
Coil–rod–coil triblock copolymers synthesized by macromolecular clicking and their compatibilizer effects in all-polymer solar cells†
Sultan
Otep
a,
Yu-Cheng
Tseng
b,
Naomasa
Yomogita
a,
Jia-Fu
Chang
b,
Chu-Chen
Chueh
 *b and
Tsuyoshi
Michinobu
*a
*b and
Tsuyoshi
Michinobu
*a
aDepartment of Materials Science and Engineering, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8552, Japan. E-mail: michinobu.t.aa@m.titech.ac.jp
bDepartment of Chemical Engineering and Advanced Research Center for Green Materials Science and Technology, National Taiwan University, Taipei 10617, Taiwan. E-mail: cchueh@ntu.edu.tw
First published on 25th November 2021
Abstract
Poly(diethynylthiophene) (PDET) synthesized via Hay coupling polycondensation retains terminal alkynes for tethering polystyrene (PS) blocks by a simple Cu-catalyzed azide–alkyne click (CuAAC) reaction. The successful synthesis of triblock copolymers was confirmed by 1H NMR, FTIR, and GPC measurements. Optical and electrochemical properties of the PDET block were conserved in the triblock copolymers, as determined from the UV-vis absorption spectra and redox potentials. Surface topography of the polymer films revealed the micrometer-scale features attributable to phase separation, which was supported by thermal analyses. The compatibilizer functions of PDET and triblock copolymer P1 were investigated and compared in all-polymer solar cells (all-PSCs). Addition of 1 wt% P1 was shown to result in an enhanced power conversion efficiency (PCE) from 5.90% to 6.24%, corresponding to a relative increase of ∼6%, whereas the addition of 1 wt% PDET decreased the resultant PCE. Notably, adding a proper compatibilizer helped reduce the device's potential loss, as evidenced by the improved Voc in the 1 wt% P1 device. Our results highlight the critical role of the coil segment in designing block copolymer-based compatibilizers for all-PSCs. Also, this study demonstrates a straightforward synthetic route for coil–rod–coil triblock copolymers that afford a compatibilizer function suitable for all-PSCs.
Introduction
Block copolymers have been conventionally synthesized by sequentially adding different monomers in living polymerization systems.1,2 In another approach, macromolecular coupling of pre-made reactive polymers by highly efficient click chemistry reactions, such as Cu-catalyzed azide–alkyne cycloaddition (CuAAC), is recently often adopted to produce various diblock copolymers3–8 and multiblock copolymers.9–12 Of these, rod–coil block copolymers are mostly based on poly(3-hexylthiophene) (P3HT) tethered to non-conjugated polymers, such as polystyrene and polyvinylpyrrolidone. This is because terminal alkyne-substituted P3HT can be prepared by Grignard metathesis polymerization and it is a suitable platform for macromolecular CuAAC coupling with azide-substituted counter polymers. On the other hand, rigid polymer backbones with terminal alkynes at both sides are required to synthesize coil–rod–coil block copolymers using a similar CuAAC coupling approach. In addition, to expand the scope of the macromolecular coupling approach, rigid conjugated polymer backbones other than P3HT are desired. In this context, we noted that poly(arylenebutadiynylene)s are readily prepared by self-polycondensation of diethynylarylene monomers and can possess two terminal alkynes if no side reactions occur during polymerization. In addition, various aromatic monomers can be designed, which enables us to expand the library of coil–rod–coil block copolymers. Furthermore, poly(arylenebutadiynylene)s were recently shown to be an effective crosslinking matrix and their charge-transport and light-harvesting properties can be tuned by the degree of crosslinking.13,14 However, no block copolymers of poly(arylenebutadiynylene)s have, to the best of our knowledge, been reported so far.In recent years, bulk heterojunction (BHJ) design has become the most prevailing and efficient system for organic photovoltaics (OPVs), which consists of an interpenetrating network formed by an electron-rich (donor) conjugated molecule and an electron-deficient (acceptor) conjugated molecule. During earlier times, acceptors were generally made from fullerene derivatives and they have realized power conversion efficiencies (PCEs) approaching 12%.15–17 However, limited by their weak light absorption, the development of fullerene-based OPVs has encountered a bottleneck. To this end, the exploitation of non-fullerene small molecules received increasing research attention and has significantly pushed the PCE to over 17% in single junction OPVs.18–20 The rapid rise of non-fullerene small molecules also encouraged the development of n-type conjugated polymers for realizing efficient all-polymer solar cells (all-PSCs), and the state-of-the-art PCEs have just recently exceeded 10%.21–34
Besides the significant efforts in developing new polymer donors and acceptors, another promising approach for optimizing PCEs is to construct a ternary BHJ blend incorporating a third component that is called “compatibilizer” or “additive”.35–37 For example, Hou et al. reported a noticeably enhanced PCE (from 5.53 to 7.07%) for a PCE12:N2200-based all-PSC by adding a high boiling point solvent of 1,8-diiodooctane (DIO).38 Additives of π-conjugated small molecules and polymers were also reported to possess similar effects for all-PSCs.39 For example, Chen et al. introduced a BDT-based conjugated polymer (J71) as a compatibilizer to improve the PCE of an all-PSC comprising PCE12 and NDI-based acceptor polymer (PNDI-2T-TR(5)) from 7.51 to 9.05%.40
We recently demonstrated the effective compatibilizer function for a series of rod–coil or coil–rod–coil block copolymers that were synthesized by the nucleophilic substitution reaction of the propagation anion of polystyrene with the bromoalkane terminals of P3HT.41–43 When the block copolymers were employed as P3HT:PCBM interfacial compatibilizers, it was found that the coil–rod–coil triblock copolymer has a superior compatibilizer effect to that of the coil–rod diblock copolymer due to the higher crystallization capability of the P3HT domain. In addition, the triblock copolymer had a higher thermal property than the diblock copolymer. Many other block copolymer-based compatibilizers were also reported by scientists in the field.44,45 However, it should be noted that the syntheses of all these block copolymers require multi-step reactions involving strict living polymerization techniques. In addition, most of these block copolymer compatibilizers were employed for fullerene and non-fullerene small molecule acceptor-based OPVs.46–50 To the best of our knowledge, no coil–rod–coil block copolymers have been applied as the interfacial compatibilizers for all-PSCs thus far, although an all-conjugated block copolymer has recently found to become an effective compatibilizer.51
Based on the above consideration,41–43 in this study, we selected poly(diethynylthiophene) (PDET) as the “rod” platform and it was successfully reacted with two equivalents of azide-substituted “coil” polystyrene under CuAAC conditions to yield a series of coil–rod–coil block copolymers, PSm-b-PDETn-b-PSm. Both PDET and PS37-b-PDET29-b-PS37 (P1 as a representative) were investigated as the compatibilizers for an all-PSC based on the PCE12:N2200 blend. It was shown that adding 1 wt% P1 into the binary blend can deliver a relative ∼6% enhancement in PCE; whereas, adding 1 wt% PDET did not provide any positive effects on the photovoltaic performance. This result clearly suggests that the coil–rod–coil triblock copolymer design provides a more prominent compatibilizer function. Besides, we also unveiled that the compatibilizer can help reduce the device's potential loss to produce a higher open-circuit voltage than the control device as benefitted from its capability of tuning the BHJ morphology of the blend film.
Results and discussion
Synthesis of block polymers
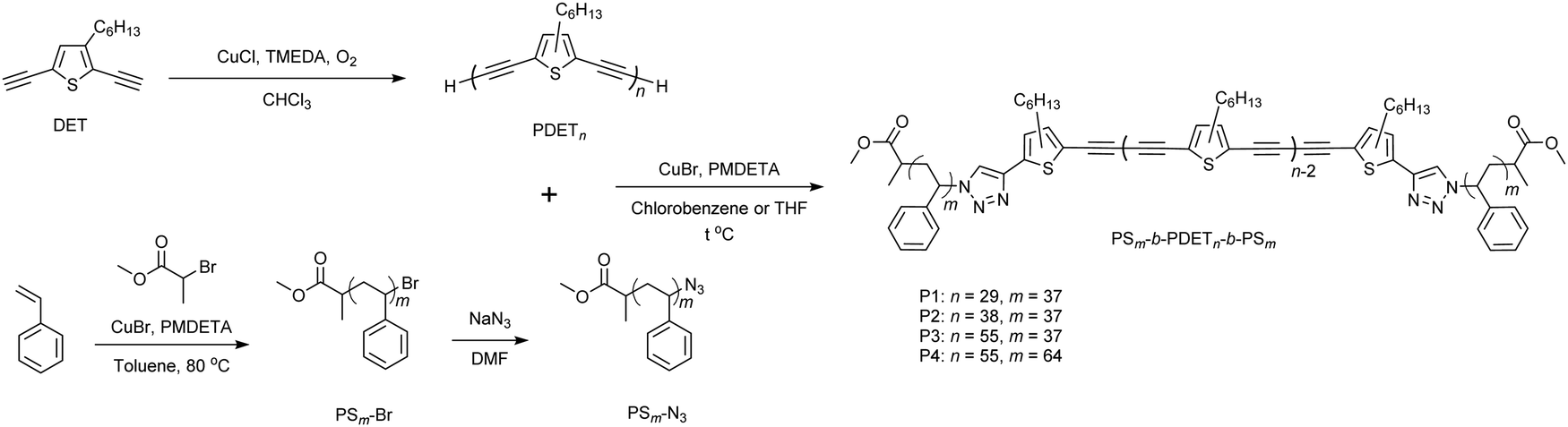
We recently reported the synthesis of poly(3-hethylthiophene-2,5-diylbutadiynylene) (PDET) by self-polycondensation of 2,5-diethynyl-3-hexylthiophene (DET) using Hay coupling with CuCl/N,N,N′,N′-tetramethylethylenediamine (TMEDA) catalysts in the presence of air at room temperature (Scheme 1).52 Due to the limited chemical stability of DET in air, the monomer was taken in a polymerization flask in the form of a n-hexane solution (174 mg in 1 mL). However, in this work, we improved the polymerization rate by increasing the monomer concentration (154 mg in 0.3 mL), which also resulted in higher number-average molecular weights (Mns). This allowed the control of the molecular weights of PDET. Polymerization was quenched after 40 and 60 min to synthesize two PDETs with the Mn values (estimated from GPC) of 8.14 kg mol−1 (named PDET38) and of 11.8 kg mol−1 (named PDET55), respectively (Table S1, ESI†). Although it was possible to synthesize PDET with a Mn value of up to 20 kg mol−1 using this method, it was decided to limit its Mn value in the range of ∼10 kg mol−1 to better match those of the PS parts of the targeted triblock copolymers (vide infra). The presence of terminal alkynes in PDET was confirmed from the 1H NMR peak at 3.49 ppm and FTIR signal at 3309 cm−1 (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H stretching). The Mn values of the polymers were estimated by comparing the 1H NMR peak areas of the terminal alkynes to those of the repeat DET units (Fig. S1 and S2, ESI†), showing a good agreement with the values determined by GPC (Table S1, ESI†). This also demonstrated that there are no noticeable side reactions at the terminal alkynes during polymerization. Therefore, PDET38 and PDET55 together with the previously reported PDET (labeled as PDET29)52 were employed for the synthesis of triblock copolymers, because the alkynes of polymer terminals are a functional group suitable for click chemistry-based post-modification reactions.
