
Electrochemical immunosensors and their recent nanomaterial-based signal amplification strategies: a review
Syazana Abdullah Lim
ab and
Minhaz Uddin Ahmed
*b
aEnvironmental and Life Sciences Programme, Faculty of Science, Universiti Brunei Darussalam, Jalan Tungku Link, Gadong, BE 1410, Brunei Darussalam
bBiosensors and Biotechnology Laboratory, Chemical Science Programme, Faculty of Science, Universiti Brunei Daruusalam, Jalan Tungku Link, Gadong, BE 1410, Brunei Darussalam. E-mail: minhaz.ahmed@ubd.edu.bn; minhazua@gmail.com; Tel: +673 888 4752
First published on 1st March 2016
Abstract
In recent years, tremendous advances have been made in biosensors based on nanoscale electrochemical immunosensors for use in the fields of agriculture, food safety, biomedicine, quality control, and environmental and industrial monitoring. One of the main challenges in biosensors is achieving an extremely low limit of detection. A current trend to address this is fabrication of highly sensitive electrochemical immunosensors through the use of nanotechnology; for example, biofunctionalization of nanomaterials that are used as a catalyst, label or biosensing transducer. This review introduces recent advances in signal amplification strategies for electrochemical immunosensing applications, with a particular focus on nanotechnology. The strategies employed and their general principles to increase sensitivity, as well as the advantages and limitations of electrochemical immunosensors developed to date are also considered.
 Syazana Abdullah Lim | Syazana A. Lim is currently a Ph.D. student in the area of nanomaterials based electrochemical immunosensors at the Universiti Brunei Darussalam under the supervision of Dr Minhaz Uddin Ahmed. She has completed her BSc and MSc from University of Reading, United Kingdom in the area of human nutrition and food technology respectively. Her doctoral research interest include the development of carbon nanostructured based state-of-the-art sensitive immunosensor to detect clinically important analytes. |
 Minhaz Uddin Ahmed | Dr Minhaz Uddin Ahmed is a Senior Lecturer of Analytical Chemistry and Biotechnology at the Universiti Brunei Darussalam (UBD). He obtained his Ph.D. in Chemical Materials Science from Japan Advanced Institute of Science and Technology (JAIST) in 2008 under the supervision of Prof. Eiichi Tamiya and later worked as a JSPS post-doctoral fellow at Osaka University, Japan (2008–2009); DARPA post-doctoral research associate at Duke University, US (2009–2010); and NSERC post-doctoral fellow at INRS-EMT, Canada (2010–2012). His research interests are in the areas of biosensors and diagnostic technologies as well as applications to nanobiotechnology, clinical and environmental chemistry. |
1. Introduction
1.1 Biosensors
According to a proposed International Union of Pure and Applied Chemistry (IUPAC) definition, “A biosensor is a self-contained integrated device which is capable of providing specific quantitative or semi-quantitative analytical information using a biological recognition element (biochemical receptor) which is in direct spatial contact with a transducer element. A biosensor should be clearly distinguished from a bioanalytical system, which requires additional processing steps, such as reagent addition. Furthermore, a biosensor should be distinguished from a bioprobe which is either disposable after one measurement, i.e. single use, or able to continuously monitor the analyte concentration”.1 In a broad context, a biosensor can be simply described as an analytical device that translates a biological interaction into a signal that can be quantified.2 A biosensor integrates two main components: (1) a bioreceptor or biorecognition element that recognizes a target analyte such as an enzyme substrate, complementary DNA, or antigen; and (2) a sensor or transducer element that detects the (bio)chemical interaction of the analyte with the bioreceptor and subsequently converts the signal into a measurable signal. The biological element may be tissue, living cells, an enzyme, antibody or antigen, and the sensor/transducer element may be an electric current, electric potential, intensity or phase of electromagnetic radiation, mass, conductance, impedance, temperature or viscosity or combination of two or more of these techniques. Biosensors are categorized into biocatalytic and bioaffinity-based biosensors. While biocatalytic biosensors mainly use enzymes as the biological mediator to catalyze a signaling biochemical reaction, bioaffinity-based biosensors monitor the binding episode itself. In bioaffinity-based biosensors, biomolecular recognition can involve specific binding proteins, lectins, receptors, nucleic acids, membranes, whole cells, antibodies or antibody-related substances.3In recent years, electrochemical immunosensors have become widely used in different sectors such as agriculture, food and medical applications, quality control, environmental and industrial surveillance as well as point-of-care devices.4–9 Correspondingly, there has been an exponential increase in the number of papers published on electrochemical immunosensors since 2000 (Fig. 1). With the aims of successful fabrication and application of immunosensors, much recent research has been focused on signal amplification strategies to obtain sensors with a low limit of detection (LOD) and thus high sensitivity. This review highlights recent advances in electrochemical immunosensors for protein detection that use signal amplification strategies. A selection of representative examples based on studies published during the past five years are described. It is worthwhile noting that although this report primarily focuses on protein-based detection protocols, DNA-based detection strategies are also ubiquitous in the field of biosensors.10–15 Many recent signal amplification strategies use nanomaterials (NMs) such as carbon nanotubes (CNTs), graphene and nanoparticles (NPs); these strategies will be discussed in detail in the following sections of this review.
 | ||
| Fig. 1 Exponential growth of the number of articles published from 2000–2014 on electrochemical immunosensors (Source: http://www.Scopus.com). | ||
1.2 Classification and properties of nanomaterials
Recently, NMs have stimulated an unprecedented amount of research interest in biosensor development because of their impressive chemical and physical properties.16 For instance, NMs are used as “molecular wires” in electrochemical sensors to promote electron transfer because biomolecules are unable to communicate directly with electrodes. Because any scientific development requires classification, this subsection will provide a brief overview of the classification and properties of NMs widely used for signal enhancement through the transduction of biomolecular recognition and binding events.NMs are defined as materials composed of particles with at least one dimension ranging from 1–100 nm and possess characteristics that are remarkably different from those of their bulk counterparts.17 Classification of NMs does not strictly adhere to their size range but is based on their number of dimensions, with classes including: zero-dimensional (0D) spheres, particles, quantum dots (QDs) and clusters; one-dimensional (1D) needle-like nanorods, nanofibers and nanowires; two-dimensional (2D) films, plates, and networks; and three-dimensional (3D) graphite,18 as depicted in Fig. 2.19 In addition, 0D NMs, in which all three dimensions are less than 100 nm, are further grouped into magnetic, metallic, semiconductor and insulating NPs based on their conducting properties. As displayed in Fig. 2, 1D NMs are a lengthened form of NPs where one of the dimensions exceeds the nanoscale range. Examples of 1D NMs are nanotubes, nanorods and nanowires. Meanwhile, 2D NMs, which possess two dimensions greater than 100 nm, have sheet-like structures and include nanofilms, nanolayers and nanocoatings. In NMs with 3D structure, all three dimensions exceed 100 nm.20
 | ||
| Fig. 2 Molecular models of different types of sp2-like hybridized carbon nanostructures with different dimensionalities: (a) buckminsterfullerene (C60); (b) nested giant fullerenes or graphitic onions; (c) carbon nanotube; (d) nanocones or nanohorns; (e) nanotoroids; (f) graphene surface; (g) 3D graphite crystal; (h) Haeckelite surface; (i) graphene nanoribbons; (j) graphene clusters; (k) helicoidal carbon nanotube; (l) short carbon chains; (m) 3D Schwarzite crystals; (n) carbon nanofoams (interconnected graphene surfaces with channels); (o) 3D nanotube networks; (p) nanoribbon 2D networks.19 | ||
The method used to synthesize a NM will influence its properties, size, shape and chemical composition.21 Therefore, knowledge of fabrication procedures represents a cornerstone for further development and application of nanotechnology. NM production processes must be completely controllable and reproducible to attain the desired properties and performance of the final integrated biosensor devices.22,23 NMs can be fabricated using top-down and bottom-up strategies. The bottom-up approach is based on the congregation of atoms or molecules to assemble NMs. This strategy includes methods such as the sol–gel technique, self-assembly, chemical vapor deposition and template-assisted electrodeposition, all of which have been widely reported in nanotechnology literature. This is because the bottom-up approach produces nanostructured materials with few defects, homogeneous chemical composition and short- and long-range order. The bottom-up approach works principally by lowering the Gibbs free energy so the resulting NMs are in a state closer to thermodynamic equilibrium than the starting materials. Conversely, in the top-down strategy, NM formation involves the use of larger starting materials, which are scaled down to nanoscale size. This strategy includes methods like lithography, deposition, and etching, and the resulting NMs often contain surface defects and suffer from internal stress.24,25
Despite NMs exhibiting a plethora of outstanding properties, ultimately only two of their attributes—high surface area and excellent electrical conductivity—markedly affect their electrochemical performance because of their configuration.23,26 Therefore, the inclusion of NMs (such as CNTs, graphene, or nanowires) in electrochemical biosensors amplifies signal response via the following mechanisms:
