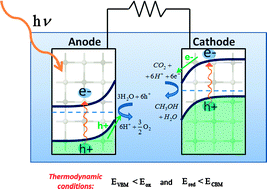
The positions of electronic band edges are one important metric for determining a material's capability to function in a solar energy conversion device that produces fuels from sunlight. In particular, the position of the valence band maximum (conduction band minimum) must lie lower (higher) in energy than the oxidation (reduction) reaction free energy in order for these reactions to be thermodynamically favorable. We present first principles quantum mechanics calculations of the band edge positions in five transition metal oxides and discuss the feasibility of using these materials in photoelectrochemical cells that produce fuels, including hydrogen, methane, methanol, and formic acid. The band gap center is determined within the framework of DFT+U theory. The valence band maximum (conduction band minimum) is found by subtracting (adding) half of the quasiparticle gap obtained from a non-self-consistent GW calculation. The calculations are validated against experimental data where possible; results for several materials including manganese(II) oxide, iron(II) oxide, iron(III) oxide, copper(I) oxide and nickel(II) oxide are presented.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