C–H stretching). The Mn values of the polymers were estimated by comparing the 1H NMR peak areas of the terminal alkynes to those of the repeat DET units (Fig. S1 and S2, ESI†), showing a good agreement with the values determined by GPC (Table S1, ESI†). This also demonstrated that there are no noticeable side reactions at the terminal alkynes during polymerization. Therefore, PDET38 and PDET55 together with the previously reported PDET (labeled as PDET29)52 were employed for the synthesis of triblock copolymers, because the alkynes of polymer terminals are a functional group suitable for click chemistry-based post-modification reactions.
 | ||
| Scheme 1 Synthetic routes for PSm-b-PDETn-b-PSm triblock copolymers, P1, P2, P3 and P4. | ||
Lutz and Matyjaszewski have showed that the synthesis of Br-functionalized polystyrene (PS-Br) is a trade-off between conversion (linked to Mn) and functionality of the terminal Br, thus leading to optimized Mn values of PS-Br to less than ∼10 kg mol−1.53 They also found that a high Mn value and high end-group functionality can be achieved simultaneously by using a high monomer-to-catalyst/initiator ratio and quenching polymerization at early stages. According to this method, PS37-Br with a Mn value of 3.97 kg mol−1 and PS64-Br with a Mn value of 6.85 kg mol−1 (estimated from GPC) were synthesized (Table S1, ESI†). The polydispersity (Đ: Mw/Mn) was 1.05 for both polymers. Their degree of polymerization (DP), estimated from the 1H NMR spectroscopy (Fig. S3 and S4, ESI†), showed a good agreement with the GPC results and the difference was merely two repeat units. The integration ratio of the 1H NMR signal corresponding to –CH–Br (4.35–4.59 ppm) against the one associated with the terminal –OCH3 (3.37–3.54 ppm) suggested that the Br-functionalization was over 90% for both polymers. An azido-functionalized polystyrene (PS-N3) was then synthesized by reacting the corresponding PS-Br with an excess sodium azide (NaN3), following the procedure reported in the literature.54 Successful formation of PS37-N3 and PS64-N3 was confirmed by 1H NMR and FTIR spectroscopies. In the 1H NMR spectra, the signal at 4.35–4.59 ppm (–CH–Br) disappeared, while a new broad peak at 3.90 ppm (–CH–N3) appeared (Fig. S5, ESI†). The integration ratio of the signals confirmed that the azido-functionality (fN3) of the polymers is in line with that of the corresponding PS-Br polymers. In addition, a strong signal at 2097 cm−1, ascribed to the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NN stretching, appeared in the FTIR spectra (Fig. S6–S9, ESI†). GPC results suggested that the Đ values of the obtained PS-N3 polymers were the same as those of the starting PS-Br polymers.
NN stretching, appeared in the FTIR spectra (Fig. S6–S9, ESI†). GPC results suggested that the Đ values of the obtained PS-N3 polymers were the same as those of the starting PS-Br polymers.
Triblock copolymers, PSm-b-PDETn-b-PSm, were then synthesized by the CuAAC click reaction between PDET and a slight excess of PS-N3 using CuBr/N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) as the catalyst.54 The reaction was conducted either in chlorobenzene or tetrahydrofuran (THF). After the reaction, the crude product mixture was thoroughly washed with acetone to remove the unreacted PS-N3. The targeted triblock copolymers were extracted with cold chloroform, leaving behind insoluble solids, which could be the products of further polycondensation and/or cross-linking of PDET.52 Thus, four triblock copolymers P1 (PS37-b-PDET29-b-PS37), P2 (PS37-b-PDET38-b-PS37), P3 (PS37-b-PDET55-b-PS37), and P4 (PS64-b-PDET55-b-PS64) were obtained in 43–53% yields. The 1H NMR peaks at 3.90 ppm ascribed to CH–N3 of PS-N3 and 3.49 ppm ascribed to CH of PDET disappeared, while the other characteristic signals were present, confirming successful macromolecular clicking (see Fig. 1a for P1). In addition, FTIR signals at 2097 cm−1 (NNN stretching) and 3309 cm−1 (C–H stretching) disappeared, which indicated that all the terminal alkynes of PDET and azide groups of PS-N3 were used up (Fig. 1b and Fig. S8–S10, ESI†). GPC measurements revealed that the Mn values of triblock copolymers were close to those expected from the reacted PS-N3 and PDET polymers (Table 1). Notably, the Đ values for the triblock copolymers decreased compared to those of the starting PDET polymers (Fig. 1c and Fig. S11, ESI†). In addition, a comparison of the 1H NMR peak integration value at 3.37–3.54 ppm (–OCH3) to those of the repeat DET units allowed the estimation of the extent of the click reaction. For example, the 1H NMR of P1 suggested that there were 31 repeat DET units per PS chain. This result indicated that 7% of the PDET29 chains were not clicked to PS, probably due to the loss of terminal alkynes under the click reaction conditions. However, due to the unlikelihood of losing both terminal alkynes on a single polymer chain, it is assumed that at most 14% of PDET29 formed the diblock copolymer with PS37. The incomplete click reactions led to the observed lower Mn value in GPC than the expected values (see P1, P3 and P4 in Table 1).
