
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransition of lithium growth mechanisms in liquid electrolytes†
Peng
Bai
*a,
Ju
Li
bc,
Fikile R.
Brushett
a and
Martin Z.
Bazant
*ade
aDepartment of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. E-mail: pengbai@mit.edu; bazant@mit.edu
bDepartment of Nuclear Science and Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA
cDepartment of Materials Science and Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA
dDepartment of Mathematics, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA
eDepartment of Materials Science and Engineering and SUNCAT Interfacial Science and Catalysis, Stanford University, Stanford, CA 94305, USA
First published on 1st September 2016
Abstract
Next-generation high-energy batteries will require a rechargeable lithium metal anode, but lithium dendrites tend to form during recharging, causing short-circuit risk and capacity loss, by mechanisms that still remain elusive. Here, we visualize lithium growth in a glass capillary cell and demonstrate a change of mechanism from root-growing mossy lithium to tip-growing dendritic lithium at the onset of electrolyte diffusion limitation. In sandwich cells, we further demonstrate that mossy lithium can be blocked by nanoporous ceramic separators, while dendritic lithium can easily penetrate nanopores and short the cell. Our results imply a fundamental design constraint for metal batteries (“Sand's capacity”), which can be increased by using concentrated electrolytes with stiff, permeable, nanoporous separators for improved safety.
Broader contextConsumer electronic devices, portable power tools, and electric vehicles have been enabled, but also constrained, by the steady improvement of lithium-ion batteries. To develop batteries with higher energy density, such as Li–O2, Li–S, and other Li metal batteries using intercalation cathodes, lithium is believed to be the ideal anode material for its extremely high theoretical specific capacity (3860 mA h g−1), low density (0.59 g cm−3) and the lowest negative electrochemical potential (−3.04 V vs. the standard hydrogen electrode). Unfortunately, lithium growth is unstable during battery recharging and leads to rough, mossy deposits, whose fresh surfaces consume the electrolyte to form solid–electrolyte interphase layers, resulting in high internal resistance, low Coulombic efficiency and short cycle life. Finger-like lithium dendrites can also short-circuit the cell by penetrating the porous separator, leading to catastrophic accidents. Controlling such hazardous instabilities requires accurately determining their mechanisms, which are more complex than the well-studied diffusion-limited growth of copper or zinc from aqueous solutions. Such fundamental understanding is critical for the success of the lithium metal anode and could provide guidance for the optimal design and operation of rechargeable lithium metal batteries. |
Introduction
The lithium metal anode is a key component of future high-energy batteries, such as Li–S and Li–O2 batteries,1 for economical and long-range electric vehicles.2 It also holds the promise to reduce the volume and weight of lithium-ion batteries by replacing the standard graphite anode, if lithium dendrites can be safely controlled during recharging to avoid internal shorts and life-threatening accidents.3 While it has been demonstrated that electrolyte additives,4–6 artificial solid electrolyte interphase (SEI) layers,7,8 and increasing the salt concentration in electrolytes,9,10 either alone or in combination, can improve the stability of lithium under small currents4–7 and low capacities,9,11 the challenge of suppressing dendrites at practical currents (>1 mA cm−2) and areal capacities (>1 mA h cm−2) remains a major obstacle for the development of rechargeable lithium metal batteries.8,12 The time is ripe for a thorough investigation of lithium growth mechanisms under these conditions, in order to establish theoretical principles and design constraints for dendrite-free charging.The prevailing understanding of lithium growth instability is largely based on the simpler case of aqueous copper electrodeposition,13–17 where dendritic fractal patterns are telltale signs of long-range diffusion-limited growth.18–20 When a current is applied to recharge the battery, cations are consumed by reduction reaction, as anions are expelled by the electric field. In a binary electrolyte, the evolution of neutral salt concentration obeys an effective diffusion equation.21 For currents exceeding diffusion limitation, the salt concentration at the electrode surface decreases to zero at a characteristic time,22,23 and uniform electroplating becomes unstable.13,14,17 This characteristic time, tSand, was first derived by Sand in 1901,22 and is now known as “Sand's time”,23 after which the scarce supply of cations preferentially deposits onto surface protrusions, leading to a self-amplifying process of dendritic growth (i.e. tip growth mode) that propagates at the velocity of bulk anion electromigration, in order to preserve electroneutrality.13,17,21,24,25
Attempts to transfer this understanding from copper to lithium have been inconclusive. In lithium/polymer-electrolyte cells,26–28 the onset time for dendritic growth exhibits similar scaling with current as Sand's time, but surprisingly, far below the diffusion-limited current.27 In lithium/liquid-electrolyte cells, decreasing the mobility and the transference number of anions by using modified separators can enhance the cycle life,29 albeit again at currents well below the diffusion-limited current. Ramified moss-like or “mossy” deposits have even been observed at a current density of 10 μA cm−2 (ref. 30) and have been observed to grow from their roots,31 rather than their tips, in contrast to all existing growth models. Moreover, the microscopic morphology of serpentine lithium filaments observed in various electrolytes,3,5,9,32–35 over a range of length scales, do not resemble the branched, fractal structure of copper dendrites. These striking discrepancies between lithium and copper metal electrodeposition have lingered for decades without a clear explanation.
