
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Less is more: on the effect of benzannulation on the solid-state emission of difluoroborates†
Iryna
Knysh
 a,
Anna
Kozakiewicz-Piekarz
b,
Andrzej
Wojtczak
b,
Damian
Plażuk
c,
Glib
Baryshnikov
de,
Rashid
Valiev
fg,
Rinat
Nasibullin
f,
Hans
Ågren
h,
Denis
Jacquemin
*i,
Borys
Ośmiałowski
*b and
Robert
Zaleśny
*a
a,
Anna
Kozakiewicz-Piekarz
b,
Andrzej
Wojtczak
b,
Damian
Plażuk
c,
Glib
Baryshnikov
de,
Rashid
Valiev
fg,
Rinat
Nasibullin
f,
Hans
Ågren
h,
Denis
Jacquemin
*i,
Borys
Ośmiałowski
*b and
Robert
Zaleśny
*a
aFaculty of Chemistry, Wrocław Univ. of Science and Technology, Wyb. Wyspiańskiego 27, PL-50370 Wrocław, Poland. E-mail: robert.zalesny@pwr.edu.pl
bFaculty of Chemistry, Nicolaus Copernicus University, Gagarina 7, PL-87100 Toruń, Poland. E-mail: borys.osmialowski@umk.pl
cLaboratory of Molecular Spectroscopy, Department of Organic Chemistry, Faculty of Chemistry, University of Łódź, Tamka 12, PL-91-403, Łódź, Poland
dLinköping University, Department of Science and Technology, Laboratory of Organic Electronics, Norrköping, SE-60174, Sweden
eDepartment of Chemistry and Nanomaterials Science, Bohdan Khmelnytsky National University, Cherkasy, Ukraine
fTomsk State University, Tomsk 634050, Russia
gDepartment of Chemistry, University of Helsinki, Helsinki FIN – 00014, Finland
hDepartment of Physics and Astronomy, Uppsala University, Box 516, SE-751 20 Uppsala, Sweden
iUniversité de Nantes, CNRS, CEISAM UMR 6230, F-44000 Nantes, France. E-mail: denis.jacquemin@univ-nantes.fr
First published on 29th September 2021
Abstract
We investigate the emission properties of four organic dyes containing a strong electron-donating N(CH3)2 group and an NBF2O-bearing heterocyclic moiety acting as the electron-accepting group. The four studied compounds differ in the number and positions of the fused benzo rings included in the heterocyclic moiety. They exhibit strong emission in solution, with fluorescence quantum yields (Φf) systematically exceeding 0.8 at least in one of the solvents used, regardless of the benzannulation architecture. The strong dipolar character, achieved by substitution with the N(CH3)2 group and benzannulation, enhances the photoinduced charge transfer and appears to be an effective strategy to tune the photophysical properties of these dyes in solution. Indeed, red-shifted absorption spectra are obtained without deteriorating the emission properties. However, the present joint theory–experiment study clearly demonstrates that such a molecular design is not effective for solid-state applications, as only one derivative still exhibits significant emission in the crystalline form, namely the most compact one. We show that the combination of benzannulation in the presence of the strong amino donor leads to substantial changes in crystal packing and that a different network of intermolecular interactions can be found in the crystal. More specifically, going from the parent compound to its benzannulated derivatives induces a stronger π⋯π stacking combined with multiple CH⋯π interactions involving the fused benzo rings and the hydrogen atoms of the dimethylamino group, which impedes efficient emission of the crystals.
1 Introduction
Solid-state emission of organic materials remains in the limelight owing to the countless technological applications of light-emissive organic devices, e.g., light-emitting diodes,1–3 field effect transistors,4,5 fluorescent sensors,6,7 and laser dyes.8,9 Development of new systems for lasing is also an important challenge in materials chemistry. A number of organic molecules are employed to that end including compounds carrying the BF2 group.8 The same moiety might be used to control the efficient emission of polarized light, but to fully exploit these possibilities a careful molecular design is needed10 especially when no asymmetric atom is present in the emissive molecule but the chirality is obtained by changing the relative orientation of two moieties.11 Likewise, efficient organics-based diodes require tweaking of the molecular structure.12 It is now well established that molecular topology is one of the key factors in the design of efficient systems for organic electronics.13–15 This popularity of organic fluorophores stems from their easy functionalization, making it possible to tune their photophysical properties using various synthetic and design strategies.16 In particular, push–pull molecules presenting an electron-donating (D) and an electron-accepting (A) substituent separated by a π-conjugated linker are very popular.17–19 Such molecules exhibit intramolecular charge transfer (ICT),20,21 leading to a red-shift of the ππ* absorption band as the strength of the D and A groups and/or the delocalizable nature of the linker increases.22,23 However, excessive ICT strength might also yield emission quenching due to the appearance of a TICT (twisted intramolecular charge transfer) or PICT (planarized intramolecular charge transfer) structure in the lowest electronic excited state, or more generally lead to a change in geometry and effective non-radiative pathways.Another approach that makes it possible to tune the optical properties of fluorescent dyes is the fusion of benzene rings, also referred to as benzannulation.24–27 This leads to extension of the π-conjugated skeleton, which results in beneficial red-shifts of the spectral signatures.28,29 In addition, benzannulation comes with increased molecular rigidity which might facilitate maximizing Φf – a highly desired feature for many technological applications. On the other hand, large, stiff planar structures tend to stack which may lead to undesirable photophysical features in the solid state. Clar proposed an empirical rule for describing the structural features of large fused polycyclic hydrocarbons30 which states that the system is driven toward a mesomeric structure that contains the largest number of fully delocalized π electrons. Consequently, fused rings might act as accepting groups, making benzannulation a handy approach for controlling the photophysical properties as a substitute for or in combination with more traditional accepting substituents like cyano groups.