 | ||
| Fig. 1 Evidence of successful synthesis of P1 by comparing (a) 1H NMR spectra, (b) FTIR spectra, and (c) GPC curves of PS37-N3, PDET29, and P1. Magnified regions of the 1H NMR spectra at 3–5 ppm are provided for PS37-N3 and P1 to visualize the peak associated with –N3. All the 1H NMR and FTIR spectra as well as the GPC curves were normalized and manually offset for the convenience of visualization. For P2, P3 and P4, see Supporting Information (Fig. S8–S11, ESI†). | ||
| Polymer | Measured Mna (kg mol−1) | Đ | Expected Mnb (kg mol−1) | Click reaction conditionsc |
|---|---|---|---|---|
| a Measured in GPC with o-dichlorobenzene at 40 °C and molecular weights estimated by comparing to polystyrene standards. b Calculated from the Mns of the starting PDET and PS-N3, assuming 100% reaction yield. c All reactions performed under an Ar atmosphere using dry solvents and pre-purified CuBr catalyst. | ||||
| P1 (n = 29, m = 37) | 13.6 | 2.04 | 14.1 | Chlorobenzene, 4 h, 80 °C |
| P2 (n = 38, m = 37) | 16.2 | 2.56 | 16.0 | THF, 7 h, 60 °C |
| P3 (n = 55, m = 37) | 18.9 | 2.34 | 19.6 | THF, 4 h, 60 °C |
| P4 (n = 55, m = 64) | 23.5 | 2.21 | 25.3 | THF, 4 h, 60 °C |
Thermal properties
Thermogravimetric analysis (TGA) showed that PDET29 had the onset of weight loss (decomposition temperature, Td) at 352 °C, while this value shifted to 261 °C for P1, which was closer to the Td of PS37-N3 (Fig. 2a and Table 2). In addition, the TGA curve of P1 had two slopes corresponding to the decomposition of PDET29 and PS37-N3. The PDET content of P1 estimated from the TGA curve was 59% by mass. This value is higher than those calculated from the 1H NMR spectra (47%) and GPC (44%). Thus, the above result must be considered with care, since the decomposition rates of PDET29 and PS37 in P1 might be different from those of the pristine polymers. For example, it was previously shown that PDET undergoes cross-linking by 1,4-addition of 1,3-butadiyne moieties at a temperature (T) above 110 °C.55 This can be clearly seen from the exothermic peaks (Texo,1 and Texo,2) in the differential scanning calorimetry (DSC) curve of PDET29 (Fig. 2b). While Texo,1 disappeared in P1, Texo,2 was present, indicating that PDET29 in P1 still underwent cross-linking at a temperature >150 °C. The sample turned completely black after heating to 250 °C, which further supports the possibility of cross-linking.55 As cross-linking was shown to negatively affect the semiconducting properties of PDET, the increase in the cross-linking initiation temperature should be beneficial for P1. PS37-N3 exhibited a glass transition temperature (Tg) of 89 °C, which was in agreement with the results reported earlier.54 In addition, decomposition of the azide group (Td,N3) was observed at 158 °C.56 The PS37 in P1 retained its Tg at nearly the same temperature. The endothermic peaks of PS37-N3 and P1 at ∼50 °C disappeared in the 2nd heating scan (Fig. S12, ESI†). | ||
| Fig. 2 Comparison of (a) TGA curves, (b) DSC curves (1st heating scan), (c) UV-vis absorption spectra and (d) CV curves of PDET29 and P1. DSC curves, UV-vis spectra and CV curves were normalized and manually offset for the convenience of visualization. | ||
| Polymer | T d (°C) | T g (°C) | T exo (°C) | E HOMO (eV) | E LUMO (eV) |
λ
solmax![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) e (nm) e (nm) |
λ
filmmaxe (nm) |
λ onset (nm) | E g,opt (eV) |
|---|---|---|---|---|---|---|---|---|---|
| a Determined from the onset of mass loss in the TGA curve. b Determined from step change in the heating cycle of the DSC curve. c Other exothermic peaks. d Determined from the onsets of oxidation and reduction peaks in CV. e UV-vis absorption maxima of CHCl3 solution (λsolmax) and the as-cast film (λfilmmax). Onset of UV-vis absorption band (λonset) of the as-cast films. Eg,opt calculated from λonset of the as-cast films. | |||||||||
| PS37-N3 | 255 | 89 | 158 | — | — | — | — | ||
| PDET29 | 352 | — | 154, 206 | −5.78 | −3.05 | 452 | 463 | 531 | 2.34 |
| P1 | 261 | ∼90 | >200 | −5.81 | −3.02 | 452 | 461 | 531 | 2.34 |
Optical and electrochemical properties
PDET29 and P1 exhibited identical light absorption spectra with an optical bandgap (Eg,opt) of 2.34 eV (Fig. 2c and Table 2). The absorption spectra of the as-cast thin films showed a notable red-shift (∼10 nm) when compared to the corresponding solution spectra, indicating substantial aggregation in thin films. Cyclic voltammograms (CVs) of the thin films of PDET29 and P1 exhibited irreversible oxidation and reduction steps. Both polymers displayed the highest occupied molecular orbital (HOMO) energy level (EHOMO) of −5.8 eV and the lowest unoccupied molecular orbital (LUMO) energy level (ELUMO) of −3.0 eV. Similar optical and electrochemical results were obtained for P2, P3 and P4 (Fig. S13, ESI†). These results were somewhat expected, since attaching PS blocks did not alter the fundamental electronic properties of the PDET backbone. In addition to this, PDET38 and PDET55 had no difference in the UV-vis absorption spectra and CV curves compared to PDET29 (Fig. S13, ESI†), suggesting that all PDETs reached an effective conjugation length.57 The identical UV-vis absorption spectra also indicated that the molecular packing motif does not change among these polymers.58AFM measurements
Tapping-mode atomic force microscopy (AFM) topographical images of the as-cast films of P1, P2, P3 and P4 on an octadecyltrimethoxysilane-modified Si/SiO2 substrate exhibited smooth surfaces (Fig. 3). When the films were annealed at a temperature slightly above Tg of the PS block (at 120 °C), distinct surface features appeared for each polymer. P1 formed isolated spherical particles with diameters of up to 200 nm and height of up to 60 nm. In case of P2 film, these spheres were even larger with a cross section of up to 500 nm and a height of up to 120 nm. Interestingly, the spheres were much smaller in the P3 film (<100 nm in diameter), while new interconnected stripes were formed. The larger area scan image (Fig. S14, ESI†) showed that this network covered the entire surface and an individual stripe spanned up to several micrometers uninterruptedly. In the P4 film, more sphere-like particles were again observed. The spheres were interconnected as in the case of P3. These changes in surface morphology suggested the possible occurrence of phase separation in these triblock copolymer films. Notably, different surface structures could be achieved by modifying the molecular weights of the polymer blocks. | ||
| Fig. 3 Tapping-mode AFM topographical images of the as-cast films of (a) P1 (magnified by a factor of 2 from the original image), (b) P2, (c) P3 and (d) P4 and the annealed films of (e) P1, (f) P2, (g) P3 and (h) P4. Films were annealed on a hotplate at 120 °C for 30 min. | ||
Stripe-like morphology of the annealed P3 film can be explained to be due to the low PS content both in terms of mass fraction (42%) and chain length (29%). Shorter PS blocks would make the self-assembly into spherical particles difficult. In contrast, the mass fraction of the PS blocks in P1, P2 and P4 was in the range of 50–60%, and about 40% of the single polymer chain length was PS. It was thus possible to form larger spheres for P1 and P2. In the case of P4, much longer PS chains could entangle and form more interconnected spherical particles. A delicate balance between the coil and rod fraction in the triblock copolymers produced unique thermodynamically stable self-assembled structures.
For comparison, AFM images of the as-cast and annealed PDET29 and P1 films were also measured (Fig. S15, ESI†). Both as-cast and annealed PDET29 films showed relatively smooth surfaces, expectedly indicating that there was no phase separation. Annealing of P1 at a temperature closer to its Tg (100 °C) did not result in phase separation as well, suggesting that temperature was not enough to mobilize the polymer chains. Interestingly, when the P1 film was rapidly heated to 200 °C, the surface roughened but the structures were different from those previously observed in the films annealed at 120 °C. The underlying surface still resembled that of the P1 film annealed at 100 °C. These findings led us to theorize that as the temperature quickly increased above Tg, the phase separation did not have enough time to occur. Instead, cross-links could form at temperatures >150 °C and stiffen the film fixing the initial surface morphology. It is worth noting that substrates are usually not heated to temperatures as high as 200 °C in the device fabrication process to avoid macro-phase separation.
Compatibilizer effect in all-PSCs
We have previously demonstrated the compatibilizer effect for a series of coil-rod-coil block copolymers in the fullerene and non-fullerene small molecule-based OPVs.41–43 We herein investigate the compatibilizer effect of the synthesized PDET29 (referred as PDET hereafter) and P1 in an all-PSC using a representative PCE12:N2200 BHJ blend and compare their differences (Fig. 4a). Details of device fabrication are described in the Experimental Section. Fig. 4b shows the current density–voltage (J–V) curves of the fabricated all-PSCs measured under AM 1.5G illumination, and the relevant photovoltaic parameters, including open-circuit voltage (Voc), short-circuit current (Jsc), fill factor (FF), and photoconversion efficiency (PCE), are summarized in Table 3. | ||
| Fig. 4 (a) Schematic configuration and the energy-level diagram of the device and the chemical structures of the studied materials. (b) The J–V and (c) EQE curves of the fabricated all-PSCs using different compatibilizers. Inset in (c) shows the relative improvement in the EQE between the control device and the 1 wt% P1 device. | ||
| V oc (V) | J SC (mA cm−2) | FF (%) | PCEmaxb (%) | |
|---|---|---|---|---|
| a PCE12:N2200 BHJ system. b The average PCEs shown in the table are based on 15 devices. | ||||
| Controla | 0.878 (0.863 ± 0.015) | 11.50 (11.30 ± 0.2) | 58.44 (57.61 ± 0.83) | 5.90 (5.61 ± 0.29) |
| 1 wt% PDET | 0.862 (0.843 ± 0.023) | 9.64 (9.47 ± 0.51) | 59.05 (58.17 ± 3.53) | 4.91 (4.65 ± 0.32) |
| 1 wt% P1 | 0.883 (0.875 ± 0.008) | 11.50 (11.37 ± 0.13) | 61.45 (59.70 ± 1.75) | 6.24 (5.93 ± 0.31) |
| 3 wt% P1 | 0.877 (0.832 ± 0.045) | 10.04 (10.10 ± 0.58) | 60.73 (57.64 ± 4.73) | 5.33 (4.84 ± 0.57) |
As seen, the control device showed a decent maximum PCE (PCEmax) of 5.90% with a Voc of 0.878 V, a Jsc of 11.50 mA cm−2, and an FF of 58.44%. After adding the compatibilizers, the performance of the derived device showed a distinctly different change. Compared to the control device, the device added with 1 wt% P1 showed an improved PCEmax of 6.24% with a slightly larger Voc of 0.883 V, a similar Jsc of 11.50 mA cm−2, and an increased FF of 61.45%. Considering the mismatched energy levels of P1 compared to those of PCE12 and N2200, such a performance enhancement suggests the potential compatibilizer effect for P1. That is, adding P1 enables a better BHJ morphology. In contrast, the device added with 1 wt% PDET gave a lousy PCEmax of 4.91% with a decreased Voc of 0.862 V, a largely decreased Jsc of 9.64 mA cm−2, and a similar FF of 59.05%. These results clearly unveil the important role of the block copolymer design on the resultant compatibilizer function. Similar to the previous results reported in the literature,43 the coil–rod–coil triblock P1 demonstrated a more prominent compatibilizer function. This highlights the critical function of the coil segment in mediating the BHJ morphology and this will be discussed later. Despite a decent compatibilizer effect, the tolerance of P1 in the BHJ blend was limited to below 1 wt%, mainly due to its insulating properties and the phase aggregation induced by the excess amount of block copolymers. The device performance decreased to a PCEmax of 5.33% with a Voc of 0.877 V, a Jsc of 10.04 mA cm−2, and an FF of 60.73% when the blending amount of P1 was increased to 3 wt%.