In this study, we aim to determine the precise conditions for short-inducing dendritic lithium to form, in order to establish design constraints for safe rechargeable metal batteries. We choose one of the most successful electrolytes for lithium-ion batteries,36 LiPF6 in the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC), and conduct two sets of experiments to investigate the mechanisms of lithium dendrite growth under various conditions. The first experiments with novel capillary cells reveal that the relatively dense “mossy” lithium growth is reaction-limited and changes to fractal “dendritic” lithium in response to electrolyte diffusion limitation. The second experiments using sandwich cells demonstrate that root-growing mossy lithium can be blocked by a nanoporous ceramic membrane, while tip-growing dendritic lithium can easily penetrate the nanopores and cause the internal short only at over-limiting currents. Our results suggest that optimizing and monitoring the intrinsic transport properties of the battery could eliminate the formation of dendritic lithium and the risk of internal shorts thereafter.
:1 mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC), and conduct two sets of experiments to investigate the mechanisms of lithium dendrite growth under various conditions. The first experiments with novel capillary cells reveal that the relatively dense “mossy” lithium growth is reaction-limited and changes to fractal “dendritic” lithium in response to electrolyte diffusion limitation. The second experiments using sandwich cells demonstrate that root-growing mossy lithium can be blocked by a nanoporous ceramic membrane, while tip-growing dendritic lithium can easily penetrate the nanopores and cause the internal short only at over-limiting currents. Our results suggest that optimizing and monitoring the intrinsic transport properties of the battery could eliminate the formation of dendritic lithium and the risk of internal shorts thereafter.
Results
Transition from mossy to dendritic lithium
To better track the growth of lithium in situ, we fabricate a lithium|electrolyte|lithium symmetric cell in a special glass capillary, whose middle part is pulled thinner (Fig. 1a). A small piece of lithium metal is pushed into each end of the capillary until it lodges in the tapering part to seal the cell. Here, we are interested in the lithium deposition (reduction reaction), so the corresponding electrode should be called cathode. However, in order to be consistent with the convention of lithium metal anodes, we assume that the electrodeposition is a recharging process and the electrode is designated as an anode. | ||
| Fig. 1 In situ observations of lithium electrodeposition in a glass capillary filled with an electrolyte solution consisting of 1 M LiPF6 in EC/DMC. (a) Photo of the capillary cell, whose middle part was pulled thinner for easier optical observation. (b) Voltage responses of the capillary cell at a deposition current density of 2.61 mA cm−2. (c–g) In situ snapshots of the growth of lithium during the electrodeposition. Red arrow in (e) points to the emergence of dendritic lithium. Red dash line in (g) labels the clear morphological difference between the pre- and post-Sand's time lithium deposits. (h) Theoretical interpretation of the growth mechanisms of lithium electrodeposition during concentration polarization. | ||
When a constant current is applied, moss-like lithium starts to deposit (Fig. 1c and d), and the salt concentration near the surface starts to decrease,37 as indicated by the gradually increasing voltage (Fig. 1b). After ∼40 min of polarization, the voltage starts to diverge upon salt depletion at the anode surface,38 and a wispy dendrite suddenly shoots out (at 2678s in Fig. 1e) in an obvious tip-growing manner, leaving behind stagnant mossy lithium. The dendrite's fractal structure remains the same after two weeks of relaxation.
The striking differences in morphology and dynamics imply two different mechanisms, switching from reaction-limited to transport-limited growth at the voltage spike. In the early stages of electrodeposition, mossy lithium mainly grows from its roots, as revealed by the movement of the tips, which barely change shape as they are pushed forward (ESI,† Fig. S1 and Movies S1, S2). Root growth has also been observed by Yamaki et al.31 below the limiting current and attributed to internal stress release beneath the SEI layer on the lithium electrode. While growing into the open electrolyte, the mossy lithium also thickens, and the process has been described vividly as “rising dough”.30 It is noteworthy that at the microscopic scale, the relatively dense moss-like structure is composed of whiskers, although the width of an individual whisker varies in different electrolytes.5,9,30,35 Such random surface growth is typical of reaction-limited deposition.19 Compared with copper electrodeposition,14 the key difference is that lithium, covered by SEI, develops whiskers and mossy structures, while copper, without SEI, forms whisker-free yet compact deposition before Sand's time.39 Due to the insulating SEI that forms on individual lithium whiskers, mossy lithium is unable to transform into a uniform metallic film through a ripening process, even under mechanical pressure. At the voltage spike, sparse lithium dendrites grow explosively from their tips with the fractal morphology of diffusion-limited aggregation,19 also shared by copper dendrites,13,14 because electrodeposition is in the same universality class.40,41 The similarity between lithium and copper dendrites implies that both metals have similar surface tension, so the formation of dendritic lithium is correlated with the lack of lithium salt to form SEI different from that of mossy lithium, or very little SEI until the concentration relaxes after the initial burst of growth.