The fluorescent dyes bearing a BF2 group, which are our focus in the present work, are popular platforms for the above described functionalizations. Among these dyes, the BODIPY family is one of the most studied ones due to the exceptional photophysical properties: large Φf, high extinction coefficients, as well as narrow absorption and emission bands.31,32 However, BODIPY dyes are known to be relatively poor for solid-state emission devices, as they suffer from aggregation caused quenching (ACQ),33,34 notably due to reabsorption of the emitted photons by the nearby molecules, and by self-quenching due to the close contacts between the molecules (e.g. π-stacking) leading to the non-radiative energy dissipation. The detrimental ACQ effects can be circumvented to some extent by changing the intermolecular packing using various methods, including (i) the formation of head-to-tail aggregates via J-type aggregation;35,36 (ii) the promotion of cross-stacking in the crystal structure;37 (iii) the formation of a supramolecular co-crystal;38,39 and (iv) the use of bulky substituents to reduce the strength of the π⋯π stacking interactions.7,40 Of course, the emission properties might greatly benefit from aggregation through the opposite process, namely, aggregation induced emission (AIE).34,41,42 The AIE phenomenon is typically caused by restriction of molecular rotational motions in solution (when the concentration of the fluorophore is close to precipitation) or in the solid state.43,44 However, AIE has been only reported in difluoroborates, though not completely undescribed.45–48
As stated above the popular approach to hamper π⋯π stacking is to introduce bulky substituents in order to increase the intermolecular distance. Notably, it was shown that an enhancement of the Φf in the solid-state of perylene diimides (PDI) can be achieved by substitution with polyhedral oligomeric silsesquioxanes (POSS) at the imide position via either rigid or flexible linkages.49 The studies of π-extended coumarin derivatives possessing either a seven-(DBU) or five-membered ring (DBN) attached to the core molecule also revealed differences in the photophysical properties due to different crystal packing.50 The difference in the packing leads to fluorescence quenching in the solid state for DBN (possessing a close parallel arrangement with long-range π⋯π stacking) and an enhancement of Φf for DBU (with discrete dimeric units built by π-systems of two adjacent molecules). In another study, the influence of the π⋯π interactions on boron difluoride derivatives exhibiting solid state NIR-light emission was achieved by D'Aléo and co-workers.51 The X-ray structure revealed differences in the packing modes of acetophenone and acetonaphthone-based BF2 complexes. The latter tends to pack in a face-to-face fashion which increases the π-overlap, whereas the former yields displaced dimer units which reduces the π-contact area. This logically translated into an increase (reduction) of Φf in the solid state for the acetophenone (acetonaphthone)-based crystal compared to the solution. Moreover, the fine tuning of the crystal packing was used to obtain two different crystals of the same BF2-carrying molecule characterised by completely different properties (TADF vs RTP – thermally activated delayed fluorescence vs room-temperature phosphorescence).52
Extensive studies of BF2 fluorescent dyes containing NBF2N,53–55 NBF2O44,56,57 or OBF2O58,59 units have been reported. Some of them show bright fluorescence in the solid state.44,51,56,60,61 We have recently shown that the optical properties of difluoroborates in solution can be tuned by combining variations of the electron-donating substituent and benzannulation of the central pyridine ring.62–64 Such a strategy made it possible to control the strength of the donor–acceptor interactions and thus the properties of the excited states. The aim of the present work is to study the photophysical properties of crystals of the four dipolar dyes shown in Fig. 1, that all exhibit bright emission in many solvents. The use of NMe2 group in the studied series was dictated by its high electron-donating strength and widespread presence in organic fluorescent dyes and this group hampers hydrogen bonding. More specifically we analyze the influence of benzannulation on the solid state (crystal) emission for perfectly homologous compounds, using a palette of experimental and computational methods in an effort to understand the subtle relationships between molecular structure, solution, and solid-state fluorescence.
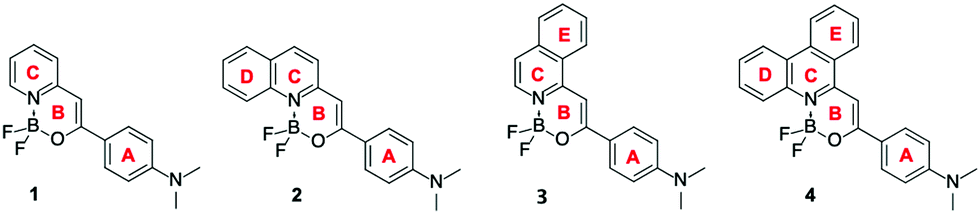 | ||
| Fig. 1 Structures and ring labelling of the compounds studied herein. | ||
2 Methods
2.1 Theory
| ΔEMP2int = ΔEHFint + ΔEMP2corr | (1) |
| ΔEHFint = ε(10)el + ΔEHLex + ΔEHFdel | (2) |
| ΔEMP2corr = ε(12)el,r + ε(20)disp + ΔE(2)ex | (3) |
| ΔEMP2corr = ΔEMP2int − ΔEHFint | (4) |
For the simulations of the optical properties, the molecular structures of the ground singlet state were optimized at the DFT level using the B3LYP95 functional and the 6-31G(d,p)96 basis set. Excitation energies were calculated in the gas phase using both the extended multiconfiguration quasidegenerate perturbation theory at the second order level (XMC-QDPT2)97 and time-dependent density functional theory (TD-DFT).98 The XMC-QDPT2 calculations were performed using the Firefly software99 with active spaces consisting of 10 electrons in 10 molecular orbitals (see Table S11, ESI†). The non-adiabatic coupling matrix elements (NACME) were calculated using Turbomole.100 The spin–orbit coupling matrix elements (SOCME) between the first singlet excited state and the lowest triplet state were calculated using the PySOC software101 based on the Breit–Pauli (BP) spin–orbit Hamiltonian with an effective charge approximation. The internal conversion (IC) and intersystem crossing (ISC) rate constants were calculated within the Herzberg–Teller approximation102,103 taking into account the anharmonicity effects.104 The IC rate constants (kic) were obtained using the algorithm published elsewhere,105 which assumes that the X–H vibrations mainly contribute to the IC process.