Fig. 4c presents the external quantum efficiencies (EQE) of these devices. Compared to the control device, the device added with 1 wt% of P1 exhibited a slightly enhanced photoresponse in the wavelength region of 400–800 nm. Inset in Fig. 4c shows the relative improvement of the EQE, which is clearly attributed to the enhanced absorption of PCE12 and N2200. On the other hand, both devices with 1 wt% PDET and 3 wt% P1 exhibited much reduced photoresponse. This result suggests that the original BHJ morphology underwent an undesired phase separation, resulting in a poor carrier collection. Notably, for the device with 3 wt% P1, an additional peak at 400–500 nm appeared and it might have arisen from the absorption of P1 (Fig. 2c). Also, the changed EQE profile compared to that of the control device again suggests that the excess P1 varied the phase aggregation of the BHJ blend.
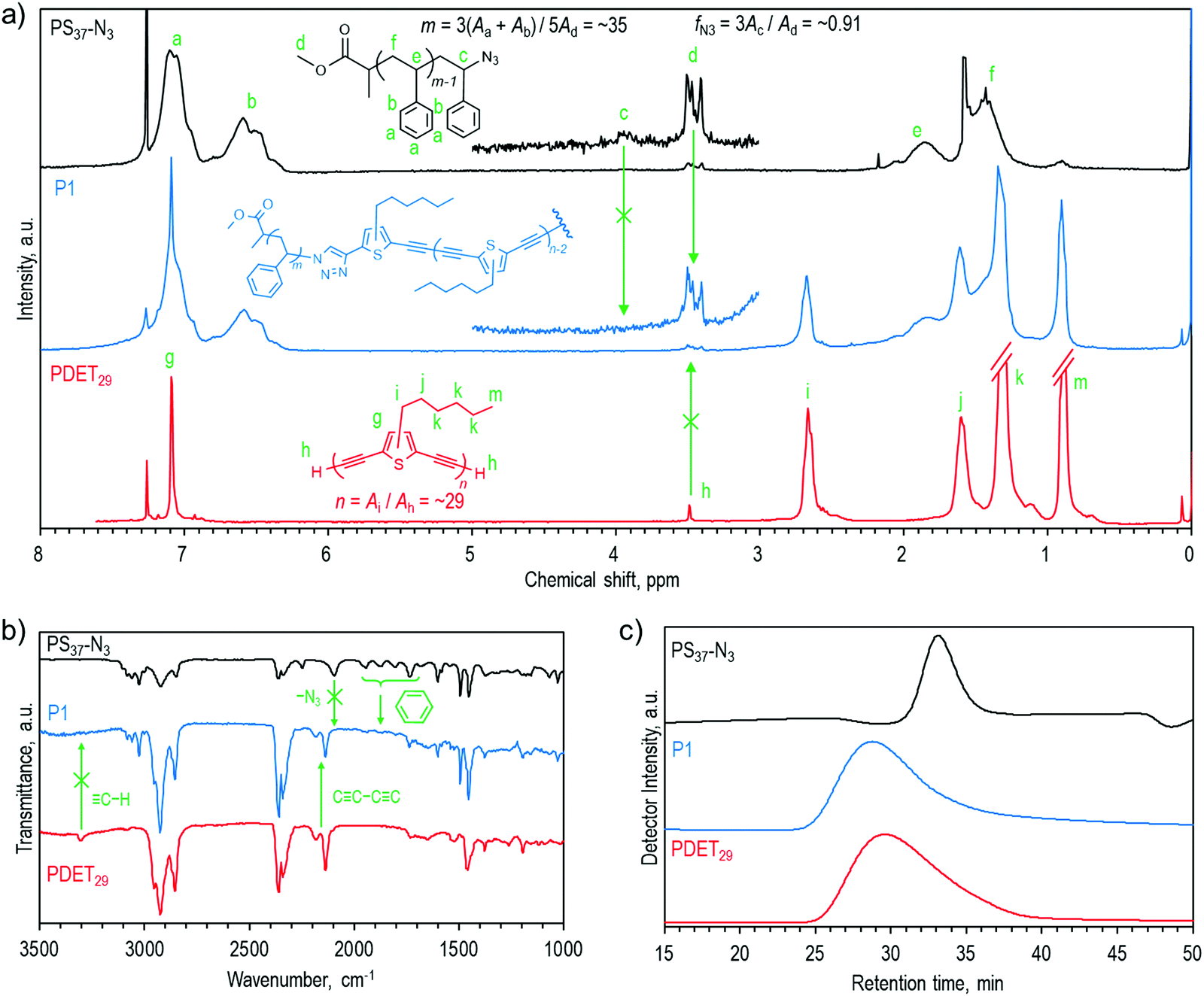
Charge recombination behaviors of these devices were investigated by plotting photocurrent density (Jph) as a function of effective voltage (Veff), as shown in Fig. 5a. Jph is defined by the equation Jph= JL − JD, where JL and JD stand for the current density under AM 1.5G illumination and under the dark conditions, respectively, and Veff is defined as Veff= V0 − Vbias, where V0 is the voltage while Jph is zero and Vbias is the applied bias. Jph generally approaches a saturated value (Jsat) at the high bias region, and under these conditions, charges will fully dissociate into free carriers and be swept out by the electric field. Under the maximum power output conditions, the charge collection probability can be evaluated using the ratio Jph,max/Jsat, and the exciton dissociation probability can be estimated using the ratio Jph,sc/Jsat under the short-circuit conditions. It thus can be expected that only a portion of photogenerated excitons dissociate into free carriers and are collected by electrodes at a certain Veff between the short-circuit conditions and maximum power output conditions, and the charge dissociation and collection probabilities can be calculated using the ratio Jph/Jsat.59 As seen, at the same Veff of 0.1 V, the estimated probabilities for the control, 1 wt% P1, and 1wt% PDET all-PSCs were 0.54, 0.60, and 0.56, respectively. The highest value observed for the 1 wt% P1 device suggests its most efficient charge dissociation and collection capability among the studied devices. Fig. 5b displays the dependence of Jsc on various light intensities (Plight). The relationship between them follows a power-law dependence with respect to the equation Jsc ∝ (Plight)α. Generally, an α value approaching 1.0 indicates that all the dissociated free carriers are collected by electrodes before bimolecular recombination, while an α value smaller than 1.0 suggests the occurrence of bimolecular recombination.60,61 As shown, all of the fabricated devices showed a linear dependence of Jsc on Plight with a logarithmic plot. Among them, the 1 wt% P1 device possessed the highest α value of 0.9896, implying its extremely low bimolecular recombination. The above result clearly supports the performance enhancement observed for the 1 wt% P1 device. Again, when considering the electronic properties and energy levels of P1, such enhancement should stem from the optimization of BHJ morphology of the photoactive blend.