To test the hypothesis of diffusion limitation, the experimental times to reach the voltage spike are used to calculate an apparent diffusion coefficient Dapp from Sand's formula,22
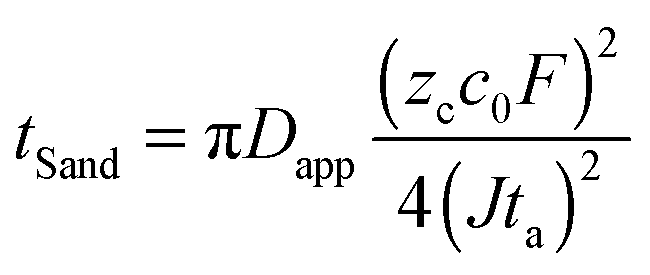 | (1) |
 | ||
| Fig. 2 Change of growth mechanism at Sand's time during concentration polarization. (a) Voltage responses of capillary cells at various deposition current densities. (b) Representative optical images of lithium deposits demonstrating the clear change of morphologies at Sand's time for various current densities. (c) Log–log plot of the experimental Sand's times for various current densities. (d) Current-dependent Sand's capacity with previous reports shown. | ||
As a new battery relevant metric, we convert Sand's time into “Sand's capacity” by multiplying with the current density. The plot of Sand's capacity versus current density (Fig. 2d) provides a simple design constraint to avoid dendritic lithium. Interestingly, most state-of-the-art lithium metal anodes do not operate in the regime of dendritic lithium identified by the capillary cell, which is already much lower than that of the sandwich cells. Since the growth mechanism switches by diffusion limitation, absolute current densities cannot be meaningfully compared across different cells. It is the relative current density, with respect to the system-specific limiting current, that controls the transition from mossy to dendritic lithium.
Blockage of mossy lithium
We then apply the knowledge of growth mechanisms from the capillary cell to investigate the ability of a nanoporous separator to block mossy and dendritic lithium in a battery relevant sandwich cell. Since the smallest known whiskers in mossy lithium are ∼1 μm thick,3 we construct the cell using an anodic aluminum oxide (AAO) membrane with submicron pores (<200 nm), to see whether the mossy lithium can be blocked (Fig. 3a). | ||
| Fig. 3 Lithium electrodeposition in sandwich cells. (a) Structure of the symmetric sandwich cell, where names of the electrodes follow the convention of lithium batteries, i.e. lithium deposits onto the anode during recharging. (b) Voltage responses of the sandwich cells, indicating the complete blockage of lithium deposits even beyond the theoretical capacity of the lower compartment at 10 mA cm−2, as well as the quick penetration of AAO and short-circuiting of the cell at 50 mA cm−2. (c and d) Scanning electron microscopy (SEM) images of the AAO/Li-deposit interface, revealing the blockage of bulky porous lithium formed in the under-limiting current conditions. (e and f) SEM images of the anode-facing side of AAO, displaying both bulky and needle-like lithium deposits formed in the over-limiting current conditions. (g and h) SEM images of the cathode-facing side of AAO, showing nanosized lithium deposits coming out of the nanopores of AAO. Inset: Magnification of the clusters of granular lithium deposits around the pores of AAO (appear as black dots). | ||
Based on the electrode separation, 300 μm, the limiting current density for the sandwich cell is approximately 20 mA cm−2. When an under-limiting current density of 10 mA cm−2 is applied, the voltage stabilizes at 0.2 V and lasts for a capacity well beyond the pure lithium limit that the compartment below the AAO can accommodate (Fig. 3a), indicating some deformation of the membrane. After dissembling the cell, an intact, free-standing AAO membrane is recovered with a dense lithium disk below it (Fig. S5, ESI†), which confirms the complete blockage of mossy lithium growth. Surprisingly, even when the AAO membrane is pressed in direct contact with the lithium metal anode, a significant amount of porous lithium can still be deposited below the AAO, as shown in Fig. 3c and d. The mossy lithium shown in the SEM images is clearly too bulky to penetrate AAO, and only forms a space-filling porous layer between the electrode and the separator, reducing the risk of short circuit below the limiting current. These results help explain why various ceramic membranes can prevent lithium short circuits,6,44,48,49 especially under normal conditions (<10 mA cm−2), where only the dense root-growing mossy lithium is developed in the cell.