1.3 Recent signal amplification strategies for electrochemical immunosensors
Electrochemical techniques are generally used in the development and design of innovative biosensors because of their ease of miniaturization, high sensitivity, low cost and compatibility with advanced microfabrication technology.27,28 Electrochemical processes commonly used to quantify the amount of an analyte of interest are cyclic voltammetry, potential step techniques like square-wave voltammetry and chronoamperometry, the rotating disk method and potentiometry. Comprehensive reviews of these techniques, excellent textbooks and specialist manuscripts on electrochemistry that deal with the theory and applications of analytical electrochemistry are available.29–32 Immunosensor signals are conventionally obtained using labels such as enzymes, electroactive molecules, redox complexes, and metal ions.28 The drawback of immunoassays that use this conventional approach is the low number of labels captured per biorecognition event, which results in low sensitivity. Thus, signal amplification has attracted considerable attention to realize highly sensitive immunosensors with low LOD.33 Many published reports on immunosensors have focused on creating innovative approaches that include integration of NMs in various amplification processes and transducer platforms to achieve low LOD. Without doubt, nanotechnology provides new signal enhancement strategies with the ability to use NMs as labels and produce NM-modified electrochemical transducers.27 Table 1 introduces recent studies that use NMs as their core strategy for signal amplification. Because NMs are widely used for signal enhancement based on different strategies, the following section describes label and label-free methods of detection using NMs. The present review focuses on CNTs, graphene-based materials and NPs because these NMs are the most commonly used in existing electrochemical immunosensors.| Nanomaterial | Electrode (E)/Label (L) | Detection method | Analyte | Limit of detection | Ref. |
|---|---|---|---|---|---|
| a CEA – carcinoembryonic antigen, AFP – alpha-fetoprotein, CA 125 – cancer antigen 125, CA 153 – cancer antigen 153, IgG – immunoglobulin G. | |||||
| Graphene | Monolithic and macroporous graphene (E) | Differential pulse voltammetry (DPV) | CEA | 90 pg mL−1 | 34 |
| Palladium–reduced graphene oxide (E) | DPV, amperometry | AFP | 700 pg mL−1 | 35 | |
| Graphene–polyaniline (E); horseradish peroxidase–graphene oxide–antibody (L) | DPV | Estradiol | 20 pg mL−1 | 36 | |
| Ionic liquid-gold NPs–graphene nanosheets (L) | Amperometry | Human apurinic/apyrimidinic endonuclease 1 | 0.04 pg mL−1 | 37 | |
| Metal nanoparticles (NPs) | Graphene-coupled quantum dots and gold NPs-labeled horseradish peroxidase (E) | Electrochemiluminescence (ECL) | Mercury(II) ion | 60 pg mL−1 | 38 |
| Poly(o-phenylenediamine)/gold (L) | DPV | CEA | 5.0 pg mL−1 | 39 | |
| Poly(vinyl ferrocene-2-aminothiophenol)gold (L) | DPV | AFP | 3.0 pg mL−1 | 40 | |
| Gold/silver/gold core/double-shell NPs (L); gold NPs–mercapto-functionalized graphene sheet (E) | Amperometry | Squamous cell carcinoma antigen | 0.18 pg mL−1 | 41 | |
| Copper-doped titanium dioxide NP (L); carboxyl-functionalized graphene oxide (E) | Square wave voltammetry, chronoamperometry | IgG | 0.052 pg mL−1 | 42 | |
| Mesoporous platinum NP (L) | DPV | CA 125 | 0.002 U mL−1 | 43 | |
| CA 153 | 0.001 U mL−1 | ||||
| CEA | 7.0 pg mL−1 | ||||
| Gold NP-modified graphene paper (E) | Impedimetry | Escherichia coli 0157:H7 | 150 CFU mL−1 | 44 | |
| Gold NPs–graphene–chitosan (E) | Cyclic voltammetry (CV) | CEA | 20 pg mL−1 | 45 | |
| Sodium nano-montmorillonite–polyaniline–gold NPs (L) | Amperometry | Squamous cell carcinoma antigen | 0.30 pg mL−1 | 46 | |
| Strepavidin functionalized silver NPs (L) | Linear sweep stripping voltammetry | AFP | 0.046 pg mL−1 | 47 | |
| Single-domain antibody-conjugated gold NPs (L) | Impedimetry | Clostridium difficile toxin A | 0.61 pg mL−1 | 48 | |
| Clostridium difficile toxin B | 0.60 pg mL−1 | ||||
| AuNPs–horseradish peroxidase (L) | LSV | Pantoea stewartii subsp. stewartii-NCPPB 44 | 7.8 × 103 CFU mL−1 | 49 | |
| AuNPs–Prussian blue, polyaniline/poly (acrylic acid) (E); Au-hybrid graphene nanocomposite (L) | Amperometry | Salbutamol | 40 pg mL−1 | 50 | |
| Ferrocene carboxylic acid–platinum NPs (L) | DPV | Procalcitonin | 6.0 pg mL−1 | 51 | |
| Polypyrrole film–Au nanocluster (E); functionalized gold nanorod (L) | CV | Ofloxacin | 30 pg mL−1 | 52 | |
| Chitosan-encapsulated silica NP hybrid film (E) | Potentiometry | Hepatitis B surface antigen | 3890 pg mL−1 | 53 | |
| Nano-gold modified planar gold electrode (E) | Potentiometry | Mouse IgG | 200 pg mL−1 | 54 | |
| Pyrolytic graphite sensor disk electrodes coated with gold NPs (E) | Rotating-disk electrode amperometry | NANOG | 0.1 pg mL−1 | 55 | |
| Prussian blue–gold hybrid nanostructure (L) | Linear sweep voltammetry | AFP | 40 pg mL−1 | 56 | |
| Carbon NPs (L) | Conductometry | Tissue polypeptide antigen | 0.28 pg mL−1 | 57 | |
| Carbon nanotubes (CNTs) | CNT/manganese dioxide (L) | Conductometry | AFP | 50 pg mL−1 | 58 |
| AuNPs–multiwalled carbon nanotubes (MWCNTs)–chitosan (E) | Amperometry | Ricin | 2100 pg mL−1 | 59 | |
| MWCNTs-chemically reduced graphene (E) | CV | IgG | 200 pg mL−1 | 60 | |
| MWCNTs–thionine–chitosan (E) | Amperometry | Chlorpyrifos | 46 pg mL−1 | 61 | |
| MWCNTs–poly(pyrrole propionic acid) (E) | Amperometry | Hormone insulin-like growth factor 1 | 30 pg mL−1 | 62 | |
| ZnO quantum dots dotted carbon nanotube (L) | ECL | Prostate specific antigen | 0.61 pg mL−1 | 63 | |
2. Nanomaterials as labels in electrochemical immunosensors
Electrochemical applications of NMs as amplifying tags can be categorized as follows:• as carriers for numerous signal molecules because of the large surface area and high loading capacity of NMs;
• as electroactive tracers because NMs are electrochemically active;
• to accumulate a large amount of sample molecules;
• as catalysts, which is attributed to the ready availability of active sites on their surfaces.
To illustrate the working principles of NMs as labels in signal amplification strategies and recent developments in this direction, selected examples are provided for each application strategy in the following sections.
2.1 Nanomaterials as nanocarriers
NMs act as excellent nanocarriers by loading and carrying numerous signal molecules, such as enzymes, organic dyes, oligonucleotides and electroactive compounds, because their large surface area can increase the number of signal molecules transported to the electrode surface for detection, resulting in a considerable amplification of electrochemical signal responses.64,65 Various NMs including gold nanoparticles (AuNPs), magnetic beads (MBs), multi-walled carbon nanotubes (MWCNTs), single-walled carbon nanotubes (SWCNTs), silica NPs, carbon spheres, graphene oxide, dendrimers and electroactive component-encapsulated NPs have been used as nanocarriers in electrochemical sensing. For example, detection signals have been amplified by using AuNPs to increase the loading of MBs in bioconjugate labels,66 an AuNP–graphene hybrid to increase antibody loading,67 reduced graphene oxide–tetrathylene pentamine to load Pb and Cu ions,68 and peptide nanowires to load electroactive ferrocene (Fc).69Horseradish peroxidase (HRP) has been extensively used as an enzymatic label in electrochemical studies and has been carried on a variety of NMs because it generates a sensitive electrochemical response through enzymatic catalytic reaction, as well as being inexpensive and convenient. Nevertheless, the practical application of HRP is hindered because of interference by dissolved oxygen reduction. Such interference can be resolved through the replacement of HRP with noble metal NPs such as AuNPs and AgNPs because trace amounts of metal ions can be electrochemically determined by stripping analysis at relatively positive potential range. Cheng and co-workers70 replaced HRP with AuNPs in a sandwich-based immunosensor using Au nanorods as a nanocarrier for loading of detection antibody and glucose oxidase. In the resulting nanobioprobe, glucose oxidase was used for catalytic deposition of AuNPs onto the Au nanorods. Amplified signal response was obtained based on simultaneous electrochemical stripping analysis of the captured Au nanorod carrier and the enzymatically produced AuNPs, allowing the sensitive detection of carcinoembryonic antigen (CEA).
For the determination of avian leukosis virus subgroup J, Ai and colleagues exploited the large surface areas of graphene quantum dots and apoferritin-encapsulated CuNPs.71 In their microcrystal encapsulation approach, a “supernova effect” of encapsulated electroactive compounds was initiated upon exposure to a releasing agent. Dissolution of core crystals released a large number of signal-generating molecules, which diffused across the capsule wall into the outer surroundings,72 resulting in an amplified signal. Graphene quantum dots (GQD) were incorporated into the sensor to increase the loading of both the antibodies and apoferritin-encapsulated CuNPs, and in turn, the apoferritin-encapsulated CuNPs increased the loading of electroactive species. This dual signal amplification strategy is illustrated in Fig. 3(a) and (b). The essential roles of graphene quantum dots and apoferritin-encapsulated CuNPs in this signal enhancement strategy are characterized by the voltammetry responses in Fig. 4. The immunosensor with graphene quantum dots displayed a larger current signal than that of the immunosensor without the quantum dots (ΔI = 19.96 μA). This phenomenon was attributed to the larger available space for conjugation of both apoferritin-encapsulated CuNPs and antibodies in the sensor with quantum dots. Interestingly, the apoferritin-encapsulated CuNP-based immunosensor exhibited a larger current peak than that of the sensor without apoferritin (ΔI = 47.74 μA). This was ascribed to the high loading capacities of apoferritin-encapsulated CuNPs allowing them to accommodate a large amount of electroactive redox species.