 | ||
| Fig. 5 (a) Jph–Veff characteristics, (b) JSC–light intensity fitting curves, and (c) normalized FTPS-EQE and the fitting curves of the fabricated all-PSCs using different compatibilizers. | ||
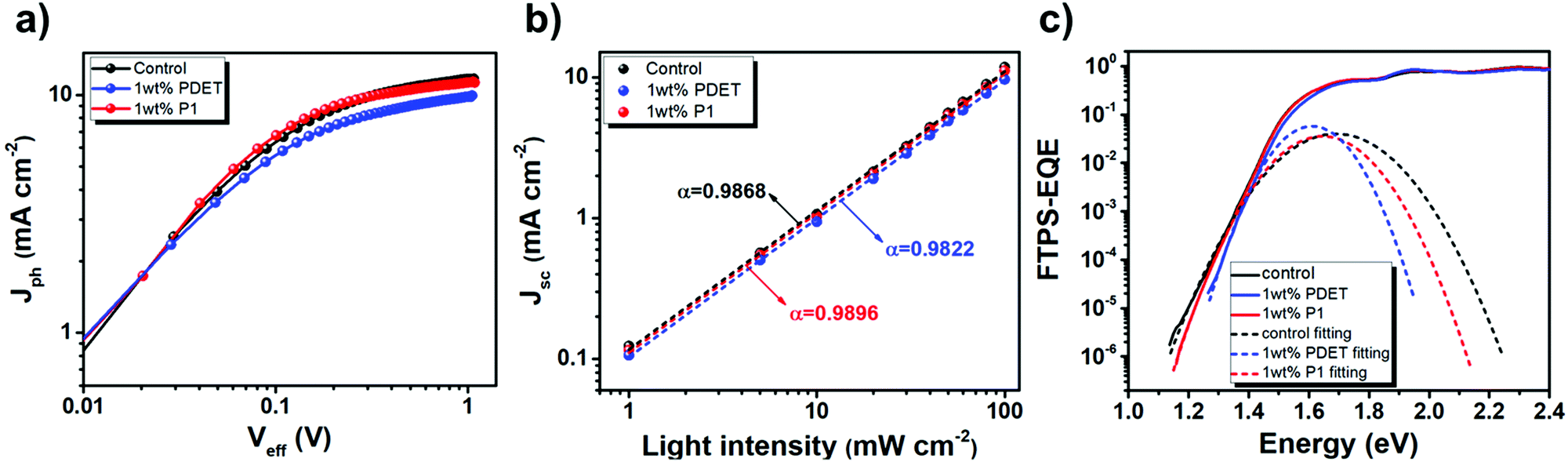
To scrutinize the possible changes of the BHJ morphology, grazing incidence wide-angle X-ray diffraction scattering (GIWAXS) measurements of the control binary blend and the ternary blends with 1wt% P1 and PDET prepared on the ZnO electron-transporting layer were carried out. Fig. 6a–c show their 2-D GIWAXS patterns and the detailed information of the peaks are depicted in Fig. 6d. In the in-plane direction, all these films showed a similar (100) diffraction peak (qxy = 0.159 Å−1, 0.150 Å−1, and 0.152 Å−1 for control, 1 wt% P1, and 1 wt% PDET films, respectively), corresponding to the lamellar stacking with a d-spacing of ∼39–41 Å. The crystal coherence length (CCL) value was then calculated using the Scherrer equation of 2π/Δq, where Δq represents the full width half maximum (FWHM) of the diffraction peak obtained by Gaussian fitting. Both films with compatibilizers delivered a lower CCL value (107.96 Å for 1 wt% P1 and 106.1 Å for 1 wt% PDET) than that (149.63 Å) of the control film. This result suggests that the addition of P1 and PDET suppresses the phase separation between the constituent components.43
 | ||
| Fig. 6 Fig. 6 2D GIWAXS patterns of (a) the binary PCE12:N2200 film and (b and c) the ternary blend films with different compatibilizers. (d) Their corresponding 1D linecuts in the out-of-plane (solid line) and in-plane (dotted line) directions. | ||
For the diffraction in the out-of-plane direction, all of these films exhibited an obvious (010) peak at ∼1.5 Å−1, representing the face-on orientation of these polymer films. The control and 1 wt% P1 films exhibited similar CCL values of 33.11 and 31.01 Å, respectively; whereas, the 1 wt% PDET film delivered the lowest CCL value of 27.24 Å. Moreover, an additional peak at qz = 1 Å−1 with different CCL values (20.45 Å, 17.46 Å, and 17.42 Å for the control, 1 wt% P1 and 1 wt% PDET films, respectively) were observed in these films. This peak can be attributed to the π–π stacking of PCE12.62 These results thus indicate that the addition of PDET largely varies the polymer packing in the out-of-plane direction, which hampers the transport of charge carriers. Notably, another peak at qz = 0.19 Å−1 was found only for the control film but not for the other films. This suggests that the lamella packing of the polymers in the out-of-plane direction diminished after adding the compatibilizers, which enabled an easier formation of the face-on orientation for the BHJ films. In brief, the addition of P1 suppresses the self-aggregation of photoactive components through facilitating their intermixing in the in-plane direction without impacting the π–π stacking in the out-of-plane direction, leading to better photovoltaic performances.
We noticed that Voc was slightly improved for the 1 wt% P1 device and this might have been associated with the reduction of potential loss. To clarify this, we measured the total energy loss (Eloss) for the control, 1 wt% P1, and 1wt% PDET devices using the Fourier transform photocurrent spectroscopy EQE (FTPS-EQE). Fig. 5c presents the normalized curves along with the fitting curves of these devices and the relevant parameters are summarized in Table S2, ESI.† In principle, the total Eloss can be divided into three contributions: (i) charge generation (ΔE2); (ii) radiative recombination (ΔE1); and (3) non-radiative recombination (ΔE3), and it can be calculated using the following equation:63,64
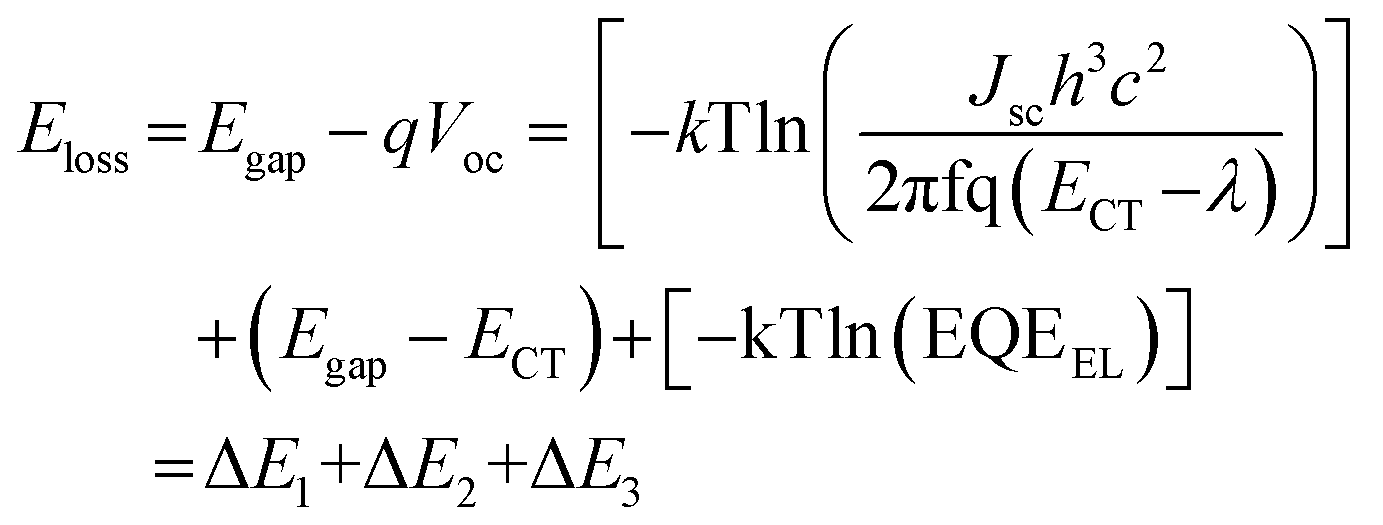
As shown in Table S2, ESI,† the ECT values for the control, 1 wt% P1, and 1 wt% PDET devices were 1.42, 1.44, and 1.46 eV, respectively, and all of them exhibited similar ΔE1 values (0.26–0.27 eV) that are inherent to the devices. The ΔE2 values for the control, 1 wt% P1, and 1 wt% PDET devices were 0.061, 0.047, and 0.056 eV, respectively. Note that the ΔE2 values have been generally recognized to be associated with the energetic offsets between the photoactive materials, and a smaller energetic offset results in a smaller ΔE2 value.65 The smallest ΔE2 value observed for the 1 wt% P1 device clearly benefits from its compatibilizer effect that better optimizes the BHJ morphology of the photoactive blend. Adding additives increased the ΔE3 value of the device, but compared to the 1wt% PDET device, the 1wt% P1 device possessed a lower ΔE3 value, indicating its superior capability to overcome the non-radiative recombination loss. Overall, the 1wt% P1 device delivered the smallest Eloss among the studied devices, confirming the advantage of the block copolymer compatibilizer in reducing the potential loss.
Conclusions
Straightforward synthesis of triblock copolymers from PDET and PS-N3 by using efficient Cu-catalyzed azide–alkyne click reaction was demonstrated in this study. The resulting triblock copolymers retained the optical and electrochemical properties of the starting PDET, which is advantageous for practical applications. Pure triblock copolymers exhibited signs of phase separation upon annealing at a temperature above the Tg of the PS block. One-step simple preparation of poly(arylenebutadiynylene)s and reliability of the azide–alkyne click reaction create a possibility for developing a variety of coil–rod–coil block copolymers. We finally explored the compatibilizer function of the starting PDET and the derived triblock copolymers (P1 as the representative) in an all-polymer solar cell based on the PCE12:N2200 blend. We found that adding 1 wt% P1 can result in a relative ∼6% enhancement in PCE, but adding 1 wt% PDET delivered a decreased performance. Besides elucidating P1's compatibilizer function on tuning the BHJ morphology of the blend film, its effect on reducing device's potential loss was also investigated. Collectively, our results provide an efficient method for synthesizing versatile coil–rod–coil triblock copolymers and demonstrate their potential for photovoltaic applications.Experimental
Materials and synthesis
All reagents and chemicals were purchased from Tokyo Chemical Industry (TCI), Kanto Chemical, Co., Inc., and Sigma-Aldrich and used as received unless otherwise stated. CuBr was purified by stirring in glacial acetic acid for 6 h, followed by filtering, washing with dry methanol and drying in a vacuum oven. Synthesis of DET monomer and PDET29 has been reported before.52PDET38 and PDET55 were synthesized similar to PDET29. PS37-Br and PS64-Br were synthesized using a modified literature procedure.3,54 The polymerizations were carried out in an Ar-filled glovebox, and the initiator amount and reaction time were varied to achieve desired molecular weights and polydispersities. PS37-N3 and PS64-N3 were prepared as reported in the literature.54C−H stretching), 2924, 2852 (C–H stretching), 2207, 2142 (CC–CC stretching), 2104 (CC stretching), 1728, 1645, 1598, 1518, 1465, 1437, 1393, 1376, 1299, 1264, 1242, 1170, 1111, 1078, 1023, 985, 954, 916, 876, 858, 837, 806, 721, 700, 657, 644, 631, 620.
A procedure same as above using a reaction time of 60 min afforded 105 mg of PDET55 (70% yield). GPC (o-DCB): Mn = 11.8 kg mol−1, PDI = 2.83. 1H NMR and FTIR spectra were identical to those of PDET38.
C stretching), 1375, 1328, 1182, 1155, 1067, 907, 840, 754.
A procedure same as above afforded 3.04 g of PS64-Br from 12 mL of styrene using the styrene/CuBr/PMDETA/MBP ratio of 200:1:1:1 (molar) and the reaction time of 9 h (27.8% yield). GPC (o-DCB, 40 °C): Mn = 6.85 kg mol−1, PDI = 1.05. 1H NMR and FTIR spectra were identical to those of PS37-Br.