As demonstrated in the capillary cell experiments, once Sand's capacity is exceeded, dendritic lithium suddenly appears. When an over-limiting current density of 50 mA cm−2 is applied to the sandwich cell, the voltage quickly increases and leads to a short circuit. As revealed by the SEM images, very thin lithium filaments can now be found among mossy deposits on the anode side (Fig. 3e and f). Clusters of granular deposits, smaller than the pores of AAO, are also clearly visible on the cathode side (Fig. 3g and h), which confirm that lithium penetration through the ceramic nanopores caused the short circuit. The stark difference between mossy and dendritic lithium deposits leads us to propose that the term “dendrite” be used more narrowly, only to describe a fractal, tip-growing deposit resulting from diffusion-limited growth, consistent with the well-studied copper and zinc dendrites. If lithium dendrite penetration in AAO were mainly opposed by surface tension, then the breakthrough voltage (where the overpotential exceeds the Young–Laplace pressure) would scale with the inverse of the pore size.50 The dendrite penetration may be further suppressed by modifying the surface charge of the nanopores,15 when over-limiting mass transfer is opposed by surface conduction.46
Sand's capacity as the safety limit
Our results reveal why the risk of dendrites increases with aging, and how to mitigate it. According to the definition of Sand's capacity, | (2) |
 | ||
| Fig. 4 Concentration-dependent Sand's behavior. Experimental (a) Sand's times and (b) Sand's capacities for 0.5 M and 2 M electrolytes, with results of 1 M electrolyte from Fig. 2 as references. | ||
Dimensionless safety criterion
In order to compare different systems and make general scaling predictions about the safety limit, we employ dimensional analysis. Following earlier definitions, L is the distance between the two electrodes, then L/2 is a characteristic length scale for electrolyte diffusion with apparent diffusivity Dapp, i.e. the distance from the electrode (where salt depletion occurs) to a reservoir at concentration c0. Let Jlim be the steady-state diffusion-limited current, and CSand be the maximum (Sand's) areal capacity for safe operation at a given current density J. If these are the only important parameters, then, simply as a consequence of their physical units, Buckingham's theorem53 states that there must exist a scaling relation![[C with combining tilde]](https://www.rsc.org/images/entities/i_char_0043_0303.gif) Sand = f(
Sand = f(![[J with combining tilde]](https://www.rsc.org/images/entities/i_char_004a_0303.gif) ) between the dimensionless Sand's capacity and the dimensionless applied current density,
) between the dimensionless Sand's capacity and the dimensionless applied current density, | (3) |
 | (4) |
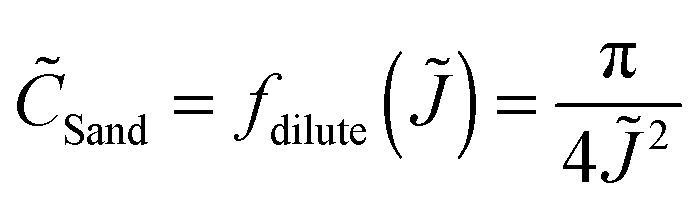 | (5) |
![[t with combining tilde]](https://www.rsc.org/images/entities/i_char_0074_0303.gif) Sand = 4tSandDappL−2 = f(), which can also be derived by solving the ambipolar diffusion equation for transient overlimiting current density > 1 (ESI†).
Sand = 4tSandDappL−2 = f(), which can also be derived by solving the ambipolar diffusion equation for transient overlimiting current density > 1 (ESI†).
The scaling function will differ for concentrated electrolytes54 (with concentration-dependent diffusivities and coupled Stefan–Maxwell fluxes) in porous separators47 (with possibly variable porosity and tortuosity, surface conduction and electro-osmotic flows), but the trend should be the same as predicted by dilute solution theory.55 When the experimental data shown in Fig. 4b are nondimensionalized with corresponding Dapp (Table S3, ESI†) and plotted in Fig. 5 as Sandversus , a reasonable data collapse is observed, similar to the dilute solution prediction, eqn (5), but with a modified scaling function, f() = 0.265−1.274, which is mainly attributable to electro-osmotic convection in the capillary cells (Fig. S4, ESI†).