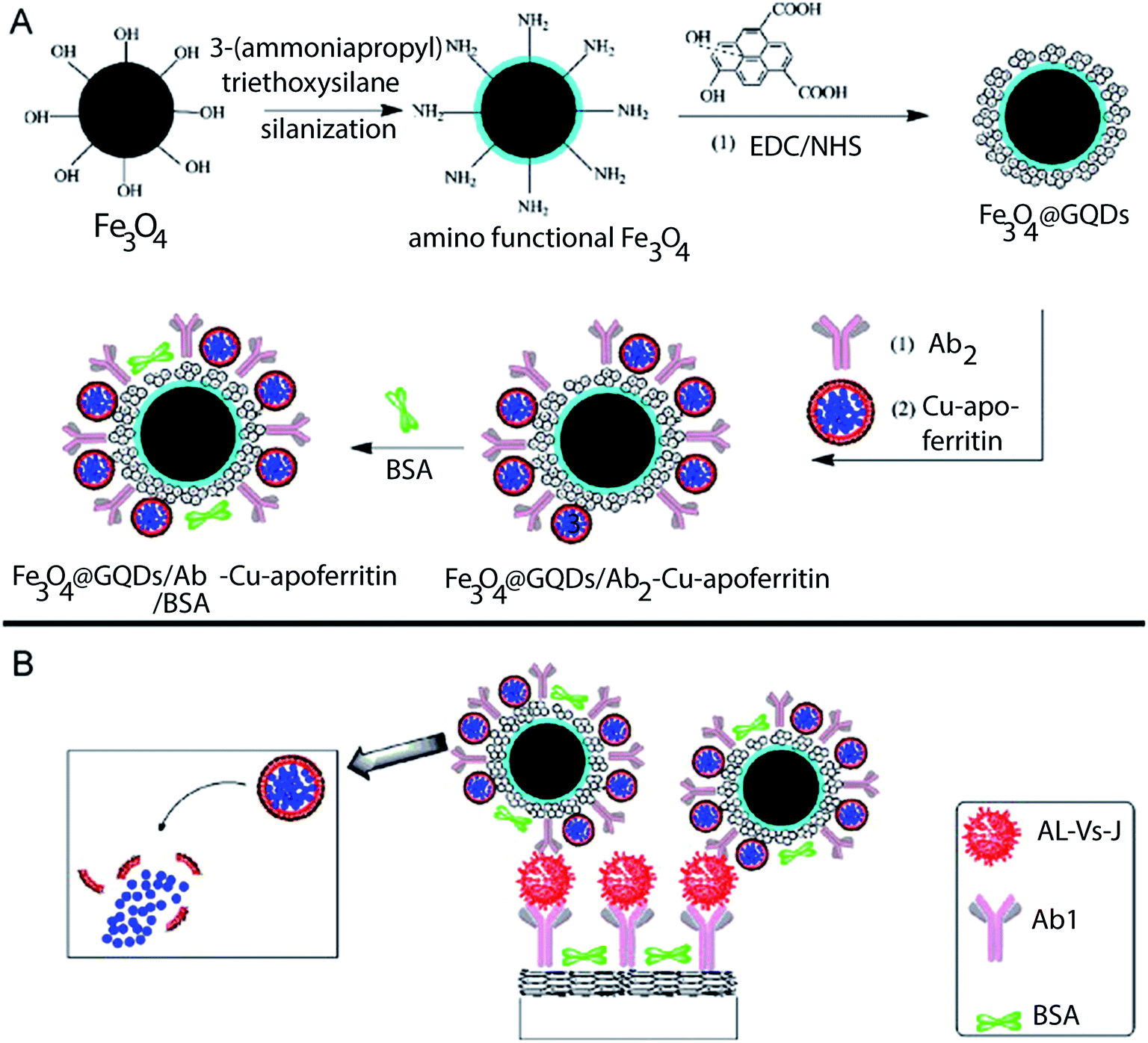 | ||
| Fig. 3 Schematic representations of (A) the preparation of Fe3O4@GQDs/Ab2–Cu-apoferritin/BSA and (B) immunosensing. EDC/NHS is (1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide)/N-hydroxysuccinimide, BSA is bovine serum albumin, and GQD is graphene quantum dots.65 | ||
 | ||
| Fig. 4 Differential pulse voltammetry responses of immunosensors with (upper curve) and without (lower curve) (A) graphene quantum dots and (B) apoferritin-encapsulated CuNPs.71 | ||
2.2 Nanomaterials as electroactive nanotracers
In electrochemical studies, NMs, particularly metal NPs, have been used as electroactive nanotracers. The NMs accumulate captured molecules and/or load signal molecules onto the electrode surfaces to increase the number of electroactive species present.73 Because metal NPs are highly stable, they need to be dissolved in acidic medium to produce metal ions and pre-concentrated by an anodic stripping-based electrochemical method for further analysis and quantification. This process generates thousands of detectable metal ions per biorecognition event and hence enhances electrochemical response.74Among noble metal NPs, AuNPs are the most commonly used for this type of signal amplification because of their simple fabrication, efficient bioconjugation, ready biofunctionalization and excellent stability.75–77 For example, Lim et al.78 used AuNPs to load a large amount of signal molecules. Upon conjugation of AuNPs with a detection antibody, a series of sandwich-type immunoreactions occurred. Then, an electrochemical response was generated by pre-oxidation of AuNPs in 0.5 M HCl at a high potential of 1.2 V for 40 s followed by immediate reduction of [AuCl4]− to Au0 and scanning in differential pulse voltammetry (DPV) mode (Fig. 5). This approach was based on the redox properties of the AuNPs in acidic medium, where target analyte human chorionic gonadotropin (hCG) in the sample was detected by quantifying the released Au ions. A decrease of DPV response was observed with increasing concentration of hCG in standard and phosphate-buffered saline (PBS) solutions. The reduction properties of Au ions to metallic Au in HCl were also investigated using a carbon screen-printed electrode (SPE) and graphene-modified SPE. Comparison of current signals for both types of electrodes revealed that the graphene-modified SPE exhibited a more intense reduction peak (19 μA) compared with that of the carbon SPE (1.7 μA).71 This showed that graphene promoted the reduction of AuNPs better than carbon because of its high surface area and outstanding electron transfer, which occurs primarily at the edge of graphene rather than at its basal plane. The immunosensor based on a graphene-modified SPE achieved a linear range from 0 to 500 pg mL−1 with a LOD of 5.0 pg mL−1.
 | ||
| Fig. 5 Schematic illustration of an electrochemical immunosensor system. (a) The primary antibody was immobilized on a graphene working electrode by physical adsorption. BSA was used to block the uncoated surface of the electrode. A sandwich-type immunoreaction was then performed. (b) Redox reaction was carried out at a high potential of 1.2 V for 40 s (termed pre-oxidation) in 0.5 M HCl to oxidize the AuNPs, which were immediately reduced and (c) scanned by differential pulse voltammetry from 1 to 0 V.78 | ||
Regardless of the sensitivity obtained by this strategy, one of its major disadvantages is high background signal because oxidation of AuNPs takes place in the potential region near to potential limit in aqueous electrolyte solutions. To alleviate this problem, AgNPs have become increasingly popular as nanotracers that can be oxidized at a more negative potential and produce a sharper peak than AuNPs.64 Furthermore, by controlling the deposition of Ag on electrode surfaces, the issue of dominating background signal can be completely circumvented.79
AuNPs have the ability to catalyze many reactions including chemical reduction of Ag ions to metallic Ag on the surface of AuNPs. Following the deposition of Ag metal onto the surface of AuNPs, quantitative analysis of Au can be carried out through the oxidation of Ag. In this scenario, AuNPs are enlarged with “silver-enhancing” solution, resulting in the formation of an Ag layer around them, and then electrochemically stripped and detected at a favorable potential range (Fig. 6).73 Another method to regulate the deposition of Ag is by controlling metal ion precipitation enzymatically. This method was recently used to detect major peanut allergen Ara h 1, a 7S vicilin-like globulin, in which the secondary antibody was labeled with alkaline phosphatase (AP) and electrochemical detection relied on enzyme-catalyzed metal precipitation followed by anodic voltammetric potential scanning.80 In principle, AP catalyzes the dephosphorylation of substrate 3-indoxyl phosphate to an indoxyl intermediate that reduces the Ag ions to metallic Ag. This process is confined to where the enzymatic label AP is attached. The enzymatically deposited Ag is then electrochemically stripped into solution and subsequently detected by anodic stripping voltammetry.79 Using this protocol, a wide linear range from 13 to 2000 ng mL−1 and LOD of 38 ng mL−1 were obtained.
 | ||
| Fig. 6 A three-step signal amplification strategy for ultrasensitive immunosensing of a cancer biomarker. Reprinted with permission from ref. 77. Copyright © 2012 American Chemical Society. | ||
To omit steps involving metal dissolution in acidic medium and pre-concentration to detect metal ions, metals ions can be directly attached to the surface of signal probes. For example, Feng et al.81 directly immobilized Zn2+ and Cd2+ on the surface of titanium phosphate nanospheres with excellent ion-exchange properties. More recently, Xu and co-workers82 further improved this approach by simplifying the strategy to immobilize metal ions on probes without the need to fabricate a template for ion-exchange to allow simultaneous detection of CEA and alpha-fetoprotein (AFP). They conjugated the detection antibodies on the surface of AuNPs through S–Au and NH–Au covalent bonds, producing NP surfaces with abundant amino groups. Next, Cu2+ and Pb2+ were separately absorbed on the NP surface through the interaction of the metal ions with the amino groups of antibody-conjugated colloidal AuNPs. The metal ion labels were then directly detected through DPV without metal pre-concentration. The observed well-defined voltammetric peaks had a close relationship with each sandwich-type immunoreaction. Simultaneous determination of CEA and AFP with linear ranges of 10–50 ng mL−1 were obtained with LODs for CEA and AFP of 5 pg mL−1 (calibration curve of y = 3.3x + 8.9) and 3 pg mL−1 (calibration curve of y = 4.9x + 14), respectively.
2.3 Magnetic nanomaterials as accumulators
The use of MBs in biosensors has shown excellent progress recently. Micro- or nanoscale MBs can be conveniently and immediately separated by an external magnetic field and used together with antibodies or proteins with a high affinity for the target. The functionalization and separation of MBs using magnetic forces can substantially improve the performance of not only immunosensors but also aptasensors (aptamer-based biosensors).83 An important issue in immunosensor development is renewal of the sensing surface, which is made more difficult by strong antigen–antibody interactions, and hinders integration of immunosensor into automatic systems.84 However, by using MBs as a reaction carrier, this problem of electrode fouling can be avoided. In addition, the performance of immunological reactions can be improved through the unique advantages of MBs.85 Signal amplification materials such as metal NPs have been integrated in MB-based immunosensors, where they function as signal tags to amplify signals.In 2011, Zhang et al.86 synthesized polyethyleneimine-functionalized MBs with electroactive thionine molecules and AuNPs alternately immobilized on their surface using an adsorption technique and in situ synthesis method, respectively. Thyroid-stimulating hormone was detected electrochemically on the AuNP-functionalized graphene sensing platform. In addition, HRP-labeled anti-thyroid-stimulating hormone antibodies were immobilized on the surface of AuNPs, which were used as signal tags for determination of the hormone with a sandwich-type immunoassay format. Effectively, the MBs aided the localization of the immunocomplexes on the electrode surface and increased the concentration of the enzyme as a tracer on the electrode, which resulted in a large electrochemical response amplified by an enzymatic reaction. The use of magnetic particles made a low LOD possible because a magnetic particle collection step was included in the assay to concentrate the sample.