NN stretching), 1943, 1942, 1870, 1867, 1802, 1799, 1738, 1736, 1601, 1583, 1540, 1492, 1452 (CC stretching), 1375, 1328, 1182, 1155, 1067, 1027, 907, 840, 754.
A procedure same as above afforded 1.90 g of PS64-N3 from 2.00 g of PS64-Br (95% yield). GPC (o-DCB, 40 °C): Mn = 6.87 kg mol−1, PDI = 1.05. 1H-NMR and FTIR spectra were identical to those of PS37-N3.
NN peak (2097 cm−1) in the FTIR spectrum. Chloroform (20 mL) was added to the collected solid and the mixture was sonicated for 30 min. Undissolved solids were removed by filtration. The filtrate was concentrated and precipitated into methanol. The solid was collected by filtration and dried under vacuum to afford 41 mg of P1 as a fluffy dark orange solid (45% yield). GPC (o-DCB): Mn = 13.6 kDa, PDI = 2.04. 1H NMR (CDCl3, ppm) δ: 6.46–7.42 (br), 7.09 (s), 3.39–3.49 (m), 2.67 (br), 1.81–1.86 (br), 1.60 (br), 1.44 (br, CH–Ph), 1.31 (br), 0.90 (br). FTIR (cm−1): 3309 (CC–H stretching), 3082, 3081, 3062, 3059, 3025, 3002, 3001, 2923, 2849 (C–H stretching), 2207, 2142 (CC–CC stretching), 1943, 1942, 1870, 1867, 1802, 1799, 1738, 1736, 1728, 1645, 1601, 1598, 1583, 1540, 1518, 1492, 1465, 1452 (CC stretching), 1437, 1393, 1376, 1328, 1299, 1264, 1242, 1182, 1170, 1155, 1111, 1078, 1067, 1027, 1023, 985, 954, 916, 907, 876, 858, 840, 837, 806, 754, 721, 700, 657, 644, 631, 620.
A procedure same as above afforded 40 mg of P2 from PDET38 (38 mg, 4.7 μmol) and PS37-N3 (48 mg, 12.0 μmol) in THF (1.3 mL) stirred at 60 °C for 7 h (53% yield). GPC (o-DCB): Mn = 16.2 kg mol−1, PDI = 2.56. 1H-NMR and FTIR spectra were identical to those of P1.
A procedure same as above afforded 30 mg of P3 from PDET55 (40 mg, 3.40 μmol) and PS37-N3 (34 mg, 8.5 μmol) in THF (1.5 mL) stirred at 60 °C for 4 h (43% yield). GPC (o-DCB): Mn = 18.9 kg mol−1, PDI = 2.34. 1H-NMR and FTIR spectra were identical to those of P1.
A procedure same as above afforded 38 mg of P4 from PDET55 (40 mg, 3.40 μmol) and PS64-N3 (58 mg, 8.5 μmol) in THF (1.5 mL) stirred at 60 °C for 4 h (42% yield). GPC (o-DCB): Mn = 23.5 kg mol−1, PDI = 2.21. 1H-NMR and FTIR spectra were identical to those of P1.
General measurements
1H NMR (300 MHz) and 13C NMR (75 MHz) were recorded using a JEOL AL-300 spectrometer using deuterated chloroform as the solvent. GPC was measured using a JASCO GULLIVER 1500 system equipped with two Shodex GPC KF-803 columns (8.0 mm I.D. × 300 mm L) at 40 °C using o-dichlorobenzene (o-DCB) as the eluent with polystyrene standards. CV was measured using a BAS electrochemical analyzer model 612C in a three-electrode cell with the Ar-bubbled dehydrated acetonitrile solution of tetra-n-butylammonium hexafluorophosphate (0.1 M) at a sweep rate of 1 mV s−1. The working, reference, and counter electrodes were glassy carbon, Ag/AgCl, and Pt wire, respectively. Polymer films were drop cast onto the working electrode from chloroform solutions. The redox potential of ferrocene/ferrocenium (Fc/Fc+) was used for calibration. Attenuated total reflection FTIR spectroscopy was measured using a JASCO FT/IR 4200 spectrometer. TGA and DSC measurements were carried out using Rigaku TG8120 and Rigaku DSC8230 instrument, respectively, under a N2 flow at a heating/cooling rate of 10 °C min−1. UV-vis absorption spectra were recorded using a JASCO V-670 spectrophotometer. Tapping-mode AFM images was taken using a Seiko Instruments SPA-400 with a stiff cantilever Seiko Instruments DF-20. Grazing incidence wide-angle X-ray scattering (GIWAXS) data of the polymer films were obtained using beamlines 17A1 with a wavelength of 1.321 Å at the National Synchrotron Radiation Research Center (NSRRC), Taiwan.Device fabrication and characterization
An inverted device structure of ITO glass/ZnO/BHJ/MoO3/Ag was fabricated in this work. The ITO glass substrates were sequentially washed with DI water, acetone, and isopropyl alcohol for 15 min each. The dried ITO glass was then subjected to plasma treatment for 10 min. The electron transporting ZnO layer was spin-coated on the ITO glass at 4000 rpm for 30 s and then dried at 180 °C for 30 min in air. The control precursor solution of PCE12:N2200 (2:1 weight ratio) at a concentration of 8 mg mL−1 was prepared in CB. Different compatibilizers with weight ratios of 1–3% were separately blended into the control precursor solution. All of the precursor solutions were vigorously stirred at 50 °C for 12 h in a N2-filled glove box, followed by annealing at 100 °C for 10 min. The thickness of the BHJ layer was ∼80–90 nm on the ZnO layer. Finally, 10 nm MoO3 and 100 nm Ag were sequentially thermally deposited onto the BHJ layer to complete device fabrication. The active area for the device is 8.5 mm2.
The current–voltage (J–V) characteristics of the devices were measured under AM 1.5G illumination (100 mW cm−2) using a Newport LCS-100 simulator, which were recorded using a computer-controlled Keithley 2400 source measurement unit (SMU). The illumination intensity was calibrated by using a Si photodiode detector with a KG-5 filter. The external quantum efficiency (EQE) was measured with a monochromatic light from a xenon lamp during illumination (QE-R, Enlitech Co., Ltd) and using a standard single crystal Si photovoltaic cell to calibrate the light intensity under the wavelength from 300 to 900 nm. The FTPS-EQE was measured using the FTPS PECT-600 instrument from Enlitech, which calculates the detailed parameter of energy loss.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
T. M. thanks financial supports from JSPS KAKENHI Grant 19H02786, JST ASTEP JPMJTM20CS, and the Heiwa Nakajima Foundation. C. C. C. gratefully thanks financial supports from Top University Project of National Taiwan University (110L7836) and the Featured Area Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (110L9006) and the Ministry of Science and Technology in Taiwan (MOST 110-2634-F-002-043, 109-2628-E-002-008-MY3, 110-2923-E-002-007-MY3, 110-2628-E-002-011-).References
- T. Higashihara, K. Sugiyama, H.-S. Yoo, M. Hayashi and A. Hirao, Combining Living Anionic Polymerization with Branching Reactions in an Iterative Fashion to Design Branched Polymers, Macromol. Rapid Commun., 2010, 31, 1031–1059 CrossRef CAS PubMed.
- G. Polymeropoulos, G. Zapsas, K. Ntetsikas, P. Bilalis, Y. Gnanou and N. Hadjichristidis, 50th Anniversary Perspective: Polymers with Complex Architectures, Macromolecules, 2017, 50, 1253–1290 CrossRef CAS.
- A. P. Vogt and B. S. Sumerlin, An Efficient Route to Macromonomers via ATRP and Click Chemistry, Macromolecules, 2006, 39, 5286–5292 CrossRef CAS.
- J. S. Kim, J. Han, Y. Kim, H. Park, J. P. Coote, G. E. Stein and B. J. Kim, Domain Structures of Poly(3-Dodecylthiophene)-Based Block Copolymers Depend on Regioregularity, Macromolecules, 2018, 51, 4077–4084 CrossRef CAS.
- C. M. Loudy, J. Allouche, A. Bousquet, C. Courrèges, H. Martinez and L. Billon, Core@Corona Functional Nanoparticle-Driven Rod-Coil Diblock Copolymer Self-Assembly, Langmuir, 2019, 35, 16925–16934 CrossRef PubMed.
- H. Erothu, J. Kolomanska, P. Johnston, S. Schumann, D. Deribew, D. T. W. Toolan, A. Gregori, C. Dagron-Lartigau, G. Portale, W. Bras, T. Arnold, A. Distler, R. C. Hiorns, P. Mokarian-Tabari, T. W. Collins, J. R. Howse and P. D. Topham, Synthesis, Thermal Processing, and Thin Film Morphology of Poly(3-hexylthiophene)-Poly(styrenesulfonate) Block Copolymers, Macromolecules, 2015, 48, 2107–2117 CrossRef CAS.
- P. M. Reichstein, J. C. Brendel, M. Drechsler and M. Thelakkat, Poly(3-hexylthiophene)- block-Poly(tetrabutylammonium-4-styrenesulfonate) Block Copolymer Micelles for the Synthesis of Polymer Semiconductor Nanocomposites, ACS Appl. Nano Mater., 2019, 2, 2133–2143 CrossRef CAS.
