
Design of biodegradable and biocompatible conjugated polymers for bioelectronics
 *a
and
Jonathan
Rivnay
*b
*a
and
Jonathan
Rivnay
*b
Abstract
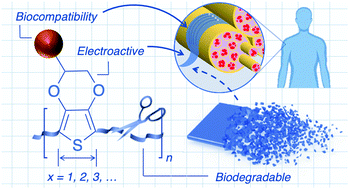
The emerging field of bioelectronics leverages the optoelectronic properties of synthetic materials to interface with living systems. The convergence of modern electronics with biology has offered lifesaving medical treatments, with applications related to drug delivery, regenerative engineering, and continuous biosignal monitoring for healthcare on the horizon. This next generation of bioelectronic technologies requires an intimate biointerface, necessitating electroactive materials which are both mechanically and physiochemically compatible. Organic systems such as conjugated polymers offer an alternative design space for electroactive materials that are mechanically compatible (flexible, stretchable, conformal) and chemically tunable through various well-established synthetic methods and can therefore be tailored for integration with biological systems. Currently, conjugated polymers utilized for bioelectronic applications consist of prominent high-performing materials emerging from adjacent organic electronic communities with slight chemical modifications, and are therefore generally not well-suited for the entire lifecycle of a biomaterial. While early investigations have demonstrated the potential of such conjugated polymers as semiconductors and conductors in vivo, their limited biodegradability and long-term biocompatibility have slowed widespread adoption and clinical translation. To aid in the development of the next generation of bioelectronic materials, this review details various synthetic strategies to endow a conjugated material with degradability and biocompatibility. Prominent examples of conjugated materials are used to illustrate design principles, current limitations, and future directions towards such electroactive materials. The main factors that need to be considered for the rational design of biodegradable and biocompatible conjugated polymers for bioelectronic applications are highlighted, with future directions emphasized.
- This article is part of the themed collection: Special issue in memoriam of Alasdair James Campbell
 Please wait while we load your content...
Please wait while we load your content...