2.2 Experiment
The description of synthesis and full characterization of compounds in solution by 1H, 11B, 13C, 15N and 19F NMR spectroscopy and elemental analysis can be found in ref. 19 and 106 (1), ref. 107 (2), ref. 63 (3) and ref. 64 (4). The X-ray data were collected at 293(2) K using an Oxford Sapphire CCD diffractometer using MoKα radiation (λ = 0.71073 Å). The structures were solved by direct methods and refined using the full-matrix least-squares method on F2 with the use of SHELX2017 program package.108 The analytical absorption corrections were applied by CrysAlis 171.38.43 package of programs Rigaku OD.109 The C-bonded hydrogen atom positions were calculated from geometry. All hydrogen atoms were constrained during refinement. A summary of the crystal data and refinement details for compounds 2–4 is given in Table S6 (ESI†). CCDC 2090001 for 2 and 2090002 for 3, and 2090003 for 4. Emission spectra of solid-state samples of 1–4 were recorded on a Fluoromax-4 (Horiba) spectrofluorometer equipped with a long pass filter and the fluorescence quantum yields were measured using the same apparatus equipped with an integrating sphere Quanta.3 Results
3.1 Photophysical properties
Previously we showed that the benzannulation position has a significant impact on the fluorescence features in solution.62–64,110 The Φf of 1–4 measured in the various solvents are listed in Table 1 (see also Table S1, ESI†). All compounds exhibit bright emission in solution, the Φf's magnitude being dependent on the nature of the solvent. The largest Φf is observed in diethyl ether (Et2O) for 1, and in methylcyclohexane (MCH) for 2–4. As shown in Table S1 (ESI†), changing the solvent polarity influences both the radiative (kr) and non-radiative (knr) constants. Illustratively, passing from MCH to dimethylformamide (DMF) kr is decreasing and knr is increasing.| Comp. | MCHb | Et2Oc | THFd | EAe | AcMef | MeOHg | MeCNh | DMFi | SSj |
|---|---|---|---|---|---|---|---|---|---|
| a Solvents in the table are ordered according to increasing magnitude of their dipole moment. b Methylcyclohexane. c Diethyl ether. d Tetrahydrofuran. e Ethyl acetate. f Acetone. g Methanol. h Acetonitrile. i Dimethylformamide. j Solid state. | |||||||||
| 1 | 0.519 | 0.868 | 0.684 | 0.699 | 0.569 | 0.454 | 0.517 | 0.382 | 0.314 |
| 2 | 0.862 | 0.747 | 0.517 | 0.614 | 0.374 | 0.308 | 0.296 | 0.187 | <0.01 |
| 3 | 0.979 | 0.848 | 0.658 | 0.663 | 0.587 | 0.499 | 0.638 | 0.561 | 0.020 |
| 4 | 1.000 | 0.999 | 0.585 | 0.655 | 0.441 | 0.471 | 0.447 | 0.323 | 0.063 |
In order to gain insight into the fluorescence properties of 1–4, and more specifically on the rates of the radiative and non-radiative decay pathways, we first performed gas-phase calculations carried out at the TD-DFT/B3LYP/6-31G(d,p) and XMC-QDPT2/6-31G(d,p) levels of theory. The results are presented in Tables S2–S5 in the ESI.† Tables S4 and S5 (ESI†) show that the TD-DFT predicts twice larger S1–T1 energy gaps and much smaller SOCME values than XMC-QDPT2. As a consequence, TD-DFT foresees internal conversion as the main deactivation pathway, while XMC-QDPT2 predicts intersystem crossing to be a competitive deactivation channel (compare Tables S3 and S5, ESI†). Despite this difference in prediction of non-radiative deactivation rate constants, both levels of theory show that the radiative rate constant dominates over the IC and ISC deactivation channels for all molecules. The prevalence of the kr over knr agrees with the experimental data measured in the least polar solvents such as MCH and Et2O (Table S1, ESI†). It is mainly due to the exponential growth of the internal conversion efficiency with the decrease of the S1–S0 energy gap.111,112 Indeed, the polar solvents affect the bathochromic shift of the emission wavelengths for all molecules (Table S1, ESI†) which results in a monotonous decrease of the fluorescence quantum yield. Thus, although the obtained theoretical data indicate non-zero values of SOCME and NACME for the coupling between the S1–T1 and S1–S0 states, respectively, the quite large values of the S1–S0 vertical transition energies and oscillator strengths for 1–4 hint that fluorescence dominates over the non-radiative processes for the single molecules.
In order to study the photophysical properties in the solid state, crystals of 1–4 were obtained by slow evaporation of the chloroform solution. The solid state fluorescence spectra are displayed in Fig. 2 and the measured Φf values are listed in Table 1. The emission bands are red-shifted in the crystal as compared to in the solution, with peaks at 1 (563 nm) < 3 (600 nm) < 4 (611 nm) < 2 (645 nm) in the solid state. More interestingly, while these measurements confirm that benzannulation significantly affects the emission properties, rather different patterns were obtained in solution and in the crystal, with very strong quenching of the fluorescence for 2–4 and with bright emission for 1 only in the latter phase. The fluorescence lifetimes of compounds 1 and 4 in the solid state are 9.64 μs and 8.93 μs, respectively. For compounds 2 and 3, exhibiting very low fluorescence quantum yields, the results are not reliable due to the spectrometer limitations. The lifetimes measured in the solid state are three orders of magnitude higher than that determined in chloroform solution.19,62–64,106
 | ||
| Fig. 2 Solid-state fluorescence of the investigated compounds. | ||
3.2 Molecular packing
To understand the fundamental origins of these striking differences between the emission measured for solutions and crystals, first we analyze the packing of each dye and the intermolecular interactions. To this end, we analyze published X-ray measurements for 1,106 and use crystal structures determined in the present work for the other three dyes, and additionally further considered a parent, unsubstituted compound of 4 (denoted as 4(H) below), which was studied by Wu and co-workers56 and is available in the CCDC database. The crystal structure information and the detailed description of the molecular packing of 2–4 are given in the ESI.† The crystals of 1, 3, and 4 correspond to orthorhombic and 2 to triclinic crystal systems with the space groups Pca21, Pbca, Pbca, and P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , respectively. The molecular packing as well as the main interactions and dimers of 1–4 selected for further studies are depicted in Fig. 3. The asymmetric unit of the structure for compound 1 consists of 2 molecules – a (red) and b (blue) in Fig. 3a. The π⋯π interactions are found between the pyridine rings C (labeling of the rings can be found in Fig. 1) of molecule a and molecule b in the 1baππ dimer. A series of C–H⋯π interactions are also present in that structure between the pyridine C and aniline A rings of both molecules, leading to that the 1abTp–1 and 1abTp–2 dimers show edge-to-face T-shape and weak (due to the angle between rings) π⋯π stacking, see Fig. 3a. Only a single molecule, a, of 2 is found in the asymmetric part of the crystal structure. The X-ray crystallographic analysis reveals several π⋯π interactions between the C and A rings. In addition, the methyl group of the aniline moiety (ring A) is involved in C–H⋯π interactions with the ring D.
, respectively. The molecular packing as well as the main interactions and dimers of 1–4 selected for further studies are depicted in Fig. 3. The asymmetric unit of the structure for compound 1 consists of 2 molecules – a (red) and b (blue) in Fig. 3a. The π⋯π interactions are found between the pyridine rings C (labeling of the rings can be found in Fig. 1) of molecule a and molecule b in the 1baππ dimer. A series of C–H⋯π interactions are also present in that structure between the pyridine C and aniline A rings of both molecules, leading to that the 1abTp–1 and 1abTp–2 dimers show edge-to-face T-shape and weak (due to the angle between rings) π⋯π stacking, see Fig. 3a. Only a single molecule, a, of 2 is found in the asymmetric part of the crystal structure. The X-ray crystallographic analysis reveals several π⋯π interactions between the C and A rings. In addition, the methyl group of the aniline moiety (ring A) is involved in C–H⋯π interactions with the ring D.
 | ||
| Fig. 3 Molecular packing of 1 (a), 2 (b), 3 (c), 4 (d), and 4(H) (e) with the indication of the main types of intermolecular interactions and key dimers that are studied herein. | ||
Both interaction types are found in the two types of head-to-tail dimers, 2aaππ–1 and 2aaππ–2, which have a parallel displaced stacked configuration (Fig. 3b). For the isoquinoline derivative 3, the asymmetric unit contains 3 molecules – a (red), b (blue), and c (green), see Fig. 3c. π⋯π-type stacks are found only between equivalent molecules leading to the 3aaππ, 3bbππ, 3ccππ dimers, see the left-hand side of Fig. 3c. The dominating π⋯π interactions take place between the B and C (C and E) rings for molecules c (a and b). C–H⋯π interactions are found between the methyl of the NMe2 groups and the A rings. For further analysis, we also consider the dimers with edge-to-face T-shape π⋯π stacking (3bbTp and 3caTp) and C–H⋯π interactions (3baCHp and 3ccCHp). For the phenanthridine derivative, 4, only one molecule type (a) is present. Two types of dimers can be evidenced: 4aaππ formed by π⋯π interactions between the C and E rings, and 4aaCHp characterized by C–H⋯π interactions between the methyl group of the aniline moiety and ring A (Fig. 3d). Finally, as can be seen in Fig. 3e, the molecular packing for 4(H) suggests only π⋯π interactions between the phenanthridine derivatives 4(H), leading to the 4(H)ππ dimer.