- H. N. Choi, H. S. Yang, J. H. Chae, T. L. Choi and I. H. Lee, Synthesis of Conjugated Rod-Coil Block Copolymers by RuPhos Pd-Catalyzed Suzuki-Miyaura Catalyst-Transfer Polycondensation: Initiation from Coil-Type Polymers, Macromolecules, 2020, 53, 5497–5503 CrossRef CAS.
- W. Wang, T. Li, T. Yu and F. Zhu, Synthesis of Multiblock Copolymers by Coupling Reaction Based on Self-Assembly and Click Chemistry Synthesis of Multiblock Copolymers by Coupling Reaction Based on Self-Assembly and Click Chemistry, Macromolecules, 2008, 41, 9750–9754 CrossRef CAS.
- X. He, L. Liang, M. Xie, Y. Zhang, S. Lin and D. Yan, Synthesis of Novel Linear PEO-b-PS-b-PCL Triblock Copolymers by the Combination of ATRP, ROP, and a Click Reaction, Macromol. Chem. Phys., 2007, 208, 1797–1802 CrossRef CAS.
- S. Reinicke and H. Schmalz, Combination of Living Anionic Polymerization and ATRP via “Click” Chemistry as a Versatile Route to Multiple Responsive Triblock Terpolymers and Corresponding Hydrogels, Colloid Polym. Sci., 2011, 289, 497–512 CrossRef CAS.
- K. Wang, L. Liang, S. Lin and X. He, Synthesis of Well-Defined ABC Triblock Copolymers with Polypeptide Segments by ATRP and Click Reactions, Eur. Polym. J., 2008, 44, 3370–3376 CrossRef CAS.
- H. Sun, I. H. Öner, T. Wang, T. Zhang, O. Selyshchev, C. Neumann, Y. Fu, Z. Liao, S. Xu, Y. Hou, A. Turchanin, D. R. T. Zahn, E. Zschech, I. M. Weidinger, J. Zhang and X. Feng, Molecular Engineering of Conjugated Acetylenic Polymers for Efficient Cocatalyst-free Photoelectrochemical Water Reduction, Angew. Chem., Int. Ed., 2019, 58, 10368–10374 CrossRef CAS PubMed.
- S. Otep, T. Michinobu and Q. Zhang, Pure Organic Semiconductor-Based Photoelectrodes for Water Splitting, Sol. RRL, 2020, 4, 1900395 CrossRef CAS.
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Efficient Organic Solar Cells Processed from Hydrocarbon Solvents, Nat. Energy, 2016, 1, 15027 CrossRef CAS.
- D. Deng, Y. Zhang, J. Zhang, Z. Wang, L. Zhu, J. Fang, B. Xia, Z. Wang, K. Lu, W. Ma and Z. Wei, Fluorination-Enabled Optimal Morphology Leads to over 11% Efficiency for Inverted Small-Molecule Organic Solar Cells, Nat. Commun., 2016, 7, 13740 CrossRef CAS PubMed.
- G. Zhang, K. Zhang, Q. Yin, X. F. Jiang, Z. Wang, J. Xin, W. Ma, H. Yan, F. Huang and Y. Cao, High-Performance Ternary Organic Solar Cell Enabled by a Thick Active Layer Containing a Liquid Crystalline Small Molecule Donor, J. Am. Chem. Soc., 2017, 139, 2387–2395 CrossRef CAS PubMed.
- Y. Lin, Y. Firdaus, M. I. Nugraha, F. Liu, S. Karuthedath, A. H. Emwas, W. Zhang, A. Seitkhan, M. Neophytou, H. Faber, E. Yengel, I. McCulloch, L. Tsetseris, F. Laquai and T. D. Anthopoulos, 17.1% Efficient Single-Junction Organic Solar Cells Enabled by n-Type Doping of the Bulk-Heterojunction, Adv. Sci., 2020, 7, 1903419 CrossRef CAS PubMed.
- Y. Cui, H. Yao, L. Hong, T. Zhang, Y. Tang, B. Lin, K. Xian, B. Gao, C. An, P. Bi and J. Hou, Organic Photovoltaic Cell with 17% Efficiency and Superior Processability, Natl. Sci. Rev., 2020, 7, 1239–1246 CrossRef CAS PubMed.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao, K. Sun, S. Yang, X. Zhang and L. Ding, 18% Efficiency Organic Solar Cells, Sci. Bull., 2020, 65, 272–275 CrossRef CAS.
- B. Fan, L. Ying, P. Zhu, F. Pan, F. Liu, J. Chen, F. Huang and Y. Cao, All-Polymer Solar Cells Based on a Conjugated Polymer Containing Siloxane-Functionalized Side Chains with Efficiency over 10%, Adv. Mater., 2017, 29, 1703906 CrossRef PubMed.
- B. Fan, L. Ying, Z. Wang, B. He, X. F. Jiang, F. Huang and Y. Cao, Optimisation of Processing Solvent and Molecular Weight for the Production of Green-Solvent-Processed All-Polymer Solar Cells with a Power Conversion Efficiency over 9%, Energy Environ. Sci., 2017, 10, 1243–1251 RSC.
- J. Yang, B. Xiao, S. W. Heo, K. Tajima, F. Chen and E. Zhou, Effects of Inserting Thiophene as a π-Bridge on the Properties of Naphthalene Diimide-alt-Fused Thiophene Copolymers, ACS Appl. Mater. Interfaces, 2017, 9, 44070–44078 CrossRef CAS PubMed.
- N. B. Kolhe, H. Lee, D. Kuzuhara, N. Yoshimoto, T. Koganezawa and S. A. Jenekhe, All-Polymer Solar Cells with 9.4% Efficiency from Naphthalene Diimide-Biselenophene Copolymer Acceptor, Chem. Mater., 2018, 30, 6540–6548 CrossRef CAS.
- N. Zhou and A. Facchetti, Naphthalenediimide (NDI) Polymers for All-Polymer Photovoltaics, Mater. Today, 2018, 21, 377–390 CrossRef CAS.
- X. Liu, Y. Zou, H. Q. Wang, L. Wang, J. Fang and C. Yang, High-Performance All-Polymer Solar Cells with a High Fill Factor and a Broad Tolerance to the Donor/Acceptor Ratio, ACS Appl. Mater. Interfaces, 2018, 10, 38302–38309 CrossRef CAS PubMed.
- A. Robitaille, S. A. Jenekhe and M. Leclerc, Poly(naphthalene diimide-alt-bithiophene) Prepared by Direct (Hetero)Arylation Polymerization for Efficient All-Polymer Solar Cells, Chem. Mater., 2018, 30, 5353–5361 CrossRef CAS.
- J. Yuan, Y. Xu, G. Shi, X. Ling, L. Ying, F. Huang, T. H. Lee, H. Y. Woo, J. Y. Kim, Y. Cao and W. Ma, Engineering the Morphology via Processing Additives in Multiple All-Polymer Solar Cells for Improved Performance, J. Mater. Chem. A, 2018, 6, 10421–10432 RSC.
- G. Wang, F. S. Melkonyan, A. Facchetti and T. J. Marks, All-Polymer Solar Cells: Recent Progress, Challenges, and Prospects, Angew. Chem., Int. Ed., 2019, 58, 4129–4142 CrossRef CAS PubMed.
- C. Lee, S. Lee, G.-U. Kim, W. Lee and B. J. Kim, Recent Advances, Design Guidelines, and Prospects of All-Polymer Solar Cells, Chem. Rev., 2019, 119, 8028–8086 CrossRef CAS PubMed.
- Z. Li, L. Ying, P. Zhu, W. Zhong, N. Li, F. Liu, F. Huang and Y. Cao, A Generic Green Solvent Concept Boosting the Power Conversion Efficiency of All-Polymer Solar Cells to 11%, Energy Environ. Sci., 2019, 12, 157–163 RSC.
- H. Yao, F. Bai, H. Hu, L. Arunagiri, J. Zhang, Y. Chen, H. Yu, S. Chen, T. Liu, J. Y. L. Lai, Y. Zou, H. Ade and H. Yan, Efficient All-Polymer Solar Cells Based on a New Polymer Acceptor Achieving 10.3% Power Conversion Efficiency, ACS Energy Lett., 2019, 4, 417–422 CrossRef CAS.
- Y. Wang, S. W. Kim, J. Lee, H. Matsumoto, B. J. Kim and T. Michinobu, Dual Imide-Functionalized Unit-Based Regioregular D-A1-D-A2 Polymers for Efficient Unipolar n-Channel Organic Transistors and All-Polymer Solar Cells, ACS Appl. Mater. Interfaces, 2019, 11, 22583–22594 CrossRef CAS PubMed.
- S. W. Kim, Y. Wang, H. You, W. Lee, T. Michinobu and B. J. Kim, Impact of Incorporating Nitrogen Atoms in Naphthalenediimide-Based Polymer Acceptors on the Charge Generation, Device Performance, and Stability of All-Polymer Solar Cells, ACS Appl. Mater. Interfaces, 2019, 11, 35896–35903 CrossRef CAS PubMed.
- A. Bonasera, G. Giuliano, G. Arrabito and B. Pignataro, Tackling Performance Challenges in Organic Photovoltaics: An Overview about Compatibilizers, Molecules, 2020, 25, 1–39 CrossRef PubMed.
- M. Saito, Y. Tamai, H. Ichikawa, H. Yoshida, D. Yokoyama, H. Ohkita and I. Osaka, Significantly Sensitized Ternary Blend Polymer Solar Cells with a Very Small Content of the Narrow-Band Gap Third Component That Utilizes Optical Interference, Macromolecules, 2020, 53, 10623–10635 CrossRef CAS.
