
EDC/NHS activation mechanism of polymethacrylic acid: anhydride versus NHS-ester†
Abstract
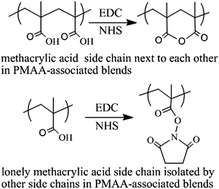
Polymer brushes of polymethacrylic acid (PMAA) and PMAA-associated polymer blends of PMAA/PNIPAM (poly-N-isopropylacrylamide) were prepared on porous silicon for further investigation of the EDC/NHS (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide/N-hydroxysuccinimide) activation mechanisms by infrared spectroscopy. When the fragmentation degree of PMAA blocks in PMAA-associated polymer blends is increased, the production of anhydride wanes from dominant to recessive, whereas a complementary product of the NHS-ester waxes from recessive to dominant. The Thorpe–Ingold effect was applied to explain the formation of anhydride: the gem-dialkyl groups of PMAA next to the carboxylic acids compress the acid side chains close to each other; thus, once the intermediate of O-acylisourea forms, it will be attacked by the intramolecular neighboring acid much faster than any other nucleophiles such as NHS and water, and therefore the six-membered ring of the anhydride will be formed. All acid side chains in PMAA standing next to each other will form an anhydride, primarily due to the Thorpe–Ingold effect, unless they are sterically hindered, whereas only isolated acid side chains form the NHS-ester. The EDC/NHS activation results for four small molecules of dicarboxylic acids in aqueous media, namely, glutaric acid and 2,2-dimethyl glutaric acid, which generate disuccinimidyl ester with high yield, and succinic acid and 2,2-dimethyl succinic acid, which remain intact, can also be explained by the Thorpe–Ingold effect. A clear understanding of the EDC/NHS activation mechanisms of PMAA will take us a step closer for resolving the mechanistic ambiguity of the carbodiimide/additive coupling reactions for amide bond formation.
 Please wait while we load your content...
Please wait while we load your content...