 | ||
| Fig. 5 Linear and logarithmic (inset) plots of dimensionless Sand's capacity versus dimensionless current density. Dashed line is the prediction of Sand's formula for dilute electrolytes, while solid line is the best fit to the experimental data. | ||
Discussion
Our results suggest that monitoring the capacity loss and transient responses to estimate transport properties and the associated Sand's capacity could enable battery management systems to avoid dendrites by adjusting the applied current or cycled capacity windows in real time, which is particularly important for rechargeable lithium metal batteries cycling at high capacities. This prediction may seem at odds with the fact that some Li–S prototypes can be cycled at a very large specific capacity for hundreds of times without signs of internal shorts. There is no contradiction, however, after accounting for capacity differences. By multiplying the specific capacity with the small loading mass of the active sulfur,56 the converted areal capacity (in units of mA h cm−2) that matters for the metal anode is actually smaller than those of mature lithium-ion batteries.56,57 For future rechargeable lithium metal batteries that possess a high specific energy with respect to the total mass, and operate at a truly large areal capacity, on-board diagnosis of the intrinsic Sand's capacity of the battery to avoid dendritic lithium may become a practical solution for safe operation, before a robust chemistry that can completely suppress the continuous consumption of electrolyte (due to the growth of lithium whiskers) is developed.Carbonate-base electrolytes, such as what we use in this work, are known to effect relatively thin lithium whiskers.32 Ether-based electrolytes, in contrast, allow lithium whiskers to grow much thicker.7–9,58,59 At a given areal capacity, thicker lithium whiskers create less surface area and therefore consume less lithium salt and solvent to develop SEI layers. In addition, fluorosulfonate species used in these electrolytes, such as lithium bis(trifluoromethane-sulphonyl)imide (LiTFSI) and lithium bis(fluorosulfonyl)imide (LiFSI), could undergo extensive reactions with lithium to form a robust LiF-rich SEI,52,59 which could also be facilitated by employing very high salt concentrations.9 However, whether the SEI layers formed in ether-based electrolytes will remain stable during cycling at larger areal capacities, and thereby retard the continuous consumption of electrolytes12,60 to retain high Coulombic efficiency and long cycle life is yet to be verified experimentally. Investigating the fundamental mechanisms alongside may help engineer better SEI in other high-voltage solvents,36,61 with which the standard graphite anode in lithium-ion batteries may be replaced by ultrathin lithium metal anodes or simply removed to double the energy density. Of course, the chemistry of SEI does not override transport processes in electrolytes. Transitions from root-growing mossy lithium to tip-growing dendritic lithium also occur in ether-based electrolytes (Fig. S7 and Movie S6, S7, ESI†).
An important implication of our study is the need for consistent terminology, not only to refer to the different lithium morphologies, but also to clarify the underlying mechanisms for rational battery design and engineering. Comparing various published work with ours, the thin needle-like lithium filaments that grow from their roots below the limiting current should be called “whiskers”, which interweave with each other to form a “mossy” structure as the capacity increases. In contrast, the widely-used term “dendrites” should be reserved for the classical branched fractal structures that grow at their tips, which only occur at diffusion limitation and cannot revert to form a mossy structure. Although individual whiskers in the mossy structure may become thinner or disconnected over many deposition/dissolution cycles, as long as the current density remains under-limiting, the root-growth mechanism will make penetrating ceramic nanopores as difficult as threading a needle. With further investigations of SEI formation on mossy lithium and its interaction with ceramic separators during cycling, an ultimate safe solution should be possible.
Conclusions
While the failure mechanisms in practical batteries with opaque separators are still challenging to investigate in situ,62 our capillary cells provide a simple and effective means to explore the hidden physics. We have demonstrated that lithium growth in liquid electrolytes follows two different mechanisms, depending on the applied current and capacity. Below Sand's capacity, reaction-limited mossy lithium mainly grows from the roots and cannot penetrate hard ceramic nanopores in a sandwich cell. Above Sand's capacity, transport-limited dendritic lithium grows at the tips and can easily cross the separator to short the cell. Our results suggest maximizing Sand's capacity by increasing the salt concentration in the electrolyte. Electrolyte degradation should also be monitored to prevent dendrites by keeping the cycled capacity below Sand's capacity. Ceramic separators with pores smaller than mossy lithium whiskers could replace conventional polyolefin separators with flexible large pores to enhance safety and cycle life, and the effect could be further reinforced with lithium salts and solvents that favor thicker columnar deposits. To the broader field of electrodeposition, our results clarify the physical connections between lithium and copper/zinc dendrites formed in liquid electrolytes. Mechanisms and mathematical models of copper/zinc dendrite growths cannot be and should not be applied to explain either the development or the suppression of lithium whiskers. Future theoretical investigations should take into account the dynamics of SEI formation during both the root-growth and tip-growth processes of lithium electrodeposition.Methods
Materials
The battery grade electrolyte (1 M LiPF6 in ethylene carbonate/dimethyl carbonate with a volume ratio of 1:1), ethylene carbonate (EC, anhydrous, 99%), dimethyl carbonate (DMC, anhydrous, ≥99%), and Whatman AAO membranes (pore size 100 nm, thickness 60 μm, diameter 13 mm) were purchased from Sigma-Aldrich, and used as received. Lithium bis(trifluoromethane-sulphonyl)imide (LiTFSI), 1,3-dioxolane (DOL) and 1,2-dimethoxyethane (DME) were purchased from BASF Corporation. Lithium bis(fluorosulfonyl)imide (LiFSI) was purchased from Oakwood Products Inc. Copper wires, stainless steel wires, and polyvinylidene fluoride (PVDF) sheets were purchased from McMaster-Carr. The glass capillaries were purchased from Narishige Co., Ltd. Lithium chips (99.9%, thickness 250 μm, diameter 15.6 mm) were purchased from MTI Corporation.