3.3 Intermolecular interaction energy decomposition
The above analysis of packing modes reveals quite different structural motifs which require further characterization. To this end, we performed quantum-chemical calculations (see Methods section) to shed light onto the strength of these interactions and unravel their physical origin. The calculated intermolecular interaction energies together with their decomposition according to the VP-EDS scheme for representative dimers (see Fig. 3 and Table S10 in the ESI†) are given in Table 2 and in Fig. 4. In that Table, we provide the interaction energies obtained with the ωB97X-D functional, MP2, and SCS-MP2. The dimer complexes with π⋯π stacking, especially in compounds 3, 4, and 4(H), have total interaction energies ΔEMP2int larger (by up to 3.1 kcal mol−1) than the corresponding ΔEwB97X-Dint energies. It is indeed the usual trend,113 that MP2 overestimates the π⋯π stacking intermolecular interaction energies. This overestimation, due to the uncoupled dispersion energy term, tends to be systematic and does not hamper the analysis of the intermolecular interaction energies of the different stacking motifs. As discussed above, there are three main interaction types in the crystals: parallel displaced π⋯π stacking, edge-to-face T-shape together with weak π⋯π stacking (T-shape + π⋯π), and C–H⋯π intermolecular interactions. The strength of the interaction energies increases in the following order C–H⋯π < T-shape + π⋯π < parallel displaced π⋯π complexes (Fig. 4). Nevertheless, for dimer 4aaCHp the total interaction energy is comparable to those determined for T-shape + π⋯π stacked dimers. We also note that the strength of the π⋯π interactions is very similar for 4 and 4(H) which, as discussed in the next Section, have very different emission properties in the solid state. The calculations highlight the key role of the electrostatic and electron correlation terms (which include the uncoupled dispersion term) for the selected dimers; these two components determine the intermolecular interaction energies for the π-stacked complexes of 2–4 and 4(H). It can be seen that the exchange repulsion term, which is a destabilizing contribution, plays a significant role in the π-stacked dimers but has a trifling impact on the complexes based on C–H⋯π interactions.| Dimer | Total interaction energies | Decomposition of MP2 energies | ||||||
|---|---|---|---|---|---|---|---|---|
| ΔEωB97X-Dint | ΔEMP2int | ΔESCS-MP2int | ΔEHFint | e (10)el | ΔEHLex | ΔEHFdel | ΔEMP2corr | |
| a All values are given in kcal mol−1. All calculations were performed using the aug-cc-pVDZ basis set. The description of intermolecular interaction energies components is given in the Methods section. | ||||||||
| 1 | ||||||||
| 1ba ππ | −11.07 | −11.20 | −8.99 | −1.85 | −7.91 | 7.58 | −1.52 | −9.35 |
| 1ab Tπ–1 | −11.66 | −11.20 | −7.91 | 2.86 | −2.92 | 7.18 | −1.40 | −14.06 |
| 1ab Tπ–2 | −13.70 | −12.90 | −10.45 | −2.58 | −6.57 | 5.7 | −1.71 | −10.32 |
| 2 | ||||||||
| 2aa ππ-1 | −25.00 | −25.27 | −18.47 | 4.61 | −8.98 | 16.68 | −3.09 | −29.88 |
| 2aa ππ–2 | −26.26 | −28.23 | −20.96 | 3.81 | −11.34 | 18.52 | −3.37 | −32.04 |
| 3 | ||||||||
| 3aa ππ | −18.21 | −20.58 | −14.81 | 5.29 | −10.45 | 18.68 | −2.94 | −25.87 |
| 3bb ππ | −17.30 | −19.53 | −14.14 | 4.58 | −9.47 | 16.53 | −2.48 | −24.11 |
| 3cc ππ | −17.39 | −19.67 | −14.43 | 3.77 | −9.08 | 15.14 | −2.29 | −23.44 |
| 3bb Tπ | −13.34 | −12.57 | −10.61 | −4.26 | −6.04 | 3.15 | −1.37 | −8.31 |
| 3ba CHπ | −5.52 | −4.76 | −3.44 | 1.09 | −1.77 | 3.34 | −0.48 | −5.85 |
| 3cc CHπ | −7.36 | −6.65 | −4.95 | 0.92 | −2.66 | 4.27 | −0.69 | −7.57 |
| 3ca Tπ | −13.88 | −13.35 | −11.35 | −4.80 | −6.58 | 3.32 | −1.54 | −8.55 |
| 4 | ||||||||
| 4aa ππ | −22.87 | −25.96 | −19.04 | 4.76 | −10.68 | 18.56 | −3.12 | −30.72 |
| 4aa CHπ | −12.10 | −11.32 | −8.98 | −0.94 | −4.94 | 5.04 | −1.04 | −10.38 |
| 4(H) | ||||||||
| 4(H) ππ | −25.74 | −28.34 | −20.55 | 6.26 | −10.03 | 19.45 | −3.16 | −34.60 |
 | ||
| Fig. 4 Decomposition of RI-MP2/aug-cc-pVDZ intermolecular interaction energy into electrostatic, exchange, delocalization, and electron correlation contributions for studied dimers of 1–4 and compound 4(H). The solid line corresponds to the sum of all components. | ||
4 Discussion
In the Introduction, we argued that the π⋯π interactions play an important role not only in determining the crystal packing but also in tuning the solid state emission. However, the C–H⋯π interaction or the stronger intermolecular hydrogen bonds may also have a considerable effect, as clearly shown by the data presented above. In what follows we relate literature conclusions to our findings of the Φf of the crystals.4.1 Literature survey
The studies of the correlation between molecular packing and photophysical properties for N-methylpyrazoline derivatives demonstrated the impact of the intermolecular interactions present in the crystal lattice.114 The comparison of relative contributions of different close contacts with their fluorescence characteristics revealed that higher relative contributions of H⋯O and H⋯N contacts yield enhanced non-radiative decay, and consequently fluorescence quenching. Similarly, the links between molecular packing and substitution effects were recently established for chalcone crystals with a focus on the balance between red emissive and photodimerization-triggered hopping behaviour.115 By changing the nature of the amino group two different crystal arrangements with anti-parallel packing have been obtained: (i) NMe2-bearing chalcone derivatives with face-to-face alignment of the D and A groups, as well as multiple C–H⋯π interactions; (ii) NPh2-substituted chalcone compounds without D–A stacking. Whilst the two compounds develop similar photophysical properties in solution, only the latter molecule is bright in the crystal. Iwasaki and co-workers reported that different alkyl chains can affect the solid-state photophysical properties of 1,3,6,8-tetraalkylpyrenes.116 Their investigation indicates that alkyl groups modify the crystal packing, which significantly affects both the emission colour and the quantum yield.As illustrated by the above examples, a dense network of weak intermolecular interactions can quench the fluorescence; however, there are many examples in which it is the interplay between strong π⋯π and other types of weaker interactions that dictates the final response. Illustratively, the influence of π⋯π interactions on the solid-state emission properties of triphenylamino benzothiazole-based fluorogens undergoing (or not) excited-state intramolecular proton transfer (ESIPT) have been studied by Padalkar et al.117 The face-to-face arrangements of dimers in the crystal structure are responsible for the presence of strong π⋯π stacking which, along with other intermolecular interactions (C–H⋯π, C–H⋯O, C–H⋯N), hinder both the ESIPT and the fluorescence processes. 7-Amino and 4-methyljulolydyl coumarin crystals have a network of hydrogen bonds and/or π⋯π stacking interactions, leading to very weak solid-state fluorescence.118 In contrast, 7-diethylamino coumarin dyes that form isolated monomer- and dimer-type stacking show intense fluorescence. Eventually, an investigation of the molecular packing of isomeric benzofuranonaphthoquinol-type fluorophores made it possible to rationalize the fluorescence quenching of phenyl substituted compounds.119 Indeed, the crystal data confirmed the presence of hydrogen bonds and strong π⋯π interactions explaining the low emission of these molecules in the solid state. Interestingly, enhanced Φf could be achieved by changing the hydroxy (–OH) to the butoxy (–OBu) group which leads to packing with reduced interaction strengths.
