, X. Cui, Y. Cui and Z. Bao, Nano Lett., 2011, 11, 2905–2911 CrossRef CAS PubMed.
- G. Yu, L. Hu, N. Liu, H. Wang, M. Vosgueritchian, Y. Yang, Y. Cui and Z. Bao, Nano Lett., 2011, 11, 4438–4442 CrossRef CAS PubMed.
- F. Meng, J. Zhao, Y. Ye, X. Zhang, S. Li, J. Jia, Z. Zhang and Q. Li, J. Mater. Chem., 2012, 22, 16277–16282 RSC.
- I. Shakir, Z. Ali, J. Bae, J. Park and D. J. Kang, Nanoscale, 2014, 6, 4125–4130 RSC.
- Y.-S. Wang, S.-M. Li, S.-T. Hsiao, W.-H. Liao, P.-H. Chen, S.-Y. Yang, H.-W. Tien, C.-C. M. Ma and C.-C. Hu, Carbon, 2014, 73, 87–98 CrossRef CAS.
- Q. Zhou, X. Ye, Z. Wan and C. Jia, J. Power Sources, 2015, 296, 186–196 CrossRef CAS.
- W.-w. Liu, X.-b. Yan, J.-w. Lang, C. Peng and Q.-j. Xue, J. Mater. Chem., 2012, 22, 17245–17253 RSC.
- A. Ramadoss, B. Saravanakumar and S. J. Kim, Nano Energy, 2015, 15, 587–597 CrossRef CAS.
- C. Zhao, K. Shu, C. Wang, S. Gambhir and G. G. Wallace, Electrochim. Acta, 2015, 172, 12–19 CrossRef CAS.
- F. Liu, S. Wang, G. Han, R. Liu, Y. Chang and Y. Xiao, J. Appl. Polym. Sci., 2014, 131, 41023 Search PubMed.
- Y. Cao, M. Zhu, P. Li, R. Zhang, X. Li, Q. Gong, K. Wang, M. Zhong, D. Wu, F. Lin and H. Zhu, Phys. Chem. Chem. Phys., 2013, 15, 19550–19556 RSC.
- Y. Huang, H. Hu, Y. Huang, M. Zhu, W. Meng, C. Liu, Z. Pei, C. Hao, Z. Wang and C. Zhi, ACS Nano, 2015, 9, 4766–4775 CrossRef CAS PubMed.
- L. Liu, Y. Yu, C. Yan, K. Li and Z. Zheng, Nat. Commun., 2015, 6, 7260 CrossRef CAS PubMed.
- P. Yu, Y. Li, X. Zhao, L. Wu and Q. Zhang, Langmuir, 2014, 30, 5306–5313 CrossRef CAS PubMed.
- X. Li, P. Sun, L. Fan, M. Zhu, K. Wang, M. Zhong, J. Wei, D. Wu, Y. Cheng and H. Zhu, Sci. Rep., 2012, 2, 395 Search PubMed.
- X. Li, R. Zhang, W. Yu, K. Wang, J. Wei, D. Wu, A. Cao, Z. Li, Y. Cheng, Q. Zheng, R. S. Ruoff and H. Zhu, Sci. Rep., 2012, 2, 870 Search PubMed.
- W. Wang, T. Yang, H. Zhu and Q. Zheng, Appl. Phys. Lett., 2015, 106, 171903 CrossRef.
- X. Lee, T. Yang, X. Li, R. Zhang, M. Zhu, H. Zhang, D. Xie, J. Wei, M. Zhong, K. Wang, D. Wu, Z. Li and H. Zhu, Appl. Phys. Lett., 2013, 102, 163117 CrossRef.
- X. Wang, Y. Qiu, W. Cao and P. Hu, Chem. Mater., 2015, 27, 6969–6975 CrossRef CAS.
- T. Yang, W. Wang, H. Zhang, X. Li, J. Shi, Y. He, Q.-s. Zheng, Z. Li and H. Zhu, ACS Nano, 2015, 9, 10867–10875 CrossRef CAS PubMed.
- Y. Wang, L. Wang, T. Yang, X. Li, X. Zang, M. Zhu, K. Wang, D. Wu and H. Zhu, Adv. Funct. Mater., 2014, 24, 4666–4670 CrossRef CAS.
- T. Yang, Y. Wang, X. Li, Y. Zhang, X. Li, K. Wang, D. Wu, H. Jin, Z. Li and H. Zhu, Nanoscale, 2014, 6, 13053–13059 RSC.