Cell fabrication and electrochemical testing
The glass capillaries were pulled 7 mm longer with a vertical type micropipette puller (PC-10, Narishige Co., Ltd). The pulled capillary was bonded onto a glass slide with silicone and then transferred into the Argon-filled glovebox. To avoid gas bubbles, the electrolyte was filled in only by the capillary effect. Then, a small piece of lithium metal was pushed into each end of the cell by a metal wire to clog at the tapering part of the capillary to seal the cell. Separation between the lithium electrodes is around 5 mm for all cells. Sandwich cells were constructed in the split test cells purchased from MTI Corporation. A piece of lithium chip was first gently pressed onto the bottom part of the cell and covered by a customized PVDF washer punched off from the PVDF sheet. Several drops of electrolyte were dispensed on the surface of lithium, which also immerse the PVDF washer. A piece of AAO was then carefully placed on top of the PVDF washer and covered by another piece of PVDF washer. A few more drops of electrolyte were dispensed on AAO until the second PVDF washer is immersed. Finally the second lithium chip was stacked on top of the second PVDF, and then covered by a stainless steel disk. The whole cell was assembled together with the upper part (spring-loaded) of the test cell. Electrochemical tests were conducted with an Arbin battery tester (BT 2043, Arbin Instruments). In situ images were captured by an optical microscope (MU500, AmScope). All experiments were performed at room temperature in an Argon-filled glovebox (Vigor Tech USA) with water and oxygen content less than 1 ppm.SEM characterization
AAO separators with lithium deposits harvested from sandwich cells were washed with DMC for three times, then fixed onto the SEM sample holders with carbon adhesive and sealed in an air-tight box before moving out of the Argon-filled glovebox. The residual DMC on the samples helps protect the lithium from the ambient atmosphere when transferring them into the chamber of the Analytical Scanning Electron Microscope (JEOL, 6010LA), which usually takes less than 10 s before the vacuum evacuation.Acknowledgements
This work is supported by Robert Bosch LLC through the MIT Energy Initiative (MITei). P. B. thanks Mr William DiNatale in the Institute for Soldier Nanotechnologies at MIT for providing the access to the micropipette puller. J. L. acknowledges support by NSF DMR-1410636. M. Z. B. acknowledges support from the Global Climate and Energy Project at Stanford University and by the US Department of Energy, Basic Energy Sciences through the SUNCAT Center for Interface Science and Catalysis. The authors thank Dr Sarah Stewart for helpful discussions.Notes and references
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 CAS.
- F. Ding, W. Xu, G. L. Graff, J. Zhang, M. L. Sushko, X. L. Chen, Y. Y. Shao, M. H. Engelhard, Z. M. Nie, J. Xiao, X. J. Liu, P. V. Sushko, J. Liu and J. G. Zhang, J. Am. Chem. Soc., 2013, 135, 4450–4456 CrossRef CAS PubMed.
- W. Y. Li, H. B. Yao, K. Yan, G. Y. Zheng, Z. Liang, Y. M. Chiang and Y. Cui, Nat. Commun., 2015, 6, 7436 CrossRef CAS PubMed.
- Y. Y. Lu, Z. Y. Tu and L. A. Archer, Nat. Mater., 2014, 13, 961–969 CrossRef CAS PubMed.
- G. Y. Zheng, S. W. Lee, Z. Liang, H. W. Lee, K. Yan, H. B. Yao, H. T. Wang, W. Y. Li, S. Chu and Y. Cui, Nat. Nanotechnol., 2014, 9, 618–623 CrossRef CAS PubMed.
- D. Aurbach, E. Zinigrad, H. Teller and P. Dan, J. Electrochem. Soc., 2000, 147, 1274–1279 CrossRef CAS.