The above-mentioned studies show that packing and photophysical properties of crystals are significantly influenced by various types of intermolecular interactions originating from the molecular structure. The increase of Φf in the solid state can be achieved (i) by suppressing hydrogen bonding (H⋯O and H⋯N contacts) and π⋯π interactions using the –OBu, methoxy (–OMe), or acetyl (–COOMe) group as a substituent rather than the –OH group; (ii) by reducing the network of hydrogen bonds and C–H⋯π interactions as well as π⋯π stacking using the NPh2 or diethylamino (NEt2) group instead of the NMe2 or amino (NH2) group. In short, the weaker the intermolecular interactions the higher the fluorescence in the solid state.
4.2 Analysis of compounds 1–4
The data obtained in the present study partially parallel the findings summarized above. We recall that the analysis of the molecular packing revealed the following similarities within the crystal structures presented in this work: the packing of 1–4 is characterized by π⋯π and CH⋯π interactions, while only π⋯π stacking is found in the 4(H) crystal. The intermolecular interaction energy calculations of the corresponding dimers (Table 2) indicated that the 1baππ dimer exhibits much weaker π⋯π interactions than any of the dimers of 2, 3, 4, and 4(H) in π⋯π stacked alignment. On the other hand, the T-shape π⋯π stacked complexes of 1, 3, and 4aaCHp exhibit similar intermolecular interaction energies to that found in the 1baππ dimer. This energetic analysis can be linked with the finding that the crystal of 1, formed by C–H⋯π and weak π⋯π interactions, possesses moderate Φf. The molecular packing of 2, 3 and 4, exhibiting strong π⋯π stacking and several CH⋯π interactions, leads to quenching of the fluorescence in the solid state. However, the strong π⋯π interactions present in the crystal of 4(H) do not have a detrimental effect on fluorescence quantum yield and green emission is observed for this crystal.56 In the case of 2, 3, and 4 one finds multiple CH⋯π interactions involving fused benzene rings and the hydrogen atoms of the dimethylamino group. It follows from our comparative analysis of (1vs.2–4 and 4vs.4(H)) that these additional interaction modes are detrimental as far as the emission of the crystals is concerned. Based on our findings we can propose that the strategy to tune photophysical properties of topologically similar dyes that aims at retaining high Φf values in the solid-state should consider (i) changing the dimethylamino group to diethylamino, which can reduce the dense network of CH⋯π interactions; and (ii) using bulky piperidine or dibutylamino moieties instead of the NMe2 group that can increase the spacing between aromatic rings, therefore decrease the energy of π⋯π interactions.5 Conclusions
In this work we have studied the emission properties of a series of four fluoroborate organic dyes. These dipolar compounds contain an electron-donating N(CH3)2 group and various NBF2O-containing heterocyclic moieties acting as electron-accepting units. The members of the series differ in the number and positions of the fused benzene rings in the heterocyclic part. These compounds exhibit strong emission in solution, with fluorescence quantum yields exceeding 0.8 for each member of the series in at least one of the solvents used, regardless of the benzannulation type. The theoretical calculations performed on the isolated structures highlight the dependence between the S1 state energy and the Φf. The fluorescence efficiency is quenched with decrease of the S1-S0 gap, which logically increases the rate constants for internal conversion. This analysis is confirmed by the experimental data: a redshift of the fluorescence maximum caused by an increase of the solvent polarity induces a drastic decrease of the Φf for all fluorophores. The increase of the dipolar character, achieved by substitution by the N(CH3)2 group and benzannulation, enhances the photoinduced charge transfer nature and is an effective strategy to tune the photophysical properties of these dyes in solution, i.e., it allows a redshift of the absorption spectra without deterioration of the emission properties. Moreover, the present work clearly demonstrates that this is not an effective strategy in the solid state, or more precisely, that more refined strategies need to be used for maximizing the emission brightness of the crystals. Indeed, only one crystal of the studied series exhibits a significant emission. The various benzannulation schemes yield substantial changes in the crystal packing and new interaction types between the molecules in the unit cells. More specifically, on passing from the parent compound to its benzannulated derivatives one finds stronger π⋯π stacking supplemented by multiple additional CH⋯π interactions involving the fused benzene rings and the hydrogen atoms of the dimethylamino group. It follows from our comparative analysis that these new interactions are detrimental as far as emission from the crystals is concerned. The strategy for tuning the photophysical properties of similar dyes, aiming at retaining high Φf values in the solid state, should avoid combinations of moieties with multiple fused rings as well as groups facilitating CH⋯π interactions.Author contributions
Conceptualization (B. O., R. Z.). Investigation (I. K., A. K., A. W., D. P., G. B., R. V., R. N., B. O., R. Z.). Methodology (I. K., D. J., B. O., R. Z.). Project administration (B. O., R. Z.). Software (H. A., G. B., R. V.). Validation (I. K., G. B., R. V., R. N., R. Z.). Visualization (I. K., A. K., A. W., G. B., R. V., R. Z.). Writing-original draft (I. K., A. W., D. J., B. O., R. Z.). Writing-review and editing (all authors).Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. O. and R. Z. acknowledge financial support from the Polish National Science Centre (Grant No. 2017/26/M/ST5/00327). A. K. and A. W. acknowledge the partial support of the Center of Excellence “Towards Personalized Medicine” operating under Excellence Initiative-Research University (NCU). G. B. thanks the Swedish Research Council (starting grant No. 2020-04600) and the Ministry of Education and Science of Ukraine (project no. 0121U107533) for support. This work has also been supported by the Academy of Finland through projects 1325369, 1315600 (R. V.). R. V. thanks the Decree of the Government of the Russian Federation N. 220 of 09 April 2010 (Agreement No. 075-15-2021-615 of 4 June 2021). The computations were enabled by resources provided by the Swedish National Infrastructure for Computing (SNIC) at the High Performance Computing Center North (HPC2N) partially funded by the Swedish Research Council through grant agreement no. 2020/3-29. Calculations were also performed at the Wroclaw Center for Networking and Supercomputing. The Wroclaw and Nantes team are indebted to the CNRS for supporting their collaboration in the framework of the ABSOLUTA-IEA grant. D. J. thanks the CCIPL computational center installed in Nantes for the generous allocation of computational time. The authors thank Lizaveta Petrusevich for the preparation of the graphical TOC entry.References
- J. Liu, H. Zhang, H. Dong, L. Meng, L. Jiang, L. Jiang, Y. Wang, J. Yu, Y. Sun, W. Hu and A. J. Heeger, Nat. Commun., 2015, 6, 10032 CrossRef CAS PubMed.