- Y. Wang, T. Yang, J. Lao, R. Zhang, Y. Zhang, M. Zhu, X. Li, X. Zang, K. Wang, W. Yu, H. Jin, L. Wang and H. Zhu, Nano Res., 2015, 8, 1627–1636 CrossRef CAS.
- Z. Kang, X. Tan, X. Li, T. Xiao, L. Zhang, J. Lao, X. Li, S. Cheng, D. Xie and H. Zhu, Phys. Chem. Chem. Phys., 2016, 18, 1992–1997 RSC.
- J. Han, X. Wang, Y. Qiu, J. Zhu and P. Hu, Carbon, 2015, 87, 206–214 CrossRef CAS.
- X. Zang, Q. Chen, P. Li, Y. He, X. Li, M. Zhu, X. Li, K. Wang, M. Zhong, D. Wu and H. Zhu, Small, 2014, 10, 2583–2588 CrossRef CAS PubMed.
- X. Li, X. Zang, Z. Li, X. Li, P. Li, P. Sun, X. Lee, R. Zhang, Z. Huang, K. Wang, D. Wu, F. Kang and H. Zhu, Adv. Funct. Mater., 2013, 23, 4862–4869 CAS.
- X. Zang, X. Li, M. Zhu, X. Li, Z. Zhen, Y. He, K. Wang, J. Wei, F. Kang and H. Zhu, Nanoscale, 2015, 7, 7318–7322 RSC.
- X. Zang, M. Zhu, X. Li, X. Li, Z. Zhen, J. Lao, K. Wang, F. Kang, B. Wei and H. Zhu, Nano Energy, 2015, 15, 83–91 CrossRef CAS.
- X. Liu, X. Sun, Z. Wang, X. Shen, Y. Wu and J.-K. Kim, ACS Appl. Mater. Interfaces, 2015, 7, 21455–21464 CAS.
- L. Zhang, M. Becton and X. Wang, Mater. Sci. Eng., A–, 2015, 620, 367–374 CrossRef.
- D. A. C. Brownson, D. K. Kampouris and C. E. Banks, J. Power Sources, 2011, 196, 4873–4885 CrossRef CAS.
- J. Zhu, D. Yang, Z. Yin, Q. Yan and H. Zhang, Small, 2014, 10, 3480–3498 CrossRef CAS PubMed.
- J. Molina, J. Fernández, C. García, A. I. del Río, J. Bonastre and F. Cases, Electrochim. Acta, 2015, 166, 54–63 CrossRef CAS.
- Y. Lee, S. H. Yu, J. Jeon, H. Kim, J. Y. Lee, H. Kim, J.-H. Ahn, E. Hwang and J. H. Cho, Carbon, 2015, 88, 165–172 CrossRef CAS.
- X.-K. Kong, C.-L. Chen and Q.-W. Chen, Chem. Soc. Rev., 2014, 43, 2841–2857 RSC.
- W. Zhang, L. Wu, Z. Li and Y. Liu, RSC Adv., 2015, 5, 49521–49533 RSC.
- J. Duan, S. Chen, M. Jaroniec and S. Z. Qiao, ACS Catal., 2015, 5, 5207–5234 CrossRef CAS.
- H. Fei, J. Dong, M. J. Arellano-Jiménez, G. Ye, N. D. Kim, E. L. G. Samuel, Z. Peng, Z. Zhu, F. Qin, J. Bao, M. J. Yacaman, P. M. Ajayan, D. Chen and J. M. Tour, Nat. Commun., 2015, 6, 8668 CrossRef CAS PubMed.
- F. Bonaccorso, A. Lombardo, T. Hasan, Z. Sun, L. Colombo and A. C. Ferrari, Mater. Today, 2012, 15, 564–589 CrossRef CAS.
- M. C. Z. Liu and H. Zhang, Chem. Soc. Rev., 2015, 44, 2584–2586 RSC.
- X. Duan, C. Wang, A. Pan, R. Yu and X. Duan, Chem. Soc. Rev., 2015, 44, 8859–8876 RSC.
- C. Tan and H. Zhang, Chem. Soc. Rev., 2015, 44, 2713–2731 RSC.
- E. Lepore, F. Bonaccorso, M. Bruna, F. Bosia, S. Taioli, G. Garberoglio, A. C. Ferrari and N. M. Pugno, 2015, arXiv:1504.06751 [cond-mat.mtrl-sci].
| This journal is © The Royal Society of Chemistry 2016 |