- J. F. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J. G. Zhang, Nat. Commun., 2015, 6, 6362 CrossRef CAS PubMed.
- L. Suo, Y.-S. Hu, H. Li, M. Armand and L. Chen, Nat. Commun., 2013, 4, 1481 CrossRef PubMed.
- M. H. Ryou, Y. M. Lee, Y. J. Lee, M. Winter and P. Bieker, Adv. Funct. Mater., 2015, 25, 834–841 CrossRef CAS.
- D. Aurbach, E. Zinigrad, Y. Cohen and H. Teller, Solid State Ionics, 2002, 148, 405–416 CrossRef CAS.
- V. Fleury, M. Rosso, J. N. Chazalviel and B. Sapoval, Phys. Rev. A: At., Mol., Opt. Phys., 1991, 44, 6693–6705 CrossRef CAS.
- J. Elezgaray, C. Leger and F. Argoul, J. Electrochem. Soc., 1998, 145, 2016–2024 CrossRef CAS.
- J.-H. Han, E. Khoo, P. Bai and M. Z. Bazant, Sci. Rep., 2014, 4, 7056 CrossRef CAS PubMed.
- R. M. Brady and R. C. Ball, Nature, 1984, 309, 225–229 CrossRef CAS.
- C. Leger, J. Elezgaray and F. Argoul, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1998, 58, 7700–7709 CrossRef CAS.
- T. A. Witten and L. M. Sander, Phys. Rev. Lett., 1981, 47, 1400–1403 CrossRef CAS.
- A.-L. s. Barabási and H. E. Stanley, Fractal concepts in surface growth, Press Syndicate of the University of Cambridge, New York, NY, USA, 1995 Search PubMed.
- A. Bunde and S. Havlin, Fractals and disordered systems, Springer, Berlin, New York, 1996 Search PubMed.
- J. S. Newman and K. E. Thomas-Alyea, Electrochemical systems, J. Wiley, Hoboken, NJ, 2004 Search PubMed.
- H. J. S. Sand, Philos. Mag., 1901, 1, 45–79 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical methods: fundamentals and applications, Wiley, New York, 2001 Search PubMed.
- D. P. Barkey and P. D. Laporte, J. Electrochem. Soc., 1990, 137, 1655–1656 CrossRef CAS.
- M. Z. Bazant, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1995, 52, 1903–1914 CrossRef CAS.
- C. Brissot, M. Rosso, J. N. Chazalviel and S. Lascaud, J. Power Sources, 1999, 81–82, 925–929 CrossRef CAS.
- M. Rosso, T. Gobron, C. Brissot, J. N. Chazalviel and S. Lascaud, J. Power Sources, 2001, 97–98, 804–806 CrossRef CAS.
- M. Rosso, C. Brissot, A. Teyssot, M. Dolle, L. Sannier, J. M. Tarascon, R. Bouchetc and S. Lascaud, Electrochim. Acta, 2006, 51, 5334–5340 CrossRef CAS.
- Z. Y. Tu, P. Nath, Y. Y. Lu, M. D. Tikekar and L. A. Archer, Acc. Chem. Res., 2015, 48, 2947–2956 CrossRef CAS PubMed.
- J. Steiger, D. Kramer and R. Moenig, Electrochim. Acta, 2014, 136, 529–536 CrossRef CAS.
- J. Yamaki, S. Tobishima, K. Hayashi, K. Saito, Y. Nemoto and M. Arakawa, J. Power Sources, 1998, 74, 219–227 CrossRef CAS.
- F. Ding, W. Xu, X. L. Chen, J. Zhang, M. H. Engelhard, Y. H. Zhang, B. R. Johnson, J. V. Crum, T. A. Blake, X. J. Liu and J. G. Zhang, J. Electrochem. Soc., 2013, 160, A1894–A1901 CrossRef CAS.
- R. R. Miao, J. Yang, Z. X. Xu, J. L. Wang, Y. Nuli and L. M. Sun, Sci. Rep., 2016, 6, 21771 CrossRef CAS PubMed.
- Z. Li, J. Huang, B. Y. Liaw, V. Metzler and J. B. Zhang, J. Power Sources, 2014, 254, 168–182 CrossRef CAS.
- R. Cao, J. Chen, K. S. Han, W. Xu, D. Mei, P. Bhattacharya, M. H. Engelhard, K. T. Mueller, J. Liu and J.-G. Zhang, Adv. Funct. Mater., 2016, 26, 3059–3066 CrossRef CAS.
- K. Xu, Chem. Rev., 2004, 104, 4303–4417 CrossRef CAS PubMed.
- H. J. Chang, A. J. Ilott, N. M. Trease, M. Mohammadi, A. Jerschow and C. P. Grey, J. Am. Chem. Soc., 2015, 137, 15209–15216 CrossRef CAS PubMed.