2.4 Nanomaterials as nanocatalysts
A NM-based strategy that takes advantage of the excellent catalytic activity of NMs, this approach involves indirect quantification of nanocatalyst using an electrocatalytic system, which is a similar concept to the use of enzymes as redox labels.75,87 However, unlike enzymes, NM nanocatalysts possess numerous active sites on their surfaces. Therefore, when NMs are employed as catalysts, larger signals are generated per electrochemical reaction, enhancing signal response.88 CNTs and noble metal NPs composed of metals such as Au, Pt and Ag have been widely implemented to enhance electrocatalytic activity in electrochemical immunosensors.For instance, in the determination of procalcitonin, a biomarker for septicemia, a conjugate of single-walled nanohorns and hollow Pt chains was used as a label to amplify signal because of its excellent electrocatalytic activity with hydrogen peroxide (H2O2).89 Exploring the exceptional electrocatalytic attributes of MWCNTs in the reduction of H2O2, Li and colleagues synthesized AuNP-functionalized magnetic MWCNTs loaded with Pb2+ for the determination of AFP.90 They reported that the label with MWCNTs generated an enhanced signal compared with that of the label without MWCNTs, and observed further signal enhancement when Pb2+ and AuNPs were also present. The inclusion of these NMs resulted in multifaceted signal amplification of the reduction of H2O2 as an analytical signal with a LOD of 0.003 pg mL−1.
Immunosensors utilizing conductometry as detection method are associated with enzymatic-catalyzed reactions involving the change in conductivity of solution through the utilization or production of charged particles. Double-codified nanogold particles were utilized as secondary antibodies in signal amplification of conductometric immunosensor to detect hepatitis B surface antigen (HBsAg).91 Conjugating double-codified nanogold particles to secondary antibodies demonstrated much larger changes in conductometric signals (with reported LOD of 10 pg mL−1 HBsAg, estimated to be 3× the standard deviation of zero-dose response) than using those without nanogold particles (LOD of HBsAg reported to be 500 pg mL−1). This observation was due to the large surface area of nanogold particles that could accommodate large amount of immobilized secondary antibody, which increased the possibility the antigen–antibody interaction and also the bioelectrocatalytic reaction of the immobilized HRP that amplified the conductometric signal response.
Of late, research interest has turned towards the collision of individual NPs pioneered by Bard and colleagues,92 who explored the electrocatalytic properties of a single PtNP and bare ultramicroelectrode. Their work was based on the high current amplification involved in a rapid electrocatalytic reaction with single NP collisions. Inspired by this excellent study, Castañeda and co-workers93 went a step further by modifying an ultramicroelectrode with a passivating polyelectrolyte multilayer (PEM) through layer-by-layer assembly and detecting the amplified current achieved by electrocatalysis between the negatively charged Pt NPs and PEM. Layer-by-layer assembly, an effective procedure developed by Decher, allows simple fabrication of multilayer films.94,95 The ionic attraction between oppositely charged molecules is the primary driving force involved as each layer of film being attached is exposed to polycationic and polyanionic solutions to fabricate a film with individual layers at desired positions. It was observed that by changing the layer number of the PEM, and thus reversing the charge, the current could effectively be turned on and off. Based on these innovations, it would be interesting to observe further development of this strategy for future electrochemical sensing applications.
2.5 Multi-step enhancement strategies
Many recent enzyme-based immunosensor studies have reported signal amplification by multiple steps using HRP, glucose oxidase and alkaline phosphatase (AP). This system requires a redox mediator, because these enzymes are not directly involved in electron exchange with the electrodes.96 The fundamental concept of multi-step amplification strategies is to increase the amount of immobilized antibodies on secondary enzyme-labelled antibodies to improve detection sensitivity.Tang et al.87 and Malhotra et al.97 reported NM-based multi-enzyme amplification strategies that use substrate recycling protocols. In their substrate recycling strategies, signal amplification is markedly improved because the shuttle analyte (e.g., enzyme substrate) is measured repeatedly with the use of an oxidizing or reducing agent (chemical approach), oxidation or reduction of the substrate on the electrode surface (electrochemical recycling), and/or enzymes (enzymatic recycling).98,99
Recently, a novel electrochemical immunoassay protocol based on catalytic recycling of product to determine apurinic/apyrimidinic endonuclease (APE-1) using a three-step signal amplification process was published.100 The first step of this process involved the biocatalysis of ascorbic acid 2-phosphate (AA-P) to produce ascorbic acid (AA) in situ by labelled biotinylated alkaline phosphatase (bio-AP) on nickel hexacyanoferrate NP-decorated Au nanochains (Ni–AuNCs). By subsequent electrochemical oxidization of the AA produced in situ by the Ni–AuNCs, the signal was additionally enhanced. Using nanochain-modified streptavidin (SA), the stoichiometry of bio-AP was further improved through the well-known specific and high-affinity interaction of SA and biotin (Fig. 7). This strongly amplified the generated signal. This three-step amplification approach showed a wide linear range of 0.01–100 pg mL−1 with a remarkably low LOD of 0.004 pg mL−1 (signal/noise = 3). The important role of each component in the three-step amplification protocol was evaluated using different labelled bioconjugates. The results revealed that bio-AP/SA/Antibody2 (Ab2)/Ni–AuNC labels yielded the highest current when compared with Ni–AuNC-labelled Ab2 and bio-AP/Ab2/Ni–AuNCs lacking the three types of signal amplification (Fig. 8). The current response was increased ten-fold for the label with NiNPs compared with that of the label without NPs, which confirmed the role of NiNPs as a nanocatalyst. An aromatic compound (denoted as PTC-NH2) prepared by reaction of 3,4,9,10-perylene tetracarboxylic dianhydride (PTCDA) and ethylenediamine was employed as the electrode material because of its amino-functionalized interface and low electrochemical background current.
 | ||
| Fig. 7 Preparation of an immunosensor with a three-step signal amplification mechanism. (A) Stepwise fabrication of the bio-AP/SA/Ab2/Ni–AuNC bioconjugate: (a) absorption of NiNPs, (b) Ab2 loading, (c) blocking with SA, and (d) binding bio-AP. (B) Molecular structure of an aromatic compound (denoted as PTC-NH2) formed by reaction of 3,4,9,10-perylene tetracarboxylic dianhydride (PTCDA) and ethylenediamine.100 | ||
 | ||
| Fig. 8 Cyclic voltammograms obtained after the sandwich immunoreaction of the immunosensor with various Ab2 bioconjugates: (A) BSA/Ab2/Ni–AuNCs, (B) bio-AP/Ab2/Ni–AuNCs, (C) bio-AP/SA/Ab2/Ni–AuNCs with APE-1 concentrations of (a) 0, (b) 20, (c) 40 and (d) 100 pg mL−1, and (D) cyclic voltammograms obtained using the immunosensors with (a) AuNCs and (b) Ni–AuNCs when incubated with 100 pg mL−1 APE-1. All voltammograms were measured in the presence of 5.0 mM AA-P.92 | ||
However, practical applications of these NM-based multi-enzyme probes are still restricted because the sensitive nature of enzymes results in problems such as leakage, denaturation and high cost. Furthermore, non-enzymatic sensors are preferred over enzymatic ones by some research groups because of their enhanced sensitivity and lower LOD.101 To overcome the limitations originating from the fragility of enzyme-based immunosensors, researchers have been experimenting using electroactive reagents as labels in place of enzymes. For example, Zhao et al.102 incorporated Au–PdNPs to catalyze the reduction of H2O2 in their enzyme-free immunosensor system along with graphene as their sensing platform for the detection of AFP. Using hybrid Au–PdNPs had increased the effective surface area for biomolecule conjugation. The sensor achieved a LOD of 5 pg mL−1 with a linear range of 50 pg mL−1 to 30 ng mL−1.