- X. Yang, G. Zhou and W.-Y. Wong, Chem. Soc. Rev., 2015, 44, 8484–8575 RSC.
- A. Zampetti, A. Minotto, B. M. Squeo, V. G. Gregoriou, S. Allard, U. Scherf, C. L. Chochos and F. Cacialli, Sci. Rep., 2017, 7, 1611 CrossRef PubMed.
- F. Cicoira and C. Santato, Adv. Funct. Mater., 2007, 17, 3421–3434 CrossRef CAS.
- C. Wang, X. Ren, C. Xu, B. Fu, R. Wang, X. Zhang, R. Li, H. Li, H. Dong, Y. Zhen, S. Lei, L. Jiang and W. Hu, Adv. Mater., 2018, 30, 1706260 CrossRef PubMed.
- L. Basabe-Desmonts, D. N. Reinhoudt and M. Crego-Calama, Chem. Soc. Rev., 2007, 36, 993–1017 RSC.
- G.-L. Fu, H. Pan, Y.-H. Zhao and C.-H. Zhao, Org. Biomol. Chem., 2011, 9, 8141–8146 RSC.
- Y. Jiang, Y.-Y. Liu, X. Liu, H. Lin, K. Gao, W.-Y. Lai and W. Huang, Chem. Soc. Rev., 2020, 49, 5885–5944 RSC.
- R. Aoki, R. Komatsu, K. Goushi, M. Mamada, S. Y. Ko, J. W. Wu, V. Placide, A. D'Aléo and C. Adachi, Adv. Opt. Mater., 2021, 9, 2001947 CrossRef CAS.
- J. Li, X. Peng, C. Huang, Q. Qi, W.-Y. Lai and W. Huang, Polym. Chem., 2018, 9, 5278–5285 RSC.
- J. Li, C. Hou, C. Huang, S. Xu, X. Peng, Q. Qi, W.-Y. Lai and W. Huang, Research, 2020, 3839160 CAS.
- Y. Xu, P. Xu, D. Hu and Y. Ma, Chem. Soc. Rev., 2021, 50, 1030–1069 RSC.
- C.-F. Liu, X. Liu, W.-Y. Lai and W. Huang, Chem. Rec., 2019, 19, 1571–1595 CrossRef CAS PubMed.
- X. Liu, M. Sang, H. Lin, C. Liu, J. Zhang, J. Yi, K. Gao, W.-Y. Lai and W. Huang, Chem. – Eur. J., 2020, 26, 3103–3112 CrossRef CAS PubMed.
- T. P. I. Saragi, T. Spehr, A. Siebert, T. Fuhrmann-Lieker and J. Salbeck, Chem. Rev., 2007, 107, 1011–1065 CrossRef CAS PubMed.
- S. P. Anthony, ChemPlusChem, 2012, 77, 518–531 CrossRef CAS.
- S. Xu, R. E. Evans, T. Liu, G. Zhang, J. N. Demas, C. O. Trindle and C. L. Fraser, Inorg. Chem., 2013, 52, 3597–3610 CrossRef CAS PubMed.
- A. M. Grabarz, A. D. Laurent, B. Jedrzejewska, A. Zakrzewska, D. Jacquemin and B. Osmiałowski, J. Org. Chem., 2016, 81, 2280–2292 CrossRef CAS PubMed.
- B. Jedrzejewska, A. Skotnicka, A. D. Laurent, M. Pietrzak, D. Jacquemin and B. Osmiałowski, J. Org. Chem., 2018, 83, 7779–7788 CrossRef CAS PubMed.
- Y.-Y. Wu, Y. Chen, G.-Z. Gou, W.-H. Mu, X.-J. Lv, M.-L. Du and W.-F. Fu, Org. Lett., 2012, 14, 5226–5229 CrossRef CAS PubMed.
- J. Chen, W. Liu, J. Ma, H. Xu, J. Wu, X. Tang, Z. Fan and P. Wang, J. Org. Chem., 2012, 77, 3475–3482 CrossRef CAS PubMed.
- E. Kim, A. Felouat, E. Zaborova, J.-C. Ribierre, J. W. Wu, S. Senatore, C. Matthews, P.-F. Lenne, C. Baffert, A. Karapetyan, M. Giorgi, D. Jacquemin, M. Ponce-Vargas, B. L. Guennic, F. Fages and A. D'Aléo, Org. Biomol. Chem., 2016, 14, 1311–1324 RSC.
- S. Redon, G. Eucat, M. Ipuy, E. Jeanneau, I. Gautier-Luneau, A. Ibanez, C. Andraud and Y. Bretonnière, Dyes Pigm., 2018, 156, 116–132 CrossRef CAS.
- Y. Avlasevich, C. Li and K. Müllen, J. Mater. Chem., 2010, 20, 3814–3826 RSC.
- K. Hanson, L. Roskop, P. I. Djurovich, F. Zahariev, M. S. Gordon and M. E. Thompson, J. Am. Chem. Soc., 2010, 132, 16247–16255 CrossRef CAS PubMed.
- K. Kurotobi, K. S. Kim, S. B. Noh, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2006, 45, 3944–3947 CrossRef CAS PubMed.
- M. J. Kuisma, A. M. Lundin, K. Moth-Poulsen, P. Hyldgaard and P. Erhart, J. Phys. Chem. C, 2016, 120, 3635–3645 CrossRef CAS PubMed.
- R. Gresser, M. Hummert, H. Hartmann, K. Leo and M. Riede, Chem. – Eur. J., 2011, 17, 2939–2947 CrossRef CAS PubMed.
- S. Yamazawa, M. Nakashima, Y. Suda, R. Nishiyabu and Y. Kubo, J. Org. Chem., 2016, 81, 1310–1315 CrossRef CAS PubMed.
- E. Clar, Aromatic Sextet, Willey, London, 1972 Search PubMed.