- N. Gasparini, A. Salleo, I. McCulloch and D. Baran, The Role of the Third Component in Ternary Organic Solar Cells, Nat. Rev. Mater., 2019, 4, 229–242 CrossRef.
- L. Ye, X. Jiao, W. Zhao, S. Zhang, H. Yao, S. Li, H. Ade and J. Hou, Manipulation of Domain Purity and Orientational Ordering in High Performance All-Polymer Solar Cells, Chem. Mater., 2016, 28, 6178–6185 CrossRef CAS.
- H. I. Kim, M. Kim, C. W. Park, H. U. Kim, H. K. Lee and T. Park, Morphological Control of Donor/Acceptor Interfaces in All-Polymer Solar Cells Using a Pentafluorobenzene-Based Additive, Chem. Mater., 2017, 29, 6793–6798 CrossRef CAS.
- S. Liu, D. Chen, W. Zhou, Z. Yu, L. Chen, F. Liu and Y. Chen, Vertical Distribution to Optimize Active Layer Morphology for Efficient All-Polymer Solar Cells by J71 as a Compatibilizer, Macromolecules, 2019, 52, 4359–4369 CrossRef CAS.
- H. Fujita, T. Michinobu, M. Tokita, M. Ueda and T. Higashihara, Synthesis and Postfunctionalization of Rod-Coil Diblock and Coil-Rod-Coil Triblock Copolymers Composed of Poly(3-hexylthiophene) and Poly(4-(4′-N,N-dihexylaminophenylethynyl)styrene) Segments, Macromolecules, 2012, 45, 9643–9656 CrossRef CAS.
- H. Fujita, T. Michinobu, S. Fukuda, T. Koganezawa and T. Higashihara, Sequentially Different AB Diblock and ABA Triblock Copolymers as P3HT:PCBM Interfacial Compatibilizers for BHJ Photovoltaics, ACS Appl. Mater. Interfaces, 2016, 8, 5484–5492 CrossRef CAS PubMed.
- Y.-A. Su, N. Maebayashi, H. Fujita, Y.-C. Lin, C.-I. Chen, W.-C. Chen, T. Michinobu, C.-C. Chueh and T. Higashihara, Development of Block Copolymers with Poly(3-hexylthiophene) Segments as Compatibilizers in Non-Fullerene Organic Solar Cells, ACS Appl. Mater. Interfaces, 2020, 12, 12083–12092 CrossRef CAS PubMed.
- J. Chen, X. Yu, K. Hong, J. M. Messman, D. L. Pickel, K. Xiao, M. D. Dadmun, J. W. Mays, A. J. Rondinone, B. G. Sumpter and S. M. Kilbey, II, Ternary Behavior and Systematic Nanoscale Manipulation of Domain Structures in P3HT/PCBM/P3HT-b-PEO Films, J. Mater. Chem., 2012, 22, 13013–13022 RSC.
- D. Kipp and V. Ganesan, Exploiting the Combined Influence of Morphology and Energy Cascades in Ternary Blend Organic Solar Cells Based on Block Copolymer Additives, Macromolecules, 2016, 49, 5137–5144 CrossRef CAS.
- D. Kipp, R. Verduzco and V. Ganesan, Block Copolymer Compatibilizers for Ternary Blend Polymer Bulk Heterojunction Solar Cells – An Opportunity for Computation Aided Molecular Design, Mol. Syst. Des. Eng., 2016, 1, 353–369 RSC.
- Y. Sun, P. Pitliya, C. Liu, X. Gong, D. Raghavan and A. Karim, Block Copolymer Compatibilized Polymer:Fullerene Blend Morphology and Properties, Polymer, 2017, 113, 135–146 CrossRef CAS.
- E. Mohammadi-Arbati and S. Agbolaghi, Efficiency above 6% in Poly(3-Hexylthiophene):Phenyl-C-Butyric Acid Methyl Ester Photovoltaics via Simultaneous Addition of Poly(3-Hexylthiophene) Based Grafted Graphene Nanosheets and Hydrophobic Block Copolymers, Polym. Int., 2019, 68, 1292–1302 CrossRef CAS.
- H. J. Kim, J. H. Kim, J. H. Ryu, Y. Kim, H. Kang, W. B. Lee, T. S. Kim and B. J. Kim, Architectural Engineering of Rod-Coil Compatibilizers for Producing Mechanically and Thermally Stable Polymer Solar Cells, ACS Nano, 2014, 8, 10461–10470 CrossRef CAS PubMed.
- M. Zhu, H. Kim, Y. J. Jang, S. Park, D. Y. Ryu, K. Kim, P. Tang, F. Qiu, D. H. Kim and J. Peng, Toward High Efficiency Organic Photovoltaic Devices with Enhanced Thermal Stability Utilizing P3HT-b-P3PHT Block Copolymer Additives, J. Mater. Chem. A, 2016, 4, 18432–18443 RSC.
- A. Kato, L.-Y. Su, Y.-C. Lin, L. Wang, W.-C. Chen, C.-C. Chueh and T. Higashihara, Naphthalene-diimide-based All-conjugated Block Copolymer as an Effective Compatibilizer to Improve the Performance and Thermal Stability of All-polymer Solar Cells, Mater. Chem. Front., 2021, 5, 7216–7227 RSC.
- S. Otep, K. Ogita, N. Yomogita, K. Motai, Y. Wang, Y.-C. Tseng, C.-C. Chueh, Y. Hayamizu, H. Matsumoto, K. Ishikawa, T. Mori and T. Michinobu, Cross-linking of Poly(arylenebutadiynylene)s and Its Effect on Charge Carrier Mobilities in Thin Film Transistors, Macromolecules, 2021, 54, 4351–4362 CrossRef CAS.
- J. F. Lutz and K. Matyjaszewski, Nuclear Magnetic Resonance Monitoring of Chain-End Functionality in the Atom Transfer Radical Polymerization of Styrene, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 897–910 CrossRef CAS.
- K. Ishizuki, H. Oka, D. Aoki, R. Goseki and H. Otsuka, Mechanochromic Polymers That Turn Green Upon the Dissociation of Diarylbibenzothiophenonyl: The Missing Piece toward Rainbow Mechanochromism, Chem. – Eur. J., 2018, 24, 3170–3173 CrossRef CAS PubMed.
- D. R. Rutherford, J. K. Stille, C. M. Elliott and V. R. Reichert, Poly(2,5-Ethynylenethiophenediylethynylenes), Related Heteroaromatic Analogues, and Poly(Thieno[3,2-b]Thiophenes). Synthesis and Thermal and Electrical Properties, Macromolecules, 1992, 25, 2294–2306 CrossRef CAS.
- G. L’abbe, Decomposition and Addition Reactions of Organic Azides, Chem. Rev., 1969, 69, 345–363 CrossRef.
- H. Bassler and K. Anna, Charge Transport in Organic Semiconductors, in Unimolecular and Supramolecular Electronics I, ed. R. M. Metzger, Springer, Berlin, Heidelberg, 2011 Search PubMed.
- F. C. Spano and C. Silva, H- and J-Aggregate Behavior in Polymeric Semiconductors, Annu. Rev. Phys. Chem., 2014, 65, 477–500 CrossRef CAS PubMed.
- L. Lu, T. Xu, W. Chen, E. S. Landry and L. Yu, Ternary Blend Polymer Solar Cells with Enhanced Power Conversion Effeiciency, Nat. Photon., 2014, 8, 716–722 CrossRef CAS.
- P. Yin, T. Zheng, Y. Wu, G. Liu, Z.-G. Zhang, Z. Cui, Y. Li and P. Shen, Achieving Efficient Thick Active Layer and Large Area Ternary Polymer Solar Cells by Incorporating a New Fused Heptacyclic Non-fullerene Acceptor, J. Mater. Chem. A, 2018, 6, 20313–20326 RSC.
- A. Yin, D. Zhang, S.-H. Cheung, S.-K. So, Z. Fu, L. Ying, F. Huang, H. Zhou and Y. Zhang, On the Understanding of Energetic Disorder, Charge Recombination and Voltage Loss in All-Polymer Solar Cells, J. Mater. Chem. C, 2018, 6, 7855–7863 RSC.
- Y. Zhang, Y. Xu, M. J. Ford, F. Li, J. Sun, X. Ling, Y. Wang, J. Gu, J. Yuan and W. Ma, Thermally Stable All-Polymer Solar Cells with High Tolerance on Blend Ratios, Adv. Energy Mater., 2018, 8, 1800029 CrossRef.
- J. Zhang, W. Liu, M. Zhang, Y. Liu, G. Zhou, S. Xu, F. Zhang, H. Zhu, F. Liu and X. Zhu, Revealing the Critical Role of HOMO Alignment on Maximizing Current Extraction and Suppressing Energy Loss in Organic Solar Cells, iScience, 2019, 19, 883–893 CrossRef CAS PubMed.
- Z. Zhou, S. Xu, J. Song, Y. Jin, Q. Yue, Y. Qian, F. Liu, F. Zhang and X. Zhu, High-efficiency Small-molecule Ternary Solar Cells with a Hierarchical Morphology Enabled by Synergizing Fullerene and Non-fullerene Acceptors, Nat. Energy, 2018, 3, 952–959 CrossRef CAS.
- S. M. Menke, N. A. Ran, G. C. Bazan and R. H. Friend, Understanding Energy Loss in Organic Solar Cells: Toward a New Efficiency Regime, Joule, 2018, 2, 25–35 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc04948h |
| This journal is © The Royal Society of Chemistry 2022 |