To achieve highly sensitive catalytic amplification, many researchers have reported strategies to recycle redox substrates to ensure a continuous increase of signal intensity. For instance, a system to detect CEA based on a ‘one-to-many’ autocatalytic strategy using thionine–cerium oxide organic–inorganic hybrid nanostructures (Thi–CeO2) as a nanocatalyst was reported recently.96 The ‘one-to-many’ autocatalytic strategy involved the chemical catalytic recycling of the self-produced reactant between AA and dehydroascorbic acid (DAA), producing an amplified electrochemical signal. The immunosensor worked by autocatalyzing the hydrolysis of the phosphate ester bond of AA-P by CeO2 NPs to produce AA as a new reactant. The produced AA was then electro-oxidized to DAA by the assembled thionine in Thi–CeO2. DAA then reduced back to AA by tris(2-carboxyethyl)phosphine. This sensor realized a linear range of 0.1 pg mL−1 to 80 ng mL−1 with a LOD of 0.08 pg mL−1 for CEA.103
Table 2 summarizes the roles of NMs in selected reported electrochemical immunosensors and compares the enhancements achieved by NMs under identical experimental conditions. Although label-based electrochemical amplification strategies are extremely sensitive with detection of target down to femtogram level and able to detect more than one analyte simultaneously, their complicated fabrication, high assay cost and long analysis time hinder their practical application.104
| Electrochemical application | Analyte | Label | Electrode | Detection method | Signal amplification strategy | Comparison of enhancement with nanomaterial, hybrid nanomaterial and without nanomaterials (under identical experimental conditions) | Limit of detection (LOD) | Linear range | Ref. | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| With NM/NM hybrid | Without NMs/single NM | Reason for enhanced signal | |||||||||
| a CEA – carcinoembryonic antigen, AFP – alpha-feto protein, HER-2 – human epidermal growth factor receptor type-2. | |||||||||||
| Nanocarrier | CEA | Au nanorods, Au nanoparticles (NPs) | Carbon nanotube (CNT)-modified screen-printed electrode | Stripping DPV | Antibody and glucose oxidase (GOD) loaded on Au nanorods. GOD was further used for catalytic deposition of AuNPs onto Au nanorod. Stripping analysis of the Au nanorod carrier and enzymatically produced AuNPs allowed sensitive detection | CNT-modified screen-printed electrode showed a three-fold increase in current signal compared with that of a carbon screen-printed electrode | High conductivity of CNTs | 4.2 pg mL−1 | 10–100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 pg mL−1 000 pg mL−1 |
70 | |
| Nanocarrier | CEA | 3,3′,5,5′-Tetramethylbenzidine (TMB) enzyme on magnetic beads (MBs) | AuNPs deposited on polydopamine film | Differential pulse voltammetry (DPV) | Electrocatalytic oxidation of ascorbic acid by TMB enzyme label after competitive binding between MB/TMB-conjugated-CEA and free-CEA | CEA/MB/TMB gave peak separation of 0.16 V and demonstrated irreversibility because of the large electron transfer distance between the electrode and the MB-supported TMB molecules | CEA/TMB gave peak separation of 0.6 V | High loading of TMB enzyme labels on MBs | 1.0 pg mL−1 | 1–10000 pg mL−1 |
71 |
| Nanocarrier | Thyroxine | Magnetic graphene spheres | CNTs/Nafion on glassy carbon | DPV | Cascade catalysis involving GOD catalyzing oxidation of glucose to generate H2O2, which can then be further catalyzed by cytochrome c (Cyt c) | Cyt c and GOD carried on graphene spheres produced a current response of Δ20.9 μA in the presence of 5 ng mL−1 of analyte | Without graphene spheres, cascade catalysis gave a current response of Δ6.7 μA at 5 ng mL−1 of analyte | Large surface area, high redox activity and the loading numerous detection antibodies by the graphene spheres | 0.015 pg mL−1 | 0.05–5000 pg mL−1 | 102 |
| Nanocatalyst | CEA | GOD–Au–Ag mesoporous NPs (anti-CEAAuAgHS-GOD) | Graphene/Prussian blue (PB) catalase on glassy carbon | DPV | Dual amplification strategy by catalytic recycling of product paired with GOD and PB artificial catalase. AgNPs and AuNPs catalyzed reduction of H2O2 produced by GOD and then catalytically reduced by PB on graphene nanosheet for second amplification | The anti-CEA-AuAgHS-GOD label was 1.42 times more sensitive than anti-CEA-AuHS-GOD and 1.09 times more sensitive than anti-CEA-AgHS-GOD | Higher conductivity of AgNPs than AuNPs, and high catalytic ability of AgNPs towards reduction of H2O2 | 1.0 pg mL−1 | 5–50000 pg mL−1 |
85 | |
| Nanocarrier and nanocatalyst | CEA | Mesoporous carbon foam (MCF) and AuNPs | Electrochemically reduced graphene oxide/chitosan film on glassy carbon | Stripping DPV | Electrochemical stripping of Ag based on enlargement of gold NPs with Ag | Antibody/Au/MCF gave current peak 64.2 μA at 0.1 ng mL−1 of CEA | Antibody/MCF gave current peak of 32.8 μA at 0.1 ng mL−1 of CEA | MCF strongly catalyzed Ag deposition to produce AgNPs on sensor because of its abundance of surface carboxyl groups | 0.024 pg mL−1 | 0.05–1000 pg mL−1 | 105 |
| Nanocatalyst | AFP | Pt hybrid mullti-walled CNT–copper oxide NPs (Pt@CuOMWCNT) | B-cyclodextrin-functionalized graphene on glassy carbon | Amperometry | Catalytic reduction of H2O2 in enzyme-free amplification strategy | Pt@CuOMWCNT gave a signal response two times higher than that of CuO–MWCNT | CuO–MWCNT provided a signal response 20 times higher than that of MWCNTs alone | High catalytic activity based on synergistic effect of Pt@CuOMWCNT towards reduction of H2O2 | 0.3 pg mL−1 | 1–20000 pg mL−1 |
103 |
| Nanocarrier and nanocatalyst | AFP | CNTs/manganese dioxide (CNT/MnO2) | Nanogold/chitosan film on glassy carbon | Linear sweep voltammetry | High catalytic reduction performance of H2O2 by manganese dioxide. Inclusion of CNTs increased surface area to allow high loading of biomolecules | CNT/MnO2 demonstrated higher anodic current than MnO2 alone with a current ratio of 87 | MnO2 alone provided a current ratio of 36 | Large surface area of CNTs allowed conjugation of numerous biomolecules and MnO2 NPs | 40 pg mL−1 | 200–100000 pg mL−1 |
106 |
| Accumulator and nanocarrier | CEA | Magnetic mesoporous NiCo2O4 nanosheet | Electrodeposited nanogold on glassy carbon | DPV | Excellent adsorption properties of the magnetic mesoporous NiCo2O4 nanosheet. The interlayer of Nafion/thionine organic molecules and nanogold allowed attachment of a large amount of horseradish peroxidase-labeled secondary anti-CEA antibody | Magnetic mesoporous NiCo2O4 nanosheets gave a LOD 10-fold lower than that of solid NiCo2O4 | Accumulation and pre-concentration of sample by the magnet, and the high surface area of magnetic mesoporous NiCo2O4 loaded a large amount of biomolecules | 0.5 pg mL−1 | 5–160000 pg mL−1 |
107 | |
3. Nanomaterials as electrode materials
Despite the high sensitivity of label-based sensors arising from the excellent characteristics of the labels, label-free methods are still preferred by many research groups because of the limitations of label-based sensors discussed in Section 2.5. Lately, interest in label-free immunosensors has surged in a drive to simplify the fabrication of immunosensors without compromising sensitivity. In label-free immunosensors, the change in surface properties after the formation of an antigen–antibody complex is detected as a transducer signal. Thus, label-free techniques rely on detection of potential change upon the formation of an immunocomplex on the surface of the electrode.108 Additionally, a new attractive research direction is the use of nanocomposites as a label-free strategy to enhance the signal from immunocomplexes. NMs can act as effective electrochemical sensing platforms because their large surface area increases mass transport, provides excellent loading capacity for receptor molecules to allow synergistic amplification of the target response, in addition, the unique biocompatible, electronic, and catalytic properties of NMs aid the translation of molecular biorecognition interactions to an electrochemical signal output.109,110 One popular approach is the inclusion of carbon NMs, namely CNTs and graphene, and metallic NPs in sensor electrodes.111,1123.1 Carbon nanotube-based electrodes
Apart from the high availability of reactive carboxyl groups that can be conveniently and efficiently functionalized and conjugated to proteins and biomolecules, an additional advantage of CNT-based materials for electrode fabrication is their inherent potential to improve catalytic redox reactions by facilitating electron transfer at low overpotential, which reduces the likelihood of electrode fouling. Studies have demonstrated that the outstanding electron transfer properties of CNTs arise from the curved structure of the tubes, which shifts the energy bands close to the Fermi level.113Voelcker and co-authors designed an elegant system exploiting the magnetic properties of iron oxide NPs (FeNPs).114 They functionalized FeNPs with SWCNTs to obtain composites that served as both an immobilization platform and magnetic immunocarrier for the detection of MS2 bacteriophage (Fig. 9). Using an external magnet, the FeNP–SWCNT composite was captured on the electrode surface and then electrografted using diazonium salt to form an antibody-modified SWCNT–NP composite. This strategy effectively eliminates the need for electrode modification. The researchers also presented another strategy in which SWCNTs were covalently coupled with a cysteamine-modified Au electrode via amide bond formation.114
 | ||
| Fig. 9 Preparation of sensing platforms based on (A) covalently bound SWCNTs and (B) SWCNTs used as magnetic immunocarriers.114 | ||
3.2 Nanoparticles in electrode fabrication
To provide a stable platform and facilitate electron transfer, metal NPs are commonly included in electrodes. Decher and co-workers demonstrated the simple fabrication of multilayer films from aqueous anionic and cationic polyelectrolytes on solid supports.95 They observed minimal changes in the adsorption behavior of the deposited films for at least 100 consecutive alternating layers. This technique was used by Liu et al.115 to assemble zinc selenide quantum dots (ZnSe QDs)/Azure I/AuNP/poly(3,4-ethylenedioxythiophene) (PEDOT) on Pt electrodes for detection of AFP. The AuNP–PEDOT composite provided a stable platform to facilitate electron transfer while the water-soluble ZnSe QDs were used to immobilize antibody and hence enhance the loading of anti-AFP. Azure I was chosen as the redox mediator because it can readily adsorbed onto the surface of the AuNP–PEDOT composite films through π–π stacking interactions. HRP was used to prevent nonspecific binding and its electrocatalytic activity for H2O2, which amplified the current signal of the antigen–antibody reaction. This system exhibited a low LOD of 0.011 pg mL−1 (signal/noise = 3) and a linear range from 5 pg mL−1 to 250 ng mL−1.To promote the electrochemical reaction process and thus amplify the signal response, it is critical to consider the roughness of the electrode surface: greater surface roughness gives enhanced electrochemical activity.101 A commonly used direct procedure to increase surface roughness is direct electrodeposition of metal NPs onto electrode surfaces, creating a more favorable microenvironment for the attachment of biomolecules.116 AuNPs are widely used for this role because of their outstanding biocompatibility with antigens and antibodies.117 For example, a system developed for simultaneous detection of CEA and AFP achieved LOD of 0.7 pg mL−1 (y = 39 + 12x) and 0.9 pg mL−1 (y = 50 + 16x), respectively (signal/noise = 3).118 This system used a reduced graphene oxide/thionine/AuNP nanocomposite as a supporting matrix for anti-CEA immobilization and reduced graphene oxide/Prussian blue/AuNP composite to immobilize anti-AFP on an indium tin oxide electrode. Nevertheless, it should be highlighted that because non-enzymatic CNT/metal nanocomposite-based sensors need to be at basic pH so hydroxyl groups can form higher oxides, such as Ni/CuOOH, their practical use in blood samples of either neutral or acidic pH remains a challenge.101
3.3 Nanomaterials as both labels and electrode materials for signal amplification
In Section 3.1 and 3.2, the main roles of NMs in electrochemical immunosensors as signal tags and in electrode fabrication for amplified signal recognition were described. Rather than using NMs for a single role, numerous reports have focused on the use of NMs as both labels and transducer platform materials to obtain sensors with enhanced sensitivity and performance. Here, a few recent representative studies are introduced as examples.Very recently, Li et al.119 obtained an amplified signal response using ultrathin Au nanowires incorporated with graphene oxide on a 3-D microfluidic paper-based electrochemical transducer. In their work, CuS was loaded on the surface of graphene oxide as a signal tag for AFP detection. They ascribed their amplified signal to the high CuS loading of the CuS–graphene oxide composite resulting from the presence of numerous oxygen-containing groups on the graphene sheets. The high CuS loading provided abundant sites for reduction of H2O2. Also, the high conductivity of graphene oxide contributed to the amplified electrochemical response because it facilitated charge transfer. To demonstrate the superior sensing ability of Au nanowires, graphene oxide and Au nanowire–graphene oxide hybrid transducer platforms were constructed and their ability to sense AFP antigen compared. The Au nanowire–graphene oxide hybrid sensor displayed enhanced sensitivity compared with that of graphene oxide alone because of its better biocompatibility through inclusion of additional surface functionalities, and high solubility and conductivity.