- R. Ziessel, G. Ulrich and A. Harriman, New J. Chem., 2007, 31, 496–501 RSC.
- J. Bañuelos, Chem. Rec., 2016, 16, 335–348 CrossRef PubMed.
- J. B. Birks, Photophysics of Aromatic Molecules, Wiley, London, 1970 Search PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- S. Kim, J. Bouffard and Y. Kim, Chem. – Eur. J., 2015, 21, 17459–17465 CrossRef CAS PubMed.
- D. Kim, U. Lee, J. Bouffard and Y. Kim, Adv. Opt. Mater., 2020, 8, 1902161 CrossRef CAS.
- Z. Xie, B. Yang, F. Li, G. Cheng, L. Liu, G. Yang, H. Xu, L. Ye, M. Hanif, S. Liu, D. Ma and Y. Ma, J. Am. Chem. Soc., 2005, 127, 14152–14153 CrossRef CAS PubMed.
- D. Yan, A. Delori, G. O. Lloyd, T. Frišci, G. M. Day, W. Jones, J. Lu, M. Wei, D. G. Evans and X. Duan, Angew. Chem., Int. Ed., 2011, 50, 12483–12486 CrossRef CAS PubMed.
- L. Sun, W. Zhu, W. Wang, F. Yang, C. Zhang, S. Wang, X. Zhang, R. Li, H. Dong and W. Hu, Angew. Chem., Int. Ed., 2017, 56, 7831–7835 CrossRef CAS PubMed.
- T. Ozdemir, S. Atilgan, I. Kutuk, L. T. Yildirim, A. Tulek, M. Bayindir and E. U. Akkaya, Org. Lett., 2009, 11, 2105–2107 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- Y. Yang, X. Su, C. N. Carroll and I. Aprahamian, Chem. Sci., 2012, 3, 610–613 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- Y. Kubota, K. Kasatani, H. Takai, K. Funabiki and M. Matsui, Dalton Trans., 2015, 44, 3326–3341 RSC.
- H. Gao, D. Xu, X. Liu, A. Han, L. Zhou, C. Zhang, Y. Yang and W. Li, RSC Adv., 2017, 7, 1348–1356 RSC.
- S. Gong, Q. Liu, X. Wang, B. Xia, Z. Liu and W. He, Dalton Trans., 2015, 44, 14063–14070 RSC.
- F. Qi, J. Lin, X. Wang, P. Cui, H. Yan, S. Gong, C. Ma, Z. Liu and W. Huang, Dalton Trans., 2016, 45, 7278–7284 RSC.
- Y. Wu, Z. Li, Q. Liu, X. Wang, H. Yan, S. Gong, Z. Liu and W. He, Org. Biomol. Chem., 2015, 13, 5775–5782 RSC.
- Y. Shao, G.-Z. Yin, X. Ren, X. Zhang, J. Wang, K. Guo, X. Li, C. Wesdemiotis, W.-B. Zhang, S. Yang, M. Zhu and B. Sun, RSC Adv., 2017, 7, 6530–6537 RSC.
- B. Ventura, Y. M. Poronik, I. Deperasińska and D. T. Gryko, Chem. – Eur. J., 2016, 22, 15380–15388 CrossRef CAS PubMed.
- A. D'Aléo, D. Gachet, V. Heresanu, M. Giorgi and F. Fages, Chem. – Eur. J., 2012, 18, 12764–12772 CrossRef PubMed.
- S. Li, L. Fu, X. Xiao, H. Geng, Q. Liao, Y. Liao and H. Fu, Angew. Chem., Int. Ed., 2021, 60, 18059–18064 CrossRef CAS PubMed.
- R. Yoshii, A. Hirose, K. Tanaka and Y. Chujo, J. Am. Chem. Soc., 2014, 136, 18131–18139 CrossRef CAS PubMed.
- X. Wang, Y. Wu, Q. Liu, Z. Li, H. Yan, C. Ji, J. Duan and Z. Liu, Chem. Commun., 2015, 51, 784–787 RSC.
- L. Zhou, D. Xu, H. Gao, C. Zhang, F. Ni, W. Zhao, D. Cheng, X. Liu and A. Han, J. Org. Chem., 2016, 81, 7439–7447 CrossRef CAS PubMed.
- D.-E. Wu, X.-L. Lu and M. Xia, New J. Chem., 2015, 39, 6465–6473 RSC.
- H. Gao, D. Xu, Y. Wang, C. Zhang, Y. Yang, X. Liu, A. Han and Y. Wang, Dyes Pigm., 2018, 150, 165–173 CrossRef CAS.
- T. Butler, W. A. Morris, J. Samonina-Kosicka and C. L. Fraser, ACS Appl. Mater. Interfaces, 2016, 8, 1242–1251 CrossRef CAS PubMed.
- W. A. Morris, T. Butler, M. Kolpaczynska and C. L. Fraser, Mater. Chem. Front., 2017, 1, 158–166 RSC.
- M. A. Potopnyk, R. Lytvyn, Y. Danyliv, M. Ceborska, O. Bezvikonnyi, D. Volyniuk and J. V. Gražulevicius, J. Org. Chem., 2018, 83, 1095–1105 CrossRef CAS PubMed.
- K. Benelhadj, J. Massue, P. Retailleau, G. Ulrich and R. Ziessel, Org. Lett., 2013, 15, 2918–2921 CrossRef CAS PubMed.
- A. Zakrzewska, R. Zalesny, E. Kolehmainen, B. Omiałowski, B. Jedrzejewska, H. Ågren and M. Pietrzak, Dyes Pigm., 2013, 99, 957–965 CrossRef CAS.
- B. Osmiałowski, A. Zakrzewska, B. Jedrzejewska, A. Grabarz, R. Zalesny, W. Bartkowiak and E. Kolehmainen, J. Org. Chem., 2015, 80, 2072–2080 CrossRef PubMed.
- A. M. Grabarz, B. Jedrzejewska, A. Zakrzewska, R. Zalesny, A. D. Laurent, D. Jacquemin and B. Osmiałowski, J. Org. Chem., 2017, 82, 1529–1537 CrossRef CAS PubMed.
- K. Morokuma, J. Chem. Phys., 1971, 55, 1236–1244 CrossRef CAS.
- T. Ziegler and A. Rauk, Theor. Chim. Acta, 1977, 46, 1–10 CrossRef CAS.
- K. Kitaura and K. Morokuma, Int. J. Quantum Chem., 1976, 10, 325–340 CrossRef CAS.
- P. S. Bagus, K. Hermann and C. W. Bauschlicher, J. Chem. Phys., 1984, 80, 4378 CrossRef CAS.
- W. J. Stevens and W. H. Fink, Chem. Phys. Lett., 1987, 139, 15–22 CrossRef CAS.
- E. D. Glendening and A. Streitwieser, J. Chem. Phys., 1994, 100, 2900 CrossRef CAS.