- C. Brissot, M. Rosso, J. N. Chazalviel and S. Lascaud, J. Electrochem. Soc., 1999, 146, 4393–4400 CrossRef CAS.
- K. I. Popov, S. S. Djokic and B. N. Grgur, Fundamental aspects of electrometallurgy, Kluwer Academic/Plenum Publishers, New York, 2002 Search PubMed.
- M. Z. Bazant, Proc. R. Soc. A, 2004, 460, 1433–1452 CrossRef.
- M. Z. Bazant, J. Choi and B. Davidovitch, Phys. Rev. Lett., 2003, 91, 045503 CrossRef PubMed.
- L. O. Valoen and J. N. Reimers, J. Electrochem. Soc., 2005, 152, A882–A891 CrossRef CAS.
- A. Nyman, M. Behm and G. Lindbergh, Electrochim. Acta, 2008, 53, 6356–6365 CrossRef CAS.
- S. Choudhury, R. Mangal, A. Agrawal and L. A. Archer, Nat. Commun., 2015, 6, 10101 CrossRef CAS PubMed.
- J. M. Huth, H. L. Swinney, W. D. Mccormick, A. Kuhn and F. Argoul, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1995, 51, 3444–3458 CrossRef CAS.
- E. V. Dydek, B. Zaltzman, I. Rubinstein, D. S. Deng, A. Mani and M. Z. Bazant, Phys. Rev. Lett., 2011, 107, 118301 CrossRef PubMed.
- A. Mani and M. Z. Bazant, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 84, 061504 CrossRef PubMed.
- Y.-k. Ahn, J. Park, D. Shin, S. Cho, S. Y. Park, H. Kim, Y. Piao, J. Yoo and Y. S. Kim, J. Mater. Chem. A, 2015, 3, 10715–10719 CAS.
- Z. Tu, Y. Kambe, Y. Lu and L. A. Archer, Adv. Energy Mater., 2014, 4, 1300654 CrossRef.
- A. Jana, D. R. Ely and R. E. Garcia, J. Power Sources, 2015, 275, 912–921 CrossRef CAS.
- D. P. Lv, Y. Y. Shao, T. Lozano, W. D. Bennett, G. L. Graff, B. Polzin, J. G. Zhang, M. H. Engelhard, N. T. Saenz, W. A. Henderson, P. Bhattacharya, J. Liu and J. Xiao, Adv. Energy Mater., 2015, 5, 1400993 CrossRef.
- X. B. Cheng, R. Zhang, C. Z. Zhao, F. Wei, J. G. Zhang and Q. Zhang, Adv. Sci., 2016, 3, 1500213 CrossRef.
- G. I. Barenblatt, Dimensional analysis, Gordon and Breach Science Publishers, New York, 1987, pp. 31–48 Search PubMed.
- S. A. Mareev, D. Y. Butylskii, A. V. Kovalenko, A. V. Petukhova, N. D. Pismenskaya, L. Dammak, C. Larchet and V. V. Nikonenko, Electrochim. Acta, 2016, 195, 85–93 CrossRef CAS.
- M. Z. Bazant, 10.626 Electrochemical Energy Systems (Spring 2014), Massachusetts Institute of Technology: MIT OpenCourseWare, http://ocw.mit.edu. License: Creative Commons BY-NC-SA.
- M. Hagen, D. Hanselmann, K. Ahlbrecht, R. Maca, D. Gerber and J. Tubke, Adv. Energy Mater., 2015, 5, 1401986 CrossRef.
- H. H. Zheng, J. Li, X. Y. Song, G. Liu and V. S. Battaglia, Electrochim. Acta, 2012, 71, 258–265 CrossRef CAS.
- J. Steiger, G. Richter, M. Wenk, D. Kramer and R. Monig, Electrochem. Commun., 2015, 50, 11–14 CrossRef CAS.
- R. R. Miao, J. Yang, X. J. Feng, H. Jia, J. L. Wang and Y. N. Nuli, J. Power Sources, 2014, 271, 291–297 CrossRef CAS.
- D. Aurbach, Y. Talyosef, B. Markovsky, E. Markevich, E. Zinigrad, L. Asraf, J. S. Gnanaraj and H. J. Kim, Electrochim. Acta, 2004, 50, 247–254 CrossRef CAS.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- K. J. Harry, D. T. Hallinan, D. Y. Parkinson, A. A. MacDowell and N. P. Balsara, Nat. Mater., 2014, 13, 69–73 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 to S7, Tables S1 to S3, Movies S1 to S7 and additional references. See DOI: 10.1039/c6ee01674j |
| This journal is © The Royal Society of Chemistry 2016 |