Of late, many reports have used Fc as an electroactive label. Fc-based derivatization is useful in analytical chemistry because of its well-established chemistry. Fc contains an iron ion that can be reversibly oxidized, which makes it attractive for use in electrochemical immunosensors.120 To enhance the signal amplification potential of Fc, various groups have used different materials, such as AuNPs, magnetic NPs, and dendrimers, to anchor numerous Fc moieties.121–123
Li et al.121 prepared dopamine-functionalized Fe3O4 and conjugated it with ferrocene carboxylic acid (FC) and secondary antibody. In their electrochemical immunosensor, the as-prepared Fe3O4 was used to detect PSA and graphene sheet was employed as a sensor platform. Signal amplification was achieved through the large amount of dopamine molecules loaded on the Fe3O4 surface enhancing the immobilization of FC and antibody on the Fe3O4 NPs. As a biosensor platform, the graphene sheets contributed to signal amplification in two ways: (i) their large surface area helped to capture numerous primary antibodies; and (ii) their good conductivity improved the detection sensitivity of FC. Using the redox amperometric current of FC as the signal, the immunosensor displayed a linear range of 0.01–40 ng mL−1 and low LOD of 2 pg mL−1.
However, Gao and Cranston124 stated that electroactive labels may not be the most sensitive labels because each electroactive label is only able to generate one electron during oxidation. They thus suggested the use of an electroactive polymer, each molecule of which is able to generate numerous electrons during electrochemical oxidation, to assist in electrochemical signal amplification and improve detection sensitivity. As proof-of-principle, polytyrosine was used conjugated with a PSA peptide and used as an electroactive signal tag in competitive electrochemical immunoassays. During oxidation, the phenol group of tyrosine lost two protons and two electrons. With a molecular weight of 10000–40000 Da (i.e., approximately 50–200 tyrosyl residues), one molecule of polytyrosine would produce 100–400 electrons when oxidized completely on an electrode surface. When conjugated to an antibody or antigen, this would dramatically enhance the electrochemical signal. MWCNTs were used as a transducer to facilitate immobilization of primary antibodies and electron transfer. The sensor obtained a LOD for PSA peptide of approximately 1 nM. Table 3 summarizes different roles of NMs in signal enhancement strategies.
| Electrochemical role | Nanomaterials | Amplification strategy | Analyte | Limit of detection | Ref. |
|---|---|---|---|---|---|
| a CEA – carcinoembryonic antigen IgG – immunoglobulin G PSA – prostate specific antigen AFP – alpha-fetoprotein. | |||||
| Electroactive tracer | Au nanoparticles (NPs) graphene | A triple signal amplification strategy combining AuNP-catalyzed Ag deposition for anodic stripping signal amplification with graphene used as electrode for rapid electron transfer | CEA | 0.12 pg mL−1 | 77 |
| Silica nanospheres AgNPs | Signal based on impedance or inhibition effect by target antigen. Conjugation of primary antibody to silica nanospheres amplified signal inhibition of electrochemical stripping signal of the AgNP–chitosan nanocomposite | IgG | 0.7 pg mL−1 | 125 | |
| Multistep-enhancement strategies | AuNP-modified Prussian blue onion-like mesoporous graphene sheets (Au@PBNPs/O-GS) | Dual amplification by catalysis of the ascorbic acid 2-phosphate to produce ascorbic acid in situ, and oxidation of ascorbic acid catalyzed by Au@PBNPs/O-GS and Au@NiNPs/O-GS nanohybrids, respectively, to obtain the higher signal responses | PSA | 7 pg mL−1 | 100 |
| AuNP-modified nickel hexacyanoferrate NP-decorated onion-like mesoporous graphene sheets (Au@NiNPs/O-GS) | Free prostate specific antigen | 3 pg mL−1 | |||
| AuNPs | Aggregation of many nanocatalysts (AuNPs) on one nanolabel (CNT) increased the number of nanocatalyst particles. Detection was based on catalytic reduction of p-nitrophenol to p-aminophenol by AuNPs on the CNT–AuNPs with subsequent redox cycling of p-aminophenol and p-quinone imine | AFP | 0.001 pg mL−1 | 105 | |
| Carbon nanotube (CNT) | |||||
| Transducer platform fabrication | Multi-walled CNT (MWCNT) | MWCNT-modified glassy carbon electrode followed by electropolymerization of poly(pyrrole propionic acid) | Hormone insulin-like growth factor 1 | 30 pg mL−1 | 126 |
| AuNPs | Incorporation of hybrid nanoparticles in electrode fabrication | Nuclear matrix protein 22 | 3 pg mL−1 | 127 | |
| PtNPs | |||||
| Au–Ag–graphene hybrid nanosheet | Signal amplification based on physical characteristics of AgNPs but acquiring the surface chemistry of AuNPs used in electrode fabrication. This was attributed to AgNPs being able to occupy the interspace between AuNPs because of the smaller relative size of AgNPs than AuNPs, which facilitated electron transfer | AFP | 0.5 pg mL−1 | 128 | |
| Sodium dodecylbenzene sulfonate-functionalized graphene sheets | Use of ionic liquid as a modifier because of its high ionic conductivity and good biocompatibility with biomolecules to increase signal output | Salbutamol | 7.0 pg mL−1 | 129 | |
| Pd NPs in functionalized mesoporous silica | |||||
4. Practical applications and future perspectives
The use of nanotechnology in the field of biosensors has allowed development of innovative electrochemical immunosensors, and opened up endless possibilities for signal amplification strategies. The incorporation of NMs in immunosensors has resulted in unprecedented success in improving sensing performance. NMs have been used as labels to increase loading of signal entities and enhance electron transfer. In addition, NMs have been incorporated in electrodes to dramatically increase the surface area of electrochemical transducers with the ultimate objective of achieving higher sensitivity. High selectivities and recoveries have been obtained in real sample analysis using NM-based sensors. In biomedical settings, ultrasensitive biosensors show huge potential in the early detection of tumor and cancer biomarkers. Their successful application to real samples will undoubtedly have an immense effect on this field.However, despite the sophisticated sensing principles and signal amplification strategies with impressive LOD being reported, their feasibility in clinical and field settings requires consideration of reliability and cost. For example, although AuNPs and graphene have been demonstrated to be excellent materials in point-of-care devices, they may not be cost-effective for practical usage. Thus, smooth transition from academic research and development to affordable products requires more time and investment. The practical usefulness of reported strategies in real-life situations is also limited because of their complicated fabrication procedures, and issues with instability and reproducibility. The main contributing factor to failure in the development and practical application of NM-based sensors is the sensor-to-sensor variability of their properties and ultimately their analytical performance. This variability results from fluctuation of the electrical, chemical and mechanical properties of fabricated NMs, such as contact resistance and graphene configuration on metal electrodes. For instance, in the synthesis of metal NP/CNT nanohybrids, it is highly desirable to fabricate well-dispersed, uniformly small metal NPs on CNT surfaces. However, it is a still challenge to maintain the original configuration and properties of CNTs and at the same time introduced groups onto CNTs via functionalization. The size, shape, structure, dispersibility and stability of NPs on CNTs can all affect CNT properties. In the case of nanowire fabrication, the inability to control the number of nanowires incorporated into a sensor and their diameter causes variation of sensing performance. Furthermore, the impressive sensitivity achieved by reported electrochemical immunosensors was obtained under optimal conditions in a laboratory; these sensitivities cannot be directly transcribed to real biological samples.
Consequently, a great deal of research still needs to be performed to eliminate matrix interference in real-sample measurements with different biological microenvironments, shorten incubation times for real-time detection of analytes, and simplify fabrication procedures. Nevertheless, with the successful development of biosensors at a laboratory scale, the opportunity to further improve them for actual field applications remains. Although NM-based biosensors have been proven to be propitious for sensitive detection, for them to be commercially successful, their sensitivity and stability in detection of real samples that are free from matrix interference still need to be evaluated.
Acknowledgements
The authors thank Graduate Research Scholarship of Universiti Brunei Darussalam for financial assistance. S.A. Lim expresses gratitude to the Ministry of Education, Brunei Darussalam for the opportunity to undertake a PhD course at Universiti Brunei Darussalam.References
- International Union of Pure and Applied Chemistry, Technical Report, 1999, vol. 71, 12, pp. 2333–2348, DOI:10.1351/pac199971122333.
- R. Reilly and T. Lee, Tech. Health Care, 2011, 19, 285–293 Search PubMed.
- R. Luppa, L. Sokoll and D. Chan, Clin. Chim. Acta, 2011, 314, 1–26 CrossRef.
- S. Ripp, M. Diclaudio and G. Sayler, in Environmental Microbiology, ed. R. Mitchell and J. Gu, John Wiley & Sons, Inc, Hoboken, NJ, USA, 2nd edn, 2010, vol. 9, pp. 213–233 Search PubMed.
- M. Ahmed, M. Hossain, M. Safavieh, Y. Wong, I. Rahman, M. Zourob and E. Tamiya, Crit. Rev. Biotechnol., 2015 DOI:10.3109/07388551.2014.992387.
- M. U. Ahmed, I. A. Saaem, P. C. Wu and A. Brown, Crit. Rev. Biotechnol., 2014, 34, 180–196 CrossRef PubMed.
- M. U. Ahmed, M. M. Hossain and E. Tamiya, Electroanalysis, 2008, 20, 616–626 CrossRef CAS.
- M. Safavieh, S. Nahar, M. Zourob and M. Ahmed, in High Throughput Screening for Food Safety Assessment, in Biosensor Technologies, Hyperspectral Imaging and Practical Applications, ed. A. K. Bhunia and S. K. Moon, Woodhead Publishing, Cambridge, Waltham, Kidlington, 1st edn, 2014, vol. 15, pp. 327–350 Search PubMed.
- M. Ahmed, A. Brown and P. Wu, in Genomic and Personalized Medicine, ed. G. A. Willard, Elsevier, London, Waltham, San Diego, 2nd edn, 2013, vol. 32, pp. 372–380 Search PubMed.