- Y. Mo, J. Gao and S. D. Peyerimhoff, J. Chem. Phys., 2000, 112, 5530 CrossRef CAS.
- P. Su and H. Li, J. Chem. Phys., 2009, 131, 014102 CrossRef PubMed.
- M. Gutowski, F. V. Duijneveldt, G. Chałasiński and L. Piela, Mol. Phys., 1987, 61, 233–247 CrossRef CAS.
- W. A. Sokalski, S. Roszak and K. Pecul, Chem. Phys. Lett., 1988, 153, 153–159 CrossRef CAS.
- S. M. Cybulski, G. Chałasiński and R. Moszyński, J. Chem. Phys., 1990, 92, 4357–4363 CrossRef CAS.
- G. Chałasiński and M. M. Szcześniak, Chem. Rev., 1994, 94, 1723–1765 CrossRef.
- G. Chałasiński and M. M. Szcześniak, Mol. Phys., 1988, 63, 205–224 CrossRef.
- F. B. van Duijneveldt, J. G. C. M. van Duijneveldt-van de Rijdt and J. H. van Lenthe, Chem. Rev., 1994, 94, 1873–1885 CrossRef CAS.
- R. Moszyński, S. Rybak, S. Cybulski and G. Chałasiński, Chem. Phys. Lett., 1990, 166, 609–614 CrossRef.
- S. Grimme, J. Chem. Phys., 2003, 118, 9095–9102 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian Revision C.01, Gaussian Inc., Wallingford CT, 2019 Search PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1994, 100, 2975–2988 CrossRef CAS.
- M. Feyereisen, G. Fitzgerald and A. Komornicki, Chem. Phys. Lett., 1993, 208, 359–363 CrossRef CAS.
- F. Weigend, M. Häser, H. Patzelt and R. Ahlrichs, Chem. Phys. Lett., 1998, 294, 143–152 CrossRef CAS.
- F. Weigend, A. Köhn and C. Hättig, J. Chem. Phys., 2002, 116, 3175–3183 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2002, 4, 4285–4291 RSC.
- H.-J. Werner, F. R. Manby and P. J. Knowles, J. Chem. Phys., 2003, 118, 8149–8160 CrossRef CAS.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery Jr, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- R. W. Gora, EDS package, Revision 2.8.3, Wrocław, Poland, 1998–2008 Search PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, W. Györffy, D. Kats, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, S. J. Bennie, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, S. J. R. Lee, Y. Liu, A. W. Lloyd, Q. Ma, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, T. F. Miller III, M. E. Mura, A. Nicklass, D. P. O'Neill, P. Palmieri, D. Peng, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson, M. Wang and M. Welborn, MOLPRO, version 2012.1, a Package of ab initio Programs, 2012, http://www.molpro.net.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Comput. Chem., 2001, 22, 976–984 CrossRef CAS.
- A. A. Granovsky, J. Chem. Phys., 2011, 134, 214113 CrossRef PubMed.
- D. P. Chong, Recent Advances in Density Functional Methods, World Scientific, 1995 Search PubMed.
- A. A. Granovsky, Firefly ver. 8, http://classic.chem.msu.su/gran/firefly/index.html.
- TURBOMOLE V7.4 2019, 2019, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com.
- X. Gao, S. Bai, D. Fazzi, T. Niehaus, M. Barbatti and W. Thiel, J. Chem. Theory Comput., 2017, 13, 515–524 CrossRef CAS PubMed.
- R. R. Valiev, V. N. Cherepanov, G. V. Baryshnikov and D. Sundholm, Phys. Chem. Chem. Phys., 2018, 20, 6121–6133 RSC.
- R. R. Valiev, V. N. Cherepanov, R. T. Nasibullin, D. Sundholm and T. Kurten, Phys. Chem. Chem. Phys., 2019, 21, 18495–18500 RSC.
- R. R. Valiev, R. T. Nasibullin, V. N. Cherepanov, G. V. Baryshnikov, D. Sundholm, H. Ågren, B. F. Minaev and T. Kurtén, Phys. Chem. Chem. Phys., 2020, 22, 22314–22323 RSC.
- R. R. Valiev, R. T. Nasibullin, V. N. Cherepanov, A. Kurtsevich, D. Sundholm and T. Kurtén, Phys. Chem. Chem. Phys., 2021, 23, 6344–6348 RSC.
- J. Bednarska, R. Zaleny, M. Wielgus, B. Jedrzejewska, R. Puttreddy, K. Rissanen, W. Bartkowiak, H. Ågren and B. Ośmiałowski, Phys. Chem. Chem. Phys., 2017, 19, 5705–5708 RSC.
- A. Zakrzewska, E. Kolehmainen, A. Valkonen, E. Haapaniemi, K. Rissanen, L. Checińska and B. Omiałowski, J. Phys. Chem. A, 2013, 117, 252–256 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2015, C71, 3–8 CrossRef PubMed.
- CrysAlisPro 171.38.43 Package of Programs. Rigaku Oxford Diffraction, 2015.
- B. Jedrzejewska, A. Grabarz, W. Bartkowiak and B. Omiałowski, Spectrochim. Acta, Part A, 2018, 199, 86–95 CrossRef CAS PubMed.
- V. G. Plotnikov, Int. J. Quantum Chem., 1979, 16, 527–541 CrossRef CAS.
- Y. M. Poronik, G. V. Baryshnikov, I. Deperasińska, E. M. Espinoza, J. A. Clark, H. Ågren, D. T. Gryko and V. I. Vullev, Chem. Commun., 2020, 3, 190 CrossRef CAS.
- S. M. Cybulski and M. L. Lytle, J. Chem. Phys., 2007, 127, 141102 CrossRef PubMed.
- B. Kupcewicz and M. Małecka, Cryst. Growth Des., 2015, 15, 3893–3904 CrossRef CAS.
- X. He, J. Zhao, Z. Tan, J. Zhao, X. Cheng and C. Zhou, CrystEngComm, 2020, 22, 3110–3114 RSC.
- T. Iwasaki, S. Murakami, Y. Takeda, G. Fukuhara, N. Tohnai, Y. Yakiyama, H. Sakurai and N. Kambe, Chem. – Eur. J., 2019, 25, 14817–14825 CrossRef CAS PubMed.
- V. S. Padalkar, D. Sakamaki, K. Kuwada, A. Horio, H. Okamoto, N. Tohnai, T. Akutagawa, K.-I. Sakai and S. Seki, Asian J. Org. Chem., 2016, 5, 938–945 CrossRef CAS.
- S.-Y. Park, M. Ebihara, Y. Kubota, K. Funabiki and M. Matsui, Dyes Pigm., 2009, 82, 258–267 CrossRef CAS.
- K. Yoshida, Y. Ooyama, H. Miyazaki and S. Watanabe, J. Chem. Soc., Perkin Trans. 2, 2002, 700–707 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Data from the X-ray and spectroscopic measurements, and results from electronic-structure calculations. CCDC 2090001–2090003. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1tc03316f |
| This journal is © The Royal Society of Chemistry 2021 |