- S. Nahara, M. Ahmed, M. Safavieh, A. Rochette, C. Toro and M. Zourob, Analyst, 2014, 140, 931–937 RSC.
- M. Safavieh, M. Ahmed, A. Ng and M. Zourob, Biosens. Bioelectron., 2014, 15, 101–106 CrossRef PubMed.
- M. Safavieh, M. Ahmed, E. Sokullu, A. Ng, L. Braescu and M. Zourob, Analyst, 2014, 139, 482–487 RSC.
- C. Tlili, E. Sokullu, M. Safavieh, M. Tolba, M. Ahmed and M. Zourob, Anal. Chem., 2013, 85, 4893–4901 CrossRef CAS PubMed.
- M. U. Ahmed, S. Nahar, M. Safavieh and M. Zourob, Analyst, 2014, 138, 907–915 RSC.
- M. U. Ahmed, Y. Yoshimura, M. M. Hossain, E. Tamiya and K. Fujimoto, BioChip J., 2011, 5, 206–221 CrossRef CAS.
- Q. Liu, Q. Zhou and G. Jiang, Trends Anal. Chem., 2014, 58, 10–22 CrossRef CAS.
- J. Wang, A. J. Wain, X. Zhu and F. Zhou, in Biosensing using Nanomaterials, ed. A. Merkoçi, John Wiley & Sons, New Jersey, 2009, vol. 4, pp. 97–135 Search PubMed.
- S. Sagadevan, Rev. Adv. Mater. Sci., 2013, 34, 44–61 CAS.
- M. Terrones, A. R. Botello-Méndez, J. Campos-Delgado, F. Lópex-Urías, Y. I. Vega-Cantú, F. J. Rodríguez-Macías, A. L. Elías, E. Muñoz-Sandoval, A. G. Cano-Márquez, J. C. Charlier and H. Terrones, Nano Today, 2010, 5, 351–372 CrossRef.
- V. V. Pokropivny and V. V. Skorokhod, Mater. Sci. Eng., C, 2007, 27, 990–993 CrossRef CAS.
- P. Suman and M. Orlandi, J. Nanopart. Res., 2010, 13, 2081–2088 CrossRef.
- I. Gebeshuber, D. Fu and J. Junas, in Encyclopedia of Nanoscience and Society, ed. D. Guston, Sage Publications, Thousand Oaks, CA, 2010, pp. 495–499, ISBN: 9781412969871 Search PubMed.
- C. Justino, T. Rocha-Santos, S. Cardoso and A. Duarte, TrAC, Trends Anal. Chem., 2013, 47, 27–36 CrossRef CAS.
- S. Logothetidis, in NanoScience and Technology, ed. S. Logothetidis, Springer, Heidelberg, Dordrecht, London, New York, 2012, vol. 1, pp. 1–20 Search PubMed.
- G. Cao and Y. Wang, Nanoscience and Nanotechnology in World Scientific Series, ed. F. Spaepen, Imperial College Press, London, 2nd edn, 2012, p. 2, ISBN: 978-981-4322-50-8 Search PubMed.
- M. García, P. Batalla and A. Escarpa, Trends Anal. Chem., 2014, 57, 6–22 CrossRef.
- G. Liu and Y. Lin, Talanta, 2007, 74, 308–317 CrossRef CAS PubMed.
- A. Warsinke, A. Benkert and F. Scheller, J. Anal. Chem., 2000, 366, 622–634 CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical methods: Fundamentals and applications, Wiley & Sons, New York, 2001 Search PubMed.
- P. T. Kissinger and W. R. Heineman, Laboratory Techniques in Electroanalytical Chemistry, Marcel Dekker, New York, 1996 Search PubMed.
- J. Newman and K. E. Thomas-Ayea, Electrochemical systems, Wiley, New York, 2004 Search PubMed.
- C. M. A. Brett and A. M. O. Brett, Electroanalysis, Oxford University Press, Oxford, 1998 Search PubMed.
- D. Han, Y. Kim, C. Kang and D. Chung, Anal. Chem., 2014, 86, 5991–5998 CrossRef CAS PubMed.
- J. Liu, J. Wang, T. Wang, D. Li, F. Xi, J. Wang and E. Wang, Biosens. Bioelectron., 2014, 65, 281–286 CrossRef PubMed.
- T. Qi, J. Liao, Y. Li, J. Peng, W. Li, B. Chu, H. Li, Y. Wei and Z. Qian, Biosens. Bioelectron., 2014, 61, 245–250 CrossRef CAS PubMed.
- J. Li, S. Liu, J. Yu, W. Lian, M. Cui, W. Xu and J. Huang, Sens. Actuators, B, 2013, 188, 99–105 CrossRef CAS.
- Z. Zhong, M. Li, Y. Qing, N. Dai, W. Guan, W. Liang and D. Wang, Analyst, 2014, 139, 6563–6568 RSC.
- F. Cai, Q. Zhu, K. Zhao, A. Deng and J. Li, Environ. Sci. Technol., 2015, 49, 5013–5020 CrossRef CAS PubMed.
- H. Chen, Z. Gao, Y. Cui, G. Chen and D. Tang, Biosens. Bioelectron., 2013, 44, 108–114 CrossRef CAS PubMed.
- Z. Liu, Q. Rong, Z. Ma and H. Han, Biosens. Bioelectron., 2014, 65, 307–313 CrossRef PubMed.
- Y. Wang, Y. Zhang, Y. Su, F. Li, H. Ma, H. Li, B. Du and Q. Wei, Talanta, 2014, 124, 60–66 CrossRef CAS PubMed.
- S. Zhang, H. Ma, L. Yan, W. Cao, T. Yan, Q. Wei and B. Du, Biosens. Bioelectron., 2014, 59, 335–341 CrossRef CAS PubMed.
- Z. Cui, D. Wu, Y. Zhang, H. Ma, H. Li, B. Du, Q. Wei and H. Ju, Anal. Chim. Acta, 2014, 807, 44–50 CrossRef CAS PubMed.
- Y. Wang, J. Ping, Z. Ye, J. Wu and Y. Ying, Biosens. Bioelectron., 2013, 49, 492–498 CrossRef CAS PubMed.
- S. Samanman, A. Numnuam, W. Limbut, P. Kanatharana and P. Thavarungkul, Anal. Chim. Acta, 2015, 853, 521–532 CrossRef CAS PubMed.
- H. Jia, P. Gao, H. Ma, Y. Li, J. Gao, B. Du and Q. Wei, Talanta, 2015, 132, 803–808 CrossRef CAS PubMed.
- D. Lin, C. Mei, A. Liu, H. Jin, S. Wang and J. Wang, Biosens. Bioelectron., 2015, 66, 177–183 CrossRef CAS PubMed.
- Z. Zhu, L. Shi, H. Feng and H. Zhou, Bioelectrochemistry, 2014, 101, 153–158 CrossRef PubMed.
- Y. Zhao, L. Liu, D. Kong, H. Kuang, L. Wang and C. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 21178–21183 CAS.
- J. Huang, Q. Lin, X. Zhang, X. He, X. Xing, W. Lian, M. Zuo and Q. Zhang, Food Res. Int., 2011, 44, 92–97 CrossRef CAS.
- P. Li, W. Zhang, Z. Zhou and L. Zhang, Clin. Biochem., 2015, 48, 156–161 CrossRef CAS PubMed.
- S. Zhang, Y. Liu, M. Lin, J. Kang, Y. Sun and H. Le, Electrochim. Acta, 2013, 90, 246–253 CrossRef.
- P. R. Liang, H. Peng and J. Qiu, J. Colloid Interface Sci., 2008, 320, 125–131 CrossRef PubMed.
- Y. Liu, International Conference Bioinformatics and Biomedical Engineering, 2011, pp. 1–4 Search PubMed.
- B. V. Chikkaveeraiah, A. Soldà, D. Choudhary, F. Maran and J. F. Rusling, Nanomedicine, 2012, 7, 957–965 CrossRef CAS PubMed.
- T. Xu and T. S. Xu, Biochem. Eng. J., 2016, 105, 36–43 CrossRef CAS.
- S. E. Ichi, F. Leon, L. Vossier, H. Marchandin, A. Errachid, J. Coste, N. Jaffrezic-Renault and C. Fournier-Wirth, Biosens. Bioelectron., 2014, 54, 378–384 CrossRef PubMed.
- M. Tu, H. Chen, Y. Wang, S. Moochhala, P. Alagappan and B. Liedberg, Anal. Chim. Acta, 2015, 853, 228–233 CrossRef CAS PubMed.
- S. Suresh, M. Gupta, G. Kumar, V. Rao, O. Kumar and P. Ghosal, Analyst, 2012, 137, 4086–4092 RSC.
- Y. Liu, Y. Liu, H. Feng, Y. Wu, L. Joshi, X. Zeng and J. Li, Biosens. Bioelectron., 2012, 35, 63–68 CrossRef CAS PubMed.
- X. Sun, Y. Cao, Z. Gong, X. Wang, Y. Zhang and J. Gao, Sensors, 2012, 12, 17247–17261 CrossRef CAS PubMed.
- V. Serafín, G. Martínez-García, L. Agüí, P. Yáñez-Sedeño and J. Pingarrón, Biosens. Bioelectron., 2014, 52, 98–104 CrossRef PubMed.
- F. Liu, W. Deng, Y. Zhang, S. Ge, J. Yu and X. Song, Anal. Chim. Acta, 2014, 818, 46–53 CrossRef CAS PubMed.
- L. Wu, E. Xiong, X. Zhang, X. Zhang and J. Chen, Nano Today, 2014, 9, 197–211 CrossRef CAS.
- R. Akter, C. Rhee and M. Rahman, Biosens. Bioelectron., 2014, 54, 351–357 CrossRef CAS PubMed.
- A. Sanaullah, B. Jeong, R. Akter, O. Han and M. Rahman, Bull. Korean Chem. Soc., 2014, 35, 2193–2196 CrossRef CAS.
- X. Ren, D. Wu, Y. Wang, Y. Zhang, D. Fan, X. Pang, Y. Li, B. Du and Q. Wei, Biosens. Bioelectron., 2015, 72, 156–159 CrossRef CAS PubMed.
- G. Yuan, C. Yu, C. Xia, L. Gao, W. Xu, W. Li and J. He, Biosens. Bioelectron., 2015, 72, 237–246 CrossRef CAS PubMed.
- Y. Ding, D. Li, B. Li, K. Zhao, W. Du, J. Zheng and M. Yang, Biosens. Bioelectron., 2013, 48, 281–286 CrossRef CAS PubMed.
- H. Cheng, G. Lai, L. Fu, H. Zhang and A. Yu, Biosens. Bioelectron., 2015, 71, 353–358 CrossRef CAS PubMed.
- X. Wang, L. Chen, X. Su and S. Ai, Biosens. Bioelectron., 2013, 47, 171–177 CrossRef CAS PubMed.
- W. Mak, K. Cheung, D. Trau, A. Warsinke, F. Scheller and R. Renneberg, Anal. Chem., 2005, 77, 2835–2841 CrossRef CAS PubMed.
- J. Lei and H. Ju, Chem. Soc. Rev., 2012, 41, 2122–2134 RSC.
- N. Sanvicens, C. Pastells, N. Pascual and M. Marco, TrAC, Trends Anal. Chem., 2009, 28, 1243–1252 CrossRef CAS.
- R. Polsky, R. Gill, L. Kaganovsky and I. Willner, Anal. Chem., 2006, 78, 2268–2271 CrossRef CAS PubMed.
- V. Upadhyayula, Anal. Chim. Acta, 2012, 715, 1–18 CrossRef CAS PubMed.
- D. Lin, J. Wu, M. Wang, F. Yan and H. Ju, Anal. Chem., 2012, 84, 3662–3668 CrossRef CAS PubMed.
- S. A. Lim, H. Yoshikawa, E. Tamiya, H. Mohd Yasin and M. Ahmed, RSC Adv., 2014, 4, 58460 RSC.
- P. Fanjul-Bolado, D. Hernández-Santos, M. González García and A. Costa-García, Anal. Chem., 2007, 79, 5272–5277 CrossRef CAS PubMed.
- R. Alves, F. Pimentel, H. Nouws, R. Marques, M. Gonzalez-Garcia, M. Oliveira and C. Delerue-Matos, Biosens. Bioelectron., 2015, 64, 19–24 CrossRef CAS PubMed.
- L. Feng, Z. Bian, J. Peng, Y. Zhu, D. Yang, L. Jiang, J. Zhu, F. Jiang and G. Yang, Anal. Chem., 2012, 84, 7810–7815 CrossRef CAS PubMed.
- T. Xu, X. Jia, X. Chen and Z. Ma, Biosens. Bioelectron., 2014, 56, 174–179 CrossRef CAS PubMed.
- M. Gijs, Microfluid. Nanofluid., 2004, 1, 22–40 CAS.
- N. Jaffrezic-Renault, C. Martelet, Y. Chevolot and C. Jean-Pierre, Sensors, 2007, 7, 589–614 CrossRef CAS.
- E. Zacco, M. Pividori, S. Alegret, R. Galve and M. Marco, Anal. Chim. Acta, 2006, 78, 1780–1788 CAS.
- B. Zhang, D. Tang, B. Liu, Y. Cui, H. Chen and G. Chen, Anal. Chim. Acta, 2012, 711, 17–23 CrossRef CAS PubMed.
- J. Tang, D. Tang, Q. Li, B. Su, B. Qiu and G. Chen, Anal. Chim. Acta, 2011, 697, 16–22 CrossRef CAS PubMed.
- J. Das, H. Kim, K. Jo, K. Park, S. Jon, K. Lee and H. Yang, Chem. Commun., 2009, 6394–6396, 10.1039/b912450k.
- F. Liu, G. Xiang, R. Yuan, X. Chen, F. Luo, D. Jiang, S. Huang, Y. Li and X. Pu, Biosens. Bioelectron., 2015, 60, 210–217 CrossRef PubMed.
- F. Li, J. Han, L. Jiang, Y. Wang, Y. Li, Y. Dong and Q. Wei, Biosens. Bioelectron., 2015, 68, 626–632 CrossRef CAS PubMed.
- H. Liu, Y. Yang, P. Chen and Z. Zhong, Biochem. Eng. J., 2009, 45, 107–112 CrossRef CAS.
- X. Xiao and A. Bard, J. Am. Chem. Soc., 2007, 129, 9610–9612 CrossRef CAS PubMed.
- A. Castañeda, T. Alligrant, J. Loussaert and R. Crooks, Langmuir, 2015, 31, 876–885 CrossRef PubMed.
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- G. Decher, J. Hong and J. Schmitt, Thin Solid Films, 1992, 210–211, 831–835 CrossRef CAS.
- N. Xia, Y. Zhang, X. Wei, Y. Huang and L. Liu, Anal. Chim. Acta, 2015, 878, 95–101 CrossRef CAS PubMed.
- R. Malhotra, V. Patel, J. Vaqué, J. Gutkind and J. Rusling, Anal. Chem., 2010, 82, 3118–3123 CrossRef CAS PubMed.
- M. Campas, M. Olteanu and J. Jean-Louis Marty, Sens. Actuators, B, 2008, 129, 263–267 CrossRef CAS.
- S. Singh, A. Srivastava, O. H. C. Ahn, G. Choi and R. Asthana, Toxicon, 2012, 60, 878–894 CrossRef CAS PubMed.
- J. Han, Y. Zhuo, Y. Chai, Y. Xiang, R. Yuan, Y. Yuan and N. Liao, Biosens. Bioelectron., 2013, 41, 116–122 CrossRef CAS PubMed.
- Z. Zhu, L. Garcia-Gancedo, A. Flewitt, H. Xie, F. Moussy and W. Milne, Sensors, 2012, 12, 5996–6022 CrossRef PubMed.
- L. Zhao, S. Li, J. He, G. Tian, Q. Wei and H. Li, Biosens. Bioelectron., 2013, 49, 222–225 CrossRef CAS PubMed.
- J. Tang, X. Chen, J. Zhou, Q. Li, G. Chen and D. Tang, Analyst, 2013, 138, 4327–4333 RSC.
- E. Polo, S. Puertas and P. Batalla, Front. Nanosci., 2012, 4, 247–267 Search PubMed.
- J. Tang, D. Tang, B. Su, J. Huang, B. Qiu and G. Chen, Biosens. Bioelectron., 2011, 26, 3219–3226 CrossRef CAS PubMed.
- Q. Li, L. Zeng, J. Wang, D. Tang, B. Liu, G. Chen and M. Wei, ACS Appl. Mater. Interfaces, 2011, 3, 1366–1373 CAS.
- D. Lin, J. Wu, H. Ju and F. Yan, Biosens. Bioelectron., 2014, 52, 153–158 CrossRef CAS PubMed.
- A. Ghindilis, P. Atanasov, M. Wilkins and E. Wilkins, Biosens. Bioelectron., 1998, 13, 113–131 CrossRef CAS PubMed.
- I. Tothill, World Mycotoxin J., 2011, 4, 361–374 CrossRef CAS.
- L. Yang, H. Zhao, S. Fan, S. Deng, Q. Lv, J. Lin and C. Peng, Biosens. Bioelectron., 2014, 57, 199–206 CrossRef CAS PubMed.
- H. Jang, S. Kim, H. Chang and J. Choi, Biosens. Bioelectron., 2015, 63, 546–551 CrossRef CAS PubMed.
- T. Qi, J. Liao, Y. Li, J. Peng, W. Li, B. Chu, H. Li, Y. Wei and Z. Qian, Biosens. Bioelectron., 2014, 61, 245–250 CrossRef CAS PubMed.
- G. Rivas, M. Rubianes, M. Rodríguez, N. Ferreyra, G. Luque, M. Pedano, S. Miscoria and C. Parrado, Talanta, 2007, 74, 291–307 CrossRef CAS PubMed.
- B. Prieto-Simón, N. Bandaru, C. Saint and N. Voelcker, Biosens. Bioelectron., 2015, 67, 642–648 CrossRef PubMed.
- K. Liu, J. Zhang, Q. Liu and H. Huang, Electrochim. Acta, 2013, 114, 448–454 CrossRef CAS.
- J. Han, Y. Zhuo, Y. Chai, R. Yuan, W. Zhang and Q. Zhu, Anal. Chim. Acta, 2012, 746, 70–76 CrossRef CAS PubMed.
- D. Wang, W. Dou, G. Zhao and Y. Chen, J. Microbiol. Methods, 2014, 106, 110–118 CrossRef CAS PubMed.
- X. Jia, Z. Liu, N. Liu and Z. Ma, Biosens. Bioelectron., 2014, 53, 160–166 CrossRef CAS PubMed.
- L. Li, L. Zhang, J. Yu, S. Ge and X. Song, Biosens. Bioelectron., 2015, 71, 108–114 CrossRef CAS PubMed.
- B. Seiwert and U. Karst, Anal. Bioanal. Chem., 2008, 390, 181–200 CrossRef CAS PubMed.
- H. Li, Q. Wei, J. He, T. Li, Y. Zhao, Y. Cai, B. Du, Z. Qian and M. Yang, Biosens. Bioelectron., 2011, 26, 3590–3595 CrossRef CAS PubMed.
- Y. Teng, W. Zhang, X. Zhang, Y. Fu, H. Liu, Z. Wang and L. Jin, Biosens. Bioelectron., 2011, 26, 4661–4666 CrossRef CAS PubMed.
- Y. Zhuo, Y. Chai, R. Yuan, L. Mao, Y. Yuan and J. Han, Biosens. Bioelectron., 2011, 26, 3838–3844 CrossRef CAS PubMed.
- Y. Gao and R. Cranston, Anal. Chim. Acta, 2010, 659, 109–114 CrossRef CAS PubMed.
- C. Yin, G. Lai, L. Fu, H. Zhang and A. Yu, Electroanalysis, 2014, 26, 409–415 CrossRef CAS.
- V. Serafín, L. Agüí, P. Yáñez-Sedeño and J. Pingarrón, Biosens. Bioelectron., 2014, 52, 98–104 CrossRef PubMed.
- H. Jia, P. Gao, H. Ma, D. Wu, B. Du and Q. Wei, Bioelectrochemistry, 2015, 101, 22–27 CrossRef CAS PubMed.
- B. Su, D. Tang, Q. Li, J. Tang and G. Chen, Anal. Chim. Acta, 2011, 692, 116–124 CrossRef CAS PubMed.
- Z. Cui, Y. Cai, D. Wu, H. Yu, Y. Li, K. Mao, H. Wang, H. Fab, Q. Wei and B. Du, Electrochim. Acta, 2012, 69, 79–85 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |